

Summary
How exercise elicits systemic metabolic benefits in both muscles and non-contractile tissues is unclear. Autophagy is a stress-induced lysosomal degradation pathway that mediates protein and organelle turnover and metabolic adaptation. Exercise activates autophagy in not only contracting muscles but also non-contractile tissues including the liver. However, the role and mechanism of exercise-activated autophagy in non-contractile tissues remain mysterious. Here we show that hepatic autophagy activation is essential for exercise-induced metabolic benefits. Plasma or serum from exercised mice is sufficient to activate autophagy in cells. By proteomic studies, we identify fibronectin (FN1), which was previously considered as an extracellular matrix protein, as an exercise-induced, muscle-secreted, autophagy-inducing circulating factor. Muscle-secreted FN1 mediates exercise-induced hepatic autophagy and systemic insulin sensitization, via the hepatic receptor α5β1 integrin and the downstream IKKα/β-JNK1-BECN1 pathway. Thus, we demonstrate that hepatic autophagy activation drives exercise-induced metabolic benefits against diabetes, via muscle-secreted soluble FN1 and hepatic α5β1 integrin signaling.
Keywords: Autophagy, exercise, muscle, liver, insulin sensitivity, fibronectin, integrin, ATG7, BECN1
Graphical Abstract
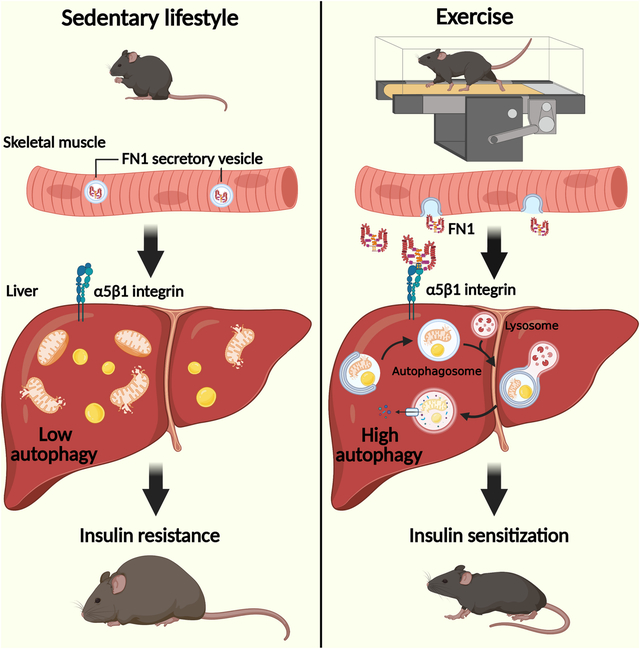
eTOC Blurb:
Kuramoto et al. uncovers a unique mechanism by which exercise elicits health benefits systemically. Contracting muscle secretes FN1/fibronectin, which activates liver autophagy, a degradative pathway for nutrient mobilization and damage removal, via hepatic α5β1 integrin receptor signaling. Activated autophagy in the non-contractile organ is essential for exercise-induced systemic metabolic benefits.
Introduction
Physical exercise exerts a broad spectrum of beneficial effects against chronic diseases, including type 2 diabetes (T2D), cardiovascular diseases and metabolic syndromes1–3. Contrarily, sedentary lifestyle and physical inactivity are causal factors of T2D development4,5. Upon muscle contraction, exercise facilitates muscle adaptation in morphology and function, and more importantly improves the functions of many non-contractile tissues systemically. However, the molecular mechanisms by which exercise elicits systemic metabolic benefits are largely unknown, limiting the potential of developing therapeutic interventions against metabolic disorders.
Autophagy is an essential lysosomal degradation pathway that breaks down and recycles damaged or unnecessary intracellular structures by fusion of autophagosome vesicles with lysosomes6–8. Autophagy occurs at a low basal level under normal conditions, and its activity is induced by a variety of stressors, such as fasting9,10. Autophagy abnormality has recently been implicated in many metabolic disorders11–14. We and others previously discovered that exercise is a potent inducer of autophagy in skeletal muscle in both rodents and humans15–17. Intriguingly, in addition to contracting muscle, we found that exercise also activates autophagy systemically in non-contractile tissues, including the liver, pancreas and adipose tissue16. This is puzzling because during exercise, non-contractile tissues do not experience the autophagy-inducing stressors as contracting muscles do, such as energetic stress18,19, oxidative stress20, and increased intracellular Ca2+ 21. Thus, how autophagy is activated in non-contractile tissues during/post exercise, and the functions of such autophagy activation, are important open questions to investigate. Physiologically, studies using two global autophagy-deficient mouse models15,16, the global Becn1/Beclin 1+/− knockout mice and Bcl2AAA mice, support that whole-body autophagy activation is key for exercise-induced improvements in exercise endurance and glucose tolerance, respectively. However, because the previous studies have only focused on the effects of exercise on autophagy in skeletal muscle6,22,23, the mechanism and the physiological importance of exercise-activated autophagy in non-contractile tissues remain mysterious.
Here we demonstrate that exercise-activated autophagy in the non-contractile liver mediates exercise-induced health benefits against T2D, via exercise-elicited, muscle-secreted fibronectin (FN1) and the hepatic FN1 receptor α5β1 integrin. Through cell biology and proteomic analyses, muscle-specific FN1 depletion, and liver-specific α5 integrin depletion in mouse models, we discover a previously uncharacterized FN1-α5β1 integrin-IKK-JNK1-BECN1 pathway that regulates exercise-induced muscle-liver communication, systemic autophagy activation, and whole-body metabolic improvement.
Results
Hepatic autophagy activation is essential for exercise-induced metabolic benefits
To study the tissue-specific function of exercise-induced autophagy, especially in non-contractile tissues, we generated a hepatocyte-specific knockdown (KD) model of the essential autophagy gene ATG7 (ATG7Δliver mice), via AAV2/8 (adeno-associated virus 2/8) delivery of shATG7 driven by the hepatocyte-specific TBG (human thyroxine-binding globulin) promoter into wild-type (WT) mice (Fig. 1A). The advantage of AAV-mediated gene KD over breeding with a Cre line is that gene inhibition occurs in adulthood, avoiding metabolic phenotypes that are developmental or in utero. Mice injected with the empty virus AAV2/8-TBG-Null were used as control. ATG7 depletion in hepatocytes leads to inhibition of autophagy, evidenced by accumulation of the autophagy cargo receptor SQSTM1/p62 and reduced lipidation of the autophagosome marker protein LC3-II in whole liver lysates (Fig. S1A). Immunohistochemistry staining revealed that SQSTM1/p62 accumulation is even, not zonal, throughout the liver parenchyma (Fig. S1B), further validating that autophagy inhibition is uniform in the liver of ATG7Δliver mice.
Figure 1. Hepatic autophagy activation is essential for exercise-induced insulin sensitization against high-fat diet (HFD).
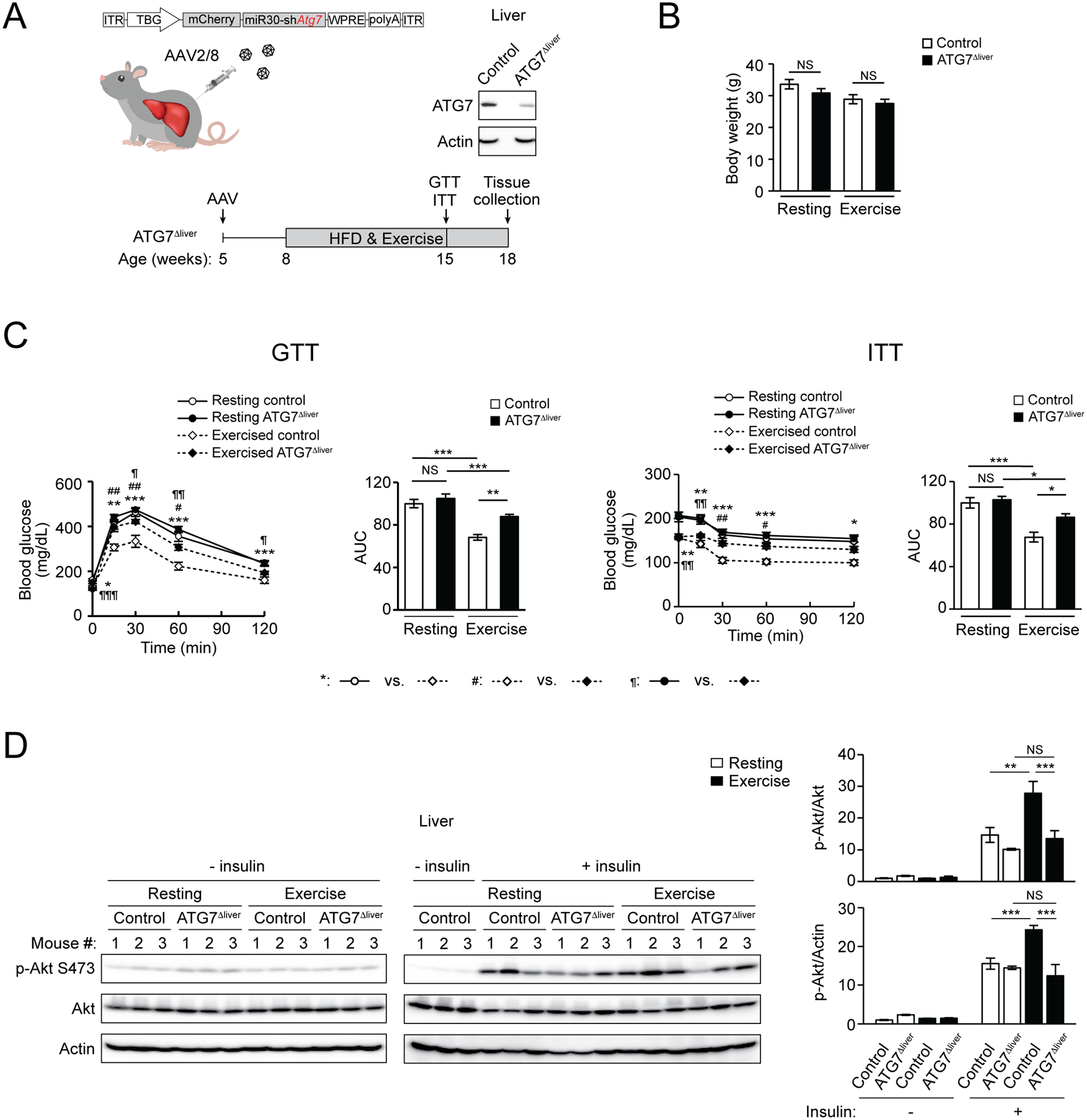
(A) Hepatic ATG7 knockdown efficiency and experimental design using ATG7Δliver mice. WT mice were intravenously (i.v.) injected with control AAV (control mice) or AAV2/8-miR30-TBG promoter-mCherry-shATG7 (ATG7Δliver mice). The remaining ATG7 expression in ATG7Δliver mice is likely derived from other non-hepatocyte cell types in the liver. (B) Comparable body weight of control and ATG7Δliver mice under HFD feeding, with or without daily exercise for 7 weeks. N=5–12. (C) Glucose tolerance test (GTT) and insulin tolerance test (ITT) of control and ATG7Δliver mice fed with HFD with or without daily 50-min treadmill exercise for 7 weeks. AUC, area under the curve. N=5–12. (D) Western blot (WB) analysis of insulin-stimulated Akt phosphorylation/activation in the liver of control and ATG7Δliver mice fed with HFD with or without daily 50-min exercise for 7 weeks, and then injected with 2 U/kg insulin 15 min prior to tissue collection. The “- insulin resting” controls were loaded twice on both gels to allow for normalization of samples to the same controls. N=3. One-way ANOVA with Tukey-Kramer test. *, #, ¶, P<0.05; **, ##, ¶¶, P<0.01; ***, ¶¶¶, P<0.001; NS, not significant.
