

Abstract
Microviridins and other ω-ester linked peptides (OEPs), collectively known as graspetides, are characterized by sidechain-sidechain linkages installed by ATP-grasp enzymes. Here we report the discovery of a family of graspetides, the gene clusters of which also encode an O-methyltransferase with homology to the protein repair catalyst protein L-isoaspartyl methyltransferase (PIMT). Using heterologous expression, we produced the ribosomally synthesized and post-translationally modified peptide (RiPP), fuscimiditide. NMR analysis of fuscimiditide revealed that the peptide contains two ester crosslinks forming a stem-loop macrocycle. Furthermore, an unusually stable aspartimide moiety is found within the loop macrocycle. We have also fully reconstituted fuscimiditide biosynthesis in vitro including formation of the ester and aspartimide moieties. The aspartimide moiety embedded in fuscimiditide hydrolyzes regioselectively to isoaspartate (isoAsp). Surprisingly, this isoAsp-containing protein is also a substrate for the PIMT homolog, thus driving any hydrolysis products back to the aspartimide form. Whereas an aspartimide is often considered a nuisance product in protein formulations, our data suggest that some RiPPs have aspartimide residues intentionally installed via enzymatic activity.
Graphical Abstract
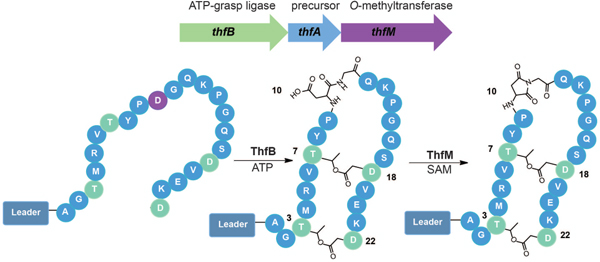
Two of the structural hallmarks of ribosomally synthesized and post-translationally modified peptides (RiPPs)1 are macrocyclization and backbone modifications. Macrocyclization occurs using backbone-backbone cyclization in the case of cyclotides/orbitides,2–4 and cyanobactins,5 backbone to sidechain linkages for lasso peptides,6–8 sactipeptides,9,10 and thiopeptides,11–13 and an expanding array of crosslinks between sidechains catalyzed by numerous enzymes.14–19 The most well-studied example of backbone modification in RiPPs is the introduction of oxazol(in)e and thiazol(in)e moieties,20–23 though other modifications such as epimerization are observed as well.24 Both macrocyclization and backbone modification serve to endow RiPPs with improved protease resistance relative to their unmodified counterparts. Many of these products are secreted into hostile, protease-rich environments, and thus require stabilization of their structure. Macrocyclization and backbone modification are also important for the bioactivity of RiPPs. Lasso peptides are comprised of one covalently bonded macrocycle and one mechanically bonded macrocycle,25 and this unique structure is required for molecular recognition of targets such as RNA polymerase.26,27 Likewise, the thiazoles and oxazoles in linear azol(in)e peptides such as klebsazolicin and phazolicin are involved in base stacking interactions in the ribosome.28,29
One well-studied example of macrocyclized RiPPs is the microviridins. The isolation of the first microviridin was published in 1990,30 and subsequent work revealed that these peptides are RiPPs.31–35 The class-defining modifications for microviridins are two sidechain-sidechain ester linkages and one amide linkage that are installed via the action of two distinct ATP-grasp enzymes.34,35 More recent work has uncovered further examples of macrocyclic peptides with only ester crosslinks installed by a single ATP-grasp enzyme, plesiocin36,37 and thuringinin.38 Given that microviridins and these newer examples all contain sidechain-sidechain ester linkages, Kim and coworkers have named this class of peptides omega ester-containing peptides (OEPs). In a recent bioinformatic survey of OEPs, Kim and coworkers further classified these peptides into 12 distinct groups based on a sequence similarity network constructed from ATP-grasp sequences.39 Ramesh et al. recently proposed 12 additional groups.40 OEP groups 1, 2, and 3 correspond to microviridins, plesiocin, and thuringinin, respectively (Fig. 1a). Characterization of examples from OEP groups 4–7,11, and 16 has been carried out revealing connectivity patterns different from groups 1–3.39–42 There has been a recent call to rename all members of this class graspetides43 because of the class-defining ATP-grasp enzyme.
Fig. 1.
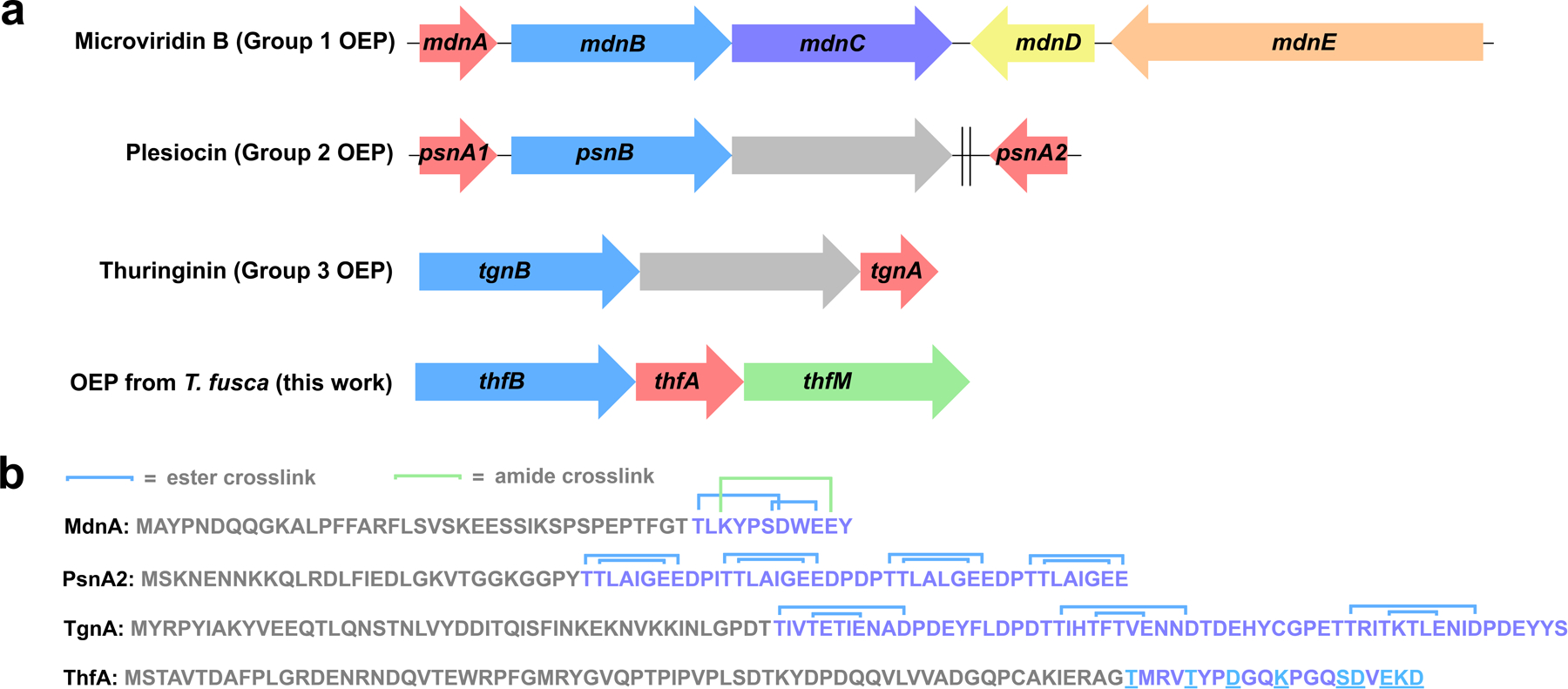
Gene clusters and precursors for graspetides. a) Biosynthetic gene clusters (BGCs) for microviridin and other omega ester peptides (OEPs). Red: precursor peptides, light blue: ester-installing ATP-grasp enzymes. In the microviridin B BGC, mdnC codes for an amide-installing ATP-grasp enzyme (purple), mdnD codes for an acetyltransferase (yellow), and mdnE codes for a protease/ABC transporter (peach). The plesiocin and thuringinin BGCs harbor a gene that encodes a membrane protein of unknown function (gray). The psnA2 gene is distal from the plesiocin BGC as indicated by the double line. BGC for the putative graspetide from T. fusca is distinguished by the presence of an O-methyltransferase gene (green). b) Precursor peptide sequences for the BGCs in part a. Putative leader and core sequences are shown in gray and purple respectively. Ester and amide connectivity is shown in the core peptide for microviridin B, plesiocin, and thuringinin, while potential crosslinkable residues are underlined in the T. fusca precursor ThfA.
Here we describe the discovery and characterization of an example of an OEP/graspetide, fuscimiditide, from the thermophilic actinobacterium Thermobifida fusca. The biosynthetic gene cluster (BGC) for fuscimiditide includes an aa precursor peptide ThfA, a single ATP-grasp enzyme ThfB, and an O-methyltransferse ThfM with homology to the protein repair catalyst protein L-isoaspartyl methyltransferase (PIMT). We show through a series of heterologous expression and in vitro enzymology experiments coupled with detailed NMR analysis that fuscimiditide is a stem-loop structure peptide macrocycle with two sidechain-sidechain ester linkages and an aspartimide modification within the backbone of the loop macrocycle. ThfM methylates an Asp residue and the methyl ester is subsequently attacked by the adjacent backbone to form an aspartimide. This is in contrast to native PIMTs which have a strong preference for isoaspartate (isoAsp) as a substrate. Whereas aspartimide is typically viewed solely as an intermediate in age-related protein damage and its subsequent repair,44–47 our data shows that the aspartimidylated peptide is the desired final product of the BGC in cellulo. While the aspartimide residue in fuscimiditide is stable for days in neutral, low ionic strength conditions, it hydrolyzes regioselectively to an isoAsp moiety under higher ionic strength or basic conditions. Remarkably, ThfM is also able to recognize isoAsp and drive the hydrolyzed material back to the aspartimidylated form, showing that ThfM has unprecedented promiscuity among PIMT enzymes.
