

Abstract
Objectives:
To investigate the association between cognitive decline and cortical atrophy in individuals with mild cognitive impairment (MCI) and chronic subsyndromal symptoms of depression (SSD) over a four-year period.
Design:
Prospective cohort study.
Setting:
Multicenter, clinic-based.
Participants:
Within the Alzheimer’s Disease Neuroimaging Initiative repository, the Neuropsychiatric Inventory was used to identify MCI individuals with stable endorsement (SSD group N=32) or no endorsement (non-SSD group N=69) of depressive symptoms across timepoints.
Measurements:
Repeated measures of cognitive outcomes, cortical atrophy, and their associations were evaluated with mixed effects models adjusting for age, education, gender, and APOE genotype.
Results:
The SSD group demonstrated accelerated decline on measures of global cognition (Alzheimer’s Disease Assessment Scale (df=421, t=2.242, p=0.025), memory (Wechsler Memory Scale-Revised Logical Memory II (df=244, t=−2.525, p=0.011), information processing speed (Trail Making Test Parts A (df=421, t=2.376, p=0.018) and B (df=421, t=2.533, p=0.012)), and semantic fluency (Category Fluency (df=424, t=−2.418, p=0.016), as well as accelerated frontal lobe (df=341, t=−2.648, p=0.008) and anterior cingulate (df=341, t=−3.786, p<0.001) atrophy. No group differences were observed for rate of decline on measures of attention, learning, and confrontation naming or for rate of atrophy in any other regions. Accelerated frontal lobe and anterior cingulate atrophy was associated with cognitive decline on measures of global cognition, information processing speed, and semantic fluency (all p <0.05), but not memory.
Conclusions:
Individuals with chronic SSD may represent an MCI subgroup that is highly vulnerable to accelerated cognitive decline, an effect that may be governed by frontal lobe and anterior cingulate atrophy.
Keywords: depressive symptoms, mild cognitive impairment, cortical atrophy, cognitive decline, frontal lobe, anterior cingulate
Introduction
Mild cognitive impairment (MCI) has been conceptualized as an antecedent to dementia onset (1). However, only 10–15% of those with MCI convert to dementia each year, posing challenges to accurate prediction of individual long-term outcomes within this population. Of note, subsyndromal symptoms of depression (SSD), depressive symptomatology below the frequency and severity for a diagnosis of major or mild depression (2), has been associated with more rapid conversion from MCI to Alzheimer’s disease (AD) dementia (3), suggesting that SSD may be an etiological risk factor or prodromal manifestation of dementia neuropathology (4). Yet, efforts to disentangle the directionality and temporal association between SSD and dementia within MCI have been limited by the relapse and remitting course of SSD. Within comorbid SSD and MCI populations, 35% of individuals’ depressive symptomatology remits, whereas 65% display a stable or worsening course over a three-year period (5). Those with stable or progressive depressive symptomatology may accumulate increased neuropathological burden over time, heightening the risk for dementia conversion. Thus, longitudinal evaluations of chronic SSD in MCI may provide an optimal framework for assessing the neurobiological correlates of depressive symptomology on cognitive decline.
In accordance with the hypothesis that chronic SSD may confer risk for neuropathology, individuals with MCI and chronic SSD have been found to display more rapid deterioration in global cognitive functioning as well as accelerated progression to AD dementia (6, 7). Lee et al. (2012) (8) found that SSD at baseline was associated with accelerated cognitive decline on measures of global cognition, language, processing speed, and executive function across a two year period. While the results indicate that SSD may independently accelerate cognitive decline, little is known about the underlying etiological processes.
Neuroimaging assessments of cortical atrophy may hold particular promise to elucidate the neurobiological mechanisms of SSD-related accelerated cognitive decline in MCI populations. Cortical atrophy in the hippocampus and entorhinal cortex has been consistently reported in MCI (9). Similarly, late life depression has been associated with cortical atrophy, particularly within the anterior cingulate, prefrontal cortex, and orbitofrontal cortex (10). Yet, few studies have evaluated the associations of chronic SSD with atrophy within MCI samples. The existent literature suggests that atrophy in the anterior cingulate and frontal lobe may be characteristic of chronic SSD in individuals with MCI (3, 8), indicating that rate of cortical atrophy may be a mechanism linking accelerated cognitive decline and depressive symptomatology.
Thus, the overarching goal of the current study was to determine if accelerated cognitive decline in MCI participants with chronic SSD is associated with rate of cortical atrophy. Based on previous investigations (6–8), we predicted that MCI participants with chronic SSD would display accelerated cognitive decline across global cognition, memory, information processing speed, and semantic fluency domains. Additionally, it was hypothesized that MCI participants with SSD would display more rapid cortical atrophy in the frontal lobe and anterior cingulate, but not regions typically associated with MCI due to AD such as the posterior cingulate, temporal lobe, hippocampus, and entorhinal cortex (9, 11). Finally, we hypothesized that SSD-related accelerated cognitive decline within the domains of global cognition, memory, information processing speed, and semantic fluency would be associated with rate of frontal lobe and anterior cingulate atrophy.
Methods and Materials
Participants
Data for the current study was obtained from the September 24, 2015 version of Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (adni.loni.usc.edu). The ADNI study is conducted in accordance with the Declaration of Helsinki and procedures were approved by the institutional review boards of all participating sites. All participants provided written informed consent at enrollment.
From the larger ADNI database, the present study included individuals with baseline MRI data who were diagnosed with MCI by a psychiatrist or neurologist at each study site and reviewed by a central review committee. Criteria for MCI diagnosis included 1) Age between 55 and 90 years; 2) Subjective memory complaint; 3) Mini-Mental Status Score >24; 4) Global Clinical Dementia Rating Scale score = 0.5; 5) Evidence of memory impairment on neuropsychological assessment (WMS-R Logical Memory II ≤8 (12), adjusted for age and education). Exclusion criteria at baseline included: 1) the presence of Major Depressive Disorder or significant symptoms of depression (Geriatric Depression Scale (GDS) >6); 2) modified Hachinski ischemia score>5; 3) significant neurological or psychiatric illness; 4) use of antidepressant drugs with anticholinergic side effects; and 5) high dose of neuroleptics or chronic sedatives or hypnotics, antiparkinsonian medication, and use of narcotic analgesics.
