

Abstract
Abnormal tau protein aggregates constitute a hallmark of Alzheimer's disease. The mechanisms underlying the initiation of tau aggregation in sporadic neurodegeneration remain unclear. Here we investigate whether a non‐human prion can seed tau aggregation. Due to their structural similarity with tau aggregates, we chose Sup35NM yeast prion domain fibrils for explorative tau seedings. Upon in vitro incubation with tau monomers, Sup35NM fibrils promoted the formation of morphologically distinct tau fibril strains. In vivo, intrahippocampal inoculation of Sup35NM fibrils accentuated tau pathology in P301S tau transgenic mice. Thus, our results provide first in vivo evidence for heterotypic cross‐species seeding of a neurodegenerative human prion‐like protein by a yeast prion. This opens up the conceptual perspective that non‐mammalian prions present in the human microbiome could be involved in the initiation of protein misfolding in neurodegenerative disorders, a mechanism for which we propose the term “trans‐seeding.”
Keywords: Alzheimer's disease, neurodegeneration, prion, seeding, Sup35, tau, trans‐seeding, yeast
1. NARRATIVE
1.1. Contextual background
Most human neurodegenerative diseases are associated with prion‐like protein aggregation. 1 In Alzheimer's disease (AD), amyloid beta (Aβ) accumulates in extracellular plaques, whereas tau protein forms intraneuronal aggregates known as tangles. 2 While the appearance of tau tangles correlates well with cognitive decline, 3 , 4 the initial cause for this disease‐defining protein aggregation remains unknown. In AD, the normally soluble tau protein forms rigid β‐sheet structured aggregates, which then can spread from cell to cell and be transmitted upon inoculation. 5 With these latter features, tau resembles classical prions. 6
Prions are proteinaceous particles with an infectious behavior, which is much more pronounced than the relatively slow cell‐to‐cell spreading of tau. The term “prion” was introduced by Prusiner for the infectious particle causing scrapie, a neurodegenerative disease that primarily affects sheep. 7 Several years later, Wickner identified Sup35 as a first non‐mammalian prion in yeast. 8 Classical prion diseases involve modifications of the prion protein PrP. 9 Like tau, PrP can misfold and aggregate into β‐sheet structured aggregates. 10 Such β‐sheet structures are made up of twisted and pleated polypeptide strands. 11 The folding of such β‐sheet templates can be “copied” by soluble molecules, which thereby adopt the rigid conformation of their β‐sheet templates. This template‐based induction of β‐sheet structure is referred to as “seeding.” 12 , 13 Thus, AD patient‐derived or synthetic tau fibrils can seed soluble tau into β‐sheet–structured aggregates in a prion‐like manner. 4
In neurodegenerative diseases, for reasons not yet understood, tau aggregates often occur in parallel with aggregates of other β‐sheet–structured prion‐like proteins, for example with Aβ in AD or α‐synuclein in Parkinson's disease (PD). 14 In this context, cross‐seeding of tau has been observed for several human prion‐like proteins, including α‐synuclein, 15 , 16 Aβ, 17 , 18 and islet amyloid polypeptide. 19 Given the demonstrated cross‐seedings of tau with various human prion‐like proteins, we hypothesized that even non‐human prion proteins might be able to seed tau aggregation, provided they feature β‐sheet structures similar to tau aggregates. Such non‐human prions might thus serve as an infectious pathogenic factor in sporadic AD. We therefore set out to analyze, as a proof of concept, whether a β‐sheet–structured yeast prion was capable of triggering tau aggregation.
The concept that non‐mammalian prions might be involved in seeding tau aggregation may seem unexpected (if not far‐fetched), given the still widely held species barrier theory for prions. 20 , 21 Nevertheless, considering the potentially significant consequences of such a scenario for public health, further exploration of this hypothesis is warranted.
1.2. Study conclusions and disease implications
In our study, the addition of minute amounts of an aggregated yeast prion domain to soluble tau promoted the formation of tau fibrils. These tau fibrils, induced by the yeast Sup35NM prion domain, differ from the tau fibrils that form in the absence of Sup35NM, pointing to a direct interaction of the yeast prion template with soluble tau. The yeast prion aggregates therefore not only accelerate tau aggregation, but also result in morphologically distinct tau fibril strains. Furthermore, the inoculation of Sup35NM yeast prion fibrils into the brains of tau transgenic mice accentuated their tau pathology at the injection site, providing first evidence for seeding of tau by a non‐mammalian prion. Our findings clearly demonstrate that a non‐mammalian prion is capable of modulating tau aggregation as it occurs in the pathogenesis of AD and other tauopathies.
Prions have recently been found to be more widely present in nature than anticipated, occuring also in bacteria, 22 , 23 plants, 24 and even in the form of a viral expression factor. 25 This opens up the perspective for a relevant contribution of non‐mammalian prions to the initiation of neurodegenerative diseases by seeding, a mechanism for which we propose the term “trans‐seeding.” Trans‐seeding comprises the aspects of β‐sheet templated seeding of human prion‐like proteins by non‐mammalian prions, pointing to a potentially infectious transmissibility across the species barrier, capable of triggering human diseases.
In fact, besides our finding of Sup35NM‐induced tau aggregation, further examples for trans‐seeding have recently been identified in the context of neurodegenerative diseases. Aβ aggregation can be induced by bacterial FapC amyloid fragments of Pseudomonas aeruginosa in vivo. 26 Aβ has recently also been shown to be seeded by Sup35NM in vitro, complementing our findings and pointing to a potential role of yeast prions in AD pathogenesis. 27 Furthermore, the PD‐defining α‐synuclein aggregates can be seeded by the bacterial prion Curli. 28
How would these lines of trans‐seeding evidence fit into our current understanding of AD pathogenesis?
RESEARCH IN CONTEXT
Systematic review: Aggregation of tau protein and its prion‐like spreading constitute characteristic hallmarks of Alzheimer's disease (AD). Why tau undergoes aggregation in AD is not yet understood. In light of the recent discoveries of prions in all forms of life, we speculate whether tau protein aggregation can be provoked by non‐mammalian prions.
Interpretation: Our proof‐of‐concept study demonstrates that the yeast Sup35NM prion domain can promote the formation of tau aggregates in vitro and in vivo. Our findings provide first evidence for cross‐species seeding of tau by a non‐mammalian prion.
Future directions: We propose to study cross‐species seeding of tau and amyloid beta by non‐mammalian (fungal, bacterial, viral) prions. The search for non‐mammalian prions as potentially harmful agents should be intensified. Further studies may then explore the presence of non‐mammalian prions in AD patients.
