

Abstract
Objective:
To study differences in cardiovascular (CV) risk factors and diseases between patients with and without genetic variants in the leptin-melanocortin pathway.
Patients and Methods:
A cross-sectional study of patients with a history of severe obesity genotyped in June 2019 as part of the Mayo Clinic Biobank study to assess differences in cardiovascular risk and diseases (CVD) assessed in March 2022 between carriers of a heterozygous variant in the leptin-melanocortin pathway and non-carriers. CV risk factors included hypertension, diabetes, dyslipidemia, and smoking. CVD includes coronary artery disease, peripheral artery disease, and cerebrovascular accidents. Patients with a history of bariatric surgery were excluded. We used logistic regression models to estimate the odds ratio (OR) and 95% confidence intervals (CI), adjusting for age, body mass index (BMI), and sex.
Results:
Among total of 168 carriers (8%) (72% females, mean [SD] age of 65.1 [14.9] years, BMI of 44.0 [7.4] kg/m2) and 2039 non-carriers (92%) (71% females, mean age of 64.9 [14.4] years, BMI of 42.9 [6.6] kg/m2), carriers had higher prevalence odds of hypertension (OR, 3.26; 95% CI, 2.31–4.61; p < .001) and reported higher number of cardiovascular risk factors compared with non-carriers participants (2.4 [1.1] vs. 2.0 [1.1]; p < .001). There were no significant differences in the adjusted odds associated with diabetes, dyslipidemia, smoking, or CVD.
Conclusion:
Despite having similar body weight and BMI, carriers of heterozygous mutations in the leptin-melanocortin pathway had higher rates of hypertension than non-carriers. These findings point to an association between hypertension and leptin-melanocortin pathway mutations.
Introduction
The prevalence of obesity and its associated cardiometabolic diseases have increased in recent decades and have become a major concern for most healthcare systems due to their considerable socioeconomic burden.1 In 2016, there were 671 million adults with obesity worldwide (12% of the world’s adult population), substantially double the burden from the preceding 30 years.2 Furthermore, as body weight increases, so do the risks of cardiovascular morbidity and mortality, including stroke, congestive heart failure, myocardial infarction, and cardiovascular death.3 As a result, obesity has been labeled as a major, modifiable risk factor for cardiovascular diseases (CVD).4
Obesity is a multifactorial disease which results from an energy-balance dysregulation. Bodyweight is regulated by complicated interactions between hereditary and environmental factors. Overall, the heritability of obesity is estimated at 40% to 70%.5 However, only around 10% of severe early-onset obesity, as early as less than 5 years of age, is caused by monogenic variants. This is mainly due to autosomal recessive mutations in genes of the leptin-melanocortin pathway. This pathway is critical for regulating food intake and body weight.6 Heterozygous variants in the leptin-melanocortin pathway are the most prevalent in approximately 6% of children with severe early-onset obesity and 2.5% of adults with obesity.7,8
The leptin–melanocortin pathway is a network of neurons in the hypothalamus that integrates information regarding peripheral energy storage, which is predominantly conveyed by the leptin hormone. The pathway contains leptin-responsive neurons that express neuropeptide Y and agouti-related protein (AgRP), as well as neurons that express proopiomelanocortin (POMC). POMC is cleaved to yield melanocyte-stimulating hormones (MSH).9 Downstream, MSH acts as an agonist to target the melanocortin 3 receptor (MC3R) and the melanocortin 4 receptor (MC4R) expressing neurons. On the other hand, AgRP is the natural antagonist of MC3R and MC4R.
Animal studies have suggested that the leptin-melanocortin pathway plays an important role in cardiovascular regulation and that obesity is linked to cardiovascular illness, with hypertension serving as the principal mediator of obesity-induced cardiovascular disease.10,11 This study aims to investigate the association between cardiovascular diseases and heterozygous variants in the leptin-melanocortin pathway in patients with obesity. Understanding the effect of these variants and cardiovascular morbidity and mortality would aid clinicians in addressing its modifiable risk factors, better serving the needs of this rapidly growing portion of the population.