Under regular diet (RD) feeding, ATG7Δliver mice have similar body weight (Fig. S1C), normal glucose tolerance (by glucose tolerance test, GTT) and slight insulin intolerance (by insulin tolerance test, ITT) (Fig. S1D), compared to control mice. However, after concurrent high-fat diet (HFD) feeding and daily treadmill exercise training, despite comparable body weight (Fig. 1B), exercise endurance (Fig. S1E), and liver and muscle tissue weight (Fig. S1F) as control mice, ATG7Δliver mice failed to show exercise-induced improvement in either glucose tolerance or insulin sensitivity (Fig. 1C), suggesting that disrupting exercise-stimulated autophagy specifically in the liver impairs exercise-induced insulin sensitization. Furthermore, hepatic insulin signaling, analyzed by insulin-stimulated phosphorylation of Akt downstream of the insulin receptor, failed to show enhancement in ATG7Δliver mice as in control mice after daily exercise (Fig. 1D). Together, these data suggest that autophagy activation in the non-contractile liver during exercise is essential for exercised-induced metabolic benefits against HFD.
Exercise induces the secretion of pro-autophagy FN1
We aimed to resolve how autophagy is activated in the liver by exercise. Using reporter cells expressing the autophagosome marker GFP-LC3, we found that when added to the culture medium, the plasma of exercised WT mice, but not that of resting WT mice, is sufficient to activate the autophagy flux, evidenced by increased accumulation of GFP-LC3 puncta when autophagosomal degradation is blocked by the lysosomal inhibitor chloroquine (Fig. 2A). In addition, the serum of exercised WT mice also elevates the autophagy flux, demonstrated by increased accumulation of red puncta in reporter cells expressing a pH-sensitive tandem-tagged mCherry-GFP-LC324,25 (Fig. S2A), where acid-sensitive GFP is quenched once delivered in lysosomes while acid-insensitive mCherry remains stable. These data suggest that the plasma or serum of exercised mice can activate autophagy non-cell autonomously. Given that exercise may reduce serum insulin levels16 and that the insulin signaling pathway inhibits autophagy10, we first studied whether insulin plays a role in autophagy activation by exercised serum. Yet even with supplementation of excess high levels of exogenous insulin (10–20 times of the endogenous circulating insulin level), exercised serum still induces the autophagy flux in cultured reporter cells expressing mCherry-GFP-LC3 (Fig. 2B), suggesting that exercise-induced systemic autophagy is not caused by reductions in circulating insulin levels. In addition, exercise potentially activates AMPK (AMP-activated protein kinase), a master regulator of energy metabolism and an autophagy inducer, in skeletal muscle, but does not do so in the liver (Fig. S2B), suggesting that exercise-induced AMPK activation is not the cause of autophagy activation in non-contractile tissues. Thus, we propose that additional new exercise-induced circulating factors mediate hepatic autophagy activation during exercise.
Figure 2. Identification of FN1 as an exercise-induced, muscle-secreted, autophagy-inducing factor.
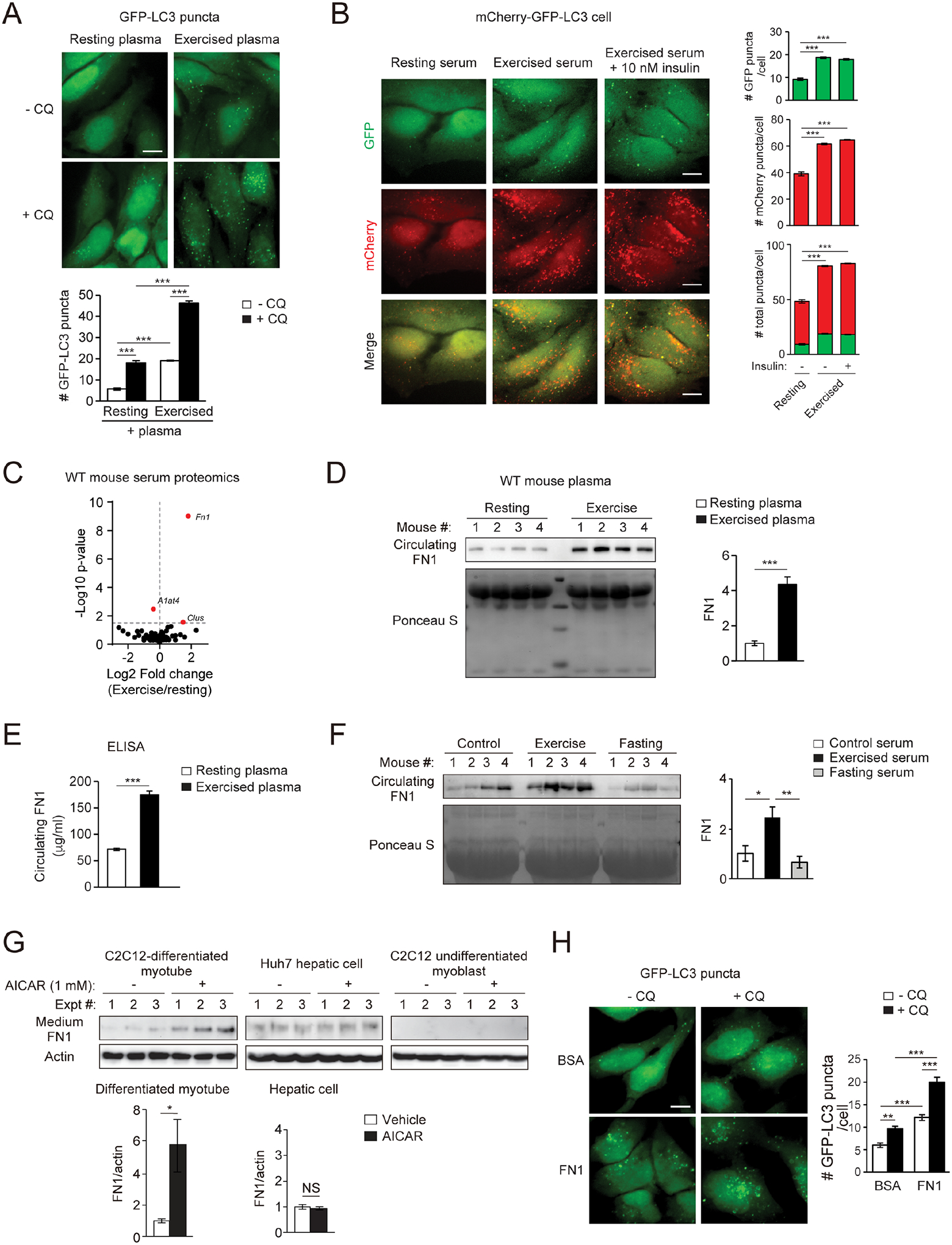
(A) Representative images and quantification of autophagosomes (GFP-LC3 puncta) in HeLa cells stably expressing the GFP-LC3 reporter cultured for 3 h in medium containing 10% plasma from WT mice at rest or after 90-min treadmill running, with or without the lysosomal inhibitor chloroquine (CQ, 10 μM). Bar, 10 μm. N=3 mice. 50 cells per mouse were analyzed. (B) Representative images and quantification of autolysosomes (red) and autophagosomes (green) in HeLa cells stably expressing tandem mCherry-GFP-LC3 cultured for 3 h in medium containing 10% serum from WT mice at rest or after 90-min treadmill running with or without supplementation of 10 nM insulin. N=3 mice (30 cells/mouse serum treatment). Bar, 10 μm. (C) Volcano plot of mass spectrometry analysis on serum of WT mice at rest or after 90-min exercise. (D) WB of circulating plasma FN1 in WT mice at rest or after 90-min exercise. N=4. (E) ELISA of plasma FN1 in WT mice at rest or after 90-min exercise. N=10. (F) WB analysis of circulating FN1 in WT mice under fed and rested conditions, or after 90-min exercise or 24-h fasting. N=4. (G) WB analysis of FN1 in the conditioned medium of C2C12-differentiated myotubes, Huh7 hepatic cells, and C2C12 undifferentiated myoblasts treated with vehicle or 1 mM AICAR (AMPK activator) for 1 h. N=3. (H) Representative images and quantification of GFP-LC3 puncta in GFP-LC3 HeLa cells treated with or without FN1 or BSA (bovine serum albumin) (100 μg/ml) and the lysosomal inhibitor chloroquine (CQ) for 3 h. Bar, 10 μm. N=50 cells/condition. (A-B, F, H), one-way ANOVA with Tukey-Kramer test. (D-E, G), t-test. *, P<0.05; **, P<0.01; ***, P<0.001; NS, not significant.