Participants were classified as SSD if at least one depressive symptom (i.e. depression/dysphoria, apathy/indifference, or loss of appetite) was endorsed at each timepoint on the informant-based Neuropsychiatric Inventory (NPI) (13). Insomnia was not considered an inclusionary symptom of SSD given that older adults have been reported to display significant variability in sleep quality over time (14). Participants were included in the non-SSD group if no depressive symptoms, as described above, were reported across all timepoints.
Procedures
Participants completed clinical, cognitive, laboratory, and neuroimaging assessments every six months during the first two study years followed by annual assessments thereafter. Neuropsychiatric Assessment
Neuropsychiatric symptoms were evaluated using the informant-based Neuropsychiatric Inventory (NPI) (13), which uses a semi-structured interview formant to assess 12 behavioral domains (delusions, hallucinations, agitation, depression, anxiety, euphoria, apathy, disinhibition, irritability, and aberrant motor behavior). If a behavior is endorsed, the informant is asked to assess the frequency on a 4-point scale (ranging 1–4) and the severity on a 3 point scale (ranging 1–3). For the current study, the the number of endorsed depressive symptoms on the NPI was used define the SSD and non-SSD groups.
Cognitive Assessment
As previously described, participants completed standard clinical neuropsychological instruments with established reliability and validity. The assessment included measures of 1) General cognitive functioning: The Alzheimer’s Disease Assessment Scale- Cognitive Scale (ADAS-Cog) total score (15); 2) Verbal Learning: Wechsler Memory Scale (WMS) Logical Memory I (12) and Rey Auditory Verbal Learning Test (RAVLT) Immediate Recall total scores; 3) Memory: WMS Logical Memory II (12) and RAVLT Delayed Recall total scores; 4) Information Processing Speed (IPS): Trail Making Test Parts A & B time to completion (16), Digit Symbol Substitution Test total correct responses (17); 5) Semantic fluency: Category fluency (animals) total correct responses (18); 6) Attention: Wechsler Adult Intelligence Scale-Third Edition (WAIS-III) Digit Span subtest total correct forwards and backwards responses (17); 7) Confrontation Naming: The Boston Naming Test-30 item (BNT) total correct responses (19).
MRI Acquisition and Analysis
MRI acquisition and analysis has been previously described (3). Briefly, high resolution T1 structural images were obtained using 1.5T General Electric (Fairfield, CT), Philips (Andover, MA), or Siemens (Washington, DC) scanners with standardized MRI protocol and acquisition parameters (adni.loni.usc.edu/methods/documents/mri-protocols/). ADNI MRI quality control was conducted at the Mayo Clinic (20). Cortical thickness quantification was conducted using the Freesurfer Version 4.4 longitudinal processing framework (http://surfer.nmr.mgh.harvard.edu/) (21, 22). Similar to prior literature (3), cortical thickness was derived from nine a priori regions of interest that have been associated with late life depression and/or MCI (9–11): frontal lobe (medial and lateral orbitofrontal, rostral and caudal middle frontal, and superior frontal), anterior cingulate (rostral and caudal regions), precuneus, cuneus, posterior cingulate, temporal lobe, parahippocampus, hippocampus, and entorhinal cortex. In exploratory analyses, we also examined five separate subregions of frontal lobe (medial and lateral orbitofrontal, rostral and caudal middle frontal, and superior frontal ) and two subregions of the anterior cingulate (rostral and caudal regions). Thickness values in the left and right hemispheres were averaged to obtain one measure for each cortical region of interest.
Statistical Analyses
SSD and non-SSD group differences in demographic characteristics (age, gender, APOE status, years of education), NPI scores, and GDS scores were analyzed using Mann-Whitney U tests for continuous measures and Fisher’s Exact test for categorical measures. The effects of missing data on demographics, APOE status, and chronic SSD were evaluated with a generalized linear mixed effects regression model (GLME) using a binomial outcome and Wald test derived p-values.
In order to assess group differences in the rate of cognitive change, we employed linear mixed effects regression for repeated measures of the 12 cognitive outcomes with each model separately fit with both a random intercept and slope and an unstructured covariance matrix for the random effects. Independent variables in the model included age, gender, years of education, APOE status (at least one APOE ε4 allele versus ε4 non-carrier), depression group (chronic SSD versus non-SSD), time since the initial observation, and an interaction between depression group and time since the initial observation.
To examine group differences in rate of cortical atrophy, we modeled repeated measures of cortical thickness in nine a priori regions of interest with linear mixed effects regression with random intercept and slope with covariates for age, gender, APOE status, depression group, time since the initial observation, and an interaction between depression group and time since the initial observation. In exploratory analyses, we re-reran the models for the five subregions of the frontal lobe and two subregions of the ACC. It should be noted that the analyses and sample are similar to previous work by our group (3), but include a slightly larger sample size.
Finally, we evaluated whether change in cortical atrophy over time in two regions, the frontal lobe and anterior cingulate (selected based on previous literature (3)), was associated with the rate of cognitive decline in measures with a significant SSD group effect. This analysis was done in two steps. In step one, subject-specific rates of cognition and atrophy were estimated separately using mixed effects regression. In step two, rates of cognitive decline were then regressed on atrophy rates, adjusting for age, gender, and APOE genotype. Steps one and two were repeated in 500 bootstrap samples to estimate the variance of the association between atrophy and cognitive decline and test for its significance. In exploratory analyses, we also tested the interaction term between atrophy and SSD to predict changes in cognition. Additionally, we assessed the correlation between baseline cortical thickness and cognition measures to evaluate for potential confounds in our models.
Model fits were assessed by examination of the residuals. All p-values were two-tailed and cognitive and cortical atrophy outcomes were adjusted for multiple comparisons using a false discovery rate (FDR) correction. Statistical analyses were conducted with the R Package (v 2.8.1, The R Foundation for Statistical Computing, http:/www.r.project.org/).
Results
Demographic Factors –
The baseline study sample included 32 participants in the SSD group and 69 participants in the non-SSD group. Median follow-up time was 3 years with a median of 6 neuropsychological assessments (with the exception of the WMS (median = 4)) and 5 MRI scans. As seen in Table 1, there were no baseline differences between SSD and non-SSD groups in regards to sociodemographics (age, gender, years of education), APOE status, or baseline MMSE score. The SSD group had significantly higher scores on the GDS and NPI than the non-SSD group.
