

Abstract
Introduction
It is unknown whether vascular and metabolic diseases assessed in early adulthood are associated with Alzheimer's disease (AD) later in life.
Methods
Association of AD with lipid fractions, glucose, blood pressure, body mass index (BMI), and smoking obtained prospectively from 4932 Framingham Heart Study (FHS) participants across nine quadrennial examinations was evaluated using Cox proportional hazard and Kaplan‐Meier models. Age‐, sex‐, and education‐adjusted models were tested for each factor measured at each exam and within three adult age groups (early = 35‐50, middle = 51‐60, and late = 61‐70).
Results
A 15 mg/dL increase in high density lipoprotein (HDL) cholesterol was associated with decreased AD risk during early (15.4%, P = 0.041) and middle (17.9%, P = 0.014) adulthood. A 15 mg/dL increase in glucose measured during middle adulthood was associated with 14.5% increased AD risk (P = 0.00029). These findings remained significant after adjusting for treatment.
Discussion
Our findings suggest that careful management of cholesterol and glucose beginning in early adulthood can lower AD risk.
Keywords: Alzheimer's disease, cardiovascular risk factors, glucose, high‐density lipoprotein cholesterol, lipids, Midlife
1. INTRODUCTION
Alzheimer's disease (AD) is the fifth leading cause of death among Americans 65 years of age or older, with a prevalence of 5.8 million cases. 1 This number is projected to nearly triple to 14 million people by 2060. 1 , 2 To date, there are no proven effective disease‐modifying therapies to prevent or slow cognitive decline from AD and related diseases. Early identification and treatment of individuals at risk for the common form of AD occurring after age 65 have been recognized as an important contributor to reductions in AD mortality and delaying the symptoms of the disease. 3
Individuals with AD commonly exhibit features of cerebrovascular disease in combination with amyloid beta (Aβ) and tau neuropathology. 4 , 5 , 6 Genetic studies of AD have identified common and rare variants associated with AD in genes involved in lipoprotein metabolism and processing related to AD. 7 , 8 Historically, cerebrovascular diseases including stroke were exclusionary factors for an AD diagnosis because of their role in vascular dementia. 9 However, pathological and epidemiological studies suggest that potentially modifiable cerebrovascular risk factors including obesity, diabetes, and high cholesterol contribute to the development of late‐life cognitive impairment and AD, 10 , 11 , 12 , 13 , 14 , 15 but not all studies agree. 16 , 17 Although vascular risk burden measured in mid‐ to late‐life (ie, ≥55 years if age) is a predictor of dementia including AD, 18 , 19 less is known about whether AD risk is associated with exposure to vascular factors in early adulthood. Addressing this question has added importance in light of the growing recognition that AD is a life‐course disease. 3 In this study, we investigated the influence of vascular risk factors measured longitudinally for an average period of more than 30 years on incident AD in the Framingham Heart Study (FHS) Offspring Cohort participants. 20 This allowed us to evaluate the effect of vascular risk exposure on incident AD based on single time‐point measurements from early, middle, and late adulthood with a goal of exploring the appropriate timing of vascular screening and interventions necessary to maximize benefits to cognitive and brain health.
2. METHODS
2.1. Study design and participants
The present study was designed to assess the association of modifiable vascular risk factors measured at time points throughout adulthood with future risk of AD among participants of the FHS, a single‐site, multigeneration, community‐based, prospective cohort study of health in Framingham, Massachusetts (Figure 1). This design addresses the question of future AD risk assessed at multiple starting points and assuming a constant exposure from that point forward. This study focused on the FHS Offspring cohort (Generation 2) participants for whom multiple longitudinal measures of vascular risk factors were available and who have been rigorously evaluated for cognitive decline and dementia since 1979. Ascertainments and other details of this cohort have been described previously. 21 In brief, at baseline, the cohort included 5124 participants who were 5‐ to 70‐years‐old (mean age of 36 years) at the first health examination (1971‐1975). These participants have been examined longitudinally every 4 years on average from 1971 until the present, with a total of 10 exams. There is minimal loss to follow‐up for FHS participants, some of whom have moved to other regions in New England and are examined in their home. All enrolled participants are tracked until death, at which time all medical and nursing home records are requested for a final review of cardiovascular and other related health events. Participant data included in this study were collected through the period ending in 2016. Physical and cognitive examinations, interviews to obtain information about general health and exposures, and collection of blood samples for blood chemistry and molecular analyses were administered at each exam cycle. Written informed consent was obtained from all study participants. The study protocol was approved by the Boston University Institutional Review Board, followed the Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) reporting guideline, and was monitored by a National Heart, Lung, and Blood Institute Observational Study Monitoring Board.
FIGURE 1.
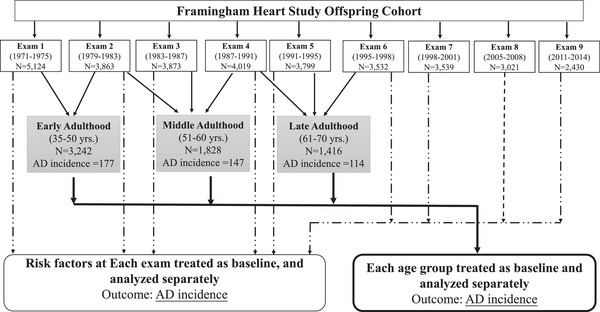
Study design
2.2. Risk factor assessment
A variety of vascular, metabolic, and physical exam measurements were obtained at each examination including blood lipid fractions (high‐density and low‐density lipoprotein cholesterol [HDL‐C and LDL‐C] total cholesterol [TC], and triglyceride [TG]), blood glucose (BG), body mass index (BMI), systolic and diastolic blood pressure (SBP and DBP), and number of cigarettes smoked per day. Protocols for variable measurement and data processing including quality control procedures have been described previously. 22 Of note, plasma LDL‐C concentration at each exam was calculated using the Friedewald formula: LDL‐C = (TC – HDL‐C) – (TG / 5). 23 If TG values were >400 mg/dL, LDL‐C was set to missing. Lipids and glucose measured at exams 1 and 2 were non‐fasting (ie, before the time participants were requested to fast before blood samples were collected), and these measurements were obtained under fasting conditions for ≈98% of participants at subsequent exams. Age and treatment history (yes or no) for hypertension, hypercholesterolemia, and diabetes at the time closest to, but not after, risk factor assessment was included in the statistical analyses. This study focused on quantitative measures rather than vascular‐related disease end points in order to capture the potential effect of modifiable processes underlying these disorders on the development of AD later in life. Apolipoprotein E (APOE) genotype information was unavailable for 941 FHS Offspring participants (18.4%), most of whom were evaluated at exams 1 and 2 only before biospecimens were collected for genetic analysis.
1. RESEARCH IN CONTEXT
Systematic Review: The authors are familiar with the literature related to methods for evaluating cognitive impairment and dementia. PubMed searches were conducted to identify other publications relevant to the evaluation of vascular risk factors for Alzheimer's disease (AD) in prospectively followed cohorts. References that support the significance of the identified associations of risk factors with AD are cited.
Interpretation: Although it is well established that risk factors for cardiovascular disease and diabetes increase the risk of AD, this report demonstrates for the first time that low high‐density lipoprotein (HDL) and elevated glucose levels measured as early as age 35 years are associated with AD later in life. These findings suggest that careful management of cholesterol and glucose levels beginning in early adulthood can lower AD risk.
Future directions: A better understanding of the molecular mechanisms underlying these associations will require experimental studies. These vascular and metabolic factors may not be AD specific and may be of similar size or even larger for other forms of dementia. Studies of other cohorts containing much larger samples of non‐AD dementias will be necessary to address this question. Further studies are also needed to enhance these findings and generalize them to non‐European ancestry populations.