To identify such factors, we performed proteomic analyses on the serum of WT mice before and after exercise, and identified a candidate, FN1 (Fig. 2C), which is previously considered as an extracellular matrix (ECM) protein primarily secreted from fibroblasts and hepatocytes under normal resting conditions26. We found that circulating FN1 is significantly increased in WT mice by exercise, demonstrated by Western blot (WB) analyses of both plasma (Fig. 2D) and serum (Fig. S2C) samples. ELISA studies also confirmed elevated post-exercise plasma FN1 levels in WT mice (Fig. 2E). Importantly, such increase in circulating FN1 is specifically induced by exercise, but not by fasting (Fig. 2F). Notably, FN1 transcription is not significantly increased in either muscle or liver after exercise (Fig. S2D), consistent with the fact that exercise-induced FN1 secretion is rapid (through immediate testing after 60–90 min of exercise), which is not sufficient to allow for mRNA transcription and protein synthesis. Using the AMPK activator AICAR to mimic exercise conditions in cell culture systems, we found that exercise-mimicking conditions induce FN1 secretion to the medium, but only in differentiated myocytes and not in hepatic cells or undifferentiated myoblasts (Fig. 2G). Furthermore, Sec6, a component of the exocyst machinery, which is a multiprotein complex facilitating plasma-membrane docking of secretory vesicles, is required for AICAR-induced FN1 secretion from myotubes (Fig. S2E). In addition, two paralogue Rab GTPase-activating proteins (RabGAPs), TBC1D1 and TBC1D4 (also known as Akt Substrate of 160 kDa, AS160), which are phosphorylated and inhibited by active AMPK and exercise27, also partially regulate FN1 secretion from differentiated myotubes in response to the exercise-mimetic stimulus AICAR (Fig. S2F). Thus, although FN1 can be secreted from multiple cell types under resting conditions, in response to exercise, its secretion is stimulated from myocytes and is triggered by AMPK activation and regulated by AMPK-modulated RabGAPs and the exocyst machinery. Furthermore, purified FN1 (within the physiological range of circulating concentrations), but not the same concentration of bovine serum albumin (BSA), induces the autophagy flux under nutrient-rich conditions, using the lysosomal inhibitor chloroquine to block autophagic degradation in GFP-LC3 reporter cells (Fig. 2H) and using tandem mCherry-GFP-LC3 reporter cells (Fig. S2G). Thus, taken together, we identified an exercise-induced circulating factor FN1 that can activate autophagy non-cell autonomously.
FN1 secreted from contracting muscle mediates exercise-induced hepatic autophagy and systemic insulin sensitization
To further study the function of muscle-derived FN1 during exercise, we generated muscle-specific FN1-depletion (FN1Δmuscle) mice, by AAV2/9-mediated delivery of FN1 shRNA driven by the muscle-specific MHCK7 promoter28 into WT or GFP-LC3 reporter mice29–32 (Fig. 3A). Muscle-specific depletion of FN1 almost completely abolished acute exercise-induced increase of circulating FN1 in FN1Δmuscle mice (Fig. 3B), suggesting that during exercise, increased circulating FN1 is indeed secreted from the muscle. Importantly, muscle-specific FN1 depletion does not affect exercise-induced muscle autophagy (Fig. 3C), muscle morphology (Fig. S3A), or maximal exercise capacity (Fig. 3D), but prevents exercise-induced autophagy in the liver (Fig. 3E). These data suggest that muscle-secreted FN1 is required for muscle-liver communication and hepatic autophagy activation during exercise.
Figure 3. FN1 secreted from contracting muscle mediates exercise-induced hepatic autophagy and systemic insulin sensitization.
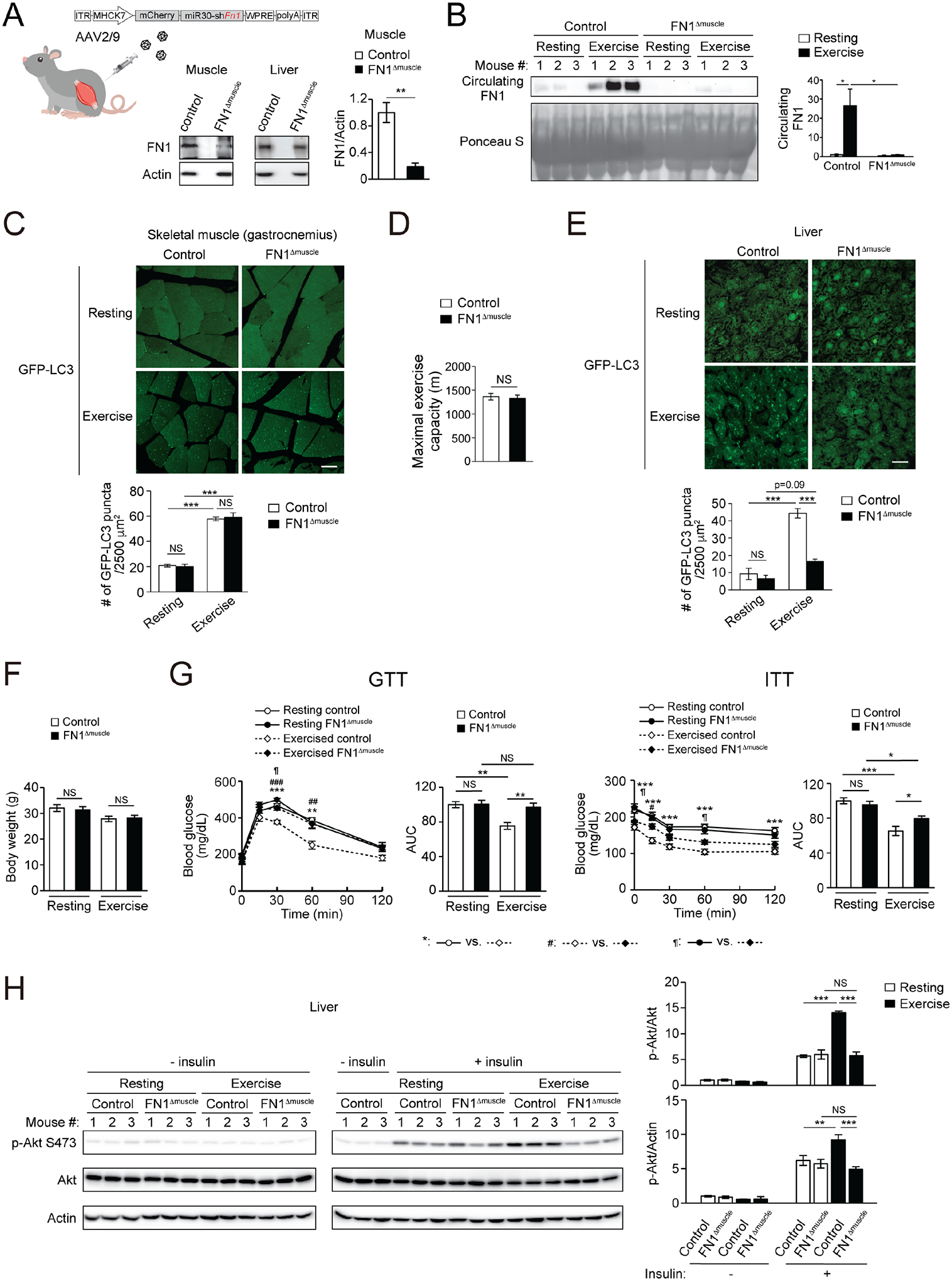
(A) FN1 levels in skeletal muscle and liver of WT mice i.v. injected with control AAV (control mice) or AAV2/9-MHCK7 promoter-mCherry-miR30-shFN1 (FN1Δmuscle mice). N=3–4. (B) Circulating FN1 levels of control mice or FN1Δmuscle mice at rest or after 90-min exercise. N=3. (C) GFP-LC3 puncta in skeletal muscle of control or FN1Δmuscle mice expressing the GFP-LC3 reporter at rest or after 90-min exercise. N=3 mice (15–16 areas/mouse). Bar, 25 μm. (D) Maximal running distance of control or FN1Δmuscle mice on a treadmill. N=6 mice. (E) GFP-LC3 puncta in the liver of control or FN1Δmuscle mice expressing GFP-LC3 at rest or after 90-min exercise. N=3 mice (12 areas/mouse). Bar, 25 μm. (F) Comparable body weight of control and FN1Δmuscle mice under HFD feeding, with or without daily exercise for 7 weeks. N=7–9. (G) GTT and ITT of control or FN1Δmuscle mice fed with HFD with or without daily 50-min exercise for 7 weeks. AUC, area under the curve. N=6–9. (H) WB analysis of insulin-stimulated Akt phosphorylation in the liver of control or FN1Δmuscle mice fed with HFD with or without daily 50-min exercise for 7 weeks, and then injected with 2 U/kg insulin 15 min prior to tissue collection. The “- insulin resting” controls were loaded twice on both gels to allow for normalization of samples to the same controls. N=3. (A, D), t-test. (B-C, E-H), one-way ANOVA with Tukey-Kramer test. *, ¶, P<0.05; **, ##, P<0.01; ***, ###, P<0.001; NS, not significant.
