

Background & Aims:
In patients with primary biliary cholangitis (PBC), the serum liver biochemistry measured during treatment with ursodeoxycholic acid—the UDCA response—accurately predicts long-term outcome. Molecular characterization of patients stratified by UDCA response can improve biological understanding of the high-risk disease, thereby helping to identify alternative approaches to disease-modifying therapy. In this study, we sought to characterize the immunobiology of the UDCA response using transcriptional profiling of peripheral blood mononuclear cell subsets.
Methods:
We performed bulk RNA-sequencing of monocytes and TH1, TH17, TREG, and B cells isolated from the peripheral blood of 15 PBC patients with adequate UDCA response (“responders”), 16 PBC patients with inadequate UDCA response (“nonresponders”), and 15 matched controls. We used the Weighted Gene Co-expression Network Analysis to identify networks of co-expressed genes (“modules”) associated with response status and the most highly connected genes (“hub genes”) within them. Finally, we performed a Multi-Omics Factor Analysis of the Weighted Gene Co-expression Network Analysis modules to identify the principal axes of biological variation (“latent factors”) across all peripheral blood mononuclear cell subsets.
Results:
Using the Weighted Gene Co-expression Network Analysis, we identified modules associated with response and/or disease status (q<0.05) in each peripheral blood mononuclear cell subset. Hub genes and functional annotations suggested that monocytes are proinflammatory in nonresponders, but antiinflammatory in responders; TH1 and TH17 cells are activated in all PBC cases but better regulated in responders; and TREG cells are activated—but also kept in check—in responders. Using the Multi-Omics Factor Analysis, we found that antiinflammatory activity in monocytes, regulation of TH1 cells, and activation of TREG cells are interrelated and more prominent in responders.
Conclusions:
We provide evidence that adaptive immune responses are better regulated in patients with PBC with adequate UDCA response.
INTRODUCTION
Primary biliary cholangitis (PBC) is an autoimmune liver disease characterized by progressive destruction of the small, intra-hepatic bile ducts, leading in many cases to cirrhosis.1 First-line treatment of PBC is with ursodeoxycholic acid (UDCA), a hydrophilic bile acid that acts mainly by displacing hydrophobic bile acids in bile and by enhancing choleresis. In patients with PBC, the serum liver biochemistry during treatment with UDCA—the UDCA response—accurately predicts long-term outcome.2 Thus, patients with adequate UDCA response are at a lower risk of disease progression, whereas those with inadequate UDCA response are at a higher risk. The latter are prioritized for second-line treatment with agents that also act mainly through choleresis: the farnesoid X receptor agonist, obeticholic acid, or the peroxisome proliferator-activated receptor agonists, bezafibrate or fenofibrate.3 Up to 50% of patients with inadequate response to UDCA also have an inadequate response to second-line treatment, however, and so remain at risk of disease progression.4 Novel therapeutic approaches—not just targeting the composition or secretion of bile—are needed for such patients. Molecular characterization of patients stratified by the UDCA response can improve biological understanding of the high-risk disease, thereby helping to identify alternative approaches to disease-modifying therapy.
We have reported gene expression analysis of liver tissue from patients with high versus low-risk PBC, which showed greater biliary epithelial cell (BEC) senescence and T-cell activation in those with high-risk disease.5 More recently, we reported serum proteomic profiling of 526 patients with PBC stratified by UDCA response, which showed reproducible elevation of proteins with well-defined roles in inflammation and immunity, such as CCL20, CXCL11, IL-4RA, and IL-18R1, in nonresponders.6 These findings highlight the potential value of profiling the immune systems of patients stratified by UDCA response. A starting point is to profile the immune cell types that are thought to be dysregulated in PBC itself.
Immune cell types implicated in the pathogenesis of PBC include monocytes; TH1, TH17, and TREG cells; CD8+ T cells, and B cells. For example, in PBC, monocytes are more sensitive to toll-like receptor signalling7; portal infiltrates consist mainly of CD4+ T cells, heavily skewed towards TH1 and TH17 cells8,9; there is insufficiency of TREG cells10; CD8+ T cells infiltrate the biliary epithelia and cause segmental apoptotic destruction of cholangiocytes11,12; and B cells contribute to disease through cross-presentation of antigen and production of autoantibodies.13,14
In the current study, guided by these observations, we used bulk RNA-sequencing (RNA-seq) to profile the transcriptomes of monocytes and TH1, TH17, TREG, and B cells from the peripheral blood of PBC patients with an adequate response to UDCA, those with inadequate response, and healthy controls. We designed the study for case-control and within-case analyses to gain insight into the immunobiology of PBC itself and that of refractory disease. For each cell type, we sought to identify networks of co-expressed genes associated with 1 or more traits. To do this, we used an approach called weighted gene co-expression network analysis (WGCNA).15 The premise of this approach (and others like it) is that genes do not function in isolation but in networks16; genes within those networks are co-expressed; and statistical methods can be used to identify those co-expressed genes.17 Association can then be determined between gene co-expression networks and traits of interest; and functional annotation of associated networks can be employed to gain biological insight into those traits.
METHODS
Participants
Cases were adults (≥18 y of age) with an established diagnosis of PBC,3 who had received≥12 months of ongoing treatment with UDCA. Response to UDCA was defined by alkaline phosphatase<1.67 times the upper limit of normal.18 Controls were age and sex-matched healthy volunteers. For all participants, exclusion criteria were other forms of liver disease (eg, viral hepatitis), decompensated cirrhosis, HCC, liver transplantation, immunosuppression, poorly controlled diabetes mellitus, pregnancy, and alcohol misuse. Decompensated cirrhosis was defined by a total bilirubin>50 μmol/L or any occurrence of variceal hemorrhage, ascites, or HE.19 Cases were recruited from 6 liver treatment centers across the UK as part of the UK-PBC Nested Cohort Study.6 Adherence to treatment with UDCA was verified in all cases. Controls were recruited from the Cambridge NIHR BioResource (https://www.cambridgebioresource.group.cam.ac.uk/). Written informed consent was obtained from each participant. The study protocol conformed to the ethical guidelines of the 1975 Declaration of Helsinki. Ethical approval for the study was from the North-West Preston Research Ethics Committee (REC reference: 14/NW/1146).