1.2.1. Potential route of infection
In AD, trans‐seeding prions could reach neurons projecting toward the brain primarily via the nasopharynx as an entry port. They may either be ingested or locally produced by the microbiome. From the nasopharyngeal cavity, prions may enter the brain similarly to viruses, as most recently exemplified by SARS‐CoV‐2. 29 Early in the course of COVID‐19, SARS‐CoV‐2 can reach the olfactory and limbic systems, disturbing the sense of smell and causing cognitive dysfunction. 30 Similarly, olfactory dysfunction often constitutes an early symptom in patients developing PD or AD. 31 , 32 , 33 In the olfactory bulb, trans‐seeding prions may drive initial tau aggregation, resulting in neuronal dysfunction and a diminished sense of smell. 34 From there, tau pathology can spread in a prion‐like manner trans‐neuronally via the olfactory nerve to the entorhinal cortex, amygdala, and the hippocampus, to eventually reach the neocortex. Such a spreading pattern of tau pathology from the olfactory bulb towards the limbic system with subsequent involvement of neocortical structures would well mirror the pathological and clinically progressive course of AD. 34 , 35
1.2.2. Incubation period
Templated seedings of neurodegenerative prion‐like proteins are typically associated with long incubation periods, even when seed and seeded protein share the same primary structure. Incubation times of up to three decades were documented for patients with iatrogenic PrPSc infection or Aβ transmission by contaminated growth hormone or gonadotropin, 36 , 37 and reached even five decades in people afflicted by cannibalism‐associated Kuru disease. 38 In the case of trans‐seeding, in which non‐mammalian seed and human prion‐like protein differ in structure, even longer incubation times would be expected. Thus, exposure to microbiotic or exogenous non‐mammalian prions early in life could well explain the sporadic manifestation of AD decades later, with typical disease onset after the seventh decade of life.
1.2.3. Heterogeneity of tauopathies
By acquiring unique conformational features and quaternary structures, tau has the ability to form distinct strains. 39 , 40 Thus, tau filaments can either resemble the filaments found in AD, as is the case with chronic traumatic encephalopathy, 39 or fold differently, as in Pick's disease. 41 Each tau strain may affect specific susceptible cells and brain regions, resulting in the evolution of different disease characteristics. As the transmission of distinct tau strains is specific to unique pathological conformations, 42 trans‐seeding by different prions could explain the formation of different tau strains and the clinical heterogeneity of tauopathies.
In summary, the concept of trans‐seeding is well in line with our current knowledge on the course of tau pathology and associated neuronal dysfunction in AD. It could explain how the clinical manifestations relate to pathophysiology and provide a plausible explanation for the anatomical specificity of early lesions associated with AD. In addition, this concept may also account well for the mixed pathology frequently observed in AD and other tauopathies, in which aggregation of different neurodegenerative prion‐like proteins co‐occurs.
1.3. Limitations and directions for future research
While our study demonstrates seeding of a neurodegenerative prion‐like protein by a yeast prion, it remains to be investigated (1) whether trans‐seeding can be provoked by various prions, (2) how it may relate to neurotoxicity, and, most crucially, (3) whether it does indeed occur in the pathophysiological context of AD and other neurodegenerative conditions.
1.3.1. Basic research
First, we propose to screen the trans‐seeding competence of various bacterial and plant prions with tau and other neurodegenerative prion‐like proteins in vitro. This will allow assessment of the frequency of this phenomenon, and which structural properties are required. Mouse models may not only help to rapidly validate the trans‐seeding competence of specific prions due to the accelerated formation of aggregates in transgenic lines, but also serve to examine potential entry routes of non‐human prions upon their intranasal and oral application.
It will be of interest to test to what extent trans‐seeded aggregates structurally match patient‐derived fibrils. Cryogenic electron microscopy of trans‐seeded fibrils collected from inoculated transgenic mice will allow the comparison of their folding patterns to characteristic patient‐derived fibrils. 40 , 41 , 43 In addition, it should be addressed, whether trans‐seeding is capable of inducing oligomeric species of tau or Aβ, as in the current understanding of AD, neuronal dysfunction is primarily mediated by small oligomeric aggregates rather than by extended fibrils of tau or Aβ. 44 , 45 , 46 In a trans‐seeding situation, such toxic oligomers might result from a surface‐catalyzed seeding mechanism. To assess oligomer formation and potential links to neurotoxicity, trans‐seeded proteins will have to be generated in larger amounts, analyzed biochemically, and assessed in genetically modified reporter cell lines. The use of algorithms capable of identifying proteins with prion properties may furthermore allow us to search for yet‐unidentified prions generated by the human microbiome. 22
1.3.2. Clinical studies and public health aspects
To make the case for a truly pathogenic role of trans‐seeding in AD, clinical studies will eventually be required to investigate the prevalence of microbiotic or exogenous prions in AD patients. Samples collected from their nasopharyngeal or oral microbiome should be screened for the presence of β‐structured proteins based on resistance to formic acid dissolution, similar to a recent study in rats, in which amyloid proteins have been successfully identified in the microbiome. 47 As minute amounts of seeds can trigger the aggregation of prion‐like proteins, 48 an initial seed may be undetectable at the time of clinical AD manifestation. Thus, asymptomatic younger subjects should be included in the screening for microbiotic prions.
Future research may also assess the presence of non‐mammalian prions in food and the environment. The Sup35 yeast prion investigated in our present study, as an example, occurs in wild yeast on grapes and in Saccharomyces cerevisiae strains of commercial dry wine yeast. 49 , 50 Epidemiological studies addressing the “AD exposome” should include such prion candidates to provide insights, which may also inform public health recommendations and policies. 51
Identification of specific trans‐seeding prions could ultimately result in preventive, diagnostic, and therapeutic approaches to neurodegenerative disorders. Measures could include the avoidance of food contaminated by natural prions or the modulation of the human microbiome, for example, through special diets, toward less prion‐generating species.
1.3.3. Conclusions
We demonstrate that a yeast prion domain can promote the formation of tau aggregation, providing first in vivo evidence for seeding of a human neurodegenerative prion‐like protein by a yeast prion. Our finding calls for further analysis of the trans‐seeding competence of fungal, bacterial, plant, and viral prions, as well as for research on the presence of such prions in the human microbiome and the environment. Given the prospect of potentially identifying non‐human prions as modifiable neurodegeneration‐triggering factors, which would undoubtedly be of significant public health interest, we deem respective research efforts warranted. The elaboration of the prion concept took more than four decades from the first description of the inoculability of scrapie by Cuillé and Chelle in 1936 52 to Prusiner's formulation of the protein‐only hypothesis and isolation of the protease‐resistant prion protein in 1982. 7 , 53 While Prusiner had already early on speculated about a role for prions in the pathogenesis of AD, 7 the prion‐like behavior of Aβ and tau was only recognized two decades later. 5 , 54 , 55 In this light, the search for a more definite conclusion on the trans‐seeding concept may well last into the next decade.
2. CONSOLIDATED STUDY DESIGN AND RESULTS
To investigate whether a non‐mammalian prion can provoke human tau aggregation, we studied the interplay of the Sup35NM yeast prion domain with tau in vitro and in vivo. Several reasons prompted us to choose the Sup35NM yeast prion domain for our trans‐seeding experiments: Sup35 prions form a parallel in‐register β‐sheet structured amyloid similar to tau aggregates, 56 which might be a prerequisite for trans‐seeding. Several yeast prions are known to promote the aggregation of other yeast prions, suggesting heterogeneous seeding competence. 57 Moreover, Sup35NM accelerates amyloid formation in a murine model of silver nitrate‐induced serum amyloid A amyloidosis. 58
First, we analyzed whether fibrils of the yeast prion domain Sup35NM are capable of promoting the aggregation of mutant tau in vitro. We chose mutant tau that harbors the P301S mutation of a familial tauopathy for the first set of experiments, as it can rapidly assemble into fibrils, even in the absence of a seed. 59 When minute amounts of Sup35NM yeast prion fibrils were added, the aggregation of P301S tau was not only accelerated, but, unexpectedly, the induced tau fibrils exhibited a particular corkscrew‐like folding pattern. This latter finding implies a structurally different conformation of Sup35NM‐induced compared to spontaneously formed P301S tau filaments.