Under RD feeding, FN1Δmuscle and control mice showed comparable body weight (Fig. S3B), GTT and ITT (Fig. S3C). However, after concurrent HFD feeding and exercise training (Fig. S3D), although control and FN1Δmuscle mice have similar body weight (Fig. 3F) and liver and muscle tissue weight (Fig. S3E), daily exercise training only improves GTT and ITT values in HFD-fed control mice, but fails to do so in HFD-fed FN1Δmuscle mice (Fig. 3G), suggesting that muscle-derived FN1 is essential for exercise-induced systemic insulin sensitization. In addition, control mice showed exercise-induced improvements in insulin signaling (insulin-stimulated Akt activation) in both muscle and liver; however, FN1Δmuscle mice only showed exercise-improved insulin signaling in the muscle (Fig. S3F), but not in the liver (Fig. 3H), suggesting that muscle-secreted FN1 is important for exercise-induced insulin sensitization in the liver, rather than in the muscle. Thus, taken together, exercise-induced muscle-secreted FN1 non-cell autonomously upregulates autophagy activity and insulin sensitivity in the non-contractile liver.
Muscle-secreted FN1 activates hepatic autophagy via the α5β1 integrin-IKKα/β-JNK1 pathway
One of the integrin family members, α5β1 integrin, is suggested as a receptor for FN126,33,34. Integrins are cell-surface receptors formed by different combinations of an α subunit and a β subunit. They function in cell adherence and signaling and contribute to cell survival and cancer cell migration35. However, their roles in the regulation of cellular and systemic metabolism are unknown. We first studied whether integrins play a role in exercise-induced autophagy. KD of either the α5 (ITGA5) or β1 (ITGB1) subunit of integrin abolishes the autophagy flux activated by serum of exercised mice in reporter cells (Fig. 4A), suggesting that the α5β1 integrin is required for exercise-induced autophagy. In addition, a neutralizing antibody of ITGB1 also blocks the autophagy flux induced by serum from exercised mice in GFP-LC3 reporter cells (Fig. S4A), further supporting that activation of the ITGB1 integrin pathway is responsible for exercised serum-induced autophagy. In contrast, KD of α5β1 integrin does not affect basal or fasting-induced autophagy flux (Fig. 4B, Fig. S4B). These data suggest that α5β1 integrin regulates exercise-induced autophagy, but is not involved in the regulation of basal or fasting-induced autophagy.
Figure 4. Muscle-secreted FN1 activates hepatic autophagy via the α5β1 integrin-IKKα/β-JNK1-BECN1 pathway.
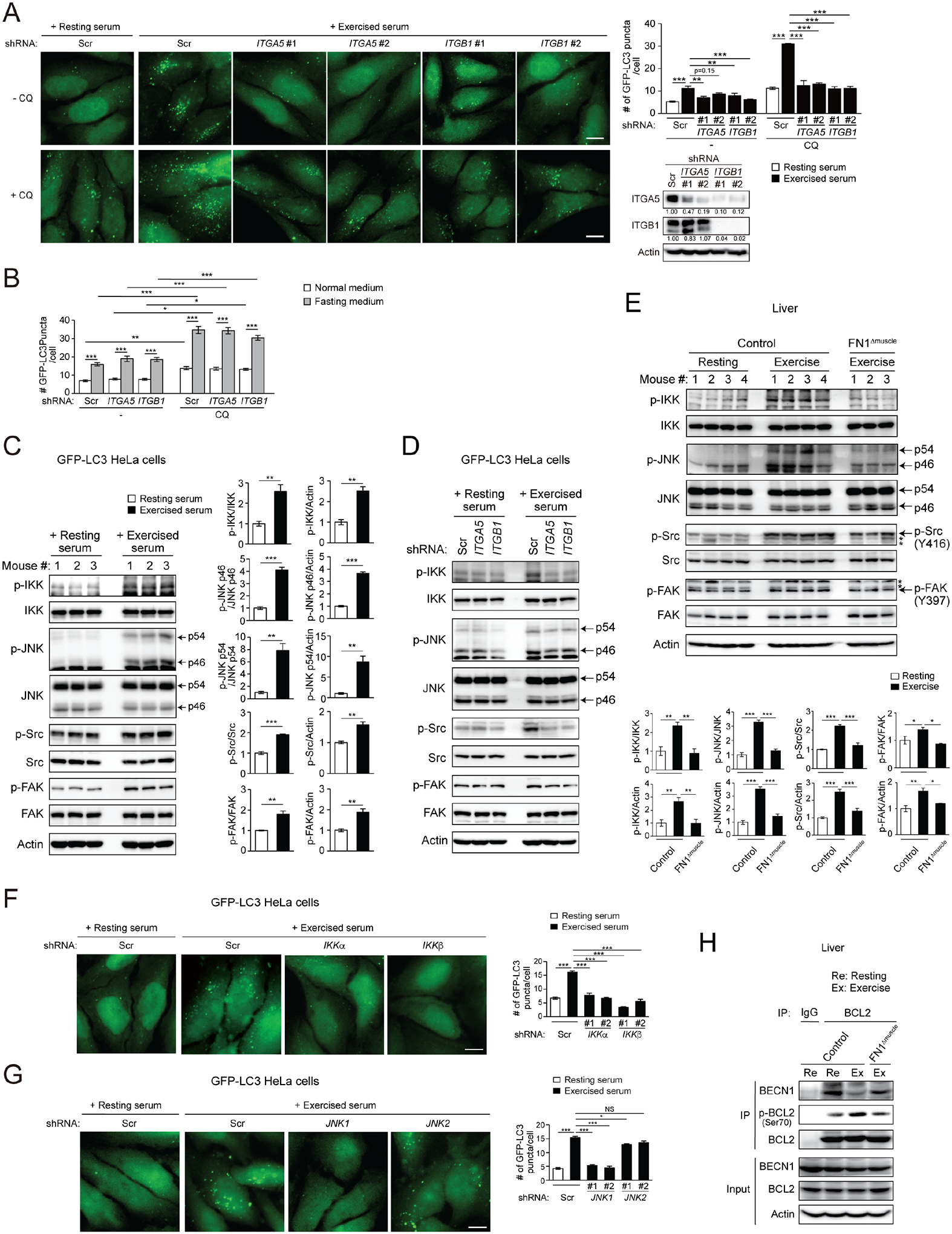
(A) Representative images and quantification of autophagosomes (GFP-LC3 puncta) in GFP-LC3 HeLa cells stably expressing scrambled (Scr), integrin α5 (ITGA5), or integrin β1 (ITGB1) shRNA cultured in medium containing 10% serum from resting or exercised WT mice in the presence or absence of the lysosomal inhibitor chloroquine (CQ) for 3 h. N=3–4 mice (50 cells/group). Bar, 10 μm. (B) Quantification of GFP-LC3 puncta in GFP-LC3 HeLa cells expressing scrambled (Scr), ITGA5 or ITGB1 shRNA cultured in normal or fasting (EBSS) medium in the presence or absence of chloroquine (CQ) for 3 h. N=50 cells. (C) WB analysis of IKK, JNK1/2, Src and FAK phosphorylation in GFP-LC3 HeLa cells cultured in medium containing 10% serum from resting or exercised WT mice for 15 min. N=3. (D) WB analysis of IKK, JNK, Src and FAK phosphorylation in GFP-LC3 HeLa cells stably expressing scrambled (Scr), integrin α5 (ITGA5), or integrin β1 (ITGB1) shRNA cultured in medium containing 10% serum from resting or exercised WT mice for 15 min. (E) WB analysis of phosphorylation of IKK, JNK, Src and FAK in the liver of control or FN1Δmuscle mice at rest or after 90-min exercise. *, non-specific band. N=3–4. (F-G) Representative images and quantification of GFP-LC3 puncta in GFP-LC3 HeLa cells stably expressing Tet-inducible scrambled (Scr), IKKα or IKKβ (F), and JNK1 or JNK2 (G) shRNA treated with Tet for 72 h and then cultured with 10% serum from resting or exercised WT mice for 3 h. N=3 mice (50 cells/group). Bar, 10 μm. (H) Co-immunoprecipitation of BECN1 with BCL2, and phosphorylation of BCL2 at Ser70, in the liver of control and FN1Δmuscle mice at rest or after exercise. (A-B, E-G), one-way ANOVA with Tukey-Kramer test. (C), t-test. *, P<0.05; **, P<0.01; ***, P<0.001; NS, not significant.