Samples
Up to 100 mL of blood was collected from each participant for the isolation of peripheral blood mononuclear cells (PBMCs). The isolation of PBMCs was undertaken on-site within 2 hours of phlebotomy according to a standard operating procedure. The PBMCs were stored overnight at −80°C and transferred on dry ice the next day to the University of Birmingham for long-term storage over liquid nitrogen. Please see Supplementary Data (Fig. S1, http://links.lww.com/HC9/A236) for further details.
Sorting of PBMC subsets
Peripheral blood mononuclear cells were sorted into subsets at the University of Birmingham. The PBMCs were first thawed as described in the Supplementary Data, http://links.lww.com/HC9/A236. CD14+ monocytes were then selected using human CD14 MicroBeads (Miltenyi Biotec, Woking, UK) according to the manufacturer’s instructions; CD14+ cell pellets were frozen at −80°C until RNA extraction. Non-CD14+ cells were resuspended in PBS and stained with live/dead marker (Zombie APC-Cy7 at 1:1000; Biolegend, San Diego, CA, USA) for 20 minutes at room temperature. Cells were washed and stained with the following fluorochrome-conjugated antibodies (BD Biosciences, UK): CD3-APC, CD4-PECF594, CD127-PECy7, CD25-BB515, CXCR3-PerCP-Cy5.5, CCR6-BV421, and CD19-PE for fluorescent-associated cell sorting (using BD Aria Fusion, BD, UK) of the immune cell subsets: (1) CD19+CD3– B cells, (2) CD3+CD4+CD25highCD127low TREG cells, (3) CD3+CD4+CCR6+CXCR3– TH17 cells, and (4) CD3+CD4+CCR6–CXCR3+ TH1 cells (Fig. S2, http://links.lww.com/HC9/A236). We used an anti-CD3 monoclonal antibody that did not induce T-cell activation (Fig. S3, http://links.lww.com/HC9/A236). All immune cell subsets were sorted into tubes containing RPMI−1640+10% FCS media kept at 4°C during the sorting period. After sorting, cells were pelleted and resuspended in RLT+βME buffer for lysis and RNA extraction.
RNA extraction
RNA was extracted from PBMC subsets at the University of Birmingham using the RNEasy Plus Micro and RNeasy Plus Mini Kits (Qiagen Ltd, Manchester, UK) for<500,000 and>500,000 sorted cells, respectively. As recommended, eluted RNA samples were stored at −80°C until RNA-seq.
RNA-sequencing
RNA-seq was completed in the Stratified Medicine Core Laboratory at the University of Cambridge. Following quality control (QC) checks, the SMARTer Stranded Total RNA-seq Pico kit from Clontech (Mountain View, CA, USA) was used to generate cDNA libraries within 30 days of RNA extraction, and the Illumina (San Diego, CA, USA) HiSeq. 2500 and HiSeq. 4000 platforms were used to sequence them.
Statistical analysis
We employed a standardized pipeline for QC and processing of reads. Read quality was assessed using FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc); alignment was performed using STAR (Spliced Transcripts Alignment to a Reference)20; and count matrices were generated using FeatureCounts in Rsubreads21 and stored as a DGEList object in edgeR22 for downstream analysis. Additional QC checks (including tests for batch effect) were done using DESeq223 and limma (Linear models for microarray data).24
For each PBMC subset, we used WGCNA to identify networks of co-expressed genes associated with disease or UDCA response status. We first identified modules, which are clusters of highly interconnected genes, each representing a gene co-expression network. By convention, each module is arbitrarily allocated the name of a color. For each module, we calculated the module eigengene (ME), module membership, and gene significance. The ME is the first principal component of the module, representative of gene expression profiles in that module; module membership is the correlation coefficient for each gene with the ME; and gene significance is the correlation coefficient for each gene with the trait of interest. Finally, we tested the association of each module with each trait (ie, all cases vs. controls, nonresponders vs. controls, responders vs. controls, and responders vs. nonresponders) by calculating the eigengene significance, based on correlation between the ME and trait of interest. We defined a significant association between a module and trait as q<0.05, where q is the false discovery rate adjusted p-value.
For each module associated with 1 or more traits, we identified hub genes and looked for enrichment of functional annotations. Hub genes are the genes with the highest connectivity within the module (ie, the genes most strongly co-expressed with the greatest number of other genes). In general, we identified hub genes as being among the 30 most connected genes in the module, as well as having module membership≥3rd quartile and gene significance≥3rd quartile. We used the Enrichr platform25 to look for the enrichment of functional annotations. For brevity and consistency, only Hallmark gene sets26 are reported in this manuscript. For each pairwise comparison, we also performed Gene Set Enrichment Analysis (GSEA)27 of normalized count data from each PBMC subset. As recommended at the GSEA platform (https://www.gsea-msigdb.org/gsea/index.jsp), we took q<0.25 to define significant enrichment.
Finally, recognizing that each PBMC subset is part of a unified immune system, we used Multi-Omics Factor Analysis (MOFA)28 for integrated analysis of all modules identified across all PBMC subsets. By this approach, we decomposed the combined data into a small number of latent factors (LFs) and used ANOVA to identify the LFs associated with trait status (ie, control, responder, or nonresponder). To characterize the LFs associated with trait status, we identified the modules with the heaviest loading on each of these factors. Please see the Supplementary Data, http://links.lww.com/HC9/A236, for further details.
RESULTS
Participants and samples
