

Abstract
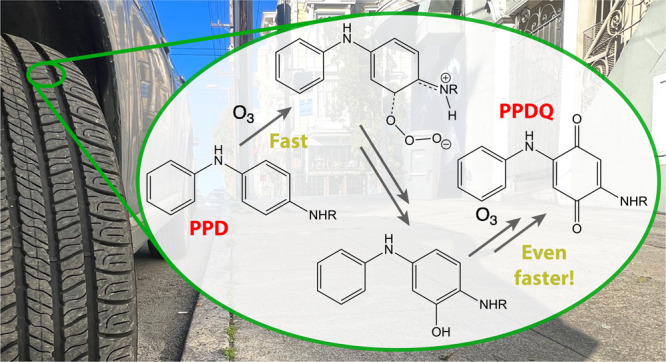
The discovery that the commercial rubber antidegradant 6PPD reacts with ozone (O3) to produce a highly toxic quinone (6PPDQ) spurred a significant research effort into nontoxic alternatives. This work has been hampered by lack of a detailed understanding of the mechanism of protection that 6PPD affords rubber compounds against ozone. Herein, we report high-level density functional theory studies into early steps of rubber and PPD (p-phenylenediamine) ozonation, identifying key steps that contribute to the antiozonant activity of PPDs. In this, we establish that our density functional theory approach can achieve chemical accuracy for many ozonation reactions, which are notoriously difficult to model. Using adiabatic energy decomposition analysis, we examine and dispel the notion that one-electron charge transfer initiates ozonation in these systems, as is sometimes argued. Instead, we find direct interaction between O3 and the PPD aromatic ring is kinetically accessible and that this motif is more significant than interactions with PPD nitrogens. The former pathway results in a hydroxylated PPD intermediate, which reacts further with O3 to afford 6PPD hydroquinone and, ultimately, 6PPDQ. This mechanism directly links the toxicity of 6PPDQ to the antiozonant function of 6PPD. These results have significant implications for development of alternative antiozonants, which are discussed.
Keywords: 6PPD, ozone, mechanism, quinone, DFT, EDA
Short abstract
Mechanistic DFT computations explain the reactivity of 6PPD toward ozone and directly link PPD function to toxicity through quinone formation.
1. Introduction
Rubber tires are essential across various sectors, including transportation and agriculture. Indeed, tire manufacturers produced 19 million tons of rubber in 2019, and continued global industrialization is expected to increase tire demand, requiring nearly 23 million tons annually by 2024.1 Maximization of the longevity of tires is a form of sustainability, reducing the annual flow of tires to landfills and other waste streams. This and other pressures have led the development of highly effective rubber additives that protect rubber from degradation during manufacture and use,2 most notably p-phenylenediamines (PPDs).3,4 Among these, 6PPD (N-(1,3-dimethylbutyl)-N′-phenyl-p-phenylenediamine) in particular has gained ubiquity in the tire industry and is included at 0.5–1.5 wt % in standard formulations.2 As a result, annual U.S. consumption of 6PPD ranges from 50 to 100 million tons, the majority of which is used in tires.5
Despite widespread use, PPD additives are known to aggravate various toxicity end points for both human and environmental health.6−11 6PPD in particular has recently gained notoriety due to the extreme aquatic toxicity of its quinone transformation product (6PPDQ) to coho salmon12−14 and other aquatic species.15−17 Though unknown for decades, 6PPDQ is produced in nearly 10% molar yield upon ozonation of 6PPD.18 While work is ongoing, this process is believed to occur at the surface of tires and tread wear particles18 before 6PPD, 6PPDQ, and other transformation products enter water systems through roadway runoff in storms.19,20
Replacement of 6PPD poses a formidable challenge as rubber compounds are susceptible to attack from numerous reactive species—peroxyl radicals, alkyl radicals, ozone—and 6PPD protects rubber compounds from each of these degradation pathways.21−23 Degradation due to peroxyl and alkyl radicals is relatively well-characterized,2 and research into alternative antioxidants was underway well before the discovery of 6PPDQ and its toxicity.24−34 The ozonation chemistry of PPDs and the development of safer antiozonants have been more elusive. Broadly speaking, it is believed that 6PPD protects tires in two distinct but overlapping ways:35 (1) kinetic scavenging that consumes O3 at the tire surface before it is able to react with the rubber3,36,37 and (2) subsequent formation of a protective film that provides a mechanical barrier against O3.36,38,39 Not observed in all PPDs,37 these protective films are comprised of PPD reaction products,38−40 although details are poorly understood. Reinvestigation of the mechanism of 6PPD ozonation is therefore imperative to understanding the kinetics of its ability to scavenge ozone, the likely products that lead to film formation, and the pathway to 6PPD quinone.
Herein, we report the first investigation into the mechanism of PPD ozonation since the discovery of 6PPDQ.12 Others have laid seeds for this work through detection of potential intermediates41,42 and developing broad-strokes proposals of the pathway.12,41,42 We continue this work through high-level computational analysis of the ozonation pathways of PPDs. Comparison of the barrier heights for various PPD ozonation pathways demonstrates that the route to the quinone is uniquely accessible, linking the toxicity of PPDs directly to their function as antiozonants. Throughout this study, mechanistic proposals are derived from ozonation mechanisms in related systems (Section 1.1), and our computational protocol follows best practices for O3 modeling (Section 1.2). We benchmark our methodology against existing methods (Section 3.1) before presenting mechanistic results for ozonation of a rubber surrogate (Section 3.3) and PPDs (Section 3.4). We conclude (Section 3.5) by discussing the implications of this work for development of alternatives to 6PPD.
1.1. Ozonation Mechanisms
While the ozonation chemistry of PPDs is underexplored, the literature contains a vast body of work on ozonation reactions in similar systems. Recent reviews and monographs provide a thorough treatment of this chemistry in a wide array of systems.43,44 Here, we provide an overview of critical aspects of ozonation chemistry in alkenes, amines, and aromatic systems, which are all relevant to understanding the reactions of O3 with rubber systems and/or PPDs.
Alkenes
Generally speaking, ozone reacts with unsaturated systems to produce scission products of the parent substrate.44 The overall reaction pathway is uncontroversial: ozone adds to olefins to form so-called primary ozonides, which may decompose or rearrange to form (secondary) ozonides, ultimately resulting in scission of the original olefin into two carbonyl products. This pathway explains the degradation of rubber upon exposure to ozone.37,38,45
Despite this consensus, two distinct proposals for the mechanism of primary ozonide formation have gained popularity: a concerted addition across the double bond (Criegee mechanism)46 and a stepwise addition, where ozone reacts with an olefin to form a biradical intermediate which may subsequently collapse to the ozonide (DeMore mechanism).47 Research increasingly indicates that the Criegee mechanism prevails in simple (i.e., less substituted, electronically symmetric) systems,48−51 while both steric and electronic effects of olefin substituents increase the activity of DeMore channels.49,52,53 Recent theoretical work indicates that solvent effects can make stepwise mechanisms dominant in systems where they otherwise would not be.54
Amines
The reactivity between amines and O3 is generally understood as a nucleophilic attack of N toward O3,55−57 and this pathway is particularly important in tertiary amines.58,59 Depending on the substitution pattern of the parent amine, reaction products may include N-oxides, nitrones, hydroxylamines, nitroso-alkanes, nitroalkanes, and dealkylated amines.57,59−61 Amines may also react with ozone through insertion into the N–H bond.62 The N lone pair plays a critical role in amine reactivity toward O3, such that more highly substituted aliphatic amines exhibit increased O3 reactivity.57 By the same token, these reactions are highly sensitive to pH, as amine protonation eliminates the reaction channel.59,63 Even in the most favorable systems and conditions, high barrier heights for N–O bond formations62 can limit these reactions relative to other available ozonation pathways.56
Aromatics
Reference to the ozonation3 chemistry of aromatic systems like anilines and phenols also enhances our understanding of its reactivity in PPDs. In both types of systems, ozonation reactions produce manifold products, and a broad array of mechanistic pathways has been suggested.64−68 Motifs common to the preceding functional groups are present here as well: primary ozonide formation,68,69 nucleophilic attack of ozone from heteroatoms like N67 or the aromatic ring,66,70 and ring cleavage reactions65,68,69 have all been reported in aromatic systems. These initial ozonation steps often produce intermediates that are themselves reactive toward O3, complicating reaction pathways.60 Electron donating groups activate reactions between aromatic carbons and O3, resulting in highly oxidized aromatic products like quinones.66,68,69,71
PPDs
Historically, work on PPD ozonation has emphasized formation of N–O oxides, analogous to ozonation of amines, resulting in dinitrone products for PPD ozonation.37,40,72−74 In the case of 6PPD, this assignment was recently repudiated on the basis of two-dimensional nuclear magnetic resonance spectroscopy,12 and quinones are now understood to form instead,42,75 representing nearly 10% of the product distribution for 6PPD.18 Beyond this, a great number of reaction products have been determined experimentally,37,40−42,72 though mechanistic details of these transformations are unclear. A few recent studies have emphasized the formation of quinone diimines (QDIs) in the various ozonation pathways of PPDs, including the formation of 6PPDQ.41,42,73 Even still, the stepwise mechanism of C–O bond formation in PPDs is unexplored to the best of our knowledge, despite the ultimate necessity of this motif in the production of 6PPDQ.
Charge Transfer Mechanisms
For each of the classes of reactions discussed above, a number of authors argue that ozonation reactions are initiated by one-electron charge transfer (CT) to form biradical, zwitterionic prereaction complexes (Figure 1). Examples for alkenes,76−79 amines,55−57,59,80 aromatics,67,69 and PPDs73,74,81,82 can be found at the indicated references. Such proposals are defended on the basis of a strong correlation between substrate ionization potential and O3 reaction rates,74,76 electron spin resonance (ESR) spectra that indicate the presence of radicals in the reaction mixture,73 or details of product distributions.59,79 Some have expressed skepticism about these CT intermediates, which have not been isolated experimentally, though theoretical literature analyzing such proposals is sparse.4,57,83 Within the CT mechanism, one-electron transfer is taken to be the rate-determining step of ozonation, so the existence of these complexes for olefins and PPDs is of present interest.
Figure 1.

Ozone charge transfer complexes for (a) natural rubber and (b) 6PPD have been proposed by a number of authors (see text).
1.2. Modeling Ozone Chemistry
Accurate modeling of the electronic structure of ozone (O3) is a formidable challenge in quantum chemistry, and many generally reliable methods have been shown to yield (sometimes catastrophically) bad predictions for ozonation chemistry.84 Beginning with some of the earliest theoretical work, O3 was generally accepted to have a biradical ground state,85,86 though subsequent results called this into question. At least one multireference study indicated that the ground state of O3 is a “regular” (i.e., closed-shell) singlet, with authors arguing that the electronic structure of O3 can be completely captured without reference to biradical character.87 Ozone is a difficult modeling problem because the truth lies somewhere between these two extremes, and a number of researchers have quantified the degree of biradical character in O3, with values ranging from 16–49%.88−92 Regardless of the exact value, it is clear that O3 is a genuinely multireference (MR) system. As a result, much of the highest quality theoretical literature on O3 chemistry resorts to one of a number of MR schemes.49,93
Nevertheless,
many single-reference (SR) approaches to O3 modeling have
been reported.50,62,94−103 Errors in these predictions can be substantial. As Wheeler et al.
have noted, generally respectable SR methodologies predict barrier
heights with discrepancies in excess of 10 kcal mol–1 for small systems like C2H2 and C2H4, and even the CCSD(T) (coupled-cluster with single,
double, and perturbative triple excitations) method gives unsatisfactory
results.99 To take just one relevant example,
these high-level methods predict divergent results for the initial
step of O3 addition to olefins.49−51 In order to
systematically approach the fully correlated limit, inclusion of explicit
triple and perturbative quadruple excitations (the CCSDT(Q) method)
is necessary.62,99,104 While Trogolo et al. report results of this quality for a number
of small systems,62 the  scaling of this method excludes its use
for the present systems.
scaling of this method excludes its use
for the present systems.
Instead, systems of present interest require treatment with either less computationally demanding wave function approaches or density functional approximations (DFAs). A number of DFA treatments of ozone chemistry have been reported.57,62,70,97,98 Generally speaking, however, out-of-the box DFA treatments are unable to predict ozone reaction energies with consistent fidelity,62 while more success can be expected for barrier heights.104 Hybrid density functionals generally exhibit the best performance for both overall thermodynamics and transition energies.62 Spin projection schemes105 are known to improve DFA results for related systems,106 and this approach has been used to model O3 previously.49,57 On the other hand, novel low-scaling wave function approaches like regularized orbital-optimized second-order Møller–Plesset perturbation theory (κ-OOMP2) effectively treat radical species in other contexts,107−109 though their use for ozonation chemistry has not been previously explored.