Furthermore, the IκB kinase (IKK) complex, composed of an α subunit (IKKα) and a β subunit (IKKβ), is recently identified as a key signaling molecule downstream of integrins. IKK is activated by mechano-stress such as stretch and detachment36, and is involved in fasting- and cell detachment-induced autophagy37–39. Because IKK activation further leads to phosphorylation of JNK1/2 (c-Jun N-terminal protein kinase 1/2), and the ability of IKK to induce autophagy is JNK-dependent38, we proposed that JNK may act downstream of IKK to transduce input from integrins into intracellular biochemical signals. Indeed, we found that serum from exercised mice, but not from resting mice, activates IKK and JNK, as well as the canonical FN1-integrin signaling molecules such as phosphorylation of Src and FAK, in autophagy reporter cells (Fig. 4C), which are dependent on ITGA5 and ITGB1 (Fig. 4D), supporting that exercise-induced serum indeed stimulates α5β1 integrin-mediated cell signals. Furthermore, treatment of purified FN1 also activates both IKK and JNK in Huh7 hepatic cells (Fig. S4C), and double knockdown of IKKα and IKKβ abolishes FN1-induced JNK activation in Huh7 cells (Fig. S4D), supporting our hypothesis that FN1-mediated JNK activation is through IKKα/β in hepatocytes. In addition, in vivo, exercise activates IKK, JNK, Src and FAK, in the liver of control mice, but fails to do so in FN1Δmuscle mice (Fig. 4E). These data suggest that exercise-induced muscle-secreted FN1 activates the hepatic integrin-IKK-JNK pathway.
We further found that KD of either IKKα or IKKβ, and KD of JNK1 but not JNK2 (Fig. 4F–G, Fig. S4E), prevent exercised serum-induced autophagosome formation, suggesting that IKKα/β and JNK1, but not JNK2, are required for exercise-induced autophagy. This is reminiscent of fasting or ceramide treatment conditions in vitro, where JNK1, but not JNK2, regulates the reversible binding between the essential autophagy protein BECN1 and its inhibitor BCL2, by phosphorylating BCL2 at sites including Ser70 and releasing BECN1 from phosphorylated BCL2 for autophagy activation40–42. We previously found that in vivo, exercise induces BECN1 dissociation from BCL2 in skeletal muscle16, and asked whether it also occurs in non-contractile tissues as a cellular mechanism of autophagy activation. Via co-immunoprecipitation, we found that exercise induces phosphorylation of BCL2 at Ser70 and decreases BECN1-BCL2 binding in the liver of control mice; however, these exercise-induced effects are largely reduced in the liver of FN1Δmuscle mice (Fig. 4H), suggesting that besides in muscles, exercise also promotes BCL2 phosphorylation and releases BECN1 from the inhibitory binding of BCL2 in non-contractile tissues, which is mediated by muscle-secreted FN1. Taken together, we conclude that muscle-secreted FN1 upregulates autophagy via activating the α5β1 integrin-IKK-JNK1 pathway.
Hepatic α5 integrin is essential for exercise-induced IKK-JNK1-BECN1 activation, hepatic autophagy, and systemic metabolic benefits
To further study the role of the α5β1 integrin signaling pathway in the regulation of exercise-induced autophagy and metabolic benefits in non-contractile tissues such as liver, we generated ITGA5Δliver mice, which lack the α5 integrin subunit specifically in the liver via AAV2/8-mediated delivery of shITGA5 driven by the TBG promoter (Fig. 5A). We targeted ITGA5 but not ITGB1 because ITGB1 is a common subunit of different integrins and ITGA5 is specific for the FN1 receptor. Similar to control mice, both ITGA5Δliver mice and autophagy-deficient ATG7Δliver mice have normal exercise-induced circulating FN1 levels (Fig. 5B). However, different from control or ATG7Δliver mice, which have normal exercise-induced activation of IKK, JNK, Src and FAK (Fig. 5C) and BECN1-BCL2 dissociation (Fig. 5D) in the liver, ITGA5Δliver mice show defective IKK, JNK, Src and FAK activation and BECN1-BCL2 dissociation in the liver after exercise, suggesting that ITGA5 functions upstream of IKK, JNK and BECN1 to mediate their activation in the liver, while ATG7 acts downstream of or in parallel with the IKK-JNK-BECN1 pathway. Depletion of either ITGA5 or ATG7 in the liver significantly reversed exercise-induced reduction in the level of the autophagy substrate SQSTM1/p62 (Fig. 5C), confirming that both ITGA and ATG7 function in exercise-induced autophagy in the liver. Whereas different from ATG7Δliver mice that are defective in both exercise- and fasting-induced hepatic autophagy, ITGA5Δliver mice show defects specifically in exercise-induced autophagy, but not fasting-induced autophagy, in the liver (Fig. 5E), which is consistent with the role of ITGA5 in autophagy in vitro (Fig. 4A–B, Fig. S4B). Metabolically, after RD feeding and 3 weeks of HFD feeding without exercise, ITGA5Δliver mice and control mice showed comparable body weight gain, GTT and ITT (Fig. S5A–C). However, after concurrent HFD feeding and daily exercise, despite comparable body weight (Fig. 5F), exercise endurance, and tissue weight (Fig. S5D–E) in control and ITGA5Δ mice, daily exercise training improves glucose tolerance, insulin tolerance and hepatic insulin signaling (insulin-stimulated Akt activation) only in control mice, but fails to do so in ITGA5Δ mice (Fig. 5G–H, Fig. S5F). These data suggest that hepatic α5β1 integrin plays an essential role in regulating exercise-induced insulin sensitization. Taken together, we demonstrate that hepatic α5β1 integrin mediates exercise-stimulated IKK-JNK activation, BECN1-BCL2 dissociation, exercise (but not fasting)-induced hepatic autophagy, and metabolic benefits of exercise.
Figure 5. Hepatic α5 integrin is essential for exercise-induced IKK-JNK1-BECN1 activation, hepatic autophagy, and systemic metabolic benefits.
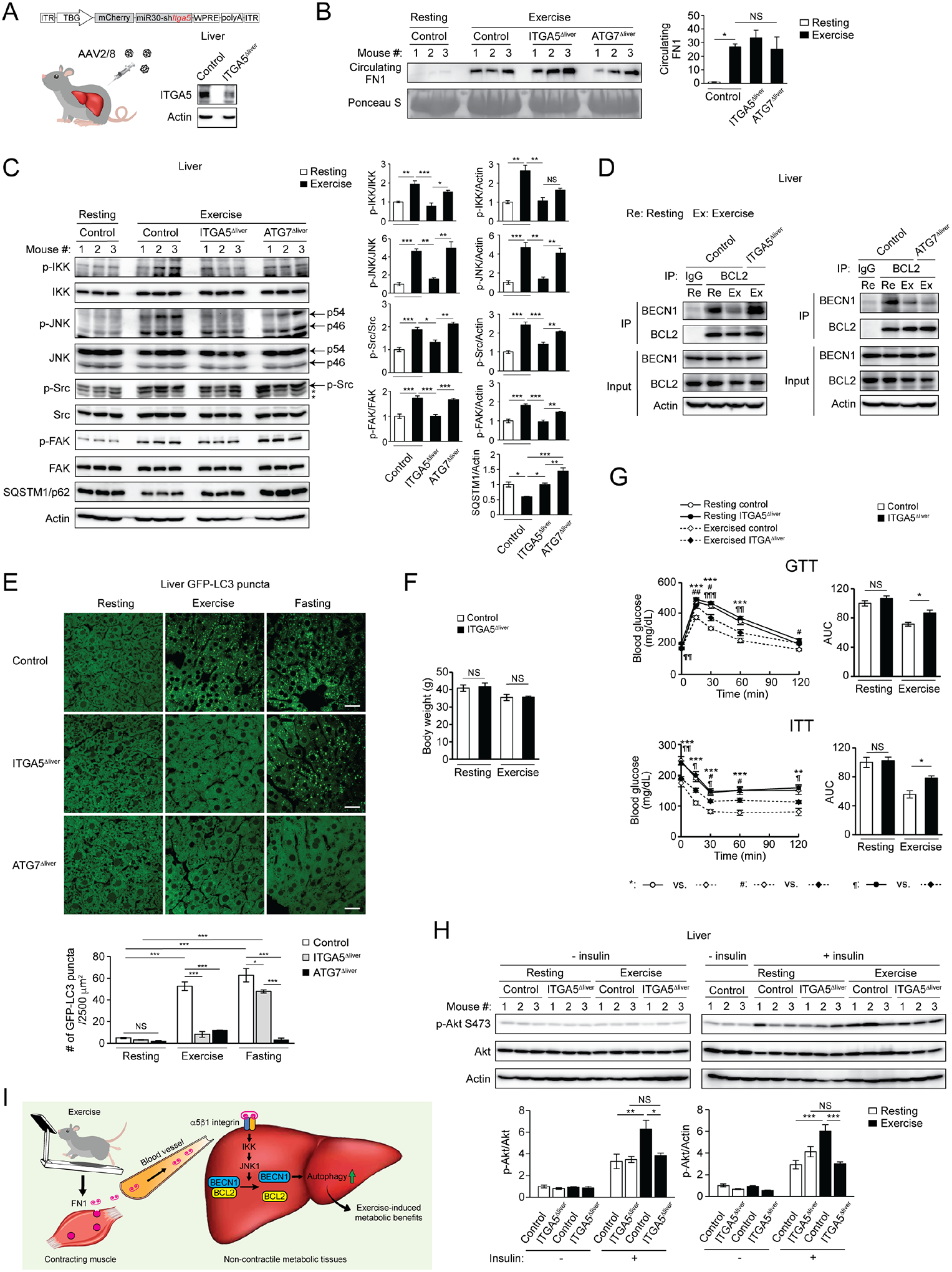
(A) ITGA5 expression in the liver of WT mice i.v. injected with control AAV (control mice) or AAV2/8-miR30-TBG promoter-mCherry-shITGA5 (ITGA5Δliver mice). The remaining ITGA5 expression in ITGA5Δliver mice is likely derived from non-hepatocyte cell types in the liver. (B) WB analysis of circulating FN1 levels in control, ITGA5Δliver and ATG7Δliver mice at rest or after 90-min exercise. N=3. (C) WB analysis of phosphorylation of IKK, JNK, Src and FAK, and levels of SQSTM1/p62, in the liver of control, ITGA5Δliver and ATG7Δliver mice at rest or after 90-min exercise. *, non-specific band. N=3. (D) Co-immunoprecipitation of BECN1 with BCL2 in the liver of control, ITGA5Δliver and ATG7Δliver mice at rest or after exercise. (E) Representative images and quantification of GFP-LC3 puncta in the liver of control, ITGA5Δliver and ATG7Δliver mice at rest, after 90-min exercise, or after 24-h fasting. Bar, 25 μm. N=3 mice (12 areas/mouse). (F) Comparable body weight of control and ITGA5Δliver mice under HFD feeding, with or without daily exercise for 7 weeks. N=5–9. (G) GTT and ITT of control or ITGA5Δliver mice fed with HFD with or without daily 50-min exercise for 7 weeks. AUC, area under the curve. N=5–9. (H) WB analysis of insulin-stimulated Akt phosphorylation in the liver of control and ITGA5Δliver mice fed with HFD with or without daily exercise for 7 weeks, and then injected with 2 U/kg insulin 15 min prior to tissue collection. The “- insulin resting” controls were loaded twice on both gels to allow for normalization of samples to the same controls. N=3. One-way ANOVA with Tukey-Kramer test. *, #, ¶, P<0.05; **, ##, ¶¶, P<0.01; ***, ¶¶¶, P<0.001; NS, not significant. (I) Proposed model of exercise-activated autophagy in non-contractile tissues. During exercise, FN1 is secreted from contracting muscle. Circulating FN1 is sensed by α5β1 integrin on non-contractile tissues (such as liver) and activates autophagy via IKK-JNK1 signaling and BECN1 release from BCL2, which drives exercise-induced systemic metabolic benefits against T2D.