We analyzed samples from 15 responders (median alkaline phosphatase 0.87×ULN, interquartile range [IQR] 0.75–1.00); 16 nonresponders (median alkaline phosphatase 3.68×ULN, IQR 3.14–4.48); and 15 disease-free controls. These groups were well-matched except for PBC status or activity (Table 1, Fig. S4, http://links.lww.com/HC9/A236). Vibration-controlled transient elastography measurements were comparable in responders and nonresponders. Only 1 participant, a responder, had cirrhosis according to the Vibration-controlled transient elastography thresholds proposed by Corpechot et al (2012).29 Cell counts are shown in Fig. S5, http://links.lww.com/HC9/A236. The median RNA integrity number (RIN) of the samples was 8.9 (IQR 8.1–9.2). Following QC checks, the mean number of raw reads per sample was 145 million; the number of mapped reads was 43 million; the percentage PCR duplicates was 37%; and the number of genes at 1 fragment per kilobase of transcript per million mapped reads, was 14,000. MA plots showed no evidence of systematic bias in the RNA-seq output (Fig. S6, http://links.lww.com/HC9/A236). We confirmed the identity of each cell subset by high expression of characteristic genes (Fig. S7, http://links.lww.com/HC9/A236). Multidimensional scaling of count data confirmed similarity within PBMC subsets and dissimilarity between them (Fig. S8, http://links.lww.com/HC9/A236).
TABLE 1.
Participant characteristics
| Nonresponders (n=16) | Responders (n=15) | Controls (n=15) | p-value | |
|---|---|---|---|---|
| Age (y) | 60 (56–66) | 65 (54–71) | 57 (31–74) | 0.2 |
| No. of women (%) | 14 (87.5%) | 15 (94%) | 15 (100%) | 0.4 |
| Disease duration (y) | 10.8 (6.7–13.8) | 8.3 (3.2–12.5) | — | 0.3 |
| UDCA dose (mg/kg/day) | 14.3 (13.2–16.0) | 11.9 (10.3–13.8) | — | 7.2×10−3 |
| ALP (U/L) | 467.5 (383.8–541.5) | 97.0 (83.5–120.0) | 64 (53–78) | 2.3×10−6 |
| ALP/ULN | 3.7 (3.1–4.5) | 0.8 (0.7–1.0) | 0.5 (0.4–0.6) | 2.3×10−6 |
| ALT (U/L) | 60.5 (47–81.5) | 31 (19.5–49.5) | 19 (16–28) | 5.3×10−3 |
| Bilirubin (μmol/L) | 12 (9.8–19.3) | 7 (6–10) | 10 (9–11) | 1.4×10−3 |
| Albumin (g/L) | 38 (36–40.3) | 45.0 (42.5–47.5) | 39 (37–40) | 4.5×10−4 |
| Platelets (109/L) | 250 (203.5–303.5) | 245 (190–322.5) | 224 (198–300) | 0.9 |
| Transient elastography (kPa) | 6.8 (5.6–8.0) | 7.6 (5.0–13.9) | — | 0.7 |
Notes: Median values (interquartile range) are shown for all parameters except for the number (percentage) of women.
Statistical tests were ANOVA for comparison of age across all 3 groups; the Kruskal-Wallis chi-square test to compare the proportion of women in each group; and a 2-tailed Mann-Whitney test to compare all other parameters in nonresponders versus responders.
Abbreviations: ALP, alkaline phosphatase; UDCA, Ursodeoxycholic acid.
Using WGCNA, we identified 19 modules (ie, networks of co-expressed genes) in monocytes (Figure 1A). Of these, 6 were correlated at q<0.05 with 1 or more traits (ie, case vs. control, nonresponder vs. control, responder vs. control, or nonresponder vs. responder) (Figure 1B). Among the other PBMC subsets, 7 of 11 modules in TH1 cells; 6 of 11 modules in TH17 cells; 5 of 9 modules in TREG cells; and 5 of 10 modules in B cells were also correlated at q<0.05 with 1 or more traits. Table 2 lists modules showing at least moderate correlation (r≥0.5) with 1 trait or another.
FIGURE 1.
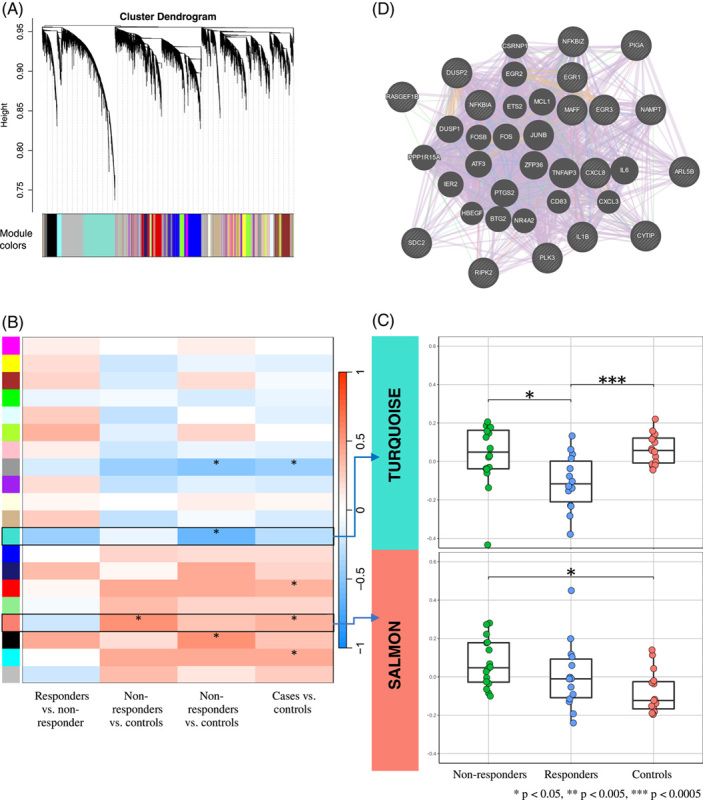
Weighted gene co-expression network analysis (WGCNA) of RNA-sequencing data from CD14 cells. (A) Dendrogram showing gene co-expression networks (‘modules’). (B) Heatmap showing the strength of correlation between each module (rows) and each trait (columns). Asterisks indicate that the module is associated with the trait at q<0.05, where q is the false discovery rate-adjusted p-value. Rows outlined in black identify modules with at least moderate correlation (r≥0.5) with 1 or more traits and enriched with Hallmark gene sets at q<0.05. (C) Boxplots of the module eigengenes (ie, the first principal component of gene expression in the module; y-axis) in each group of participants (x-axis). Asterisks indicate the statistical difference between the module eigengene significance (based on the correlation between the module eigengene and trait of interest) and the trait comparison groups. Asterisks indicate the corresponding p values. (D) GeneMANIA protein-protein interaction plot showing protein-coding–hub genes in the salmon module for nonresponders versus controls. Protein-coding–hub genes are shown with cross-hatched circles of a uniform size, while those that were added as relevant genes by GeneMANIA are shown with solid circles whose size is proportional to the number of interactions they have. Lines correspond to the type of interactions. Purple: Genes known to be co-expressed in existing gene databases. Pink: Proteins known to be linked. Turquoise: Genes present in a shared annotated pathway. Blue: Genes expressed in the same tissue. Orange: Predicted functional relationships between genes. Green: Predicated genetic interactions. Olive: Shared protein domains. Abbreviations: WGCNA, Weighted gene co-expression network analysis.
TABLE 2.
Modules of co-expressed genes
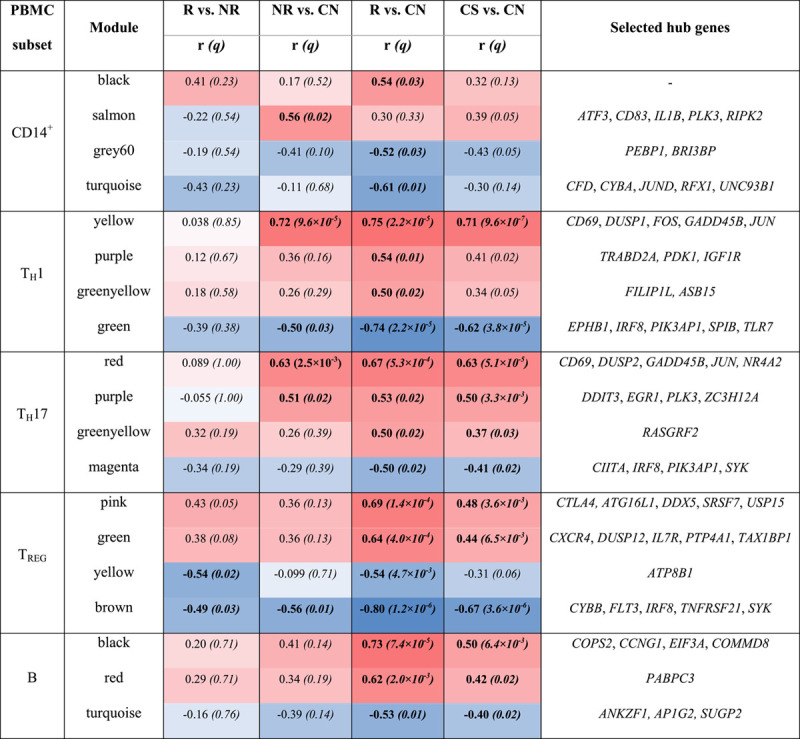
Notes: Modules are shown that were at least moderately correlated (r≥0.5) with one or more traits.
Bold indicates correlation at q<0.05.
Selected hub genes are those which readily describe the module.
Abbreviations: NR, non-responder; PBMC, peripheral blood mononuclear cell; R, responder.
In monocytes, the salmon module was positively correlated with nonresponders versus controls (r=0.56, q=0.023). This module was strongly enriched for TNF signalling (Table 3). Hub genes (ie, the most highly connected genes in the module) included CD83, an activation marker for Ag-presenting cells; RIPK2, a potent activator of NF-кB; and IL1B, a key mediator of inflammation (Table 2, Figure 1D). In contrast, the turquoise module was negatively correlated with responders versus controls (r=−0.61, q=7.7×10−3). Hub genes in this module included JUND, an activator protein (AP)-1 transcription factor; and CYBA, which is essential for the generation of superoxide in phagocytes. As shown in Figure 1C, genes in the salmon module were upregulated in nonresponders compared with controls (p = 1.2×10−3), whereas those in the turquoise module were downregulated in responders compared with controls (p = 3.8×10−4). This suggests a predominance of activated and proinflammatory monocytes in nonresponders, in contrast to a predominance of inactivated monocytes in responders.
TABLE 3.
Pathway enrichment
| Enrichr | ||||||
|---|---|---|---|---|---|---|
| PBMC subset | Module | Comparison group direction of effect in WGCNA | Hallmark pathway | p | q | |
| CD14+ | Salmon | — | — | TNFα signalling through NF-кB | 1.75×10−52 | 7.89×10−51 |
| — | — | Apoptosis | 3.45×10−6 | 3.58×10−5 | ||