Results
A graspetide-like BGC in T. fusca harbors an uncharacterized methyltransferase
Our lab has previously worked with Thermobifida fusca, a thermophilic actinobacterium, in our studies on fuscanodin, a lasso peptide.48 In a closely related organism, Thermobifida cellulosilytica, there is a lasso peptide biosynthetic gene cluster (BGC) with an O-methyltransferase in addition to the minimal biosynthetic genes for lasso peptides.49 We searched T. fusca for similar O-methyltransferases and found one associated with an ATP-grasp enzyme and a putative RiPP precursor. In a previous bioinformatic survey of ATP-grasp enzyme prevalence, the colocalization of an ATP-grasp enzyme, an O-methyltransferase, and a precursor peptide was noted in actinobacteria.50 To our knowledge, there have been no investigations into the products of these BGCs. The putative precursor peptide in T. fusca, the gene for which will be referred to as thfA, is 89 aa long and enriched in Thr, Ser, Asp, and Glu residues at its C-terminus, consistent with it being an graspetide precursor (Fig. 1b). In this BGC, the precursor is found in between the gene encoding the ATP-grasp enzyme (ThfB) and the methyltransferase (ThfM) (Fig. 1a). The pattern of residues at the C-terminus of ThfA is different from the patterns in any of the 12 groups of OEPs delineated by Kim and coworkers (Fig. 1b).39 This fact, coupled with our curiosity about the function of the O-methyltransferase, prompted us to study the thfBAM BGC in more detail.
Heterologous expression of Thf BGC.
To determine whether ThfB and ThfM were able to modify the ThfA precursor peptide, we developed a coexpression system in E. coli. An N-terminal His-tag was added to ThfA and the gene was placed under an IPTG-inducible T7 promoter. The biosynthetic enzymes ThfB and ThfM were cloned individually under an IPTG-inducible T5 promoter. Utilizing E. coli BL21 (DE3) ΔslyD as the host strain, ThfA was heterologously expressed and purified with a yield of ~3.3 mg/L (Supplementary Fig. 1). Following purification, LC-MS analysis of the precursor revealed an observed mass of 11,361 Da corresponding to the full protein with cleavage of the N-terminal methionine (Fig. 2a). Next, we sought to determine the extent of the PTMs installed by ThfB and ThfM and whether there was a specific order of these modifications. Coexpression of the precursor ThfA and ATP-grasp ThfB yielded an observed average mass of 11,325 Da indicating a loss of 36 Da corresponding to a 2-fold dehydrated product; minor amounts of 11,361 Da (unmodified) and 11,343 Da (one dehydration) were also detected (Fig. 2a). On the other hand, coexpression with the O-methyltransferase ThfM did not yield any modification on ThfA (Supplementary Fig. 2). When all three components of the biosynthetic pathway were coexpressed, we expected to see a product corresponding to doubly dehydrated ThfA with addition of a methyl group (11,339 Da). Surprisingly, the major product of the heterologous expression was a 3-fold dehydrated precursor with an observed mass of 11,307 Da (Fig. 2a). These experiments established that the ATP-grasp ThfB was responsible for installing two dehydrations, while the O-methyltransferase ThfM installed a third dehydration (Fig. 2a). Notably, ThfM only recognizes the doubly dehydrated ThfA that has been modified by ThfB, establishing a strict order of modification.
Fig. 2.
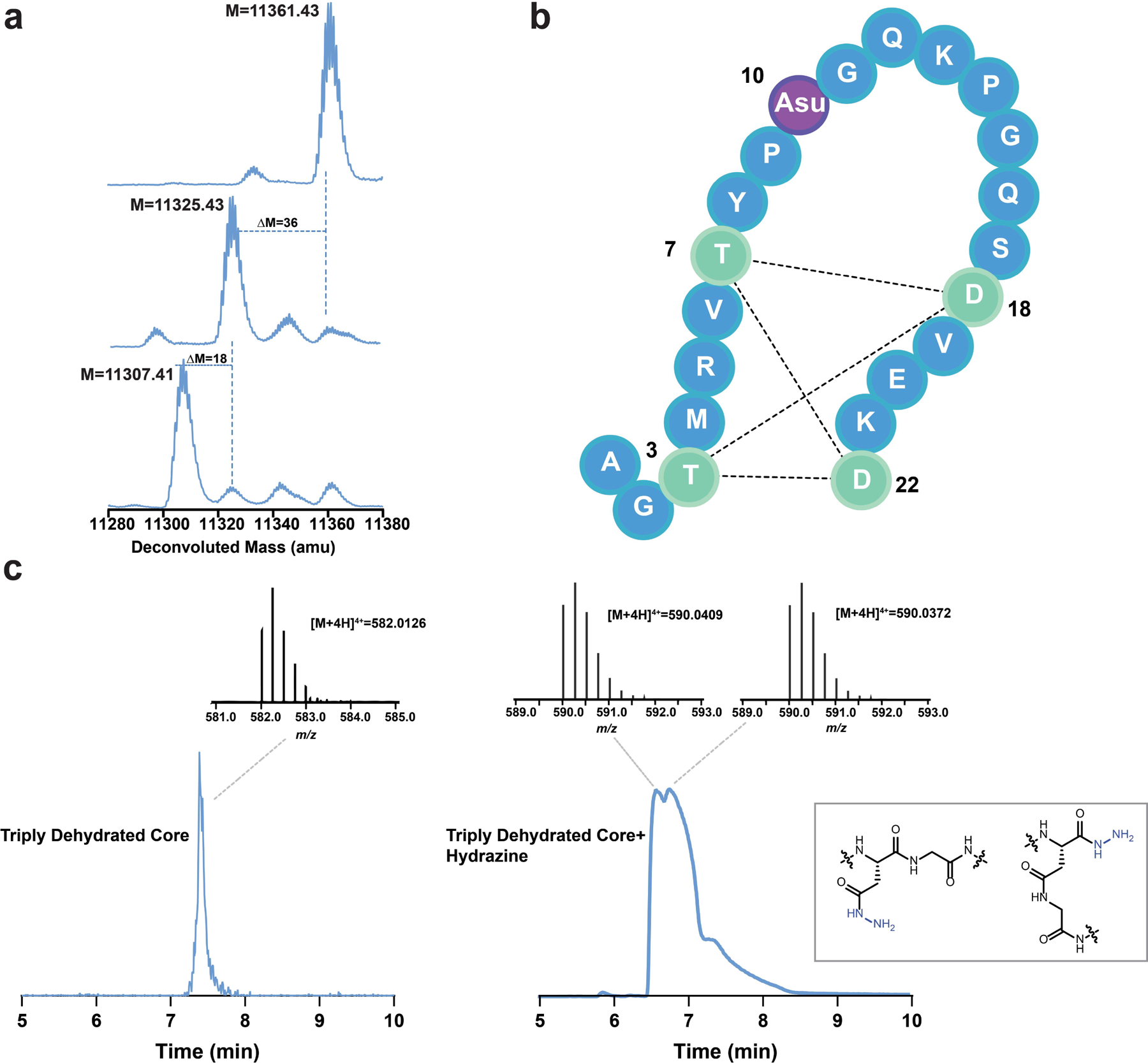
Posttranslational modifications (PTMs) on ThfA. a) Deconvoluted mass spectra of unmodified ThfA (top), ThfA co-expressed with ATP-grasp enzyme ThfB (middle), and ThfA co-expressed with ThfB and methyltransferase ThfM (bottom). ThfB dehydrates ThfA twice, while ThfM adds a third dehydration. b) The three dehydrations on ThfA are localized to the C-terminal 22 aa, the putative core peptide. MS/MS analysis (Fig. S5) indicates a macrocyclic diester structure stretching from T3 to D22 with the third dehydration being an aspartimide at D10 (Asu). c) Left: extracted ion current (EIC) chromatogram and mass spectrum of 22 aa triply dehydrated ThfA core peptide starting material. Right: EIC chromatogram and mass spectra of products of hydrazine addition to the triply dehydrated ThfA core peptide. Two distinct species with masses corresponding to the peptide plus a single hydrazide are observed (structures shown in inset), consistent with hydrazine addition to two different locations on aspartimide.
Characterization of PTMs on ThfA.
In order to identify the posttranslational modification (PTMs) in doubly and triply dehydrated ThfA, we examined the ThfA sequence for potential trypsin cleavage sites. Digestion of the 2-fold dehydrated ThfA yielded a C-terminal 22 amino acid peptide containing 2 dehydrations and 3 missed trypsin cleavages (Supplementary Fig. 3). Similarly, digestion of the 3-fold dehydrated ThfA yielded the same fragment with 3 dehydrations marking the digested product as the putative core peptide of ThfA (Supplementary Fig. 3). Following identification of the core sequence, we sought to gather more evidence regarding the chemical nature of the installed PTMs. Since ATP-grasp ligases are known to catalyze the formation of esters and/or amides in graspetides, we reasoned that stability studies conducted at varying pHs could differentiate between these two possibilities. Therefore, we incubated doubly and triply dehydrated ThfA in 1x PBS buffer for 21 hours at 25 °C at a range of pHs; pH 3–11 for the doubly dehydrated species and pH 7–11 for the triply dehydrated species. The 2-fold dehydrated ThfA was stable at pH 3–10; partial hydrolysis was only observed at pH 11 indicating that the installed PTMs were likely ester linkages (Supplementary Fig. 4). Meanwhile, hydrolysis of the third dehydration (installed by ThfM) was observed at pH 7–11 suggesting a more labile modification (Supplementary Fig. 4).
Following these stability studies, we performed MS/MS analysis of doubly and triply dehydrated ThfA core peptide. In previous work on OEPs, the ester linkages were cleaved during MS/MS analysis, allowing for fragmentation patterns that assist in position determination of the ester forming residues.36,38 However, we did not observe cleavage of doubly dehydrated ThfA core peptide under collision-induced dissociation (CID) conditions. Instead, we observed minimal fragmentation yielding the b2, y20, and y21 ions in addition to several fragments corresponding to internal peptide cleavages between Pro9 and Pro14 of the core peptide (Supplementary Fig. 5).51 The lack of fragmentation beyond the observed b/y ions suggested the involvement of Thr at position 3 and Asp in position 22 (Fig. 2b). Moreover, the peptide fragmentation pattern coupled with the fact that there were 3 missed trypsin cleavages indicated macrocyclization of the peptide (Supplementary Fig. 3). The triply dehydrated ThfA core exhibited an identical fragmentation pattern except for one internal cleavage fragment between Asp10 and Gly11, which was no longer observed.