Therefore, overall, we establish a model for exercise-induced autophagy in non-contractile tissues such as liver, in which hepatic autophagy activation drives exercise-induced metabolic benefits against T2D, through muscle-secreted FN1 and the FN1-α5β1 integrin-IKK-JNK1-BECN1 pathway (Fig. 5I).
Discussion
Autophagy is an evolutionarily conserved catabolic pathway to recycle and mobilize nutrients as building blocks for anabolic processes and as fuels for energy production under stress conditions. Its tissue-specific functions in different physiological and pathological conditions are yet to be revealed. In this study, our findings demonstrate the metabolic function and mechanism of exercise-activated autophagy in non-contractile tissues (liver), as an essential pathway by which exercise mediates its health benefits.
We found that muscle-derived soluble FN1 is a crucial regulator of exercise-induced hepatic autophagy, as well as hepatic and systemic insulin sensitization against HFD feeding. Muscle is an endocrine organ that releases a variety of myokines. Of note, a number of proteomic studies have contributed to the profiling of the muscle cell secretome43–46; however, none of the existing myokines have been identified as an effective inducer of autophagy post exercise. For example, interleukin-6 (IL-6), a reported exercise-induced myokine47, has controversial roles on autophagy activity. Both inhibitory and stimulatory effects of IL-6 on autophagy have been reported48–54. The autophagy-activating effect of IL-6 is primarily observed under basal (resting) conditions, but after exercise, IL-6 seems to inhibit autophagy and autophagy gene expression in hepatocytes50, likely via regulating the BECN1 inhibitor BCL248. In addition, irisin, another previously reported myokine that can lead to browning of white adipose tissue55, is not identified in our proteomics study. We reasoned that it is partly due to the small level of increase in circulating irisin (by 19%) even after high-intensity exercise56, compared to more than 2-fold increase in FN1. Thus, discovery and characterization of new factors and pathways that can activate autophagy systemically is helpful for understanding organ crosstalk during exercise. Our findings reveal such previously uncharacterized function of the FN1-integrin pathway, which was mainly studied in ECM formation and tumorigenesis, in regulating exercise-induced muscle-liver communication.
Our study also reveals that muscle-secreted FN1, but not nutrient deprivation (fasting), signals through α5β1 integrin on target non-contractile tissues to activate autophagy. Notably, α5β1 integrin is also recently reported to be involved in autophagy induction upon Group A Streptococcus infection57. Thus, α5β1 integrin may regulate autophagy activation in response to a variety of stresses under nutrient-rich conditions.
Overall, our study demonstrates that hepatic autophagy activated by contracting muscle-secreted FN1 drives exercise-induced insulin sensitization. In the study, we focus on the liver as an example of non-contractile tissues. It is possible that exercise-induced autophagy in other non-contractile tissues, such as adipose tissue, also contributes to the metabolic improvement of exercise, given that liver-specific depletion of ATG7 or ITGA5 does not completely abolish exercise-induced insulin sensitization. Thus, in the future, it is worthwhile to study the role of the α5β1 integrin-IKK-JNK-autophagy axis in other non-muscle metabolic tissues, to expand on the understanding of systemic autophagy activation and whole-body metabolism during exercise.
Limitations of the Study
One important question arising from our work is whether the findings in exercised mice are translatable to humans. In the future, it is important to collaborate with experts on population and exercise science in humans to address whether exercise (for example, treadmill running) induces circulating FN1, and if so, whether exercise-induced FN1 increase is defective in people with T2D and associated with the severity of diabetes progression. In addition, we speculate that simply overexpressing FN1 in muscle is not sufficient to activate hepatic autophagy, based on two findings: first, transcriptional upregulation of FN1 is not detected in muscle after exercise (Fig. S2D), and second, FN1 release requires the activation of AMPK and the secretory mechanism/pathway in the muscle (Fig. 2G and S2E–F). Although we found that AMPK-modulated RabGAPs and the exocyst machinery are required to stimulate FN1 secretion from differentiated myocytes, the regulatory link between AMPK activation and FN1 secretion is largely unknown. Future studies will focus on revealing the molecular mechanisms by which exercise induces FN1 secretion in contracting muscle.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for reagents and resources should be directed and will be fulfilled by the lead contact, Congcong He (congcong.he@northwestern.edu).
Materials availability
The plasmids used in this study are available from the lead contact.
Data and code availability
All original data for creating all graphs in the paper are provided in Data S1.
This study did not generate/analyze datasets/code.
Any additional information required to reanalyze the data reported in this paper is available from the Lead Contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse models
All mouse care and procedures were performed in accordance with animal experimental protocols approved by the Northwestern University Institutional Animal Care and Use Committee (IACUC). All mice were housed on a 14-hr/10-hr light/dark cycle with ad libitum access to chow diet and water. GFP-LC3 mice were described previously63. C57BL/6J mice (JAX#000664) were acquired from the Jackson Laboratory. All experiments were performed with sex- and age-matched mice. Diet-induced obese mice were generated by high-fat diet (D12492, Research Diets Inc) feeding. Regular diet-fed mice were analyzed at 8 weeks of age, and high-fat diet-fed mice were analyzed at 16 weeks of age. Exercise studies and autophagy analyses were performed in both male and female mice. To induce autophagy by starvation, mice were fasted for 24 hr with free access to water.
Cells
C2C12 myoblasts, Huh7 cells, HeLa cells, and HEK293T cells were cultured in culture medium: Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine serum (FBS), 100 units/ml penicillin, and 100 μg/ml streptomycin. For C2C12 myoblast differentiation to myotubes, fully confluent C2C12 myoblasts (day 0) were cultured in DMEM containing 2% horse serum, 100 units/ml penicillin and 100 μg/ml streptomycin, and refed with fresh medium every other day. On day 6 after differentiation, cells were utilized for experiments. For tetracycline-inducible shRNA knockdown, cells were treated with tetracycline at 1 μg/ml for 72 hr, then used for experiments. For starvation-induced autophagy, HeLa cells stably expressing GFP-LC364 were washed with PBS and cultured in EBSS starvation medium (1 × EBSS [E7510, MilliporeSigma], 2.2 g/L sodium bicarbonate) for 3 hr with or without 10 μM chloroquine (14194, Cayman Chemical). For FN1 treatment, HeLa cells stably expressing GFP-LC3 were treated with 25–100 μg/ml FN1 (F2006, MilliporeSigma) or BSA (A1595, MilliporeSigma) for 3 hr in nutrient-rich medium (Opti-MEM [31985, Thermo Fisher Scientific]/DMEM = 3:1, 5% FBS, 1 × non-essential amino acids [11140, Thermo Fisher Scientific], 2 mM glutamine). Huh7 cells were pre-cultured in DMEM containing 0.5% FBS for 6 hr and then treated with 100 μg/ml FN1 for 5 or 15 min.