Next, we investigated, whether even wild‐type tau, which is known to have a low aggregation propensity, can be seeded by the addition of Sup35NM fibrils. Without seeds, only sparse tau filaments formed after 16 days of incubation. In contrast, Sup35NM aggregates prompted the formation of wild‐type tau filaments within only 3 days of incubation, again with fibrils harboring a characteristic corkscrew‐like shape. We then used short fragments of these Sup35NM‐induced tau fibrils, which we obtained by sonification, to again seed soluble wild‐type tau. Strikingly, these fragmented tau fibrils were also able to act in a prion‐like manner, with the next generation of tau fibrils again showing the “original” characteristic corkscrew‐like pattern. These in vitro findings demonstrate, that the yeast Sup35NM prion domain can promote tau aggregation and provoke a characteristic fibril pattern.
To study the interaction of tau and Sup35NM in vivo, we inoculated pre‐aggregated Sup35NM fibrils into the hippocampus of 3‐month‐old mice expressing P301S mutant tau in their neurons. 60 After 3 months of incubation, we found a strong accentuation of tau pathology at the inoculation site. This focal increase of tau aggregation was significant compared to the contralateral, non‐inoculated hippocampus, as well as compared to controls.
In summary, our findings provide solid first evidence for the induction of tau aggregation by a non‐mammalian prion protein.
3. DETAILED METHODS AND RESULTS
3.1. Methods
3.1.1. Schematic of background and main conclusions
Background, principal experimental approach, results, and main conclusions of this paper are summarized in Figure S1 in supporting information.
3.1.2. Expression, purification, and fibrillization of Sup35NM
Sup35NM containing a carboxy‐terminal His‐tag was expressed using the plasmid p1404, 61 which was a kind gift from Prof. Reed B. Wickner (Laboratory of Biochemistry and Genetics, National Institutes of Health, Bethesda, Maryland, USA). E. coli Rosetta (DE3) pLysS was used as expression host and transformed via heat shock. 62 A transformed colony was incubated overnight at 37°C in a 50 mL LB‐ampicillin‐kanamycin flask, then 20 mL of growing culture were added to a flask containing 2L Terrific Broth‐ampicillin‐kanamycin medium. Induction was performed with 1 mM IPTG at an optical absorption at 600 nm wavelength (A600) of ≈1. Cells were harvested by centrifugation after 5 hours of IPTG induction, and lysed by sonication in lysis buffer (50 mM Tris‐HCL, 1 mM phenylmethylsulfonyl fluoride, 8 M urea, pH 8.0). The lysate was cleared at 12,000 g for 20 minutes at 4°C, and the supernatant applied to a 1 mL HisTrap (GE Healthcare) column pre‐equilibrated with lysis buffer. The column was washed (50 mM Tris‐HCl, 1 mM DTT, 8 M urea, 300 mM NaCl, and 20 mM imidazole) until absorption at 280 nm reached zero. Sup35NM was eluted with 250 mM imidazole. Purity of Sup35NM in collected fractions was analyzed by 4% to 12% sodium dodecyl sulfate polymerase gel electrophoresis. It was then loaded onto a HiLoad 16/60 Superdex 75 PG (GE Healthcare) column equilibrated with 7 M urea, 50 mM Tris (pH 7.5), and 150 mM NaCl. Fractions containing Sup35NM were assessed via Western blotting using an anti‐His‐Tag antibody (Anti‐6X His tag antibody [HRP], Abcam), pooled, concentrated, and dialyzed against phosphate‐buffered saline (PBS) using a Slide‐A‐Lyzer 10K dialysis cassette (Thermo Fisher Scientific). Although the C‐terminal His‐tag used for purifying Sup35NM can change Sup35NM fibril structure, 63 both untagged and His‐tagged Sup35NM share a β‐sheet structure similar to the one present in tau fibrils. 56 , 64 Sup35NM was concentrated to 0.271 mg/mL using an Amicon ultra‐centrifugal filter with a 10K cutoff (Millipore), and incubated at 4°C under slight agitation for 3 days to enable fibrillization. Aliquots of fibrillized samples were stored at –80°C.
3.1.3. In vitro seedings
For in vitro seedings, recombinant 2N4R wild‐type tau (ab84700, Abcam) and recombinant 2N4R P301S mutant tau (009‐001‐U01, Rockland Immunochemicals Inc.) were used together with the Sup35NM fibrils (described in Section 3.1.2) and ‐5TyrSup35NM monomers (GenScript).
The buffer of tau monomers was exchanged to Dulbecco's phosphate‐buffered saline (DPBS) without calcium and magnesium (D8537, Merck KGaA) using Microspin G‐50 columns (GE27‐5330‐01, GE Healthcare).
Sup35NM seeds were generated from filaments by sonication (cycle: 0.2 seconds, amplitude: 100%, 4 × 20 seconds with 2 minutes breaks in between) in a UP200St tube sonicator (Hielscher Ultrasonics GmbH) and filtered with 0.22 μm filter centrifuge tubes to exclude longer filaments.
Tau monomers at a final concentration of 0.1 mg/mL were mixed with 0.027 mg/mL of seeds in DPBS containing 0.02% sodium azide and 0.02 mg/mL heparin (H3393, Merck KGaA). Samples were incubated at 37°C on an overhead rotator at 30 RPM. Seeding experiments were replicated three times.
Seeds from Sup35NM induced 2N4R wild‐type tau filaments were generated by sonication in a similar manner as described above (cycle: 0.2 seconds, amplitude: 100%, 4 × 5 minutes with 5 minute breaks in between). Tau seeds were diluted 1:3 in 2N4R wild‐type tau monomers for re‐seeding experiments.
3.1.4. Transmission electron microscopy
In vitro samples were negatively stained and studied by transmission electron microscopy (TEM). 4 μL of sample were adsorbed for 60 seconds on glow‐discharged copper grids coated with 2% parlodion and a continuous carbon film. Samples were stained with 2% uranyl acetate solution.
Labelling of tau filaments with immuno‐gold was adapted from Goedert et al. 65 Samples were adsorbed on TEM grids as described above. Grids were laid face down on a drop of 0.1% gelatin (G7041, Merck KGaA) in DPBS, incubated for 10 minutes, transferred to a droplet of HT7 anti‐tau antibody (MN1000, Thermo Fisher Scientific, 1:20 dilution in DPBS), and incubated for 60 minutes. After washing in gelatin solution, grids were stained with anti‐mouse antibody conjugated to 10 nm gold beads (EM.GAF10). Uranyl acetate was used for negative stains.
Samples were imaged with FEI Tecnai T12 (operated at 120 kV) and FEI Tecnai G2 Spirit (operated at 80 kV) transmission electron microscopes (Thermo Fisher Scientific) equipped with TVIPS TemCam‐F416 (Tietz Video and Image Processing Systems GmbH) and EMSIS VELETA (EMSIS GmbH) cameras, respectively.