Following the line of evidence indicating that the third PTM is labile and the lack of observable MS/MS fragmentation between Asp10 and Gly11 in triply dehydrated core, we hypothesized that the dehydration installed by ThfM could be an aspartyl succinimide (Asu), also known as aspartimide (Fig. 2b). ThfM belongs to a family of O-methyltransferases with homology to protein repair catalysts protein L-isoaspartyl methyltransferases (PIMTs). The usual role of PIMTs is salvaging aging proteins that have acquired backbone isoAsp residues via spontaneous asparagine deamidation or aspartate dehydration with subsequent hydrolysis (Supplementary Fig. 6). The PIMT enzyme specifically methylates isoAsp, which is followed by non-enzymatic formation of an aspartyl succinimide. The succinimide can open to either Asp or isoAsp, with repeated cycles of catalysis leading to reversion of most of the protein to its Asp form. To test whether triply dehydrated ThfA core contained an aspartimide, we incubated the doubly and triply dehydrated core peptides with a 2 M hydrazine solution in water at room temperature for 30 mins. Given the selective reactivity of hydrazine with succinimides under the abovementioned conditions, a mass shift of +32 was expected.52,53 LC-MS analysis of the reaction products indicated partial hydrolysis of doubly dehydrated core owing to increased basicity of the reaction solution, while triply dehydrated core exhibited a mass change of +32. Hydrazine trapping of the putative aspartimide yielded two distinct peaks corresponding to opening of the succinimide on two different sides (Fig. 2c). MS/MS analysis of hydrazine reacted core yielded the abovementioned fragmentation pattern with an additional +32 mass change corresponding to hydrazide addition on a residue between Pro9 and Lys13 (Supplementary Fig. 7). Taken together, this MS evidence points to the formation of aspartimide at Asp10 in triply dehydrated ThfA core. Since ThfA is a ribosomal peptide, it will be composed solely of L-amino acids. However, Asu residues can slowly epimerize.54 We carried out Marfey’s analysis on the triply dehydrated ThfA and observed only L-amino acids (Supplementary Figs. 8, 9).
Additionally, the secondary structures of unmodified linear core peptide (obtained by solid-phase peptide synthesis) as well as doubly and triply dehydrated core peptide (obtained by co-expression followed by tryptic digest) were analyzed using CD spectroscopy. Linear core peptide displayed the signature of a random coil, while both dehydrated peptides exhibited β character presumably endowed by the ester crosslinking (Supplementary Fig. 10a). We also analyzed the entire precursor ThfA using CD and were surprised to see that this protein exhibited a random coil structure (Supplementary Fig. 10b) given its stability during heterologous expression in E. coli.
To further confirm the presence of 2 ester linkages, we executed an alanine scan of all residues with side chains capable of forming amides or esters. Swaps to alanine of Thr3, Thr7, Asp18, and Asp22 in the core peptide led to products that were no longer doubly dehydrated (Supplementary Fig. 11) strongly suggesting the involvement of these residues in ester cross-linkages. Thus, the macrocycle formed in doubly and triply dehydrated ThfA is predicted to span 20 aa from Thr3 to Asp22; two different linkage patterns are possible (Supplementary Fig. 12). As discussed above, the linkage pattern observed in this macrocycle differs from any of the known or predicted classes of graspetides.39–42
NMR structures of doubly and triply dehydrated ThfA core peptide.
Following preliminary structural analysis of the doubly and triply dehydrated peptides, COSY, TOCSY, NOESY, HSQC, and HMBC spectra were acquired on the doubly dehydrated ThfA core peptide in 98:2 H2O:D2O at 25°C (Supplementary Figs. 13–16). The TOCSY and NOESY spectra were used for sequential assignment of amino acid protons in addition to providing key pieces of evidence as to amino acid interactions in three-dimensional space (Supplementary Table 2). In the NOESY spectra, strong NOEs could be observed between the amide proton of Asp18 and the amide proton of Thr7, the amide proton of Asp22 and the α-proton of Thr3, and the amide proton of Thr7 and the α-proton of Asp18 (Supplementary Fig. 17, Supplementary Table 5). Additionally, a pronounced downfield shift was observed for the β-proton of Thr3 and Thr7 as compared to its average statistical value55 (Supplementary Table 2). HMBC correlations were observed between the β-proton of Thr3 and Thr7 and the Asp22 and Asp18 side chain carbonyl carbons, respectively. Strikingly, the Asp18 and Asp22 side chain carbonyls were shifted upfield to 169.0 ppm and 169.5 ppm indicating esterification (Supplementary Fig. 18, Supplementary Table 2). Mutagenesis results coupled with NOE and HMBC correlations confirmed that ThfB installed two sidechain-sidechain ester linkages: Thr3-Asp22 and Thr7-Asp18. Following assignment, CYANA 2.1 was used to calculate the 20 lowest energy conformers of the ester linked peptide. The peptide was shown to form a stem-loop structure with anti-parallel β character in the stem, thereby validating the secondary structure observed by CD spectroscopy (Fig. 3a, Supplementary Fig. 19). The modeled CYANA structure shows that the peptide formed a bis-macrocyclic structure, where the stem macrocycle was composed of 34 backbone atoms, while the loop macrocycle contained 32. The Tyr8 to Ser17 residues, corresponding to the loop of the stem-loop structure, exhibited few NOEs to non-adjacent residues, suggesting that this macrocycle did not extensively fold back upon the stem. This structure differs substantially from microviridins, however group 2 and 3 OEPs are predicted to also exhibit stem-loop elements in their structure, albeit with different macrocycle sizes.36,38 Notably, doubly dehydrated ThfA has a larger loop than any of the experimentally characterized graspetides (Supplementary Fig. 20). This work represents a comprehensive NMR analysis of an ATP-grasp modified stem-loop structure.
Fig. 3.
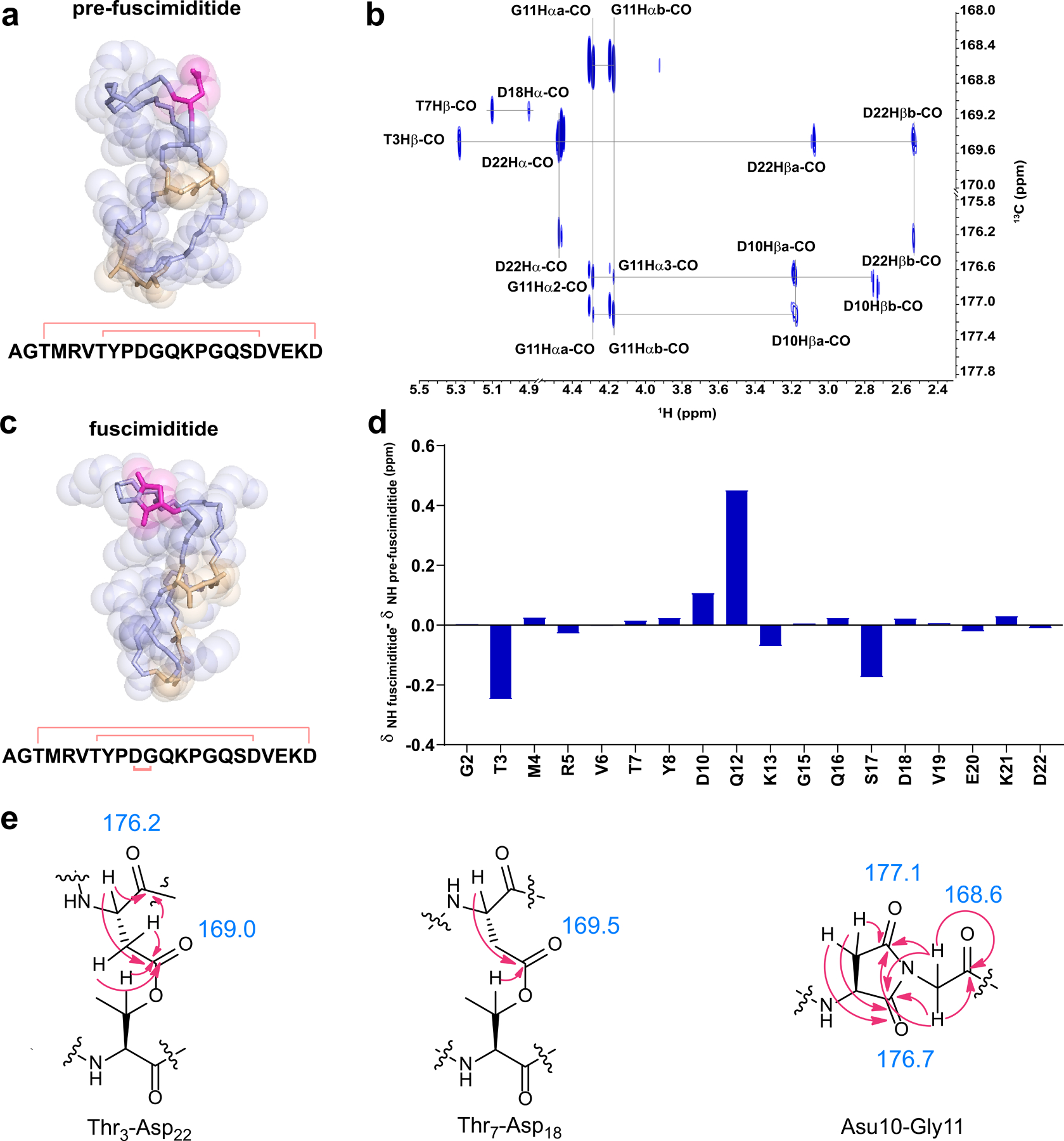
NMR structures of pre-fuscimiditide and fuscimiditide. a) Top structural model of pre-fuscimiditide, the ThfA core peptide with two ester linkages, as computed by CYANA 2.1. Ester linkages (wheat) connect T3 to D22 and T7 to D18 forming a stem-loop structure comprised of two edge-to-edge macrocycles. The peptide backbone is colored light blue; the D10 sidechain is in magenta. An overlay of the top 20 structures is in Supplementary Fig. 19. b) Portion of the 1H-13C HMBC spectrum of fuscimiditide, ThfA core peptide with two esters and an aspartimide. Key correlations that show the ester and succinimide connectivity are labeled. c) Top structural model of fuscimiditide. The color scheme is as in part a; the succinimide moiety is now colored magenta. An overlay of the top 20 structures is in Supplementary Fig. 19. d) Chemical shift deviations of amide protons between pre-fuscimiditide and fuscimiditide. A1, P9, G11, and P14 are not shown because of a lack of amide protons on these residues. e) Structures of ester and aspartimide linkages in pre-fuscimiditide with HMBC correlations from part b) shown. The chemical shifts of important carbonyl carbons are listed.