2. Computational Methods
Methods of Electron Correlation
In the present study, we consider the ωB97X-V110 and ωB97M-V111 density functional approximations (DFAs), as well as regularized orbital-optimized second-order Møller–Plesset perturbation theory (κ-OOMP2)107 in our modeling of O3 reactivity. These density functionals were chosen on the basis that hybrids outperform other classes of DFAs for O3 modeling62 and that ωB97X-V and ωB97M-V, specifically, have been shown to yield highly accurate predictions for thermochemistry and reaction barrier heights in other contexts.112 We also explore the use of κ-OOMP2107 as a low-scaling wave function method for modeling ozonation reactions. Though unexplored for O3, κ-OOMP2 is able to successfully treat strong correlation in other systems when combined with spin projection,106 as discussed further below.
Exploring Potential Energy Surfaces
Single-point energies were evaluated using either the def2-TZVPP113 or def2-QZVPPD114 basis set, as indicated in the text below. Exchange–correlation integrals were evaluated using fine-mesh (99,590) Lebedev integration grids.115,116 Self-consistent field (SCF) iterations were converged to at least 1 × 10–8 a.u. in all cases, and a tighter threshold of 1 × 10–10 a.u. was achieved where possible. Unrestricted reference states and stability analysis were used to confirm ground electronic state configurations were obtained. Where included, molecular orbitals were obtained as intrinsic bonding orbitals (IBOs),117 using a recently reported implementation.118
All geometries utilized in this work were optimized using the ωB97X-V density functional with the def2-TVZPP basis, and stationary points were obtained using gradient and energy thresholds of 3 × 10–4 and 1 × 10–6 a.u., respectively. Yamaguchi’s approximate spin projection (AP) method105 was used to address spin contamination in both DFA and κ-OOMP2 computations (Section S1), and free energies at T = 298.15 K were obtained using the quasi rigid rotor-harmonic oscillator scheme with a frequency cutoff of 100 cm–1.119 While algorithms for structural optimization on spin projection surfaces have been reported,49,120 all geometric properties and harmonic vibrational frequencies have been obtained on the (spin-contaminated) singlet energy surfaces. Initial guesses for transition state optimizations were obtained using either the freezing string method121,122 or constrained optimizations near the expected transition structure. Harmonic frequencies for each optimized structure were determined through diagonalization of the full Hessian matrix, and stationary points were characterized as local minima or transition states based on the presence or absence of a single imaginary frequency in this analysis. Intrinsic reaction coordinate (IRC) analysis123,124 was used to determine which local minima were associated with each transition structure. Coordinates for all such reactant, product, and transition structures are found in the SI. All computations were completed using the Q-Chem package.125
Energy Decomposition Analysis
The adiabatic energy decomposition analysis (EDA) of Head-Gordon and co-workers126 was used to evaluate the formation of charge transfer (CT) complexes. Interactions between fragments defined as substrate radical cations (X.+) and ozone radical anion (O3.–) were treated using a hierarchy of mathematical constraints that model frozen electrostatics (FRZ), orbital polarization (POL), and CT contributions to overall binding energies. A full description of this formalism and the components of the different potential energy surfaces may be found elsewhere.126−128 Incremental energy contributions are defined as
| 1 |
| 2 |
| 3 |
where EFRZ, EPOL, and EFULL are the minimum energies on the frozen, polarization, and unconstrained PESs, respectively, and Eα are the electronic energies of the radical ion fragments in unconstrained computations. Free energies were not computed for use with EDA predictions.
For present purposes, it is critical to note that EPOL contains all terms of the standard (e.g., Born–Oppenheimer, nonrelativistic, etc.) physical model but explicitly forbids CT between fragments. By evaluating the energetics of radical ionic fragments on this surface relative to an unconstrained computation, we can determine the feasibility of CT complex formation.
Choice of Model System
It has long been known that the reactions of O3 with both tire rubber and PPDs in tires are surface phenomena.36 Indeed, absent the presence of strain, which exposes deeper layers in the rubber to the atmosphere, cracking of the rubber surface does not occur.129 In this, experimental evidence demonstrates that the diffusion of PPDs to the rubber surface is rapid enough to prevent rubber ozonation, even given their relatively small concentration.38,39,130 We are therefore ultimately interested in understanding these chemistries at the rubber/air interface, rather than deep in the matrix of the tire. Even in the latter case, it is likely the general electrostatic “solvent” effects would be negligible due to the low permittivity of rubber (ε ≈ 3).131 We therefore report in vacuo energies for all transformations in this work, as these should nicely approximate the chemistry at the interface of environmental interest.
The structural optimization procedures outlined above are computationally expensive, bordering on intractability for 6PPD and for large polymeric units of natural rubber (cis-polyisoprene, NR). We have therefore used surrogate molecules for each of these compounds in our exploration of the potential energy surfaces (PESs) of ozone reaction and the characterization of stationary points. Specifically, we have used 2-methyl-2-butene (2M2B, 1) and 4-aminodiphenylamine (4ADPA, 2) as stand-ins for natural rubber and 6PPD, respectively (Figure 2). We have also modeled select key PPD reaction steps using N-methyl-N′-phenyl-p-phenylenediamine (MePPD, 3) to account for the known effects of N-alkylation on PPD ozonation rates.3,4 Our in vacuo model of reactions of these substrates omits any effects from the tire matrix, but we expect these effects to be small on the basis of the arguments in the preceding paragraph.
Figure 2.

Substrates used in modeling ozone reactivity. Primary exploration of potential energy surfaces was completed using 1 and 2, as surrogates for NR and 4. Select modeling was completed for compound 3 to better understand experimental results for ozonation kinetics.
3. Results and Discussion
3.1. Accurate Modeling of Ozone Chemistry
As noted in Section 1.2, accurate modeling of ozonation chemistry is challenging, and model validation is therefore critical. The majority of experimental work on ozonation reactions has been conducted in aqueous or organic solvents, complicating comparison of this work to other systems (e.g., rubber). In the absence of experimental results in an appropriate environment, high-level theoretical results represent the best benchmark for model development. Recent work by Trogolo et al. has provided a nearly exact set of such benchmarks for ozone chemistry,62 which we use to assess the model of the present work. Their results consist of CCSDT(Q) energies extrapolated to the complete basis set (CBS) limit for three cycloadditions (C2H4, C2H2, HCN), two insertion reactions (HCl, NH3), two linear additions (N(CH3)3 and Br–), and three ozone scission reactions (O3, N(CH3)3O3, BrO3–). Benchmark comparisons to these high-level CC reaction energies and barrier heights for the ωB97X-V and ωB97M-V density functionals as well as the κ-OOMP2 approach are found in Table 1, and comparisons for vdW complexes are found in Table S1.
Table 1. Errors in Spin-Contaminated (SC) and Approximate Projection (AP) Single-Point Energies for Benchmark Ozonation Reactionsb.

Computed indirectly via addition of partial reaction channels. See ref (62) for details.
For the κ-OOMP2 (κ = 1.45) results, AP is applied to reactions that produce 1O2, as this is the only species that exhibited spin polarization for this method. Reference values are CCSDT(Q) results extrapolated to the CBS limit obtained from ref (62). Highlighted boxes represent the best performing methodology for a given parameter at a given basis set truncation.
Hybrid Density Functionals
As in previous work,49 significant spin contamination was found on the singlet DFA potential energy surfaces (PESs) for reactive oxygen species, which we addressed through Yamaguchi’s approximate projection scheme (Section S1).105 Comparison of the results from spin contaminated (SC) and approximate projection (AP) energies highlights the complexities of strong correlation in ozone systems. Specifically, these data are conclusive that SC predictions of barrier heights consistently outperform their AP counterparts, while AP schemes are necessary in order to achieve accurate predictions of reaction energies for ozone reactions (Table 1). These corrections are especially important when 1O2 is a product, as SC errors can approach 20 kcal mol–1. These errors are so egregious that SC predictions for both ωB97X-V and ωB97M-V reactions energies are qualitatively incorrect in some cases, even predicting the wrong sign for the thermicity of Br– ozone addition and then BrO3– dissociation to BrO– and 1O2.
The discrepancy between performance for ΔErxn and ΔETS is troubling, and we discuss the theoretical basis for this in Section S1.3. Here, we note simply that the best performance on ΔETS is achieved using spin-contaminated structures, where def2-QZVPPD RSMDs for ωB97X-V and ωB97M-V are 1.38 and 1.23 kcal mol–1, respectively. Both DFAs even predict energies within the bounds of chemical accuracy for about half SC transition structures. AP schemes are necessary to achieve tolerable predictions of ΔErxn for these functionals, resulting in RMSDs of 2.29 and 2.46 kcal mol–1 for reactions that do not produce 1O2. AP-ωB97M-V energies are particularly accurate for O3 scission reactions, and chemical accuracy is achieved in all cases. For ωB97X-V, AP errors are 1.48–2.58 kcal mol–1 in magnitude.
Wave Function Approaches
Recent developments in electronic structure theory have led to orbital optimization techniques that provide tractable alternatives to DFAs on the one hand and high-level CC or multireference approaches on the other. Orbital-optimized MP2 (OOMP2) has proven particularly promising, and regularization schemes like κ-OOMP2 have been shown to perform well across a variety of radical and closed shell systems.107−109 The strength of the regularizer κ is known to impact the quality of results.108,109 Benchmark predictions evaluated at various values of κ (Table S2) indicate κ = 1.45 provides the best results for the present systems, in line with a previous recommendation.107 With this method, only 1O2 exhibits symmetry breaking, so we only apply the AP scheme for reactions that produce this species. All other κ-OOMP2 values in Table 1 are uncorrected.
Using the def2-QZVPPD basis, this method yields RMSDs for ΔErxn and ΔETS of 5.12 and 2.10 kcal mol–1 relative to CCSDT(Q)/CBS results. In some cases, predictions for individual entries are nearly exact, while disagreement in the reaction energy for N(CH3)3 is in excess of 10 kcal mol–1. As for the DFAs, AP is necessary to achieve reasonable agreement for O3 scission reactions, but even here it exhibits errors in excess of 1 kcal mol–1.
Method Selection
Overall, the ωB97X-V and ωB97M-V DFAs perform surprisingly well for these systems, exceeding even the good expectations for hybrid density functionals established in previous work.62 Each of these methods outperforms κ-OOMP2, where a regularizer balancing the effects of static and dynamic correlation could not be achieved (Section S1.2). Based on these results, DFAs are used for the analysis that follows. The discrepancy between ωB97X-V and ωB97M-V performance is smaller than the overall errors of each of these methods relative to CCSDT(Q), such that the choice between these two methods is unlikely to make a material difference. On the basis of performance with the truncated def2-TZVPP basis set, which we use for systems below, we focus on results from ωB97X-V. The results in Table 1 lead us to expect this method to produce results accurate within 1-sigma errors of 1.25 kcal mol–1. Though not the primary purpose of this work, we emphasize here the importance of establishing an effective protocol for DFT modeling of ozone transition states and reaction energies, which are notoriously difficult problems in electronic structure theory.
3.2. EDA of Radical Ion Complexes
As documented in Section 1.1, a number of authors argue that explicit X → O3 one-electron transfer initiates ozonation chemistry for a wide variety of substrates X. We have used adiabatic energy decomposition analysis (EDA) to analyze this reactivity for 2M2B (1) and 4ADPA (2) as representative examples of alkenes/unsaturated polymers and PPDs, respectively.
CT complexes between 1 and O3 on the series of ωB97X-V/def2-TZVPP EDA surfaces indicate that these structures are highly unstable relative to neutral fragments (Figure 3). The initial promotion to ionized fragments in this system is energetically expensive, with ΔEIF = 145.2 kcal mol–1. This result is physically reasonable on the basis of experimental results132 for the first ionization energy of 1 and the electron affinity of ozone, 200.4 and 48.5 kcal mol–1, respectively, yielding ΔEIFexpt = 151.9 kcal mol–1. Interactions between these ionized fragments on the FRZ and POL surfaces are unable to compensate for the high promotion energy, yielding structures that are destabilized by 50.3 and 40.5 kcal mol–1, respectively, relative to the neutral fragments. Upon relaxation of all constraints, spontaneous charge transfer results in a neutral van der Waals (vdW) complex between 1 and O3 (Figure 3, inset). The inability of this approach to locate a structural minimum on the [1]·+[O3]·– surface indicates that this complex cannot form spontaneously, and the vdW complex will preferentially form instead.