Table 1:
Demographic and clinical characteristics of SSD groups at baseline (n=101, df=99)
| Non-SSD (n=69) | SSD (n=32) | Mann-Whitney U or Fisher’s Exact p-value | |
|---|---|---|---|
| Age, years | 73.71±7.99 | 75.84±7.59 | 0.278 |
| Gender (M:F) | 47:22 | 24:8 | 0.640 |
| APOE Status (ε4:non-ε4) | 35:34 | 19:13 | 0.521 |
| Education, years | 15.93±3.01 | 15.62±2.86 | 0.547 |
| MMSE | 27.19±1.90 | 27.09±1.69 | 0.742 |
| GDS Total Score | 1.28±1.24 | 2.19±1.51 | 0.004 |
| NPI Total Score | 0.43±0.83 | 5.41±4.05 | <0.001 |
Note: Group differences were analyzed using Mann-Whitney U tests for continuous measures and Fisher’s Exact test for categorical measures. All values represent mean±standard deviation unless otherwise noted. MMSE= Mini Mental Status Exam, GDS = Geriatric Depression Scale; NPI =Neuropsychiatric Inventory
Baseline Cognitive Performance –
There were no baseline differences in cognitive performance between SSD and non-SSD groups with the exception of poorer performance of the SSD group on one measure, RAVLT Immediate Recall (Table 2)
Table 2:
Linear mixed effects regression results of the relationship between SSD in MCI and cognitive performance baseline (df=95)
| Non-SSD (n=69) | SSD (n=32) | B Estimate | Standard Error | t-value | p-value | FDR p-value | |
|---|---|---|---|---|---|---|---|
| Alzheimer’s Disease Assessment Scale | 10.72±4.31 | 12.47±3.71 | 1.277 | 0.802 | 1.592 | 0.115 | 0.614 |
| WMS Logical Memory I | 7.57±3.16 | 6.75±3.34 | −0.676 | 0.649 | −1.042 | 0.300 | 0.614 |
| RAVLT Immediate Recall | 36.54±11.17 | 28.72±8.07 | −5.669 | 1.979 | −2.864 | 0.005* | 0.067 |
| WMS Logical Memory II | 4.30±3.01 | 3.47±2.29 | −0.768 | 0.632 | −1.216 | 0.227 | 0.614 |
| RAVLT Delayed Recall | 2.94±3.52 | 2.19±1.96 | 0.309 | 0.593 | 0.521 | 0.603 | 0.784 |
| Trail Making Test A | 41.91±24.09 | 45.09±20.16 | 1.638 | 4.291 | 0.382 | 0.703 | 0.831 |
| Trail Making Test B | 117.17±68.22 | 151.91±84.69 | 16.753 | 14.731 | 1.372 | 0.258 | 0.614 |
| Digit Symbol Substitution Test | 39.99±10.46 | 35.88±9.98 | −3.401 | 2.331 | −1.459 | 0.148 | 0.614 |
| Category Fluency (animals) | 16.00±5.32 | 15.69±4.43 | −0.124 | 1.056 | −0.118 | 0.907 | 0.929 |
| WAIS-III Digit Span Forwards | 8.67±1.98 | 8.06±2.14 | −0.334 | 0.398 | −0.840 | 0.403 | 0.655 |
| WAIS-III Digit Span Backwards | 6.10±2.10 | 6.22±2.21 | −0.034 | 0.383 | −0.089 | 0.929 | 0.929 |
| Boston Naming Test | 26.07±3.41 | 24.44±4.33 | −0.778 | 0.795 | −0.978 | 0.331 | 0.614 |
Note: Longitudinal cognitive data was regressed on age, gender, years of education, APOE status, SSD group, time interval from baseline, and the interaction between SSD group and time interval from baseline using linear mixed models.
p<0.05
SSD=Subsyndromal Depression; WMS = Wechsler Memory Scale, RAVLT= Rey Auditory Verbal Learning Test, WAIS-III= Wechsler Adult Intelligence Scale-Third Edition
Rate of Cognitive Decline in SSD –
As compared to the non-SSD group, the SSD group displayed significantly accelerated cognitive decline on 5 out of the 12 cognitive measures in the fully adjusted models (Table 3): ADAS-Cog, Trail Making Test Parts A and B, WMS Logical Memory Delayed Recall, and Category Fluency. With FDR-correction, the results for the Trail Making Test Parts A and B, WMS Logical Memory Delayed Recall, and Category Fluency were at the p=0.05 level.
Table 3:
Linear mixed effects regression results of the relationship between SSD in MCI and change in cognitive performance across the four year interval
| Interaction Between SSD and Time | ||||||
|---|---|---|---|---|---|---|
| Df | β Estimate | Standard Error | t-value | p-value | FDR p-value | |
| Alzheimer’s Disease Assessment Scale | 421 | 1.580 | 0.705 | 2.242 | 0.025* | 0.061 |
| WMS Logical Memory I | 245 | −0.300 | 0.287 | −1.046 | 0.297 | 0.445 |
| RAVLT Immediate Recall | 423 | 0.290 | 0.719 | 0.403 | 0.687 | 0.687 |
| WMS Logical Memory II | 244 | −0.698 | 0.274 | −2.525 | 0.011* | 0.054 |
| RAVLT Delayed Recall | 421 | 0.116 | 0.248 | 0.466 | 0.641 | 0.687 |
| Trail Making Test A | 421 | 4.681 | 1.970 | 2.376 | 0.018* | 0.054 |
| Trail Making Test B | 411 | 12.150 | 4.796 | 2.533 | 0.012* | 0.054 |
| Digit Symbol Substitution Test | 391 | −0.782 | 0.895 | −0.874 | 0.383 | 0.503 |
| Category Fluency (animals) | 424 | −0.855 | 0.354 | −2.418 | 0.016* | 0.054 |
| WAIS-III Digit Span Forwards | 392 | −0.190 | 0.154 | −1.233 | 0.218 | 0.374 |
| WAIS-III Digit Span Backwards | 390 | −0.133 | 0.164 | −0.809 | 0.419 | 0.503 |
| Boston Naming Test | 423 | −0.466 | 0.375 | −1.242 | 0.215 | 0.374 |
Notes: Longitudinal cognitive data was regressed on age, gender, years of education, APOE status, SSD group, time interval from baseline, and the interaction between SSD group and time interval from baseline using linear mixed models.
