

Abstract
Homeostasis in the blood system is maintained by the balance between self‐renewing stem cells and nonstem cells. To promote self‐renewal, transcriptional regulators maintain epigenetic information during multiple rounds of cell division. Mutations in such transcriptional regulators cause aberrant self‐renewal, leading to leukemia. MOZ, a histone acetyltransferase, and MLL, a histone methyltransferase, are transcriptional regulators that promote the self‐renewal of hematopoietic stem cells. Gene rearrangements of MOZ and MLL generate chimeric genes encoding fusion proteins that function as constitutively active forms. These MOZ and MLL fusion proteins constitutively activate transcription of their target genes and cause aberrant self‐renewal in committed hematopoietic progenitors, which normally do not self‐renew. Recent progress in the field suggests that MOZ and MLL are part of a transcriptional activation system that activates the transcription of genes with nonmethylated CpG‐rich promoters. The nonmethylated state of CpGs is normally maintained during cell divisions from the mother cell to the daughter cells. Thus, the MOZ/MLL‐mediated transcriptional activation system replicates the expression profile of mother cells in daughter cells by activating the transcription of genes previously transcribed in the mother cell. This review summarizes the functions of the components of the MOZ/MLL‐mediated transcriptional activation system and their roles in the promotion of self‐renewal.
Keywords: histone acetyltransferase, histone methyltransferase, leukemia, MLL, MORF, MOZ, protein complex, self‐renewal, transcription, unmethylated CpG
MOZ, a histone acetyltransferase, and MLL, a histone methyltransferase, are transcriptional regulators that promote the self‐renewal of hematopoietic stem cells. They activate the transcription of genes with nonmethylated CpG‐rich promoters via the AF4/ENL/P‐TEFb complex to replicate the expression profile of the mother cell in the daughter cells. Constitutively active mutants of MOZ and MLL confer self‐renewing ability to committed hematopoietic progenitors that normally do not self‐renew, causing refractory leukemia.

Abbreviations
- AEP
AF4 family/ENL family/P‐TEFb
- BRPF1
bromodomain‐PHD finger protein 1
- CMP
common myeloid progenitors
- DPF
double PHD finger
- EBD
ENL‐binding domain
- GMP
granulocyte/macrophage progenitors
- H15
histones H1‐ and H5‐like domain
- HMT
histone methyltransferase
- HSC
hematopoietic stem cells
- MBM
MENIN‐binding motif
- MEAF6
MYST/Esa1‐associated factor 6
- MLL‐r
MLL‐rearranged
- MOZ
monocytic leukemia zinc finger
- MPP
multipotent progenitors
- PRC1
polycomb repressive complex 1
- PZP
PHD fingers linked by a zinc knuckle
- RBM
RNA polymerase II‐binding motif
- RNAP2 non‐P
RNAP2 with nonphosphorylated C‐terminal heptapeptide motifs
- RNAP2 Ser5‐P
RNAP2 with Ser 5‐phosphorylated C‐terminal heptapeptide motifs
- TBP
TATA‐binding protein
Introduction
Multicellular organisms achieve cellular homeostasis by maintaining the balance between the self‐renewing proliferation of stem cells and the non‐self‐renewing proliferation of nonstem cells. During development when the cell population is rapidly expanding, these two types of proliferation appear to be strictly controlled. The maintenance of epigenetic information likely plays a critical role in self‐renewal. Epigenetic information is mainly transmitted by chemical modifications of DNAs and histones. For example, the nonmethylated state of CpGs in CpG islands is maintained during cell divisions and triggers gene expression in daughter cells after each cell division to replicate the expression profile of the mother cells. The transcription event transmits epigenetic information such as the trimethylation of histone H3 lysine 36 (H3K36me3), which is recognized by transcriptional regulators in every interphase of the cell cycle. By reading and acting on epigenetic information, cells replicate the expression of genes similar to that in the mother cell, thereby promoting self‐renewal. Mutations in the transcriptional regulators of this transcriptional activation system cause aberrant self‐renewal in nonstem cells, thereby leading to cancer.
The mixed lineage leukemia (MLL) gene (also known as ALL‐1, KMT2A, HRX, HTRX, or MLL1) encodes a transcriptional maintenance factor for Hox gene expression during development. MLL is a histone methyltransferase (HMT) that induces the methylation of histone H3 lysine 4 (H3K4) via its SET domain. MLL rearrangements that occur via chromosomal translocations cause aggressive leukemia (Fig. 1) [1, 2, 3], accounting for approximately 5–10% of all acute leukemia cases and most infant acute leukemia cases [4]. In addition, the clinical outcomes for MLL‐rearranged (MLL‐r) leukemia patients are typically unfavorable [5]. Therefore, the development of better therapeutic strategies is urgently needed.
Fig. 1.
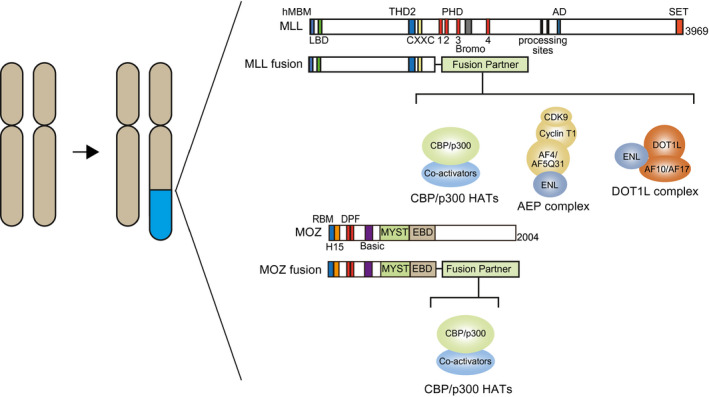
Gene rearrangements of MLL and MOZ cause leukemia. Gene rearrangements via chromosomal translocations generate chimeric genes encoding MLL and MOZ fusion proteins. MLL fuses with CBP/p300 HAT‐containing complexes and ENL‐containing complexes such as the AEP and DOT1L complexes (e.g., MLL‐CBP, MLL‐AF4, and MLL‐AF10). MOZ fuses CBP/p300 HAT‐containing complexes (e.g., MOZ‐CBP and MOZ‐TIF2). Structures of MLL and MOZ fusion proteins are shown. hMBM, high‐affinity Menin‐binding motif; LBD, LEDGF‐binding domain; THD2, trithorax homology domain 2; CXXC, CXXC domain; PHD, plant homeodomain; Bromo, bromodomain; AD, activation domain; SET, SET HMT domain; RBM, RNA polymerase II‐binding motif; H15, histone H1/5‐like domain; MYST, MYST HAT domain; Basic, basic domain; EBD, ENL‐binding domain.
MLL gene rearrangements generate chimeric genes encoding MLL fusion proteins, which function as constitutively active transcriptional machinery that causes the sustained expression of genes that are normally expressed in immature progenitors such as hematopoietic stem cells (HSCs). To date, more than 80 MLL fusion partners have been identified, including the components of the AF4 family/ENL family/P‐TEFb (AEP), DOT1L, and CBP/p300 histone acetyltransferase (HAT) complexes (Fig. 1) [4]. Similarly, gene rearrangements of the monocytic leukemia zinc finger (MOZ) gene (also known as MYST3 or KAT6A) generate chimeric genes encoding the fusion proteins of MOZ and the CBP/p300 HAT complex components causing aggressive leukemia [6]. MLL and MOZ have recently been revealed as components of a common transcriptional activation system that promotes self‐renewal [7]. This review focuses on the functional cooperation between MOZ and MLL in the development of leukemia.
MOZ is a HAT required for development and hematopoiesis
MOZ belongs to the MYST HAT family that acetylates the histones—H3, H4, H2A, and H2B in vitro [8, 9, 10, 11]. Particularly, the MOZ HAT acetylates lysine 14 of histone H3 (H3K14) and lysine residues 5, 8, 12, and 16 of histone H4 (H4K5/8/12/16) in vitro [12, 13]. During embryogenesis, the acetylation of lysine 9 of histone H3 (H3K9) was remarkably reduced in Moz‐knockout embryos [14], indicating that MOZ either directly acetylates H3K9 or indirectly influences its acetylation in vivo. In addition, MOZ is critically required for embryogenesis as evidenced by in utero death of homozygous knockout mice with severe hematopoietic defects [15, 16]. MOZ plays essential roles in the activation and maintenance of Hox expression by counteracting the polycomb repressive complex 1 (PRC1) to confer segmental identity [14, 17]. Hox genes, such as Hoxa9, are necessary for the repopulating ability of HSCs [18, 19, 20]. MOZ maintains Hoxa9 expression during hematopoiesis to support the expansion of hematopoietic progenitors [16, 21].
MOZ forms a core complex with BRPF1, ING5, and MEAF6 to target specific chromatin
MOZ and its homolog MORF (also known as MYST4 or KAT6B) form a biochemically stable complex with three other core components, including bromodomain‐PHD finger protein 1 (BRPF1), MYST/Esa1‐associated factor 6 (MEAF6), and inhibitor of growth 5 (ING5) (Fig. 2A) [11, 13]. MOZ/MORF proteins contain an RNA polymerase II‐binding motif (RBM), histones H1‐ and H5‐like domain (H15), double PHD finger (DPF), basic domain, catalytic MYST HAT domain, and an ENL‐binding domain (EBD) (Fig. 2A). MOZ/MORF complexes contain various chromatin reader modules that facilitate their association with particular chromatin [22]. The DPF of MOZ specifically recognizes acetylated H3K14 (H3K14ac) and unmodified arginine 2 of histone H3 (H3R2un) via its first and second PHD fingers (PHD1 and PHD2), respectively (Fig. 2A) [23]. PHD1 recognizes a range of different acylation modifications, including crotonylation, butyrylation, and propionylation, among which it has the highest affinity for crotonylation [24, 25]. In the case of MORF, DPF‐mediated binding to H3K14ac further promotes the acetylation of lysine 23 of histone H3 (H3K23ac) [26].