Methods
Study design, study population, and sample selection
We conducted a cross-sectional study on participants of the Mayo Clinic Biobank (n=56,862) 12 who had a history of severe obesity (body mass index [BMI] ≥ 40 mg/km2) according to relevant International Classification of Diseases, Ninth Revision, Clinical Modification (ICD-9) 278.01 or had bariatric surgery, assessed in March 2022, who were genotyped for variants in the hypothalamic leptin-melanocortin pathway (n = 3,002) and evaluated the differences in the prevalence of cardiovascular risk factors and CVD between carriers of heterozygous pathogenic variants, likely pathogenic variants, and variants of undetermined significance in genes in the leptin-melanocortin pathway and non-carriers. A total of 30 genes (Supplemental Table 1) previously related to the leptin-melanocortin pathway were sequenced by Sema4 Laboratories (Stamford, CT, USA) and are detailed in Supplementary Material.7 The study was approved by the institutional review board (IRB) of Mayo Clinic (IRB 18–010159). All participants provided prior written authorization for research use of their medical records and biological samples, including use for genotyping studies. We excluded deceased patients (n=217) and patients with any history of bariatric procedure (n=557), which can influence the prevalence of cardiovascular disease or risk factors.13 We also excluded patients with homozygous variants and compound heterozygous (n=19), presenting a different phenotype than with a heterozygous variant (Figure 1).
Figure 1.
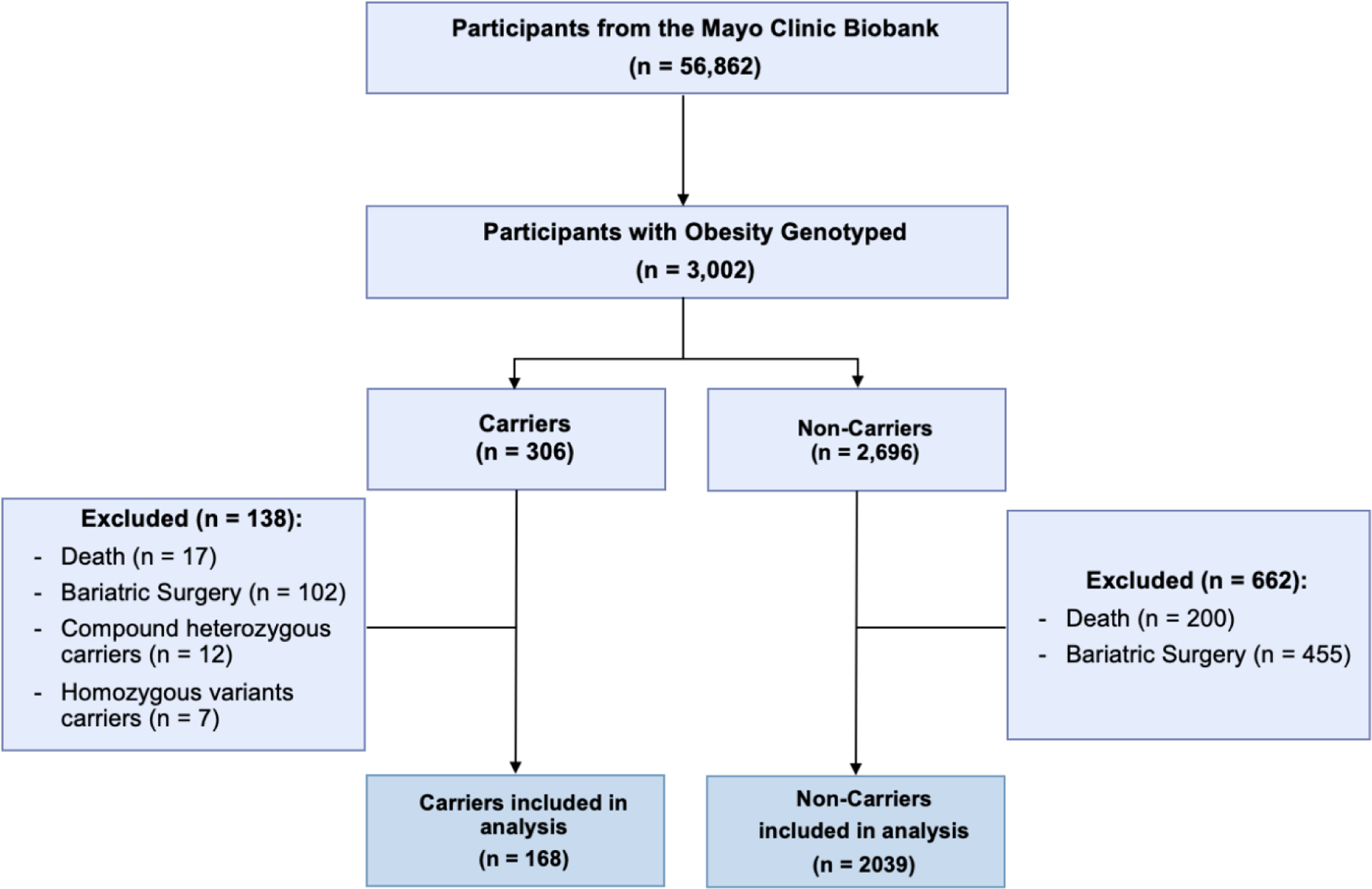
Participant Flow-Chart
Outcome variables
Up to February 2022, the following demographic data were extracted from the electronic medical record: sex, age, height, and weight as last entered in the electronic medical record (EMR). The following demographic data were abstracted from the electronic medical record: sex, age, height, and weight last recorded in the EMR until February 2022. Data on comorbidities were collected by a computerized hospital system according to relevant ICD-9 or ICD-10 codes (Supplemental Table 2)14. The comorbidities investigated were all forms of cardiovascular risk factors: (1) hypertension, (2) diabetes, (3) dyslipidemia, and (4) smoking. The cardiovascular diseases (hereafter referred to as cardiovascular disease [CVD]) included: (1) coronary artery disease (CAD), (2) peripheral artery disease, and (3) history of cerebrovascular accident (CVA). Coronary artery disease included angina and non-fatal myocardial infarction and CVA was defined as having any history of ischemic stroke, hemorrhagic stroke, or transient ischemic attack.
