

Abstract
Objective:
This study aims to investigate the role of the hyperglycemia-induced increase in tumor necrosis factor-α (TNF-α) and interferon-γ (IFN-γ) in the ubiquitination and degradation of platelet endothelial cell adhesion molecule-1 (PECAM-1) in the diabetic retina.
Methods:
Type-I diabetes was induced in rats by the injection of streptozotocin, with age-matched non-diabetic rats as controls. Primary rat retinal microvascular endothelial cells were grown in normal or high glucose media for six days, or in normal glucose media for 24 hours with addition of TNF-α and/or IFN-γ. PECAM-1, TNF-α, IFN-γ, and ubiquitin levels were assessed using western blotting, immunofluorescence, and immunoprecipitation assays. Additionally, proteasome activity was assessed both in vivo and in vitro.
Results:
Under hyperglycemic conditions, total ubiquitination levels in the retina and RRMECs, and PECAM-1 ubiquitination levels in RRMECs, were significantly increased. Additionally, TNF-α and IFN-γ levels were significantly increased under hyperglycemic conditions. PECAM-1 levels in RRMECs treated with TNF-α and/or IFN-γ were significantly decreased. Moreover, there was a significant decrease in proteasome activity in the diabetic retina, in hyperglycemic RRMECs, and in RRMECs treated with TNF-α or IFN-γ.
Conclusion:
TNF-α and IFN-γ may contribute to the hyperglycemia-induced loss of PECAM-1 in retinal endothelial cells, possibly by upregulating PECAM-1 ubiquitination.
Keywords: Diabetic retinopathy, PECAM-1, TNF-α, IFN-γ, Ubiquitination
Introduction
Hyperglycemia is a causative and independent factor that can affect the homeostasis of retinal vascular endothelial cells, and can trigger mechanisms leading to the development of diabetic retinopathy (DR) 1. DR is a major complication of diabetes, and the leading cause of blindness among working-age adults worldwide 2, 3. Hyperglycemia-induced microvascular alterations are a hallmark of DR, which include an increase in vascular permeability 4, microaneurysms 5, cotton-wool spots 6, abnormal neovascularization 7, and capillary blockage8. The molecular mechanisms leading to the development of DR are still under investigation, including the hyperglycemia-induced loss of platelet endothelial cell adhesion molecule-1 (PECAM-1), a protein that plays essential roles in the maintenance and proper function of the endothelium.
PECAM-1 is a cell surface protein that belongs to the immunoglobulin (Ig) superfamily with an immunoreceptor tyrosine-based inhibitory motif (ITIM) domain 9. The structure of PECAM-1 consists of six extracellular Ig domains, a short transmembrane domain, and a large and complex cytoplasmic tail 10. PECAM-1 is highly expressed on endothelial cells, as well as on platelets, neutrophils, monocytes, and some T-cells 10. Moreover, multiple mechanisms by which endothelial cells are regulated, maintained, and enabled to perform their function involve PECAM-1, such as cell-to-cell adhesion 11, maintenance of the blood barrier 12–14, leukocyte transmigration 15–17, cell survival 18–20, mechanotransduction 18, 21–23, angiogenesis 24, and endothelial cell signaling 25–27. Therefore, a loss of PECAM-1 can have detrimental effects on endothelial cell integrity and function.
Hyperglycemia can activate multiple signaling pathways and cellular mechanisms that can participate in DR pathogenesis, including inflammation 28, 29. Inflammation is a process by which a response to an injury or stress is initiated via complex signaling cascades. The identification of DR as an inflammatory disease has been recognized due to the multiple inflammatory processes involved in the development and progression of the disease 29, 30. The binding of pathogens or advanced glycation end products (AGEs) to their receptor can trigger the synthesis of inflammatory molecules that have detrimental effects on the vascular endothelium31. Moreover, studies have reported the upregulation of multiple inflammatory cytokines and chemokines, including tumor necrosis factor-α (TNF-α) 32, 33 and interferon-γ (IFN-γ) 33, in the vitreous and plasma of diabetic patients and animal models.
TNF-α and IFN-γ are potent proinflammatory cytokines that participate in the inflammatory response of DR. TNF-α is expressed initially as a transmembrane protein, and upon its cleavage, is released into the plasma forming a homotrimer that is its active form34, 35. Studies have revealed a role for TNF-α in the blood-retinal barrier (BRB) breakdown and cellular apoptosis that occur in diabetic conditions in the retina 36. Moreover, TNF-α can regulate the gene expression of a plethora of proteins that are involved in inflammation and apoptosis 37, and is achieved through the complex signaling pathways initiated when TNF-α interacts and binds with its receptors, TNFR1 and TNFR2 34, 37. IFN-γ is a pleiotropic, N-glycosylated, homodimer cytokine that has immunomodulatory, antiviral, and antitumor functions38, 39. In addition, IFN-γ can regulate the gene expression of proteins involved in inflammation and immunity via its interaction with the IFN-γ receptor, and the activation of the Jak-STAT1 signaling pathways 40, 41. Furthermore, IFN-γ has been shown to increase endothelial cell permeability via the activation of p38 MAP kinase and the rearrangement of the actin cytoskeleton 42.
TNF-α and IFN-γ have been shown to decrease PECAM-1 expression at the transcriptional level 43–47. However, to our knowledge, no reports have investigated the posttranslational regulation of PECAM-1 under hyperglycemic conditions in the retina, or the role of inflammatory mediators in this process. We have recently reported in the diabetic rat retina a loss of PECAM-1 48 and decreased PECAM-1/β-catenin interactions49, with a possible cleavage of PECAM-1 by matrix metalloproteinases (MMPs). Additionally, we observed decreased PECAM-1 levels in the plasma of diabetic rats 48, which partially could occur due to intracellular degradation via the ubiquitin-proteasome pathway. The current study was conducted to examine the posttranslational modification of PECAM-1 in retinal endothelial cells under hyperglycemic conditions, and the role that inflammatory mediators have in this process.
Methods
Animals
Male Wistar rats (Envigo, Indianapolis, IN, purchased at 100-120 g) were used in a model of type I diabetes, and were provided food and water ad libitum. The experimental protocols used in this study were approved by the Institutional Animal Care and Use Committee of LSUHSC-Shreveport.
Animal model of type I diabetes in rats
Age-matched rats were injected intraperitoneally with either vehicle (sodium citrate buffer, control) or streptozotocin (STZ, diabetic, Sigma-Aldrich, St. Louis, MO), 30 mg/kg/day, for three consecutive days 48, 50, 51. Glucose levels and body weight were monitored, and hyperglycemia was defined as rats having a non-fasting glucose value of >300 mg/dl, with control rats having a non-fasting glucose value of ~150 mg/dl. Eight weeks post STZ injections, rats were anesthetized with ketamine/pentobarbital (100 and 50 mg/kg, respectively); femoral artery cannulations were used for blood collection, and eyes were enucleated to collect retinas for further analysis. To collect plasma, blood was centrifuged at 10,000 g for 10 minutes, at 4 °C, and plasma was collected and stored at −80 °C until used.
Cell culture and treatments
For in vitro experiments, primary rat retinal microvascular endothelial cells (RRMECs, Cell Biologics, Chicago, IL) were grown under either normal glucose conditions (NG, 5 mM), high glucose conditions (HG, 25 mM), or with mannitol as an osmolar control (20 mM mannitol + 5 mM glucose), for six days using Dulbecco’s modified eagle media (DMEM) containing 10% fetal bovine serum. For cytokine treatments, TNF-α (20 ng/ml, MilliporeSigma, Burlington, MA) and IFN-γ (30 ng/ml, MilliporeSigma, Burlington, MA) were added to confluent RRMECs grown under NG conditions for 24 hours. For treatment with MMPs, MMP-2 and MMP-9 (R&D Systems, Minneapolis, MN) were activated using 4-aminophenylmercuric acetate (APMA, Sigma-Aldrich, St. Louis, MO) prior to their use. Briefly, MMP-2 and MMP-9 were incubated (1 hour or 8 hours, respectively) in 2.5 mM APMA at 37 °C, after which, activated MMP-2 (at 50, 100, 200, and 400 ng/ml), or MMP-9 (at 50, 100, 200, and 500 ng/ml) were added to confluent RRMECs grown under NG conditions for 24 hours.
In vitro apoptosis assay
Apoptosis in RRMECs treated with TNF-α and/or IFN-γ was determined using the Annexin V-FITC and propidium iodide (PI) apoptosis assay kit according to the manufacturer’s instructions (abcam, Cambridge, MA). Briefly, RRMECs were plated on glass coverslips, and at 100% confluency were treated with the cytokines at the indicated concentration for 24 hours. Samples were then incubated with 0.5 μg/ml Annexin V-FITC and 2 μg/ml PI for 5 minutes at room temperature in the dark. Cells were washed with the Annexin V binding buffer provided by the kit and fixed with 4% paraformaldehyde. Mounting media containing DAPI was added, and the cells were imaged using a NIKON E600FN fluorescent microscope and analyzed using ImageJ (NIH). As a positive control for apoptosis, staurosporin (1 μM) was added to confluent RRMECs for 4 and 8 hours, followed by the apoptosis assay. Additionally, permeabilized and fixed cells were stained with PI to show positive labeling of nuclei.
Western blotting
For western blot analyses, RRMECs or retinas were collected in ice-cold radioimmunoprecipitation assay (RIPA, Sigma-Aldrich, St. Louis, MO) buffer with the addition of protease inhibitors (Sigma-Aldrich, St. Louis, MO). Protein concentrations were assessed using the bicinchoninic acid assay (BCA, Thermo Fisher Scientific, Waltham, MA). Laemmli buffer was added to samples under reducing and denaturing conditions, and equal amounts of protein were loaded on 8-12% SDS-polyacrylamide gels. Proteins were transferred into nitrocellulose membranes, blocked with a blocking buffer (Protein free block, Thermo Fisher Scientific, Waltham, MA), and primary antibodies were added overnight at 4 °C (PECAM-1, 1:1500; Santa Cruz Biotechnology, Dallas, TX; TNF-α, 1:1000; Bioss antibodies, Woburn, MA; IFN-γ, 1:1000; abcam, Cambridge, MA). HRP-conjugated secondary antibodies (Jackson ImmunoResearch, 1:10000, West Grove, PA) were added for one hour at room temperature. An internal housekeeping protein was used to ensure equal loading and proper transfer (β-actin, 1:5000; Sigma-Aldrich), and plasma samples were normalized to total protein using a Ponceau total protein stain (Sigma-Aldrich). A ChemiDoc XRS gel imaging system (Bio-Rad, Hercules, CA) was used to detect the specific bands, and the bands were quantified by densitometry using ImageJ (National Institutes of Health (NIH), Bethesda, MD, USA).