Fig. 2.
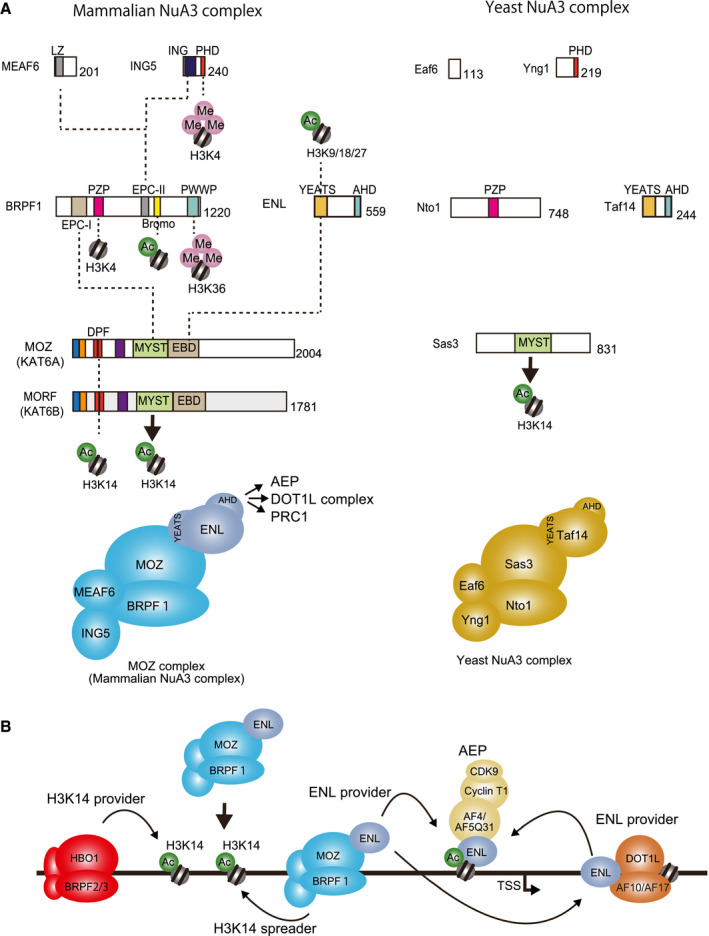
Structures and functions of the MOZ complex. (A) Structures and functions of the human MOZ and yeast NuA3 complexes. Domain structures are common between the mammalian NuA3 complex (MOZ complex) and the yeast NuA3 complex. Interactions are indicated by dotted lines. LZ, leucine zipper; ING, ING N‐terminal domain; EPC‐I/II, enhancer of polycomb (EPC)‐like domain I/II; PZP, PHD‐zinc knuckle‐PHD module; PWWP, PWWP domain; YEATS, YEATS domain; AHD, ANC1 homology domain. (B) Spreading of H3K14ac marks and loading of ENL onto the chromatin by the MOZ complex. The MOZ and HBO1 complexes provide and spread H3K14ac marks. The MOZ and DOT1L complexes provide ENL to establish AEP on chromatin.
In addition to its HAT activity, the MYST HAT domain also has DNA‐binding ability [27]. By having both the reader and writer modules for the acylation of H3K14, MOZ/MORF complexes likely spread H3K14ac marks from the founder H3K14ac mark, which is presumably introduced by another MYST HAT termed HBO1 (also known as KAT7) (Fig. 2B) [28, 29, 30]. MOZ/MORF proteins associate with BRPF1 through the MYST HAT domain (Fig. 2A) [7, 11]. BRPF1 is required for the transcriptional maintenance of Hox genes during development [10] and serves as a scaffold that tethers each MOZ/MORF complex component to its target chromatin. BRPF1 binds to MOZ/MORF through the N‐terminal portion of its EPC homology domain (EPC‐I) and to ING5/MEAF6 through its C‐terminal portion (EPC‐II) [10, 11, 31]. A module composed of two PHD fingers linked by a zinc knuckle [PZP (PHD‐Zn knuckle‐PHD) domain] associates with DNA and unmethylated histone H3 lysine 4 [31, 32] and is required for the efficient acetylation of nucleosomes by MOZ. The bromodomain of BRPF1 binds to acetylated histones [10]. The PWWP domain preferentially associates with histones H2A/B over histones H3/4 and specifically recognizes the di/trimethylated histone H3 lysine 36 (H3K36me2/3) [10, 33]. BRPF1 is retained on the metaphase chromatin via its PWWP domain, whereas MOZ is dissociated [10], suggesting that it plays important roles in the marking of the target chromatin of MOZ/MORF complexes for transcriptional reactivation in the next G1 phase.
BRPF family proteins also associate with HBO1 [30, 31, 34]. Notably, BRPF2/3 preferentially associates with HBO1, whereas BRPF1 associates with MOZ/MORF, indicating the nonredundant roles of the BRPF family members [30, 34]. HBO1 acetylates histone H3 when associated with BRPF family proteins. However, when associated with the structurally similar JADE family proteins, HBO1 acetylates histone H4. Thus, BRPF/JADE family proteins play a decisive role in the selection of target histone tails by the MYST HATs [31]. Genes of the BRPF family are required for proper skeletal and hematopoietic development as they maintain segment‐specific Hox expression [10, 30, 35, 36, 37] in a manner similar to MOZ [14, 15, 16].
ING5 is a chromatin reader protein containing a PHD finger and specifically associates with di/trimethylated histone H3 lysine 4 (H3K4me2/3) to promote histone acetylation [13, 31, 38]. MEAF6 is the smallest subunit of the MOZ/MORF complexes and is shared by the HBO1 complex [13]. In summary, MOZ/MORF complexes are formed on BRPF1 to recruit ING5 and MEAF6. Each subunit contains unique chromatin reader modules that render association with particular target chromatin segments to spread acetylation marks.
MOZ complex associates with ENL and RNAP2, and MLL
ENL (also known as MLLT1) is a frequent fusion partner of MLL [1]. Our group recently investigated the factors that interact with ENL on chromatin by using affinity purification from the ENL‐bound chromatin fractions of HEK293T cells transiently expressing ENL [7, 39]. This method allowed the copurification of ENL‐associated factors in their chromatin‐bound form and identified MOZ/MORF complex components as ENL binders, along with known ENL‐associated factors (e.g., AEP and DOT1L complexes) (Fig. 2B) [7]. Domain mapping analysis indicated that ENL binds to MOZ through its YEATS domain, which is known to specifically bind to acetylated histone H3 lysine 9/18/27 (H3K9/18/27ac) [40, 41, 42, 43]. Presumably, the MOZ complex recruits ENL to the target chromatin and functions as an ENL provider to the DOT1L complex, which subsequently loads the ENL protein onto chromatin to build an AEP complex (Fig. 2B). The MOZ domain responsible for ENL association was mapped to the ENL‐binding domain (EBD) [7]. The yeast NuA3 complex, presumably the yeast counterpart of MOZ/MORF complexes, contains a YEATS domain‐containing component (i.e., Taf14) [11, 44], which had been lacking in previously characterized mammalian MOZ/MORF complexes. Thus, we can conclude that ENL is the missing component that completes the human NuA3 complex nucleated by MOZ/MORF (Fig. 2A). Further studies will provide insight into the details of the involvement of ENL in the NuA3 complex.
Furthermore, the MOZ complex associates with RNA polymerase II (RNAP2). The MOZ complex specifically binds to the promoters of MYC and HOXA9 in HEK293T cells. Domain mapping analysis of the structure responsible for association with these promoters revealed that 84 residues located in the N‐terminal region are the major determinant for target recognition. Subsequent proteomic analysis revealed that this structure specifically associated with the RNAP2 complex and was thus named the RNAP2‐binding motif (RBM) (Fig. 3A). Interestingly, RNAP2 with nonphosphorylated C‐terminal heptapeptide motifs (RNAP2 non‐P) specifically bound to the RBM of MOZ, whereas its Ser 5‐phosphorylated form (RNAP2 Ser5‐P) did not, indicating that MOZ associated with RNAP2 whose transcription was not yet initiated [7]. Overall, these findings indicate that MOZ targets the RNAP2 complex in the early phase of transcription.
Fig. 3.