Having established the ester connectivity in doubly dehydrated ThfA core peptide, we sought to use NMR to identify the location of the additional dehydration installed by the O-methyltransferase (Supplementary Figs. 21–23). Our MS and hydrazine addition data discussed above (Supplementary Figs. 5,7) suggested that the Asp10 side chain was being converted to an aspartimide residue. In the TOCSY and NOESY spectra for triply dehydrated ThfA core peptide, the amide proton of Gly11 could no longer be observed. This suggests the existence of a stable aspartimide linking the side chain of Asp10 and the backbone of Gly11 (Supplementary Table 3). More directly, HMBC correlations were observed for the Gly11 α-protons and the Asp10 backbone and side chain carbonyls (Fig. 3b, 3e) confirming that O-methyltransferase catalysis facilitates the formation of the peptide aspartimide. The correlations and chemical shift values we observed for the succinimide moiety are in good agreement with published data on model peptides. Specifically, one hallmark of aspartimide residues is the large difference observed in the chemical shifts of the two β protons.56 We observe this in triply dehydrated ThfA core peptide; the two β proton chemical shifts differ by 0.5 ppm. Another hallmark of aspartimide moieties is a downfield shift of both the backbone and sidechain carbonyl resonances relative to Asp and an upfield shift of the β carbon resonance,56 all of which we observe. Upon confirmation of the aspartimide residue within the triply dehydrated peptide, we decided to name the peptide fuscimiditide which reflects both the organism that encodes it in its genome as well as its unique aspartimide group. The doubly dehydrated ThfA core peptide containing only esters will be referred to as pre-fuscimiditide (Fig. 3a). Utilizing proton assignments and NOESY constraints, fuscimiditide was modeled to be a stem-loop structure with the aspartimide positioned in the loop macrocycle (Fig. 3c). Notably, the aspartimide lies within a 6 aa stretch, PDGQKP, making this region especially rich in residues containing backbone constrained side chains. When comparing chemical shift deviations between pre-fuscimiditide and fuscimiditide, the largest chemical shift deviations were observed for the Gly11 and Gln12 α-protons and the Gln12 amide proton (Fig. 3d, Supplementary Table 6). The aspartimide had an especially strong influence on the chemical environment of the Gln12 backbone as indicated by the downfield shift of its amide proton by 0.45 ppm.
Fuscimiditide contains an unusually stable aspartimide.
As discussed above, aspartimides are generally observed as intermediates in protein repair. However, our data strongly suggests that the aspartimidylated peptide may be the final intended product of the fuscimiditide BGC. In fuscimiditide, the aspartimide moiety was able to withstand purification from cells (even under denaturing conditions), 1 h tryptic digests, and acquisition of multiple NMR spectra, prompting us to study its stability in more depth. Incubation of fuscimiditide in water, at RT, for three days resulted in less than 1% hydrolysis indicating that the aspartimide was stable in low ionic strength conditions. However, as discussed above, incubation of the peptide in PBS at varying pH values resulted in more rapid hydrolysis of the aspartimide (Supplementary Fig. 4), while the ester linkages remained intact. Hydrolysis products were also observed in tryptic digests that were carried out for ~16 hours in 50 mM ammonium bicarbonate (Fig. 4a). During reverse-phase HPLC purification of the hydrolyzed fuscimiditide, two hydrolysis products with different retention times were observed. The minor peak (~5% of the hydrolysate) had a retention time identical to pre-fuscimiditide, whereas the major peak (~95% of the hydrolysate) eluted differently (Supplementary Fig. 24). The most obvious explanation for these two peaks is the formation of a mixture of Asp and isoAsp in the backbone of fuscimiditide upon hydrolysis (Fig. 4b). Our data suggest that the aspartimide of fuscimiditide regioselectively opens primarily to the isoAsp form.
Fig. 4.
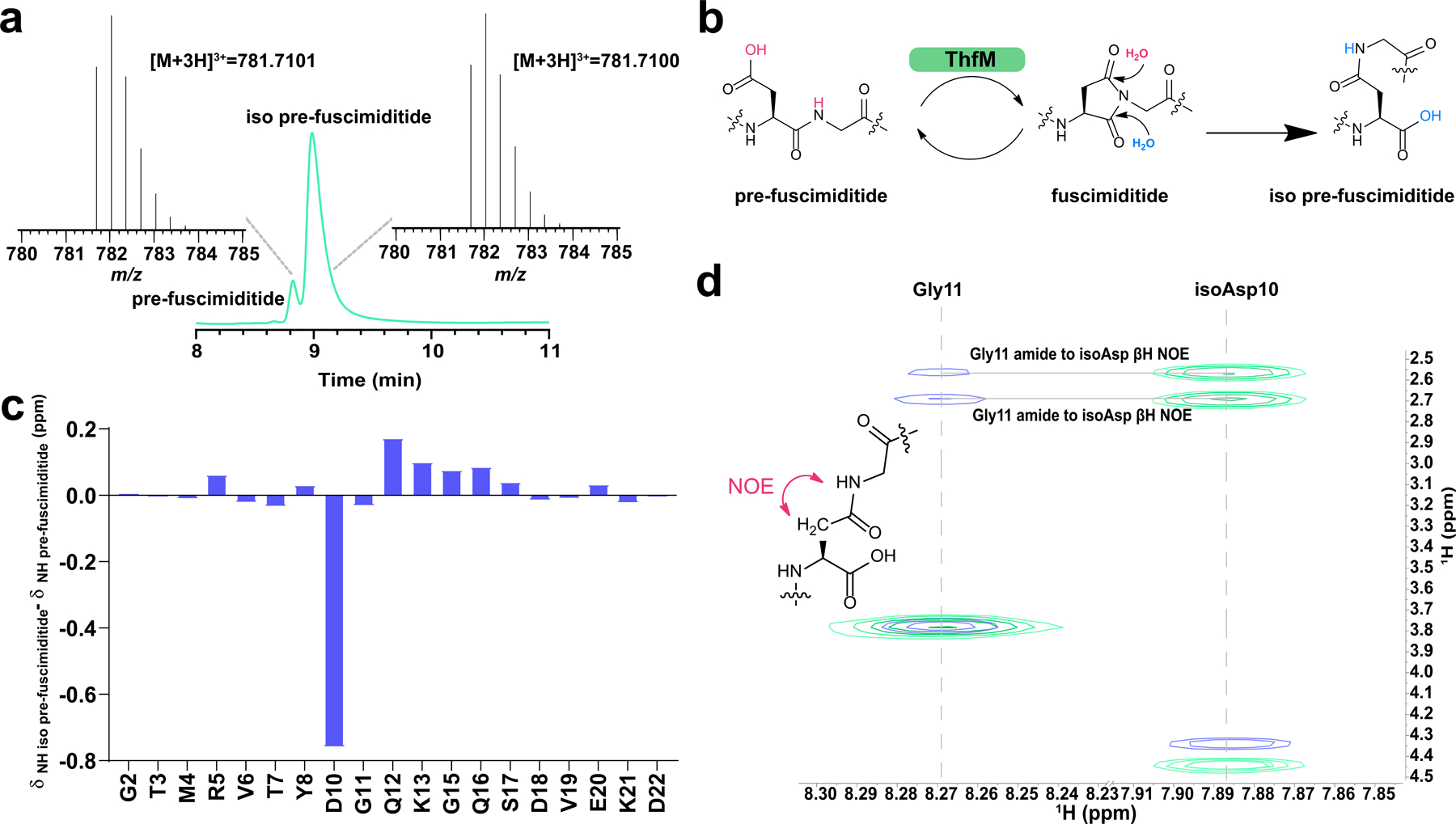
Characterization of the major fuscimiditide hydrolysis product, iso pre-fuscimiditide. a) HPLC trace of the products of fuscimiditide hydrolysis. The small peak at 8.8 min corresponds to the retention time of pre-fuscimiditide, but the major peak elutes later. These two peaks were collected and have identical masses according to LC-MS (insets). b) The aspartimide moiety in fuscimiditide (center) can hydrolyze to either Asp (left) or isoAsp (right). Succinimide formation enabled by ThfM catalysis is also shown. c) Chemical shift deviations of amide protons between pre-fuscimiditide and iso pre-fuscimiditide. d) Overlay of TOCSY (green) and NOESY (blue) spectra for iso pre-fuscimiditide showing connectivity of the D10 sidechain to G11 main chain. NOESY crosspeaks between the β protons of D10 and the amide proton of G11 are observed.
Intrigued by the regioselectivity of fuscimiditide hydrolysis, we sought NMR confirmation that the preferred product had isoAsp in its backbone. In the TOCSY and NOESY spectra (Supplementary Figs. 25, 26) of the putative isoAsp containing peptide, there was a distinct upfield shift of 0.713 ppm observed for the Asp10 amide proton (Fig. 4c, Supplementary Tables 4, 6). Additionally, strong NOEs were observed between the amide proton of Gly11 and the two distinct β-protons of Asp10 (Fig. 4d). In contrast, a single NOE was observed between the amide proton of Gly11 and the α-proton of Asp10 for pre-fuscimiditide (Supplementary Fig. 27). These NOEs indicate that the native peptide bond was no longer intact, and it was replaced by a newly formed β-peptidyl linkage. We will refer to this major hydrolysis product of fuscimiditide as iso pre-fuscimiditide to signify the presence of the isoAsp residue in its backbone (Fig. 4b). Marfey’s analysis was carried out on iso pre-fuscimiditide and, as was the case for fuscimiditide (Supplementary Fig. 9), no evidence of epimerization at the isoAsp residue was observed (Supplementary Fig. 28).