Plasma levels of IFN-γ
The levels of IFN-γ in plasma collected from control (non-diabetic) and diabetic rats were determined using rat IFN-γ quantikine® enzyme-linked immunosorbent assay (ELISA) (R&D Systems, Minneapolis, MN) according to the manufacturer’s instructions.
Immunofluorescence (IF) labeling
RRMECs were fixed with ice-cold methanol for 5 minutes, permeabilized using 0.1% Triton-X100 in phosphate buffered saline (PBS), and blocked with 5% bovine serum albumin (BSA) and 10 % FBS for one hour at RT. The anti-PECAM-1 antibody (1:100) was added to the cells, and incubated at 4 °C overnight. Fluorescently labeled secondary antibodies (1:1000) were added for one hour at room temperature, and a mounting medium containing DAPI (Vector Labs, Burlingame, CA, USA) was used. Images were taken with a NIKON E600FN fluorescent microscope and analyzed using ImageJ (NIH)52. Secondary antibody staining without a primary antibody was used to ensure specificity.
In vitro protein ubiquitination assay
Retinas and RRMECs were collected and lysed in a cold lysis buffer (50 mM Tris-HCl, 150 mM NaCl, 1% Triton X-100, 0.1% SDS, 1 mM EDTA, PH 7.6) containing protease inhibitors (Sigma, St. Louis, MO). The primary antibody for the protein of interest was incubated with SureBeads magnetic beads (Bio-rad, Hercules, CA) for 10 minutes at room temperature. Equal amounts of retinal or cellular lysate were added to the antibody-beads complex and incubated for 1 hour at room temperature. Immunocomplexes were washed and resuspended in 1X Laemmli buffer by heating the sample to 75 °C for 15 minutes, and were subjected to SDS-PAGE western blot analysis. Ubiquitin was detected by incubating the membranes with an anti-ubiquitin antibody (Enzo-life Sciences, Farmingdale, NY), and blots were further incubated with an antibody against the protein of interest to act as an assay efficiency and loading control.
In silico analysis of MMP cleavage sites
The predicted cleavage sites of TNF-α by MMPs were identified using a Protease Specificity Prediction Server (PROSPER; Monash University, Victoria, Australia 53). A rat TNF-α protein amino acid sequence (UniProt Consortium, Wellcome Trust Genome Campus, Hinxton, UK) was used to confirm TNF-α as a substrate for MMPs.
Proteasome activity assay
RRMECs grown under NG and HG conditions, and retinas from non-diabetic and diabetic rats, were collected in 0.5% NP-40 lysis buffer without the addition of protease inhibitors. Cells and retinas were homogenized, centrifuged at 16,000 g at 4 °C for 15 minutes, and the supernatants were collected. Proteasome activity under hyperglycemic conditions was assessed in the retinas and RRMECs using a proteasome activity assay kit (abcam) following the manufacturer’s protocol. Fluorescence was measured with an Epoch microplate spectrophotometer (BioTek Instruments, Winooski, VT), with the addition or absence of an MG132 proteasome inhibitor included with the kit.
Statistical analysis
Statistical comparisons were conducted using GraphPad Prism software (La Jolla, CA). Student t-tests or one-way analysis of variance (ANOVA) followed by Student-Newman-Keuls post-hoc correction were performed to compare the means of groups. P < 0.05 was considered statistically significant, and the data presented express the mean ± standard error.
Results
The inflammatory mediators TNF-α and IFN-γ are upregulated under hyperglycemic conditions
Studies have suggested a role for inflammation in the pathogenesis and progression of DR. TNF-α and IFN-γ are potent inflammatory mediators that participate in initiating the inflammatory response, and lead to the deleterious effects on endothelial cell homeostasis and function37, 54, 55. TNF-α levels did not change when comparing non-diabetic and diabetic retinas, from whole retinal homogenates (Figure 1A). However, TNF-α western blot mean band intensities were significantly increased in the plasma collected from diabetic rats (120% increase, p<0.05, Figure 1B) and in RRMECs (46% increase, p<0.05, Figure 1C) grown under hyperglycemic conditions. Additionally, IFN-γ western blot mean band intensities from diabetic retinal tissues, and plasma IFN-γ concentrations collected from diabetic rats, were significantly increased (by 35% and 224%, respectively, p<0.05, Figure 2A, 2B) when compared to retinal tissues and plasma collected from control rats. Moreover, IFN-γ western blot mean band intensities were significantly increased in RRMECs grown under HG and mannitol treatment conditions (by 80% and 42%, respectively, Figure 2C). No statistical significance was found between HG and mannitol treatments in TNF-α and IFN-γ levels in RRMECs (Figure 1C, 2C).
Figure 1. TNF-α levels under hyperglycemic conditions.
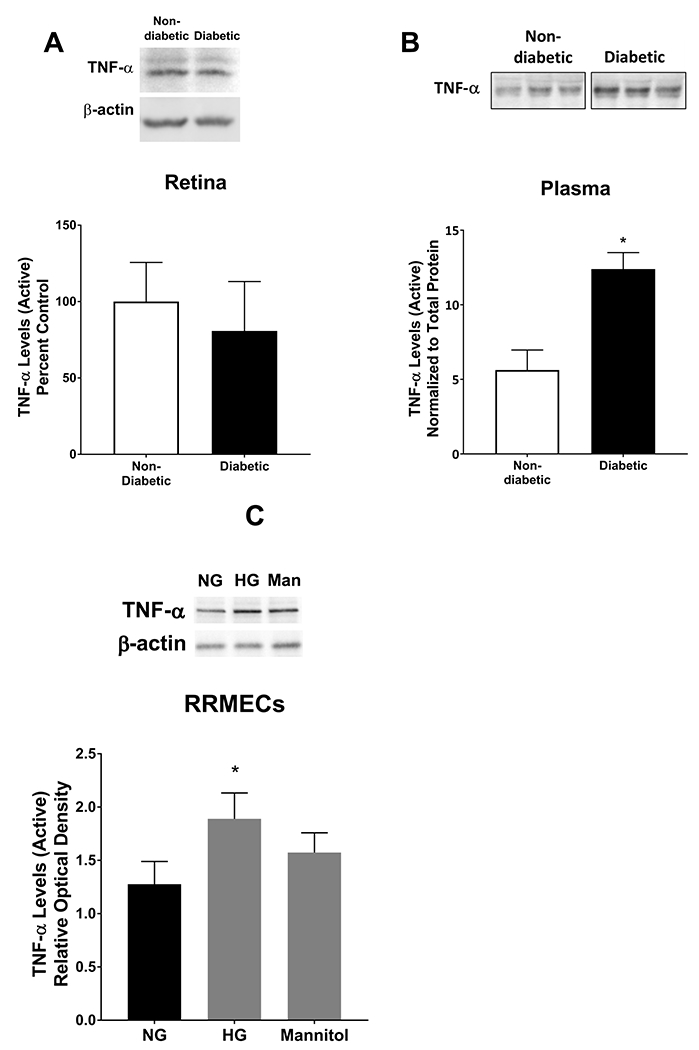
TNF-α levels in retinas, plasma, and RRMECs were determined using western blotting techniques. A) TNF-α levels in retinas collected from diabetic rats were not altered when compared to retinas collected from non-diabetic rats. However, there was a significant increase in TNF-α levels in plasma (B) collected from diabetic rats when compared to controls. C) TNF-α in RRMECs grown under hyperglycemic (HG) conditions had significantly higher levels when compared to RRMECs grown under NG conditions, and no significant change observed with mannitol (Man). Western blotting data is a comparison of the relative TNF-α/β-actin or TNF-α/total protein (Plasma) band densities.*p<0.05, N=5-7 per group in vivo and N=3 per group in vitro.
Figure 2. IFN-γ levels under hyperglycemic conditions.
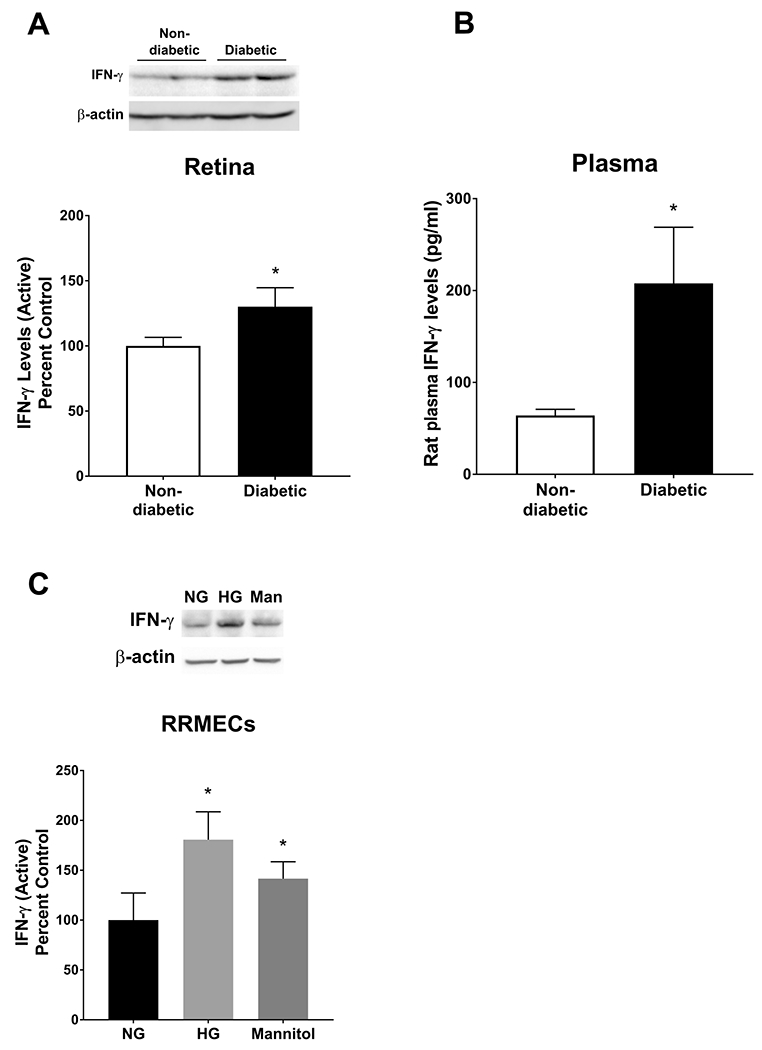
IFN-γ levels were significantly increased in retinas and plasma collected from diabetic rats (A, B) and in RRMECs grown under hyperglycemic conditions (HG) and mannitol (Man) (C) when compared to controls. Western blotting data is a comparison of the relative IFN-γ/β-actin band densities. *p<0.05, N=5-7 per group in vivo and N=3-6 per group in vitro.