Figure 3.
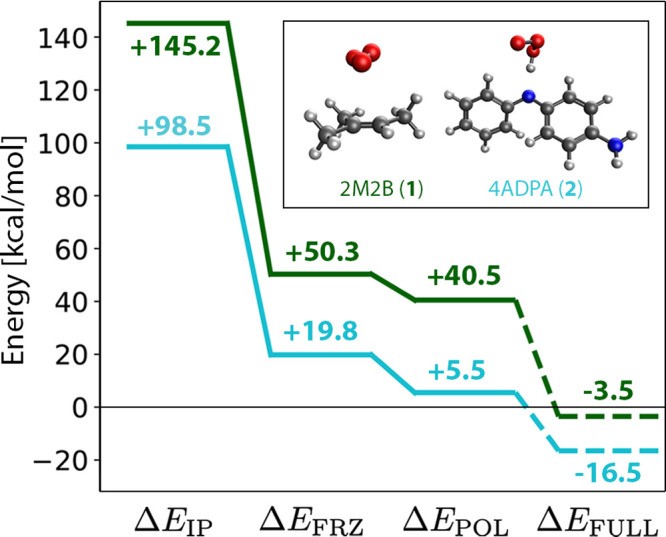
Energy decomposition analysis for CT complexes of 2M2B (1, top line) and 4ADPA (2, bottom line) with O3. Structural minima for biradical zwitterionic CT complexes could not be found on unconstrained surfaces, where spontaneous charge transfer (indicated by dashed lines) resulted in charge-neutral fragments and the structures shown in the inset. The zero of energy corresponds to the optimal geometries of isolated neutral fragments.
The situation is similar for 2, which we take to be representative of other PPDs where these CT complexes have been repeatedly proposed.73,74 Ionization energies for PPDs are generally lower than those for olefins,73 and here we compute ΔEIFcomp = 98.5 kcal mol–1 for 2. This lower energetic penalty results in CT complexes that are less energetically unfavorable than those for 1, with ΔEFRZ and ΔEPOL of 19.8 and 5.5 kcal mol–1, respectively (Figure 3). They are nonetheless still above the zero of energy corresponding to neutral fragments, and so here too relaxation of the CT constraint results in spontaneous electron transfer to afford neutral fragments. Upon subsequent geometry optimization cycles, the neutralized O3 molecule abstracts a proton from 2 as depicted in the inset of Figure 3. While the energy cost to form [2]·+[O3]·– on the POL surface is relatively small, it is still unbound. Spontaneous reversion to neutral fragments upon relaxation of the constraints used to force formation of the CT complex indicates that such complexes do not form for these systems.
Taken together, the results of Figure 3 indicate that CT complexes like those described above do not form in the ozonation reactions of 1 and 2. Inasmuch as these compounds are representative of olefins and PPDs more broadly, this conclusion carries over, and one-electron charge transfer should not be relied upon to explain the kinetics of ozone chemistry for olefins and PPDs, as is common (cf. Section 1.1).
3.3. Mechanisms of Natural Rubber Ozonation
Having eliminated the possibility of a CT mechanism for 1, two possibilities remain: concerted (Criegee) and stepwise (DeMore) addition of O3 across the 1 double bond. Conceptually, two distinct DeMore pathways exist for 1, corresponding to initial reactions forming secondary (1a) and tertiary (1b) carbon radicals (Figure 4[a]). Each of these structures may then collapse to the primary ozonide (1c). Therefore, we expect at least three distinct transition structures on the 1–O3 PES.
Figure 4.

Mechanistic possibilities for the ozonation of 2-methyl-2-butene (1) to form primary ozonide 1c. In addition to the symmetric addition of O3 to 1 (ΔG298.15 K‡ = 15.5 kcal mol–1) and the true DeMore pathway through 1b (ΔG298.15 K = 14.7 kcal mol–1), an asymmetric addition (ΔG298.15 K‡ = 13.2 kcal mol–1) without an isolable DeMore intermediate was identified.
The Criegee mechanism for addition of O3 to 1, where the symmetric formation of two C–O bonds was unambiguously confirmed with intrinsic reaction coordinate (IRC) analysis,123,124 possesses a barrier height of 15.5 kcal mol–1 on the ωB97X-V/def2-TZVPP surface (Figure 4[b]). Analysis of the DeMore structures 1a and 1b was more complicated. While a structure corresponding to 1a with a single imaginary frequency corresponding to the formation of the C2–O3 bond was identified, IRC on this structure identified it as a saddle point connecting the vdW complex and primary ozonide 1c. Hence, this structure actually represents an asymmetric pseudo-Criegee structure, with a barrier of 13.2 kcal mol–1. A similar structure that initially forms the C3–O3 bond was also identified. These structures are DeMore-like in the asymmetric formation of C–O bonds, but they do not result in true DeMore intermediates, i.e. local minima on the ωB97X-V/def2-TZVPP surface. Nevertheless, asymmetric addition to the double bond in 1 is more favorable than symmetric addition by approximately 2.3 kcal mol–1 at the ωB97X-V/def2-TZVPP level.
In addition to these three transition structures, a true DeMore pathway exists for the tertiary radical intermediate 1b, where IRC analysis does identify a local minimum for this species. The ωB97X-V/def2-TZVPP barrier height for 1 → 1b is 14.7 kcal mol–1 (Figure 4[b]), meaning this will be a minor pathway relative to the pseudo-Criegee structures reported above. A second transition structure (ΔG‡ = 0.2 kcal mol–1) was identified for the collapse of 1b → 1c. It is possible that solvation effects could stabilize this pathway, as has been observed in other systems,54 but we do not explore this here as it is irrelevant to the rubber system of interest. Though the differences between these barrier heights are on the order of the tolerance of our ωB97X-V model (Section 3.1 and Table 1) and we have omitted any effects of the tire matrix, these values provide an estimate of the degree of O3 reactivity necessary to protect rubber in tires.
Overall, the lowest energy pathway for 1 ozonation is an asymmetric Criegee addition, which possesses a barrier height of a mere 13.2 kcal mol–1 at the ωB97X-V/def2-TZVPP level. This and other small barrier heights for this system underscore one difficulty of developing rubber antiozonants. Candidate molecules must possess even smaller transition structure energies in order to effectively scavenge O3. As seen below, PPDs are among the few molecules that achieve this threshold of reactivity.
3.4. Mechanistic Pathways for PPDs
PPDs are highly reactive substrates in oxygen and ozone chemistry, and a number of potential pathways for the reaction of ozone with these compounds can be imagined.41,42 This panoply of reaction intermediates is reminiscent of the ozone chemistry of aniline67 and phenolic systems,64 complicating efforts to identify the mechanism of quinone formation. While a few overarching frameworks have been suggested,12,41 no stepwise mechanisms for 6PPD quinone formation have been proposed. We begin the analysis of PPD ozonation with discussion of the kinetics and thermodynamics of ozonation steps for 4ADPA (2) in Section 3.4.1, demonstrating that the particularly high activity of PPDs toward O3 stems from direct interactions with the PPD ring system. We then consider the effects of N-alkylation on the kinetics of key ozonation steps in Section 3.4.2. Finally, we discuss proposals that PPD quinones form through quinone diimines (QDIs) in Section 3.4.3.
3.4.1. Ozonation of 4ADPA
Charge transfer mechanisms aside (Section 3.2), we consider a number of initial steps for the reactivity between 4-ADPA (2) and O3, including linear addition of O3 to N atoms, insertion of O3 into N–H bonds, and both Criegee (concerted) and Demore (stepwise) addition to the aromatic ring (Figure 5). In all cases, a number of regioisomers and conformers were considered. While energetic data for each of these can be found in the SI, here we present transformation energies for only the most stable conformers and select other structures of interest.
Figure 5.

Selected reaction pathways for PPDs and ozone. In general, each structure represents a collection of regioisomers. Two sequential additions of ozone to the PPD ring system can afford hydroquinone g (R2 = OH), which is readily oxidized to quinone q. R1 = H, Me, and 1,3-dimethylbutyl correspond to 4-ADPA (2), N-Me-PPD (3), and 6PPD (4), respectively.
Of considered steps, direct interaction between PPD amines and O3 corresponds to the least favorable transition structures, contrary to historical and prevailing opinion (Figure 6).37,40,72−74 Insertions into the N–H bond exhibited lower barrier heights than N additions, but even here ΔG298.15 K‡ = 30.3 and 26.9 kcal mol–1 for terminal and central amines 2a, respectively (Figures 5 and 6). Such high barriers are qualitatively consistent with the high value of the benchmark quality result for NH3 insertion (ΔE‡0 K = 23.1 kcal mol–1) from Trogolo et al.62 Experimental evidence also supports the conclusion that N–O bond formation is not the major path of ozone consumption in PPDs. Rate constants for PPD ozonation in CCl4 are generally on the order of 5–20 × 106 M–1 s,74,133 while those for aliphatic amines (where only the N–O addition/N–H insertion pathways exist) in aqueous solution increase from 9.3 × 104 to 4.1 × 106 M–1 s–1 with alkyl substitution.59,80,134 The influence of solvent can be roughly determined by reference to results for triethylamine (the upper bound in this range) in CCl4, where the rate constant is 2.3 × 104 M–1 s–1.133 If the primary pathway of PPD ozonation involved N–O interaction, we would expect a rate constant below this final value. That ozonation rate constants for PPDs are at least 2 orders of magnitude faster indicates the presence of more reactive ozonation channels.
Figure 6.

Free energy barrier heights (T = 298.15 K) for PPD ozonation pathways compared to the minimum barrier pathway for 2M2B ozonation (1, solid black lines). Transition structures are shown for 4-ADPA transformations. For N–H insertion reactions, solid and dashed lines correspond to insertion at terminal and central N atoms, respectively. DeMore (DM) additions of O3 to the rings of 4-ADPA (2), 4-ADPA–OH (2g), and MePPD (3) are the only transformations that are kinetically competitive with 1 ozonation. DeMore TSs with secondary H-abstraction interactions show additional stabilization.
Instead, direct interaction with the PPD ring results in more stable—thereby, more accessible—transition structures. Among these interactions, concerted addition of ozone to the ring system, which has been proposed for other electron-rich aromatics,68,69 to form primary ozonide 2b is the least favorable. The barrier for the minimum energy Criegee transition structure (ΔG298.15 K‡ = 25.9 kcal mol–1) is on the same order as that for N–H insertion. However, asymmetric addition to the ring system is significantly more accessible, with barrier heights ranging from 14.2–19.0 kcal mol–1. Conformational details of these transition and product structures explain the range of ΔG‡ values in these systems and provide significant insight into the reactivity of PPDs and likely fate of these structures.
Each of the most stable DeMore transition structures in 2 is stabilized by a secondary interaction between the substrate and O3. This additional stabilization comes either from a second C–O interaction or interaction between the O3 terminus and the amine hydrogens. These latter conformations are particularly stable, and the lowest energy structure exhibits a barrier height of ΔG298.15 K‡ = 14.2 kcal mol–1, on par with the minimum barrier for 1 within the uncertainty of our ωB97X-V/def2-TZVPP treatment (see Table 1). IRC computations indicate a single C–O bond formation, but the resulting intermediate is primed for amine H abstraction, a reaction that affords the hydroperoxy imine, 2d. In both types of DeMore transition structures—those set up for primary ozonide formation and for H abstraction—4ADPA ozonation pathways initiated through interaction with the C atoms of the PPD ring are significantly more accessible than interactions with PPD amines.
These transition structures indicate that transformations from various isomers of DeMore adduct 2c to primary ozonide 2b and hydroperoxy-imine 2d represent the most significant early stage intermediates of 2 ozonation. As a result, the reactivity of these compounds determines the distribution of the products for the overall reaction between 2 and ozone. 1,2-Primary ozonide 2b is expected to decompose into open-chain scission products64,67,69 similar to the wide variety of experimentally determined PPD ozonation products.42 While not the focus of the present study, the wax-like structure of these products (especially in the 6PPD system, which has a longer alkyl tail) makes them potential candidates for the film formation mechanism of PPDs in tires.37 For present purposes, the opening of the PPD ring is the most salient feature of these products, making them poor candidates for the formation of 2 quinone, and we do not consider them further.