Production of fuscimiditide in T. fusca
Having determined the structure of fuscimiditide from heterologous expression, we returned to T. fusca, the organism that harbors the fuscimiditide BGC in its genome, to determine whether it produces any compounds related to fuscimiditide. T. fusca was grown for 7 days in liquid medium (see Supplementary Methods for culture conditions), and both the spent medium and the cell lysate were analyzed by LC-MS. A mass corresponding to fully matured fuscimiditide lacking its two N-terminal amino acids (ie fuscimiditide ΔN2) was detected with high accuracy (4.3 ppm error from the predicted mass, Supplementary Table 8) in the cell lysate (Supplementary Fig. 29). The removal of the N-terminal residues results in the formation of a symmetrical stem-loop structure with no overhanging amino acids. No masses corresponding to fuscimiditide species were detected in the culture medium. The fuscimiditide ΔN2 species is produced at much lower level in the native host than in the heterologous expression experiments described above. Moreover, fuscimiditide ΔN2 coelutes with several other species in the cell lysate, complicating its isolation for further characterization. We cannot exclude the possibility that this material in T. fusca has undergone further rearrangements or modifications relative to the fuscimiditide produced heterologously. Of note is that species corresponding to the starting material/hydrolysis products (ie pre-fuscimiditide and iso pre-fuscimiditide) were not detected in the T. fusca lysate.
Role of leader peptide in fuscimiditide maturation.
Having established the structure of fuscimiditide, pre-fuscimiditide, and the hydrolysis product iso pre-fuscimiditide, we turned our attention to investigating the enzymology of its biosynthesis in more detail. First, we asked whether the leader peptide was required for ThfB activity. In both lanthipeptides and lasso peptides, N-terminal portions of the leader peptide can be removed without affecting modification of the resulting peptide.57,58 In several other RiPPs such as cyanobactins, lanthipeptides, and microviridins, the leader peptide can be supplied in trans or can be fused to the N-terminus of the modifying enzymes.59–61 Since the fuscimiditide precursor ThfA was observed to be a random coil, we reasoned that large N-terminal truncations of the leader peptide may be tolerated without having an effect on peptide cyclization. To test the role of the lengthy (67 aa, without the added His tag) leader and to identify potential key residues, a series of truncation variants were created by removing 10 amino acid segments from the N-terminus (Supplementary Fig. 30). Removal of more than 20 N-terminal residues abolished modification by the ATP-grasp ThfB. The intolerance of truncations larger than 20 residues indicated that much of the leader peptide was necessary for installation of the ester linkages by ThfB.
To further probe the importance of the leader peptide, we searched it for conserved motifs. In microviridins, the highly conserved PFFARFL α-helical motif at the N-terminus is key in ATP-grasp activation.62 We generated a list of ThfA homologs by BLAST (Supplementary Fig. 31a), and upon alignment identified a conserved region, RPFG at positions 25–28 of ThfA (Supplementary Fig. 31b), at the beginning of the minimal leader peptide. We tested the significance of the RPFG motif by executing an alanine scan. However, all variants were successfully modified by both ATP-grasp ThfB and O-methyltransferase ThfM (Supplementary Fig. 31c).
In vitro reconstitution of fuscimiditide biosynthesis.
We next turned our attention to in vitro reconstitution of fuscimiditide biosynthesis. We purified unmodified N-terminal 6xHis tagged ThfA precursor both by native and denaturing methods. Following the strict order of reaction observed in heterologous expression experiments (Fig. 2a), 10 μM ThfA was first incubated with 1 μM ThfB in the presence of a 1000-fold excess of ATP, MgCl2, and DTT in 100 mM sodium phosphate buffer, pH 6.8 at 37 °C. DTT was used to prevent dimerization of ThfA (Supplementary Fig. 32), which contains one Cys residue in its leader region. The reaction was monitored every hour by LC-MS; after 5 h nearly all the starting material was consumed, and the major product was doubly dehydrated ThfA (Fig. 5a). Overnight incubation of the reaction mixture did not yield an appreciable change in the extent of reaction. ThfA purified under native and denaturing conditions performed equally well in these reactions. These results indicate that ThfB can modify ThfA in vitro, albeit at a relatively low turnover rate which could be due to tight binding of the enzyme to the matured product. Additionally, it was previously reported that the reaction efficiency of ATP-grasp catalyzed reactions can be increased ~20-fold in the presence of monovalent cations such as K+.63 We repeated the reaction in the presence of 100 mM KCl and monitored the formation of the 2-fold dehydrated product every 10 mins; the reaction was ~86% complete in 1 hr (Supplementary Fig. 33). Under both reaction conditions (with and without KCl) we observed minimal singly dehydrated ThfA (Fig. 5a), suggesting that once one ester forms, the second one forms rapidly. This result is in contrast to the proposed distributive mechanism for ester installation in plesiocin and thuringinin.36,38
Fig. 5.
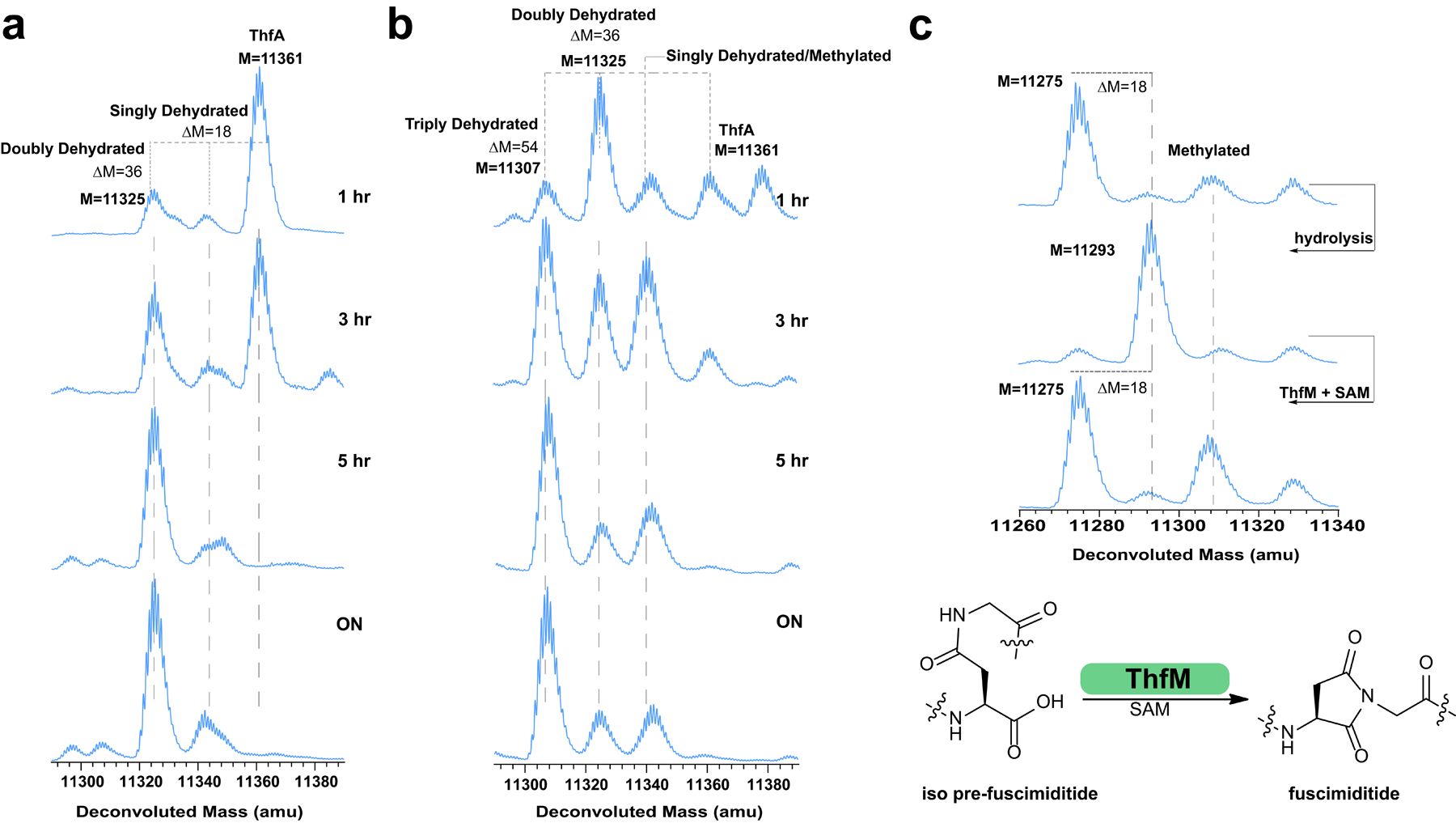
Analysis of ThfB and ThfM activity in vitro. a) Time course of the reaction between ThfA and ThfB. Unmodified ThfA (M = 11361) is converted primarily to the doubly esterified product over the course of 5 h. The single ester product does not accumulate, suggesting a model in which the formation of the first ester enables the formation of the second ester. b) Time course of the reaction of ThfA with ThfB and ThfM. A triply dehydrated product is the major species after 5 h indicating that ThfM can facilitate aspartimide formation in vitro. The isotopic patterns of singly esterified ThfA and methylated, doubly esterified ThfA overlap. c) Triply dehydrated ThfA C62A (top) was hydrolyzed to its doubly esterified form, comprised primarily of the isoAsp hydrolysate (see Fig. 4). Upon treatment with ThfM and SAM, this protein was dehydrated again to its fully modified form. Some putatively methylated protein is observed as well. These results show that doubly esterified ThfA with isoAsp within its loop macrocycle is a substrate for ThfM (bottom).