MMP-2 but not MMP-9 increases TNF-α levels
The cleavage of TNF-α and its release to the plasma is an important step of its activation34, and could possibly be performed by MMPs. We have previously reported upregulated MMP-2 and MMP-9 levels in RRMECs grown under hyperglycemic conditions 48. Thus, we wanted to investigate whether the increase in MMPs can affect TNF-α expression. Treating RRMECs with MMP-2 (50-400 ng/ml, 24 hours) significantly increased active TNF-α western blot mean band intensities (by > 2-fold increases, p<0.05, Figure 3A); however, MMP-9 treatment did not modulate active TNF-α levels in RRMECs (Figure 3B). Moreover, we performed an in silico analysis to identify possible MMP cleavage sites on TNF-α (PROSPER), and identified possible cleavage sites for MMP-2, MMP-3 and MMP-9 (Figure 3C).
Figure 3. MMP-2, but not MMP-9, increased TNF-α in RRMECs.
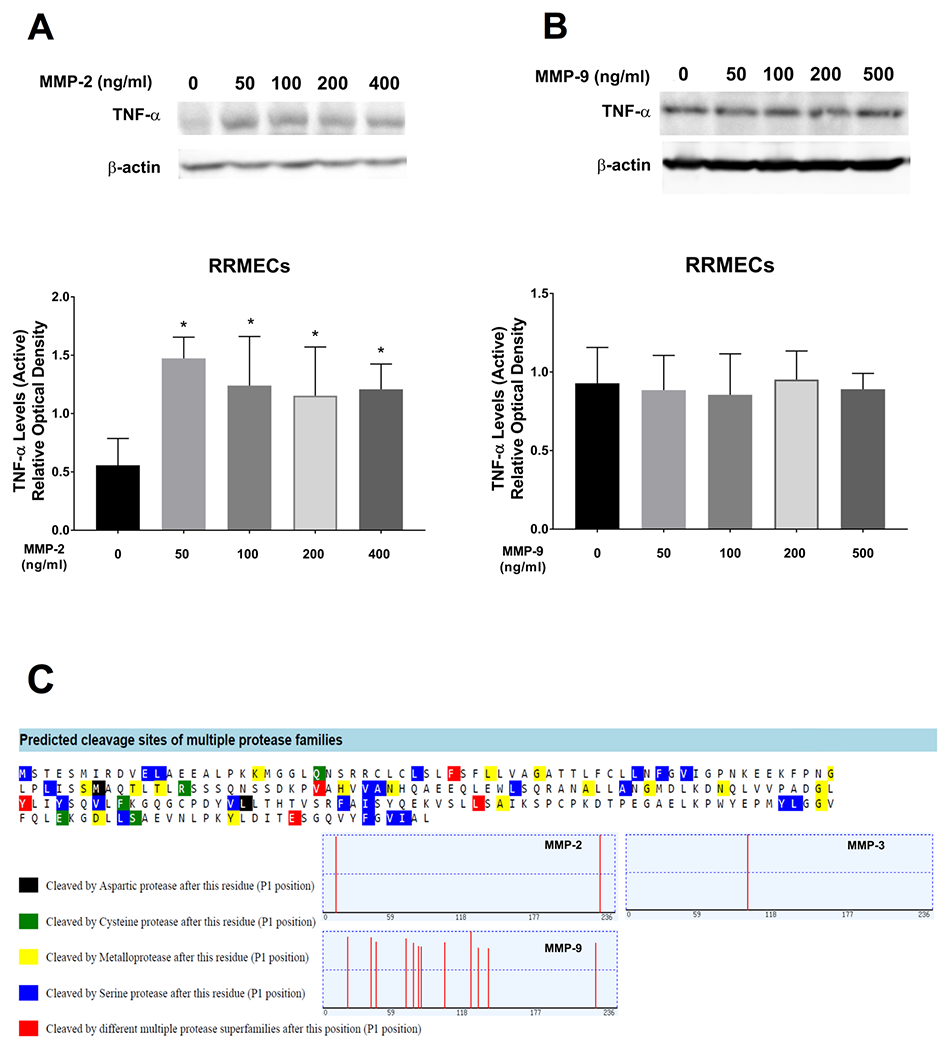
A) TNF-α levels were significantly increased in RRMECs treated with MMP-2, but no change in TNF-α levels (B) was observed in RRMECs treated with MMP-9. C) In silico analysis (Prosper) to predict TNF-α cleavage sites by MMPs revealed cleavage sites for MMP-2, MMP-3, and MMP-9. Western blotting data is a comparison of the relative TNF-α/β-actin band densities.*p<0.05, N=3 per group.
PECAM-1 levels are significantly decreased with TNF-α and IFN-y treatments
To study the effects of TNF-α and IFN-γ treatments on PECAM-1 levels in retinal endothelial cells, confluent RRMECs in normoglycemic media were treated with TNF-α (5, 10, 20, and 50 ng/ml) for 24 hours, with a significant loss of PECAM-1 observed at the 20 and 50 ng/ml concentrations (Figure 4A). Additionally, TNF-α (20 ng/ml), IFN-γ (30 ng/ml), or a combination of both cytokines were added to RRMECs in NG media for 24 hours. PECAM-1 mean band intensities were significantly decreased with TNF-α treatment (40% decrease, p<0.05), IFN-γ treatment (57% decrease, p<0.05), TNF-α and IFN-γ combined treatment (55% decrease, p<0.05), as well as in RRMECs grown under HG conditions (50% decrease, p<0.05) (Figure 4B). Moreover, immunofluorescence staining of RRMECs treated with TNF-α, IFN-γ, or both, showed a decrease in PECAM-1 intensity, with the effects of TNF-α and IFN-γ not additive (Figure 4C). To investigate whether PECAM-1 loss was due to apoptosis56 with cytokine treatments, we performed an apoptosis assay using the Annexin V-FITC PI kit, and found no change between the treated and untreated RRMECs (Figure 5A), with positive controls shown in Figure 5B.
Figure 4. TNF-α and IFN-γ significantly decreased PECAM-1 levels in RRMECs.
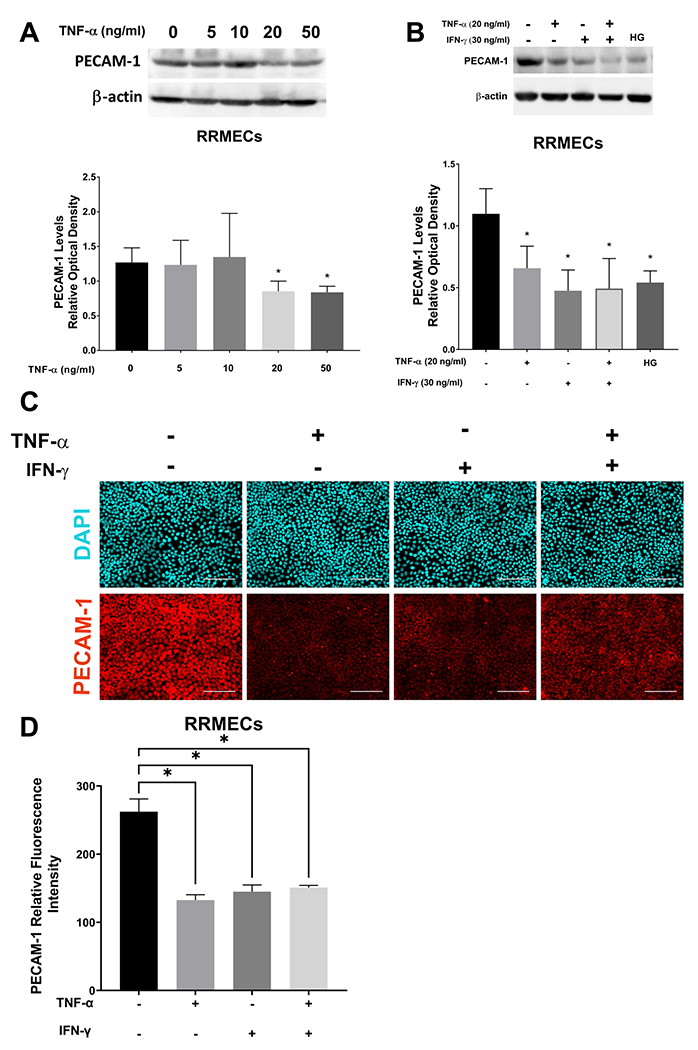
A) TNF-α (5, 10, 20, 50 ng/ml) treated RRMECs (24 hours) had a significant PECAM-1 loss at 20 and 50 ng/ml. B) TNF-α (20 ng/ml) and IFN-γ (30 ng/ml) treatments for 24 hours significantly decreased PECAM-1 levels in RRMECs, as did hyperglycemia (HG) following 6 days of treatment. C) Representative immunostaining images showing decreased PECAM-1 levels with TNF-α and IFN-γ treatments. D) Quantification of PECAM-1 fluorescence intensity with TNF-α and IFN-γ treatments. Western blotting data is a comparison of the relative PECAM-1/β-actin band densities. Scale bar = 50 μm, *p<0.05, N=3 per group.
Figure 5. TNF-α and IFN-γ treatment on RRMECs did not induce apoptosis.