= p≤0.05
SSD=Subsyndromal Depression; WMS= Wechsler Memory Scale, RAVLT= Rey Auditory Verbal Learning Test, WAIS-III= Wechsler Adult Intelligence Scale-Third Edition
Baseline Cortical Thickness –
At baseline, the SSD group demonstrated lower temporal lobe and entorhinal cortex cortical thickness (Table 4). No other significant differences were observed. Rate of Cortical Atrophy in SSD – In the fully adjusted models, the SSD group demonstrated accelerated atrophy in the frontal and anterior cingulate relative to the non-SSD group (Table 5). Significant findings were also observed in the majority of subregions of the anterior cingulate and frontal lobe including the rostral anterior cingulate (df=341, β=−0.029, S.E=0.011, t=−2.782, p=0.006); caudal anterior cingulate (df=341, β=−0.042, S.E=0.011, t=−3.652, p<0.001), medial orbitofrontal (df=341, β=−0.023, S.E=0.008, t=−2.780, p=0.006), rostral middle frontal (df=341, β=−0.018, S.E=0.007, t=−2.459, p=0.014), and superior frontal (df=341, β=−0.021, S.E=0.009, t=−2.269, p=0.024). No significant group differences were observed in the lateral orbitofrontal (df=341, β=−0.019, S.E=0.010, t=−1.897, p=0.059) and caudal middle frontal gyri (df=341, β=−0.017, S.E=0.009, t=−1.855, p=0.064). All significant results persisted with multiple comparisons correction (FDR p<0.05).
Table 4:
Linear mixed effects regression results of the relationship between SSD in MCI and cortical thickness in a priori regions of interest at baseline (df=95)
| β Estimate | Standard Error | t-value | p-value | FDR p-value | |
|---|---|---|---|---|---|
| Frontal Lobe | −0.050 | 0.034 | −1.460 | 0.148 | 0.542 |
| Anterior Cingulate | −0.001 | 0.551 | 0.017 | 0.987 | 0.987 |
| Precuneus | −0.038 | 0.040 | −0.947 | 0.832 | 0.659 |
| Cuneus | 0.006 | 0.026 | 0.212 | 0.532 | 0.915 |
| Posterior Cingulate | −0.051 | 0.044 | −1.164 | 0.247 | 0.659 |
| Temporal Lobe | −0.119 | 0.045 | −2.655 | 0.009* | 0.051 |
| Parahippocampus | −0.030 | 0.067 | −0.451 | 0.653 | 0.896 |
| Hippocampus | −98.919 | 107.474 | −0.920 | 0.356 | 0.659 |
| Entorhinal Cortex | −0.279 | 0.102 | −2.729 | 0.008* | 0.051 |
Note: Longitudinal cortical atrophy was regressed on age, gender, APOE status, SSD group, time interval from baseline, and the interaction between SSD group and time interval from baseline using linear mixed models.
= p<0.05
SSD=Subsyndromal Depression
Table 5:
Linear mixed effects regression results of the relationship between SSD in MCI and change in cortical thickness in a priori regions of interest over the four-year study interval (df=341)
| Interaction Between SSD and Time | |||||
|---|---|---|---|---|---|
| β Estimate | Standard Error | t-value | p-value | FDR p-value | |
| Frontal Lobe | −0.020 | 0.007 | −2.648 | 0.008* | 0.040 |
| Anterior Cingulate | −0.036 | 0.009 | −3.786 | <0.001* | <0.001 |
| Precuneus | −0.009 | 0.007 | −1.238 | 0.217 | 0.380 |
| Cuneus | −0.003 | 0.005 | −0.626 | 0.532 | 0.760 |
| Posterior Cingulate | −0.015 | 0010 | −1.518 | 0.130 | 0.320 |
| Temporal Lobe | −0.018 | 0.010 | −1.706 | 0.089 | 0.300 |
| Parahippocampus | −0.012 | 0.010 | −1.212 | 0.226 | 0.380 |
| Hippocampus | −5.924 | 14.530 | −0.408 | 0.684 | 0.860 |
| Entorhinal Cortex | 0.001 | 0.019 | 0.042 | 0.967 | 0.970 |
Note: Longitudinal cortical atrophy was regressed on age, gender, APOE status, SSD group, time interval from baseline, and the interaction between SSD group and time interval from baseline using linear mixed models.
= p<0.05
SSD=Subsyndromal Depression
Relation Between Rate of Change in Cognition and Cortical Thickness –
Accelerated frontal lobe atrophy was associated with more rapid decline on the ADAS-Cog, Trail Making Test Part B, and Category fluency (Table 6, Figure 1). Accelerated anterior cingulate lobe atrophy was associated with faster decline on the ADAS-Cog, Trail Making Test Part A and B, and Category fluency (Table 6, Figure 1). No significant interactions between rate of atrophy and SSD group were observed across cognitive measures (all p>0.05). There were significant baseline correlations between frontal lobe cortical thickness and Trail Making Test Parts A &B (Table 6), so these models were re-analyzed statistically adjusting for this association. The association between rate of frontal lobe atrophy and decline in Trail Making Test Part A remained non-significant (β=−0.430, S.E=1.519, t=−0.283, p=0.777) and Trail Making Test Part B was no longer statistically significant after this adjustment (β=−1.406, S.E=1.037, t=−1.356, p=0.179).