We further probed the reconstitution of the full biosynthetic pathway by adding O-methyltransferase ThfM (1 μM) simultaneously with ATP-grasp ThfB in the abovementioned reaction mixture (lacking KCl) in addition to SAM (0.4 mM, 40-fold excess relative to ThfA). Conversion to ~75% triply dehydrated product was observed in 5 h with minimal difference upon overnight incubation of the reaction (Fig. 5b). We also set up in vitro reactions to test whether ThfM alone could function or if it needed ThfB in the reaction. In accordance with our heterologous expression experiments, a reaction that included unmodified ThfA (10 μM) and ThfM (1 μM) with SAM (0.4 mM) led to no reaction. We also tested whether doubly dehydrated ThfA was a substrate for ThfM. Under native protein purification conditions, however, doubly dehydrated His-tagged ThfA co-elutes with a roughly equimolar amount of untagged ThfB (Supplementary Fig. 32a) Therefore, we repurified doubly dehydrated ThfA with an additional denaturing step to isolate it from ThfB (Supplementary Fig. 32b). When doubly dehydrated ThfA was incubated with ThfM in the presence and absence of SAM, formation of the 3-fold dehydrated product was strictly dependent on the presence of SAM and proceeded to ~50% completion within 1 h (Supplementary Fig. 34). This experiment shows that the ThfM enzyme can function on its own, as long as it provided a doubly dehydrated ThfA substrate, and that ThfM acts more rapidly than does ThfB.
Heterologous expression experiments demonstrated that most of the leader peptide of ThfA was needed for the ATP-grasp ThfB to function, while the O-methyltransferase ThfM only functioned on the doubly esterified precursor. With a functioning in vitro system, we could ask whether the leader peptide was required for the modification of the core peptide by ThfM. This experiment is not possible with heterologous expression due to the requirement for the leader peptide in the ThfB reaction. Reactions were set up with pre-fuscimiditide (ie doubly dehydrated core peptide without a leader (10 μM), ThfM (1 μM) and SAM (0.4 mM) in the same buffer described above. Both methylation and dehydration were observed on pre-fuscimiditide in this reaction, demonstrating that the leader peptide is not required for the proper function of ThfM (Supplementary Fig. 35). This experiment establishes that while the leader peptide is strictly required for esterification by ThfB, the leader peptide is dispensable for aspartimidylation by ThfM, a result analogous to what is observed in aspartimidylated lasso peptides.49
Further probing ThfM activity in vitro, we attempted modification of ThfA with a D77A substitution. This position corresponds to Asp10 of fuscimiditide, the site of aspartimidylation. We set up a reaction with ThfA D77A, ThfB, and ThfM with ATP and SAM in the same concentrations used above. As expected, overnight incubation of this reaction led to a product with only two dehydrations, further validating Asp10 as the site of aspartimidylation (Supplementary Fig. 36). Additionally, we noted above that fuscimiditide can hydrolyze into two distinct forms (Fig. 4a) with either Asp or isoAsp in the backbone. To explore the possibility of dual substrate recognition by ThfM, we hydrolyzed 3-fold dehydrated ThfA precursor to yield a mixture of full-length precursors corresponding to pre-fuscimiditide and iso pre-fuscimiditide. A C62A variant of ThfA incapable of disulfide bond formation (discussed below in more detail) was used for these experiments. Following hydrolysis, we incubated ThfM with this mixture under the abovementioned conditions. Intriguingly, overnight incubation yielded >90% conversion to the 3-fold dehydrated product (Fig. 5c). This result suggests the exciting possibility that ThfM is a promiscuous PIMT homolog that can drive both hydrolysis products, containing either Asp or isoAsp, back to the aspartimide.
Under the conditions established for the in vitro reconstitution of fuscimiditide, the rate of the ATP-grasp catalyzed reaction was fairly slow; it took several hours for ThfB to execute ~10 turnovers of ThfA (Fig. 5a). As discussed above, heterologous expression of doubly dehydrated ThfA purified under native conditions pulled down an equimolar amount of untagged ATP-grasp ThfB. Our initial thought was that this pulldown could be caused by disulfide bond formation between a Cys residue in ThfB with a Cys residue in the precursor ThfA. Therefore, we generated a C62A substitution in ThfA. However, ATP-grasp pulldown was still observed in the absence of Cys (Supplementary Fig. 37). This type of association between a RiPP product and its maturation enzyme has been observed before between microcin B17 and its maturation machinery.64 Ultimately, we were able to separate the 2-fold dehydrated precursor from ThfB by repurifying the complexes in the presence of urea (Supplementary Fig. 32b). Having the C62A ThfA variant allowed us to probe its interaction with the ATP-grasp ThfB via biolayer interferometry (BLI). To facilitate this experiment, a maltose binding protein (MBP) fusion to ThfB was purified to use as the analyte in the BLI experiments (Supplementary Fig. 38). Upon testing the unmodified and doubly dehydrated versions of C62A ThfA, strong affinity to ATP-grasp ThfB was observed for both versions. The Kd value for unmodified ThfA binding to ThfB was 28 nM, while doubly dehydrated ThfA exhibited a similar value of 18 nM indicating strong affinity to the substrate as well as the matured product (Supplementary Fig. 39). This tight binding between the enzyme and its product likely accounts for the low turnover numbers observed in the in vitro enzymology experiments described above.
Discussion
Here, we have described the discovery, biosynthesis, and NMR structure of fuscimiditide, a stem-loop bicyclic peptide and a novel example of graspetide RiPPs. The fuscimiditide BGC includes an O-methyltransferase gene that installs a stable aspartimide residue within the loop macrocycle. Though we have focused on this single example of an aspartimide-modified graspetide, bioinformatic analyses reveal that this class of RiPP BGCs is widespread, though primarily found in actinobacteria. Supplementary Fig. 40 shows a sampling of 50 putative precursors with homology to the fuscimiditide precursor ThfA. The BGC for each of these precursors also includes ATP-grasp and O-methyltransferase enzymes. These precursors are all enriched in Ser, Thr, Asp, and Glu residues near their C-termini, and all of them have an Asp residue that is the putative site for aspartimidylation. Of interest is that this Asp residue is mostly followed by either Gly or Ser. The architecture of these BGCs is also well-conserved with all examples exhibiting an A-B-M gene order except for fuscimiditide, in which the gene order is B-A-M as discussed above.
The aspartimide installed by O-methyltransferase ThfM differentiates fuscimiditide from other characterized graspetides. Normally aspartimide functions as either an intermediate in protein repair or as a nuisance product in solid-phase peptide synthesis or protein pharmaceutical formulation.44,45,65 Our data strongly supports the idea that, at least in cellulo, the aspartimidylated product fuscimiditide, is the final, intended product of the BGC. The three PTMs of fuscimiditide (two esters and one aspartimide) are all confined within the final 20 aa of the precursor peptide ThfA, so it is logical to think that there may be a protease that removes the leader peptide of ThfA and releases the macrocycle. We did not observe any obvious candidates for proteases in the immediate vicinity of the fuscimiditide BGC. We note that the fuscimiditide product generated by heterologous expression and tryptic digestion has two additional N-terminal amino acids relative to the fuscimiditide ΔN2 product observed in T. fusca. This result suggests that T. fusca harbors either a specific protease for leader removal or generalized peptidases that degrade the leader peptide. Recent work on class IV lanthipeptides has shown that multiple proteases not colocalized with the BGC can function in leader peptide removal.66,67 In general, the proteolysis of leader peptides is understudied in the graspetide field; a C39 peptidase-ABC transporter fusion protein has been hypothesized to serve this role for microviridins.18,31,62 If a similar system for leader peptide cleavage and secretion of fuscimiditide is present in the T. fusca genome, the final state of its aspartimide residue will be dictated by the environment into which it is secreted. While we expect the aspartimide to be long-lived in acidic or neutral conditions, it will rapidly hydrolyze to the isoAsp form, iso pre-fuscimiditide, in basic conditions. Another intriguing possibility is that posttranslationally modified ThfA remains in the cell. In this case, even if the aspartimide hydrolyzes, it will be dehydrated again due to the promiscuous activity of ThfM.
While this paper has focused on detailed characterization of the structure and biosynthesis of fuscimiditide, we have not studied its bioactivity in great depth. One possible speculation about the bioactivity of fuscimiditide and other aspartimide-modified graspetides is based on the observation that many members of this class serve as protease inhibitors.32,37 However, in an initial test we did not observe any inhibition of trypsin or elastase by fuscimiditide. The addition of an electrophilic moiety like the succinimide to such inhibitors could convert them into suicide inhibitors that covalently attach to an enzyme target. Similarly, the electrophilic aspartimide could be the site of a further modification by a nucleophilic molecule. In addition to graspetides as described here, this class of Asp-modifying O-methyltransferases has also been found in lasso peptide BGCs47 as well as in lanthipeptide BGCs.68 While all three examples of these RiPP modifying methyltransferases start off with Asp methylation and formation of aspartimide, the fate of the aspartimide residue differs. In the lanthipeptide case, the aspartimide moiety hydrolyzes to give a mixture of Asp and isoAsp residues in the lanthipeptide backbone. For the fuscimiditide case described here and the lasso peptide case, the macrocycle-embedded aspartimide moieties are more stable. When fuscimiditide hydrolyzes, it does so in a regioselective fashion yielding ~95% of the isoAsp backbone peptide. The opposite result is observed for the lasso peptides cellulonodin-2 and lihuanodin, both of which open exclusively to the Asp backbone form. The authors of the lanthipeptide work suggest that an aspartimide-modified lanthipeptide may be the desired product, which is more consistent with what we observe here. Taken together, these works demonstrate a unique function for the well-studied PIMT family of enzymes. This work also suggests further studies into the stability, bioactivity, and structural implications of aspartimide residues found in macrocycles.
Methods
Expression and purification of ThfA, ThfB, and ThfM.
His-tagged versions of ThfA, ThfB, and ThfM were generated using standard recombinant DNA techniques (see SI Methods for full details). Non His-tagged variants of ThfB and ThfM were also generated for coexpression with His-tagged ThfA (see SI Methods for cloning details). All proteins (ThfA coexpressed with ThfB, ThfM, or both, as well as ThfA, ThfB, and ThfM alone) were expressed in E. coli BL21(DE3) ΔslyD. E. coli harboring the plasmid of interest were cultured at 37 °C until the culture reached an OD600 of 0.5 at which point the culture was induced with 1 mM IPTG. The culture was further incubated at 37 °C for 4 h. The cells were collected by centrifugation, lysed by sonication, and His-tagged proteins were purified using Ni-NTA resin under either native or denaturing conditions (see SI methods for details). Purified proteins were desalted into phosphate buffered saline (PBS) with 10% glycerol and frozen at −80 °C until needed.