METHOD DETAILS
AAV delivery of pAAV-miR30-shRNA
The AAV plasmids pAAV-MHCK7-miR30-mCherry-shFn1, pAAV-TBG-miR30-mCherry-shItga5 and pAAV-TBG-miR30-mCherry-shAtg7, and the AAV virus AAV2/9-shFn1, AAV2/8-shItga5 and AAV2/8-shAtg7, were custom-produced by VectorBuilder Inc. Control AAV2/8-TBG-PI-Null-bGH virus (#105536-AAV8) was purchased from Addgene. The virus titer of AAV2/9-shFn1, AAV2/8-shItga5, AAV2/8-shAtg7, and AAV2/8-Null were 3.19 × 1013 viral genomes (VG)/ml, 3.61 × 1013 VG/ml, 4.60 × 1013 VG/ml, and 1.70 × 1013 VG/ml, respectively. Viral aliquots were stored at −80°C until tail intravenous (i.v.) injection. 5-week-old mice were i.v. injected with AAV virus at 1 × 1012 VG/mouse for muscle-specific Fn1 knockdown and at 1 × 1011 VG/mouse for liver-specific Itga5 or Atg7 knockdown.
Treadmill exercise
Three weeks after AAV injection, exercise was performed using a 10° uphill Exer 3/6 open treadmill (Columbus Instruments). The acute exercise (total 90 min) was performed as described previously16 with some modifications65. Briefly, for acclimation, on day 1, mice ran for 5 min at 8 m/min, and on day 2, mice ran for 5 min at 8 m/min followed by another 5 min at 10 m/min. On day 3, mice were subject to acute running exercise starting at 12 m/min for 40 min. After 40 min, the treadmill speed was increased at the rate of 1 m/min every 10 min for 30 min and then every 5 min for 20 min.
Long-term exercise and maximal running distance measurement were performed as described previously16. Briefly, for long-term exercise, 3 weeks after AAV injection, mice were exercised for 50 min/day, 5 days/week at 17 m/min, following 2-day acclimation to treadmill running as described in acute exercise. ITGA5ΔLiver mice were HFD-pretreated for 3 weeks before long-term exercise training. Maximal running distance was measured following the acute exercise protocol as described above with some modifications: after 90-min acute exercise, the treadmill speed was increased at the rate of 1 m/min every 5 min till mice were exhausted.
Mass spectrometry analysis
Sera were collected from 3 resting or 3 exercised WT mice, and were pooled. High abundance serum proteins, albumin and IgG were removed from pooled serum samples using Proteome Purify 2 (MIDR002–020, R&D systems) according to the manufacturer’s protocol. Mass spectrometry analysis was performed in the Northwestern Proteomics Core Facility.
GTT and ITT
GTT and ITT were performed as previously described65,66. Briefly, mice were fasted for 6 hr and 4 hr prior to GTT and ITT, respectively. Glucose was intraperitoneally (i.p.) injected at 1.5 g/kg body weight. Insulin (I0516, MilliporeSigma) was i.p. injected at 0.75 U/kg or 0.5 U/kg. Blood was collected from the tail vein, and the blood glucose levels were measured using glucose meters (Counter Next EZ; Ascensia Diabetes Care). For in vivo insulin stimulation to examine Akt phosphorylation levels, mice were i.p. injected with insulin (1.5 U/kg) after 4 hr of fasting, and tissues were collected 15 minutes after insulin injection.
Fibronectin (FN1) ELISA
Plasma were collected using heparinized tubes (MV-CB300, Braintree Sci). Plasma FN1 levels were determined by ELISA (ab210967, Abcam) according to the manufacturer’s protocol.
Immunohistochemistry (IHC)
Liver sections of 4 μm thickness were cut from PFA-fixed paraffin-embedded blocks of mouse liver. The sections were stained with anti-SQSTM1/p62 antibody (#23214, Cell Signaling Technology) according to the manufacturer’s protocol.
Function-blocking antibody assay
GFP-LC3 HeLa cells were plated in 8-well chamber slides, and were incubated with serum from resting or exercised mice, and 20 μg/ml control mouse IgG (#10400C, Thermo Fisher Scientific) or anti-ITGB1 monoclonal antibody (AIIB2 clone) (#605–930, Thermo Fisher Scientific), with or without 10 μM chloroquine for 3 h. After treatment, the cells were fixed with 4% PFA/PBS for 30 min and subjected to microscopy.
Quantitative PCR (qPCR)
Total RNA was isolated from the mouse liver and skeletal muscle using the Trizol Reagent, and 2 μg of total RNA was reverse-transcribed using High-Capacity cDNA Reverse Transcription kit (4368814, Thermo Fisher Scientific) according to the manufacturer’s protocol. qPCR was performed using PowerUP SYBR Green Master Mix and LightCycler 480 Real-Time PCR system (Roche). Relative gene expression was quantified using the −2^(ΔΔCt) method and normalized to Rplp0.
Co-immunoprecipitation
Mouse liver was lysed in Triton X-100 cell lysis buffer (1% Triton X-100, 20 mM HEPES [pH7.4], 150 mM NaCl, 1 mM EDTA, 5 mM sodium fluoride, 1mM sodium orthovanadate, 1 mM sodium pyrophosphate, 1 mM β-Glycerophosphate, 1 × Halt protease inhibitor cocktail [78438, Thermo Fisher Scientific]). The lysate was centrifuged at 15,000 × g for 10 min at 4°C, and the supernatant was collected and pre-cleaned by incubating with Protein A/G beads (sc-2003, Santa Cruz Biotechnology) for 1 hr at 4°C. Then 6 μg total protein was incubated with 40 μl of anti-BCL2 antibody-conjugated agarose beads (sc-7382 AC, Santa Cruz Biotechnology) or mouse IgG isotype control (10400C, Thermo Fisher Scientific) with Protein A/G beads for 14–16 hr at 4°C. After beads were washed for 4 times with Triton X-100 cell lysis buffer, immunoprecipitated proteins were eluted in Laemmli buffer and detected by immunoblotting.
Plasmids
mCherry-EGFP-LC3 was subcloned into the EcoRI and the BamHI restriction enzyme sites of the pCDH-CMV-MCS-EF1-Puro lentivector using In-Fusion cloning kit (6389110, Takara Bio). Double-strand oligos encoding shRNAs against the target genes were cloned into the AgeI and EcoRI restriction enzyme sites of the pLKO.1-TRC cloning vector (Addgene plasmid #10878) or Tet-pLKO-puro (Addgene plasmid #21915). A scramble shRNA lentivector (Addgene plasmid #1864) was used as a negative control.
Lentivirus production and infection
HEK293T cells were transfected with a lentiviral expression vector together with a pCMV-VSV-G plasmid (Addgene plasmid #8454) and a psPAX2 plasmid (Addgene plasmid #12260) using the lipofectamine 3000 transfection reagent (L3000, Thermo Fisher Scientific). Thirty-six hours after transfection, the medium containing the lentivirus was collected and filtered. Cells were infected with lentivirus by incubation in the lentivirus-containing medium with 8 μg/ml polybrene.
AICAR treatment
Cells were treated with 1 mM AICAR (10010241, Cayman Chemical) in DMEM for 1 hr. Then media were collected and denatured in Laemmli buffer. Cells were also collected and lysed in Laemmli buffer. The samples were used for immunoblotting.
Immunoblotting
Cells were washed with PBS and lysed with Laemmli buffer, and heated at 95°C for 5 min. Tissues were lysed using RIPA lysis buffer (20 mM Tris-HCl [pH 7.4], 150 mM NaCl, 1% NP-40, 0.1% sodium lauryl sulfate [SDS], 0.5% sodium deoxycholate, 1 mM EDTA, 5 mM sodium fluoride, 1 mM sodium orthovanadate, 1 mM sodium pyrophosphate, 1 mM β-Glycerophosphate, 1 × Halt protease inhibitor cocktail [78438, Thermo Fisher Scientific]). After centrifugation at 15, 000 × g for 10 min at 4°C, supernatants were collected and protein concentrations were determined using a BCA protein assay kit (23225, Thermo Fisher Scientific). Proteins were denatured in Laemmli buffer by heating at 95°C for 5 min. Samples containing equivalent protein amounts were resolved by SDS-PAGE and transferred to polyvinylidene difluoride (PVDF) membrane. After membrane blocking by 0.5% skim milk or 5% BSA in Tris-buffered saline containing 0.05% Tween-20 (TBS-T), the proteins were probed with primary antibody overnight at 4°C. The membrane was then washed with TBS-T for 10 min 3 times, and incubated with HRP-conjugated secondary antibody for 1 hr. After membrane washing with TBS-T, specific bands were visualized by chemiluminescence using the ChemiDoc MP Imaging system (Bio-Rad). The band intensities were quantified using ImageJ software.