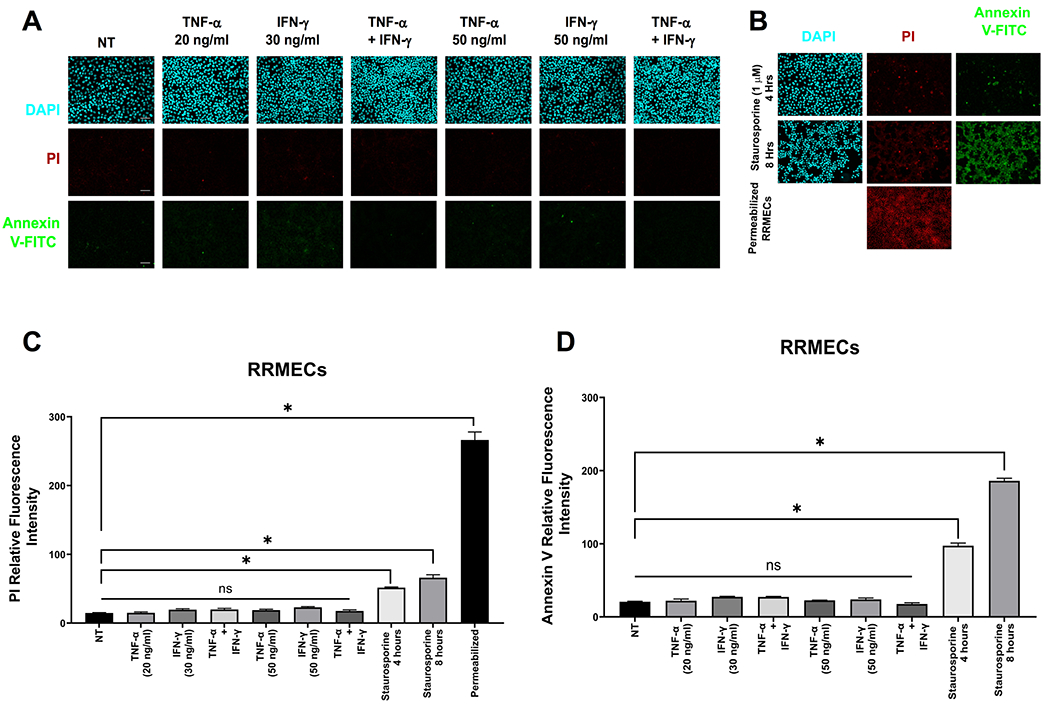
A) Apoptosis assays (propidium iodide (PI) and annexin V) of RRMECs treated with TNF-α and IFN-γ showing no change from non-treated (NT) cells. B) Positive controls for apoptosis. C) Quantification of propidium iodide (PI) fluorescence intensity with TNF-α and IFN-γ treatments. D) Quantification of annexin V fluorescence intensity with TNF-α and IFN-γ treatments. Scale bar = 50 μm, *p<0.05, N=3 per group.
PECAM-1 ubiquitination levels are significantly increased under hyperglycemic conditions
Ubiquitination of proteins is an important posttranslational regulatory mechanism by which ubiquitin is added to a protein by a multi-step enzymatic action57. Polyubiquitinated proteins can then be targeted for degradation by proteasomes. Total protein ubiquitination was significantly increased in the diabetic retina when compared to controls (37% increase, p<0.05, Figure 6A), and in RRMECs grown under HG or mannitol treatment (by 39% and 22%, respectively, p<0.05, Figure 6B). Moreover, PECAM-1 ubiquitination levels were significantly increased in RRMECs grown under HG conditions (81% increase, p<0.05, Figure 6C).
Figure 6. Total ubiquitination and PECAM-1 ubiquitination levels were significantly increased under hyperglycemic conditions.
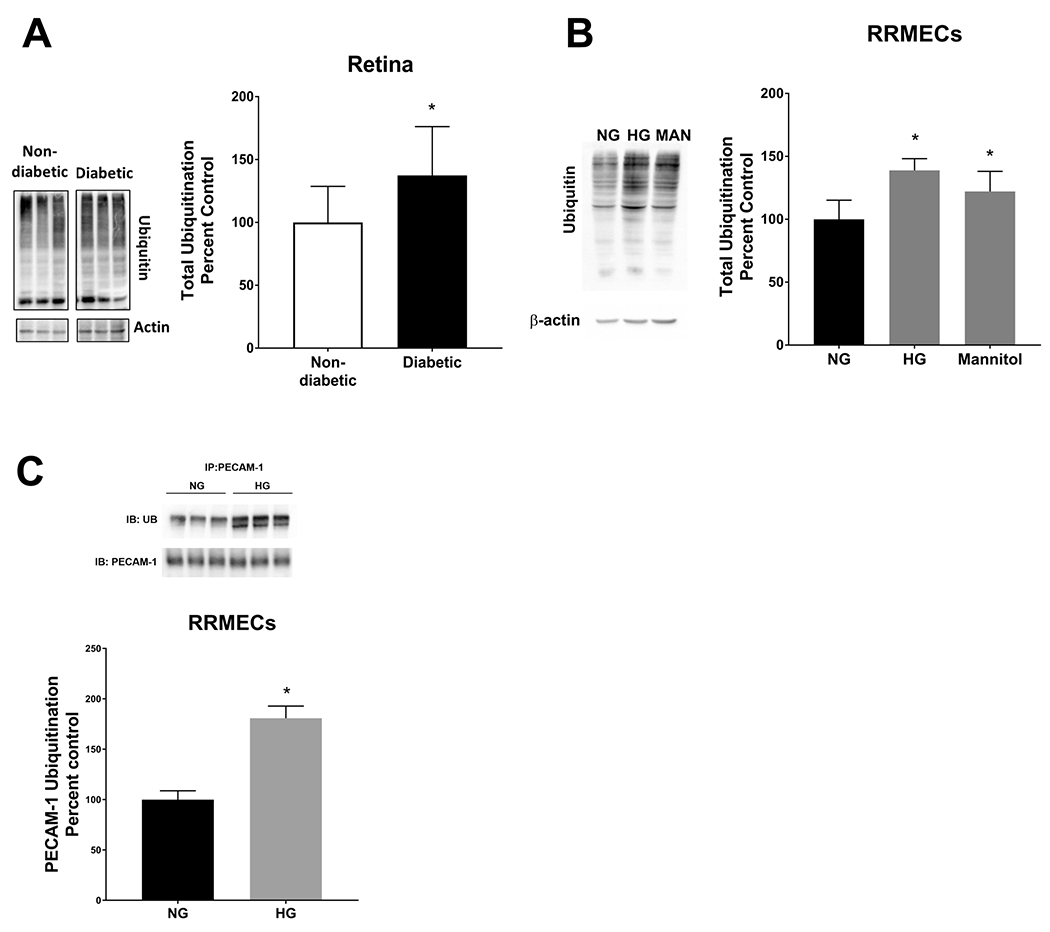
Western blot analyses showed a significant increase in total ubiquitination levels in the retina (A) and RRMECs (B) under hyperglycemic conditions and mannitol (Man) when compared to cells grown under normoglycemic (NG) conditions. C) PECAM-1 ubiquitination levels were significantly increased in RRMECs grown under hyperglycemic (HG). Western blotting data is a comparison of the relative ubiquitin/β-actin band densities. *p<0.05, N=11-12 per group in vivo and N=3-6 per group in vitro.
The treatment of RRMECs with TNF-α or IFN-γ significantly increases total ubiquitination and PECAM-1 ubiquitination levels
Inflammation can trigger several mechanisms leading to endothelial cell dysfunction. Therefore, we investigated whether the inflammatory mediators TNF-α or IFN-γ can induce the ubiquitination of proteins, including PECAM-1, in RRMECs. As shown in Figure 7A, total ubiquitination levels were significantly increased in RRMECs treated with TNF-α (22% increase, p<0.05), IFN-γ (28% increase, p<0.05), or both (20%, p<0.05). Additionally, PECAM-1 ubiquitination was significantly increased in RRMECs treated with TNF-α or IFN-γ (Figure 7B).
Figure 7. TNF-α and IFN-γ treatments significantly increased total and PECAM-1 ubiquitination levels in RRMECs.
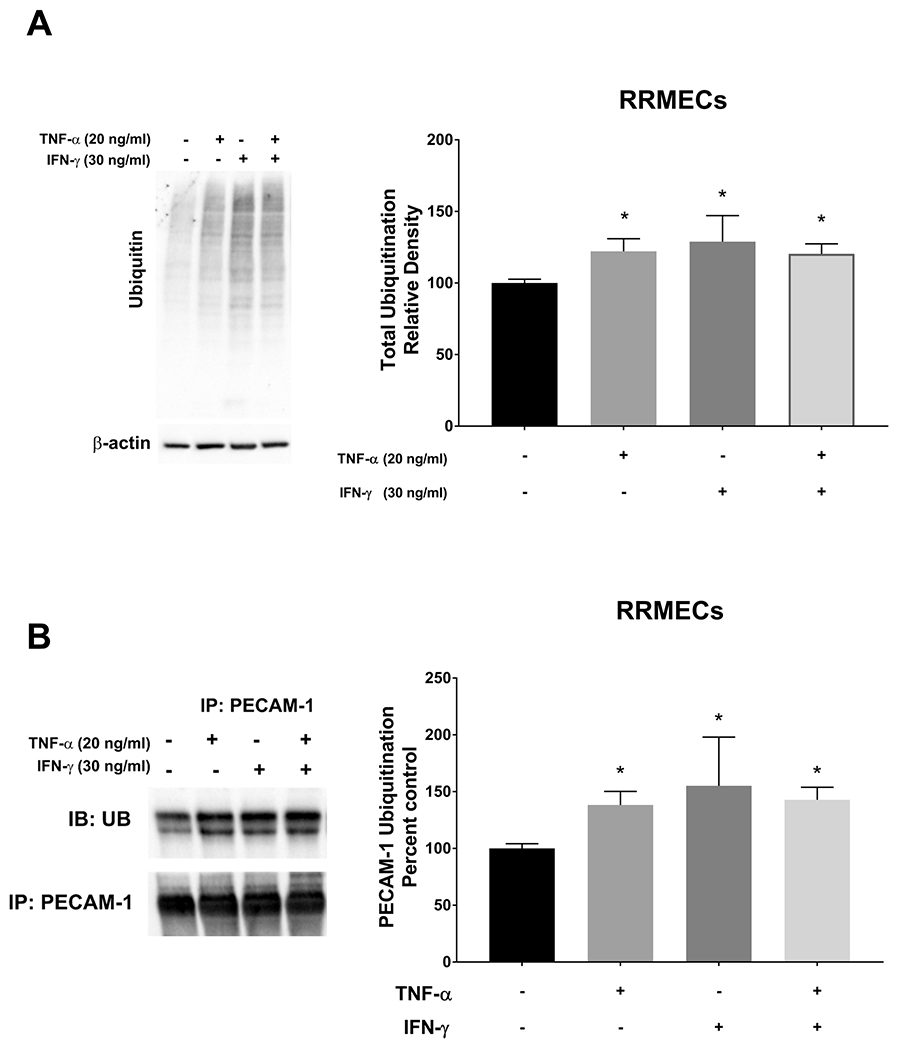
A) Total ubiquitination levels in RRMECs treated with TNF-α or IFN-γ were significantly increased. B) PECAM-1 ubiquitination levels were significantly increased with TNF-α or IFN-γ treatments in RRMECs. Western blotting data is a comparison of the relative ubiquitin/β-actin band densities. *p<0.05, N=3 per group.