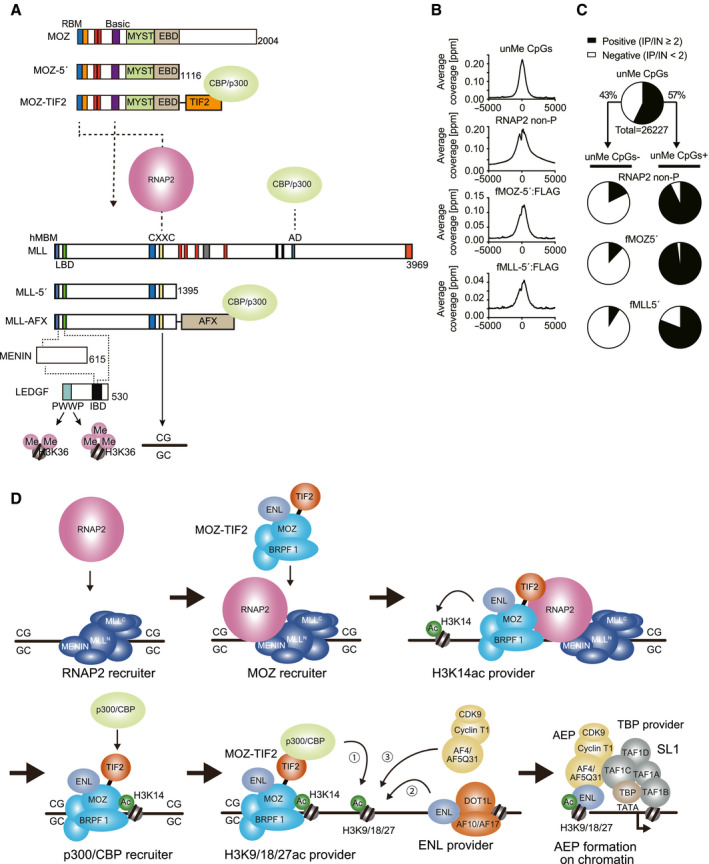
Mechanism of MOZ‐TIF2‐mediated leukemic transformation. (A) Structures and functions of MOZ and MLL proteins. MOZ‐TIF2 and MLL‐AFX associate with CBP/p300 HATs. MOZ fusion and MLL fusion complexes target similar chromatin where unmethylated CpGs and H3K36me2/3 marks are enriched. IBD, integrase‐binding domain; RNAP2, RNA polymerase II. (B) Average distribution of MOZ‐5′ and MLL‐5′ proteins at transcription start sites (TSSs). ChIP‐seq analysis of HEK293T cells transiently expressing FLAG‐tagged MOZ‐5′ and MLL‐5′ revealed their localization at CpG‐rich promoters with RNA polymerase II with nonphosphorylated C‐terminal heptapeptide motifs (RNAP2 non‐P). (C) Colocalization of MOZ and MLL proteins with RNAP2 non‐P at CpG‐rich promoters in HEK293T cells. Most unmethylated CpG‐rich promoters (black) are also positive for the ChIP signals of MOZ‐5′, MLL‐5′ and RNAP2 non‐P. (D) Mechanism of MOZ‐TIF2‐mediated gene activation. The MLL complex recruits RNA polymerase II to CpG‐rich promoters where the MOZ‐TIF2 complex is recruited to spread H3K14ac marks. The MOZ‐TIF2 complex recruits CBP/p300 HATs to produce H3K9/18/27ac marks on which an AEP complex is built. The AEP complex resultantly activates transcription via SL1 and P‐TEFb.
MOZ associates with the MLL complex via its basic domain (Fig. 3A) [7, 21]. MLL knockout in HEK293T cells resulted in the reduced presence of RNAP2 and MOZ at the promoters of MYC and CDKN2C [7], suggesting that MLL promotes the recruitment of RNAP2 and MOZ. MLL binds to the promoter proximal regions by recognizing nonmethylated CpGs via its CXXC domain [45, 46, 47, 48]. The CXXC domain also mediates the recruitment of RNAP2 non‐P [7]. Consequently, MOZ is colocalized with MLL and RNAP2 non‐P at CpG‐rich promoters in a genome‐wide manner (Fig. 3B,C).
Notably, MOZ associates with various sequence‐specific transcription factors (TFs), such as AML1 (also known as RUNX1), PU.1 (also known as SPI1), and the tumor suppressor TP53 [12, 16, 49, 50, 51]. MLL also associates with PU.1 and C/EBPα [52]. PU.1 recruits MOZ and MLL proteins to upregulate the expression of CSF1R (also known as FMS or M‐CSFR), which plays a critical role in leukemogenesis [52, 53]. Thus, MOZ and MLL likely target a unique chromatin via these sequence‐specific TFs, in addition to CpG‐rich promoters.
MOZ‐TIF2 activates MLL/AEP‐mediated transcriptional activation system
Chromosomal translocations generate chimeric genes encoding fusion proteins of MOZ/MORF and CBP/p300 HATs, inducing acute myeloid leukemia (Fig. 1) [8, 54, 55, 56, 57]. MOZ also fuses with TIF2 (also known as NCOA2) [58, 59], which in turn associates with CBP/p300 HATs (Fig. 3A) [60]. Thus, CBP/p300 or its associated proteins are the preferred fusion partners of MOZ.
MLL also fuses with CBP/p300 HATs [61, 62] and its associated factors (e.g., AFX) (Figs 1B and 3A) [63]. The wild‐type MLL protein interacts with CBP/p300 HATs via its activation domain (AD). Association with CBP/p300 HATs is presumably promoted by the copresence of sequence‐specific TFs such as MYB and CREB [64, 65, 66, 67]. Thus, the constitutive recruitment of CBP/p300 HATs to MOZ/MLL target promoters is a common mechanism during oncogenic transformation.
The structures required for leukemic transformation by MOZ fusions are mostly studied on MOZ‐TIF2 [7, 60, 68, 69], using ex vivo myeloid progenitor transformation assays [70, 71]. The association of MOZ‐TIF2 with CBP/p300 via its TIF2 portion is critical for leukemic transformation [60]. The HAT activity of the MYST domain as requirement for leukemogenesis was first questioned [60] but was later confirmed by extensive mutant analyses [68]. The MYST domain contains multiple binding surfaces for BRPF1, the association with which is critical for leukemogenesis [7, 68]. However, the DPF of MOZ was dispensable for leukemic transformation [7, 60, 69]. The RBM was critically required for leukemogenesis [7, 69] and was shown to be responsible for the association with RNAP2 [7] and PRMT1 [69]. Taken together, these results indicate that the MOZ‐TIF2 complex transforms hematopoietic progenitors through the functions of the RBM and MYST domains of MOZ and the CBP/p300‐binding domain of TIF2 (Fig. 3D). The promoter targeting ability is mainly conferred by the RBM, whereas the MYST domain likely reinforces chromatin association mediated by multiple chromatin reader modules [7].
Recruited by the TIF2 portion, CBP/p300 HATs induce the acetylation of histone H3K9/18/27 (Fig. 3D) [72, 73, 74]. The ENL family proteins (i.e., ENL and AF9) specifically bind to acetylated histone H3 lysine 9/18/27 marks [40, 41, 43] and further recruit multiple functionally distinct transcriptional regulators, including AEP, the DOT1L complex, and PRC1 through their ANC1 homology domain (AHD) (Fig. 2A) [39, 75, 76]. AEP is a transcriptional coactivator as it initiates transcription through the SL1 complex, presumably by loading the TATA‐binding protein (TBP) to the TATA element of the promoters [67]. The acetylation of histone H3 lysine 9/18/27 near the target promoters induces the recruitment of AEP and the subsequent transcriptional activation [7]. Complexes similar to AEP have been purified and characterized as transcription elongation factors and are referred to as super elongation complex [77, 78, 79]. Thus, AEP is a transcriptional activator that promotes both transcription initiation and elongation. AEP components are the most frequent fusion partners of MLL found in leukemia patients (Fig. 1) [4]. The majority of MLL fusions induce oncogenic transformation by constitutively recruiting AEP to the MLL target chromatin [80]. As the MOZ and MLL portions retained in leukemic fusion proteins (i.e., MOZ‐5′ and MLL‐5′, respectively) target active CpG‐rich promoters (Fig. 3B,C), MOZ and MLL fusions are speculated to target the same promoters. Accordingly, the partner‐swap mutants of MOZ and MLL fusions, such as MOZ‐ENL and MOZ‐AFX, activate Hoxa9 expression and transform hematopoietic progenitors [7]. Thus, MOZ and MLL fusions participate in a common mechanism to transform hematopoietic progenitors by recruiting AEP to CpG‐rich promoters and by causing aberrant and constitutive gene expression in leukemia cells.
MOZ and MLL fusions constitutively activate previously transcribed CpG‐rich promoters
MLL fusion forms a complex with MENIN through its MENIN‐binding motif (MBM) (Fig. 3A) [81, 82]. The MLL/MENIN complex further associates with LEDGF through their LEDGF‐binding domain (LBD) [83]. LEDGF has a PWWP domain that binds to the di/trimethylated histone H3 lysine 36 [84, 85]. H3K36me3 marks are deposited on the chromatin of transcribed regions by SETD2 HMT in a transcription‐coupled manner [86]. Hence, LEDGF preferentially binds to the transcriptionally active chromatin.
The minimum module required for the recognition of MLL target chromatin is constituted by the PWWP and CXXC domains [48]. The PWWP domain of LEDGF could be functionally replaced by that of BRPF1. An artificial fusion construct comprising the PWWP, CXXC, and AHD domains functioned as an oncogenic transcriptional machinery and induced leukemia in mouse models. Because the PWWP and CXXC domains are sufficient for the stable association with the MLL target promoters, the target chromatin of the MLL fusion complex is a broad range of previously transcribed CpG‐rich promoters [48]. MENIN is required for the transcriptional activation of genes regulated by wild‐type MLL [82, 87, 88]. MOZ proteins (i.e., wild‐type MOZ and MOZ fusions) target promoters bound by the wild‐type MLL/MENIN complex and RNAP2 [7]. Thus, both MOZ fusions and MLL fusions target previously transcribed CpG‐rich promoters and constitutively activate transcription via AEP (Fig. 3D).