2.3. Cognitive assessment and dementia diagnosis
Surveillance of cognitive impairment and incident dementia in the Offspring cohort began in 1979 at the second health examination when the cohort was still relatively young (mean age = 44 years) to establish a dementia‐free cohort. The Mini–Mental State Examination (MMSE) was administered starting at the fifth health examination (ie, 1991‐1995) to monitor change in cognitive status. Beginning in 1999, all surviving Generation 2 participants were invited for an in‐depth cognitive examination. Change in cognitive status or performance, report by the participant or a family member of subjective cognitive complaints, referral by a physician or by ancillary investigators of the FHS, or outside medical records indicative of cognitive complaints triggered referral to a dementia diagnostic panel consisting of at least one neurologist and one neuropsychologist. Surveillance details and consensus diagnostic procedures have been described previously. 24 , 25 Among 989 participants who were underwent a dementia diagnosis panel review (Table S1 in supporting information), 271 participants (167 female, 104 male) diagnosed with AD dementia (AD without stroke: n = 225; AD with stroke: n = 22; mixed dementia AD + vascular dementia: n = 24) were included in the analysis as cases. Participants judged to be cognitively normal after dementia review (n = 366, a number consistent with the low threshold needed to be flagged for dementia review) or who were not flagged for dementia review (n = 4,501) were eligible as controls. Participants who were assigned a diagnosis of non‐AD dementia (n = 106), uncertain dementia (n = 13), or who had an unknown diagnosis (n = 45), were excluded from the analyses of AD. Age at AD onset was defined as the earliest recorded date of cognitive impairment. AD cases without a recorded age at onset or diagnosis date (n = 6) or with a diagnosis after 2016 (n = 4) were removed.
2.4. Statistical analysis
Individuals with prevalent dementia at the time of risk factor measurement were excluded from analyses. AD cases were followed until the date of AD diagnosis before December 31, 2016. Participants who were judged to be unimpaired by cognitive screening tests or to have mild cognitive impairment that did not progress to AD during the follow‐up period were censored at either the date of death or the end date of the most recent exam before December 31, 2016. The follow‐up time for censored participants was measured in days from the date of the exam at which a blood sample was obtained for risk factor measurement to the censoring date.
The association of vascular risk factors with incident AD was evaluated using two analytical designs. In one approach, Cox proportional hazards regression models were used to assess the association of each risk factor measurement separately with incident AD. For each risk factor, these analyses were carried out separately for the exam 1 to 9 value of the risk factors, spanning ≈40 years based on 4‐year exam cycles. The base model included one risk factor and covariates for age at exam, sex, education, and family ID as a random effect to adjust for relatedness. Additional models were tested for each risk factor including terms for condition‐specific treatment at the time of the risk factor measurement and APOE ε4 carrier status, noting that APOE genotype is a confounder because it is correlated with measures of lipid fractions. 26
Prior to analysis, triglyceride measurements were log2 transformed to remove skewness in the distribution. Cox analyses were performed after confirming that assumptions of proportional hazards were met.
Because the range in ages of participants at each exam is 50 years or more (Table 1, Figure S1) and to focus on discrete time periods in life, in a second approach we performed another set of analyses that evaluated the association of risk factors with AD within three age ranges: early adulthood (ages 35‐50), middle adulthood (ages 51‐60), and late adulthood (ages 61‐70), noting that these descriptors for the stages of adulthood are used for convenience purposes only. To minimize the variation in follow‐up time within each age group, influences of secular trends and test measurement across different exams, and at the same time to maximize the number of participants and AD cases in particular in each group, we selected participants’ risk factor data that were available from the earliest of exams 1 to 2, 2 to 4, and 4 to 6 for the early adulthood, middle adulthood, and late adulthood groups, respectively (Table S2). Participants were included in each age group for which they were examined during the corresponding baseline period. The association of each risk factor with AD incidence was evaluated using Cox proportional‐hazards regression analyses and tested a similar set of models as described earlier. Analyses within age groups and for each exam were repeated for the outcome of all cause dementia, including 271 AD cases and 106 participants with a diagnosis of non‐AD dementia (Table S1).
TABLE 1.
Number of participants and incident AD cases at each exam in the FHS Offspring cohort
| Examination | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | Between 7 and 8 | 8 | Between 8 and 9 | 9 | 10 c | |
| Exam cycle | 1971–1975 | 1979–1983 | 1983–1987 | 1987–1991 | 1991–1995 | 1995–1998 | 1998–2001 | 2005–2008 | 2011–2014 |
2015– Present |
||
| Attendees a | 5124 | 3863 | 3873 | 4019 | 3799 | 3532 | 3539 | 3021 | 2430 | |||
| Age range | 5–70 | 17–77 | 18–77 | 22–81 | 26–84 | 29–86 | 33–90 | 40–93 | 46–98 | |||
| Mean age | 36 | 44 | 48 | 52 | 55 | 59 | 62 | 67 | 71 | |||
| No. incident AD cases b | 0 | 2 | 3 | 2 | 9 | 22 | 33 | 36 | 56 | 53 | 28 | 17 |
702 participants with missing education information are included in the table but were excluded from the analysis.
10 AD cases with missing age at onset (not included in the table) were excluded from the analysis.
Data for exam 10 were not included in the analysis because they are incomplete; AD cases shown were adjudicated prior to December 31, 2016.
Kaplan‐Meier survival curves were generated for each age group to compare the onset of AD symptoms among three groups defined by clinically meaningful cutoffs for risk factors showing significant associations with AD in the age group analyses. The groupings for the risk factors evaluated in this manner are as follows: HDL‐C (<45 mg/dL, 45‐64 mg/dL, ≥65 mg/dL), LDL‐C (<130 mg/dL, 130‐169 mg/dL, ≥170 mg/dL), TG (<150 mg/dL, 150‐199 mg/dL, ≥200 mg/dL), BG (<100 mg/dL, 100‐125 mg/dL, and ≥126 mg/dL), and DBP (<80 mm, 80‐89 mm, and ≥90 mm). 27 , 28 , 29 , 30 All statistical analyses were performed using R version 3.6.3.
3. RESULTS
3.1. Characteristics of subjects included in analyses
Among 5124 subjects in the FHS offspring cohort enrolled at the first examination, measurement data for at least one vascular risk factor at one or more of the nine examinations were available for 4932 subjects who were followed for a total of 186,537 person‐years (mean = 37.6 years). The numbers of participants and incident AD cases in each exam for each age group are shown in Table S2. The mean follow‐up period for participants in the early, middle, and late adulthood age groups was 35.2 years, 25.8 years, and 18.5 years, respectively (Table 2). As participants grew older, they tended to have higher triglyceride and glucose levels, higher systolic and diastolic blood pressure, and lower HDL‐C levels, as well were more likely to be treated for diabetes, hypertension, and dyslipidemia. The number of lifetime incident AD cases was similar among participants assessed in early adulthood (n = 177), middle adulthood (n = 172), and late adulthood (n = 192), which is expected because of the high overlap of subjects across age groups, despite the decreasing mean follow‐up time.
TABLE 2.