Retinal proteasome activity is significantly decreased under hyperglycemic and inflammatory conditions
Proteins that are modified with ubiquitin can become targeted for degradation by proteasomes. Previous studies have shown a decreased activity of proteasome 20S under hyperglycemic conditions in the kidney 58. To investigate the effect of hyperglycemia on proteasome activity in the retina and RRMECs, retinas collected from diabetic rats were subjected to a proteasome activity assay, where a significant decrease in proteasome activity was found when compared to non-diabetic retinas (81% decrease, p<0.05, Figure 8A). Moreover, RRMECs grown under HG conditions had a significantly lower proteasome activity than in RRMECs grown under NG conditions (73% decrease, p<0.05, Figure 8B), with similar decreases with the inflammatory cytokines TNF-α, IFN-γ, or both combined in NG conditions (50%, 66%, and 80% decrease, respectively, p<0.05, Figure 8B).
Figure 8. Proteasome activity is decreased under hyperglycemic and inflammatory conditions.
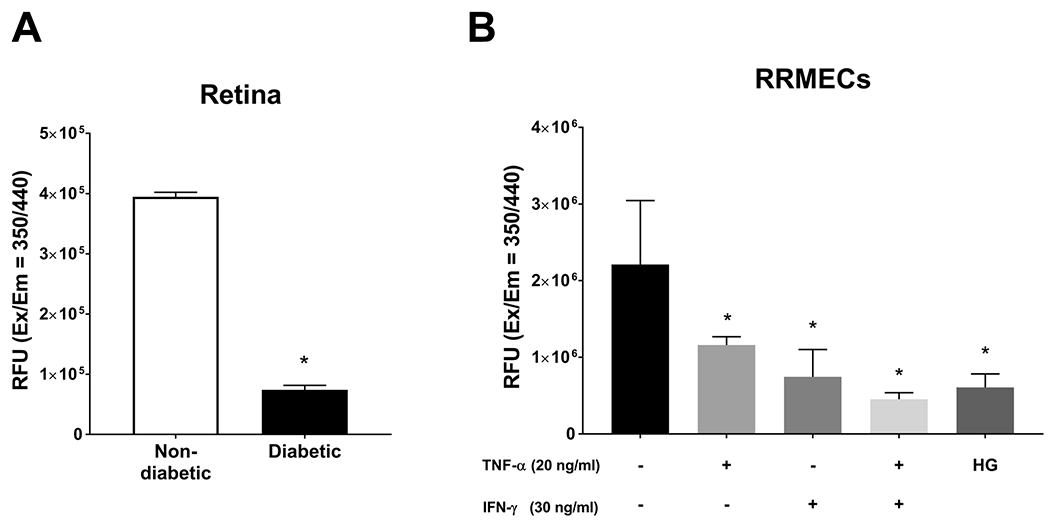
A) Proteasome activity in retinas collected from non-diabetic and diabetic rats expressed as relative fluorescent units (RFU). B) Proteasome activity in RRMECs grown under hyperglycemic conditions, or treated with TNF-α, IFN-γ or both. *p<0.05, N=4-5 per group in vivo and N=3 per group in vitro.
Discussion and conclusion
Diabetic retinopathy is a disease in which hyperglycemia-induced alterations in the retinal vasculature lead to retinal dysfunctions; these alterations include an increase in vascular permeability, endothelial cell dysfunction, alterations in endothelial cell signaling, and leukocyte plugging. Interestingly, a loss of PECAM-1 has been shown to contribute to similar pathways, that is, increased vascular permeability 13, endothelial cell apoptosis 19, 59, inhibition of leukocyte transmigration 44, 60, and altered endothelial cell signaling 21, 27, 56, 61. A loss of PECAM-1 from retinal endothelial cells under hyperglycemic conditions has been reported previously, in retinas collected from diabetic db/db mice 62, and in retinal endothelial cells grown under HG conditions (25 mM, 72 hours) 63. However, these two reports did not determine the mechanism of the loss. In a recently published study from our lab, we investigated the molecular mechanisms of PECAM-1 loss in retinas collected from type 1 diabetic rats, and in RRMECs grown under hyperglycemic conditions, and found a possible role for MMPs, particularly MMP-2, in the hyperglycemia-induced loss of PECAM-1 from retinal endothelial cells 48.
One of the novel findings we reported in our previous study was that the mediators responsible for PECAM-1 loss may in part be circulating in the diabetic plasma, as treatments of RRMECs with plasma collected from diabetic rats led to a significant decrease in PECAM-1 levels 48. Moreover, circulating full-length PECAM-1 levels were significantly decreased in the diabetic plasma, which could indicate a cleavage-independent decrease in PECAM-1 48 through a different mechanism, mediated possibly by the inflammatory mediators TNF-α and IFN-γ. Additionally, we have shown in our previous study the effect of various osmolar solutions on PECAM-1 levels (mannitol, sodium chloride, or dextran), and saw a partial decrease in PECAM-1 levels with each. In this report, with mannitol as osmotic control in vitro, we found a trend for increased expression of TNF-α, and IFN-γ levels and total ubiquitination levels were significantly increased in RRMECs. Plasma hyperosmolarity can result from increased blood glucose levels in diabetes64–66, and this change in osmolarity can initiate multiple pathological pathways and inflammation64. Multiple inflammatory cytokines such as TNF-α, IL6, IL1β, and IL8 are upregulated with hyperosmolar stress64, thus, the interplay between hyperglycemia, hyperosmolar stress, and inflammation could be an important pathway contributing to PECAM-1 loss and endothelial dysfunction in DR.
In this current study, we investigated whether inflammation can affect PECAM-1 levels in the diabetic retina by possibly inducing its posttranslational modification via ubiquitin and its subsequent degradation by proteasomes. To our knowledge, no reports have investigated the posttranslational modifications of PECAM-1 under hyperglycemic conditions in the retina, or how inflammation can mediate this process. Thus, our study provides the following novel findings: (1) PECAM-1 ubiquitination levels were significantly increased in RRMECs grown under HG conditions; (2) TNF-α and IFN-γ treatments significantly decreased PECAM-1 levels in RRMECs accompanied by a significant increase in PECAM-1 ubiquitination; (3) MMP-2 significantly increased TNF-α levels in RRMECs; and (4) proteasome activity in the retina and RRMECs were significantly decreased under hyperglycemic and inflammatory conditions, which as discussed later, could lead to an accumulation of ubiquitinated PECAM-1.
The increase in TNF-α and IFN-γ levels observed in the diabetic plasma and hyperglycemic RRMECs indicate a glucose-induced inflammatory response. Previous studies have suggested DR as an inflammatory disease after observations in the diabetic retina that included leukocyte accumulation67, increased vascular permeability, altered vascular flow, and increased ICAM-1 immunoreactivity28. Additionally, the loss of retinal PECAM-1, which can protect endothelial cells from an inflammatory response 68, 69, can further enhance the inflammatory insult. Interestingly, our data demonstrate an increase in active TNF-α levels in RRMECs treated with MMP-2, which in turn, is increased by hyperglycemia. TNF-α has been shown previously to modulate the levels of MMPs 70, 71; however, the role of MMPs in the regulation of TNF-α levels in retinal endothelial cells is poorly understood. Additionally, since enzymatic cleavage of TNF-α is an important step for its activation, the role of MMPs in this process was investigated. Studies have demonstrated the processing of pro-TNF-α by MMPs to produce the mature TNF-α 72, 73. We have identified several cleavage sites of TNF-α by MMP-2, MMP-3, and MMP-9 using in silico analysis. This finding could explain the increase in the levels of active TNF-α upon MMP-2 treatment, and suggests a mechanism by which MMP-2 could cleave the pro-form of TNF-α to produce the active form. Additionally, since there are possible predicted MMP-9 cleavage sites on TNF-α, but no increase in its active level with MMP-9 treatment, potential cleavage by MMP-9 may not be effective in producing active TNF-α.
The ubiquitin-proteasome system (UPS) is a principal and highly regulated system for protein degradation and turnover. Misfolded and abnormal proteins are cleared by the UPS; thus, it prevents the accumulation of harmful proteins in the cell, which could lead to proteotoxicity and cell death. The addition of multiple residues of ubiquitin, a 76 amino acid polypeptide, to proteins by a multistep enzymatic process targets these proteins for degradation 57. Additionally, UPS is important in the inflammatory response, where it participates in the activation of nuclear factor kappa B (NF-κB), and the regulation of inflammatory genes 74. Although the UPS has been implicated in neurodegenerative diseases such as Alzheimer’s disease and Parkinson’s disease 75, and in cancer 76, its role in DR has not been fully understood. Angiotensin II, which is upregulated in the diabetic retina 77, has been shown to promote the downregulation of synaptophysin, a major protein that is expressed heavily at the synaptic vesicles in the two plexiform layers of the retina, and has important roles in visual signal transmission 78 via synaptophysin-mediated ubiquitination and subsequent degradation 79. Thus, UPS dysfunction can contribute to the development and progression of DR, possibly via downregulating proteins that are important to retinal function and health.
TNF-α has been shown to induce the ubiquitination of connexin43 in human corneal fibroblasts, leading to its downregulation via the UPS pathway 80. The mechanism by which TNF-α can promote the ubiquitination and subsequent degradation of proteins is not fully understood. We hypothesize that TNF-α and/or IFN-γ alters PECAM-1 ubiquitination and protein levels possibly via NF-κB activation, or via improper folding of PECAM-1 due to endoplasmic reticulum (ER) stress as a result of inflammation; however, further studies need to be conducted to fully understand the mechanisms involved.
We have identified multiple possible PECAM-1 ubiquitination sites, such as lysine 384, by performing an in silico analysis using UbPred, a software that predicts possible ubiquitination sites in protein sequences81. Moreover, a recent study has identified PECAM-1 as a target for the anaphase-promoting complex cadherin-1 E3 ubiquitin ligase82, with ubiquitination possibly leading to the targeting of PECAM-1 to be degraded by proteasomes. Interestingly, our data indicated a decrease in proteasome activity under hyperglycemic conditions, and with the treatments of TNF-α and/or IFN-γ. Studies have suggested that the hyperglycemia-induced decrease in proteasome activity in the kidney is a result of the 20S proteasome subunit modification by AGEs 58. However, no studies have investigated the effect of hyperglycemia and prolonged TNF-α or IFN-γ exposure on RRMEC proteasome activity. The decrease in proteasome activity accompanied by an increase in PECAM-1 ubiquitination can lead to the accumulation of ubiquitinated PECAM-1 in RRMECs, which can affect its cellular function by inducing proteotoxicity. It should be noted, however, that although a decrease in proteasome activity was observed in our study, protein degradation is unlikely to be eliminated. Our assay used to assess proteasome activity utilized the chymotrypsin-like activity of the 20S subunit, which contains three catalytic sites that degrade different protein residues 83: (1) the chymotrypsin-like catalytic site that cleaves large hydrophobic protein residues; (2) the trypsin-like catalytic site that degrades smaller basic protein residues; and (3) the caspase-like catalytic site that degrades acidic protein residues. Our data indicated a decrease in the chymotrypsin-like activity of the proteasome under hyperglycemic and inflammatory conditions; however, further studies need to be conducted to investigate the effect of hyperglycemia and inflammation on the other two catalytic sites. In addition to the ubiquitin-proteosome pathway, a second pathway for the degradation of dysfunctional proteins is the lysosome-autophagy pathway, which is a highly regulated mechanism that eliminates aggregated or aggregation-prone proteins from the cell to prevent oxidative and metabolic stress84. Studies have shown that endothelial cells can utilize this mechanism under various pathological states85, 86. Thus, it is important to investigate the various possible mechanisms for PECAM-1 degradation, and how they may promote the progression of DR.