Table 6:
Associations between change in cortical thickness and change in cognition over time
| Cognitive Measure | Cortical Thickness Region | Baseline Cognition & Thickness Correlation | Association Between Change in Cortical Thickness and Change in Cognition | |||
|---|---|---|---|---|---|---|
| Rho (p-value) | β Estimate | Standard Error | t-value | p-value | ||
| Alzheimer’s Disease Assessment Scale | Frontal Lobe | −0.156 (0.120) | −0.993 | 0.362 | −2.743 | 0.007* |
| WMS Logical Memory II | Frontal Lobe | 0.033 (0.886) | 0.114 | −1.803 | 1.390 | 0.168 |
| Trail Making Test A | Frontal Lobe | −0.277 (0.005*) | −2.376 | 1.318 | 2.376 | 0.075 |
| Trail Making Test B | Frontal Lobe | −0.303 (0.002*) | −5.639 | 1.795 | −3.142 | 0.002* |
| Category Fluency (animals) | Frontal Lobe | 0.187 (0.062) | 0.247 | 0.111 | 2.225 | 0.028* |
| Alzheimer’s Disease Assessment Scale | Anterior Cingulate | −0.149 (0.139) | −1.119 | 0.378 | −2.960 | 0.004* |
| WMS Logical Memory II | Anterior Cingulate | −0.078 (0.439) | 0.110 | 0.073 | 1.507 | 0.134 |
| Trail Making Test A | Anterior Cingulate | 0.003 (0.977) | −2.581 | 1.127 | −2.290 | 0.024* |
| Trail Making Test B | Anterior Cingulate | −0.100 (0.321) | −5.253 | 1.528 | −3.438 | 0.001* |
| Category Fluency (animals) | Anterior Cingulate | 0.108 (0.286) | 0.207 | 0.103 | 2.010 | 0.046* |
Notes: This analysis was done in two steps. In step one, subject-specific rates of cognition and atrophy were estimated separately using mixed effects regression. In step two, rates of cognitive decline were then regressed on atrophy rates, adjusting for age, gender, and APOE genotype. Steps one and two were repeated in 500 bootstrap samples to estimate the variance of the association between atrophy and cognitive decline and test for its significance. Please refer to Tables 3 and 5 for degrees of freedom in each of the models for cognitive and cortical atrophy analyses, respectively.
= p<0.05
WMS= Wechsler Memory Scale
Figure 1:
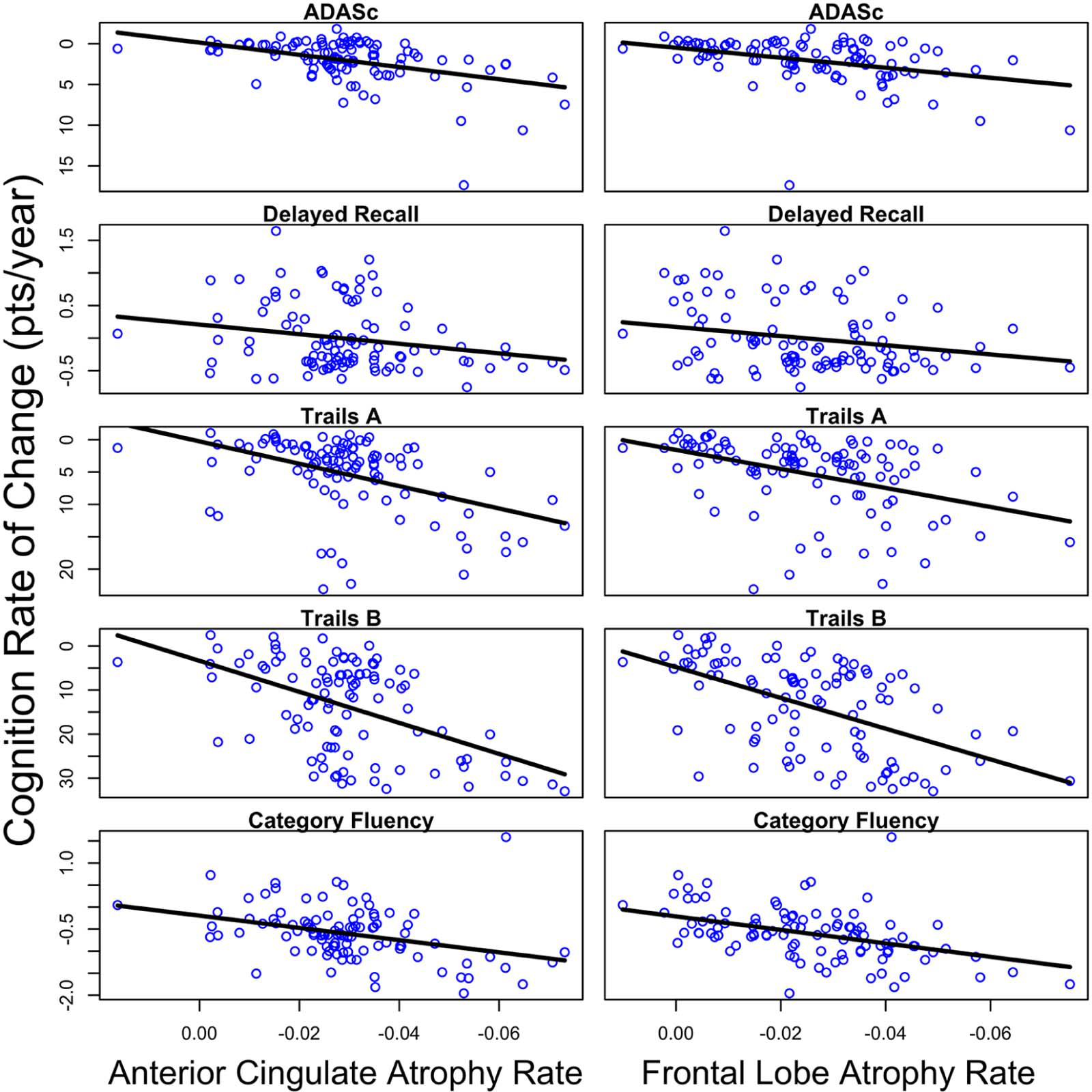
Linear mixed effects regression results of the relationship between change in cortical thickness (column 1: anterior cingulate; column 2: frontal lobe) and cognitive decline across the four-year study interval. All p<0.05.
Discussion
This study evaluated the association between rate of cortical atrophy and cognitive decline in participants with MCI and chronic SSD over an extended four-year period. Our results demonstrate that MCI participants with chronic SSD display 1) accelerated cognitive decline longitudinally on measures of global cognition, memory, information processing speed, and semantic fluency, 2) accelerated rate of frontal lobe and anterior cingulate atrophy over time, and 3) cognitive decline on measures of global cognition, information processing speed, and semantic fluency was associated with frontal lobe and anterior cingulate atrophy independent of SSD.
Consistent with the literature indicating that depression is a risk factor for dementia (6, 7) within this MCI sample chronic SSD was associated with accelerated decline on measures of global cognition, memory, information processing speed, and semantic fluency. The findings persisted over and above adjustment for other well-established cognitive decline risk factors including age, education, and APOE status (23), indicating that the association of chronic SSD with cognition is independent of these factors. The current results support the literature suggesting that late life depressive symptomatology is associated with executive dysfunction and cognitive slowing (24). Moreover, our data demonstrates that chronic SSD is also associated with cognitive decline in the broader domains of global cognition, memory, and semantic fluency, which have been less commonly assessed. Finally, by examining an MCI sample, the findings indicate that chronic SSD may accelerate cognitive decline within an already cognitively vulnerable population and may serve as a useful predictor for future cognitive deterioration with this population.