Characteristics of study participants at baseline
| Group (age range in years) a | |||
|---|---|---|---|
| Characteristics | Early adulthood (35–50) | Middle adulthood (51–60) | Lateadulthood (61–70) |
| Period of baseline risk factor measurement | 1971–1983 | 1979–1991 | 1987–1998 |
| Total, No. | 3224 | 1943 | 1577 |
| Age, mean (SD), y | 41.0 (4.3) | 54.0 (2.5) | 63.5 (2.3) |
| Female, No. (%) | 1,683 (52.2) | 995 (51.2) | 812 (51.5) |
| Education, No. (%) | |||
| Missing/unknown | 292 (9.0) | 79 (4.1) | 76 (18.9) |
| High school did not graduate | 477 (14.8) | 405 (20.8) | 298 (29.7) |
| High school graduate | 834 (25.9) | 545 (28.0) | 469 (24.1) |
| Some college | 818 (25.4) | 491 (25.3) | 380 (22.4) |
| College graduate | 803 (24.9) | 423 (21.8) | 354 (4.8) |
| Follow‐up time, mean (SD), y | 35.2 (8.9) | 25.8 (8.2) | 18.5 (6.8) |
| AD incident, No. (%) | 177 (5.5) | 172 (8.8) | 192 (12.2) |
| Male | 64 (4.2) | 60 (6.3) | 68 (8.9) |
| Female | 113 (6.7) | 112 (11.3) | 124 (15.3) |
| APOE genotype (N) | 2,761 | 1,799 | 1,518 |
| Cerebrovascular risk factors | |||
| LDL cholesterol, mean (SD), mg/dL | 133.3 (36.0) | 146.0 (36.5) | 134.0 (33.8) |
| HDL cholesterol, mean (SD), mg/dL | 51.1 (16.0) | 49.5 (15.3) | 49.4 (15.7) |
| Triglycerides, mean (SD), mg/dL | 109.0 (82.5) | 133.2 (107.1) | 153.0 (109.5) |
| Total cholesterol, mean (SD), mg/dL | 205.6 (37.7) | 225.3 (37.8) | 213.2 (38.8) |
| Blood glucose, mean (SD), mg/dL | 101.3 (15.4) | 100.8 (26.5) | 105.3 (33.7) |
| Systolic blood pressure, mean (SD), mm Hg | 123.9 (16.0) | 129.1 (17.2) | 134.9 (19.2) |
| Diastolic blood pressure, mean (SD), mm Hg | 79.5 (10.5) | 81.1 (9.3) | 77.5 (9.7) |
| Treatment | |||
| Treated for diabetes, No. (%) | 25 (0.8) | 52 (2.7) | 98 (6.2) |
| Treated for hypertension, No. (%) | 153 (4.7) | 401 (20.6) | 538 (34.1) |
| Treated for dyslipidemia, No. (%) | 17 (0.5) | 42 (2.2) | 171 (10.8) |
Participants are included in each age group for which they were examined during the corresponding baseline period as defined in Table S1.
3.2. Association of AD with risk factors by exam
AD risk was inversely associated with HDL‐C level measured at the first (hazard ratio [HR] = 0.87 [0.76–1.00] P = 0.045), second (HR = 0.83 [0.72–0.97], P = 0.016), sixth (HR = 0.82 [0.72–0.93], P = 0.0021), and seventh (HR = 0.79 [0.68–0.92], P = 0.0024) examinations at which the mean age was 36, 44, 59, and 62, respectively (Table 3). The associations at these four exams remained significant after adjusting for dyslipidemia treatment (Table S3 in supporting information). Similarly, AD risk was associated with triglyceride levels measured at the first (HR = 1.34 [1.12–1.59] P = .0010), second (HR = 1.27 [1.10–1.47], P = 0.0014), fifth (HR = 1.19 [1.23–1.39], P = 0.024), sixth (HR = 1.20 [1.03–1.40], P = 0.022) and seventh (HR = 1.33 [1.10–1.59], P = 0.0027) examinations and these findings were not meaningfully altered after adjusting for dyslipidemia treatment. BG was significantly associated with AD incidence at every exam it was measured (Exam1: HR = 1.12 [1.01–1.26], P = 0.041; Exam2: HR = 1.12 [1.04–1.20], P = 0.0020; Exam3: HR = 1.09 [1.01–1.16], P = 0.022; Exam4: HR = 1.11 [1.05–1.17], P = 0.00040; Exam5: HR = 1.07 [1.01–1.13], P = 0.020; Exam6: HR = 1.09 [1.03–1.15], P = 0.0041; Exam7: HR = 1.13 [1.05–1.12], P = 0.00060; Exam8: HR = 1.14 [1.04–1.26], P = 0.0058; Exam9: HR = 1.39 [1.06–1.82], P = 0.017) (Table 3). In general, these results were attenuated after adjustment for treatment for diabetes. AD was not associated with LDL‐C, BMI, smoking or blood pressure measured at any single examination (Table S3 in supporting information).
TABLE 3.
Association of plasma HDL‐C, triglyceride, LDL‐C, and glucose measured at each longitudinal exam with incident AD adjusted for age and sex
| HDL‐C | Triglyceride b | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Exam | No. events a | N | Beta | HR c | Lower 95% CL | Upper 95% CL | P‐Value | N | beta | HR c | Lower 95% CL | Upper 95% CL | P‐value |
| 1 | 236 | 4200 | −0.14 | 0.87 | 0.76 | 1.00 | 0.045 | 4213 | 0.29 | 1.34 | 1.12 | 1.59 | 0.0010 |
| 2 | 217 | 3630 | −0.18 | 0.83 | 0.72 | 0.97 | 0.016 | 3630 | 0.24 | 1.27 | 1.10 | 1.47 | 0.0014 |
| 3 | 208 | 3449 | −0.06 | 0.95 | 0.83 | 1.08 | 0.40 | 3461 | 0.15 | 1.16 | 0.97 | 1.39 | 0.16 |
| 4 | 217 | 3605 | −0.10 | 0.91 | 0.80 | 1.03 | 0.14 | 3620 | 0.11 | 1.12 | 0.95 | 1.31 | 0.18 |
| 5 | 208 | 3479 | −0.08 | 0.92 | 0.81 | 1.05 | 0.21 | 3490 | 0.18 | 1.19 | 1.23 | 1.39 | 0.024 |
| 6 | 186 | 3261 | −0.20 | 0.82 | 0.72 | 0.93 | 0.0021 | 3269 | 0.18 | 1.20 | 1.03 | 1.40 | 0.022 |
| 7 | 156 | 3106 | −0.24 | 0.79 | 0.68 | 0.92 | 0.0024 | 3112 | 0.28 | 1.33 | 1.10 | 1.59 | 0.0027 |
| 8 | 85 | 2739 | −0.01 | 1.01 | 0.81 | 1.26 | 0.90 | 2740 | 0.16 | 1.18 | 0.85 | 1.63 | 0.32 |
| 9 | 14 | 2148 | −0.09 | 0.91 | 0.55 | 1.50 | 0.71 | 2149 | 0.28 | 1.32 | 0.65 | 2.66 | 0.44 |
| Diastolic blood pressure | Glucose | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Exam | Number events a | N | beta | HR d | Lower 95% CL | Upper 95% CL | p‐Value | N | beta | HR c | Lower 95% CL | Upper 95% CL | p‐Value |
| 1 | 236 | 4279 | 0.05 | 1.05 | 0.93 | 1.19 | 0.44 | 4113 | 0.12 | 1.12 | 1.01 | 1.26 | 0.041 |
| 2 | 217 | 3712 | −0.02 | 0.98 | 0.87 | 1.10 | 0.70 | 3561 | 0.11 | 1.12 | 1.04 | 1.20 | 0.0020 |
| 3 | 208 | 3577 | 0.07 | 1.07 | 0.94 | 1.22 | 0.29 | 3364 | 0.08 | 1.09 | 1.01 | 1.16 | 0.022 |
| 4 | 217 | 3707 | 0.10 | 1.10 | 0.97 | 1.26 | 0.15 | 3468 | 0.10 | 1.11 | 1.05 | 1.17 | 0.00040 |
| 5 | 208 | 3512 | −0.007 | 0.99 | 0.84 | 1.18 | 0.94 | 3449 | 0.06 | 1.07 | 1.01 | 1.13 | 0.020 |
| 6 | 186 | 3299 | 0.04 | 1.04 | 0.91 | 1.20 | 0.54 | 3216 | 0.08 | 1.09 | 1.03 | 1.15 | 0.0041 |
| 7 | 162 | 3255 | 0.002 | 1.00 | 0.83 | 1.21 | 0.98 | 3065 | 0.12 | 1.13 | 1.05 | 1.12 | 0.00060 |
| 8 | 85 | 2818 | −0.12 | 0.89 | 0.71 | 1.11 | 0.29 | 2688 | 0.14 | 1.14 | 1.04 | 1.26 | 0.0058 |
| 9 | 14 | 2253 | −0.08 | 0.92 | 0.55 | 1.53 | 0.75 | 2105 | 0.33 | 1.39 | 1.06 | 1.82 | 0.017 |
Maximum number incident AD cases shown among subjects with for diastolic blood pressure measurements; numbers may be slightly less for other factors (see Table S3).