In conclusion, we have found a significant increase in PECAM-1 ubiquitination in the diabetic retina and in RRMECs grown under hyperglycemic conditions that could explain its loss in the diabetic retina and RRMECs. Moreover, TNF-α and IFN-γ, which were upregulated in the diabetic retina and plasma and in hyperglycemic and hyperosmolar RRMECs, significantly decreased PECAM-1 levels, which is possibly due to increased ubiquitination and subsequent degradation of PECAM-1. Thus, we have identified a novel mechanism of hyperglycemia-induced PECAM-1 loss in retinal endothelial cells via the ubiquitin-proteasome pathway, with a possible role for the inflammatory mediators TNF-α and IFN-γ in this mechanism. The hypothesized mechanism for the role of HG and inflammation on PECAM-1 ubiquitination is summarized in Figure 9. The role of the hyperglycemia-induced release of cytokines in the diabetic retina on PECAM-1 ubiquitination, and the possible subsequent degradation, can also occur to other important proteins in the retina, and can be an important mechanism by which hyperglycemia can induce damage to the retina. Thus, the study and understanding of this mechanism and its consequences can provide therapeutic targets by which the pathogenesis and progression of DR can be controlled at an early stage.
Figure 9.
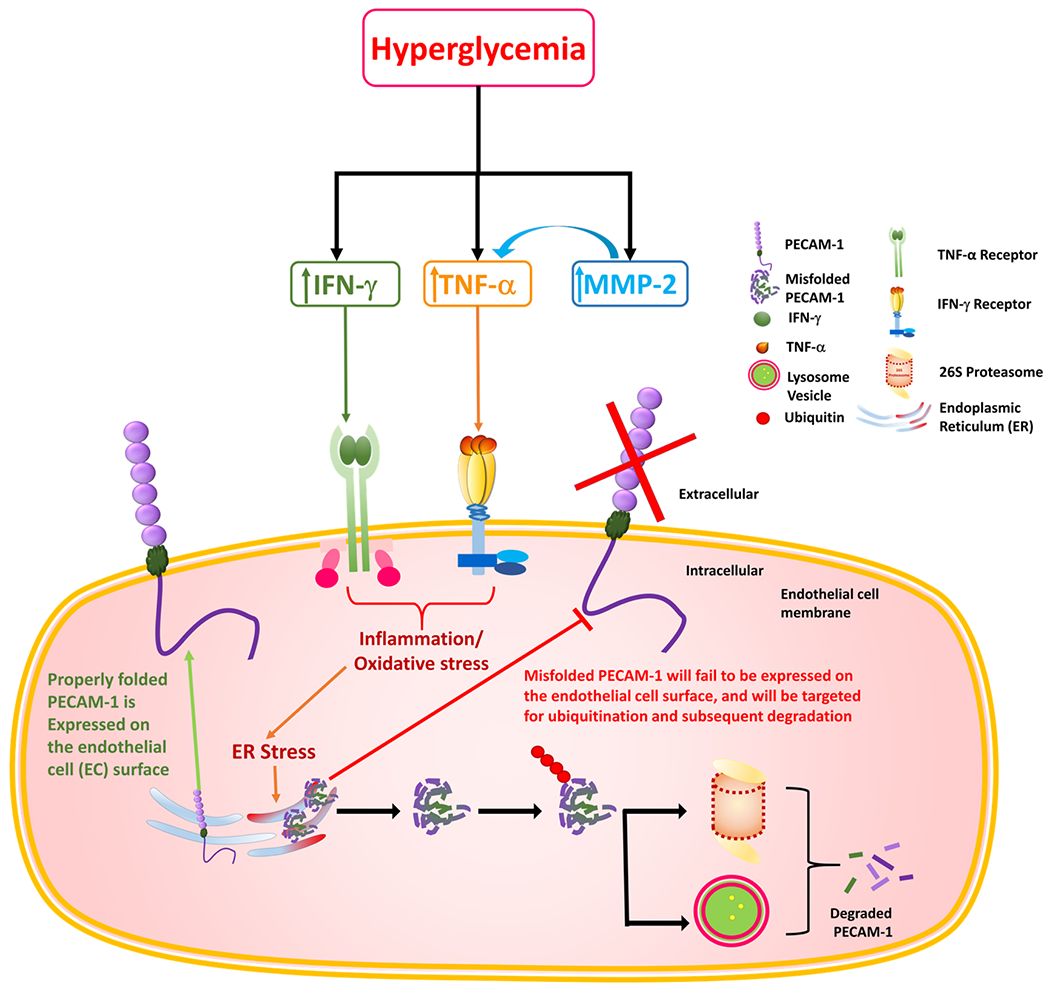
A graphical representation of the possible mechanisms involved in hyperglycemia-induced inflammation and PECAM-1 ubiquitination and degradation.
Perspective:
The hyperglycemia-induced increase in the inflammatory mediators TNF-α and IFN-γ may play a role in PECAM-1 loss in the diabetic retina, possibly by increasing the ubiquitination and degradation of PECAM-1. These findings can provide a better understanding of the mechanisms by which PECAM-1 is being lost from the diabetic retina, and can provide improved therapeutic targets for the management and treatment of DR.
Acknowledgments
We would like to thank Dr. Christopher Pattillo and his lab members for providing the ChemiDoc XRS gel imaging system and guidance with its use.
Grant support:
This work was supported by funding from the National Institute of Health (NIH) EY025632 and an American Heart Association (AHA) predoctoral fellowship.
List of Abbreviations
- AGEs
Advanced glycation end products
- ANOVA
Analysis of variance
- APMA
4-aminophenylmercuric acetate
- BRB
Blood-retinal barrier
- BSA
Bovine serum albumin
- DMEM
Dulbecco’s modified eagle media
- DR
Diabetic retinopathy
- ELISA
Enzyme-linked immunosorbent assay
- ER
Endoplasmic reticulum
- FITC
Fluorescein isothiocyanate
- HG
High glucose
- ICAM-1
Intercellular Adhesion Molecule 1
- IF
Immunofluorescence
- IFN-γ
Interferon-gamma
- Ig
Immunoglobulin
- ITIM
Immunoreceptor tyrosine-based inhibitory motif
- MAN
Mannitol
- MMP
Matrix metalloproteinases
- NF-κB
Nuclear factor kappa B
- NG
Normal glucose
- NIH
National institute of health
- PBS
Phosphate buffer saline
- PECAM-1
Platelet endothelial cell adhesion molecule-1
- PI
Propidium iodide
- RFU
Relative fluorescence unit
- RIPA
Radio immunoprecipitation assay
- RRMECs
Rat retinal microvascular endothelial cells
- STZ
Streptozotocin
- TNF-α
Tumor necrosis factor-alpha
- UPS
Ubiquitin-proteasome system
Contributor Information
Randa S. Eshaq, Louisiana State University Health Sciences Center-Shreveport, LA, Department of Molecular and Cellular Physiology
Norman R. Harris, Louisiana State University Health Sciences Center-Shreveport, LA, Department of Molecular and Cellular Physiology
Data availability statement:
The data used to support the findings of this study are available.