Formation of 2d, on the other hand, is a promising step toward the quinone, forming a necessary C–O bond and preserving the ring structure. This species is primed for the loss of hydroperoxy radical (HOO•) to form alkoxy radical 2e,67,69,70 which may abstract H• from HOO• producing 1O2. This reaction affords hydroxyl imine 2f, which may rearomatize to form hydroxyl-4ADPA (2g) with catalytic amounts of H+.67 While potentially a minor pathway, it is also possible that alkoxy radical 2e immediately recombines with HOO• and loses H2O to promptly form the quinone 2q, analogous to aqueous systems.71 Due to the difficulty of identifying transition structures, we do not report barrier heights for these transformations. Still, overall reaction energies (Table 2 and Figure 7) indicate that each individual step is thermodynamically feasible. The citations for each step also provide literature precedent for each of these transformations in similar systems, though other pathways from 2d to 2g can be imagined.66,68,69
Table 2. Barrier Heights and Reaction Free Energies (T = 298.15 K) for PPD Ozonation on Both SC- and AP-ωB97X-V/def2-TZVPP Surfacesa.
| SC |
AP |
|||
|---|---|---|---|---|
| reaction | ΔG‡ | ΔGrxn | ΔG† | ΔGrxn |
| 1 → 1c | 13.2 | –51.1 | 15.4 | –42.5 |
| 2 → 2a | 26.9 | –8.0 | 38.4 | 0.7 |
| 2 → 2b | 25.9 | –16.5 | 34.6 | –7.9 |
| 2 → 2c | 14.2 | 9.06 | 19.5 | 17.7 |
| 2c → 2d | – | –19.6 | – | –19.7 |
| 2d → 2f | – | –17.7 | – | –6.7 |
| 2f → 2g | – | –27.8 | – | N/A |
| 2g → 2c′ | 11.9 | 8.3 | 17.1 | 14.9 |
| 3 → 3a | 24.6 | –10.3 | 14.0 | –1.7 |
| 3 → 3c | 13.6 | 8.6 | 18.8 | 16.0 |
| 2h → 2i | 19.8 | –36.0 | 28.4 | –27.4 |
| 2h → 2j | 18.9 | 3.4 | 20.7 | 12.4 |
Figure 7.
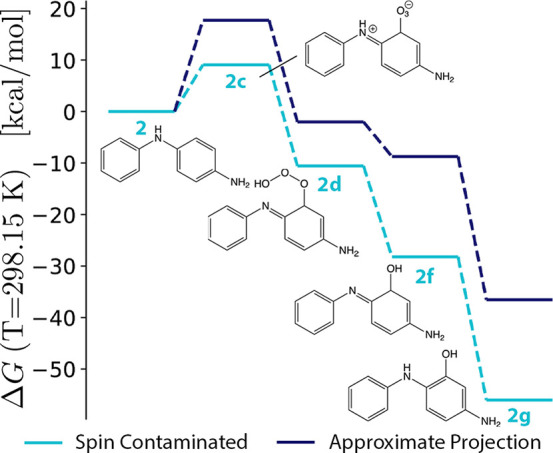
Reaction energies (ΔGrxn,298.15 K) for steps from 4ADPA (2) to 4ADPA–OH (2g) at the SC- and AP-ωB97X-V/def2-TZVPP levels. Following the initial ozonation step to form 2c, both methods indicate a strongly exothermic reaction cascade.
Regardless of the particular steps, production of hydroxyl-4ADPA 2g through direct 2–O3 interaction is the salient feature of this mechanistic proposal. This motif is analogous to ozonation of anilines,67 catechols,66 and pyrazoles,70 where hydroxylated aromatics are significant products. Furthermore, recent experimental work identified the presence of 6PPD’s analogue of 2g in snow samples collected along major roadways.42 These hydroxyl intermediates aid the function of PPDs in tires, as these derivatives possess similar reactivity toward O3 as their parent compounds.60 Indeed, addition of ozone to 2g to form hydroxylated DeMore intermediate 2c′ proceeds with a minimum barrier height of 11.9 kcal mol–1, lower than the minimum barrier for both 1 and 2. Because 2g is even more reactive toward O3 than 2, an initial ozonation of 2 to form 2g “commits” the substrate to a second ozonation. Each of the reaction pathways available to 2c exists for 2c′ (where the prime indicates a hydroxylated species), which may decompose into scission products through primary ozonide 2b′ or form hydroquinone 2g′ through 2d′ (Figure 5). This hydroquinone can slowly oxidize to 4ADPA quinone 2q at ambient conditions135 or more rapidly through further reaction with O3.68,71 Though not shown in Figure 5, adduct 2c′ may alternatively abstract the ipso H from the ring to form the quinone, similar to the mechansim proposed for phenolates.68
Overall, this cascade of reactions results in a molecular mechanism for the formation of 4ADPA quinone (2q) from 2 through a series of kinetically and thermodynamically favorable transformations. While the reactions in Figure 5 do not account for all of the experimentally determined products of PPD ozonation42 and are not intended to be exhaustive, they demonstrate a viable path to the quinone that is linked to the high activity of PPDs toward O3. The initial nucleophilic attack of O3 by the PPD ring has not been previously proposed but critically occurs with a barrier height close to that for 2M2B ozonation (Section 3.3). Additionally, consumption of two molar equivalents of O3 is consistent with experimental results.18 Thus, this channel allows kinetic scavenging of ozone, even for aryl PPD 2.
3.4.2. Effects of N-Alkylation
It is well-known that alkyl-aryl PPDs (like 6PPD) are more reactive toward ozone than aryl PPDs like our surrogate compound 2.3,4,35 Still, inclusion of the 6PPD alkyl chain into our model system would result in a nearly 50% increase in the size of our system as well as introducing a significant degree of conformational flexibility. We therefore model the kinetic effects of alkylation using N-Me-PPD (3), which is more computationally tractable and should capture the lion’s share of the relevant effects.
Many authors attribute increased ozonation rates to more stable interactions with the nitrogen atom,35 but we have already noted that these are not significant reaction channels for 2. N–H insertion pathways remain uncompetitive in 3. Unsurprisingly, alkylation of the terminal amine does not affect that barrier height for insertion into the central N–H bond significantly, and we have ΔG298.15 K‡ = 26.5 kcal mol–1 for this reaction. Methylation of the terminal nitrogen does increase the reactivity of its N–H bond considerably, resulting in an insertion barrier of 24.6 kcal mol–1 (cf. the barrier of 30.3 kcal mol–1 in 2), but this is still relatively large.
Instead, the experimental effects of alkylation can be attributed to additional stability in the interactions between ozone and the ring system, mediated through the N atom’s connection to the π-system. The DeMore-plus-H-abstraction transition structures that lead to hydroperoxy imine 3d at the methylated amine are again particularly stable, with a minimum barrier height of ΔG298.15 K‡ = 13.6 kcal mol–1 relative to isolated fragments. This stabilization can be understood with reference to the “lone pair” (LP) orbital on the terminal N atom in 3 (Figure 8[a]). Hyperconjugation of the methyl C–H density supports interactions through the ring π-system in 3, resulting in significant delocalization of the amine LP through the ring. In the transition structure for O3 addition (Figure 8[b]), this orbital clearly exhibits the nascent interaction between the floating O3 and the amine H, even though IRC confirms the single C3–O bond formation. Even for 3, the minimum predicted barrier height of 13.6 kcal mol–1 is larger than that for 1 (ΔG298.15 K = 13.2 kcal mol–1), although this difference is well within the 1.25 kcal mol–1 1-sigma error of our method (Table 1). In any case, these results underscore how difficult it is to identify molecules with lower O3 barrier heights than 1 and related compounds.
Figure 8.
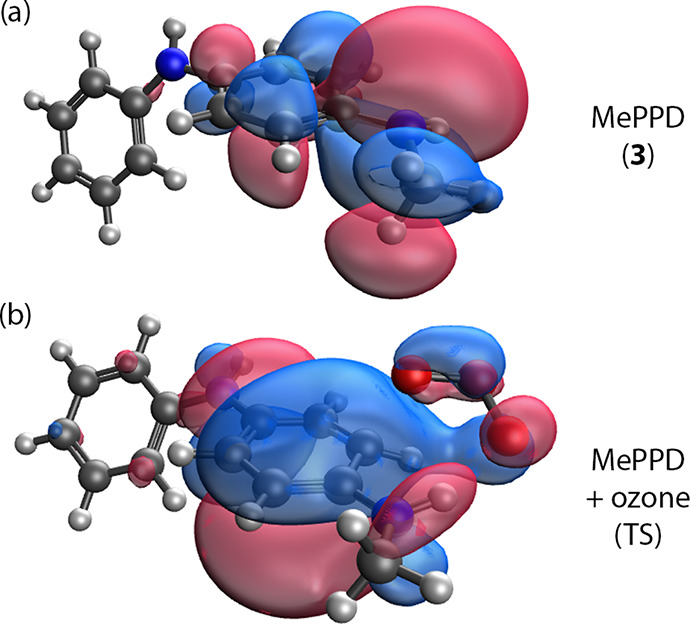
Intrinsic bonding orbitals (IBOs) for (a) MePPD (3), showing delocalization of the N lone pair stabilized by methyl hyperconjugation and (b) DeMore-like TS for 3 with a secondary interaction between the amine H and O3.
The role of amine H atoms in achieving this high degree of ozone reactivity has gone unrecognized in previous work on PPD systems. While barrier heights for certain other DeMore structures (like the precursors to 1,2-primary ozonides) are competitive, stabilization from H-abstraction leads to particularly active reaction channels. As seen above (Section 3.4.1), these channels open a path to quinone formation, and there is an inextricable link between the activity of 6PPD as an antiozonant and its toxicity through the quinone.
3.4.3. Pathways through the Quinone Diimine
While the preceding discussion provides strong evidence for one pathway to PPD quinones, alternative mechanisms are present in the literature. Indeed, recent experimental evidence has been interpreted to suggest quinone formation proceeds through the 6PPD quinone diimine (QDI).41 Specifically, mass spectrometry of the ozonation products of 6PPD results in a peak with an integrated mass of 283.1798 Da, which the authors attributed to the [M + H]+ ion of 6PPD QDI–OH. This interpretation is supported by a body of work detailing the in situ formation of 6PPD QDI itself,22,23 and others have proposed similar chemistry.42 We evaluate the possibility of 6PPD QDI ozonation to form 6PPDQ through the QDI with reference to the model 4-ADPA QDI (2h), assuming once again this system captures the fundamental chemistry.
Analogous to both 2 and 3 as discussed in Sections 3.4.1 and 3.4.2 above, 2h possesses transition structures corresponding to both Criegee (concerted) and DeMore-like (stepwise) addition of O3 to the QDI ring system (Figure 9). These two pathways are similarly facile in the QDI system. Quantitatively, the minimum barrier for concerted and stepwise addition to 2h is 19.8 kcal mol–1 and 18.9 kcal mol–1, respectively. It is noteworthy that the absence of amine H atoms in 2h corresponds to the absence of extremely low barrier ozonation pathways as were seen for 2 and 3, underscoring the importance of H-abstraction for PPD ozonation. Instead, in the case of 2h DeMore adducts, the most stable DeMore transitions are accompanied instead by secondary interactions with an additional C atom in the PPD ring. Hence, as far as ring interactions with 2h are concerned, the significant pathways will progress through primary ozonides, ultimately resulting in scission products. In particular, it is difficult to see how these structures could produce quinones.
Figure 9.

Reaction pathways for ring ozonation in 4ADPA QDI (2h). Neither primary ozonide 2i nor DeMore intermediate 2j is likely to result in products conducive to quinone formation.
It therefore seems more likely that the experimentally determined41 6PPD QDI–OH forms through the oxidation of 6PPD–OH (4g) to the QDI, rather than the hydroxylation of the QDI. These considerations also indicate that the QDI is unlikely to be a precursor to the quinone. All of the results of the present study suggest, instead, that a direct attack of the 6PPD ring initiating the pathway to the quinone occurs through 6PPD–OH, which should exhibit increased reactivity toward ozone than even the parent 6PPD.
3.5. Implications for Nontoxic PPD Design
The preceding mechanistic work highlights the vulnerability of rubber to degradation by ozone and explains the distinct ability of PPDs to afford protection against this degradation. As identified through analysis of key, rate-determining steps in the mechanism of PPD ozonation, critical features of this chemistry that result in the formation of highly toxic 6PPD quinone should inform ongoing work to identify replacements for 6PPD.