|
NR vs. CN | mTORC1 signalling | 4.78×10−6 | 3.58×10−5 | ||
| — | — | KRAS signalling up | 4.78×10−6 | 3.58×10−5 | ||
| — | — | IL-2/STAT5 signalling | 2.56×10−6 | 1.51×10−4 | ||
| — | — | IFNγ response | 2.69×10−6 | 1.51×10−4 | ||
| TH1 | Yellow |



|
NR vs. CN | TNFα signalling via NF-кB | 2.35×10−39 | 1.08×10−37 |



|
R vs. CN | p53 pathway | 2.36×10−4 | 3.63×10−3 | ||



|
CS vs. CN | Apoptosis | 5.30×10−4 | 6.10×10−3 | ||
| Green | — | — | — | — | — | |
| — | IL-2/STAT5 signalling | 2.32×10−4 | 2.77×10−3 | — | ||



|
R vs. CN | IFNγ response | 2.46×10−4 | 2.77×10−3 | ||


|
NR vs. CN | IL-6/JAK/STAT3 signalling | 1.83×10−3 | 9.94×10−3 | ||


|
CS vs. CN | TNFα signalling through NF-кB | 1.99×10−3 | 9.94×10−3 | ||
| — | — | KRAS signalling up | 1.99×10−3 | 9.94×10−3 | ||
| TH17 | Purple |


|
NR vs. CN | TNFα signalling through NF-кB | 3.35×10−6 | 8.71×10−5 |


|
R vs. CN | — | — | — | ||


|
CS vs. CN | — | — | — | ||
| Red |


|
NR vs. CN | TNFα signalling via NF-кB | 2.04×10−30 | 9.40×10−29 | |


|
CS vs. CN | IFNγ response | 3.72×10−6 | 4.27×10−5 | ||


|
R vs. CN | IL-2/STAT5 signalling | 1.52×10−5 | 1.40×10−4 | ||
| Magenta |


|
R vs. CN | KRAS signalling up | 1.13×10−9 | 2.08×10−8 | |
| — | — | IFNγ response | 6.48×10−5 | 4.79×10−4 | ||
| — | — | IL-6/JAK/STAT3 signalling | 1.40×10−4 | 8.60×10−4 | ||
| TREG | Green |


|
R vs. CN | TNFα signalling through NF-кB | 1.74×10−2 | 6.43×10−1 |
| Brown |


|
R vs. NR | KRAS signalling up | 1.44×10−6 | 6.77×10−5 | |


|
NR vs. CN | IL-6/JAK/STAT3 signalling | 8.10×10−6 | 1.27×10−4 | ||



|
R vs. CN | IL-2/STAT5 signalling | 1.33×10−5 | 1.36×10−4 | ||


|
CS vs. CN | TNFα signalling through NF-кB | 4.27×10−5 | 2.87×10−4 | ||
| B | Black |