Measurements on the electron micrographs were done with the imageJ distribution Fiji (https://fiji.sc/). For Sup35NM seeded 2N4R wild‐type tau, periodicity of the wave pattern was used to calculate mean and standard deviation.
3.1.5. Mice
Homozygous human P301S mutant tau transgenic mice 59 were used as host mice for all seeding experiments. Animal experiments were approved by the official local Committee for Animal Care and Animal Use of the Canton of Basel (License Nr. BS2471).
3.1.6. Seed inoculation into P301S tau transgenic mice
Three‐month‐old P301S tau mice were anesthetized with a mixture of ketamine (80 mg/kg) and xylazine (8 mg/kg) and placed on a heating pad to maintain body temperature during surgery. Mice were injected in the right hippocampus (A/P, −2.5 mm from bregma; L, − 2.0 mm; D/V, −1.8 mm) using a Hamilton syringe. Each mouse received a unilateral stereotaxic injection of 2.5 μL volume, at a speed of 1.25 μL/min. After injection, the needle was kept in place for an additional 3 minutes. The surgical area was cleaned with saline and the incision sutured. Mice were monitored until recovery from anesthesia, provided with analgesic medication, and checked regularly after surgery.
The following seeds were used for inoculation: Brains collected from C57BL/6 mice (B6 mice) were diluted 1:9 in PBS and homogenized using an Ultra‐Turax T8 homogenizer (IKA Labortechnik) followed by brief sonication (Bandelin SONOPULS, 90% power, 10% cycle, 10‐second pulses). Homogenates were centrifuged at 4000 g for 20 minutes at 4°C to remove debris and aliquots of supernatant were stored at –80°C for later usage as B6‐brain homogenates (B6‐bh). ‐5TyrSup35NM was obtained from GenScript, diluted in PBS, and inoculated at a concentration of 0.3 mg/mL. Sup35NM was generated as described (Section 3.1.2) and inoculated in PBS at a concentration of 0.3 mg/mL.
3.1.7. Immunohistochemical analysis
Mice were sacrificed at the age of 6.3 ± 0.5 months, before developing hindering motor palsy (see Table S3 in supporting information). To this end, animals were deeply anesthetized and brains were dissected and post‐fixed overnight. After paraffin embedding, 4 μm thick coronal sections were prepared. Sections were silver‐impregnated following the method of Gallyas 66 and counterstained by hematoxylin and eosin staining (H&E). For tau immunohistochemistry, the monoclonal antibody AT8 (Thermo Scientific; stock solution diluted 1:1000) was used as described previously. 67 Secondary antibodies were obtained from Vector Laboratories (Vectastain ABC kit).
FIGURE 1.
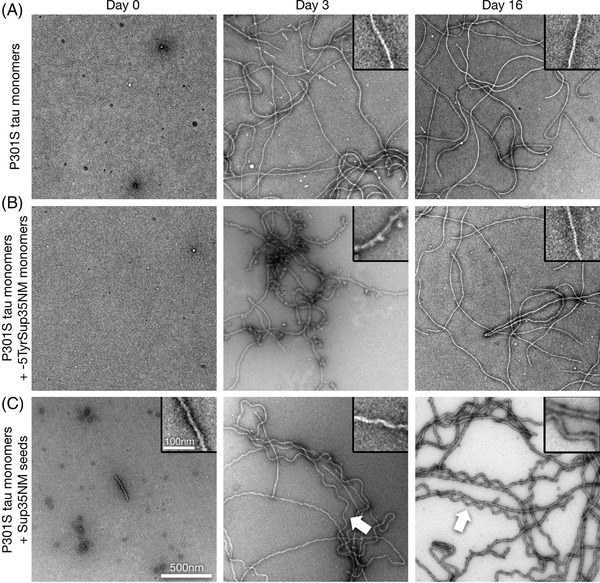
Sup35NM seeds P301S mutant tau aggregation in vitro. A‐C, Transmission electron microscope images of negatively stained preparations of human 2N4R P301S mutant tau monomer aggregates, formed under low heparin conditions (A), and after addition of ‐5TyrSup35NM monomers (B), or Sup35NM seeds (C), at days 0, 3, and 16. Note the occurrence of corkscrew‐shaped tau fibrils at days 3 and 16 upon seeding with Sup35NM fibrils (arrows, C). Scale bar for all images: 500 nm, for all magnified insets: 100 nm
For neuronal quantification of the CA3 region, a total of three tissue sections were analyzed (at levels –2.6, –2.4, and –2.2 mm from bregma) per mouse and staining. Bregma levels were determined by visual comparison to the Mouse Brain Atlas. 68 The number of AT8 positive and Gallyas silver‐stained neurons was counted by two raters (M.F., A.M.) on 10X magnified, randomly coded images, taken with a BX43 upright microscope (Olympus). The mean of the neuron numbers scored by the two raters was used for statistical analysis.
For a few stainings, only two sections next to the indicated bregma levels were well preserved and usable for quantification (four sections lacking for AT8 stainings, three sections lacking for the Gallyas stainings). Due to fixation artifacts, which interfered with immunohistochemistry but not with Gallyas silver staining, AT8 immunohistochemistry was not quantifiable for one mouse of the ‐5TyrSup35NM group, and two mice of the Sup35NM group.
3.1.8. Statistical analysis
First, we estimated the mean neuron count per mouse in the ipsilateral CA3 field of P301S tau transgenic mice inoculated with either B6‐bh, ‐5TyrSup35NM, or Sup35NM. This comparison between study groups was done using a one‐way analysis of variance. P‐values were adjusted for multiple comparisons using Tukey's test.
Next, we compared the ipsilateral (inoculated) to the contralateral (non‐inoculated) side of Sup35NM injected mice. To estimate the number of AT8‐ or Gallyas stain–positive neurons in their hippocampal CA3 fields, the mean neuron counts per mouse of the ipsilateral and contralateral sides were compared by paired t‐tests. A P‐value < .05 was considered significant. All evaluations were done using the statistical software R (www.r‐project.org). Mean values and standard deviations are given in the respective figure legends.
3.2. Results
3.2.1. In vitro seeding of tau by Sup35NM
To study the potential of the yeast Sup35 prion for trans‐seeding tau protein, we expressed the Sup35NM prion domain in E. coli and aggregated it at 4°C into fibrils (Figure S2 in supporting information). The combined N‐terminal (N) and middle (M) domains of Sup35 are sufficient to form Sup35NM fibrils with prion properties. 69 Sup35NM is highly aggregation prone, only staying monomeric when purified under denaturing conditions, which require a high concentration of reducing agents. 70 Therefore, recombinant tyrosine‐deleted Sup35NM protein (termed ‐5TyrSup35NM), previously described as a less aggregating Sup35NM mutant, 71 was used as a non‐aggregated seeding control for in vitro and in vivo seeding experiments.