In addition to accelerated cognitive decline, chronic SSD in MCI participants was also associated with more rapid frontal lobe and anterior cingulate atrophy independent of age, education, and APOE status. Late life depression has been consistently associated with structural and functional abnormalities in these two regions including lower task-related activation (25), cortical thickness (26), white matter integrity (27), and cerebral perfusion (28). The current results confirm previous findings (3) by demonstrating that even depressive symptomatology below the threshold for clinical diagnosis may be associated with accelerated. Thus, chronic SSD may represent an etiological risk factor or biomarker for accelerated frontal lobe and anterior cingulate atrophy within MCI populations.
In the current study, no significant group differences in rate of atrophy were observed in the regions most commonly associated with MCI and AD including the posterior cingulate, precuneus, temporal lobe, parahippocampus, hippocampus, and entorhinal cortex (9, 11). Atrophy in these regions may be consistent across the sample since all participants had a diagnosis of MCI. In contrast, accelerated degradation in the frontal lobe and anterior cingulate was uniquely associated with chronic SSD, indicating that depressive symptomatology may reflect cerebral pathology distinct from typical AD pathology (9, 11).
Finally, our results demonstrated that accelerated cognitive decline in global cognition, information processing speed, and semantic fluency was associated with rate of frontal lobe and anterior cingulate atrophy. The frontal lobe and anterior cingulate act in concert with one another and subcortical regions to govern attentional regulation, task switching, and response sequencing (29, 30), processes fundamental to performance in the affected cognitive domains. Our results suggest that more rapid frontal lobe and anterior cingulate atrophy may be an underlying mechanism of SSD-related accelerated cognitive decline in MCI.
In contrast to global cognition, information processing speed, and semantic fluency, decline in memory performance was not significantly associated with cortical atrophy in either the frontal lobe or the anterior cingulate. Successful memory retrieval is governed by concurrent activation of the frontal and medial temporal lobes (31). Within MCI populations, there is evidence to suggest that in addition to temporal atrophy, disconnection between the entorhinal cortex and higher cortical regions, may underlie impaired memory recall (32). Thus, joint examinations of structural neuroimaging with functional connectivity may distinguish neural network that can be targeted to prevent or attenuate memory decline in comorbid chronic SSD and MCI populations.
While the etiology of advanced atrophy in chronic SSD has yet to be fully defined, several potential mechanisms have been proposed. In particular, white matter damage, which has been commonly reported in late life depression (27), has been linked with secondary grey matter degeneration, hypometabolism, and conversion from MCI to dementia (33). Additionally, depressive symptomatology in geriatric populations has been associated with reduced cerebral blood within the frontal lobe and anterior cingulate (28). Hypoperfusion in these regions may induce neuronal degeneration (34), resulting in cortical atrophy. Depressive symptomatology and chronic stress have been associated with hypercortisolemia (35), which may diminish neurotrophin levels and accelerate atrophy in vulnerable brain regions (36). Of note, a prior postmortem study detected higher levels of inflammation in the frontal lobe and anterior cingulate within depressed individuals (37). Finally, it has been proposed that depressive symptomatology may contribute to neurodegeneration by increasing β amyloid and tau-protein accumulation (38), which may extend damage to regions typically preserved in MCI such as the frontal lobe and anterior cingulate.
The results of the current study must be considered within the strengths and limitations of the study design. The strengths of the study include the relatively large sample size with repeated neuropsychological and neuroimaging data over a four-year period. Furthermore, the ADNI dataset provides a well-characterized sample and includes strong adherence to clinical MCI diagnosis guidelines and thorough quality control for neuroimaging data. However, the ADNI cohort is limited to amnestic MCI, and it is possible that effects related to SSD may be different in a more heterogeneous MCI sample. Another limitation of the current sample is the relatively high educational obtainment, which limits the generalizability of the results. Also, the current study was restricted to subsyndromal symptoms of depression because more significant depressive symptomology is an exclusion criterion for the ADNI study. It is possible that the cognitive and neuropathological profiles of individuals with major and minor depression may differ from those in the current study. Additionally, at baseline, the SSD group displayed poorer performance on one measure of learning and reduced entorhinal and temporal lobe cortical thickness in comparison with the non-SSD group, indicating that they may have more advanced disease severity in some brain regions. Thus, future studies with longer follow-up periods should examine individuals without depressive symptoms at baseline to determine if they display a similar pattern of cortical atrophy over time. There was also a significant association between baseline frontal lobe thickness and Trail Making Test Parts A & B. Inclusion of this significant baseline correlation changed the pattern of results for Trail Making Test Part B, indicating that baseline associations between cognition and cortical thickness may exert an influence on longitudinal decline for some measures of cognition. Finally, prior studies have reported reductions in white matter integrity and cerebral perfusion in older adults with depressive symptomatology (27, 28). Thus, future studies may benefit from incorporation of multi-modal neuroimaging techniques, including indices of white matter integrity, cerebral blood blow, and amyloid beta accumulation, in effort to more fully elucidate the neurobiological mechanisms underlying cognitive decline in MCI patient with chronic SSD.
Overall, the current study indicates that chronic SSD was associated with accelerated cognitive decline and cortical atrophy within a sample of individuals carrying an MCI diagnosis. Moreover, SSD-related accelerated decline in global cognition, information processing speed, and semantic fluency domains was associated with rate of frontal lobe and anterior cingulate atrophy. Our results suggest that individuals with chronic SSD may represent an MCI subgroup with elevated risk for cognitive deterioration and potential conversion to dementia. Thus, monitoring depressive symptoms over time, rather than at a single occurrence, may be efficacious for identifying cognitively vulnerable individuals (39). Additionally, our results indicate that frontal lobe and anterior cingulate atrophy may be viable biomarkers for predicting further cognitive decline, underscoring the potential importance of treating SSD in effort to preserve cognition.