|
R vs. CN | TNFα signalling through NF-кB | 2.60×10−4 | 1.09×10−2 |


|
CS vs. CN | — | — | — | ||
Notes: Enrichment of Hallmark pathways was evaluated using the platform, Enrichr.
Pathways enriched at q<0.05 are shown.
The direction of effect for the comparison groups is highlighted.
 indicates positive correlation,
indicates positive correlation, indicates negative correlation.
indicates negative correlation.
q is the FDR corrected p-value.
▴▴indicates moderate correlation ▴▴▴indicates strong correlation.
Abbreviation: FDR, false discovery rate; NR, non-responder.
In TH1 cells, the yellow module showed a strong positive correlation with both responders (r=0.75, q=2.2×10−5) and nonresponders (r=0.72, q=9.6×10−5) versus controls (Table 2). This module was enriched for TNF signalling and contained hub genes such as CD69, an activation marker; JUN and FOS, which form the mitogenic AP-1 transcription factor; and GADD45B, which activates MAP kinases. Conversely, the green module showed a strong negative correlation with responders versus controls (r=−0.74, q=2.2×10−5) and a moderate negative correlation with nonresponders versus controls (r=−0.5, q=0.03). This module was enriched for IL-2, IFNγ, and IL-6 signalling. Hub genes included IRF8, a transcription factor that promotes TH1 cells.30 Genes in the yellow module were upregulated in both responders and nonresponders, whereas those in the green module were downregulated in responders more than in nonresponders (Fig. S10, http://links.lww.com/HC9/A236). This suggests that TH1 cells are activated in all patients with PBC (irrespective of UDCA response), with stronger regulation of these cells in responders.
In TH17 cells, the red module was correlated with responders (r=0.67, q=5.30×10−4) as well as nonresponders (r=0.63, q=2.46×10−3) versus controls (Table 2). It was enriched for TNF, IFNγ, and IL-2 signalling; hub genes included JUN, GADD45B, and NR4A2. The purple module was also correlated with both responders (r=0.53, q=0.015) and nonresponders (r=0.51, q=0.024) versus controls. Hub genes in this module included EGR1, which induces transcription of T-bet31 and ZC3H12A, which is critical for IL-17-mediated inflammation.32 In contrast, the magenta module was negatively correlated with responders versus controls (r=−0.50, q=0.015). It was enriched for KRAS, IL-6, and IFNγ signalling; and contained the hub gene, PIK3AP1, which is required for the generation of pathogenic TH17 cells.33 Genes in the red and purple modules were upregulated in responders and nonresponders, whereas those in the magenta module were downregulated in responders (Fig. S11, http://links.lww.com/HC9/A236). These findings suggest that TH17 cells are activated in all PBC patients—but may be better regulated in responders.
In TREG cells, the pink module was correlated with responders versus controls (r=0.69, q=1.4×10−4). Hub genes in this module notably included the immune checkpoint, CTLA4. The green module was also correlated with responders versus controls (r=0.64, q=4.0 ×10−4). It was enriched for TNF signalling and contained the hub genes, CXCR4 and IL7R. Conversely, the brown module showed a strong negative correlation with responders versus controls (r=−0.80, q=1.17×10−6); moderate negative correlation with nonresponders versus controls (r=−0.56, q=0.011); and a moderate negative correlation with responders versus nonresponders (r=−0.49, q=0.026) (Fig. S12, http://links.lww.com/HC9/A236). This module was enriched for KRAS, IL-6, IL-2 signalling, and TNF signalling; and contained the hub gene, IRF8, the identity-keeper for suppressive TH1-like TREG cells34 (Fig. S12H, http://links.lww.com/HC9/A236). Genes in the pink and green modules were upregulated in responders versus controls, whereas those in the brown module were downregulated in responders and nonresponders versus controls and responders versus nonresponders. These observations suggest that TREG cells are more active—but also kept in check—in responders.
In B cells, the black module showed a strong correlation with responders versus controls (r=0.73, q=7.37×10−5). This module was enriched with genes involved in TNF signalling.
Gene set enrichment analysis
We found that GSEA was broadly consistent with WGCNA. In monocytes, for example, GSEA showed positive enrichment of proinflammatory pathways in both responders and nonresponders versus controls—but also showed positive enrichment of MYC signalling and oxidative phosphorylation in responders versus nonresponders. In TH1 cells, GSEA showed positive enrichment of TNF signalling in nonresponders versus controls (implying activation)—but negative enrichment of IL-2 and IL-6 signalling in responders and nonresponders versus controls, and negative enrichment of IFNγ signalling in responders versus controls (implying regulation) (Fig. S15, http://links.lww.com/HC9/A236). In TREG cells, GSEA showed positive enrichment of MYC signalling (which encourages the proliferation of TREG cells) in responders versus controls—but negative enrichment of TGFβ and IL-2 signalling in both responders and nonresponders versus controls (Fig. S17, http://links.lww.com/HC9/A236). Thus, GSEA reiterated a balance between proinflammatory and immunoregulatory processes in PBC, favoring the latter in responders. Please see Supplementary Data (Fig. S14-S18, http://links.lww.com/HC9/A236) for further details.
Multi-omics factor analysis
In WGCNA and GSEA, we observed an overarching theme of immune cell activation counterbalanced by regulation. To confirm this, we used MOFA to identify the principal axes of variation across all PBMC subsets. We identified 7 latent factors accounting for most of the variance in each dataset (Figure 2A). Of these, LF1 and LF4 were associated with trait status at p = 2.8×10−7 and p = 0.0045, respectively. LF1 was active in all cell types. It was most active in TREG cells, however, accounting for 27% of the total variance observed in this PBMC subset, compared with 18% in TH17 cells, 14% in TH1 cells, 9% in CD14+ cells, and 8% in B cells. LF4 was active only in B cells, accounting for 34% of the variance in this PBMC subset. The modules with the heaviest loading on LF1 or LF4 are listed in Table 4. Broadly, LF1 reaffirmed a balance between the activation and regulation of immune processes across PBMC subsets, favoring regulation. Thus, LF1 showed a strong correlation with the pink and green modules in TREG cells, representing suppressive activity; a negative correlation with the turquoise module in monocytes and green module in TH1 cells, representing immune suppression; and only a modest correlation with the yellow module in TH1 cells and red module in TH17 cells, representing proinflammatory activity. Notably, LF1 showed a stronger association with responders, implying stronger regulation of innate and adaptive immune responses in this group of patients. The modules accounting for LF4 were enriched with genes involved in TNF signalling.
FIGURE 2.
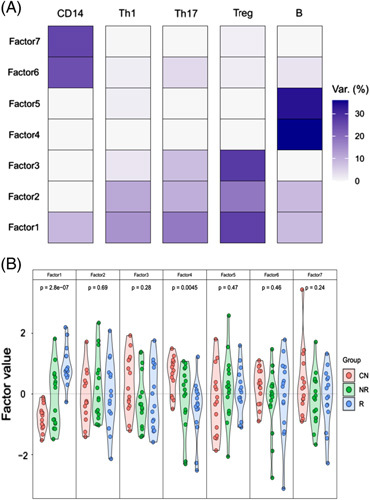
Multi-omics latent factor analysis (MOFA) of modules identified by weighted gene co-expression network analysis (WGCNA) for all cell types. (A) Heatmap to show the fitted MOFA model, displaying the percentage of variance explained for each factor (rows) in each cell type. (B) Violin plots representing the distribution of the factor values for each patient group and each latent factor (1–7). ANOVA was used to identify latent factors associated with trait status; the p-value for each latent factor is shown. Abbreviations: MOFA, Multi-omics latent factor analysis; WGCNA, Weighted gene co-expression network analysis.
TABLE 4.
Modules associated with LF1 and LF4
| Latent factor | PBMC subset | Module | Weight | r | p |
|---|---|---|---|---|---|
| 1 | CD14+ | Black | 0.61 | 0.56 | 5.4×10−5 |