MOZ and MLL fusion proteins confer self‐renewing ability to committed hematopoietic progenitors
Homeostasis of the hematopoietic system is achieved hierarchically, where HSCs are at the top of the hierarchy. Because only HSCs can self‐renew, the size of the hematopoietic system is strictly controlled. In fact, in hematopoietic reconstitution experiments wherein hematopoietic cells are transplanted into lethally irradiated mice, one HSC could reconstitute the entire hematopoietic system in the recipient mice; however, repopulation could not be achieved even with 50 multipotent progenitors (MPPs) (Fig. 4A) [89]. This result indicates that non‐HSC hematopoietic progenitors irreversibly lose a fraction of their identity on every cell division, presumably due to the incomplete replication of epigenetic information. Therefore, non‐HSC hematopoietic progenitors quickly lose the identity of immature progenitors, as they proliferate. In this way, non‐HSC hematopoietic progenitors are programmed to differentiate or senesce.
Fig. 4.
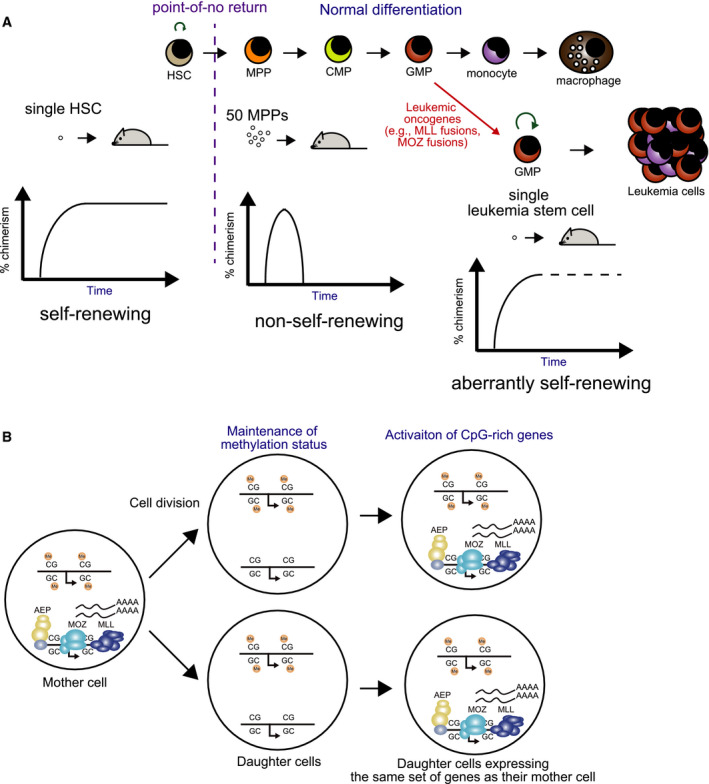
Mechanisms of self‐renewal. (A) Self‐renewal in normal hematopoiesis and leukemogenesis. One HSC can reconstitute the hematopoietic system, whereas 50 MPPs cannot achieve the same repopulation. The transition from HSC to MPP is a point‐of‐no return where cells lose their self‐renewing ability. Leukemic oncogenes such as MLL fusions and MOZ fusions confer self‐renewing ability to committed progenitors and induce leukemia. HSC, hematopoietic stem cell; MPP, multipotent progenitor; CMP, common myeloid progenitor; GMP, granulocyte/macrophage progenitor. (B) Self‐renewal mechanism mediated by the MLL/MOZ/AEP‐mediated transcriptional activation system. After each cell division, the MLL‐MOZ and AEP complexes activate CpG‐rich promoters that were previously transcribed in the mother cell to promote self‐renewal.
MOZ and MLL fusions overcome this ‘programmed differentiation’ by conferring self‐renewing ability to non‐HSC hematopoietic progenitors [90, 91, 92]. Consequently, committed progenitors, such as common myeloid progenitors (CMPs) and granulocyte/macrophage progenitors (GMPs), transduced with MOZ or MLL fusion genes indefinitely expand and induce full‐blown leukemia in mouse models [60, 70]. MOZ and MLL fusion proteins promote self‐renewal by reactivating previously transcribed CpG‐rich promoters [7]. Transcriptional activation deposits active epigenetic marks (i.e., H3K36me3), thereby leading to further activation by MOZ and MLL fusion proteins in the next G1 phase (Fig. 4B). By this process, committed progenitors, which normally do not self‐renew, acquire the ability to self‐renew and result in leukemia.
Drugs targeting the components of the MLL/MOZ/AEP‐mediated transcriptional activation system can be used for both MLL‐ and MOZ‐rearranged leukemias
A transcriptional activation system mediated by AEP, MOZ, and MLL promotes self‐renewal by conferring an expression profile similar to that of the mother cell to the daughter cells (Fig. 5) [39, 48, 93]. The mechanism of the MLL/MOZ/AEP‐mediated transcriptional activation system is as follows: First, the MLL complex binds to previously active CpG‐rich promoters. Second, it recruits RNAP2, to which the MOZ complex binds and spreads H3K14ac marks on the nearby chromatin. The MLL complex subsequently recruits CBP/p300 HATs to induce H3K9/18/27ac marks. ENL associates with H3K9/18/27ac marks to recruit AEP. Subsequently, AEP activates transcription initiation via the SL1 complex and promotes transcription elongation via P‐TEFb. Transcription triggers SETD2‐mediated H3K36 methylation and DOT1L‐mediated H3K79 methylation to promote the promoter re‐entry of the MLL complex [48] and inhibit SIRT1‐mediated transcriptional repression [94], respectively. Some inhibitors for the components of this transcriptional activation system are currently being developed with promising results [95, 96, 97, 98, 99]. Inhibitors targeting any components of the MLL/MOZ/AEP‐mediated transcriptional activation system have the potential to be novel therapeutic strategies for not only MLL‐r leukemias but also other non‐MLL‐r leukemias such as MOZ‐rearranged leukemias.
Fig. 5.
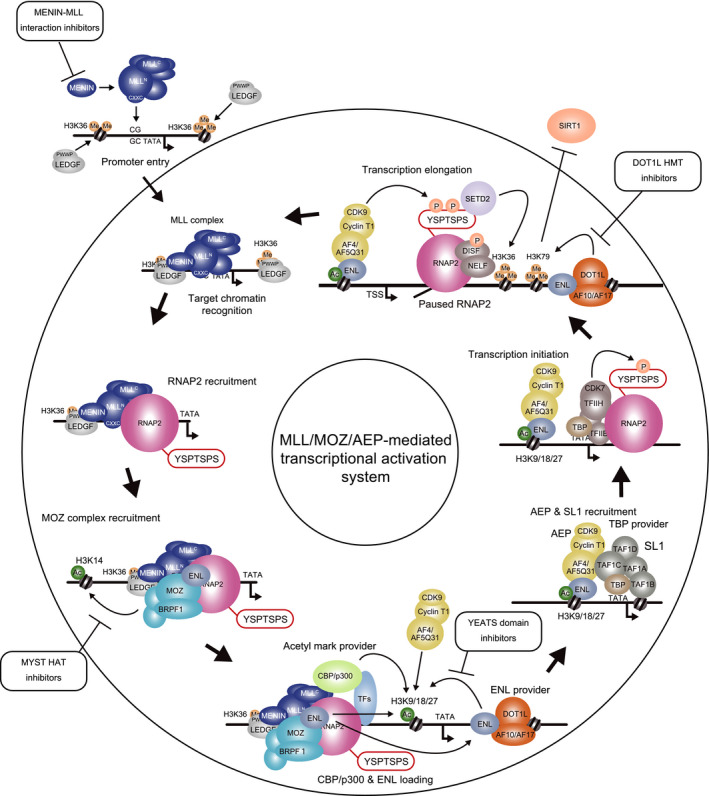
Summary of the MLL/MOZ/AEP‐mediated transcriptional activation system. MLL forms a complex with MENIN to stably associate with LEDGF on CpG‐rich promoters and recruit RNA polymerase II (RNAP2) thereto. The MOZ complex targets the same promoters via association with MLL and RNAP2 to spread H3K14ac marks nearby. MLL and TFs such as MYB recruit CBP/p300 HATs to make the H3K9/18/27ac marks, on which ENL is recruited by the DOT1L complex and an AEP complex is assembled. Leukemic oncogenes generated by the mutations of MLL and MOZ constitutively activate this step. The AEP complex activates transcription via SL1 and P‐TEFb. Transcribing RNAP2 provides H3K39me3 marks via SETD2 HMT, which further promotes LEDGF recruitment. The DOT1L complex provides the H3K79 methylation mark, which counteracts with transcriptional repressors such as SIRT1. YSPTSPS: The heptapeptide repeat in the C‐terminal domain of RNAP2.
MENIN‐MLL interaction inhibitors
Because MLL fusion proteins bind to MENIN forming a stable complex on the target chromatin [83], the inhibition of the interaction between MLL and MENIN would specifically attenuate the oncogenic property of the MLL fusion protein [81]. In MLL/MENIN complexes, the MENIN‐binding motif (MBM) of MLL specifically binds to a pocket‐like structure of MENIN [81, 100]. Grembecka et al. developed specific MENIN‐MLL interaction inhibitors that fit into this pocket [95, 96, 97, 101]. Krivtsov et al. [98] developed another type of MENIN‐MLL interaction inhibitor that demonstrated high levels of efficacy in preclinical models.