The mechanistic work in this report has dispelled a number of reports in the literature that suggest the ozonation of both alkene and PPD systems is initialized by one-electron transfer from the subtrate to ozone.76−79 This provides theoretical backing to the perhaps obvious conclusion that predicting ozonation capacity is more complicated than determining ionization potentials, even if this parameter is relatively predictive within a single class of molecules.74 Instead, the subtlety of the mechanistic results above indicate that atomistic details of reactivity must be taken into account.
Under the previous paradigm that ozonation of PPDs is facilitated through their N atoms,37,40,72 prevention of quinone formation through deactivation of the aromatic ring system is a logical approach to reduce the toxicity of 6PPD. The results above, however, demonstrate direct attack by the aromatic ring is significantly more favorable, suggesting that such approaches will be unsuccessful, as they will poison the antiozonant capacity of 6PPD. The discovery of the importance of secondary interaction with amine H atoms for stabilizing transition structures suggests the necessity of these atoms also has bearing on PPD design. Specifically, while compounds that lack the potential for these reactions may still exhibit reactivity toward O3 (e.g., 6PPD QDI42), these reactions are unlikely to afford kinetic protection against rubber ozonation.
Present results indicate that activation of the PPD ring significantly increases activity toward O3 (Figure 6). This is seen first in the minor increase in reactivity upon N-alkylation, which has been known for some time,3,4,35 and then by the significant increase upon hydroxylation. Though ring activation improves antiozonant performance, it also facilitates the reactions that can ultimately produce PPD quinones. This is perhaps the most important result of this study, in that it provides a direct, atomistic link between the function of 6PPD as an antiozonant and its aquatic toxicity through the quinone.12−17 While this suggests an obvious difficulty in continued use of the PPDs on the market today, all is not lost. It may be possible to trap intermediates in the reaction prior to formation of the (hydro)quinone, but the scheme above indicates that this will likely reduce the molar equivalents of O3 consumed by the PPD. Alternatively, the mechanistic insights of the present work explain both the rapid reactivity of PPDs toward O3 and demonstrate the channel of formation for the toxic quinone. These provide principles for rational design of effective antiozonants that inform ongoing efforts to develop nontoxic alternatives to 6PPD in tires.
Acknowledgments
We thank Howard Colvin, Michael Dodd, Frederick Ignatz–Hoover, Edward Kolodziej, Jamie McNutt, and Jessica Ray for helpful conversations related to ozone chemistry, polymer chemistry, and the toxicity of 6PPD quinone. This work was supported by the U.S. Department of Energy, Office of Science, Office of Advanced Scientific Computing, and Office of Basic Energy Sciences, via the Scientific Discovery through Advanced Computing (SciDAC) program. Additional funding was provided by the United States Department of Agriculture (USDA), Agricultural Research Services.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.est.2c08717.
S1: Further analysis of computational models; S2: Conformer specifications; Figure S1: Definition of conformer labeling for PPD DeMore adducts; Figure S2: Definition of conformer labeling for PPD-OH DeMore adducts; Figure S3: Definition of conformer labeling for PPD primary ozonides; Figure S4: Definition of conformer labeling for hydroxylated PPDs; Table S1: Errors in spin-contaminated (SC) and approximate projection (AP) singlepoint energies for benchmark van der Waals (vdW) complexation energies; and Table S2: Effect of regularizer strength (κ) on performance of κ-OOMP2 for benchmark ozonation reactions (PDF)
Absolute energies for determining transition and reaction energies (XLSX)
Stationary point geometreis (ZIP)
Mention of trade names or commercial products in this report is solely for the purpose of providing specific information and does not imply recommendation or endorsement by the USDA. USDA is an equal opportunity provider and employer.
The authors declare no competing financial interest.
Supplementary Material
References
- Rapra S.Global tire manufacturing output to grow 3.4% year-on-year to 2024. 2019.https://www.smithers.com/resources/2019/jun/global-tire-manufacturing-output-to-grow-by-2024 (accessed 2021-01-04).
- Huntink N. M.Durability of rubber products: Development of new antidegradants for long-term protection; Twente University Press: Enschede, 2003; pp 1–207. [Google Scholar]
- Cox W. L. Chemical antiozonants and factors affecting their utility. Rubber Chem. Technol. 1959, 32, 364–378. 10.5254/1.3542401. [DOI] [Google Scholar]
- Layer R. W. Reaction of ozone with p-phenylenediamine and related compounds. Rubber Chem. Technol. 1966, 39, 1584–1592. 10.5254/1.3547073. [DOI] [Google Scholar]
- Bălan S.; Brushia R.; Buck T.; Doherty A.-C.; Ernst M.; Garland M.; Grant K.; Harris K.. Product – Chemical Profile for Motor Vehicle Tires Containing N-(1,3-Dimethylbutyl)-N’-phenyl-p-phenylenediamine (6PPD); 2021.
- Chung K.-T.; Murdock C. A.; Stevens S.; Li Y.-S.; Wei C.-I.; Huang T.-S.; Chou M. W. Mutagenicity and toxicity studies of p-phenylenediamine and its derivatives. Toxicol. Lett. 1995, 81, 23–32. 10.1016/0378-4274(95)03404-8. [DOI] [PubMed] [Google Scholar]
- El-Ansary E.; Ahmed M.; Clague H. Systemic toxicity of para-phenylenediamine. Lancet 1983, 321, 1341. 10.1016/S0140-6736(83)92456-X. [DOI] [PubMed] [Google Scholar]
- Anuradha D. S.; Arora S.; Mehrotra S.; Arora A.; Kar P. Acute Renal Failure Following para-Phenylenediamine (PPD) Poisoning: A Case Report and Review. Renal Failure 2004, 26, 329–332. 10.1081/JDI-200026722. [DOI] [PubMed] [Google Scholar]
- Yamano T.; Shimizu M. Skin sensitization potency and cross-reactivity of p-phenylenediamine and its derivatives evaluated by non-radioactive murine local lymph node assay and guinea-pig maximization test. Contact Dermatitis 2009, 60, 193–8. 10.1111/j.1600-0536.2008.01500.x. [DOI] [PubMed] [Google Scholar]
- Bharali M. K.; Dutta K. Testicular toxicity of para-phenylenediamine after subchronic topical application in rat. Int. J. Environ. Health Res. 2012, 22, 270–278. 10.1080/09603123.2011.634388. [DOI] [PubMed] [Google Scholar]
- Seydi E.; Fatahi M.; Naserzadeh P.; Pourahmad J. The effects of para-phenylenediamine (PPD) on the skin fibroblast cells. Xenobiotica 2019, 49, 1143–1148. 10.1080/00498254.2018.1541264. [DOI] [PubMed] [Google Scholar]
- Tian Z.; Zhao H.; Peter K. T.; Gonzalez M.; Wetzel J.; Wu C.; Hu X.; Prate J.; Mudrock E.; Hettinger R.; Cortina A. E.; Biswas R. G.; Kock F. V.; Soong R.; Jenne A.; Du B.; Hou F.; He H.; Lundeen R.; Gillbreath A.; Sutton R.; Scholz N. L.; Davis J. W.; Dodd M. C.; Simpson A.; MvIntyre J. K.; Kolodziej E. P. A ubiquitous tire rubber-derived chemical induces acute mortality in coho salmon. Science 2021, 371, 185–189. 10.1126/science.abd6951. [DOI] [PubMed] [Google Scholar]
- Blair S. I.; Barlow C. H.; McIntyre J. K. Acute cerebrovascular effects in juvenile coho salmon exposed to roadway runoff. Canadian Journal of Fisheries and Aquatic Sciences 2021, 78, 103–109. 10.1139/cjfas-2020-0240. [DOI] [Google Scholar]
- Tian Z.; Gonzalez M.; Rideout C. A.; Zhao H. N.; Hu X.; Wetzel J.; Mudrock E.; James C. A.; McIntyre J. K.; Kolodziej E. P. 6PPD-Quinone: Revised toxicity assessment and quantification with a commercial standard. Environ. Sci. Technol. Lett. 2022, 9, 140–156. 10.1021/acs.estlett.1c00910. [DOI] [Google Scholar]
- Hiki K.; Asahina K.; Kato K.; Yamagishi T.; Omagari R.; Iwasaki Y.; Watanabe H.; Yamamoto H. Acute Toxicity of a Tire Rubber-Derived Chemical, 6PPD Quinone, to Freshwater Fish and Crustacean Species. Environ. Sci. Technol. Lett. 2021, 8, 779–784. 10.1021/acs.estlett.1c00453. [DOI] [Google Scholar]
- Varshney S.; Gora A. H.; Siriyappagouder P.; Kiron V.; Olsvik P. A. Toxicological effects of 6PPD and 6PPD quinone in zebrafish larvae. J. Hazard. Mater. 2022, 424, 127623. 10.1016/j.jhazmat.2021.127623. [DOI] [PubMed] [Google Scholar]
- Brinkmann M.; Montgomery D.; Selinger S.; Miller J. G.; Stock E.; Alcaraz A. J.; Challis J. K.; Weber L.; Janz D.; Hecker M.; Wiseman S. Acute toxicity of the tire rubber-derived chemical 6PPD-quinone to four fishes of commercial, cultural, and ecological importance. Environ. Sci. Technol. Lett. 2022, 9, 333–338. 10.1021/acs.estlett.2c00050. [DOI] [Google Scholar]
- Hu X.; Zhao H. N.; Tian Z.; Peter K. T.; Dodd M. C.; Kolodziej E. P. Transformation product formation upon heterogeneous ozonation of the tire rubber antioxidant 6ppd (N-(1,3-dimethylbutyl)-N′-phenly-p-phenylenediamine. Environ. Sci. Technol. Lett. 2022, 9, 413–419. 10.1021/acs.estlett.2c00187. [DOI] [Google Scholar]
- Challis J. K.; Popick H.; PRajapati S.; Harder P.; Giesy J. P.; McPhedran K.; Brinkmann M. Occurrences of tire rubber-derived contaminants in cold-climate urban runoff. Environ. Sci. Technol. Lett. 2021, 8, 961–967. 10.1021/acs.estlett.1c00682. [DOI] [Google Scholar]
- Johannessen C.; Helm P.; Lashuk B.; Yargeau V.; Metcalfe C. D. The tire wear compounds 6PPD-quinone and 1,3-diphenylguanidine in an urban watershed. Arch. Environ. Contam. Toxicol. 2022, 82, 171–179. 10.1007/s00244-021-00878-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong S. W.; Lin C.-Y. Improving flex fatigue and dynamic ozone crack resistance through antidegradamts. Rubber World 2000, 36–41. [Google Scholar]
- Li G.-Y.; Koenig J. FTIR imaging of oxidation of polyisoprene. 2. The role of N-phenyl-N′-dimethylbutyl-p-phenylenediamine antioxidant. Polym. Degrad. Stab. 2003, 81, 377–385. 10.1016/S0141-3910(03)00109-5. [DOI] [Google Scholar]
- Gatial A.; Plovková J.; Kortisová I.; Breza M. On the dehydrogenation of N,N′-substituted p-phenylenediamine antioxidants. II. N-Phenyl-N′-(α-methylbenzyl)-p-phenylenediamine (SPPD). Vib. Spectrosc. 2007, 44, 1–8. 10.1016/j.vibspec.2006.06.011. [DOI] [Google Scholar]