| — | Turquoise | −0.66 | −0.74 | 8.1×10−9 | |
| 1 | TH1 | Yellow | 0.44 | 0.51 | 3.0×10−4 |
| — | Green | −0.56 | −0.62 | 4.7×10−6 | |
| 1 | TH17 | Red | 0.45 | 0.55 | 9.7×10−5 |
| — | Greenyellow | 0.63 | 0.69 | 1.4×10−7 | |
| 1 | TREG | Brown | −0.57 | −0.61 | 6.2×10−6 |
| — | Green | 0.66 | 0.74 | 5.8×10−9 | |
| — | Pink | 0.88 | 0.86 | 3.4×10−14 | |
| 1 | B | Red | 0.39 | 0.60 | 1.6×10−5 |
| — | Black | 0.46 | 0.76 | 2.5×10−9 | |
| 4 | B | Black | −0.65 | −0.86 | 3.4×10−12 |
| — | Brown | −0.91 | −0.91 | <2.2×10−16 | |
| — | Turquoise | 0.93 | 0.94 | <2.2×10−16 |
Notes: Modules with the greatest loading on latent factors 1 or 4 are shown.
The weight measures how strongly the module relates to the latent factor (scale from −1 to 1).
r is the correlation coefficient between the modules and the latent factor, with the associated p-value.
DISCUSSION
In this study, we profiled the transcriptomes of monocytes and TH1, TH17, TREG, and B cells from the peripheral blood of patients with PBC with an adequate response to UDCA, those with inadequate response, and healthy controls to gain insight into the immunobiology of the UDCA response. Our data suggest that: (1) monocytes are proinflammatory in nonresponders, but antiinflammatory in responders; (2) TH1 and TH17 cells are activated in all PBC patients, but there is stronger regulation of these cell types in responders; (3) TREG cells show greater activation counterbalanced by greater regulation in responders; and (4) B cells show greater activation in responders (Figure 3). Our data imply stronger regulation of immune responses in patients with a well-controlled disease.
FIGURE 3.
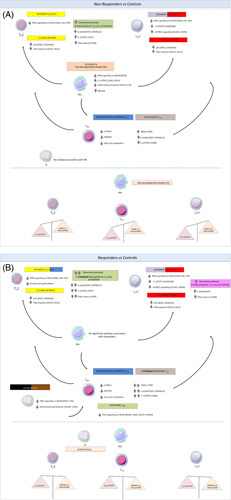
UDCA nonresponders show immunological differences compared to responders. Schematic illustrating the significant pathways correlated with nonresponders versus controls (A) and responders versus controls (B) overall highlighting the key findings of this study: (1) that monocytes are activated and proinflammatory in nonresponders; (2) TH1 and TH17 cells are activated in nonresponders and responders, but there is stronger regulation of both in the latter; (3) TREG cells exhibit greater activation counterbalanced by greater regulation in responders; and (4) B cells are activated in responders.
Previous studies have shown that transcriptional profiling of PBMC subsets can provide insight into the pathogenesis of immune traits.35–38 This is the first study, however, to use transcriptional profiling of PBMC subsets to identify networks of genes associated with UDCA response or nonresponse in PBC. We employed WGCNA, a well-established method for identifying gene networks associated with traits of interest. The major advantage of WGCNA is its use of a weighted approach to identify indirect as well as direct relationships between genes, enabling robust and informative gene clustering. Strong correlations between genes are emphasized at the expense of weak correlations, which helps to identify the most highly connected (and thus most representative) genes within each cluster—so-called “hub genes”. We then used MOFA for integrated analysis of the modules identified across all PBMC subsets. MOFA can be viewed as a statistically rigorous generalization of principal component analysis to multi-omics data, which identifies the principal axes of variation across distinct data modalities in terms of latent factors. Its advantage over corresponding methods is that MOFA quantifies the proportion of variance explained by each source of variability (ie, each latent factor) in each data modality, enabling the identification of factors shared across multiple data modalities, as well as those unique to just one.
Previous studies have shown the hyper-reactivity of monocytes in PBC.7 Consistent with this, our data suggest that monocytes are proinflammatory in nonresponders. For example, the salmon module—enriched with proinflammatory gene sets—was correlated with nonresponders. In contrast, GSEA showed positive enrichment of MYC signalling and oxidative phosphorylation in responders compared with nonresponders. In monocytes, MYC signalling promotes M2 polarization,39 while oxidative phosphorylation is the primary source of energy in M2 macrophages.40 Thus, our findings also suggest that monocytes might be antiinflammatory in responders. One possibility, albeit speculative, is that persistent cholestasis (and the inflammatory signals associated with this) might influence the behavior of monocytes—and both, in turn, might influence adaptive immune responses.
Our data suggest that TH1 cells are activated in all PBC patients, irrespective of UDCA response, but they are regulated better in responders. Thus, in TH1 cells, the yellow module—enriched for TNF signalling, which enhances T-cell proliferation41—was correlated with both responders and nonresponders. Conversely, the green module—enriched for IFNγ signalling, which promotes TH1 cell differentiation42,43—was negatively correlated just with responders. We made comparable observations in TH17 cells: activation in all patients but stronger regulation in responders. Consistent with this, we found evidence for greater activity of TREG cells in responders. For example, in TREG cells, the green module containing hub genes, such as CXCR4 and IL7R, was correlated with responders. Paradoxically, the brown module—enriched for IL-2 signalling, which causes expansion of TREG cells—was negatively correlated with responders versus nonresponders, and responders versus controls. This might represent negative feedback, however, which keeps activated TREG cells in check. We might expect such feedback to be more prominent in those with well-controlled disease.
Also paradoxical, our results suggest that there is greater activation of B cells in responders. Thus, the black module, enriched for TNF signalling, showed the strongest correlation with responders. B cells can promote regulatory T-cell differentiation44; and in a murine model of PBC, B-cell depletion exacerbates cholangitis.45 Therefore, it is plausible that B cells might have a protective role in PBC. This requires further exploration, especially the relationship between B and TREG cells in this condition.
Collectively, our findings suggest that immune responses are better contained in responders than nonresponders. One possibility is that UDCA has direct immunomodulatory effects on the immune cells tested in this study; this seems unlikely given the breadth of effects observed. Our findings might however be consistent with the BEC senescence hypothesis. Thus, when UDCA fails to ameliorate BEC senescence, proinflammatory signals from the liver continue to drive the monocyte and TH1/TH17 cell activation seen in this study. Conversely, when UDCA-induced choleresis is effective, BEC senescence is diminished, proinflammatory signals are curtailed, and regulatory mechanisms supervene. Our findings in MOFA support this concept in that LF1 showed a stronger association with responders, favored immunoregulatory over proinflammatory activity, and was most active in TREG cells. This has implications for drug development in PBC: there is clearly an important role for anti-cholestatic therapies that diminish BEC senescence—but there may also be a role for agents that target the immune processes downstream of BEC senescence.