Log2 transformed values for betas and HRs.
Hazard ratio (HR) and 95% confidence interval for each 15 mg/dL increase.
Hazard ratio (HR) and 95% confidence interval for each 10 mm increase.
3.3. Association of AD with risk factors by age group
Analysis by age group showed that incident AD was negatively associated with HDL‐C measured in early adulthood (HR = 0.85 [0.72–0.99] per 15 mg/dL, P = 0.041) and middle adulthood (HR = 0.82 [0.70–0.96] per 15 mg/dL, P = 0.014), but the effect was attenuated in the late adulthood group (HR = 0.89 [0.77–1.02] per 15 mg/dL, P = 0.11) (Table 4). Specifically, a 15 mg/dL increase in HDL‐C corresponds to a reduction in AD risk of 15.4% and 17.9% in the early and middle adulthood groups, respectively. As shown in Table S4 in the supporting information, this association remained significant with a similar effect size in the middle adulthood group when adjusted for dyslipidemia treatment (P = 0.022). BG measured in middle adulthood was significantly associated with AD at nearly the same magnitude of effect before (HR = 1.15, 1.06‐1.23; P = 0.00029) and after (HR = 1.18, 1.08‐1.29; P = 0.00036) adjusting for diabetes treatment (Table 4, Table S4). Among persons in this age group, for every 15 mg/dL increase in BG, risk of AD increases by 14.5%. TG level was significantly associated with AD only in the early adulthood group before (HR = 1.33, 1.02‐1.57; P = 0.0013) as well as after (HR = 1.30, 1.10‐1.54; P = 0.0018) adjusting for dyslipidemia treatment. Diastolic blood pressure was significantly associated with AD in late adulthood before (HR = 1.14, 1.01‐1.29; P = 0.041) and after (HR = 1.14, 1.00‐1.29; P = 0.044) adjusting for treatment. AD was not associated with LDL‐C, total cholesterol, BMI, smoking, and systolic blood pressure in any stage of adulthood (P > 0.05). In models adjusting for APIOE ε4 status, only glucose measured in middle adulthood (HR = 1.14, 1.06‐1.23; P = 0.00034) and TG measured in early adulthood (HR = 1.21, 1.02‐1.43; P = 0.030) remained significantly associated with AD (Table S5). The association with glucose in late adulthood became significant when APOE ε4 status was added to the model (HR = 1.08, 1.01‐1.15; P = 0.030).
TABLE 4.
Association of blood lipids and glucose with incident AD by age group adjusted for age and sex
| Group (age range in years) | |||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Early adulthood) (35–50) | Middle adulthood (51–60) | Late adulthood (61–70) | |||||||||||||||||||
| Risk factor | N | N events | Beta | HR a | Lower 95% CL | Upper 95% CL | P‐value | N | N events | Beta | HR a | Lower 95% CL | Upper 95% CL | P‐value | N | N events | Beta | HR a | Lower 95% CL | Upper 95% CL | P‐value |
| HDL‐C | 2875 | 156 | −0.17 | 0.85 | 0.72 | 0.99 | 0.041 | 1803 | 163 | −0.20 | 0.82 | 0.70 | 0.96 | 0.014 | 1463 | 176 | −0.12 | 0.89 | 0.77 | 1.02 | 0.11 |
| TG b | 2879 | 157 | 0.28 | 1.33 | 1.02 | 1.57 | 0.0013 | 1809 | 163 | 0.11 | 1.12 | 0.93 | 1.35 | 0.22 | 1470 | 176 | 0.05 | 1.05 | 0.89 | 1.23 | 0.57 |
| BG | 2820 | 154 | 0.07 | 1.07 | 0.92 | 1.25 | 0.39 | 1792 | 163 | 0.14 | 1.15 | 1.06 | 1.23 | 0.00029 | 1427 | 171 | 0.06 | 1.07 | 1.00 | 1.14 | 0.056 |
| DBP | 2931 | 163 | 0.05 | 1.05 | 0.91 | 1.20 | 0.53 | 1863 | 170 | −0.05 | 0.95 | 0.83 | 1.09 | 0.48 | 1501 | 181 | 0.13 | 1.14 | 1.01 | 1.29 | 0.041 |
Abbreviations: HDL‐C, high‐density lipoprotein cholesterol; TG, triglyceride; BG, blood glucose; DPB, diastolic blood pressure.
aHazard ratio (HR) and 95% confidence interval for a 15 mg/dL increase of HDL‐C and BG, and a 10 mm increase in DBP.
Log2 transformed values for betas and HRs.
3.4. Association of risk factors with all‐cause dementia
Risk factor analyses within age groups yielded a pattern of results for HDL‐C, TG, and BG levels that was similar to those observed for AD incidence only, but they were less significant especially for both lipid measures (Table S6). The associations of all‐cause dementia incidence with TG in early adulthood (HR = 1.23, 1.03‐1.48; P = 0.021, BG in middle adulthood (HR = 1.13, 1.05‐1.21; P = 0.00057), and DBP in late adulthood (HR = 1.15, 1.01‐1.31; P = 0.038) were attenuated (Table S7). However, the incidence of all‐cause dementia was not associated with HDL in any age group.
3.5. Survival analysis results
Kaplan‐Meier analysis showed that future development of AD was significantly and progressively higher and likely to occur earlier among individuals with BG in the pre‐diabetic (100‐126 mg/dL) and diabetic (>126 mg/dL) ranges compared to those with normal BG (<100 mg/dL) in early adulthood (P = 0.0030, Figure 2A, left panel) and middle adulthood (P = 0.04, Figure 2B, left panel), but not in late adulthood (P = 0.51, Figure 2C, left panel). The comparison was slightly more significant when combining those with prediabetic and diabetic BG levels in the early adulthood (P = 0.0012, Figure 2A, right panel) and middle adulthood (P = 0.0094, Figure 2B, right panel) groups. A similar trend for AD‐free survival was also observed in the comparison of individuals with normal (<130 mg/dL), elevated (130‐169 mg/dL), and clinically abnormal (≥170 mg mg/dL) LDL‐C levels (P = 0.02) in early adulthood (Figure S2A). Similar results were observed in the same age group when comparing those with normal or elevated LDL‐C to those with high LDL‐C (P = 0.02, Figure S2B in supporting information). These comparisons were not significant for HDL‐C (Figure S3 in supporting information) or TG (Figure S4 in supporting information) in any age group, but borderline significant for individuals with normal DSP (<80 mm) compared to those with elevated (80‐89 mm) and abnormal (≥90 mm) DBP in early adulthood (Figure S5, right panel).
FIGURE 2.