Statistical Analysis
We calculated proportions for categorical variables and means (± standard deviation [SD]) for continuous variables according to carrier or non-carrier status. We used two-sample independent t-test for continuous variables and Fisher’s exact test for categorical variables to compare baseline characteristics between groups. We used logistic regression to estimate the prevalence odds ratio (OR) and its 95% confidence intervals (CI). Logistic regression models were constructed to assess the association between carriers or non-carriers with cardiovascular risk factors and CVD, univariate logistic regression, and multivariate logistic regression adjusting for age and BMI (model 1); and age, sex, and BMI (model 2). Statistical significance was set at 2-sided p < .05. We used JMP®, Version 14.3.0 (SAS Institute Inc., Cary, NC, 1989–2019) to perform the statistical analysis. This study followed the Strengthening the Reporting of Observational Studies in Epidemiology (STROBE) reporting guideline (Supplementary Material).
Results
We included 168 carriers (8%) (72% females and mean [SD] age of 65.1 [14.9] years) and 2039 non-carriers (92%) (70.9% females and mean age of 64.9 [14.4] years) (Table 1). In this final cohort, we identified 119 variants in 11 different genes from the leptin-melanocortin pathway, nine genes upstream to the MC4R (LEPR, MAGEL2, PCSK1, POMC, PROK2, RAB23, RAI 1, SH2B1, and SRC1), and two downstream (SIM1 and MC4R) (Supplemental Table 3). In comparison with non-carriers, carriers did not have significantly higher body mass index (mean [SD], 44.0 [7.4] kg/m2 vs. 42.9 [6.6] kg/m2; p = 0.07; calculated as weight in kilograms divided by height in meters squared),
Table 1.
Demographics of carriers and non-carriers of a heterozygous variant in the leptin-melanocortin pathway.
| All (n = 2207) |
Carriers (n = 168) |
Non-carriers (n = 2039) |
p-value | |
|---|---|---|---|---|
| Sex, females | 1567 (71.0%) | 121 (72.0%) | 1446 (70.9%) | .79 |
| Race, white | 2098 (95%) | 158 (94%) | 1940 (95%) | .46 |
| Age, years | 64.9 (14.4) | 65.1 (14.9) | 64.9 (14.4) | .87 |
| Weight, kg | 119.6 (20.8) | 120.9 (21.1) | 119.5 (20.8) | .39 |
| BMI, kg/m 2 | 43.0 (6.6) | 44.0 (7.4) | 42.9 (6.6) | .07 |
Abbreviations: BMI, body mass index.
Data are presented as mean and standard deviation (SD).