First, we analyzed whether Sup35NM fibrils were capable of directly inducing tau aggregation in vitro. The naturally unfolded tau protein does not easily aggregate in vitro without co‐factors such as heparin. Hence, we incubated either aggregation‐prone recombinant human 2N4R P301S tau monomers (Figure 1) or 2N4R wild‐type tau monomers (Figure 2) in the presence of very low levels of heparin (0.02 mg/mL) either unseeded (Figure 1A, Figure 2A), with non‐aggregated ‐5TyrSup35NM monomers (Figure 1B, Figure 2B), or with Sup35NM fibril fragments (obtained by sonication of Sup35NM fibrils; Figures 1C and 2C).
FIGURE 2.
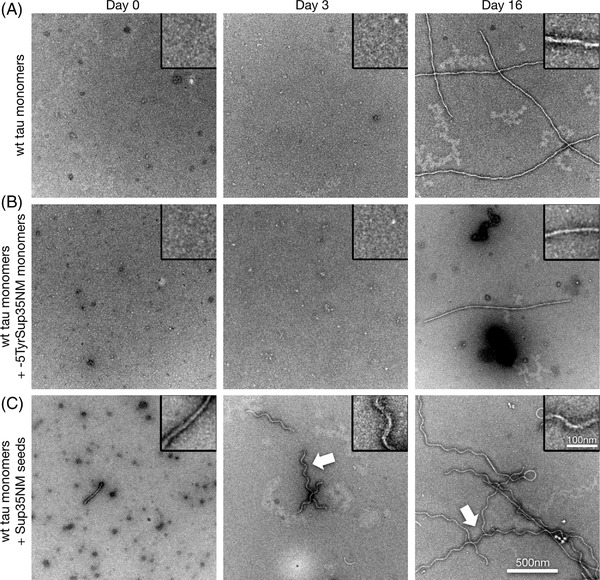
Sup35NM seeds wild‐type tau aggregation in vitro. A‐C, Transmission electron microscope images of negatively stained preparations of human 2N4R wild‐type tau monomer aggregation under low heparin conditions (A), and after addition of ‐5TyrSup35NM monomers (B), or Sup35NM seeds (C), at days 0, 3, and 16. Note the occurrence of corkscrew‐shaped tau fibrils at days 3 and 16 upon seeding with Sup35NM fibrils (arrows, C). Scale bar for all images: 500 nm, for all magnified insets: 100 nm
Seeding of P301S tau monomers with Sup35NM fibril fragments resulted in the formation of tau filaments displaying a distinctly wavy, slightly skewed corkscrew‐like pattern (Figure 1C), which was only rarely seen in unseeded or ‐5TyrSup35NM incubated tau samples. As this corkscrew‐like P301S tau fibril pattern was somewhat reminiscent of the curly tau fibrils previously described upon seedings with mutant tau seeds, 72 we assume that it might correspond to a specific tau fibril strain.
As expected, unseeded P301S tau monomers aggregated into a wide variety of filaments (Figure 1A). P301S tau monomers kept with ‐5TyrSup35NM monomers formed similar filaments as the unseeded sample. However, after 3 days of incubation these filaments showed clumpy protein decorations that were no longer visible after 16 days (Figure 1B). This suggested a transient interaction of the tau monomers with ‐5TyrSup35NM, which itself also formed fibrils after 16 days when kept without tau monomers (Figure S3 in supporting information).
Strikingly, Sup35NM seeds also provoked rapid aggregation of 2N4R wild‐type tau monomers into fibrils (Figure 2C). These fibrils again exhibited a characteristic corkscrew‐like pattern, with clear preference toward regular wave patterns with a periodicity of ≈143 nm (± 12 nm, n = 87; Figure 2C) when observed in negative stain TEM images. Compared to non‐seeded (Figure 2A) or ‐5TyrSup35NM exposed wild‐type tau monomers (Figure 2B), fibril formation was promoted by Sup35NM seeds, and corkscrew‐like fibrils formed already at day 3 (Figure 2C). Immuno‐gold labelling of the Sup35NM‐induced corkscrew‐like fibrils confirmed that they consist of tau, and not of minute amounts of Sup35NM seeds (Figure S4 in supporting information).
Next, we created short seeds out of Sup35NM‐induced human wild‐type 2N4R tau filaments by sonication, in order to seed 2N4R wild‐type tau monomers. After 3 days we observed rapid formation of tau filaments (Figure 3A), which in turn could be used for seeding a next generation of fibrils (Figure 3B). While the aggregation accelerating effect of the seeds stayed intact, the distinct wave pattern, which was well preserved in the first generation of re‐seeding, lost its exclusivity during the subsequent seeding generation, where morphologies as seen previously in heparin‐induced samples occurred more often. This observed dilution could be attributable to the fact that Sup35NM‐induced fibrils occur first due to the aggregation‐promoting effect of Sup35NM. At later time points it is possible that the interaction of tau monomers with heparin alone results in the formation of fibrils as well (as seen for the negative control without Sup35NM seeds; Figure 2A). These heparin‐induced tau fibrils could then act as templates as well, and therefore the morphology of Sup35NM‐induced fibrils becomes less dominant.
FIGURE 3.
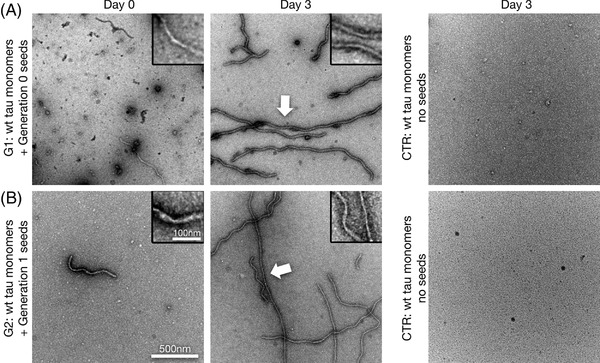
Re‐seedings of tau monomers by Sup35NM‐induced wild‐type tau filaments. A, B, Transmission electron microscope images of negatively stained preparations of human 2N4R wild‐type tau monomer aggregation under low heparin conditions at time points day 0 and day 3, using corkscrew‐like 2N4R wild‐type tau filaments (Generation 0 seeds) previously induced by Sup35NM seeds. This resulted in a next generation (Generation 1 [G1]) of corkscrew‐like filaments (arrow, A). Re‐seeding with G1 filaments again induced corkscrew‐like tau aggregates in G2 (arrow, B). Scale bar for all images: 500 nm, for all magnified insets: 100 nm
3.2.2. In vivo seeding of tau by Sup35NM
Next, we asked whether Sup35NM fibrils have the potential to accentuate tau pathology in vivo. To this end, we inoculated pre‐aggregated Sup35NM fibrils into P301S mice, which express human full‐length 0N4R tau with the P301S tau mutation present in hereditary forms of frontotemporal dementia and parkinsonism linked to chromosome 17 (FTDP‐17TAU). This widely used tauopathy model is particularly suitable for rapid in vivo tau seeding. 60 P301S tau mice develop slowly progressive intraneuronal tau inclusions, which become accentuated upon the inoculation of β‐sheet–structured tau seeds. 59 , 61 Sup35NM, ‐5TyrSup35NM, and brain homogenate of non‐transgenic C57BL/6 mice (termed B6‐bh), were used as seeding material for comparison. We inoculated 2.5 μL of Sup35NM pre‐formed fibrils, or ‐5TyrSup35NM, or B6‐bh solutions into the right hippocampus of 3‐month‐old P301S tau transgenic host mice. In Sup35NM inoculated P301S tau mice, 3 months post‐inoculation, a strong accentuation of AT8 immunohistochemistry and Gallyas silver stain positive tau pathology was noted at the hippocampal inoculation site and along the injection canal (Figure 4A‐D, Figure 5). The accentuation of tau pathology in the CA3 field upon Sup35NM injection was consistent comparing these mice to B6‐bh‐injected P301S control mice (Figure 4A, C, Table S1 in supporting information). As Sup35NM only remains non‐aggregated when high amounts of detergents are present, that is, conditions incompatible with in vivo experiments including stereotaxic inoculations, we used the less aggregating ‐5TyrSup35NM mutant as negative control. Indeed, in these mice only a mild, non‐significant increase in tau pathology was noted compared to B6‐bh inoculated mice (Figure 4A, C, Table S1). This observation paralleled our in vitro findings of a limited short‐term interaction between ‐5TyrSup35NM and P301S tau that did not lead to obvious changes in tau fibril structure. Moreover, our experiment also confirmed the strong aggregation propensity of Sup35NM, which partially persisted even with the ‐5Tyr deletion. 73
FIGURE 4.