Cumulative probability of Alzheimer's disease (AD)–free survival among individuals stratified into three (left panel) or two (right panel) clinically defined cutoffs for blood glucose (BG) concentration who were followed starting in (A) early adulthood (ages 35‐50 years) (B) middle adulthood (ages 51‐60 years), and (C) late adulthood (ages 61‐70 years). The number of years of follow‐up and number of surviving individuals in each BG level group are shown below the x‐axis. Lighter shades of color indicate standard deviation of point estimates
4. DISCUSSION
The association of vascular and metabolic risk factors including diabetes, obesity, high blood pressure, and elevated serum cholesterol levels with cognitive decline and AD is widely reported. 10 , 11 , 12 , 13 , 14 , 31 These observations contribute to the rationale for the U.S. Study to Protect Brain Health Through Lifestyle Intervention to Reduce Risk (U.S. POINTER), a 2‐year clinical trial recently launched by the Alzheimer's Association to evaluate whether healthy lifestyle interventions that target risk factors can protect cognitive function in adults 60 to 79 years of age (https://alz.org/us‐pointer/overview.asp). In this study, we demonstrated that HDL‐C, glucose, and TG measured in early adulthood (age 35‐50 years), HDL‐C and glucose measured in middle adulthood (51‐60 years), and diastolic blood pressure measured in late adulthood (61‐70 years) are significantly associated with incident AD up to several decades later. Other vascular risk factors including BMI, LDL‐C, systolic blood pressure, and smoking were not associated with AD in analyses considering measurements of the entire cohort at an individual exam or measurements of individuals within age groups. To our knowledge, this is the first report of an association of AD with HDL‐C, triglyceride, and glucose levels measured in cognitively normal individuals during early to middle adulthood. These findings are consistent with a previous study showing that the cumulative number of vascular risk factors in midlife but not late life is associated with late‐life brain amyloid measured by positron emission tomography (PET) imaging. 32
Our findings that link cholesterol fractions and pre‐diabetic glucose level in persons as young as age 35 to high AD risk decades later suggest that an intervention targeting cholesterol and glucose management starting in early adulthood can help maximize cognitive health in later life. This idea is supported by previous studies of Framingham Offspring study participants that showed that elevated coronary heart disease risk and metabolic syndrome were associated with lower cognitive performance at age 55. 18 , 19 However, our results do not distinguish whether the influences of these risk factors on the development of AD may be particularly damaging during early adulthood and midlife or reflect longer accumulated risk exposure. Our data showing only modest attenuation of treatment on the effect of these factors on AD incidence and weaker association among persons older than 60 years are consistent with the idea that exposure earlier in life may better explain our findings; however, secular trends in treatment for managing cholesterol and glucose could also have contributed to the observed patterns.
Prior studies examining the relationship between HDL‐C and risk of AD are inconclusive. Those with the highest baseline HDL‐C in elderly individuals (>65 years) had reduced AD risk, 33 and higher baseline HDL‐C protected against cognitive impairment and brain volume reductions 20 years later, 34 but other studies found no association between HDL‐C and cognitive impairment, noting that HDL was measured in subjects generally after age 65 who were followed for a period ranging from 2 to 12 years. 22 , 35 , 36 , 37 , 38 , 39 One of the negative reports was a study of 1026 members of the FHS Original Cohort (ie, including the parents of many subjects in the current study) among whom there were only 77 incident AD cases during the 10‐ to 11‐year follow‐up period after HDL‐C was measured when the participants had a mean age of 76.1 years. 22 HDL‐C concentration was also not associated with risk of AD or vascular dementia after accounting for other vascular risk factors in the large community‐based prospective 3C study of persons ≥65‐years‐old at baseline with up to 13 years of follow‐up. 15 These and other longitudinal studies 37 , 40 yield inconsistent results that depend largely on the age at which cholesterol was measured and length of follow‐up between cholesterol and cognitive assessments. Indeed, studies with a follow‐up period ≥10 years found significant associations between HDL‐C levels or apolipoprotein A1 (which is the major component of HDL particles in plasma) and AD risk, 34 , 40 whereas studies with <10 years of follow‐up did not. 37 , 39 Furthermore, association of AD risk with HDL‐C level is evident in studies with baseline measurements taken at middle age (<70 years), 40 , 41 , 42 but not in those with HDL‐C baseline measures in older subjects (≥70 years). 38 , 39 Taken together, these studies and our findings suggest that HDL may exert its greatest influence on AD risk starting in as early as age 35.
A variety of evidence supports a mechanistic role of HDL in AD pathogenesis. A recent Mendelian randomization meta‐analysis showed that reduced HDL levels might be potential causal risk factors of the development of AD, 43 but other Mendelian randomization studies do not support this conclusion. but other MR studies do not support this conclusion. 44 , 45 In vitro studies demonstrated that HDL reduces vascular Aβ accumulation 46 and attenuates Aβ‐induced endothelial inflammation. 47 Reed et al. reported that low HDL‐C and high LDL‐C are associated with elevated cerebral Aβ measured using Pittsburgh compound B and PET in living non‐demented individuals with a mean age of 78 years. 48 Other properties of HDL that are relevant to AD include its ability to reduce accumulation and increase transport of Aβ through the vasculature, inhibit Aβ‐induced endothelial activation, delay Aβ fibrillization, and induce nitric oxide (NO) production. 46 , 47 The cerebrovascular benefits of HDL in healthy humans may partly explain our finding of a protective association of circulating HDL levels against AD risk. 49 A role for HDL in AD is also supported by studies that have identified associations with genes that encode HDL biogenesis proteins and HDL protein components including APOE, ABCA1, PON1, CLU, and APOAI. 7 , 8 , 50 , 51 , 52 , 53 , 54 , 55
The functional relationship between APOE and HDL 56 likely accounts for the attenuated association of AD risk with HDL‐C level in a model including APOE ε4 carrier status, and is consistent with findings showing that the effect of the APOE ε4 allele on AD risk is mediated in part through its impact on reducing HDL‐C. 43 However, because cholesterol levels are modifiable, understanding the mechanisms through which HDL and related proteins modulate Aβ could offer new approaches to slowing Aβ deposition and thus reduce the incidence of AD including among APOE ε4 carriers.
Although the association of AD with diabetes 32 , 57 , 58 , 59 and impaired glucose levels 60 , 61 is well established, to our knowledge this is the first evidence of the association of AD with glucose levels measured in cognitively normal individuals throughout adulthood beginning as early as age 35. Of interest, our findings are consistent with results from a prospective cross‐sectional study of 799 men and women with an average age of 50 showing association of increased glucose levels with lower cognitive function. 62 Schrijvers et al. observed an increased risk of AD associated with fasting glucose levels among 3319 Rotterdam Study participants within a 3‐year follow‐up period starting at baseline, but this association was not evident (in fact, an inverse relationship) after longer follow‐up periods. 63 Notably, the mean age of subjects at baseline in that study (71.8 years) was more than 25 years older than the age of FHS participants at which we observed the deleterious effects of increased glucose on cognition. By contrast, results of another study suggest that higher glucose levels may be a risk factor for dementia, even among persons older than age 70 without diabetes. 64 That study is consistent with our findings showing association of increased glucose in middle age with dementia more generally.