- Suffield R. M.; Dillman S. H.; Haworth J. E. Evaluation of antioxidant performance of a natural product in polyolefins. J. Vinyl Addit. Technol. 2004, 10, 52–56. 10.1002/vnl.20007. [DOI] [Google Scholar]
- Dopico-García M. S.; Castro-López M. M.; López-Vilariño J. M.; González-Rodríguez M. V.; Valentão P.; Andrade P. B.; García-Garabal S.; Abad M. J. Natural extracts as potential source of antioxidants to stabilize polyolefins. J. Appl. Polym. Sci. 2011, 119, 3553–3559. 10.1002/app.33022. [DOI] [Google Scholar]
- Cerruti P.; Malinconico M.; Rychly J.; Matisova-Rychla L.; Carfagna C. Effect of natural antioxidants on the stability of polypropylene films. Polym. Degrad. Stab. 2009, 94, 2095–2100. 10.1016/j.polymdegradstab.2009.07.023. [DOI] [Google Scholar]
- Ambrogi V.; Cerruti P.; Carfagna C.; Malinconico M.; Marturano V.; Perrotti M.; Persico P. Natural antioxidants for polypropylene stabilization. Polym. Degrad. Stab. 2011, 96, 2152–2158. 10.1016/j.polymdegradstab.2011.09.015. [DOI] [Google Scholar]
- Peltzer M. A.; Navarro R.; López J. C. C.; Jiménez A. Evaluation of the melt stabilization performance of hydroxytyrosol (3,4-dihydroxy-phenylethanol) in polypropylene. Polym. Degrad. Stab. 2010, 95, 1636–1641. 10.1016/j.polymdegradstab.2010.05.021. [DOI] [Google Scholar]
- Samper M. D.; Fages E.; Fenollar O.; Boronat T.; Balart R. The potential of flavonoids as natural antioxidants and UV light stabilizers for polypropylene. J. Appl. Polym. Sci. 2013, 129, 1707–1716. 10.1002/app.38871. [DOI] [Google Scholar]
- Tátraaljai D.; Kirschweng B.; Kovács J.; Földes E.; Pukánszky B. Processing stabilisation of PE with a natural antioxidant, curcumin. Eur. Polym. J. 2013, 49, 1196–1203. 10.1016/j.eurpolymj.2013.02.018. [DOI] [Google Scholar]
- Xin M.; Ma Y.; Xu K.; Chen M. Dihydromyricetin: An effective non-hindered phenol antioxidant for linear low-density polyethylene stabilisation. J. Therm. Anal. Calorim. 2013, 114, 1167–1175. 10.1007/s10973-013-3169-1. [DOI] [Google Scholar]
- Tátraaljai D.; Földes E.; Pukánszky B. Efficient melt stabilization of polyethylene with quercetin, a flavonoid type natural antioxidant. Polym. Degrad. Stab. 2014, 102, 41–48. 10.1016/j.polymdegradstab.2014.02.010. [DOI] [Google Scholar]
- Doudin K.; Al-Malaika S.; Sheena H.; Tverezovskiy V.; Fowler P. New genre of antioxidants from renewable natural resources: Synthesis and characterisation of rosemary plant-derive antioxidants and their performance in polyolefins. Polym. Degrad. Stab. 2016, 130, 126–134. 10.1016/j.polymdegradstab.2016.05.030. [DOI] [Google Scholar]
- Xia H.; Gao H.; Zhang Y.; Wang Z.; Song L.; Liu L.; Tian X.; Huang X.; Yu Q. Natural antioxidant from bamboo leaves for the processing stability of polypropylene. J. Therm. Anal. Calorim. 2021, 146, 1657–1665. 10.1007/s10973-020-10115-0. [DOI] [Google Scholar]
- Huntink N. M.; Datta R. N.; Noordermeer J. W. M. Addressing Durability of Rubber Compounds. Rubber Chem. Technol. 2004, 77, 476–511. 10.5254/1.3547833. [DOI] [Google Scholar]
- Erickson E.; Berntsen R.; Hill E.; Kusy P. The reaction of ozone with SBR rubbers. Rubber Chem. Technol. 1959, 32, 1062–1079. 10.5254/1.3542467. [DOI] [Google Scholar]
- Razumovskii S.; Batashova L. Mechanism of protection against ozone by N-phenyl-N′-isopropyl-p-phenylenediamine. Rubber Chem. Technol. 1970, 43, 1340–1348. 10.5254/1.3547334. [DOI] [Google Scholar]
- Andries J.; Ross D.; Diem H. Ozone attack and antiozonant protection of vulcanized natural rubber. A surface study by attenuated total reflectance spectroscopy. Rubber Chem. Technol. 1975, 48, 41–49. 10.5254/1.3545038. [DOI] [Google Scholar]
- Andries J.; Rhee C.; Smith R.; Ross D.; Diem H. A surface study of ozone attack and antiozonant protection of carbon black loaded natural rubber compounds. Rubber Chem. Technol. 1979, 52, 823–837. 10.5254/1.3535244. [DOI] [Google Scholar]
- Lattimer R.; Hooser E.; Diem H.; Layer R.; Layer R.; Rhee C. Mechanisms of ozonation of N,N′-di-(1-methylheptyl)-p-phenylenediamine. Rubber Chem. Technol. 1980, 53, 1170–1190. 10.5254/1.3535087. [DOI] [Google Scholar]
- Klöckner P.; Seiwert B.; Wagner S.; Reemtsma T. Organic markers of tire and road wear particles in sediments and soils: Transformation products of major antiozonants as promising candidates. Environ. Sci. Technol. 2021, 55, 11723–11732. 10.1021/acs.est.1c02723. [DOI] [PubMed] [Google Scholar]
- Seiwert B.; Nihemaiti M.; Troussier M.; Weyrauch S.; Reemtsma T. Abiotic oxidative transformation of 6-PPD and 6-PPD quinone from tires and occurrence of their products in snow from urban roads and in municipal wastewater. Water Res. 2022, 212, 118122. 10.1016/j.watres.2022.118122. [DOI] [PubMed] [Google Scholar]
- von Sonntag C.; von Gunten U.. Chemistry of Ozone in Water and Wastewater Treatment: From Basic Principles to Applications; IWA Publishing: London, 2012; 10.2166/9781780400839. [DOI] [Google Scholar]
- Fisher T. J.; Dussault P. H. Alkene ozonolysis. Tetrahedron 2017, 73, 4233–4258. 10.1016/j.tet.2017.03.039. [DOI] [Google Scholar]
- Zheng T.; Zheng X.; Zhan S.; Zhou J.; Liao S. Study on the ozone aging mechanism of natural rubber. Polym. Degrad. Stab. 2021, 186, 109514. 10.1016/j.polymdegradstab.2021.109514. [DOI] [Google Scholar]
- Criegee R. Mechanism of ozonolysis. Angew. Chem. Chem., Int. Ed. Engl. 1975, 14, 745–752. 10.1002/anie.197507451. [DOI] [Google Scholar]
- DeMore W. Arrhenius constants for the reactions of ozone with ethylene and acetylene. Int. J. Chem. Kinet. 1969, 1, 209–220. 10.1002/kin.550010207. [DOI] [Google Scholar]
- Gillies J.; Gillies C. W.; Suenram R.; Lovas F. The ozonolysis of ethylene. microwave Spectrum, molecular structure, and dipole moment of ethylene primary ozonide (1, 2, 3-trioxolane). J. Am. Chem. Soc. 1988, 110, 7991–7999. 10.1021/ja00232a007. [DOI] [Google Scholar]
- Saito T.; Nishihara S.; Kataoka Y.; Nakanishi Y.; Kitagawa Y.; Kawakami T.; Yamanaka S.; Okumura M.; Yamaguchi K. Multireference character of 1,3-dipolar cycloaddition of ozone with ethylene and acrylonitrile. J. Phys. Chem. A 2010, 114, 12116–12123. 10.1021/jp108302y. [DOI] [PubMed] [Google Scholar]
- Krisyuk B. E.; Maiorov A. V. Competition between the concerted and nonconcerted addition of ozone to a double bond. Russ. J. Phys. Chem. B 2011, 5, 790–796. 10.1134/S1990793111090065. [DOI] [Google Scholar]
- Gadzhiev O. B.; Ignatov S. K.; Krisyuk B. E.; Maiorov A. V.; Gangopadhyay S.; Masunov A. E. Quantum chemical study of the initial step of ozone addition to the double bond of ethylene. J. Phys. Chem. A 2012, 116, 10420–10434. 10.1021/jp307738p. [DOI] [PubMed] [Google Scholar]
- Dowideit P.; von Sonntag C. Reaction of ozone with ethene and its methyl- and chlorine-substituted derivatives in aqueous solution. Environ. Sci. Technol. 1998, 32, 1112–1119. 10.1021/es971044j. [DOI] [Google Scholar]
- Kiryukhin D.; Krisyuk B.; Maiorov A. Kinetics and mechanism of ozone addition to tetrafluoroethylene and hexafluoropropylene. Russ. Chem. Bull. 2021, 70, 132–139. 10.1007/s11172-021-3067-9. [DOI] [Google Scholar]
- Kumar M.; Shee J.; Rudshteyn B.; Reichman D. R.; Friesner R. A.; Miller C. E.; Francisco J. S. Multiple stable isoprene–ozone complexes reveal complex entrance channel dynamics in the isoprene + ozone reaction. J. Am. Chem. Soc. 2020, 142, 10806–10813. 10.1021/jacs.0c02360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailey P. S.; Keller J. E. Ozonation of amines. III. t-Butylamine. J. Org. Chem. 1968, 33, 2680–2684. 10.1021/jo01271a014. [DOI] [Google Scholar]
- Bailey P. S.; Southwick L. M.; Carter T. P. Ozonation of nucleophiles. 8. Secondary amines. J. Org. Chem. 1978, 43, 2657–2662. 10.1021/jo00407a021. [DOI] [Google Scholar]
- Shen Q.; Liu Y. D.; Zhong R. Degradation mechanisms of simple aliphatic amines under ozonation: A DFT study. Environ. Sci.: Processes Impacts 2021, 23, 480–490. 10.1039/D0EM00476F. [DOI] [PubMed] [Google Scholar]
- Bailey P. S.; Lerdal D. A.; Carter T. P. Ozonation of nucleophiles. 9. Tertiary amines. J. Org. Chem. 1978, 43, 2662–2664. 10.1021/jo00407a022. [DOI] [Google Scholar]
- Lim S.; McArdell C. S.; von Gunten U. Reactions of aliphatic amines with ozone: Kinetics and mechanisms. Water Res. 2019, 157, 514–528. 10.1016/j.watres.2019.03.089. [DOI] [PubMed] [Google Scholar]
- Hübner U.; von Gunten U.; Jekel M. Evaluation of the persistence of transformation products from ozonation of trace organic compounds – A critical review. Water Res. 2015, 68, 150–170. 10.1016/j.watres.2014.09.051. [DOI] [PubMed] [Google Scholar]
- McCurry D. L.; Quay A. N.; Mitch W. A. Ozone promotes chloropicrin formation by oxidizing amines to nitro compounds. Environ. Sci. Technol. 2016, 50, 1209–1217. 10.1021/acs.est.5b04282. [DOI] [PubMed] [Google Scholar]
- Trogolo D.; Arey J. S.; Tentscher P. R. Gas-phase ozone reactions with a structurally diverse set of molecules: Barrier heights and reaction energies evaluated by coupled cluster and density functional theory calculations. J. Phys. Chem. A 2019, 123, 517–536. 10.1021/acs.jpca.8b10323. [DOI] [PubMed] [Google Scholar]
- Corrodi H.; Hardegger E. Die konfiguration des colchicins und verwandter verbindungen. Helv. Chem. Acta 1955, 38, 2030–2033. 10.1002/hlca.19550380743. [DOI] [Google Scholar]
- Mvula E.; von Sonntag C. Ozonolysis of phenols in aqueous solution. Org. Biomol. Chem. 2003, 1, 1749–1756. 10.1039/b301824p. [DOI] [PubMed] [Google Scholar]
- Hammes F.; Salhi E.; Köster O.; Kaiser H.-P.; Egli T.; von Gunten U. Mechanistic and kinetic evaluation of organic disinfection by-product and assimilable organic carbon (AOC) formation during the ozonation of drinking water. Water Res. 2006, 40, 2275–2286. 10.1016/j.watres.2006.04.029. [DOI] [PubMed] [Google Scholar]
- Pillar-Little E. A.; Camm R. C.; Guzman M. I. Catechol oxidation by ozone and hydroxyl radicals at the air–water interface. Environ. Sci. Technol. 2014, 48, 14352–14360. 10.1021/es504094x. [DOI] [PubMed] [Google Scholar]