The genetic ablation of MENIN in non‐MLL‐r cells resulted in decreased HOX expression [82, 87, 88], indicating that MENIN‐MLL interaction is required for the function of the wild‐type MLL (Fig. 5). Because the MOZ‐TIF2 complex targets the chromatin in a wild‐type MLL‐dependent manner (Fig. 3D), MENIN‐MLL interaction inhibitors attenuate the oncogenic property of MOZ‐TIF2 [7]. MENIN‐MLL interaction inhibitors also exhibit antitumor effects on leukemia with NPM1 mutations [97, 102, 103]. These findings indicate that MENIN‐MLL interaction inhibitors have a therapeutic potential against non‐MLL‐r leukemia that is dependent on wild‐type MLL.
DOT1L HMT inhibitors
DOT1L functions as an ENL provider to establish AEP on the chromatin [39]. The methylation of lysine 79 of histone H3 by DOT1L inhibits transcriptional repressors, such as SIRT1, to maintain the self‐renewal property of leukemia stem cells (Fig. 5) [94]. Daigle et al. [99, 104] have developed DOT1L HMT inhibitors that effectively inhibited the continuous proliferation of MLL‐r leukemia cells in preclinical models. MLL fusion proteins constitutively recruit AEP in a MENIN‐dependent manner, which cooperatively activates transcription with DOT1L to promote self‐renewal [39]. Accordingly, the combination of DOT1L HMT and MENIN‐MLL interaction inhibitors has demonstrated synergistic antitumorigenic effects [39, 105].
MYST HAT inhibitors
HAT inhibitors for MYST HATs have been developed by Baell et al. [106] and shown to inhibit MYC‐induced lymphoma. HBO1 has been identified as a therapeutic vulnerability of leukemia stem cells by genetic screening [107, 108]. Thus, the inhibitors of these MYST HATs may have potentials as novel drugs for hematological malignancies dependent on the MLL/MOZ/AEP‐mediated transcriptional activation system.
YEATS domain inhibitors
Although it is still in the early phase of drug development, several compounds have been generated to inhibit the interaction between the YEATS domain of the ENL family proteins and acetylated histone H3 [109, 110, 111, 112, 113]. These compounds target the critical point of the MLL/MOZ/AEP‐mediated transcriptional activation system (Fig. 5) and are thus expected to be effective for the disease relying on this transcriptional activation system.
Conclusions
Cells acquire various biological abilities during cancer development, the majority of which are considered as hallmarks of cancer [114], including sustained proliferative signaling and resistance to cell death. However, some mutations in leukemia patients do not fall into the known hallmarks of cancer. Structural and functional analyses have revealed that the gene rearrangements of MLL and MOZ generate constitutively active transcriptional machinery that promotes self‐renewal [7, 48]. Consequently, non‐HSC hematopoietic progenitors acquire the ability to self‐renew and develop leukemia [90, 92].
A transcriptional activation system mediated by MLL, MOZ, and AEP replicates the active epigenetic/transcriptional status of CpG‐rich promoters to faithfully reactivate the promoters that were previously active in the mother cell (Figs 4B and 5). This system is presumed to be highly active in HSCs but is progressively suppressed during differentiation in normal hematopoietic cells (Fig. 4A). The aberrant activation of this transcriptional activation system results in efficient replication of epigenetic information, thereby causing aberrant self‐renewal. These results indicate that promoting self‐renewal is another hallmark of cancer. Several molecularly targeted drugs targeting the components of this transcriptional activation system have been developed (Fig. 5) and will hopefully provide therapeutic benefits to patients of these refractory cancers in near future, followed by the development of newer drugs that function with similar mechanisms.
Conflict of interest
AY received research funding from Dainippon Sumitomo Pharma Co. Ltd.
Author contributions
The author is the sole contributor to this work and has approved the final version of the manuscript for publication.
Acknowledgements
I thank all the laboratory members for critical reading of the manuscript. I also thank all the members of the Shonai Regional Industry Promotion Center for their administrative support. This work was supported by the Japan Society for the Promotion of Science KAKENHI grant (19H03694) and the Yamagata Prefectural Government of the City of Tsuruoka.
References
- 1. Tkachuk DC, Kohler S & Cleary ML (1992) Involvement of a homolog of Drosophila trithorax by 11q23 chromosomal translocations in acute leukemias. Cell 71, 691–700. [DOI] [PubMed] [Google Scholar]
- 2. Gu Y, Nakamura T, Alder H, Prasad R, Canaani O, Cimino G, Croce CM & Canaani E (1992) The t(4;11) chromosome translocation of human acute leukemias fuses the ALL‐1 gene, related to Drosophila trithorax, to the AF‐4 gene. Cell 71, 701–708. [DOI] [PubMed] [Google Scholar]
- 3. Djabali M, Selleri L, Parry P, Bower M, Young BD & Evans GA (1992) A trithorax‐like gene is interrupted by chromosome 11q23 translocations in acute leukaemias. Nat Genet 2, 113–118. [DOI] [PubMed] [Google Scholar]
- 4. Meyer C, Burmeister T, Groger D, Tsaur G, Fechina L, Renneville A, Sutton R, Venn NC, Emerenciano M, Pombo‐de‐Oliveira MS et al. (2018) The MLL recombinome of acute leukemias in 2017. Leukemia 32, 273–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Tamai H & Inokuchi K (2010) 11q23/MLL acute leukemia: update of clinical aspects. J Clin Exp Hematop 50, 91–98. [DOI] [PubMed] [Google Scholar]
- 6. Yang XJ & Ullah M (2007) MOZ and MORF, two large MYSTic HATs in normal and cancer stem cells. Oncogene 26, 5408–5419. [DOI] [PubMed] [Google Scholar]
- 7. Miyamoto R, Okuda H, Kanai A, Takahashi S, Kawamura T, Matsui H, Kitamura T, Kitabayashi I, Inaba T & Yokoyama A (2020) Activation of CpG‐rich promoters mediated by MLL drives MOZ‐rearranged leukemia. Cell Rep 32, 108200. [DOI] [PubMed] [Google Scholar]
- 8. Kitabayashi I, Aikawa Y, Yokoyama A, Hosoda F, Nagai M, Kakazu N, Abe T & Ohki M (2001) Fusion of MOZ and p300 histone acetyltransferases in acute monocytic leukemia with a t(8;22)(p11;q13) chromosome translocation. Leukemia 15, 89–94. [DOI] [PubMed] [Google Scholar]
- 9. Champagne N, Pelletier N & Yang XJ (2001) The monocytic leukemia zinc finger protein MOZ is a histone acetyltransferase. Oncogene 20, 404–409. [DOI] [PubMed] [Google Scholar]
- 10. Laue K, Daujat S, Crump JG, Plaster N, Roehl HH, Tubingen Screen C, Kimmel CB, Schneider R & Hammerschmidt M (2008) The multidomain protein Brpf1 binds histones and is required for Hox gene expression and segmental identity. Development 135, 1935–1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Ullah M, Pelletier N, Xiao L, Zhao SP, Wang K, Degerny C, Tahmasebi S, Cayrou C, Doyon Y, Goh SL et al. (2008) Molecular architecture of quartet MOZ/MORF histone acetyltransferase complexes. Mol Cell Biol 28, 6828–6843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kitabayashi I, Aikawa Y, Nguyen LA, Yokoyama A & Ohki M (2001) Activation of AML1‐mediated transcription by MOZ and inhibition by the MOZ‐CBP fusion protein. EMBO J 20, 7184–7196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Doyon Y, Cayrou C, Ullah M, Landry AJ, Cote V, Selleck W, Lane WS, Tan S, Yang XJ & Cote J (2006) ING tumor suppressor proteins are critical regulators of chromatin acetylation required for genome expression and perpetuation. Mol Cell 21, 51–64. [DOI] [PubMed] [Google Scholar]
- 14. Voss AK, Collin C, Dixon MP & Thomas T (2009) Moz and retinoic acid coordinately regulate H3K9 acetylation, Hox gene expression, and segment identity. Dev Cell 17, 674–686. [DOI] [PubMed] [Google Scholar]
- 15. Thomas T, Corcoran LM, Gugasyan R, Dixon MP, Brodnicki T, Nutt SL, Metcalf D & Voss AK (2006) Monocytic leukemia zinc finger protein is essential for the development of long‐term reconstituting hematopoietic stem cells. Genes Dev 20, 1175–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Katsumoto T, Aikawa Y, Iwama A, Ueda S, Ichikawa H, Ochiya T & Kitabayashi I (2006) MOZ is essential for maintenance of hematopoietic stem cells. Genes Dev 20, 1321–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sheikh BN, Downer NL, Phipson B, Vanyai HK, Kueh AJ, McCarthy DJ, Smyth GK, Thomas T & Voss AK (2015) MOZ and BMI1 play opposing roles during Hox gene activation in ES cells and in body segment identity specification in vivo. Proc Natl Acad Sci USA 112, 5437–5442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lawrence HJ, Christensen J, Fong S, Hu YL, Weissman I, Sauvageau G, Humphries RK & Largman C (2005) Loss of expression of the Hoxa‐9 homeobox gene impairs the proliferation and repopulating ability of hematopoietic stem cells. Blood 106, 3988–3994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Lawrence HJ, Helgason CD, Sauvageau G, Fong S, Izon DJ, Humphries RK & Largman C (1997) Mice bearing a targeted interruption of the homeobox gene HOXA9 have defects in myeloid, erythroid, and lymphoid hematopoiesis. Blood 89, 1922–1930. [PubMed] [Google Scholar]