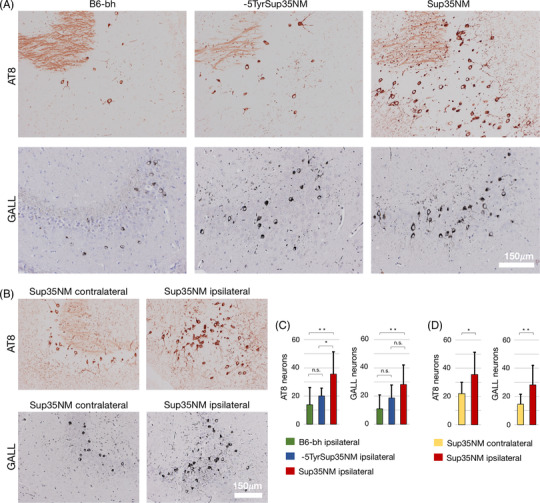
Sup35NM seeds tau aggregation in P301S tau transgenic mice. A‐D, Increase in AT8 immunohistochemistry (AT8) and Gallyas staining (GALL) positive neurons in the unilateral CA3 field after intrahippocampal Sup35NM fibril inoculation in P301S tau transgenic mice. AT8 and Gallyas positive neurons comparing P301S tau mice inoculated with C57BL/6 mouse brain homogenates (B6‐bh), ‐5TyrSup35NM solution, and Sup35NM fibrils (A, C; one‐way analysis of variance, followed by Tukey's test), and corresponding histology (Bregma level –2.6 mm). Comparison between non‐injected (contralateral) and injected (ipsilateral) side (B, D; paired t‐tests), and representative histological findings (Bregma level –2.4 mm). *P < .05, **P < .01, n.s. = non‐significant; indicated is the mean number of positive CA3 neurons per section ± standard deviation. For detailed quantitative data see Table S2 in supporting information
FIGURE 5.
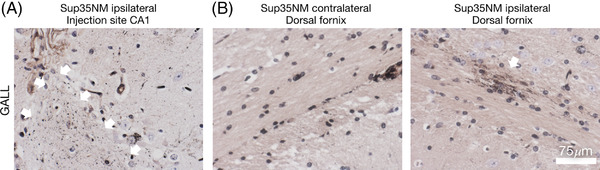
Sup35NM induces focal tau pathology. Focal tau pathology induced around hippocampal inoculation canal and ipsilateral dorsal fornix upon Sup35NM inoculation. A, B, Gallyas stains. Note the characteristic focal grain‐like tau aggregates in proximity of the hippocampal injection canal upon Sup35NM inoculation (A, arrows indicate tau pathology along the inoculation canal). Granular tau pathology develops in the ipsilateral dorsal fornix of a SUP35NM‐seeded P301S mouse (B, right; arrow), but is absent in the contralateral fornix (B, left)
Furthermore, quantitative estimates confirmed an increase in hippocampal tau pathology in the CA3 sector of the Sup35NM inoculated ipsilateral hippocampus compared to the contralateral (non‐seeded) side (Figure 4B, D, Table S2 in supporting information). Similar to the seeding effects of brain extracts from tauopathy patients in P301S host mice, 74 we noted the induction of Gallyas positive grains in the dorsal fornix in a subset of Sup35NM seeded mice, mostly in close proximity to the inoculation canal (Figure 5A, B). These findings are compatible with a robust in vivo seeding effect of Sup35NM and a mild, non‐significant residual seeding effect of the less aggregation prone ‐5TyrSup35NM mutant.
AUTHOR CONTRIBUTIONS
David T. Winkler conceived and designed the study; Martin Flach, Cedric Leu, Henning Stahlberg, and David T. Winkler designed research; Martin Flach performed the Sup35NM expression and purification; Martin Flach and Zhiva Skachokova performed the in vivo experiments and light microscopy; Cedric Leu performed the in vitro experiments and electron microscopy; Martin Flach, Cedric Leu, Alfonso Martinisi, Henning Stahlberg, and David T. Winkler analyzed data; David T. Winkler drafted the manuscript. All authors discussed the results and commented on the manuscript. All authors read and approved the final manuscript.
CONFLICTS OF INTEREST
The authors declare no competing interests.
Supporting information
Supporting information.
ACKNOWLEDGMENTS
We thank Prof. Reed Wickner, Laboratory of Biochemistry and Genetics, NIH, Bethesda, Maryland, USA, for his advice and for providing the Sup35NM plasmid, Dr. Michel Goedert, MRC, Laboratory of Molecular Biology, Cambridge CB2, UK, for his advice and for providing the P301S tau transgenic mice, and Andreas Schötzau (www.eudox.ch) for statistical advice. This work was in part supported by the Swiss National Science Foundation (32323B_123812 and 310030_169486 to DTW, 310031_188548 and CRSII5_177195 to HS, and 31003A_152846 to MT), the Mach‐Gaensslen Foundation, the Science Fund of the Department of Neurology of the University Hospital Basel, and the Synapsis Foundation Switzerland (to DTW, HS, and MT).
Open Access Funding provided by Universitat Basel.
Flach M, Leu C, Martinisi A, et al. Trans‐seeding of Alzheimer‐related tau protein by a yeast prion. Alzheimer's Dement. 2022;18:2481–2492. 10.1002/alz.12581
Martin Flach and Cedric Leu contributed equally to this study.