Cardiovascular Risk Factors by Carrier Status
Dyslipidemia was the most common cardiovascular risk factor for carriers (129 [76%]) and non-carriers (1575 [77.2%]). Carriers had a higher prevalence of hypertension compared with non-carriers (120 [71.4%] vs. 885 [43.4%]; p < .001) (Table 2). Overall, the mean (SD) number of reported cardiovascular risk factors per patient was higher in carriers compared with non-carriers’ participants (2.4 [1.1] vs. 2.0 [1.1]; p < .001). In the univariate logistic regression, carriers have statistically significantly higher odds of prevalent hypertension than non-carriers (Table 2, univariate analysis: OR, 3.26; 95% CI, 2.31–4.61; p < .001). There was no significant change when the model was further adjusted for age and BMI (Table 2, model 1), and sex (Table 2, model 2). Carriers have a nearly statistically significantly higher odds of diabetes than non-carriers (Table 2, univariate analysis: OR, 1.35; 95% CI, 0.99 – 1.86; p = .05). However, it was no longer significant after adjusting for age, sex, and BMI. There were no differences in the odds for dyslipidemia or smoking status between groups.
Table 2.
Distribution and prevalence odds ratio of cardiovascular risk factors and cardiovascular diseases among carriers of a heterozygous variant in the leptin-melanocortin pathway.
| All (n = 2207) |
Carriers (n = 168) |
Univariate OR (95% CI) |
p-value | Model 1a OR (95% CI) | p-value | Model 2b OR (95% CI) |
p-value | |
|---|---|---|---|---|---|---|---|---|
| Cardiovascular Risk Factors | ||||||||
| Hypertension, yes | 1005 (45.5%) | 120 (71.4%) | 3.26 (2.31 – 4.61) | <.001 | 3.31 (2.33 – 4.71) | <.001 | 3.32 (2.34 – 4.73) | <.001 |
| Diabetes, yes | 1000 (45.3%) | 88 (52.4%) | 1.35 (0.99 – 1.86) | .05 | 1.32 (0.95 – 1.83) | .09 | 1.34 (0.96 – 1.86) | .08 |
| Dyslipidemia, yes | 1704 (77%) | 129 (76%) | 0.97 (0.67 – 1.41) | .89 | 0.95 (0.63 – 1.43) | .82 | 0.96 (0.63 – 1.45) | .84 |
| Smoke, yes | 915 (41.4%) | 64 (38.1%) | 0.85 (0.62 – 1.18) | .36 | 0.84 (0.61 – 1.17) | .32 | 0.86 (0.61 – 1.19) | .35 |
| Cardiovascular Diseases | ||||||||
| CAD, yes | 611 (27.7%) | 45 (26.8%) | 0.95 (0.73 – 1.49) | .78 | 0.89 (0.60 – 1.31) | .55 | 0.91 (0.62 – 1.35) | .66 |
| PAD, yes | 717 (32.5%) | 48 (28.5%) | 0.95 (0.52 – 1.72) | .35 | 0.74 (0.50 – 1.09) | .13 | 0.75 (0.51 – 1.10) | .22 |
| CVA, yes | 248 (11.2%) | 17 (10.1%) | 0.86 (0.52 – 1.48) | .63 | 0.86 (0.50 – 1.46) | .57 | 0.87 (0.50 – 1.49) | .61 |
Abbreviations: CAD, coronary artery disease; CVA, cerebrovascular accident; CVD, cardiovascular disease; PAD, peripheral arterial disease.
Adjusted for age and body mass index.
Adjusted for age, sex and body mass index.
Cardiovascular Disease by Carrier Status
Overall, 27.7% of individuals had coronary artery disease, 32.5% had peripheral artery disease, and 11.2% had a history of cerebrovascular accidents. There was no significant difference in the rate of CVD across groups (Table 2). Overall, the mean number of reported cardiovascular diseases per patient was similar in carriers compared with non-carriers participants (0.65 [0.9] vs. 0.71 [0.9]; p = 0.38). In the multivariable-adjusted models, carriers did not have statistically significantly higher odds of any CVD compared with non-carriers (Table 2).
Discussion
In this study of patients with obesity with and without leptin-melanocortin pathway heterozygous genetic variants, we found that carriers had an increased prevalence of hypertension. Adjustment for age, sex, and BMI, potentially influencing cardiovascular risk factors, did not attenuate this relationship. However, despite the increased number of risk factors, there was no consistent relationship between leptin-melanocortin pathway variants and CVD.