- 20. Thorsteinsdottir U, Mamo A, Kroon E, Jerome L, Bijl J, Lawrence HJ, Humphries K & Sauvageau G (2002) Overexpression of the myeloid leukemia‐associated Hoxa9 gene in bone marrow cells induces stem cell expansion. Blood 99, 121–129. [DOI] [PubMed] [Google Scholar]
- 21. Paggetti J, Largeot A, Aucagne R, Jacquel A, Lagrange B, Yang XJ, Solary E, Bastie JN & Delva L (2010) Crosstalk between leukemia‐associated proteins MOZ and MLL regulates HOX gene expression in human cord blood CD34+ cells. Oncogene 29, 5019–5031. [DOI] [PubMed] [Google Scholar]
- 22. Klein BJ, Lalonde ME, Cote J, Yang XJ & Kutateladze TG (2014) Crosstalk between epigenetic readers regulates the MOZ/MORF HAT complexes. Epigenetics 9, 186–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Qiu Y, Liu L, Zhao C, Han C, Li F, Zhang J, Wang Y, Li G, Mei Y, Wu M et al. (2012) Combinatorial readout of unmodified H3R2 and acetylated H3K14 by the tandem PHD finger of MOZ reveals a regulatory mechanism for HOXA9 transcription. Genes Dev 26, 1376–1391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Xiong X, Panchenko T, Yang S, Zhao S, Yan P, Zhang W, Xie W, Li Y, Zhao Y, Allis CD et al. (2016) Selective recognition of histone crotonylation by double PHD fingers of MOZ and DPF2. Nat Chem Biol 12, 1111–1118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Klein BJ, Simithy J, Wang X, Ahn J, Andrews FH, Zhang Y, Cote J, Shi X, Garcia BA & Kutateladze TG (2017) Recognition of histone H3K14 acylation by MORF. Structure 25, 650–654.e2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Klein BJ, Jang SM, Lachance C, Mi W, Lyu J, Sakuraba S, Krajewski K, Wang WW, Sidoli S, Liu J et al. (2019) Histone H3K23‐specific acetylation by MORF is coupled to H3K14 acylation. Nat Commun 10, 4724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Holbert MA, Sikorski T, Carten J, Snowflack D, Hodawadekar S & Marmorstein R (2007) The human monocytic leukemia zinc finger histone acetyltransferase domain contains DNA‐binding activity implicated in chromatin targeting. J Biol Chem 282, 36603–36613. [DOI] [PubMed] [Google Scholar]
- 28. Kueh AJ, Dixon MP, Voss AK & Thomas T (2011) HBO1 is required for H3K14 acetylation and normal transcriptional activity during embryonic development. Mol Cell Biol 31, 845–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kueh AJ, Eccles S, Tang L, Garnham AL, May RE, Herold MJ, Smyth GK, Voss AK & Thomas T (2020) HBO1 (KAT7) does not have an essential role in cell proliferation, DNA replication, or histone 4 acetylation in human cells. Mol Cell Biol 40, e00506‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mishima Y, Miyagi S, Saraya A, Negishi M, Endoh M, Endo TA, Toyoda T, Shinga J, Katsumoto T, Chiba T et al. (2011) The Hbo1‐Brd1/Brpf2 complex is responsible for global acetylation of H3K14 and required for fetal liver erythropoiesis. Blood 118, 2443–2453. [DOI] [PubMed] [Google Scholar]
- 31. Lalonde ME, Avvakumov N, Glass KC, Joncas FH, Saksouk N, Holliday M, Paquet E, Yan K, Tong Q, Klein BJ et al. (2013) Exchange of associated factors directs a switch in HBO1 acetyltransferase histone tail specificity. Genes Dev 27, 2009–2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Klein BJ, Muthurajan UM, Lalonde ME, Gibson MD, Andrews FH, Hepler M, Machida S, Yan K, Kurumizaka H, Poirier MG et al. (2016) Bivalent interaction of the PZP domain of BRPF1 with the nucleosome impacts chromatin dynamics and acetylation. Nucleic Acids Res 44, 472–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Vezzoli A, Bonadies N, Allen MD, Freund SM, Santiveri CM, Kvinlaug BT, Huntly BJ, Gottgens B & Bycroft M (2010) Molecular basis of histone H3K36me3 recognition by the PWWP domain of Brpf1. Nat Struct Mol Biol 17, 617–619. [DOI] [PubMed] [Google Scholar]
- 34. Feng Y, Vlassis A, Roques C, Lalonde ME, Gonzalez‐Aguilera C, Lambert JP, Lee SB, Zhao X, Alabert C, Johansen JV et al. (2016) BRPF3‐HBO1 regulates replication origin activation and histone H3K14 acetylation. EMBO J 35, 176–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hibiya K, Katsumoto T, Kondo T, Kitabayashi I & Kudo A (2009) Brpf1, a subunit of the MOZ histone acetyl transferase complex, maintains expression of anterior and posterior Hox genes for proper patterning of craniofacial and caudal skeletons. Dev Biol 329, 176–190. [DOI] [PubMed] [Google Scholar]
- 36. You L, Yan K, Zou J, Zhao H, Bertos NR, Park M, Wang E & Yang XJ (2015) The chromatin regulator Brpf1 regulates embryo development and cell proliferation. J Biol Chem 290, 11349–11364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. You L, Li L, Zou J, Yan K, Belle J, Nijnik A, Wang E & Yang XJ (2016) BRPF1 is essential for development of fetal hematopoietic stem cells. J Clin Invest 126, 3247–3262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Champagne KS, Saksouk N, Pena PV, Johnson K, Ullah M, Yang XJ, Cote J & Kutateladze TG (2008) The crystal structure of the ING5 PHD finger in complex with an H3K4me3 histone peptide. Proteins 72, 1371–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Okuda H, Stanojevic B, Kanai A, Kawamura T, Takahashi S, Matsui H, Takaori‐Kondo A & Yokoyama A (2017) Cooperative gene activation by AF4 and DOT1L drives MLL‐rearranged leukemia. J Clin Invest 127, 1918–1931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Erb MA, Scott TG, Li BE, Xie H, Paulk J, Seo HS, Souza A, Roberts JM, Dastjerdi S, Buckley DL et al. (2017) Transcription control by the ENL YEATS domain in acute leukaemia. Nature 543, 270–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wan L, Wen H, Li Y, Lyu J, Xi Y, Hoshii T, Joseph JK, Wang X, Loh YE, Erb MA et al. (2017) ENL links histone acetylation to oncogenic gene expression in acute myeloid leukaemia. Nature 543, 265–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Li Y, Sabari BR, Panchenko T, Wen H, Zhao D, Guan H, Wan L, Huang H, Tang Z, Zhao Y et al. (2016) Molecular coupling of histone crotonylation and active transcription by AF9 YEATS domain. Mol Cell 62, 181–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Li Y, Wen H, Xi Y, Tanaka K, Wang H, Peng D, Ren Y, Jin Q, Dent SY, Li W et al. (2014) AF9 YEATS domain links histone acetylation to DOT1L‐mediated H3K79 methylation. Cell 159, 558–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. John S, Howe L, Tafrov ST, Grant PA, Sternglanz R & Workman JL (2000) The something about silencing protein, Sas3, is the catalytic subunit of NuA3, a yTAF(II)30‐containing HAT complex that interacts with the Spt16 subunit of the yeast CP (Cdc68/Pob3)‐FACT complex. Genes Dev 14, 1196–1208. [PMC free article] [PubMed] [Google Scholar]
- 45. Ayton PM, Chen EH & Cleary ML (2004) Binding to nonmethylated CpG DNA is essential for target recognition, transactivation, and myeloid transformation by an MLL oncoprotein. Mol Cell Biol 24, 10470–10478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Allen MD, Grummitt CG, Hilcenko C, Min SY, Tonkin LM, Johnson CM, Freund SM, Bycroft M & Warren AJ (2006) Solution structure of the nonmethyl‐CpG‐binding CXXC domain of the leukaemia‐associated MLL histone methyltransferase. EMBO J 25, 4503–4512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Cierpicki T, Risner LE, Grembecka J, Lukasik SM, Popovic R, Omonkowska M, Shultis DD, Zeleznik‐Le NJ & Bushweller JH (2010) Structure of the MLL CXXC domain‐DNA complex and its functional role in MLL‐AF9 leukemia. Nat Struct Mol Biol 17, 62–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Okuda H, Kawaguchi M, Kanai A, Matsui H, Kawamura T, Inaba T, Kitabayashi I & Yokoyama A (2014) MLL fusion proteins link transcriptional coactivators to previously active CpG‐rich promoters. Nucleic Acids Res 42, 4241–4256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Bristow CA & Shore P (2003) Transcriptional regulation of the human MIP‐1alpha promoter by RUNX1 and MOZ. Nucleic Acids Res 31, 2735–2744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Pelletier N, Champagne N, Stifani S & Yang XJ (2002) MOZ and MORF histone acetyltransferases interact with the Runt‐domain transcription factor Runx2. Oncogene 21, 2729–2740. [DOI] [PubMed] [Google Scholar]