- Tekle-Röttering A.; von Sonntag C.; Reisz E.; Eyser C. V.; Lutze H. V.; Türk J.; Naumov S.; Schmidt W.; Schmidt T. C. Ozonation of anilines: Kinetics, stoichiometry, product identification and elucidation of pathways. Water Res. 2016, 98, 147–159. 10.1016/j.watres.2016.04.001. [DOI] [PubMed] [Google Scholar]
- Tentscher P. R.; Bourgin M.; von Gunten U. Ozonation of para-substituted phenolic compounds yields p-benzoquinones, other cyclic α,β-unsaturated ketones, and substituted catechols. Environ. Sci. Technol. 2018, 52, 4763–4773. 10.1021/acs.est.8b00011. [DOI] [PubMed] [Google Scholar]
- Mvula E.; Naumov S.; von Sonntag C. Ozonolysis of lignin models in aqueous solution: Anisole, 2,3-dimethoxybenzene, 1,4-dimethoxybenzene, and 1,3,5-trimethoxybenzene. Environ. Sci. Technol. 2009, 43, 6275–6282. 10.1021/es900803p. [DOI] [PubMed] [Google Scholar]
- Tekle-Röttering A.; Lim S.; Reisz E.; Lutze H. V.; Abdighahroudi M. S.; Willach S.; Schmidt W.; Tentscher P. R.; Rentsch D.; McArdell C. S.; Schmidt T. S.; von Gunten U. Reactions of pyrrole, imidazole, and pyrazole with ozone: kinetics and mechanisms. Environ. Sci.: Water Res. Technol. 2020, 6, 976–992. 10.1039/C9EW01078E. [DOI] [Google Scholar]
- Ramseier M. K.; Gunten U. v. Mechanisms of phenol ozonation—kinetics of formation of primary and secondary reaction products. Ozone: Sci. Eng. 2009, 31, 201–215. 10.1080/01919510902740477. [DOI] [Google Scholar]
- Lattimer R. P.; Hooser E. R.; Layer R. W.; Rhee C. K. Mechanisms of ozonation of N-(1,3-Dimethylbutyl)-N′-phenyl-p-phenylenediamine. Rubber Chem. Technol. 1983, 56, 431–439. 10.5254/1.3538136. [DOI] [Google Scholar]
- Cataldo F.; Faucette B.; Huang S.; Ebenezer W. On the early reaction stages of ozone with N,N′-substituted p-phenylenediamines (6PPD, 77PD) and N,N′,N″-substituted-1,3,5-triazine “Durazone®”: An electron spin resonance (ESR) and electronic absorption spectroscopy study. Polym. Degrad. Stab. 2015, 111, 223–231. 10.1016/j.polymdegradstab.2014.11.011. [DOI] [Google Scholar]
- Cataldo F. Early stages of p-phenylenediamine antiozonants reaction with ozone: Radical cation and nitroxyl radical formation. Polym. Degrad. Stab. 2018, 147, 132–141. 10.1016/j.polymdegradstab.2017.11.020. [DOI] [Google Scholar]
- Wang W.; Cao G.; Zhang J.; Wu P.; Chen Y.; Chen Z.; Qi Z.; Li R.; Dong C.; Cai Z. Beyond substituted p-phenylenediamine antioxidants: Prevalence of their quinone derivative in PM2.5. Environ. Sci. Technol. 2022, 56, 10629–10637. 10.1021/acs.est.2c02463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.-M.; Zhu Q. Olefinic ozonation electron transfer mechanism. J. Org. Chem. 1997, 62, 5934–5938. 10.1021/jo970297s. [DOI] [Google Scholar]
- Cataldo F. Ozone interaction with conjugated polymers—I. Polyacetylene. Polym. Degrad. Stab. 1998, 60, 223–231. 10.1016/S0141-3910(97)00068-2. [DOI] [Google Scholar]
- Cataldo F. Ozone interaction with conjugated polymers—II. Polyphenylacetylene. Polym. Degrad. Stab. 1998, 60, 233–237. 10.1016/S0141-3910(97)00069-4. [DOI] [Google Scholar]
- Schank K.; Beck H.; Buschlinger M.; Eder J.; Heisel T.; Pistorius S.; Wagner C. Ozonation of 1,1,2,2-tetraphenylethene revisited: Evidence for electron-transfer oxygenations. Helv. Chim. Acta 2000, 83, 801–826. . [DOI] [Google Scholar]
- Muñoz F.; von Sonntag C. The reactions of ozone with tertiary amines including complexing agents nitrilotriacetic acid (NTA) and ethylenediaminetetraacetic acid (EDTA) in aqueous solution. J. Chem. Soc., Perkin Tans. 2000, 2, 2029–2033. 10.1039/b004417m. [DOI] [Google Scholar]
- Qi C.-Z.; Wang T.-M.; Zhang X.-M. Protection mechanisms of p-phenylenediamine-type antiozonants. Rubber Chem. Technol. 1998, 71, 722–729. 10.5254/1.3538500. [DOI] [Google Scholar]
- Rapta P.; Vargová A.; Polovková J.; Gatial A.; Omelka L.; Majzlík P.; Breza M. A variety of oxidation products of antioxidants based on N,N′-substituted p-phenylenediamines. Polym. Degrad. Stab. 2009, 94, 1457–1466. 10.1016/j.polymdegradstab.2009.05.003. [DOI] [Google Scholar]
- Lee M.; Zimmermann-Steffans S. G.; Arey J. S.; Fenner K.; von Gunten U. Development of prediction models for the reactivity of organic compounds with ozone in aqueous solution by quantum chemical calculations: The role of delocalized and localized molecular orbitals. Environ. Sci. Technol. 2015, 49, 9925–9935. 10.1021/acs.est.5b00902. [DOI] [PubMed] [Google Scholar]
- Pabst M.; Köhn A.; Gauss J.; Stanton J. F. A worrisome failure of the CC2 coupled-cluster method when applied to ozone. Chem. Phys. Lett. 2010, 495, 135–140. 10.1016/j.cplett.2010.06.023. [DOI] [Google Scholar]
- Goddard W. A.; Dunning T. H.; Hunt W. J.; Hay P. J. Generalized valence bond description of bonding in low-lying states of molecules. Acc. Chem. Res. 1973, 6, 368–376. 10.1021/ar50071a002. [DOI] [Google Scholar]
- Hay P. J.; Dunning T. H.; Goddard W. A. Configuration interaction studies of O3 and O3+. Ground and excited states. J. Chem. Phys. 1975, 62, 3912–3924. 10.1063/1.430306. [DOI] [Google Scholar]
- Kalemos A.; Mavridis A. Electronic structure and bonding of ozone. J. Chem. Phys. 2008, 129, 054312. 10.1063/1.2960629. [DOI] [PubMed] [Google Scholar]
- Glezakou V.-A.; Elbert S. T.; Xantheas S. S.; Ruedenberg K. Analysis of bonding patterns in the valence isoelectronic series O3, S3, SO2, and OS2 in terms of oriented quasi-atomic molecular orbitals. J. Phys. Chem. A 2010, 114, 8923–8931. 10.1021/jp105025d. [DOI] [PubMed] [Google Scholar]
- Miliordos E.; Ruedenberg K.; Xantheas S. S. Unusual inorganic biradicals: A theoretical analysis. Angew. Chem. 2013, 125, 5848–5851. 10.1002/ange.201300654. [DOI] [PubMed] [Google Scholar]
- Miliordos E.; Xantheas S. S. On the bonding nature of ozone (O3) and its sulfur-substituted analogues SO2, OS2, and S3: Correlation between their biradical character and molecular properties. J. Am. Chem. Soc. 2014, 136, 2808–2817. 10.1021/ja410726u. [DOI] [PubMed] [Google Scholar]
- Dunning T. H.; Xu L. T.; Takeshita T. Y.; Lindquist B. A. Insights into the electronic structure of molecules from generalized valence bond theory. J. Phys. Chem. A 2016, 120, 1763–1778. 10.1021/acs.jpca.5b12335. [DOI] [PubMed] [Google Scholar]
- Braïda B.; Galembeck S. E.; Hiberty P. C. Ozone and other 1,3-dipoles: Toward a quantitative measure of diradical character. J. Chem. Theory Comput. 2017, 13, 3228–3235. 10.1021/acs.jctc.7b00399. [DOI] [PubMed] [Google Scholar]
- Penotti F. E.; Cooper D. L. Combining rival π-space descriptions of O3 and of SO2. Int. J. Quantum Chem. 2016, 116, 718–730. 10.1002/qua.25094. [DOI] [Google Scholar]
- Anglada J. M.; Crehuet R.; Bofill J. M. The Ozonolysis of Ethylene: A Theoretical Study of the Gas-Phase Reaction Mechanism. Chem. – Eur. J. 1999, 5, 1809–1822. . [DOI] [Google Scholar]
- Cremer D.; Crehuet R.; Anglada J. The ozonolysis of acetylene–A quantum chemical investigation. J. Am. Chem. Soc. 2001, 123, 6127–6141. 10.1021/ja010166f. [DOI] [PubMed] [Google Scholar]
- Cremer D.; Kraka E.; Crehuet R.; Anglada J.; Gräfenstein J. The ozone–acetylene reaction: Concerted or non-concerted reaction mechanism? A quantum chemical investigation. Chem. Phys. Lett. 2001, 347, 268–276. 10.1016/S0009-2614(01)01032-6. [DOI] [Google Scholar]
- Chan W.-T.; Hamilton I. Mechanisms for the ozonolysis of ethene and propene: Reliability of quantum chemical predictions. J. Chem. Phys. 2003, 118, 1688. 10.1063/1.1531104. [DOI] [Google Scholar]
- Chan W.-T.; Wang C.; Goddard J. D. Concerted and stepwise reaction mechanisms for the addition of ozone to acetylene: A computational study. J. Phys. Chem. A 2007, 111, 4792–4803. 10.1021/jp0633188. [DOI] [PubMed] [Google Scholar]
- Wheeler S. E.; Ess D. H.; Houk K. N. Thinking out of the black box: Accurate barrier heights of 1,3-dipolar cycloadditions of ozone with acetylene and ethylene. J. Phys. Chem. A 2008, 112, 1798–1807. 10.1021/jp710104d. [DOI] [PubMed] [Google Scholar]
- Asgharzade S.; Vahedpour M. Mechanism and thermodynamics of multichannel 1:1 ammonia and ozone tropospheric oxidation reaction. Prog. React. Kinet. Mech. 2013, 38, 266–282. 10.3184/146867813X13738207456659. [DOI] [Google Scholar]
- Mousavipour S. H.; Mortazavi M.; Hematti O. Multichannel RRKM-TST and direct-dynamics CVT study of the reaction of hydrogen sulfide with ozone. J. Phys. Chem. A 2013, 117, 6744–6756. 10.1021/jp404738d. [DOI] [PubMed] [Google Scholar]
- Lee R.; Coote M. L. Mechanistic insights into ozone-initiated oxidative degradation of saturated hydrocarbons and polymers. Phys. Chem. Chem. Phys. 2016, 18, 24663–24671. 10.1039/C6CP05064F. [DOI] [PubMed] [Google Scholar]
- Cardona A. L.; Rivela C. B.; Gibilisco R. G.; Blanco M. B.; Ventura O. N.; Teruel M. Experimental and theoretical kinetic studies of the ozonolysis of select allyl sulfides (H2C = CHCH2SR, R = CH3, CH3CH2): The effect of nascent OH radicals. J. Phys. Chem. A 2022, 126, 6751–6761. 10.1021/acs.jpca.2c04547. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Tishchenko O.; Gour J. R.; Li W.; Lutz J. J.; Piecuch P.; Truhlar D. G. Thermochemical kinetics for multireference systems: Addition reactions of ozone. J. Phys. Chem. A 2009, 113, 5786–5799. 10.1021/jp811054n. [DOI] [PubMed] [Google Scholar]
- Yamaguchi K.; Jensen F.; Dorigo A.; Houk K. N. A spin correction procedure for unrestricted Hartree-Fock and Møller-Plesset wavefunctions for singlet diradicals and polyradicals. Chem. Phys. Lett. 1988, 149, 537–542. 10.1016/0009-2614(88)80378-6. [DOI] [Google Scholar]
- Lee J.; Head-Gordon M. Two single-reference approaches to singlet biradicaloid problems: Complex, restricted orbitals and approximate spin-projection combined with regularized orbital-optimized Møller-Plesset perturbation theory. J. Chem. Phys. 2019, 150, 244106. 10.1063/1.5097613. [DOI] [PubMed] [Google Scholar]
- Lee J.; Head-Gordon M. Regularized orbital-optimized second-order Møller–Plesset perturbation theory: A reliable fifth-order-scaling electron correlation model with orbital energy dependent regularizers. J. Chem. Theory Comput. 2018, 14, 5203–5219. 10.1021/acs.jctc.8b00731. [DOI] [PubMed] [Google Scholar]