Few previous studies have documented the prevalence of cardiovascular health in subjects with obesity and genetic variants in the leptin-melanocortin pathway.10,15–17 One of these reports showed that adults with obesity who were heterozygous for complete loss-of-function variants in the MC4R gene had a lower prevalence of hypertension than adults with overweight or obesity with a normal MC4R genetic sequence.16 According to their findings, MC4R-deficient participants, were substantially shielded against the predicted link between obesity and high blood pressure, mainly due to impaired sympathetic nervous system activation. However, in a cohort of adults with a history of severe obesity and heterozygous variants in the leptin–melanocortin pathway, carriers had a higher prevalence of hypertension despite having a similar body weight and BMI. More study in humans is needed to fully understand how MC4R-expressing brain pathways regulate pressor and depressor vascular effects, as seen in animal models.10,11,18
Excess weight gain and adiposity predict the development of hypertension, and the association between BMI and blood pressure appears to be almost linear in many studies.19–21 The main physiological mechanism attributed has been the activation of the sympathetic nervous system, which appears to be mediated in part by increased levels of leptin, stimulation of POMC neurons, and subsequent activation of the central nervous system melanocortin 4 receptors.22,23 The higher prevalence of hypertension in carriers of a heterozygous variant in the leptin-melanocortin pathway may be explained by an increased leptin-mediated sympathetic stimulation to organs such as the kidney, heart, and adrenal glands.
Importantly, long-term obesity does not appear to be required to elevate blood pressure, as the association between adiposity and hypertension is seen in childhood.24 Few studies on the effects of long-term obesity have discovered that a longer duration relates to a heightened incidence of diabetes and mortality but have not found an association with arterial hypertension or hyperlipidemia.25–27. Furthermore, the Framingham Heart Study data indicate that the mechanism through which excess adiposity may raise blood pressure is essentially rapid and that long-term exposure to obesity does not enhance the risk of developing hypertension beyond the level of BMI obtained.28 Patients with genetic variants in the leptin-melanocortin pathway may have experience obesity since childhood; however, the exact duration of the syndrome in this cohort has not been documented. More research is needed to determine the long-term risk of obesity in people with genetic variants and CVD.
In addition, previous studies have suggested that insulin resistance and hyperinsulinemia are key factors to high blood pressure.29,30 However, a cause-and-effect relationship has not been fully elucidated, and obesity may be the primary factor relating to these conditions. The metabolic consequences of insulin resistance appear to combine synergistically with increased sympathetic nervous system activity to aggravate hypertension in patients with obesity. This link between hypertension and insulin resistance or hyperinsulinemia caused by excess adiposity is mediated by renal sodium retention, vascular anatomy-function, and ion transport. The central melanocortin pathways that promote sympathetic activation and hypertension may also be responsible for pancreatic insulin production and hyperinsulinemia.31,32 Children with MC4R deficiency have shown higher rates of hyperinsulinemia than children with a normal MC4R genetic sequence.33 Here, we have observed a nearly significantly higher odds of diabetes in adults with obesity and heterozygous variants in the leptin-melanocortin pathway. In contrast to hypertension, the length of obesity is a significant risk factor for type 2 diabetes, regardless of BMI level.25 Timing was not evaluated in our sample; more prospective cohort studies are required to understand the effect of childhood obesity in individuals with a leptin-melanocortin variation.
The strengths of this study include the homogeneity in demographic characteristics between both groups, exclusion of patients with the bariatric procedure, recency and availability of information of the electronic medical record, high quality of genotype information, and the power given by a high number of carriers with a heterozygous variant in the leptin-melanocortin pathway. Previous studies with varying methodology and sample sizes have reported under-recognition and poor coding practices for obesity.34,35 Despite the underrepresentation of the condition using the coding system, there is a low false-positive rate for severe obesity, which allows us to examine the differences in cardiovascular risk factors and diseases in patients with severe obesity.