Peripheral glucose homeostasis has been implicated in the pathogenesis of AD. 65 Longitudinal increases in fasting plasma glucose levels are associated with higher brain glucose concentrations. Higher brain glucose levels are correlated with more severe plaques and tangles in AD brains along with impaired glucose metabolism in brain due to reduced glycolytic flux. 66 In addition, high blood glucose (hyperglycemia) may contribute to the increased risk of AD by exacerbating astrocyte‐mediated neuroinflammation and neuronal injury caused by disease‐associated agents, for example, fibrillar Aβ42. 67 Hyperglycemia can result in the glycation of proteins and dysregulate the innate immune system through glycation and oxidation of macrophage migration inhibitory factor (MIF), a process that is accelerated in both early and late‐stage AD brains. 68 Furthermore, high glucose can render cells, including those in the brain, insulin resistant, which may worsen mental functioning. It is also linked to reduced insulin uptake (resulting in lowered glucose metabolism) in areas of the brain associated with AD. 69 Finally, insulin has an important role in neuronal signaling, and thus insulin resistance can inhibit signaling between brain cells and affect memory. 70
This study has several notable strengths including the quadrennial measurement of risk factors in a community‐based sample of nearly 5000 participants beginning in early adulthood who were followed ≈38 years on average. Several caveats should also be considered. First, all participants in the FHS Offspring cohort are white, which limits generalizability to other populations. Second, although we applied two approaches to estimate the association of vascular risk factors measured across adulthood with incident AD, each design has limitations. The age‐stratified approach addresses the age question more directly than the exam‐by‐exam approach, which considers all cohort members who survived to each exam as one group. However, this approach has less power because of divided samples, as well as issues of combining risk factors measured at different exams (possible measurement bias) and introducing correlation because some individuals were included in more than one age group, although participants are independent within each age group. In the exam‐by‐exam approach, participants have the same follow‐up time for each exam and risk factors were measured in a relatively narrow period (≈2‐4 years) among participants at each exam. However, because the age range is relatively large among participants at each exam, our results might not accurately reflect age‐specific trends, even though the age of survivors at each exam is progressively increasing. Both approaches have uneven power across exams and age groups due to a progressively smaller follow‐up period, although we endeavored to balance the number of AD incident cases in the analyses within age groups. This issue may explain, for example, negative findings in previous studies and less significant results in our study for HDL in the oldest age group, which had the shortest follow‐up. Despite these concerns, the overall similarity of results from both approaches suggests that our findings are relatively robust. Third, we were unable to assess the association of fasting measures of lipids and glucose in early adulthood because blood samples were not collected under fasting conditions at the first two examinations of the Offspring cohort. However, although non‐fasting measurements may be slightly more abnormal, significant associations of HDL‐C, triglyceride, and glucose measured at exams 1 and 2 with AD risk are unlikely spurious because the onset of AD symptoms did not occur until several decades later on average. Another limitation of our findings is that they are based on analyses of one‐time measurements of risk factors. Studies that allow for time‐course changes of risk factors on the AD risk in late life may better explain the dynamic relationship between vascular risk factors on cognitive and brain health in late life; however, a time‐dependent Cox analysis generally requires complete data for subjects followed for a fixed period, is complicated by death which is a competing risk, only addresses relatively short‐term effects, and does not adequately adjust for confounders (eg, treatment) that are measured repeatedly during follow‐up. 71 , 72 Our findings showing similar but attenuated associations of all‐cause dementia with triglyceride and glucose levels, and no association with HDL‐C level, should be interpreted cautiously because of the heterogeneous etiologies of dementia. Finally, although we tested models that included a term for treatment of abnormal cholesterol levels and blood pressure, and for diabetes (adjustment for which had little impact on our results), we did not distinguish specific medications or account for duration of their use.
5. CONCLUSIONS
Our findings showing that HDL‐C, triglyceride, and glucose levels measured in early to middle adulthood are significantly associated with incident AD several decades later, which suggests that early intervention to maintain healthy HDL, triglyceride, and glucose levels may improve cognition and lower AD risk in addition to the benefits of promoting vascular and metabolic health. The effects of these risk factors, particularly glucose level, may not be AD specific and may contribute to incidence of other forms of dementia. Studies combining multiple prospective cohorts with long follow‐up periods that collectively contain much larger samples of non‐AD dementias than the FHS will be necessary to address this question.
DISCLOSURES
Received support for the present manuscript:
Xiaoling Zhang: U19‐AG068753
Ting Fang Alvin Ang: 1R01AG049899‐01 National Institute on Aging (NIA) R01 grant to Boston University (BU)
Sherral Devine: NIA support to my institution
Joseph Massaro: National Institutes of Health (NIH)/National Heart, Lung, and Blood Institute (NIH/NHLBI). Support was made to my institution (Boston University School of Public Health) as part of my salary
Kathryn L. Lunetta: U19‐AG068753
Rhoda Au: NIA (multiple grants), Alzheimer's Drug Discovery Foundation, American Heart Association, Gates Ventures ‐ all funding goes to my institution
Lindsay A Farrer: NIH support to Boston University: U19‐AG068753, 2R01‐AG048927, U01‐AG062602. Payments for all grants made to institution
In the past 36 months, authors have received grants or contracts from any entity
Xiaoling Zhang: NIH grants U19‐AG068753, U01‐AG072577. Payments for all grants made to my institution
Qiushan Tao: 04/2021 – 03/2022. Pilot grant – “Investigating the relationship between loneliness and inflammatory markers in Framingham Heart Study.” Supported by Framingham Heart Study Brain Aging Program (FHS‐BAP, NIH: U19‐AG068753.). The payments were made to my institution
Jesse Mez: All payments made to institution. Funding is from NIH (NIA, NINDS) and DOD
Joseph Massaro: NIH/NHLBI. Support was made to my institution (Boston University School of Public Health) as part of my salary
Kathryn L. Lunetta: R01AG067457 R01 HL092577 U19‐AG068753 18SFRN34230127 18SFRN34150007 U01‐AG058654 R01‐AG048927 R01CA228156 R01HL105756 R01 CA202981 U19 AG063893
Rhoda Au: NIA (multiple grants), Alzheimer's Drug Discovery Foundation, American Heart Association, Gates Ventures ‐ all funding goes to my institution
Lindsay A Farrer: NIH grants U19‐AG068753, U01‐AG072577. Payments for all grants made to institution
In the past 36 months, no authors have received royalties or licenses
In the past 36 months, authors have received consulting fees
Rhoda Au: Signant Health, Biogen, GSK, Nova Nordisk, ‐ paid to me
Lindsay A Farrer Mass Mutual Insurance ‐ to institution
In the past 36 months, authors have received payment or honoraria for lectures, presentations, speakers bureaus, manuscript writing, or educational events
Sanford Auerbach: Lecture for a CME program, payment made to total: $2,000
Rhoda Au: Eisai ‐ to be paid to me
In the past 36 months, no authors have received payment for expert testimony
In the past 36 months, no authors have received support for attending meetings and/or travel
In the past 36 months, no authors have had any patents planned, issued, or pending
In the past 36 months, no authors have participated on a Data Safety Monitoring Board or Advisory Board
In the past 36 months, no authors have held a leadership or fiduciary role in any other board, society, committee, or advocacy group (paid or unpaid)
In the past 36 months, no authors have held stock or stock options in entities related to the current manuscript and/or area of research included in this manuscript or related area of research
In the past 36 months, authors have received equipment, materials, drugs, medical writing, gifts, or other services
Rhoda Au: Gift from Robert Thomas, M.D., High Lantern and Gates Ventures, paid to my institution.
In the past 36 months, no authors have had other financial or non‐financial interests
All other authors have nothing to disclose: Tong, Andrew Chang
Supporting information
Supporting information
ACKNOWLEDGMENTS
This work was supported by the Framingham Heart Study's National Heart, Lung, and Blood Institute contract N01‐HC‐25195; the Wing‐Tat Lee Foundation and Evans Center for Interdisciplinary Biomedical Research at Boston University School of Medicine; and National Institutes of Health grants U19‐AG068753, RF1‐AG057519, 2R01‐AG048927, U01‐AG062602, R01‐NS017950, R01‐AG016495, R01‐AG008122, R01‐AG033040; R01‐AG049810, RF1‐AG054156, RF1‐AG062109; and VMF‐14‐318524 from the Alzheimer's Association. We express our gratitude to the staff and participants of the Framingham Heart Study.
Zhang X, Tong T, Chang A, et al. Midlife lipid and glucose levels are associated with Alzheimer's disease. Alzheimer's Dement. 2023;19:181–193. 10.1002/alz.12641
Xiaoling Zhang and Lindsay A. Farrer authors contributed equally to the supervision of this work.