We acknowledge the limitations of this study. The cross-sectional study design enabled us to identify associations but not causality. Even so, our study provides insight into the immunobiology of well-controlled PBC, highlighting regulatory mechanisms that could be explored for therapeutic potential. A frequent criticism of transcriptional profiling is that the genome-wide correlation between expression levels of mRNA and protein is poor. Correlation between mRNA and protein is stronger for differentially expressed genes, however, supporting the view that differential gene expression has biological meaning. We did not profile CD8+ T cells in the current study, even though CD8+ T cells are involved in the destruction of BECs in PBC.46 There were technical constraints on the number of PBMC subsets that could be sorted, however, so we prioritized T-helper and B-cell subsets to study immunoregulatory phenotypes. We acknowledge the importance of profiling CD8+ T cells (and other immune cell types, such as NK cells) in future work. Finally, we profiled circulating immune cells rather than liver infiltrates. We acknowledge that the immunopathogenic functionality and spatial array of effector and regulatory populations within inflammatory infiltrates in the liver are important to determine response to therapy in PBC. Transcriptional profiling of these populations has only recently become tractable, however, using novel approaches such as spatial transcriptomics. We look forward to using these approaches in the future.
In conclusion, this is the first study to profile the transcriptomes of PBMC subsets in patients with PBC stratified by UDCA response. Our data suggest immunological differences between responders and nonresponders—and suggest that immune cells are better restrained in PBC patients with well-controlled disease.
Supplementary Material
AUTHOR CONTRIBUTIONS
Victoria Mulcahy, Evaggelia Liaskou, Gideon M. Hirschfield, Heather J. Cordell, and George F. Mells contributed to study design; Jonathan Badrock coordinated recruitment and sample handling; Rebecca L. Jones, Stephen D. Ryder, Simon M. Rushbrook, Douglas Thorburn, Simon D. Taylor-Robinson, David E. Jones, Gideon M. Hirschfield, and George F. Mells recruited participants; Evaggelia Liaskou and GC performed experiments; Victoria Mulcahy, Jose-Ezequiel Martin, and Prasanti Kotagiri performed data analysis; Victoria Mulcahy, Jose-Ezequiel Martin, Evaggelia Liaskou, and George F. Mells performed data interpretation; Victoria Mulcahy, Evaggelia Liaskou, and George F. Mells drafted the manuscript. All authors contributed intellectually to the manuscript, critically revised the manuscript, and approved the final version.
Victoria Mulcahy and Evaggelia Liaskou are joint first authors;
Gideon M. Hirschfield and George F. Mells are joint senior authors
ACKNOWLEDGMENTS
The authors thank the staff at the National Institute for Health Research (NIHR) Cambridge BioResource for their assistance in recruiting healthy volunteers. Simon D. Taylor-Robinson thanks the British National Institute for Health Research Biomedical Facility at Imperial College London for infrastructure support. Evaggelia Liaskou thanks the Flow Cytometry Platform at the University of Birmingham for support with cell sorting experiments. Finally, they thank all participants in the study for giving up their time and for providing samples. This paper presents independent research supported by the NIHR Birmingham Biomedical Research Centre at the University Hospitals Birmingham NHS Foundation Trust and the University of Birmingham. The views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR, or the Department of Health and Social Care.
FUNDING INFORMATION
The project was funded by a Stratified Medicine award from the Medical Research Council (MRC), UK (MR/L001489/1). Victoria Mulcahy is funded by a Clinical Research Training Fellowship from the MRC, UK (MR/S021744/1). Evaggelia Liaskou is funded by the National Institute for Health Research (NIHR) Birmingham Biomedical Research Centre (BRC) (BRC-1215-20009). George F. Mells was funded by a Post-doctoral Fellowship from the NIHR Rare Diseases—Translational Research Consortium (RD-TRC), UK, and is now funded by a Clinical Academic Research Partnership from the MRC, UK (MR/T023848/1). George F. Mells and Richard N. Sandford have received funding support for UK-PBC from Intercept Pharmaceuticals. The University of Cambridge has received salary support for Richard N. Sandford from the National Health Service (NHS) in the East of England through the Clinical Academic Reserve. Gideon M. Hirschfield is supported by the Lily and Terry Horner Chair in Autoimmune Liver Disease Research.
CONFLICTS OF INTEREST STATEMENTS
Gideon M. Hirschfield has consulted for Intercept, Genfit, Cymabay, GSK, Falk, and Caliditas. George F. Mells and Richard N. Sandford have received funding support for UK-PBC from Intercept Pharmaceuticals. Simon M Rushbrook advises Falk.
Footnotes
Abbreviations: ALP, Alkaline phosphatase; FDR, False discovery rate; GSEA, Gene set enrichment analysis; GWAS, Genome-wide association studies; MOFA, Multi-omics factor analysis; NR, non-responder; PBC, Primary biliary cholangitis; PBMC, Peripheral blood mononuclear cell; QC, Quality control; R, responder; RIN, RNA integrity number; STAR, Spliced transcripts alignment to a reference; TLR, Toll-like receptor; UDCA, Ursodeoxycholic acid; ULN, Upper limit of normal; WGCNA, Weighted gene co-expression network analysis
Supplemental Digital Content is available for this article. Direct URL citations are provided in the HTML and PDF versions of this article on the journal's website, www.hepcommjournal.com.
Contributor Information
Victoria Mulcahy, Email: vlm39@medschl.cam.ac.uk.
Evaggelia Liaskou, Email: E.Liaskou@bham.ac.uk.
Jose-Ezequiel Martin, Email: jem220@medschl.cam.ac.uk.
Prasanti Kotagiri, Email: pk488@cam.ac.uk.
Jonathan Badrock, Email: j.badrock@ucl.ac.uk.
Rebecca L. Jones, Email: rebecca.jones36@nhs.net.
Simon M Rushbrook, Email: simon.rushbrook@nnuh.nhs.uk.
Stephen D. Ryder, Email: stephen.ryder@nuh.nhs.uk.
Douglas Thorburn, Email: douglas.thorburn@nhs.net.
Simon D. Taylor-Robinson, Email: s.taylor-robinson@imperial.ac.uk.
Graeme Clark, Email: grc@medschl.cam.ac.uk.
Heather J. Cordell, Email: heather.cordell@newcastle.ac.uk.
Richard N. Sandford, Email: rns13@cam.ac.uk.
David E. Jones, Email: David.Jones@newcastle.ac.uk.
Gideon M. Hirschfield, Email: gideon.hirschfield@uhn.ca.
George F. Mells, Email: gfm26@medschl.cam.ac.uk.
REFERENCES
- 1.Lleo A, Wang GQ, Gershwin ME, Hirschfield GM. Primary biliary cholangitis. Lancet. 2020;396:1915–1926. [DOI] [PubMed] [Google Scholar]
- 2.Carbone M, Sharp SJ, Flack S, Paximadas D, Spiess K, Adgey C, et al. The UK-PBC risk scores: Derivation and validation of a scoring system for long-term prediction of end-stage liver disease in primary biliary cholangitis. Hepatology. 2016;63:930–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.European Association for the Study of the Liver. Electronic address eee, European Association for the Study of the L. EASL Clinical Practice Guidelines: The diagnosis and management of patients with primary biliary cholangitis. J Hepatol. 2017;67:145–172. [DOI] [PubMed] [Google Scholar]
- 4.D’Amato D, De Vincentis A, Malinverno F, Vigano M, Alvaro D, Pompili M, et al. Real-world experience with obeticholic acid in patients with primary biliary cholangitis. JHEP Rep. 2021;3:100248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hardie C, Green K, Jopson L, Millar B, Innes B, Pagan S, et al. Early molecular stratification of high-risk primary biliary cholangitis. EBioMedicine. 2016;14:65–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Barron-Millar B, Ogle L, Mells G, Flack S, Badrock J, Sandford R, et al. The Serum proteome and ursodeoxycholic acid response in primary biliary cholangitis. Hepatology. 2021;74:3269–3283. [DOI] [PubMed] [Google Scholar]
- 7.Mao TK, Lian ZX, Selmi C, Ichiki Y, Ashwood P, Ansari AA, et al. Altered monocyte responses to defined TLR ligands in patients with primary biliary cirrhosis. Hepatology. 2005;42:802–808. [DOI] [PubMed] [Google Scholar]