REFERENCES
- 1. Goedert M . Alzheimer's and Parkinson's diseases: the prion concept in relation to assembled Abeta, tau, and alpha‐synuclein. Science. 2015;349:1255555. [DOI] [PubMed] [Google Scholar]
- 2. Alzheimer's disease facts and figures. Alzheimer's Dementn. 2016;12:459‐509. [DOI] [PubMed] [Google Scholar]
- 3. Arriagada PV, Growdon JH, Hedley‐Whyte ET, Hyman BT. Neurofibrillary tangles but not senile plaques parallel duration and severity of Alzheimer's disease. Neurology. 1992;42:631‐639. [DOI] [PubMed] [Google Scholar]
- 4. Goedert M, Eisenberg DS, Crowther RA. Propagation of tau aggregates and neurodegeneration. Annu Rev Neurosci. 2017;40:189‐210. [DOI] [PubMed] [Google Scholar]
- 5. Clavaguera F, Bolmont T, Crowther RA, et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol. 2009;11:909‐913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Aguzzi A, De Cecco E. Shifts and drifts in prion science. Science. 2020;370:32‐34. [DOI] [PubMed] [Google Scholar]
- 7. Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216:136‐144. [DOI] [PubMed] [Google Scholar]
- 8. Wickner RB. [URE3] as an altered URE2 protein: evidence for a prion analog in Saccharomyces cerevisiae. Science. 1994;264:566‐569. [DOI] [PubMed] [Google Scholar]
- 9. Prusiner SB. Molecular biology of prion diseases. Science. 1991;252:1515‐1522. [DOI] [PubMed] [Google Scholar]
- 10. Scheckel C, Aguzzi A. Prions, prionoids and protein misfolding disorders. Nat Rev Genet. 2018;19:405‐418. [DOI] [PubMed] [Google Scholar]
- 11. Nowick JS. Exploring beta‐sheet structure and interactions with chemical model systems. Acc Chem Res. 2008;41:1319‐1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Jarrett JT. Seeding “one‐dimensional crystallization” of amyloid: a pathogenic mechanism in Alzheimer's disease and scrapie? Cell. 1993;73:1055‐1058. [DOI] [PubMed] [Google Scholar]
- 13. Jucker M, Walker LC. Propagation and spread of pathogenic protein assemblies in neurodegenerative diseases. Nature Neurosci. 2018;21:1341‐1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Guo T, Noble W, Hanger DP. Roles of tau protein in health and disease. Acta Neuropathol. 2017;133:665‐704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Giasson BI, Forman MS, Higuchi M, et al. Initiation and synergistic fibrillization of tau and alpha‐synuclein. Science. 2003;300:636‐640. [DOI] [PubMed] [Google Scholar]
- 16. Guo JL, Covell DJ, Daniels JP, et al. Distinct alpha‐synuclein strains differentially promote tau inclusions in neurons. Cell. 2013;154:103‐117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Guo JP, Arai T, Miklossy J, McGeer PL. Abeta and tau form soluble complexes that may promote self aggregation of both into the insoluble forms observed in Alzheimer's disease. Proc Natl Acad Sci U S A. 2006;103:1953‐1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Vasconcelos B, Stancu IC, Buist A, et al. Heterotypic seeding of tau fibrillization by pre‐aggregated Abeta provides potent seeds for prion‐like seeding and propagation of tau‐pathology in vivo. Acta Neuropathol. 2016;131:549‐569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Arya S, Claud SL, Cantrell KL, Bowers MT. Catalytic prion‐like cross‐talk between a key Alzheimer's disease tau‐fragment R3 and the type 2 diabetes peptide IAPP. ACS Chem Neurosci. 2019;10:4757‐4765. [DOI] [PubMed] [Google Scholar]
- 20. Hagiwara Ki, Hara H, Hanada K. Species‐barrier phenomenon in prion transmissibility from a viewpoint of protein science. J Biochem. 2013;153:139‐145. [DOI] [PubMed] [Google Scholar]
- 21. Tessier PM, Lindquist S. Unraveling infectious structures, strain variants and species barriers for the yeast prion [PSI+]. Nat Struct Mol Biol. 2009;16:598‐605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Yuan AH, Hochschild A. A bacterial global regulator forms a prion. Science. 2017;355:198‐201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Chapman MR, Robinson LS, Pinkner JS, et al. Role of Escherichia coli curli operons in directing amyloid fiber formation. Science. 2002;295:851‐855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Chakrabortee S, Kayatekin C, Newby GA, Mendillo ML, Lancaster A, Lindquist S. Luminidependens (LD) is an Arabidopsis protein with prion behavior. Proc Natl Acad Sci U S A. 2016;113:6065‐6070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Nan H, Chen H, Tuite MF, Xu X. A viral expression factor behaves as a prion. Nat Commun. 2019;10:359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Javed I, Zhang Z, Adamcik J, et al. Accelerated amyloid beta pathogenesis by. Bacterial Amyloid FapC Advan Sci. 2020;7:2001299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Koloteva‐Levine N, Aubrey LD, Marchante R, et al. Amyloid particles facilitate surface‐catalyzed cross‐seeding by acting as promiscuous nanoparticles. Proc Natl Acad Sci U S A. 2021;118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Sampson TR, Challis C, Jain N, et al. A gut bacterial amyloid promotes α‐synuclein aggregation and motor impairment in mice. Elife. 2020;9:e53111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Meinhardt J, Radke J, Dittmayer C, et al. Olfactory transmucosal SARS‐CoV‐2 invasion as a port of central nervous system entry in individuals with COVID‐19. Nature Neurosci. 2021;24:168‐175. [DOI] [PubMed] [Google Scholar]
- 30. Zubair AS, McAlpine LS, Gardin T, Farhadian S, Kuruvilla DE, Spudich S. Neuropathogenesis and neurologic manifestations of the coronaviruses in the age of coronavirus disease 2019: a review. JAMA Neurol. 2020;77:1018‐1027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Risacher SL, Tallman EF, West JD, et al. Olfactory identification in subjective cognitive decline and mild cognitive impairment: association with tau but not amyloid positron emission tomography. Alzheimers Dement (Amst). 2017;9:57‐66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Palta P, Chen H, Deal JA, et al. Olfactory function and neurocognitive outcomes in old age: the atherosclerosis risk in communities neurocognitive study. Alzheimers Dement. 2018;14:1015‐1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hawkes CH, Del Tredici K, Braak H. Parkinson's disease: the dual hit theory revisited. Ann N Y Acad Sci. 2009;1170:615‐622. [DOI] [PubMed] [Google Scholar]
- 34. Ulrich J. Alzheimer changes in nondemented patients younger than sixty‐five: possible early stages of Alzheimer's disease and senile dementia of Alzheimer's type. Ann Neurol. 1985;17:273‐277. [DOI] [PubMed] [Google Scholar]
- 35. Braak H, Braak E. Neuropathological stageing of Alzheimer's‐related changes. Acta Neuropathol (Berl). 1991;82:239‐259. [DOI] [PubMed] [Google Scholar]
- 36. Jaunmuktane Z, Mead S, Ellis M, et al. Evidence for human transmission of amyloid‐beta pathology and cerebral amyloid angiopathy. Nature. 2015;525:247‐250. [DOI] [PubMed] [Google Scholar]
- 37. Purro SA, Farrow MA, Linehan J, et al. Transmission of amyloid‐beta protein pathology from cadaveric pituitary growth hormone. Nature. 2018;564:415‐449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Collinge J, Whitfield J, McKintosh E, et al. Kuru in the 21st century–an acquired human prion disease with very long incubation periods. Lancet. 2006;367:2068‐2074. [DOI] [PubMed] [Google Scholar]