REFERENCES
- 1. 2020 Alzheimer's disease facts and figures. Alzheimers Dement. 2020. [DOI] [PubMed] [Google Scholar]
- 2. Matthews KA, Xu W, Gaglioti AH, et al. Racial and ethnic estimates of Alzheimer's disease and related dementias in the United States (2015‐2060) in adults aged >/= 65 years. Alzheimers Dement. 2019;15:17‐24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Livingston G, Huntley J, Sommerlad A, et al. Dementia prevention, intervention, and care: 2020 report of the Lancet Commission. Lancet. 2020;396:413‐446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Schneider JA, Arvanitakis Z, Bang W, Bennett DA. Mixed brain pathologies account for most dementia cases in community‐dwelling older persons. Neurology. 2007;69:2197‐2204. [DOI] [PubMed] [Google Scholar]
- 5. Haroutunian V, Schnaider‐Beeri M, Schmeidler J, et al. Role of the neuropathology of Alzheimer disease in dementia in the oldest‐old. Arch Neurol. 2008;65:1211‐1217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Savva GM, Wharton SB, Ince PG, et al. Age, neuropathology, and dementia. N Engl J Med. 2009;360:2302‐2309. [DOI] [PubMed] [Google Scholar]
- 7. Jansen IE, Savage JE, Watanabe K, et al. Genome‐wide meta‐analysis identifies new loci and functional pathways influencing Alzheimer's disease risk. Nat Genet. 2019;51:404‐413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kunkle BW, Grenier‐Boley B, Sims R, et al. Genetic meta‐analysis of diagnosed Alzheimer's disease identifies new risk loci and implicates Abeta, tau, immunity and lipid processing. Nat Genet. 2019;51:414‐430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM. Clinical diagnosis of Alzheimer's disease: report of the NINCDS‐ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer's disease. Neurology. 1984;34:939‐944. [DOI] [PubMed] [Google Scholar]
- 10. Kivipelto M, Helkala EL, Hanninen T, et al. Midlife vascular risk factors and late‐life mild cognitive impairment: a population‐based study. Neurology. 2001;56:1683‐1689. [DOI] [PubMed] [Google Scholar]
- 11. Qiu C, Winblad B, Fratiglioni L. The age‐dependent relation of blood pressure to cognitive function and dementia. Lancet Neurol. 2005;4:487‐499. [DOI] [PubMed] [Google Scholar]
- 12. Kivimaki M, Luukkonen R, Batty GD, et al. Body mass index and risk of dementia: analysis of individual‐level data from 1.3 million individuals. Alzheimers Dement. 2018;14:601‐609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang J, Chen C, Hua S, et al. An updated meta‐analysis of cohort studies: diabetes and risk of Alzheimer's disease. Diabetes Res Clin Pract. 2017;124:41‐47. [DOI] [PubMed] [Google Scholar]
- 14. Snowdon DA. Aging and Alzheimer's disease: lessons from the nun study. Gerontologist. 1997;37:150‐156. [DOI] [PubMed] [Google Scholar]
- 15. Schilling S, Tzourio C, Soumare A, et al. Differential associations of plasma lipids with incident dementia and dementia subtypes in the 3C study: a longitudinal, population‐based prospective cohort study. PLoS Med. 2017;14:e1002265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Reitz C, Tang MX, Luchsinger J, Mayeux R. Relation of plasma lipids to Alzheimer disease and vascular dementia. Arch Neurol. 2004;61:705‐714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bosch J, O'Donnell M, Swaminathan B, Lonn EM, Sharma M, Dagenais G, et al. Effects of blood pressure and lipid lowering on cognition: results from the HOPE‐3 study. Neurology. 2019;92:e1435‐e46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bangen KJ, Armstrong NM, Au R, Gross AL. Metabolic syndrome and cognitive trajectories in the framingham offspring study. J Alzheimers Dis. 2019;71:931‐943. [DOI] [PubMed] [Google Scholar]
- 19. Armstrong NM, Bangen KJ, Au R, Gross AL. Associations between midlife (but not late‐life) elevated coronary heart disease risk and lower cognitive performance: results from the framingham offspring study. Am J Epidemiol. 2019;188:2175‐2187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Feinleib M, Kannel WB, Garrison RJ, McNamara PM, Castelli WP. The framingham offspring study. Design and preliminary data. Prev Med. 1975;4:518‐525. [DOI] [PubMed] [Google Scholar]
- 21. Kannel WB, Feinleib M, McNamara PM, Garrison RJ, Castelli WP. An investigation of coronary heart disease in families. The Framingham offspring study. Am J Epidemiol. 1979;110:281‐290. [DOI] [PubMed] [Google Scholar]
- 22. Tan ZS, Seshadri S, Beiser A, et al. Plasma total cholesterol level as a risk factor for Alzheimer disease: the Framingham study. Arch Intern Med. 2003;163:1053‐1057. [DOI] [PubMed] [Google Scholar]
- 23. Friedewald WT, Levy RI, Fredrickson DS. Estimation of the concentration of low‐density lipoprotein cholesterol in plasma, without use of the preparative ultracentrifuge. Clin Chem. 1972;18:499‐502. [PubMed] [Google Scholar]
- 24. Seshadri S, Beiser A, Kelly‐Hayes M, et al. The lifetime risk of stroke: estimates from the Framingham study. Stroke. 2006;37:345‐350. [DOI] [PubMed] [Google Scholar]
- 25. Seshadri S, Wolf PA. Lifetime risk of stroke and dementia: current concepts, and estimates from the Framingham study. Lancet Neurol. 2007;6:1106‐1114. [DOI] [PubMed] [Google Scholar]
- 26. Kamboh MI, Aston CE, Hamman RF. The relationship of APOE polymorphism and cholesterol levels in normoglycemic and diabetic subjects in a biethnic population from the San Luis Valley. Colorado Atherosclerosis. 1995;112:145‐159. [DOI] [PubMed] [Google Scholar]
- 27. Nielson C, Lange T, Hadjokas N. Blood glucose and coronary artery disease in nondiabetic patients. Diabetes Care. 2006;29:998‐1001. [DOI] [PubMed] [Google Scholar]
- 28. Whelton PK, Carey RM, Aronow WS, et al. ACC/AHA/AAPA/ABC/ACPM/AGS/APhA/ASH/ASPC/NMA/PCNA guideline for the prevention, detection, evaluation, and management of high blood pressure in adults: a report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. J Am Coll Cardiol. 2017;71:e127‐e248. [DOI] [PubMed] [Google Scholar]
- 29. Lowenstern A, Li S, Navar AM, et al. Does clinician‐reported lipid guideline adoption translate to guideline‐adherent care? An evaluation of the Patient and Provider Assessment of Lipid Management (PALM) registry. Am Heart J. 2018;200:118‐124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Clinic. C . Cholesterol numbers: what do they mean. 2020.