Our study also has several potential limitations. First, the observational design precludes reaching causal conclusions regarding the findings. Second, given the complexity of the CVD causal pathways, there may be a significant amount of unmeasured confounding and mediation within our analysis’s data. Third, comorbidities were defined by ICD codes, and further studies with current clinical guidelines should be performed. Fourth, we did not include data on the duration between the onset and diagnosis of any risk factors or diseases that may affect the association between risk factors and heterozygous variants in the leptin-melanocortin pathway considering obesity duration. Fifth, because our group excluded deceased participants, there may have been a survival bias, which might explain why there were no changes in CVD outcomes despite the increased prevalence of hypertension. It has been described as a protective effect of obesity in congestive heart failure, renal failure, and diabetes.36,37 The most common explanation for the paradox is selection bias, as large population-based studies show that younger individuals with obesity have a higher risk of death than normal-weight patients.38 Despite the risk of survival bias in our study, we have only compared patients with obesity, and there were no differences in the mortality rate between carriers and non-carriers. Finally, most participants from the Mayo Clinic Biobank resided in the Upper Midwest regions of the USA (Minnesota, Iowa, Illinois, or Wisconsin), Florida, or Georgia. The self-reported race among biobank participants was 90% White, and the generalizability of these results to other populations may be limited. There have been described important differences in CVD prevalence among different races. However, it is widely recognized that ethnic and racial minorities participate in and are recruited for fewer cardiovascular research and trials than white people.39 Future prospective studies should evaluate cardiovascular risk factors and diseases among different populations.
Conclusion:
In conclusion, data from this cross-sectional study indicate that patients with obesity and heterozygous variants in the leptin-melanocortin pathway may not be protected from hypertension as previously thought. Despite the fact that carriers of an MC4R variant did not have an increase in the incidence of cardiovascular disease or death, our findings support a practice of primary hypertension prevention in adults with heterozygous variants and obesity. Further studies are needed to understand the mechanisms that mediate the changes in the sympathetic neural activity in humans with obesity who are affected by a leptin-melanocortin variant.
Supplementary Material
Funding:
Dr. Acosta is supported by NIH (NIH K23-DK114460), The Mayo Clinic Biobank, and Rhythm Pharmaceuticals for the genotyping studies. The Mayo Clinic Biobank is supported by the Mayo Clinic Center for Individualized Medicine.
Abbreviations:
- AgRP
agouti-related protein
- BMI
body mass index
- CAD
coronary artery disease
- CI
confidence intervals
- CV
cardiovascular
- CVA
cerebrovascular accident
- CVD
cardiovascular diseases
- EMR
electronic medical record
- ICD-9
International Classification of Diseases, Ninth Revision, Clinical Modification
- IRB
institutional review board
- MC3R
melanocortin 3 receptor
- MC4R
melanocortin 4 receptor
- MSH
melanocyte-stimulating hormones
- OR
odds ratio
- POMC
proopiomelanocortin
- SD
standard deviation
- STROBE
Strengthening the Reporting of Observational Studies in Epidemiology
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Credit statement:
LC, AA, and JO co-conceptualized and co-designed the study, drafted the initial data, and critically reviewed the manuscript. AC, DS, ADLR, WG, and FF coordinated and supervised data collection, and critically reviwed the manuscript. MDH, AM, and JB provided critical feedback on study design and critically reviwed the manuscript. All authors approved the final manuscript as submitted and agree to be accountable for all aspects of the work.
Disclosure: Dr. Acosta is a stockholder in Gila Therapeutics, Phenomix Sciences; he served as a consultant for Rhythm Pharmaceuticals, General Mills.