- 51. Rokudai S, Aikawa Y, Tagata Y, Tsuchida N, Taya Y & Kitabayashi I (2009) Monocytic leukemia zinc finger (MOZ) interacts with p53 to induce p21 expression and cell‐cycle arrest. J Biol Chem 284, 237–244. [DOI] [PubMed] [Google Scholar]
- 52. Aikawa Y, Yamagata K, Katsumoto T, Shima Y, Shino M, Stanley ER, Cleary ML, Akashi K, Tenen DG & Kitabayashi I (2015) Essential role of PU.1 in maintenance of mixed lineage leukemia‐associated leukemic stem cells. Cancer Sci 106, 227–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Aikawa Y, Katsumoto T, Zhang P, Shima H, Shino M, Terui K, Ito E, Ohno H, Stanley ER, Singh H et al. (2010) PU.1‐mediated upregulation of CSF1R is crucial for leukemia stem cell potential induced by MOZ‐TIF2. Nat Med 16, 580–585. 1p following 585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Borrow J, Stanton VP Jr, Andresen JM, Becher R, Behm FG, Chaganti RS, Civin CI, Disteche C, Dube I, Frischauf AM et al. (1996) The translocation t(8;16)(p11;p13) of acute myeloid leukaemia fuses a putative acetyltransferase to the CREB‐binding protein. Nat Genet 14, 33–41. [DOI] [PubMed] [Google Scholar]
- 55. Chaffanet M, Gressin L, Preudhomme C, Soenen‐Cornu V, Birnbaum D & Pebusque MJ (2000) MOZ is fused to p300 in an acute monocytic leukemia with t(8;22). Genes Chromosomes Cancer 28, 138–144. [DOI] [PubMed] [Google Scholar]
- 56. Kojima K, Kaneda K, Yoshida C, Dansako H, Fujii N, Yano T, Shinagawa K, Yasukawa M, Fujita S & Tanimoto M (2003) A novel fusion variant of the MORF and CBP genes detected in therapy‐related myelodysplastic syndrome with t(10;16)(q22;p13). Br J Haematol 120, 271–273. [DOI] [PubMed] [Google Scholar]
- 57. Panagopoulos I, Fioretos T, Isaksson M, Samuelsson U, Billstrom R, Strombeck B, Mitelman F & Johansson B (2001) Fusion of the MORF and CBP genes in acute myeloid leukemia with the t(10;16)(q22;p13). Hum Mol Genet 10, 395–404. [DOI] [PubMed] [Google Scholar]
- 58. Carapeti M, Aguiar RC, Goldman JM & Cross NC (1998) A novel fusion between MOZ and the nuclear receptor coactivator TIF2 in acute myeloid leukemia. Blood 91, 3127–3133. [PubMed] [Google Scholar]
- 59. Liang J, Prouty L, Williams BJ, Dayton MA & Blanchard KL (1998) Acute mixed lineage leukemia with an inv(8)(p11q13) resulting in fusion of the genes for MOZ and TIF2. Blood 92, 2118–2122. [PubMed] [Google Scholar]
- 60. Deguchi K, Ayton PM, Carapeti M, Kutok JL, Snyder CS, Williams IR, Cross NC, Glass CK, Cleary ML & Gilliland DG (2003) MOZ‐TIF2‐induced acute myeloid leukemia requires the MOZ nucleosome binding motif and TIF2‐mediated recruitment of CBP. Cancer Cell 3, 259–271. [DOI] [PubMed] [Google Scholar]
- 61. Taki T, Sako M, Tsuchida M & Hayashi Y (1997) The t(11;16)(q23;p13) translocation in myelodysplastic syndrome fuses the MLL gene to the CBP gene. Blood 89, 3945–3950. [PubMed] [Google Scholar]
- 62. Ida K, Kitabayashi I, Taki T, Taniwaki M, Noro K, Yamamoto M, Ohki M & Hayashi Y (1997) Adenoviral E1A‐associated protein p300 is involved in acute myeloid leukemia with t(11;22)(q23;q13). Blood 90, 4699–4704. [PubMed] [Google Scholar]
- 63. So CW & Cleary ML (2002) MLL‐AFX requires the transcriptional effector domains of AFX to transform myeloid progenitors and transdominantly interfere with forkhead protein function. Mol Cell Biol 22, 6542–6552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Ernst P, Wang J, Huang M, Goodman RH & Korsmeyer SJ (2001) MLL and CREB bind cooperatively to the nuclear coactivator CREB‐binding protein. Mol Cell Biol 21, 2249–2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. De Guzman RN, Goto NK, Dyson HJ & Wright PE (2006) Structural basis for cooperative transcription factor binding to the CBP coactivator. J Mol Biol 355, 1005–1013. [DOI] [PubMed] [Google Scholar]
- 66. Goto NK, Zor T, Martinez‐Yamout M, Dyson HJ & Wright PE (2002) Cooperativity in transcription factor binding to the coactivator CREB‐binding protein (CBP). The mixed lineage leukemia protein (MLL) activation domain binds to an allosteric site on the KIX domain. J Biol Chem 277, 43168–43174. [DOI] [PubMed] [Google Scholar]
- 67. Okuda H, Kanai A, Ito S, Matsui H & Yokoyama A (2015) AF4 uses the SL1 components of RNAP1 machinery to initiate MLL fusion‐ and AEP‐dependent transcription. Nat Commun 6, 8869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Shima H, Yamagata K, Aikawa Y, Shino M, Koseki H, Shimada H & Kitabayashi I (2014) Bromodomain‐PHD finger protein 1 is critical for leukemogenesis associated with MOZ‐TIF2 fusion. Int J Hematol 99, 21–31. [DOI] [PubMed] [Google Scholar]
- 69. Cheung N, Fung TK, Zeisig BB, Holmes K, Rane JK, Mowen KA, Finn MG, Lenhard B, Chan LC & So CW (2016) Targeting aberrant epigenetic networks mediated by PRMT1 and KDM4C in acute myeloid leukemia. Cancer Cell 29, 32–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Lavau C, Szilvassy SJ, Slany R & Cleary ML (1997) Immortalization and leukemic transformation of a myelomonocytic precursor by retrovirally transduced HRX‐ENL. EMBO J 16, 4226–4237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Okuda H & *Yokoyama A (2017) Myeloid progenitor transformation assay. Bio‐protocol 7 (23):e2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Henry RA, Kuo YM & Andrews AJ (2013) Differences in specificity and selectivity between CBP and p300 acetylation of histone H3 and H3/H4. Biochemistry 52, 5746–5759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Jin Q, Yu LR, Wang L, Zhang Z, Kasper LH, Lee JE, Wang C, Brindle PK, Dent SY & Ge K (2011) Distinct roles of GCN5/PCAF‐mediated H3K9ac and CBP/p300‐mediated H3K18/27ac in nuclear receptor transactivation. EMBO J 30, 249–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Weinert BT, Narita T, Satpathy S, Srinivasan B, Hansen BK, Scholz C, Hamilton WB, Zucconi BE, Wang WW, Liu WR et al. (2018) Time‐resolved analysis reveals rapid dynamics and broad scope of the CBP/p300 acetylome. Cell 174, 231–244 e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Mueller D, Bach C, Zeisig D, Garcia‐Cuellar MP, Monroe S, Sreekumar A, Zhou R, Nesvizhskii A, Chinnaiyan A, Hess JL et al. (2007) A role for the MLL fusion partner ENL in transcriptional elongation and chromatin modification. Blood 110, 4445–4454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Leach BI, Kuntimaddi A, Schmidt CR, Cierpicki T, Johnson SA & Bushweller JH (2013) Leukemia fusion target AF9 is an intrinsically disordered transcriptional regulator that recruits multiple partners via coupled folding and binding. Structure 21, 176–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Lin C, Smith ER, Takahashi H, Lai KC, Martin‐Brown S, Florens L, Washburn MP, Conaway JW, Conaway RC & Shilatifard A (2010) AFF4, a component of the ELL/P‐TEFb elongation complex and a shared subunit of MLL chimeras, can link transcription elongation to leukemia. Mol Cell 37, 429–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. He N, Liu M, Hsu J, Xue Y, Chou S, Burlingame A, Krogan NJ, Alber T & Zhou Q (2010) HIV‐1 Tat and host AFF4 recruit two transcription elongation factors into a bifunctional complex for coordinated activation of HIV‐1 transcription. Mol Cell 38, 428–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Sobhian B, Laguette N, Yatim A, Nakamura M, Levy Y, Kiernan R & Benkirane M (2010) HIV‐1 Tat assembles a multifunctional transcription elongation complex and stably associates with the 7SK snRNP. Mol Cell 38, 439–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Takahashi S & Yokoyama A (2020) The molecular functions of common and atypical MLL fusion protein complexes. Biochim Biophys Acta Gene Regul Mech 1863, 194548. [DOI] [PubMed] [Google Scholar]
- 81. Yokoyama A, Somervaille TC, Smith KS, Rozenblatt‐Rosen O, Meyerson M & Cleary ML (2005) The menin tumor suppressor protein is an essential oncogenic cofactor for MLL‐associated leukemogenesis. Cell 123, 207–218. [DOI] [PubMed] [Google Scholar]