HPLC and mass spectrometry.
An HPLC system (Agilent 1200 series) was used for purification of fuscimiditide, pre-fuscimiditide, and iso pre-fuscimiditide, the 22 aa products of tryptic digests of modified ThfA. A semi-prep column (Zorbax 300SB-C18, 9.5 mm x 250 mm, 5 μm particle size, Agilent) was used for all purification steps; gradient details are in the SI Methods. For LC-MS analysis, an Agilent 6530 QTOF mass spectrometer was used downstream of an Agilent 1260 HPLC system. Two different analytical columns were used: for analysis of peptides, a Zorbax 300SB-C18, 2.1 mm x 50 mm, 3.5 μm particle size (Agilent) was used. For analysis of intact proteins such as ThfA and its modified variants, a XBridge Protein BEH C4 (2.1 mm x 50 mm, 3.5 μm particle size, Waters) was used. Mass spectra were analyzed using MassHunter Bioconfirm software.
NMR and structure determination.
NMR spectra were collected on a Bruker Avance III 800 MHz spectrometer. TOCSY spectra were acquired at 80 ms mixing time while NOESY spectra were acquired at both 150 ms and 700 ms mixing times. Proton-carbon HSQC and HMBC experiments were carried out at natural carbon abundance. The HMBC experiments were optimized at both 5 Hz and 10 Hz coupling constants. All spectra were assigned manually and integrated using MestReNova software; the 150 ms NOESY spectra were used to generate distance restraints. CYANA 2.1 was used to calculate 20 structural models. The structures were energy-minimized using Avogadro. The structural ensembles of fuscimiditide and pre-fuscimiditide have been deposited (PDB codes 7LIF and 7LI2, respectively).
Reconstitution of fuscimiditide biosynthesis in vitro.
Purified His-tagged ThfA, ThfB, and ThfM were used directly in in vitro assays. Assays with ThfA and ThfB were set up as follows: 10 μM ThfA, 1 μM ThfB, 10 mM ATP, 10 mM MgCl2, 10 mM DTT in 100 mM sodium phosphate buffer. Assays with ThfA, ThfB, and ThfM contained the above plus 1 μM ThfM and 400 μM SAM. In addition, assays were carried out with ThfA/modified ThfA and ThfM. These assays contained 10 μM ThfA/modified ThfA, 1 μM ThfM, 400 μM SAM, 10 mM MgCl2, 10 mM DTT in 100 mM sodium phosphate buffer. Reactions were carried out on a 50 μL scale in PCR tubes and incubated at 37 °C in a thermocycler. For time course analysis, a 10 μL was removed and quenched by the addition 1 μL of 10% formic acid. These samples were directly analyzed by LC-MS.
Supplementary Material
Acknowledgments
We thank I. Pelczer (Princeton University NMR Facility) for help with acquiring NMR spectra, H. Schroeder for assistance with tandem mass spectrometry, and R. Cohen and X. Wang (Merck & Co.) for their advice on conducting Marfey’s analysis. This work was supported by National Institutes of Health Grant GM107036 and a grant from Princeton University School of Engineering and Applied Sciences (Focused Research Team on Precision Antibiotics). L.C. was supported by an NSF Graduate Research Fellowship Program under Grant DGE-1656466. J.D.K. was supported in part by training grant T32 GM7388.
Footnotes
Competing Interests
The authors declare no competing interests.
Data Availability Statement
The coordinates for fuscimiditide and pre-fuscimiditide has been deposited in the Protein Data Bank (PDB) with accession codes 7LIF and 7LI2, respectively. Coordinates has also been deposited to the Biological Magnetic Resonance Bank (BMRB); the accession numbers are 30851 for fuscimiditide and 30849 for pre-fuscimiditide. Raw mass spectrometry data underlying figures will be provided upon request due to the large file sizes for this data. All other data is present in the main text or the supplementary information.
References
- 1.Arnison PG et al. Ribosomally synthesized and post-translationally modified peptide natural products: Overview and recommendations for a universal nomenclature. Nat. Prod. Rep 30, 108–160 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tan NH & Zhou J Plant cyclopeptides. Chem. Rev 106, 840–895 (2006). [DOI] [PubMed] [Google Scholar]
- 3.Gunasekera S, Daly NL, Anderson MA & Craik DJ Chemical synthesis and biosynthesis of the cyclotide family of circular proteins. IUBMB Life 58, 515–524 (2006). [DOI] [PubMed] [Google Scholar]
- 4.Cascales L & Craik DJ Naturally occurring circular proteins: Distribution, biosynthesis and evolution. Org. Biomol. Chem 8, 5035–5047 (2010). [DOI] [PubMed] [Google Scholar]
- 5.Lee J, Mcintosh J, Hathaway BJ & Schmidt EW Using marine natural products to discover a protease that catalyzes peptide macrocyclization of diverse substrates. J. Am. Chem. Soc 131, 2122–2124 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hegemann JD, Zimmermann M, Xie X & Marahiel MA Lasso Peptides: An Intriguing Class of Bacterial Natural Products. Acc. Chem. Res 48, 1909–1919 (2015). [DOI] [PubMed] [Google Scholar]
- 7.Maksimov MO & Link AJ Prospecting genomes for lasso peptides. J. Ind. Microbiol. Biotechnol 41, 333–344 (2014). [DOI] [PubMed] [Google Scholar]
- 8.Cheung-Lee WL & Link AJ Genome mining for lasso peptides: past, present, and future. J. Ind. Microbiol. Biotechnol 46, 1371–1379 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kawulka K et al. Structure of subtilosin A, an antimicrobial peptide from Bacillus subtilis with unusual posttranslational modifications linking cysteine sulfurs to α-carbons of phenylalanine and threonine. J. Am. Chem. Soc 125, 4726–4727 (2003). [DOI] [PubMed] [Google Scholar]
- 10.Kawulka KE et al. Structure of Subtilosin A, A Cyclic Antimicrobial Peptide from Bacillus subtilis with Unusual Sulfur to α-Carbon Cross-Links: Formation and Reduction of α-Thio-α-Amino Acid Derivatives. Biochemistry 43, 3385–3395 (2004). [DOI] [PubMed] [Google Scholar]
- 11.Brown LCW, Acker MG, Clardy J, Walsh CT & Fischbach MA Thirteen posttranslational modifications convert a 14-residue peptide into the antibiotic thiocillin. Proc. Natl. Acad. Sci. U. S. A 106, 2549–2553 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kelly WL, Pan L & Li C Thiostrepton biosynthesis: Prototype for a new family of bacteriocins. J. Am. Chem. Soc 131, 4327–4334 (2009). [DOI] [PubMed] [Google Scholar]
- 13.Bagley MC, Dale JW, Merritt EA & Xiong X Thiopeptide antibiotics. Chem. Rev 105, 685–714 (2005). [DOI] [PubMed] [Google Scholar]
- 14.Willey JM & van der Donk WA Lantibiotics : Peptides of Diverse Structure and Function. Annu. Rev. Microbiol 61, 477–501 (2007). [DOI] [PubMed] [Google Scholar]
- 15.Knerr PJ & van der Donk WA Discovery, Biosynthesis, and Engineering of Lantipeptides. Annu. Rev. Microbiol 81, 479–505 (2012). [DOI] [PubMed] [Google Scholar]
- 16.Bierbaum G & Sahl H Lantibiotics : Mode of Action , Biosynthesis and Bioengineering. Curr. Pharm. Biotechnol 10, 2–18 (2009). [DOI] [PubMed] [Google Scholar]
- 17.Schramma KR, Bushin LB & Seyedsayamdost MR Structure and biosynthesis of macrocyclic peptide containing an unprecedented lysine-to-tryptophan crosslink. Nat. Chem 7, 431–437 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Okino T, Matsuda H, Murakami M & Yamaguchi K New microviridins, elastase inhibitors from the blue-green alga Microcystis aeruginosa. Tetrahedron 51, 10679–10686 (1995). [Google Scholar]
- 19.Li K, Condurso HL, Li G, Ding Y & Bruner SD Structural basis for precursor protein-directed ribosomal peptide macrocyclization. Nat. Chem. Biol 12, 973–979 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liao R et al. Thiopeptide Biosynthesis Featuring Ribosomally Synthesized Precursor Peptides and Conserved Posttranslational Modifications. Chem. Biol 16, 141–147 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Walsh CT, Malcolmson SJ & Young TS Three Ring Posttranslational Circuses: Insertion of Oxazoles, Thiazoles, and Pyridines into Protein-Derived Frameworks. ACS Chem. Biol 7, 429–442 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ireland C, Scheuer PJ Ulicyclamide and Ulithiacyclámide, Two New Small Peptides from a Marine Tunicate. J. Am. Chem. Soc 102, 5688 (1980). [Google Scholar]
- 23.Morris RP et al. Ribosomally synthesized thiopeptide antibiotics targeting elongation factor Tu. J. Am. Chem. Soc 131, 5946–5955 (2009). [DOI] [PubMed] [Google Scholar]
- 24.Freeman MF et al. Metagenome Mining Reveals Polytheonamides as Posttranslationally Modified Ribosomal Peptides. Science 338, 387–391 (2012). [DOI] [PubMed] [Google Scholar]
- 25.Maksimov MO, Pan SJ & Link AJ Lasso peptides: Structure, function, biosynthesis, and engineering. Nat. Prod. Rep 29, 996–1006 (2012). [DOI] [PubMed] [Google Scholar]
- 26.Braffman NR et al. Structural mechanism of transcription inhibition by lasso peptides microcin J25 and capistruin. Proc. Natl. Acad. Sci. U. S. A 116, 1273–1278 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wilson KA et al. Structure of microcin J25, a peptide inhibitor of bacterial RNA polymerase, is a lassoed tail. J. Am. Chem. Soc 125, 12475–12483 (2003). [DOI] [PubMed] [Google Scholar]