- 8.Shimoda S, Ishikawa F, Kamihira T, Komori A, Niiro H, Baba E, et al. Autoreactive T-cell responses in primary biliary cirrhosis are proinflammatory whereas those of controls are regulatory. Gastroenterology. 2006;131:606–618. [DOI] [PubMed] [Google Scholar]
- 9.Yang CY, Ma X, Tsuneyama K, Huang S, Takahashi T, Chalasani NP, et al. IL-12/Th1 and IL-23/Th17 biliary microenvironment in primary biliary cirrhosis: implications for therapy. Hepatology. 2014;59:1944–1953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lan RY, Cheng C, Lian ZX, Tsuneyama K, Yang GX, Moritoki Y, et al. Liver-targeted and peripheral blood alterations of regulatory T cells in primary biliary cirrhosis. Hepatology. 2006;43:729–737. [DOI] [PubMed] [Google Scholar]
- 11.Liaskou E, Hirschfield GM, Gershwin ME. Mechanisms of tissue injury in autoimmune liver diseases. Semin Immunopathol. 2014;36:553–568. [DOI] [PubMed] [Google Scholar]
- 12.Zhao SX, Li WC, Fu N, Zhou GD, Liu SH, Jiang LN, et al. Emperipolesis mediated by CD8(+) T cells correlates with biliary epithelia cell injury in primary biliary cholangitis. J Cell Mol Med. 2020;24:1268–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chung BK, Guevel BT, Reynolds GM, Gupta Udatha DB, Henriksen EK, Stamataki Z, et al. Phenotyping and auto-antibody production by liver-infiltrating B cells in primary sclerosing cholangitis and primary biliary cholangitis. J Autoimmun. 2017;77:45–54. [DOI] [PubMed] [Google Scholar]
- 14.Lleo A, Bowlus CL, Yang GX, Invernizzi P, Podda M, Van de Water J, et al. Biliary apotopes and anti-mitochondrial antibodies activate innate immune responses in primary biliary cirrhosis. Hepatology. 2010;52:987–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Langfelder P, Horvath S. Eigengene networks for studying the relationships between co-expression modules. BMC Syst Biol. 2007;1:54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hartwell LH, Hopfield JJ, Leibler S, Murray AW. From molecular to modular cell biology. Nature. 1999;402:C47–C52. [DOI] [PubMed] [Google Scholar]
- 17.van Dam S, Vosa U, van der Graaf A, Franke L, de Magalhaes JP. Gene co-expression analysis for functional classification and gene-disease predictions. Brief Bioinform. 2018;19:575–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Nevens F, Andreone P, Mazzella G, Strasser SI, Bowlus C, Invernizzi P, et al. A placebo-controlled trial of obeticholic acid in primary biliary cholangitis. N Engl J Med. 2016;375:631–643. [DOI] [PubMed] [Google Scholar]
- 19.European Association for the Study of the Liver. Electronic address eee, European Association for the Study of the L. EASL Clinical Practice Guidelines for the management of patients with decompensated cirrhosis. J Hepatol. 2018;69:406–460. [DOI] [PubMed] [Google Scholar]
- 20.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Liao Y, Smyth GK, Shi W. The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res. 2019;47:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010;26:139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq. 2. Genome Biol. 2014;15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015;43:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics. 2013;14:128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Liberzon A, Birger C, Thorvaldsdottir H, Ghandi M, Mesirov JP, Tamayo P. The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst. 2015;1:417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, et al. Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc Natl Acad Sci U S A. 2005;102:15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Argelaguet R, Velten B, Arnol D, Dietrich S, Zenz T, Marioni JC, et al. Multi-Omics Factor Analysis-a framework for unsupervised integration of multi-omics data sets. Mol Syst Biol. 2018;14:e8124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Corpechot C, Carrat F, Poujol-Robert A, Gaouar F, Wendum D, Chazouilleres O, et al. Noninvasive elastography-based assessment of liver fibrosis progression and prognosis in primary biliary cirrhosis. Hepatology. 2012;56:198–208. [DOI] [PubMed] [Google Scholar]
- 30.Lehtonen A, Lund R, Lahesmaa R, Julkunen I, Sareneva T, Matikainen S. IFN-alpha and IL-12 activate IFN regulatory factor 1 (IRF-1), IRF-4, and IRF-8 gene expression in human NK and T cells. Cytokine. 2003;24:81–90. [DOI] [PubMed] [Google Scholar]
- 31.Shin HJ, Lee JB, Park SH, Chang J, Lee CW. T-bet expression is regulated by EGR1-mediated signaling in activated T cells. Clin Immunol. 2009;131:385–394. [DOI] [PubMed] [Google Scholar]
- 32.Tanaka H, Arima Y, Kamimura D, Tanaka Y, Takahashi N, Uehata T, et al. Phosphorylation-dependent Regnase-1 release from endoplasmic reticulum is critical in IL-17 response. J Exp Med. 2019;216:1431–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Deason K, Troutman TD, Jain A, Challa DK, Mandraju R, Brewer T, et al. BCAP links IL-1R to the PI3K-mTOR pathway and regulates pathogenic Th17 cell differentiation. J Exp Med. 2018;215:2413–2428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lee GR. IRF8: identity-keeper for suppressive Th1-like Treg cells. Cell Mol Immunol. 2018;15:1080–1081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Batliwalla FM, Baechler EC, Xiao X, Li W, Balasubramanian S, Khalili H, et al. Peripheral blood gene expression profiling in rheumatoid arthritis. Genes Immun. 2005;6:388–397. [DOI] [PubMed] [Google Scholar]
- 36.Bomprezzi R, Ringner M, Kim S, Bittner ML, Khan J, Chen Y, et al. Gene expression profile in multiple sclerosis patients and healthy controls: identifying pathways relevant to disease. Hum Mol Genet. 2003;12:2191–2199. [DOI] [PubMed] [Google Scholar]
- 37.Barnes MG, Aronow BJ, Luyrink LK, Moroldo MB, Pavlidis P, Passo MH, et al. Gene expression in juvenile arthritis and spondyloarthropathy: pro-angiogenic ELR+ chemokine genes relate to course of arthritis. Rheumatology (Oxford). 2004;43:973–979. [DOI] [PubMed] [Google Scholar]
- 38.Tuller T, Atar S, Ruppin E, Gurevich M, Achiron A. Common and specific signatures of gene expression and protein-protein interactions in autoimmune diseases. Genes Immun. 2013;14:67–82. [DOI] [PubMed] [Google Scholar]
- 39.Pello OM, De Pizzol M, Mirolo M, Soucek L, Zammataro L, Amabile A, et al. Role of c-MYC in alternative activation of human macrophages and tumor-associated macrophage biology. Blood. 2012;119:411–421. [DOI] [PubMed] [Google Scholar]
- 40.Kelly B, O’Neill LA. Metabolic reprogramming in macrophages and dendritic cells in innate immunity. Cell Res. 2015;25:771–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mehta AK, Gracias DT, Croft M. TNF activity and T cells. Cytokine. 2018;101:14–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Watanabe M, Moon KD, Vacchio MS, Hathcock KS, Hodes RJ. Downmodulation of tumor suppressor p53 by T cell receptor signaling is critical for antigen-specific CD4(+) T cell responses. Immunity. 2014;40:681–691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Diehl S, Anguita J, Hoffmeyer A, Zapton T, Ihle JN, Fikrig E, et al. Inhibition of Th1 differentiation by IL-6 is mediated by SOCS1. Immunity. 2000;13:805–815. [DOI] [PubMed] [Google Scholar]
- 44.Chien CH, Chiang BL. Regulatory T cells induced by B cells: a novel subpopulation of regulatory T cells. J Biomed Sci. 2017;24:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Dhirapong A, Lleo A, Yang GX, Tsuneyama K, Dunn R, Kehry M, et al. B cell depletion therapy exacerbates murine primary biliary cirrhosis. Hepatology. 2011;53:527–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yang GX, Wu Y, Tsukamoto H, Leung PS, Lian ZX, Rainbow DB, et al. CD8 T cells mediate direct biliary ductule damage in nonobese diabetic autoimmune biliary disease. J Immunol. 2011;186:1259–1267. [DOI] [PMC free article] [PubMed] [Google Scholar]