Acknowledgments:
Data collection and sharing for this project was funded by the Alzheimer’s Disease Neuroimaging Initiative (ADNI) (National Institutes of Health Grant U01 AG024904). ADNI is funded by the National Institute on Aging, the National Institute of Biomedical Imaging and Bioengineering, and through generous contributions from the following: Abbott, AstraZeneca AB, Bayer Schering Pharma AG, Bristol-Myers Squibb, Eisai Global Clinical Development, Elan Corporation, Genentech, GE Healthcare, GlaxoSmithKline, Innogenetics, Johnson and Johnson, Eli Lilly and Co., Medpace, Inc., Merck and Co., Inc., Novartis AG, Pfizer Inc, F. Hoffman-La Roche, Schering-Plough, Synarc, Inc., as well as non-profit partners the Alzheimer’s Association and Alzheimer’s Drug Discovery Foundation, with participation from the U.S. Food and Drug Administration. Private sector contributions to ADNI are facilitated by the Foundation for the National Institutes of Health (www.fnih.org). The grantee organization is the Northern California Institute for Research and Education, and the study is coordinated by the Alzheimer’s Disease Cooperative Study at the University of California, San Diego. ADNI data are disseminated by the Laboratory for Neuroimaging at the University of California, Los Angeles. This research was also supported by National Institutes of Health grants P30 AG010129, K01 AG030514, and the Dana Foundation. Data analysis was supported in part by the following grant from the National Institutes of Health: R01 MH098062.
Footnotes
Disclosures: Dr. Gonzales, Mr. Insel, Dr. Mattsson, Dr. Mueller, Dr. Sacuiu, and Mr. Bickford reported no biomedical financial interests or potential conflicts of interest.
During the past 2 years, Dr. Nelson has received lecture honoraria from Bristol Myers Squibb (Canada), Otsuka (Asia); he has been an advisor or consultant to Corcept, Genentech, Janssen, Lundbeck, Otsuka, and Sunovion; and has received research support from Avid and NIMH. Dr. Tosun has received research support from The National Institute of Mental Health, Avid radiopharmaceuticals, Michael J Fox Foundation, Alzheimer’s Association, and California Department of Public Health.
Dr. Weiner receives grant funding from the National Institutes of Health, the Department of Defense, the Alzheimer’s Association, the Veterans Administration, and the California Department of Public Health. Dr. Weiner has served on the Scientific Advisory Boards for Pfizer, BOLT International, Neurotrope Bioscience, Alzheon, Inc., Alzheimer’s Therapeutic Research Institute (ATRI), Eli Lilly, University of Pennsylvania’s Neuroscience of Behavior Initiative, National Brain Research Centre (NBRC), India, Dolby Family Ventures, LP, and ADNI. Dr. Weiner has provided consulting to Synarc, Pfizer, Janssen, Alzheimer’s Drug Discovery Foundation (ADDF), Neurotrope Bioscience, Avid Radiopharmaceuticals, Clearview Healthcare Partners, Perceptive Informatics, Smartfish AS, Araclon, Merck, Biogen Idec, BioClinica, and Genentech. He holds stock options with Alzheon, Inc. The following entities have provided funding for Dr. Weiner’s academic travel; Pfizer, Neuroscience School of Advanced Studies (NSAS), Kenes, Intl., ADRC, UCLA, UCSD; ADCS, Sanofi-Aventis Groupe, University Center Hospital, Toulouse, Araclon, AC Immune, Nutricia, Eli Lilly, New York Academy of Sciences (NYAS), National Brain Research Center, India for Johns Hopkins Medicine, Consortium for Multiple Sclerosis Centers (CMSC), Northwestern University, Fidelity Biosciences Research Initiative, University of Pennsylvania, The Alzheimer’s Association, Merck, ADPD, Alzheimer’s Drug Discovery Foundation (ADDF), Tokyo University, Kyoto University, Weill-Cornell University, Rockafeller University, Memorial Sloan-Kettering Cancer Center, and Biogen Idec.
During the past 2 years, Dr. Mackin has received research support from The National Institute of Mental Health, Johnson and Johnson, and Avid.
*Data used in preparation of this article were obtained from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) database (www.loni.usc.edu/ADNI). As such, the investigators within the ADNI contributed to the design and implementation of ADNI and/or provided data but did not participate in analysis or writing of this report. ADNI investigators include (complete listing available at http://adni.loni.usc.edu/study-design/ongoing-investigations/
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Albert MS, DeKosky ST, Dickson D, et al. : The diagnosis of mild cognitive impairment due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement 2011; 7:270–279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Judd LL, Rapaport MH, Paulus MP, et al. : Subsyndromal symptomatic depression: a new mood disorder? J Clin Psychiatry 1994; 55 Suppl:18–28 [PubMed] [Google Scholar]
- 3.Sacuiu S, Insel PS, Mueller S, et al. : Chronic depressive symptomatology in mild cognitive impairment is associated with frontal atrophy rate which hastens conversion to Alzheimer dementia. Am J Geriatr Psychiatry 2016; 24:126–135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Steffens DC: Depressive symptoms and mild cognitive impairment in the elderly: an ominous combination. Biol Psychiatry 2012; 71:762–764 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mackin RS, Insel P, Aisen PS, et al. : Longitudinal stability of subsyndromal symptoms of depression in individuals with mild cognitive impairment: relationship to conversion to dementia after 3 years. Int J Geriatr Psychiatry 2012; 27:355–363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gabryelewicz T, Styczynska M, Luczywek E, et al. : The rate of conversion of mild cognitive impairment to dementia: predictive role of depression. Int J Geriatr Psychiatry 2007; 22:563–567 [DOI] [PubMed] [Google Scholar]