References
- 1.Klein R; Klein BE; Moss SE; Cruickshanks KJ, Relationship of hyperglycemia to the long-term incidence and progression of diabetic retinopathy. Arch Intern Med 1994, 154 (19), 2169–78. [PubMed] [Google Scholar]
- 2.Leasher JL; Bourne RR; Flaxman SR; Jonas JB; Keeffe J; Naidoo K; Pesudovs K; Price H; White RA; Wong TY; Resnikoff S; Taylor HR; Study, V. L. E. G. o. t. G. B. o. D., Global Estimates on the Number of People Blind or Visually Impaired by Diabetic Retinopathy: A Meta-analysis From 1990 to 2010. Diabetes Care 2016, 39 (9), 1643–9. [DOI] [PubMed] [Google Scholar]
- 3.Eshaq RS; Aldalati AMZ; Alexander JS; Harris NR, Diabetic retinopathy: Breaking the barrier. Pathophysiology 2017, 24 (4), 229–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Antonetti DA; Lieth E; Barber AJ; Gardner TW, Molecular mechanisms of vascular permeability in diabetic retinopathy. Semin Ophthalmol 1999, 14 (4), 240–8. [DOI] [PubMed] [Google Scholar]
- 5.Akram MU; Khalid S; Khan SA, Identification and classification of microaneurysms for early detection of diabetic retinopathy. Pattern Recognition 2013, 46 (1), 107–116. [Google Scholar]
- 6.Ashton N; Harry J, The Pathology of Cotton Wool Spots and Cytoid Bodies in Hypertensive Retinopathy and Other Diseases. Trans Ophthalmol Soc U K 1963, 83, 91–114. [PubMed] [Google Scholar]
- 7.Ishibazawa A; Nagaoka T; Yokota H; Takahashi A; Omae T; Song YS; Takahashi T; Yoshida A, Characteristics of Retinal Neovascularization in Proliferative Diabetic Retinopathy Imaged by Optical Coherence Tomography Angiography. Invest Ophthalmol Vis Sci 2016, 57 (14), 6247–6255. [DOI] [PubMed] [Google Scholar]
- 8.Schröder S; Palinski W; Schmid-Schönbein GW, Activated monocytes and granulocytes, capillary nonperfusion, and neovascularization in diabetic retinopathy. Am J Pathol 1991, 139 (1), 81–100. [PMC free article] [PubMed] [Google Scholar]
- 9.Chistiakov DA; Orekhov AN; Bobryshev YV, Endothelial PECAM-1 and its function in vascular physiology and atherogenic pathology. Exp Mol Pathol 2016, 100 (3), 409–15. [DOI] [PubMed] [Google Scholar]
- 10.Watt SM; Gschmeissner SE; Bates PA, PECAM-1: Its Expression and Function as a Cell Adhesion Molecule on Hemopoietic and Endothelial Cells. 2009. [DOI] [PubMed] [Google Scholar]
- 11.Albelda SM; Muller WA; Buck CA; Newman PJ, Molecular and cellular properties of PECAM-1 (endoCAM/CD31): a novel vascular cell-cell adhesion molecule. J Cell Biol 1991, 114 (5), 1059–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Flynn KM; Michaud M; Canosa S; Madri JA, CD44 regulates vascular endothelial barrier integrity via a PECAM-1 dependent mechanism. Angiogenesis 2013, 16 (3), 689–705. [DOI] [PubMed] [Google Scholar]
- 13.Graesser D; Solowiej A; Bruckner M; Osterweil E; Juedes A; Davis S; Ruddle NH; Engelhardt B; Madri JA, Altered vascular permeability and early onset of experimental autoimmune encephalomyelitis in PECAM-1-deficient mice. J Clin Invest 2002, 109 (3), 383–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fernandez-Martin L; Marcos-Ramiro B; Bigarella CL; Graupera M; Cain RJ; Reglero-Real N; Jimenez A; Cernuda-Morollon E; Correas I; Cox S; Ridley AJ; Millan J, Crosstalk between reticular adherens junctions and platelet endothelial cell adhesion molecule-1 regulates endothelial barrier function. Arterioscler Thromb Vasc Biol 2012, 32 (8), e90–102. [DOI] [PubMed] [Google Scholar]
- 15.Woodfin A; Reichel CA; Khandoga A; Corada M; Voisin MB; Scheiermann C; Haskard DO; Dejana E; Krombach F; Nourshargh S, JAM-A mediates neutrophil transmigration in a stimulus-specific manner in vivo: evidence for sequential roles for JAM-A and PECAM-1 in neutrophil transmigration. Blood 2007, 110 (6), 1848–56. [DOI] [PubMed] [Google Scholar]
- 16.Liao F; Huynh HK; Eiroa A; Greene T; Polizzi E; Muller WA, Migration of monocytes across endothelium and passage through extracellular matrix involve separate molecular domains of PECAM-1. J Exp Med 1995, 182 (5), 1337–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dasgupta B; Dufour E; Mamdouh Z; Muller WA, A novel and critical role for tyrosine 663 in platelet endothelial cell adhesion molecule-1 trafficking and transendothelial migration. J Immunol 2009, 182 (8), 5041–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kakei Y; Akashi M; Shigeta T; Hasegawa T; Komori T, Alteration of cell-cell junctions in cultured human lymphatic endothelial cells with inflammatory cytokine stimulation. Lymphat Res Biol 2014, 12 (3), 136–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bird IN; Taylor V; Newton JP; Spragg JH; Simmons DL; Salmon M; Buckley CD, Homophilic PECAM-1(CD31) interactions prevent endothelial cell apoptosis but do not support cell spreading or migration. J Cell Sci 1999, 112 ( Pt 12), 1989–97. [DOI] [PubMed] [Google Scholar]
- 20.Gao C; Sun W; Christofidou-Solomidou M; Sawada M; Newman DK; Bergom C; Albelda SM; Matsuyama S; Newman PJ, PECAM-1 functions as a specific and potent inhibitor of mitochondrial-dependent apoptosis. Blood 2003, 102 (1), 169–79. [DOI] [PubMed] [Google Scholar]
- 21.Osawa M; Masuda M; Kusano K; Fujiwara K, Evidence for a role of platelet endothelial cell adhesion molecule-1 in endothelial cell mechanosignal transduction: is it a mechanoresponsive molecule? J Cell Biol 2002, 158 (4), 773–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Conway DE; Breckenridge MT; Hinde E; Gratton E; Chen CS; Schwartz MA, Fluid shear stress on endothelial cells modulates mechanical tension across VE-cadherin and PECAM-1. Curr Biol 2013, 23 (11), 1024–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Collins C; Guilluy C; Welch C; O’Brien ET; Hahn K; Superfine R; Burridge K; Tzima E, Localized tensional forces on PECAM-1 elicit a global mechanotransduction response via the integrin-RhoA pathway. Curr Biol 2012, 22 (22), 2087–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cao G; Fehrenbach ML; Williams JT; Finklestein JM; Zhu JX; Delisser HM, Angiogenesis in platelet endothelial cell adhesion molecule-1-null mice. In Am J Pathol, United States, 2009; Vol. 175, pp 903–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ilan N; Cheung L; Pinter E; Madri JA, Platelet-endothelial cell adhesion molecule-1 (CD31), a scaffolding molecule for selected catenin family members whose binding is mediated by different tyrosine and serine/threonine phosphorylation. J Biol Chem 2000, 275 (28), 21435–43. [DOI] [PubMed] [Google Scholar]
- 26.Privratsky JR; Paddock CM; Florey O; Newman DK; Muller WA; Newman PJ, Relative contribution of PECAM-1 adhesion and signaling to the maintenance of vascular integrity. J Cell Sci 2011, 124 (Pt 9), 1477–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Newman PJ; Newman DK, Signal transduction pathways mediated by PECAM-1: new roles for an old molecule in platelet and vascular cell biology. Arterioscler Thromb Vasc Biol 2003, 23 (6), 953–64. [DOI] [PubMed] [Google Scholar]
- 28.Adamis AP, Is diabetic retinopathy an inflammatory disease? Br J Ophthalmol 2002, 86 (4), 363–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Tang J; Kern TS, Inflammation in diabetic retinopathy. Prog Retin Eye Res 2011, 30 (5), 343–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rübsam A; Parikh S; Fort PE, Role of Inflammation in Diabetic Retinopathy. Int J Mol Sci 2018, 19 (4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ramasamy R; Yan SF; Schmidt AM, Receptor for AGE (RAGE): signaling mechanisms in the pathogenesis of diabetes and its complications. Ann N Y Acad Sci 2011, 1243, 88–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Boss JD; Singh PK; Pandya HK; Tosi J; Kim C; Tewari A; Juzych MS; Abrams GW; Kumar A, Assessment of Neurotrophins and Inflammatory Mediators in Vitreous of Patients With Diabetic Retinopathy. Invest Ophthalmol Vis Sci 2017, 58 (12), 5594–5603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu H; Hwang DK; Song X; Tao Y, Association between Aqueous Cytokines and Diabetic Retinopathy Stage. J Ophthalmol 2017, 2017, 9402198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kalliolias GD; Ivashkiv LB, TNF biology, pathogenic mechanisms and emerging therapeutic strategies. Nat Rev Rheumatol 2016, 12 (1), 49–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Smith RA; Baglioni C, The active form of tumor necrosis factor is a trimer. J Biol Chem 1987, 262 (15), 6951–4. [PubMed] [Google Scholar]
- 36.Huang H; Gandhi JK; Zhong X; Wei Y; Gong J; Duh EJ; Vinores SA, TNFalpha is required for late BRB breakdown in diabetic retinopathy, and its inhibition prevents leukostasis and protects vessels and neurons from apoptosis. Invest Ophthalmol Vis Sci 2011, 52 (3), 1336–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sedger LM; McDermott MF, TNF and TNF-receptors: From mediators of cell death and inflammation to therapeutic giants - past, present and future. Cytokine Growth Factor Rev 2014, 25 (4), 453–72. [DOI] [PubMed] [Google Scholar]
- 38.Gresser I, Biologic effects of interferons. J Invest Dermatol 1990, 95 (6 Suppl), 66S–71S. [DOI] [PubMed] [Google Scholar]
- 39.Ealick SE; Cook WJ; Vijay-Kumar S; Carson M; Nagabhushan TL; Trotta PP; Bugg CE, Three-dimensional structure of recombinant human interferon-gamma. Science 1991, 252 (5006), 698–702. [DOI] [PubMed] [Google Scholar]
- 40.Hu X; Ivashkiv LB, Cross-regulation of signaling pathways by interferon-gamma: implications for immune responses and autoimmune diseases. Immunity 2009, 31 (4), 539–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li W; Nagineni CN; Hooks JJ; Chepelinsky AB; Egwuagu CE, Interferon-gamma signaling in human retinal pigment epithelial cells mediated by STAT1, ICSBP, and IRF-1 transcription factors. Invest Ophthalmol Vis Sci 1999, 40 (5), 976–82. [PubMed] [Google Scholar]
- 42.Ng CT; Fong LY; Sulaiman MR; Moklas MA; Yong YK; Hakim MN; Ahmad Z, Interferon-Gamma Increases Endothelial Permeability by Causing Activation of p38 MAP Kinase and Actin Cytoskeleton Alteration. J Interferon Cytokine Res 2015, 35 (7), 513–22. [DOI] [PubMed] [Google Scholar]
- 43.Romer LH; McLean NV; Yan HC; Daise M; Sun J; DeLisser HM, IFN-gamma and TNF-alpha induce redistribution of PECAM-1 (CD31) on human endothelial cells. J Immunol 1995, 154 (12), 6582–92. [PubMed] [Google Scholar]
- 44.Rival Y; Del Maschio A; Rabiet MJ; Dejana E; Duperray A, Inhibition of platelet endothelial cell adhesion molecule-1 synthesis and leukocyte transmigration in endothelial cells by the combined action of TNF-alpha and IFN-gamma. J Immunol 1996, 157 (3), 1233–41. [PubMed] [Google Scholar]