- 31. Gottesman RF, Schneider AL, Zhou Y, et al. Association between midlife vascular risk factors and estimated brain amyloid deposition. JAMA. 2017;317:1443‐1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Akomolafe A, Beiser A, Meigs JB, et al. Diabetes mellitus and risk of developing Alzheimer disease: results from the Framingham study. Arch Neurol. 2006;63:1551‐1555. [DOI] [PubMed] [Google Scholar]
- 33. Reitz C, Tang MX, Schupf N, Manly JJ, Mayeux R, Luchsinger JA. A summary risk score for the prediction of Alzheimer disease in elderly persons. Arch Neurol. 2010;67:835‐841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Armstrong NM, An Y, Beason‐Held L, et al. Predictors of neurodegeneration differ between cognitively normal and subsequently impaired older adults. Neurobiol Aging. 2019;75:178‐186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Formiga F, Ferrer A, Chivite D, Pinto X, Cuerpo S, Pujol R. Serum high‐density lipoprotein cholesterol levels, their relationship with baseline functional and cognitive status, and their utility in predicting mortality in nonagenarians. Geriatr Gerontol Int. 2011;11:358‐364. [DOI] [PubMed] [Google Scholar]
- 36. Marcum ZA, Walker R, Bobb JF, Sin MK, Gray SL, Bowen JD, et al. Serum cholesterol and incident Alzheimer's disease: findings from the adult changes in thought study. J Am Geriatr Soc. 2018;66:2344‐2352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Li G, Shofer JB, Kukull WA, et al. Serum cholesterol and risk of Alzheimer disease: a community‐based cohort study. Neurology. 2005;65:1045‐1050. [DOI] [PubMed] [Google Scholar]
- 38. Mielke MM, Xue QL, Zhou J, Chaves PH, Fried LP, Carlson MC. Baseline serum cholesterol is selectively associated with motor speed and not rates of cognitive decline: the Women's Health and Aging Study II. J Gerontol A Biol Sci Med Sci. 2008;63:619‐624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Yaffe K, Barrett‐Connor E, Lin F, Grady D. Serum lipoprotein levels, statin use, and cognitive function in older women. Arch Neurol. 2002;59:378‐384. [DOI] [PubMed] [Google Scholar]
- 40. Saczynski JS, White L, Peila RL, Rodriguez BL, Launer LJ. The relation between apolipoprotein A‐I and dementia: the Honolulu‐Asia aging study. Am J Epidemiol. 2007;165:985‐992. [DOI] [PubMed] [Google Scholar]
- 41. Shih YH, Tsai KJ, Lee CW, et al. Apolipoprotein C‐III is an amyloid‐beta‐binding protein and an early marker for Alzheimer's disease. J Alzheimers Dis. 2014;41:855‐865. [DOI] [PubMed] [Google Scholar]
- 42. Bates KA, Sohrabi HR, Rainey‐Smith SR, et al. Serum high‐density lipoprotein is associated with better cognitive function in a cross‐sectional study of aging women. Int J Neurosci. 2017;127:243‐252. [DOI] [PubMed] [Google Scholar]
- 43. Zhang X, Tian Q, Liu D, et al. Causal association of circulating cholesterol levels with dementia: a mendelian randomization meta‐analysis. Transl Psychiatry. 2020;10:145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ostergaard SD, Mukherjee S, Sharp SJ, et al. Associations between potentially modifiable risk factors and Alzheimer disease: a mendelian randomization study. PLoS Med. 2015;12:e1001841. discussion e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Proitsi P, Lupton MK, Velayudhan L, et al. Genetic predisposition to increased blood cholesterol and triglyceride lipid levels and risk of Alzheimer disease: a Mendelian randomization analysis. PLoS Med. 2014;11:e1001713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Robert J, Button EB, Yuen B, et al. Clearance of beta‐amyloid is facilitated by apolipoprotein E and circulating high‐density lipoproteins in bioengineered human vessels. Elife. 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Robert J, Button EB, Stukas S, et al. High‐density lipoproteins suppress Abeta‐induced PBMC adhesion to human endothelial cells in bioengineered vessels and in monoculture. Mol Neurodegener. 2017;12:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Reed B, Villeneuve S, Mack W, DeCarli C, Chui HC, Jagust W. Associations between serum cholesterol levels and cerebral amyloidosis. JAMA Neurol. 2014;71:195‐200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Button EB, Gilmour M, Cheema HK, Martin EM, Agbay A, Robert J, et al. Vasoprotective functions of high‐density lipoproteins relevant to Alzheimer's disease are partially conserved in apolipoprotein B‐depleted plasma. Int J Mol Sci. 2019;20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Lupton MK, Proitsi P, Lin K, et al. The role of ABCA1 gene sequence variants on risk of Alzheimer's disease. J Alzheimers Dis. 2014;38:897‐906. [DOI] [PubMed] [Google Scholar]
- 51. Koldamova R, Fitz NF, Lefterov I. ATP‐binding cassette transporter A1: from metabolism to neurodegeneration. Neurobiol Dis. 2014;72(Pt A):13‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Contu L, Carare RO, Hawkes CA. Knockout of apolipoprotein A‐I decreases parenchymal and vascular beta‐amyloid pathology in the Tg2576 mouse model of Alzheimer's disease. Neuropathol Appl Neurobiol. 2019;45:698‐714. [DOI] [PubMed] [Google Scholar]
- 53. Corder EH, Saunders AM, Strittmatter WJ, et al. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science. 1993;261:921‐923. [DOI] [PubMed] [Google Scholar]
- 54. Erlich PM, Lunetta KL, Cupples LA, et al. Polymorphisms in the PON gene cluster are associated with Alzheimer disease. Hum Mol Genet. 2006;15:77‐85. [DOI] [PubMed] [Google Scholar]
- 55. Farrer LA, Cupples LA, Haines JL, et al. Effects of age, sex, and ethnicity on the association between apolipoprotein E genotype and Alzheimer disease. A meta‐analysis. APOE and Alzheimer disease meta analysis consortium. JAMA. 1997;278:1349‐1356. [PubMed] [Google Scholar]
- 56. Huang Y, Mahley RW. Apolipoprotein E: structure and function in lipid metabolism, neurobiology, and Alzheimer's diseases. Neurobiol Dis. 2014;72(Pt A):3‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ohara T, Doi Y, Ninomiya T, et al. Glucose tolerance status and risk of dementia in the community: the Hisayama study. Neurology. 2011;77:1126‐1134. [DOI] [PubMed] [Google Scholar]
- 58. Calderon‐Garciduenas L, de la Monte SM. Apolipoprotein E4, gender, body mass index, inflammation, insulin resistance, and air pollution interactions: recipe for Alzheimer's disease development in Mexico city young females. J Alzheimers Dis. 2017;58:613‐630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Marseglia A, Dahl Aslan AK, Fratiglioni L, Santoni G, Pedersen NL, Xu W. Cognitive trajectories of older adults with prediabetes and diabetes: a population‐based cohort study. J Gerontol A Biol Sci Med Sci. 2018;73:400‐406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Weinstein G, Maillard P, Himali JJ, Beiser AS, Au R, Wolf PA, et al. Glucose indices are associated with cognitive and structural brain measures in young adults. Neurology. 2015;84:2329‐2337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Neergaard JS, Dragsbaek K, Hansen HB, Henriksen K, Christiansen C, Karsdal MA. Late‐life risk factors for all‐cause dementia and differential dementia diagnoses in women: a prospective cohort study. Medicine (Baltimore). 2016;95:e3112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Masley SC, Roetzheim R, Clayton G, Presby A, Sundberg K, Masley LV. Lifestyle markers predict cognitive function. J Am Coll Nutr. 2017;36:617‐623. [DOI] [PubMed] [Google Scholar]
- 63. Schrijvers EM, Witteman JC, Sijbrands EJ, Hofman A, Koudstaal PJ, Breteler MM. Insulin metabolism and the risk of Alzheimer disease: the Rotterdam study. Neurology. 2010;75:1982‐1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Crane PK, Walker R, Hubbard RA, et al. Glucose levels and risk of dementia. N Engl J Med. 2013;369:540‐548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Thambisetty M, Jeffrey Metter E, Yang A, et al. Glucose intolerance, insulin resistance, and pathological features of Alzheimer disease in the Baltimore longitudinal study of aging. JAMA Neurol. 2013;70:1167‐1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. An Y, Varma VR, Varma S, et al. Evidence for brain glucose dysregulation in Alzheimer's disease. Alzheimers Dement. 2018;14:318‐329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Bahniwal M, Little JP, Klegeris A. High glucose enhances neurotoxicity and inflammatory cytokine secretion by stimulated human astrocytes. Curr Alzheimer Res. 2017;14:731‐741. [DOI] [PubMed] [Google Scholar]
- 68. Kassaar O, Pereira Morais M, Xu S, et al. Macrophage migration inhibitory factor is subjected to glucose modification and oxidation in Alzheimer's. Disease Sci Rep. 2017;7:42874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Willette AA, Bendlin BB, Starks EJ, et al. Association of insulin resistance with cerebral glucose uptake in late middle‐aged adults at risk for Alzheimer disease. JAMA Neurol. 2015;72:1013‐1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kellar D, Craft S. Brain insulin resistance in Alzheimer's disease and related disorders: mechanisms and therapeutic approaches. Lancet Neurol. 2020;19:758‐766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Dekker FW, de Mutsert R, van Dijk PC, Zoccali C, Jager KJ. Survival analysis: time‐dependent effects and time‐varying risk factors. Kidney Int. 2008;74:994‐997. [DOI] [PubMed] [Google Scholar]
- 72. Ngwa JS, Cabral HJ, Cheng DM, et al. A comparison of time dependent cox regression, pooled logistic regression and cross sectional pooling with simulations and an application to the Framingham Heart Study. BMC Med Res Methodol. 2016;16:148. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information