Data Access: Dr. Acosta has full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
References
- 1.Heymsfield SB, Wadden TA. Mechanisms, pathophysiology, and management of obesity. New England Journal of Medicine 2017; 376(3): 254–66. [DOI] [PubMed] [Google Scholar]
- 2.Abarca-Gómez L, Abdeen ZA, Hamid ZA, et al. Worldwide trends in body-mass index, underweight, overweight, and obesity from 1975 to 2016: a pooled analysis of 2416 population-based measurement studies in 128· 9 million children, adolescents, and adults. The lancet 2017; 390(10113): 2627–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Van Gaal LF, Mertens IL, Christophe E. Mechanisms linking obesity with cardiovascular disease. Nature 2006; 444(7121): 875–80. [DOI] [PubMed] [Google Scholar]
- 4.Eckel RH, Kahn R, Robertson RM, Rizza RA. Preventing cardiovascular disease and diabetes: a call to action from the American Diabetes Association and the American Heart Association. Circulation 2006; 113(25): 2943–6. [DOI] [PubMed] [Google Scholar]
- 5.Bray MS, Loos RJ, McCaffery JM, et al. NIH working group report—using genomic information to guide weight management: from universal to precision treatment. Obesity 2016; 24(1): 14–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Farooqi IS, O’Rahilly S. Mutations in ligands and receptors of the leptin–melanocortin pathway that lead to obesity. Nature Clinical Practice Endocrinology & Metabolism 2008; 4(10): 569–77. [DOI] [PubMed] [Google Scholar]
- 7.van der Klaauw Agatha A, Farooqi IS. The Hunger Genes: Pathways to Obesity. Cell 2015; 161(1): 119–32. [DOI] [PubMed] [Google Scholar]
- 8.Larsen LH, Echwald SM, Sørensen TI, Andersen T, Wulff BS, Pedersen O. Prevalence of mutations and functional analyses of melanocortin 4 receptor variants identified among 750 men with juvenile-onset obesity. The Journal of Clinical Endocrinology & Metabolism 2005; 90(1): 219–24. [DOI] [PubMed] [Google Scholar]
- 9.Schwartz MW, Woods SC, Porte D, Seeley RJ, Baskin DG. Central nervous system control of food intake. Nature 2000; 404(6778): 661–71. [DOI] [PubMed] [Google Scholar]
- 10.Tallam LS, Stec DE, Willis MA, da Silva AA, Hall JE. Melanocortin-4 receptor–deficient mice are not hypertensive or salt-sensitive despite obesity, hyperinsulinemia, and hyperleptinemia. Hypertension 2005; 46(2): 326–32. [DOI] [PubMed] [Google Scholar]
- 11.Ni X-P, Butler AA, Cone RD, Humphreys MH. Central receptors mediating the cardiovascular actions of melanocyte stimulating hormones. Journal of hypertension 2006; 24(11): 2239–46. [DOI] [PubMed] [Google Scholar]
- 12.Olson JE, Ryu E, Hathcock MA, et al. Characteristics and utilisation of the Mayo Clinic Biobank, a clinic-based prospective collection in the USA: cohort profile. BMJ open 2019; 9(11): e032707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wiggins T, Guidozzi N, Welbourn R, Ahmed AR, Markar SR. Association of bariatric surgery with all-cause mortality and incidence of obesity-related disease at a population level: A systematic review and meta-analysis. PLoS medicine 2020; 17(7): e1003206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Quan H, Sundararajan V, Halfon P, et al. Coding algorithms for defining comorbidities in ICD-9-CM and ICD-10 administrative data. Medical care 2005: 1130–9. [DOI] [PubMed]
- 15.Sayk F, Heutling D, Dodt C, et al. Sympathetic function in human carriers of melanocortin-4 receptor gene mutations. The Journal of Clinical Endocrinology & Metabolism 2010; 95(4): 1998–2002. [DOI] [PubMed] [Google Scholar]
- 16.Greenfield JR, Miller JW, Keogh JM, et al. Modulation of blood pressure by central melanocortinergic pathways. New England Journal of Medicine 2009; 360(1): 44–52. [DOI] [PubMed] [Google Scholar]
- 17.Simonds SE, Pryor JT, Ravussin E, et al. Leptin mediates the increase in blood pressure associated with obesity. Cell 2014; 159(6): 1404–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kuo JJ, da Silva AA, Tallam LS, Hall JE. Role of adrenergic activity in pressor responses to chronic melanocortin receptor activation. Hypertension 2004; 43(2): 370–5. [DOI] [PubMed] [Google Scholar]
- 19.Hall JE. The kidney, hypertension, and obesity. Hypertension 2003; 41(3): 625–33. [DOI] [PubMed] [Google Scholar]
- 20.Garrison RJ, Kannel WB, Stokes J III, Castelli WP. Incidence and precursors of hypertension in young adults: the Framingham Offspring Study. Preventive medicine 1987; 16(2): 235–51. [DOI] [PubMed] [Google Scholar]