- 45.Stewart RJ; Kashour TS; Marsden PA, Vascular endothelial platelet endothelial adhesion molecule-1 (PECAM-1) expression is decreased by TNF-alpha and IFN-gamma. Evidence for cytokine-induced destabilization of messenger ribonucleic acid transcripts in bovine endothelial cells. J Immunol 1996, 156 (3), 1221–8. [PubMed] [Google Scholar]
- 46.Shaw SK; Perkins BN; Lim YC; Liu Y; Nusrat A; Schnell FJ; Parkos CA; Luscinskas FW, Reduced Expression of Junctional Adhesion Molecule and Platelet/Endothelial Cell Adhesion Molecule-1 (CD31) at Human Vascular Endothelial Junctions by Cytokines Tumor Necrosis Factor-α Plus Interferon-γ Does Not Reduce Leukocyte Transmigration Under Flow. In Am J Pathol, 2001; Vol. 159, pp 2281–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Neubauer K; Lindhorst A; Tron K; Ramadori G; Saile B, Decrease of PECAM-1-gene-expression induced by proinflammatory cytokines IFN-γ and IFN-α is reversed by TGF-β in sinusoidal endothelial cells and hepatic mononuclear phagocytes. BMC Physiology 2008, 8 (1), 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Eshaq RS; Harris NR, Loss of Platelet Endothelial Cell Adhesion Molecule-1 (PECAM-1) in the Diabetic Retina: Role of Matrix Metalloproteinases. Invest Ophthalmol Vis Sci 2019, 60 (2), 748–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Eshaq RS; Harris NR, Hyperglycemia-induced ubiquitination and degradation of β-catenin with the loss of platelet endothelial cell adhesion molecule-1 in retinal endothelial cells. Microcirculation 2020, 27 (2), e12596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wright WS; McElhatten RM; Harris NR, Increase in retinal hypoxia-inducible factor-2alpha, but not hypoxia, early in the progression of diabetes in the rat. Exp Eye Res 2011, 93, 437–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Leskova W; Watts MN; Carter PR; Eshaq RS; Harris NR, Measurement of retinal blood flow rate in diabetic rats: disparity between techniques due to redistribution of flow. Invest Ophthalmol Vis Sci 2013, 54 (4), 2992–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Rasban WS ImageJ, U. S. National Institutes of Health: Bethesda, Maryland, USA, 1997–2012. [Google Scholar]
- 53.Song J; Tan H; Perry AJ; Akutsu T; Webb GI; Whisstock JC; Pike RN, PROSPER: an integrated feature-based tool for predicting protease substrate cleavage sites. PLoS One 2012, 7 (11), e50300–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Opperman CM; Sishi BJ, Tumor necrosis factor alpha stimulates p62 accumulation and enhances proteasome activity independently of ROS. Cell Biol Toxicol 2015, 31 (2), 83–94. [DOI] [PubMed] [Google Scholar]
- 55.Myśliwiec M; Balcerska A; Zorena K; Myśliwska J; Lipowski P; Raczyńska K, The role of vascular endothelial growth factor, tumor necrosis factor alpha and interleukin-6 in pathogenesis of diabetic retinopathy. Diabetes Res Clin Pract 2008, 79 (1), 141–6. [DOI] [PubMed] [Google Scholar]
- 56.Ilan N; Mohsenin A; Cheung L; Madri JA, PECAM-1 shedding during apoptosis generates a membrane-anchored truncated molecule with unique signaling characteristics. FASEB J 2001, 15 (2), 362–72. [DOI] [PubMed] [Google Scholar]
- 57.Kleiger G; Mayor T, Perilous journey: a tour of the ubiquitin-proteasome system. Trends Cell Biol 2014, 24 (6), 352–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Queisser MA; Yao D; Geisler S; Hammes HP; Lochnit G; Schleicher ED; Brownlee M; Preissner KT, Hyperglycemia impairs proteasome function by methylglyoxal. Diabetes 2010, 59 (3), 670–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Limaye V; Li X; Hahn C; Xia P; Berndt MC; Vadas MA; Gamble JR, Sphingosine kinase-1 enhances endothelial cell survival through a PECAM-1-dependent activation of PI-3K/Akt and regulation of Bcl-2 family members. In Blood, United States, 2005; Vol. 105, pp 3169–77. [DOI] [PubMed] [Google Scholar]
- 60.Schenkel AR; Chew TW; Muller WA, Platelet endothelial cell adhesion molecule deficiency or blockade significantly reduces leukocyte emigration in a majority of mouse strains. In J Immunol, United States, 2004; Vol. 173, pp 6403–8. [DOI] [PubMed] [Google Scholar]
- 61.Wee JL; Jackson DE, The Ig-ITIM superfamily member PECAM-1 regulates the “outside-in” signaling properties of integrin alpha(IIb)beta3 in platelets. Blood 2005, 106 (12), 3816–23. [DOI] [PubMed] [Google Scholar]
- 62.Cheung AK; Fung MK; Lo AC; Lam TT; So KF; Chung SS; Chung SK, Aldose reductase deficiency prevents diabetes-induced blood-retinal barrier breakdown, apoptosis, and glial reactivation in the retina of db/db mice. Diabetes 2005, 54 (11), 3119–25. [DOI] [PubMed] [Google Scholar]
- 63.Cao Y; Feng B; Chen S; Chu Y; Chakrabarti S, Mechanisms of endothelial to mesenchymal transition in the retina in diabetes. Invest Ophthalmol Vis Sci 2014, 55 (11), 7321–31. [DOI] [PubMed] [Google Scholar]
- 64.Brocker C; Thompson DC; Vasiliou V, The role of hyperosmotic stress in inflammation and disease. Biomol Concepts 2012, 3 (4), 345–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Stookey JD, High prevalence of plasma hypertonicity among community-dwelling older adults: results from NHANES III. J Am Diet Assoc 2005, 105 (8), 1231–9. [DOI] [PubMed] [Google Scholar]
- 66.Gual P; Le Marchand-Brustel Y; Tanti J, Positive and negative regulation of glucose uptake by hyperosmotic stress. Diabetes Metab 2003, 29 (6), 566–75. [DOI] [PubMed] [Google Scholar]
- 67.Pécsvarády Z; Fisher TC; Darwin CH; Fabók A; Maqueda TS; Saad MF; Meiselman HJ, Decreased polymorphonuclear leukocyte deformability in NIDDM. Diabetes Care 1994, 17 (1), 57–63. [DOI] [PubMed] [Google Scholar]
- 68.Privratsky JR; Newman DK; Newman PJ, PECAM-1: conflicts of interest in inflammation. Life Sci 2010, 87 (3–4), 69–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Privratsky JR; Tourdot BE; Newman DK; Newman PJ, The anti-inflammatory actions of platelet endothelial cell adhesion molecule-1 do not involve regulation of endothelial cell NF-kappa B. J Immunol 2010, 184 (6), 3157–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhou L; Yan C; Gieling RG; Kida Y; Garner W; Li W; Han YP, Tumor necrosis factor-alpha induced expression of matrix metalloproteinase-9 through p21-activated kinase-1. BMC Immunol 2009, 10, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhang B; Yan L; Tsang PC; Moses MA, Matrix metalloproteinase-2 (MMP-2) expression and regulation by tumor necrosis factor alpha (TNFalpha) in the bovine corpus luteum. Mol Reprod Dev 2005, 70 (2), 122–32. [DOI] [PubMed] [Google Scholar]
- 72.Gearing AJ; Beckett P; Christodoulou M; Churchill M; Clements JM; Crimmin M; Davidson AH; Drummond AH; Galloway WA; Gilbert R, Matrix metalloproteinases and processing of pro-TNF-alpha. J Leukoc Biol 1995, 57 (5), 774–7. [DOI] [PubMed] [Google Scholar]
- 73.Gearing AJ; Beckett P; Christodoulou M; Churchill M; Clements J; Davidson AH; Drummond AH; Galloway WA; Gilbert R; Gordon JL, Processing of tumour necrosis factor-alpha precursor by metalloproteinases. Nature 1994, 370 (6490), 555–7. [DOI] [PubMed] [Google Scholar]
- 74.Chen ZJ, Ubiquitin signalling in the NF-kappaB pathway. Nat Cell Biol 2005, 7 (8), 758–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Gong B; Radulovic M; Figueiredo-Pereira ME; Cardozo C, The Ubiquitin-Proteasome System: Potential Therapeutic Targets for Alzheimer’s Disease and Spinal Cord Injury. Front Mol Neurosci 2016, 9, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Soave CL; Guerin T; Liu J; Dou QP, Targeting the ubiquitin-proteasome system for cancer treatment: discovering novel inhibitors from nature and drug repurposing. Cancer Metastasis Rev 2017, 36 (4), 717–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Wilkinson-Berka JL, Diabetes and retinal vascular disorders: role of the renin-angiotensin system. Expert Rev Mol Med 2004, 6 (15), 1–18. [DOI] [PubMed] [Google Scholar]
- 78.Dhingra NK; Ramamohan Y; Raju TR, Developmental expression of synaptophysin, synapsin I and syntaxin in the rat retina. Brain Res Dev Brain Res 1997, 102 (2), 267–73. [DOI] [PubMed] [Google Scholar]
- 79.Ozawa Y; Kurihara T; Sasaki M; Ban N; Yuki K; Kubota S; Tsubota K, Neural degeneration in the retina of the streptozotocin-induced type 1 diabetes model. Exp Diabetes Res 2011, 2011, 108328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Kimura K; Nishida T, Role of the ubiquitin-proteasome pathway in downregulation of the gap-junction protein Connexin43 by TNF-{alpha} in human corneal fibroblasts. Invest Ophthalmol Vis Sci 2010, 51 (4), 1943–7. [DOI] [PubMed] [Google Scholar]
- 81.Radivojac P; Vacic V; Haynes C; Cocklin RR; Mohan A; Heyen JW; Goebl MG; Iakoucheva LM, Identification, analysis, and prediction of protein ubiquitination sites. Proteins 2010, 78 (2), 365–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Liu J; Yao Q; Xiao L; Li F; Ma W; Zhang Z; Xie X; Yang C; Cui Q; Tian Y; Zhang C; Lai B; Wang N, APC/Cdh1 targets PECAM-1 for ubiquitination and degradation in endothelial cells. J Cell Physiol 2020, 235 (3), 2521–2531. [DOI] [PubMed] [Google Scholar]
- 83.Nussbaum AK; Dick TP; Keilholz W; Schirle M; Stevanović S; Dietz K; Heinemeyer W; Groll M; Wolf DH; Huber R; Rammensee HG; Schild H, Cleavage motifs of the yeast 20S proteasome beta subunits deduced from digests of enolase 1. Proc Natl Acad Sci U S A 1998, 95 (21), 12504–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kenney DL; Benarroch EE, The autophagy-lysosomal pathway: General concepts and clinical implications. Neurology 2015, 85 (7), 634–45. [DOI] [PubMed] [Google Scholar]
- 85.Guo F; Li X; Peng J; Tang Y; Yang Q; Liu L; Wang Z; Jiang Z; Xiao M; Ni C; Chen R; Wei D; Wang GX, Autophagy regulates vascular endothelial cell eNOS and ET-1 expression induced by laminar shear stress in an ex vivo perfused system. Ann Biomed Eng 2014, 42 (9), 1978–88. [DOI] [PubMed] [Google Scholar]
- 86.Maes H; Kuchnio A; Peric A; Moens S; Nys K; De Bock K; Quaegebeur A; Schoors S; Georgiadou M; Wouters J; Vinckier S; Vankelecom H; Garmyn M; Vion AC; Radtke F; Boulanger C; Gerhardt H; Dejana E; Dewerchin M; Ghesquière B; Annaert W; Agostinis P; Carmeliet P, Tumor vessel normalization by chloroquine independent of autophagy. Cancer Cell 2014, 26 (2), 190–206. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data used to support the findings of this study are available.