- 7.Royall DR,Palmer RF: Alzheimer’s disease pathology does not mediate the association between depressive symptoms and subsequent cognitive decline. Alzheimers Dement 2013; 9:318–325 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee GJ, Lu PH, Hua X, et al. : Depressive symptoms in mild cognitive impairment predict greater atrophy in Alzheimer’s disease-related regions. Biol Psychiatry 2012; 71:814–821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Petersen RC,Jack CR Jr.: Imaging and biomarkers in early Alzheimer’s disease and mild cognitive impairment. Clin Pharmacol Ther 2009; 86:438–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bora E, Harrison BJ, Davey CG, et al. : Meta-analysis of volumetric abnormalities in cortico-striatal-pallidal-thalamic circuits in major depressive disorder. Psychol Med 2012; 42:671–681 [DOI] [PubMed] [Google Scholar]
- 11.Apostolova LG,Thompson PM: Mapping Progressive Brain Structural Changes in Early Alzheimer’s Disease and Mild Cognitive Impairment. Neuropsychologia 2008; 46:1597–1612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wechsler D: Manual for the Wechsler Memory Scale - Revised, San Antonio, TX, The Psychological Corporation, 1987 [Google Scholar]
- 13.Kaufer DI, Cummings JL, Ketchel P, et al. : Validation of the NPI-Q, a brief clinical form of the Neuropsychiatric Inventory. J Neuropsychiatry Clin Neurosci 2000; 12:233–239 [DOI] [PubMed] [Google Scholar]
- 14.Buysse DJ, Cheng Y, Germain A, et al. : Night-to-night sleep variability in older adults with and without chronic insomnia. Sleep Med 2010; 11:56–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rosen WG, Mohs RC, Davis KL: A new rating scale for Alzheimer’s disease. Am J Psychiatry 1984; 141:1356–1364 [DOI] [PubMed] [Google Scholar]
- 16.Reitan RM,Wolfson D: The Halstead–Reitan Neuropsychological Test Battery: Theory and Clinical Interpretation, 2nd edition, Tucson, AZ, Neuropsychology Press, 1983 [Google Scholar]
- 17.Wechsler D: Wechsler Adult Intelligence Scale–Revised, San Antonio, TX, Psychological Corporation, 1981 [Google Scholar]
- 18.Morris J, Heyman A, Mohs R, et al. : The consortium to establish a registry for Alzheimer’s disease (CERAD): I. Clinical and neuropsychological assessment of Alzheimer’s disease. Neurology 1989; 39:1159–1165 [DOI] [PubMed] [Google Scholar]
- 19.Kaplan E, Goodglass H,Weintraub S: Boston Naming Test-Revised, Philadelphia, PA, Lea & Febiger, 1983 [Google Scholar]
- 20.Jack CR, Bernstein MA, Fox NC, et al. : The Alzheimer’s disease neuroimaging initiative (ADNI): MRI methods. J Magn Reson Imaging 2008; 27:685–691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fischl B, Salat DH, Busa E, et al. : Whole brain segmentation: automated labeling of neuroanatomical structures in the human brain. Neuron 2002; 33:341–355 [DOI] [PubMed] [Google Scholar]
- 22.Reuter M, Schmansky NJ, Rosas HD, et al. : Within-subject template estimation for unbiased longitudinal image analysis. NeuroImage 2012; 61:1402–1418 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.McCullagh CD, Craig D, McIlroy SP, et al. : Risk factors for dementia. Adv Psychiatr Treat 2001; 7:24–31 [Google Scholar]
- 24.Herrmann LL, Goodwin GM, Ebmeier KP: The cognitive neuropsychology of depression in the elderly. Psychol Med 2007; 37:1693–1702 [DOI] [PubMed] [Google Scholar]
- 25.Aizenstein HJ, Butters MA, Wu M, et al. : Altered functioning of the executive control circuit in late-life depression: episodic and persistent phenomena. Am J Geriatr Psychiatry 2009; 17:30–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mackin RS, Tosun D, Mueller SG, et al. : Patterns of reduced cortical thickness in late-life depression and relationship to psychotherapeutic response. Am J Geriatr Psychiatry 2013; 21:794–802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Bae JN, MacFall JR, Krishnan KRR, et al. : Dorsolateral prefrontal cortex and anterior cingulate cortex white matter alterations in late-life depression. Biol Psychiatry 2006; 60:1356–1363 [DOI] [PubMed] [Google Scholar]
- 28.Ishizaki J, Yamamoto H, Takahashi T, et al. : Changes in regional cerebral blood flow following antidepressant treatment in late-life depression. Int J Geriatr Psychiatry 2008; 23:805–811 [DOI] [PubMed] [Google Scholar]
- 29.Badre D: Cognitive control, hierarchy, and the rostro-caudal organization of the frontal lobes. Trends Cogn Sci 2008; 12:193–200 [DOI] [PubMed] [Google Scholar]
- 30.Botvinick MM, Braver TS, Barch DM, et al. : Conflict monitoring and cognitive control. Psychol Rev 2001; 108:624–652 [DOI] [PubMed] [Google Scholar]
- 31.Cabeza R,Nyberg L: Neural bases of learning and memory: functional neuroimaging evidence. Curr Opin Neurol 2000; 13:415–421 [DOI] [PubMed] [Google Scholar]
- 32.Stoub TR, Stebbins GT, Leurgans S, et al. : Hippocampal disconnection contributes to memory dysfunction in individuals at risk for Alzheimer’s disease. Proc Natl Acad Sci 2006; 103:10041–10045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Agosta F, Pievani M, Sala S, et al. : White matter damage in Alzheimer disease and its relationship to gray matter atrophy. Radiology 2011; 258:853–863 [DOI] [PubMed] [Google Scholar]
- 34.Bennett SA, Tenniswood M, Chen J-H, et al. : Chronic cerebral hypoperfusion elicits neuronal apoptosis and behavioral impairment. Neuroreport 1998; 9:161–166 [DOI] [PubMed] [Google Scholar]
- 35.Checkley S: The neuroendocrinology of depression and chronic stress. Br Med Bull 1996; 52:597–617 [DOI] [PubMed] [Google Scholar]
- 36.Dwivedi Y: Involvement of brain-derived neurotrophic factor in late-life depression. Am J Geriatr Psychiatry 2013; 21:433–449 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Thomas A, Ferrier IN, Kalaroa Rajesh N, et al. : Cell adhesion molecule expression in the dorsolateral prefrontal cortex and anterior cingulate cortex in major depression in the elderly. Br J Psychiatry 2002; 181:129–134 [DOI] [PubMed] [Google Scholar]
- 38.Rapp MA, Schnaider-Beeri M, Purohit DP, et al. : Increased neurofibrillary tangles in patients with Alzheimer disease with comorbid depression. Am J Geriatr Psychiatry 2008; 16:168–174 [DOI] [PubMed] [Google Scholar]
- 39.David ND, Lin F,Porsteinsson AP: Trajectories of neuropsychiatric symptoms and cognitive decline in mild cognitive impairment. Am J Geriatr Psychiatry 2016; 24:70–80 [DOI] [PMC free article] [PubMed] [Google Scholar]