- 21.Burt VL, Cutler JA, Higgins M, et al. Trends in the prevalence, awareness, treatment, and control of hypertension in the adult US population: data from the health examination surveys, 1960 to 1991. Hypertension 1995; 26(1): 60–9. [DOI] [PubMed] [Google Scholar]
- 22.Hall JE, da Silva AA, do Carmo JM, et al. Obesity-induced hypertension: role of sympathetic nervous system, leptin, and melanocortins. Journal of Biological Chemistry 2010; 285(23): 17271–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bravo PE, Morse S, Borne DM, Aguilar EA, Reisin E. Leptin and hypertension in obesity. Vascular health and risk management 2006; 2(2): 163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sorof J, Daniels S. Obesity hypertension in children: a problem of epidemic proportions. Hypertension 2002; 40(4): 441–7. [DOI] [PubMed] [Google Scholar]
- 25.Abdullah A, Stoelwinder J, Shortreed S, et al. The duration of obesity and the risk of type 2 diabetes. Public health nutrition 2011; 14(1): 119–26. [DOI] [PubMed] [Google Scholar]
- 26.Sakurai Y, Teruya K, Shimada N, et al. Association between duration of obesity and risk of non-insulin-dependent diabetes mellitus: the Sotetsu Study. American journal of epidemiology 1999; 149(3): 256–60. [DOI] [PubMed] [Google Scholar]
- 27.Wannamethee SG, Shaper AG. Weight change and duration of overweight and obesity in the incidence of type 2 diabetes. Diabetes care 1999; 22(8): 1266–72. [DOI] [PubMed] [Google Scholar]
- 28.Tanamas S, Backholer K, Wong E, et al. Duration of obesity and incident hypertension in adults from the Framingham Heart Study. International Journal of Epidemiology 2015; 44(suppl_1): i74–i5. [DOI] [PubMed] [Google Scholar]
- 29.Hall JE, Summers RL, Brands MW, Keen H, Alonso-Galicia M. Resistance to metabolic actions of insulin and its role in hypertension. American journal of hypertension 1994; 7(8): 772–8. [DOI] [PubMed] [Google Scholar]
- 30.Sasaki N, Ozono R, Higashi Y, Maeda R, Kihara Y. Association of insulin resistance, plasma glucose level, and serum insulin level with hypertension in a population with different stages of impaired glucose metabolism. Journal of the American Heart Association 2020; 9(7): e015546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Fan W, Dinulescu DM, Butler AA, Zhou J, Marks DL, Cone RD. The central melanocortin system can directly regulate serum insulin levels. Endocrinology 2000; 141(9): 3072–9. [DOI] [PubMed] [Google Scholar]
- 32.Lotta LA, Mokrosiński J, de Oliveira EM, et al. Human gain-of-function MC4R variants show signaling bias and protect against obesity. Cell 2019; 177(3): 597–607. e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Farooqi IS, Keogh JM, Yeo GS, Lank EJ, Cheetham T, O’Rahilly S. Clinical spectrum of obesity and mutations in the melanocortin 4 receptor gene. New England Journal of Medicine 2003; 348(12): 1085–95. [DOI] [PubMed] [Google Scholar]
- 34.Bardia A, Holtan SG, Slezak JM, Thompson WG. Diagnosis of obesity by primary care physicians and impact on obesity management. Mayo Clinic Proceedings; 2007: Elsevier; 2007. p. 927–32. [DOI] [PubMed] [Google Scholar]
- 35.Mocarski M, Tian Y, Smolarz BG, McAna J, Crawford A. Use of International Classification of Diseases, Ninth Revision Codes for Obesity: Trends in the United States from an Electronic Health Record-Derived Database. Popul Health Manag 2018; 21(3): 222–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gupta PP, Fonarow GC, Horwich TB. Obesity and the obesity paradox in heart failure. Canadian Journal of Cardiology 2015; 31(2): 195–202. [DOI] [PubMed] [Google Scholar]
- 37.Vashistha T, Mehrotra R, Park J, et al. Effect of age and dialysis vintage on obesity paradox in long-term hemodialysis patients. American journal of kidney diseases 2014; 63(4): 612–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Stovitz SD, Banack HR, Kaufman JS. Structural bias in studies of cardiovascular disease: let’s not be fooled by the “obesity paradox”. Canadian Journal of Cardiology 2018; 34(5): 540–2. [DOI] [PubMed] [Google Scholar]
- 39.Yancy CW, Benjamin EJ, Fabunmi RP, Bonow RO. Discovering the full spectrum of cardiovascular disease: Minority Health Summit 2003: executive summary. Circulation 2005; 111(10): 1339–49. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.