- Shee J.; Loipersberger M.; Hait D.; Lee J.; Head-Gordon M. Revealing the nature of electron correlation in transition metal complexes with symmetry breaking and chemical intuition. J. Chem. Phys. 2021, 154, 194109. 10.1063/5.0047386. [DOI] [PubMed] [Google Scholar]
- Rettig A.; Shee J.; Lee J.; Head-Gordon M. Revisiting the orbital energy-dependent regularization of the orbital-optimized second-order Møller–Plesset theory. J. Chem. Theory Comput. 2022, 18, 5382–5392. 10.1021/acs.jctc.2c00641. [DOI] [PubMed] [Google Scholar]
- Mardirossian N.; Head-Gordon M. ωB97X-V: A 10-parameter, range-separated hybrid, generalized gradient approximation density functional with nonlocal correlation, designed by a survival-of-the-fittest strategy. Phys. Chem. Chem. Phys. 2014, 16, 9904–9924. 10.1039/c3cp54374a. [DOI] [PubMed] [Google Scholar]
- Mardirossian N.; Head-Gordon M. ωB97M-V: A combinatorially optimized, range-separated hybrid, meta-GGA density functional with VV10 nonlocal correlation. J. Chem. Phys. 2016, 144, 214110. 10.1063/1.4952647. [DOI] [PubMed] [Google Scholar]
- Mardirossian N.; Head-Gordon M. Thirty years of density functional theory in computational chemistry: An overview and extensive assessment of 200 density functionals. Mol. Phys. 2017, 115, 2315–2372. 10.1080/00268976.2017.1333644. [DOI] [Google Scholar]
- Weigend F.; Ahlrichs R. Balanced basis sets of split valence, triple zeta valence, and quadruple valence quality for H to Rn: Design and assessment of accuracy. Phys. Chem. Chem. Phys. 2005, 7, 3297–3305. 10.1039/b508541a. [DOI] [PubMed] [Google Scholar]
- Weigend F.; Furche F.; Ahlrichs R. Gaussian basis sets of quadruple zeta valence quality for atoms H-Kr. J. Chem. Phys. 2003, 119, 12753–12762. 10.1063/1.1627293. [DOI] [Google Scholar]
- Lebedev V. I. Values of the nodes and weights of ningth to seventeenth order gauss-markov quadrature formulae invariant under the octahedron group with inversion. USSR Computational Mathematics and Mathematical Physics 1975, 15, 44–51. 10.1016/0041-5553(75)90133-0. [DOI] [Google Scholar]
- Lebedev V. I. Quadratures on a sphere. USSR Computational Mathematics and Mathematical Physics 1976, 16, 10–24. 10.1016/0041-5553(76)90100-2. [DOI] [Google Scholar]
- Knizia G. Intrinsic atomic orbitals: An unbiased bridge between quantum theory and chemical concepts. J. Chem. Theory Comput. 2013, 9, 4834–4843. 10.1021/ct400687b. [DOI] [PubMed] [Google Scholar]
- Gimferrer M.; Aldossary A.; Salvador P.; Head-Gordon M. Oxidation State Localized Orbitals: A Method for Assigning Oxidation States Using Optimally Fragment-Localized Orbitals and a Fragment Orbital Localization Index. J. Chem. Theory Comput. 2022, 18, 309–322. 10.1021/acs.jctc.1c01011. [DOI] [PubMed] [Google Scholar]
- Li Y.-P.; Gomes J.; Mallikarjun Sharada S.; Bell A. T.; Head-Gordon M. Improved force-field parameters for QM/MM simulations of the energies of adsorption for molecules in zeolites and a free rotor correction to the rigid rotor harmonic oscillator model for adsorption enthalpies. J. Phys. Chem. C 2015, 119, 1840–1850. 10.1021/jp509921r. [DOI] [Google Scholar]
- Kitagawa Y.; Saito T.; Ito M.; Shoji M.; Koizumi K.; Yamanaka S.; Kawakami T.; Okumura M.; Yamaguchi K. Approximately spin-projected geometry optimization method and its application to di-chromium systems. Chem. Phys. Lett. 2007, 442, 445–450. 10.1016/j.cplett.2007.05.082. [DOI] [Google Scholar]
- Behn A.; Zimmerman P. M.; Bell A. T.; Head-Gordon M. Efficient exploration of reaction paths via a freezing string method. J. Chem. Phys. 2011, 135, 224108. 10.1063/1.3664901. [DOI] [PubMed] [Google Scholar]
- Mallikarjun Sharada S.; Zimmerman P. M.; Bell A. T.; Head-Gordon M. Automated transition state searches without evaluating the Hessian. J. Chem. Theory Comput. 2012, 8, 5166–5174. 10.1021/ct300659d. [DOI] [PubMed] [Google Scholar]
- Fukui K. Formulation of the reaction coordinate. J. Phys. Chem. 1970, 74, 4161–4163. 10.1021/j100717a029. [DOI] [Google Scholar]
- Schmidt M. W.; Gordon M. S.; Dupuis M. The intrinsic reaction coordinate and the rotational barrier in silaethylene. J. Am. Chem. Soc. 1985, 107, 2585–2589. 10.1021/ja00295a002. [DOI] [Google Scholar]
- Epifanovsky E.; Gilbert A. T. B.; Feng X.; Lee J.; Mao Y.; Mardirossian N.; Pokhilko P.; White A. F.; Coons M. P.; Dempwolff A. L.; Gan Z.; Hait D.; Horn P. R.; Jacobson L. D.; Kaliman I.; Kussmann J.; Lange A. W.; Lao K. U.; Levine D. S.; Liu J.; McKenzie S. C.; Morrison A. F.; Nanda K. D.; Plasser F.; Rehn D. R.; Vidal M. L.; You Z.-Q.; Zhu Y.; Alam B.; Albrecht B. J.; Aldossary A.; Alguire E.; Andersen J. H.; Athavale V.; Barton D.; Begam K.; Behn A.; Bellonzi N.; Bernard Y. A.; Berquist E. J.; Burton H. G. A.; Carreras A.; Carter-Fenk K.; Chakraborty R.; Chien A. D.; Closser K. D.; Cofer-Shabica V.; Dasgupta S.; de Wergifosse M.; Deng J.; Diedenhofen M.; Do H.; Ehlert S.; Fang P.-T.; Fatehi S.; Feng Q.; Friedhoff T.; Gayvert J.; Ge Q.; Gidofalvi G.; Goldey M.; Gomes J.; González-Espinoza C. E.; Gulania S.; Gunina A. O.; Hanson-Heine M. W. D.; Harbach P. H. P.; Hauser A.; Herbst M. F.; Hernández Vera M.; Hodecker M.; Holden Z. C.; Houck S.; Huang X.; Hui K.; Huynh B. C.; Ivanov M.; Jász A.; Ji H.; Jiang H.; Kaduk B.; Kähler S.; Khistyaev K.; Kim J.; Kis G.; Klunzinger P.; Koczor-Benda Z.; Koh J. H.; Kosenkov D.; Koulias L.; Kowalczyk T.; Krauter C. M.; Kue K.; Kunitsa A.; Kus T.; Ladjánszki I.; Landau A.; Lawler K. V.; Lefrancois D.; Lehtola S.; Li R. R.; Li Y.-P.; Liang J.; Liebenthal M.; Lin H.-H.; Lin Y.-S.; Liu F.; Liu K.-Y.; Loipersberger M.; Luenser A.; Manjanath A.; Manohar P.; Mansoor E.; Manzer S. F.; Mao S.-P.; Marenich A. V.; Markovich T.; Mason S.; Maurer S. A.; McLaughlin P. F.; Menger M. F. S. J.; Mewes J.-M.; Mewes S. A.; Morgante P.; Mullinax J. W.; Oosterbaan K. J.; Paran G.; Paul A. C.; Paul S. K.; Pavošević F.; Pei Z.; Prager S.; Proynov E. I.; Rák A.; Ramos-Cordoba E.; Rana B.; Rask A. E.; Rettig A.; Richard R. M.; Rob F.; Rossomme E.; Scheele T.; Scheurer M.; Schneider M.; Sergueev N.; Sharada S. M.; Skomorowski W.; Small D. W.; Stein C. J.; Su Y.-C.; Sundstrom E. J.; Tao Z.; Thirman J.; Tornai G. J.; Tsuchimochi T.; Tubman N. M.; Veccham S. P.; Vydrov O.; Wenzel J.; Witte J.; Yamada A.; Yao K.; Yeganeh S.; Yost S. R.; Zech A.; Zhang I. Y.; Zhang X.; Zhang Y.; Zuev D.; Aspuru-Guzik A.; Bell A. T.; Besley N. A.; Bravaya K. B.; Brooks B. R.; Casanova D.; Chai J.-D.; Coriani S.; Cramer C. J.; Cserey G.; DePrince A. E.; DiStasio R. A.; Dreuw A.; Dunietz B. D.; Furlani T. R.; Goddard W. A.; Hammes-Schiffer S.; Head-Gordon T.; Hehre W. J.; Hsu C.-P.; Jagau T.-C.; Jung Y.; Klamt A.; Kong J.; Lambrecht D. S.; Liang W.; Mayhall N. J.; McCurdy C. W.; Neaton J. B.; Ochsenfeld C.; Parkhill J. A.; Peverati R.; Rassolov V. A.; Shao Y.; Slipchenko L. V.; Stauch T.; Steele R. P.; Subotnik J. E.; Thom A. J. W.; Tkatchenko A.; Truhlar D. G.; Van Voorhis T.; Wesolowski T. A.; Whaley K. B.; Woodcock H. L.; Zimmerman P. M.; Faraji S.; Gill P. M. W.; Head-Gordon M.; Herbert J. M.; Krylov A. I. Software for the frontiers of quantum chemistry: An overview of developments in the Q-Chem 5 package. J. Chem. Phys. 2021, 155, 084801. 10.1063/5.0055522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao Y.; Horn P. R.; Head-Gordon M. Energy decomposition analysis in an adiabatic picture. Phys. Chem. Chem. Phys. 2017, 19, 5944–5958. 10.1039/C6CP08039A. [DOI] [PubMed] [Google Scholar]
- Horn P. R.; Mao Y.; Head-Gordon M. Defining the contributions of permanent electrostatics, Pauli repulsion, and dispersion in density functional theory calculations of intermolecular interaction energies. J. Chem. Phys. 2016, 144, 114107. 10.1063/1.4942921. [DOI] [PubMed] [Google Scholar]
- Mao Y.; Loipersberger M.; Horn P. R.; Das A.; Demerdash O.; Levine D. S.; Veccham S. P.; Head-Gordon T.; Head-Gordon M. From intermolecular interaction energies and observable shifts to component contributions and back again: A tale of variational energy decomposition analysis. Annu. Rev. Phys. Chem. 2021, 72, 641–666. 10.1146/annurev-physchem-090419-115149. [DOI] [PubMed] [Google Scholar]
- Ambelang J.; Kline R.; Lorenz O.; Parks C.; Wadelin C.; Shelton J. R. Antioxidants and antiozonants for general purpose elastomers. Rubber Chem. Technol. 1963, 36, 1497–1541. 10.5254/1.3539652. [DOI] [Google Scholar]
- Ignatz-Hoover F.; To B. H.; Datta R. N.; Hoog A. J. D.; Huntink N. M.; Talma A. G. Chemical additives migration in rubber. Rubber Chem. Technol. 2003, 76, 747–768. 10.5254/1.3547765. [DOI] [Google Scholar]
- Curtis H. L.; McPherson A. T.. Dielectric constant, power factor, and resistivity of rubber and gutta-percha. Bur. Stand. (U. S.), Technol. Pap 1925; Vol. 19, pp 669–722, 10.6028/nbst.8302 [DOI]
- Lias S. G.; Bartmess J. E.; Liebman J. F.; Holmes J. L.; Levin R. D.; Mallard W. G. In NIST Chemistry WebBook, NIST Standard Reference Database Number 69; Linstrom P., Mallrd W., Eds.; Chapter Ion Energetics Data. National Institute of Standards and Technology: Gaithersburg, MD. https://webbook.nist.gov/ (accessed 2022-10-31).
- Razumovskii S.; Zaikov G. E.. Ozone and Its Reactions with Organic Compounds; Elsevier: New York, NY, 1984. [Google Scholar]
- Pryor W. A.; Giamalva D. H.; Church D. F. Kinetics of ozonation. 2. Amino acids and model compounds in water and comparisons to rates in nonpolar solvents. J. Am. Chem. Soc. 1984, 106, 7094–7100. 10.1021/ja00335a038. [DOI] [Google Scholar]
- Owsik I.; Kolarz B. The oxidation of hydroquinone to p-benzoquinone catalysed by Cu(II) ions immobilized on acrylic resins with aminoguanidyl groups: Part 1. J. Mol. Catal. A: Chem. 2002, 178, 63–71. 10.1016/S1381-1169(01)00299-0. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Citations
- Lias S. G.; Bartmess J. E.; Liebman J. F.; Holmes J. L.; Levin R. D.; Mallard W. G. In NIST Chemistry WebBook, NIST Standard Reference Database Number 69; Linstrom P., Mallrd W., Eds.; Chapter Ion Energetics Data. National Institute of Standards and Technology: Gaithersburg, MD. https://webbook.nist.gov/ (accessed 2022-10-31).