- 39. Falcon B, Zivanov J, Zhang W, et al. Novel tau filament fold in chronic traumatic encephalopathy encloses hydrophobic molecules. Nature. 2019;568:420‐423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Fitzpatrick AWP, Falcon B, He S, et al. Cryo‐EM structures of tau filaments from Alzheimer's disease. Nature. 2017;547:185‐190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Falcon B, Zhang W, Murzin AG, et al. Structures of filaments from Pick's disease reveal a novel tau protein fold. Nature. 2018;561:137‐140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Vaquer‐Alicea J, Diamond MI, Joachimiak LA. Tau strains shape disease. Acta neuropathologica. 2021;142:57‐71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Zhang W, Tarutani A, Newell KL, et al. Novel tau filament fold in corticobasal degeneration, a four‐repeat tauopathy. BioRxiv. 2019:811703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Ashe KH. The biogenesis and biology of amyloid beta oligomers in the brain. Alzheimers Dement. 2020;16:1561‐1567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Martinisi A, Flach M, Sprenger F, Frank S, Tolnay M, Winkler DT. Severe oligomeric tau toxicity can be reversed without long‐term sequelae. Brain. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lo CH, Lim CK‐W, Ding Z, et al. Targeting the ensemble of heterogeneous tau oligomers in cells: a novel small molecule screening platform for tauopathies. Alzheimer's Dement. 2019;15:1489‐1502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Christensen LFB, Alijanvand SH, Burdukiewicz M, et al. Identification of amyloidogenic proteins in the microbiomes of a rat Parkinson's disease model and wild‐type rats. Protein Sci. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Eisele YS, Bolmont T, Heikenwalder M, et al. Induction of cerebral beta‐amyloidosis: intracerebral versus systemic Abeta inoculation. Proc Natl Acad Sci U S A. 2009;106:12926‐12931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Halfmann R, Jarosz DF, Jones SK, Chang A, Lancaster AK, Lindquist S. Prions are a common mechanism for phenotypic inheritance in wild yeasts. Nature. 2012;482:363‐338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kelly AC, Busby B, Wickner RB. Effect of domestication on the spread of the [PIN+] prion in Saccharomyces cerevisiae. Genetics. 2014;197:1007‐1024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Finch CE, Kulminski AM. The Alzheimer's disease exposome. Alzheimers Dement. 2019;15:1123‐1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Cuillé JC, Chelle PL. La maladie dite tremblante du mouton est‐elle inoculable? Comptes Rendus de l’ Académie des sciences (Paris). 1936;203:1552‐1554. [Google Scholar]
- 53. Bolton DC, McKinley MP, Prusiner SB. Identification of a protein that purifies with the scrapie prion. Science. 1982;218:1309‐1311. [DOI] [PubMed] [Google Scholar]
- 54. Meyer‐Luehmann M, Coomaraswamy J, Bolmont T, et al. Exogenous induction of cerebral beta‐amyloidogenesis is governed by agent and host. Science. 2006;313:1781‐1784. [DOI] [PubMed] [Google Scholar]
- 55. Kane MD, Lipinski WJ, Callahan MJ, et al. Evidence for seeding of beta ‐amyloid by intracerebral infusion of Alzheimer's brain extracts in beta ‐amyloid precursor protein‐transgenic mice. J Neurosci. 2000;20:3606‐3611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Gorkovskiy A, Thurber KR, Tycko R, Wickner RB. Locating folds of the in‐register parallel beta‐sheet of the Sup35p prion domain infectious amyloid. Proc Natl Acad Sci U S A. 2014;111:E4615‐E4622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Derkatch IL, Bradley ME, Hong JY, Liebman SW. Prions affect the appearance of other prions: the story of [PIN(+)]. Cell. 2001;106:171‐182. [DOI] [PubMed] [Google Scholar]
- 58. Lundmark K, Westermark GT, Olsen A, Westermark P. Protein fibrils in nature can enhance amyloid protein A amyloidosis in mice: cross‐seeding as a disease mechanism. Proc Natl Acad Sci U S A. 2005;102:6098‐6102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Allen B, Ingram E, Takao M, et al. Abundant tau filaments and nonapoptotic neurodegeneration in transgenic mice expressing human P301S tau protein. J Neurosci. 2002;22:9340‐9351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Ahmed Z, Cooper J, Murray TK, et al. A novel in vivo model of tau propagation with rapid and progressive neurofibrillary tangle pathology: the pattern of spread is determined by connectivity, not proximity. Acta Neuropathol. 2014;127:667‐683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Gorkovskiy A, Thurber KR, Tycko R, Wickner RB. Locating folds of the in‐register parallel β‐sheet of the Sup35p prion domain infectious amyloid. Proc Natl Acad Sci. 2014;111:E4615‐E4622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Froger A, Hall JE. Transformation of plasmid DNA into E. coli using the heat shock method. J Vis Exp. 2007:253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Bousset L, Luckgei N, Kabani M, et al. Prion amyloid polymorphs—the tag might change it all. Front Mole Biosci. 2020;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Shewmaker F, Wickner RB, Tycko R. Amyloid of the prion domain of Sup35p has an in‐register parallel beta‐sheet structure. Proc Natl Acad Sci U S A. 2006;103:19754‐19759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Goedert M, Spillantini MG, Cairns NJ, Crowther RA. Tau proteins of Alzheimer's paired helical filaments: abnormal phosphorylation of all six brain isoforms. Neuron. 1992;8:159‐168. [DOI] [PubMed] [Google Scholar]
- 66. Gallyas F. Silver staining of Alzheimer's neurofibrillary changes by means of physical development. Acta Morphol Acad Sci Hung. 1971;19:1‐8. [PubMed] [Google Scholar]
- 67. Ozcelik S, Sprenger F, Skachokova Z, et al. Co‐expression of truncated and full‐length tau induces severe neurotoxicity. Mol Psychiatry. 2016;21:1790‐1798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Franklin KBJ, Paxinos G. The Mouse Brain in Stereotaxic Coordinates. 3rd ed. Academic Press Elsevier; 2007. [Google Scholar]
- 69. Sparrer HE, Santoso A, Szoka FC Jr, Weissman JS. Evidence for the prion hypothesis: induction of the yeast [PSI+] factor by in vitro‐ converted Sup35 protein. Science. 2000;289:595‐599. [DOI] [PubMed] [Google Scholar]
- 70. Glover JR, Kowal AS, Schirmer EC, Patino MM, Liu JJ, Lindquist S. Self‐seeded fibers formed by Sup35, the protein determinant of [PSI+], a heritable prion‐like factor of S. cerevisiae. Cell. 1997;89:811‐819. [DOI] [PubMed] [Google Scholar]
- 71. Gonzalez Nelson AC, Paul KR, Petri M, et al. Increasing prion propensity by hydrophobic insertion. PLoS One. 2014;9:e89286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Frost B, Ollesch J, Wille H, Diamond MI. Conformational diversity of wild‐type tau fibrils specified by templated conformation change. J Biol Chem. 2009;284:3546‐3551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Ross ED, Edskes HK, Terry MJ, Wickner RB. Primary sequence independence for prion formation. Proc Natl Acad Sci U S A. 2005;102:12825‐12830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Boluda S, Iba M, Zhang B, Raible KM, Lee VM, Trojanowski JQ. Differential induction and spread of tau pathology in young PS19 tau transgenic mice following intracerebral injections of pathological tau from Alzheimer's disease or corticobasal degeneration brains. Acta Neuropathol. 2015;129:221‐237. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.