- 82. Yokoyama A, Wang Z, Wysocka J, Sanyal M, Aufiero DJ, Kitabayashi I, Herr W & Cleary ML (2004) Leukemia proto‐oncoprotein MLL forms a SET1‐like histone methyltransferase complex with menin to regulate Hox gene expression. Mol Cell Biol 24, 5639–5649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Yokoyama A & Cleary ML (2008) Menin critically links MLL proteins with LEDGF on cancer‐associated target genes. Cancer Cell 14, 36–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. van Nuland R, van Schaik FM, Simonis M, van Heesch S, Cuppen E, Boelens R, Timmers HM & van Ingen H (2013) Nucleosomal DNA binding drives the recognition of H3K36‐methylated nucleosomes by the PSIP1‐PWWP domain. Epigenetics Chromatin 6, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Eidahl JO, Crowe BL, North JA, McKee CJ, Shkriabai N, Feng L, Plumb M, Graham RL, Gorelick RJ, Hess S et al. (2013) Structural basis for high‐affinity binding of LEDGF PWWP to mononucleosomes. Nucleic Acids Res 41, 3924–3936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Wagner EJ & Carpenter PB (2012) Understanding the language of Lys36 methylation at histone H3. Nat Rev Mol Cell Biol 13, 115–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Hughes CM, Rozenblatt‐Rosen O, Milne TA, Copeland TD, Levine SS, Lee JC, Hayes DN, Shanmugam KS, Bhattacharjee A, Biondi CA et al. (2004) Menin associates with a trithorax family histone methyltransferase complex and with the hoxc8 locus. Mol Cell 13, 587–597. [DOI] [PubMed] [Google Scholar]
- 88. Milne TA, Briggs SD, Brock HW, Martin ME, Gibbs D, Allis CD & Hess JL (2002) MLL targets SET domain methyltransferase activity to Hox gene promoters. Mol Cell 10, 1107–1117. [DOI] [PubMed] [Google Scholar]
- 89. Yamamoto R, Morita Y, Ooehara J, Hamanaka S, Onodera M, Rudolph KL, Ema H & Nakauchi H (2013) Clonal analysis unveils self‐renewing lineage‐restricted progenitors generated directly from hematopoietic stem cells. Cell 154, 1112–1126. [DOI] [PubMed] [Google Scholar]
- 90. Cozzio A, Passegue E, Ayton PM, Karsunky H, Cleary ML & Weissman IL (2003) Similar MLL‐associated leukemias arising from self‐renewing stem cells and short‐lived myeloid progenitors. Genes Dev 17, 3029–3035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Krivtsov AV, Twomey D, Feng Z, Stubbs MC, Wang Y, Faber J, Levine JE, Wang J, Hahn WC, Gilliland DG et al. (2006) Transformation from committed progenitor to leukaemia stem cell initiated by MLL‐AF9. Nature 442, 818–822. [DOI] [PubMed] [Google Scholar]
- 92. Huntly BJ, Shigematsu H, Deguchi K, Lee BH, Mizuno S, Duclos N, Rowan R, Amaral S, Curley D, Williams IR et al. (2004) MOZ‐TIF2, but not BCR‐ABL, confers properties of leukemic stem cells to committed murine hematopoietic progenitors. Cancer Cell 6, 587–596. [DOI] [PubMed] [Google Scholar]
- 93. Yokoyama A, Lin M, Naresh A, Kitabayashi I & Cleary ML (2010) A higher‐order complex containing AF4 and ENL family proteins with P‐TEFb facilitates oncogenic and physiologic MLL‐dependent transcription. Cancer Cell 17, 198–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Chen CW, Koche RP, Sinha AU, Deshpande AJ, Zhu N, Eng R, Doench JG, Xu H, Chu SH, Qi J et al. (2015) DOT1L inhibits SIRT1‐mediated epigenetic silencing to maintain leukemic gene expression in MLL‐rearranged leukemia. Nat Med 21, 335–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. Borkin D, He S, Miao H, Kempinska K, Pollock J, Chase J, Purohit T, Malik B, Zhao T, Wang J et al. (2015) Pharmacologic inhibition of the Menin‐MLL interaction blocks progression of MLL leukemia in vivo. Cancer Cell 27, 589–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Grembecka J, He S, Shi A, Purohit T, Muntean AG, Sorenson RJ, Showalter HD, Murai MJ, Belcher AM, Hartley T et al. (2012) Menin‐MLL inhibitors reverse oncogenic activity of MLL fusion proteins in leukemia. Nat Chem Biol 8, 277–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Klossowski S, Miao H, Kempinska K, Wu T, Purohit T, Kim E, Linhares BM, Chen D, Jih G, Perkey E et al. (2019) Menin inhibitor MI‐3454 induces remission in MLL1‐rearranged and NPM1‐mutated models of leukemia. J Clin Invest 130, 981–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Krivtsov AV, Evans K, Gadrey JY, Eschle BK, Hatton C, Uckelmann HJ, Ross KN, Perner F, Olsen SN, Pritchard T et al. (2019) A Menin‐MLL inhibitor induces specific chromatin changes and eradicates disease in models of MLL‐rearranged leukemia. Cancer Cell 36, 660–673, e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Daigle SR, Olhava EJ, Therkelsen CA, Majer CR, Sneeringer CJ, Song J, Johnston LD, Scott MP, Smith JJ, Xiao Y et al. (2011) Selective killing of mixed lineage leukemia cells by a potent small‐molecule DOT1L inhibitor. Cancer Cell 20, 53–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Huang J, Gurung B, Wan B, Matkar S, Veniaminova NA, Wan K, Merchant JL, Hua X & Lei M (2012) The same pocket in menin binds both MLL and JUND but has opposite effects on transcription. Nature 482, 542–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Shi A, Murai MJ, He S, Lund G, Hartley T, Purohit T, Reddy G, Chruszcz M, Grembecka J & Cierpicki T (2012) Structural insights into inhibition of the bivalent menin‐MLL interaction by small molecules in leukemia. Blood 120, 4461–4469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Kuhn MW, Song E, Feng Z, Sinha A, Chen CW, Deshpande AJ, Cusan M, Farnoud N, Mupo A, Grove C et al. (2016) Targeting chromatin regulators inhibits leukemogenic gene expression in NPM1 mutant leukemia. Cancer Discov 6, 1166–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Uckelmann HJ, Kim SM, Wong EM, Hatton C, Giovinazzo H, Gadrey JY, Krivtsov AV, Rucker FG, Dohner K, McGeehan GM et al. (2020) Therapeutic targeting of preleukemia cells in a mouse model of NPM1 mutant acute myeloid leukemia. Science 367, 586–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Daigle SR, Olhava EJ, Therkelsen CA, Basavapathruni A, Jin L, Boriack‐Sjodin PA, Allain CJ, Klaus CR, Raimondi A, Scott MP et al. (2013) Potent inhibition of DOT1L as treatment of MLL‐fusion leukemia. Blood 122, 1017–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Dafflon C, Craig VJ, Mereau H, Grasel J, Schacher Engstler B, Hoffman G, Nigsch F, Gaulis S, Barys L, Ito M et al. (2017) Complementary activities of DOT1L and Menin inhibitors in MLL‐rearranged leukemia. Leukemia 31, 1269–1277. [DOI] [PubMed] [Google Scholar]
- 106. Baell JB, Leaver DJ, Hermans SJ, Kelly GL, Brennan MS, Downer NL, Nguyen N, Wichmann J, McRae HM, Yang Y et al. (2018) Inhibitors of histone acetyltransferases KAT6A/B induce senescence and arrest tumour growth. Nature 560, 253–257. [DOI] [PubMed] [Google Scholar]
- 107. MacPherson L, Anokye J, Yeung MM, Lam EYN, Chan YC, Weng CF, Yeh P, Knezevic K, Butler MS, Hoegl A et al. (2020) HBO1 is required for the maintenance of leukaemia stem cells. Nature 577, 266–270. [DOI] [PubMed] [Google Scholar]
- 108. Au YZ, Gu M, De Braekeleer E, Gozdecka M, Aspris D, Tarumoto Y, Cooper J, Yu J, Ong SH, Chen X et al. (2021) KAT7 is a genetic vulnerability of acute myeloid leukemias driven by MLL rearrangements. Leukemia 35, 1012–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Asiaban JN, Milosevich N, Chen E, Bishop TR, Wang J, Zhang Y, Ackerman CJ, Hampton EN, Young TS, Hull MV et al. (2020) Cell‐based ligand discovery for the ENL YEATS domain. ACS Chem Biol 15, 895–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Garnar‐Wortzel L, Bishop TR, Kitamura S, Milosevich N, Asiaban JN, Zhang X, Zheng Q, Chen E, Ramos AR, Ackerman CJ et al. (2021) Chemical inhibition of ENL/AF9 YEATS domains in acute leukemia. ACS Cent Sci 7, 815–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Christott T, Bennett J, Coxon C, Monteiro O, Giroud C, Beke V, Felce SL, Gamble V, Gileadi C, Poda G et al. (2019) Discovery of a selective inhibitor for the YEATS domains of ENL/AF9. SLAS Discov 24, 133–141. [DOI] [PubMed] [Google Scholar]
- 112. Moustakim M, Christott T, Monteiro OP, Bennett J, Giroud C, Ward J, Rogers CM, Smith P, Panagakou I, Diaz‐Saez L et al. (2018) Discovery of an MLLT1/3 YEATS domain chemical probe. Angew Chem Int Ed Engl 57, 16302–16307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Li X, Li XM, Jiang Y, Liu Z, Cui Y, Fung KY, van der Beelen SHE, Tian G, Wan L, Shi X et al. (2018) Structure‐guided development of YEATS domain inhibitors by targeting pi‐pi‐pi stacking. Nat Chem Biol 14, 1140–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Hanahan D & Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144, 646–674. [DOI] [PubMed] [Google Scholar]