- 28.Travin DY et al. Structure of ribosome-bound azole-modified peptide phazolicin rationalizes its species-specific mode of bacterial translation inhibition. Nat. Commun 10, 4563 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Metelev M et al. Klebsazolicin inhibits 70S ribosome by obstructing the peptide exit tunnel. Nat. Chem. Biol 13, 1129–1136 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Ishitsuka MO, Kusumi T, Kakisawa H, Kaya K & Watanabe MM Microviridin: A Novel Tricyclic Depsipeptide from the Toxic Cyanobacterium Microcystis viridis. J. Am. Chem. Soc 112, 8180–8182 (1990). [Google Scholar]
- 31.Ahmed MN et al. Phylogenomic Analysis of the Microviridin Biosynthetic Pathway Coupled with Targeted Chemo-Enzymatic Synthesis Yields Potent Protease Inhibitors. ACS Chem. Biol 12, 1538–1546 (2017). [DOI] [PubMed] [Google Scholar]
- 32.Ziemert N, Ishida K, Weiz A, Hertweck C & Dittmann E Exploiting the natural diversity of microviridin gene clusters for discovery of novel tricyclic depsipeptides. Appl. Environ. Microbiol 76, 3568–3574 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ziemert N, Ishida K, Liaimer A, Hertweck C & Dittmann E Ribosomal synthesis of tricyclic depsipeptides in bloom-forming cyanobacteria. Angew. Chemie - Int. Ed 47, 7756–7759 (2008). [DOI] [PubMed] [Google Scholar]
- 34.Philmus B, Christiansen G, Yoshida WY & Hemscheidt TK Post-translational modification in microviridin biosynthesis. Chembiochem 9, 3066–3073 (2008). [DOI] [PubMed] [Google Scholar]
- 35.Philmus B, Guerrette JP & Hemscheidt TK Substrate specificity and scope of MvdD, a GRASP-like ligase from the microviridin biosynthetic gene cluster. ACS Chem. Biol 4, 429–434 (2009). [DOI] [PubMed] [Google Scholar]
- 36.Lee H, Park Y & Kim S Enzymatic Cross-Linking of Side Chains Generates a Modified Peptide with Four Hairpin-like Bicyclic Repeats. Biochemistry 56, 4927–4930 (2017). [DOI] [PubMed] [Google Scholar]
- 37.Lee C, Lee H, Park JU & Kim S Introduction of Bifunctionality into the Multidomain Architecture of the ω-Ester-Containing Peptide Plesiocin. Biochemistry 59, 285–289 (2020). [DOI] [PubMed] [Google Scholar]
- 38.Roh H, Han Y, Lee H & Kim S A Topologically Distinct Modified Peptide with Multiple Bicyclic Core Motifs Expands the Diversity of Microviridin-Like Peptides. ChemBioChem 20, 1051–1059 (2019). [DOI] [PubMed] [Google Scholar]
- 39.Lee H, Choi M, Park JU, Roh H & Kim S Genome Mining Reveals High Topological Diversity of ω-Ester-Containing Peptides and Divergent Evolution of ATP-Grasp Macrocyclases. J. Am. Chem. Soc 142, 3013–3023 (2020). [DOI] [PubMed] [Google Scholar]
- 40.Ramesh S et al. Bioinformatics-Guided Expansion and Discovery of Graspetides. ACS Chem. Biol 16, 2787–2797 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kaweewan I, Nakagawa H & Kodani S Heterologous expression of a cryptic gene cluster from Marinomonas fungiae affords a novel tricyclic peptide marinomonasin. Appl. Microbiol. Biotechnol 105, 7241–7250 (2021). [DOI] [PubMed] [Google Scholar]
- 42.Unno K & Kodani S Heterologous expression of cryptic biosynthetic gene cluster from Streptomyces prunicolor yields novel bicyclic peptide prunipeptin. Microbiol. Res 244, 126669 (2021). [DOI] [PubMed] [Google Scholar]
- 43.Montalbán-López M et al. New developments in RiPP discovery, enzymology and engineering. Nat. Prod. Rep 38, 130 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Murray ED & Clarke S Metabolism of a synthetic L-isoaspartyl-containing hexapeptide in erythrocyte extracts: Enzymatic methyl esterification is followed by nonenzymatic succinimide formation. J. Biol. Chem 261, 306–312 (1986). [PubMed] [Google Scholar]
- 45.McFadden PN & Clarke S Conversion of isoaspartyl peptides to normal peptides: Implications for the cellular repair of damaged proteins. Proc. Natl. Acad. Sci. U. S. A 84, 2595–2599 (1987). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ingrosso D et al. Increased methyl esterification of altered aspartyl residues erythrocyte membrane proteins in response to oxidative stress. Eur. J. Biochem 267, 4397–4405 (2000). [DOI] [PubMed] [Google Scholar]
- 47.Young AL, Carter WG, Doyle HA, Mamula MJ & Aswad DW Structural Integrity of Histone H2B in Vivo Requires the Activity of Protein L-Isoaspartate O-Methyltransferase, a Putative Protein Repair Enzyme. J. Biol. Chem 276, 37161–37165 (2001). [DOI] [PubMed] [Google Scholar]
- 48.Koos JD & Link AJ Heterologous and in Vitro Reconstitution of Fuscanodin, a Lasso Peptide from Thermobifda fusca. J. Am. Chem. Soc 141, 928–935 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cao L, Beiser M, Koos JD, Orlova M, Elashal HE, Schroder HV & Link AJ Cellulonodin-2 and Lihuanodin: Lasso Peptides with an Aspartimide Post-Translational Modification. J. Am. Chem. Soc 143, 11690–11702 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Iyer LM, Abhiman S, Burroughs AM, Aravind L Amidoligases with ATP-grasp, glutamine synthetase-like and acetyltransferase-like domains: synthesis of novel metabolites and peptide modifications of proteins. Mol. BioSyst 5, 1636–1660 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Vaisar T & Urban J Probing Proline Effect in CID of Protonated Peptides. J. Mass Spectrom 31, 1185–1187 (1996). [DOI] [PubMed] [Google Scholar]
- 52.Someya CI, Inoue S, Irran E, Krackl S & Enthaler S New Binding Modes of 1-Acetyl- and 1-Benzoyl-5-hydroxypyrazolines - Synthesis and Characterization of O,O′-Pyrazoline- and N,O-Pyrazoline-Zinc Complexes. Eur. J. Inorg. Chem 2011, 2691–2697 (2011). [Google Scholar]
- 53.Klaene JJ, Ni W, Alfaro JF & Zhou ZS Detection and Quantitation of Succinimide in Intact Protein via Hydrazine Trapping and Chemical Derivatization. J. Pharm. Sci 103, 3033–3042 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Geiger T, Clarke S. Deamidation, Isomerization, and Racemization at Asparaginyl and Aspartyl Residues in Peptides. J. Biol. Chem 262, 785–794 (1987). [PubMed] [Google Scholar]
- 55.Ulrich EL et al. BioMagResBank 36, 402–408 (2008). [Google Scholar]
- 56.Grassi L et al. Complete NMR Assignment of Succinimide and Its Detection and Quantification in Peptides and Intact Proteins. Anal. Chem 89, 11962–11970 (2017). [DOI] [PubMed] [Google Scholar]
- 57.Cheung WL, Pan SJ & Link AJ Much of the Microcin J25 Leader Peptide is Dispensable. J. Am. Chem. Soc 25, 2514–2515 (2010). [DOI] [PubMed] [Google Scholar]
- 58.Xie L et al. Lacticin 481: In Vitro Reconstitution of Lantibiotic Synthetase Activity. Science 303, 679–681 (2004). [DOI] [PubMed] [Google Scholar]
- 59.Oman TJ, Knerr PJ, Bindman NA, Vela JE & Donk WA Van Der. An Engineered Lantibiotic Synthetase That Does Not Require a Leader Peptide on its Substrate. J. Am. Chem. Soc 134, 6952–6955 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Koehnke J et al. Structural analysis of leader peptide binding enables leader-free cyanobactin processing. Nat. Chem. Biol 11, 558–563 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Reyna-González E, Schmid B, Petras D, Süssmuth RD & Dittmann E Leader Peptide-Free In Vitro Reconstitution of Microviridin Biosynthesis Enables Design of Synthetic Protease-Targeted Libraries. Angew. Chemie - Int. Ed 55, 9398–9401 (2016). [DOI] [PubMed] [Google Scholar]
- 62.Weiz AR et al. Leader Peptide and a Membrane Protein Scaffold Guide the Biosynthesis of the Tricyclic Peptide Microviridin. Chem. Biol 18, 1413–1421 (2011). [DOI] [PubMed] [Google Scholar]
- 63.Pederick JL, Thompson AP, Bell SG & Bruning JB D-Alanine – D-alanine ligase as a model for the activation of ATP-grasp enzymes by monovalent cations. J. Biol. Chem 295, 7894–7904 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ghilarov D et al. Architecture of Microcin B17 Synthetase: An Octameric Protein Complex Converting a Ribosomally Synthesized Peptide into a DNA Gyrase Poison. Mol. Cell 73, 749–762 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Neumann K, Farnung J, Baldauf S & Bode JW Prevention of aspartimide formation during peptide synthesis using cyanosulfurylides as carboxylic acid-protecting groups. Nat. Commun 1–10 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ren H et al. Discovery and Characterization of a Class IV Lanthipeptide with a Nonoverlapping Ring Pattern. ACS Chem. Biol 15, 1642–1649 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hegemann JD, Sussmuth RD RSC Chem. Biol 1, 110–127 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Acedo JZ et al. O -Methyltransferase-Mediated Incorporation of a β-Amino Acid in Lanthipeptides. J. Am. Chem. Soc 141, 16790–16801 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The coordinates for fuscimiditide and pre-fuscimiditide has been deposited in the Protein Data Bank (PDB) with accession codes 7LIF and 7LI2, respectively. Coordinates has also been deposited to the Biological Magnetic Resonance Bank (BMRB); the accession numbers are 30851 for fuscimiditide and 30849 for pre-fuscimiditide. Raw mass spectrometry data underlying figures will be provided upon request due to the large file sizes for this data. All other data is present in the main text or the supplementary information.