Microscopy
Paraformaldehyde-fixed tissues were sectioned at 10 μm thickness using cryomicrotome. Cells were plated on cover glass and fixed with 4% paraformaldehyde. Fixed frozen tissue sections and fixed cells were washed with PBS and mounted in a ProLong Diamond Antifade mountant (P36961, Thermo Fisher Scientific). Fluorescence images were acquired using a wide-field Nikon Ti microscope or a spinning-disk confocal microscope (CSU-W1 spinning disk field scanning confocal system [Yokogawa Electric Corp.] with Hamamatsu Flash 4 camera [Hamamatsu Photonics] mounted to a Nikon Ti2 microscope [Nikon]).
QUANTIFICATION AND STATISTICAL ANALYSIS
All data were shown as mean ± standard errors of the mean (SEM). Data were analyzed by Student’s t-test for two groups, and by one-way analysis of variance (ANOVA) followed by Tukey-Kramer test or Dunnett’s test for more than two groups using BellCurve for Excel (Social Survey Research Information Co.). The difference with a P-value of < 0.05 was considered statistically significant.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| anti-BCL2 | Proteintech | Cat#12789-1-AP; RRID:AB_2227948 |
| anti-LC3B | Novus Biologicals | Cat#NB100-2200; RRID:AB_10003146 |
| Contlol mouse IgG | Thermo Fisher Scientific | Cat#10400C; RRID:AB_2532980 |
| anti-ITGB1 (AIIB2 clone) | Thermo Fisher Scientific | Cat#605-930 |
| anti-fibronectin/FN1 | Thermo Fisher Scientific | Cat#PA5-29578; RRID:AB_2547054 |
| anti-p-BCL2 (Ser70) | Santa Cruz Biotechnology | Cat#sc-293128 |
| anti-BECN1 | Santa Cruz Biotechnology | Cat#sc-48341; RRID:AB_626745 |
| anti-integrin α5 (ITGA5) | Santa Cruz Biotechnology | Cat#sc-376199; RRID:AB_10987904 |
| anti-integrin β1 (ITGB1) | Santa Cruz Biotechnology | Cat#sc-374429; RRID:AB_11012020 |
| anti-p-FAK (Tyr397) | Santa Cruz Biotechnology | Cat#sc81493; RRID:AB_1125825 |
| anti-Sec6 | Santa Cruz Biotechnology | Cat#sc-374054; RRID:AB_10916711 |
| Horseradish peroxidase (HRP)-conjugated anti-β-actin | Santa Cruz Biotechnology | Cat#sc-47778 HRP; RRID:AB_2714189 |
| anti-α-tubulin | Santa Cruz Biotechnology | Cat#sc-53029; RRID:AB_793541 |
| anti-SQSTM1/p62 | Cell Signaling Technology | Cat#23214; RRID:AB_2798858 |
| anti-Akt (pan) | Cell Signaling Technology | Cat#4691; RRID:AB_915783 |
| anti-phospho-Akt (Ser473) | Cell Signaling Technology | Cat#4060; RRID:AB_2315049 |
| anti-IKKα | Cell Signaling Technology | Cat#11930; RRID:AB_2687618 |
| anti-IKKβ | Cell Signaling Technology | Cat#8943; RRID:AB_11024092 |
| anti-phospho-IKKα/β (Ser176/180) | Cell Signaling Technology | Cat#2697; RRID:AB_2079382 |
| anti-JNK | Cell Signaling Technology | Cat#9252; RRID:AB_2250373 |
| anti-phospho-JNK (Thr183/185) | Cell Signaling Technology | Cat#4668; RRID:AB_823588 |
| anti-JNK1 | Cell Signaling Technology | Cat#3708; RRID:AB_1904132 |
| anti-JNK2 | Cell Signaling Technology | Cat#9258; RRID:AB_2141027 |
| anti-FAK | Cell Signaling Technology | Cat#3285; RRID:AB_2269034 |
| anti-phospho-Src (Tyr416) | Cell Signaling Technology | Cat#6943; RRID:AB_10013641 |
| anti-Src | Cell Signaling Technology | Cat#2123; RRID:AB_2106047 |
| anti-TBC1D1 | Cell Signaling Technology | Cat#4629; RRID:AB_1904162 |
| anti-AS160 | Cell Signaling Technology | Cat#2670; RRID:AB_2199375 |
| anti-ATG7 | Cell Signaling Technology | Cat#8558; RRID:AB_10831194 |
| anti-AMPK | Cell Signaling Technology | Cat#2532; RRID:AB_330331 |
| anti-phospho-AMPK (Thr172) | Cell Signaling Technology | Cat#2535; RRID:AB_331250 |
| HRP-conjugated anti-mouse IgG light-chain specific | Cell Signaling Technology | Cat#91196 |
| HRP-conjugated anti-rabbit IgG conformation specific | Cell Signaling Technology | Cat#5127; RRID:AB_10892860 |
| HRP-conjugated anti-rat IgG | Cell Signaling Technology | Cat#7077; RRID:AB_10694715 |
| HRP-conjugated anti-rabbit IgG | Jackson ImmunoResearch | Cat#111-035-033; RRID:AB_2313567 |
| HRP-conjugated anti-mouse IgG | Jackson ImmunoResearch | Cat#115-035-003; RRID:AB_10015289 |
| Bacterial and Virus Strains | ||
| Adeno-associated virus (AAV): AAV2/9-shFn1 | This paper | N/A |
| AAV: AAV2/8-shItga5 | This paper | N/A |
| AAV: AAV2/8-shAtg7 | This paper | N/A |
| AAV: AAV2/8-TBG-PI-Null-bGH | Gift of James M. Wilson lab | Addgene viral prep Cat#105536-AAV8; RRID:Addgene_105536 |
| Lentivirus: mCherry-EGFP-LC3 | This paper | N/A |
| Lentivirus: shRNA (pLKO.1) | This paper | N/A |
| Lentivirus: shRNA (tet-pLKO) | This paper | N/A |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Insulin | Millipore Sigma | Cat#I0516-5ML; CAS: 11070-73-8 |
| Heparinized microvette | Braintree Scientific | Cat#MV-CB300 16443 |
| anti-BCL2 antibody-conjugated agarose beads | Santa Cruz Biotechnology | Cat#sc-7382 AC |
| Protein A/G PlusAgarose | Santa Cruz Biotechnology | Cat#sc-2003 |
| Lipofectamine 3000 | Thermo Fisher Scientific | Cat#L3000150 |
| Polybrene | Millipore Sigma | Cat#TR-1003-G |
| Fibronectin (FN1) | Millipore Sigma | Cat#F2006; EC: 289-149-2 |
| Bovine Serum Albumin (BSA) solution | Millipore Sigma | Cat#A1595; CAS: 9048-46-8 |
| AICAR | Cayman Chemical | Cat#10010241; CAS:2627-69-2 |
| Chloroquine (phosphate) | Cayman Chemical | Cat#14194; CAS:50-63-5 |
| Halt Protease Inhibitor Cocktail | Thermo Fisher Scientific | Cat#78438 |
| ProLong Diamond Antifade Mountant | Thermo Fisher Scientific | Cat#P36961 |
| Critical Commercial Assays | ||
| Proteome Purify 2 Mouse Serum Protein | R&D systems | Cat#MIDR002-020 |
| Mouse Fibronectin ELISA Kit | Abcam | Cat#ab210967 |
| High-Capacity cDNA Reverse Transcription Kit | Thermo Fisher Scientific | Cat#4368814 |
| PowerUP SYBR Green Master Mix | Thermo Fisher Scientific | Cat#A25742 |
| BCA protein assay kit | Thermo Fisher Scientific | Cat#23225 |
| Experimental Models: Cell Lines | ||
| Mouse: C2C12 cells | ATCC | Cat#CRL-1772 |
| Human: HeLa cells | ATCC | Cat#CCL-2 |
| Human: 293T cells | ATCC | Cat#CRL-3216 |
| Human: Huh7 cells | JCRB cell bank | Cat#JCRB0403 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6J mice | The Jackson Laboratory | Cat#000664; RRID:IMSR_JAX:000664 |
| Mouse: GFP-LC3 mice: Tg(CAG-EGFP/Map1lc3b)53Nmz | RIKEN BioResource Research Center | Cat#RBRC00806 |
| Oligonucleotides | ||
| Target sequences for shRNA, see Table S1 | This paper | N/A |
| Primers for qPCR, see Table S2 | This paper | N/A |
| Recombinant DNA | ||
| pCDH-CMV-MCS-EF1-Puro | System Biosciences | Cat#CD510B-1 |
| pCDH-mCherry-EGFP-LC3 | This paper | N/A |
| pLKO.1-TRC cloning vector | Gift of David Root lab58 | Addgene plasmid Cat# 10878; RRID:Addgene_10878 |
| Tet-pLKO-puro | Gift of Dmitri Wiederschain lab59 | Addgene plasmid # 21915; RRID:Addgene_21915 |
| pLKO.1 scramble shRNA | Gift of David Sabatini lab60 | Addgene plasmid # 1864; RRID:Addgene_1864 |
| tet-pLKO-puro-Scrambled | Gift of Charles Rudin lab61 | Addgene plasmid # 47541; RRID:Addgene_47541 |
| pCMV-VSV-G | Gift of Bob Weinberg lab62 | Addgene plasmid # 8454; RRID:Addgene_8454 |
| psPAX2 | Gift of Didier Trono lab | Addgene plasmid # 12260; RRID:Addgene_12260 |
| pAAV-MHCK7-miR30-mCherry-shFn1 | This paper | N/A |
| pAAV-TBG-miR30-mCherry-shItga5 | This paper | N/A |
| pAAV-TBG-miR30-mCherry-shAtg7 | This paper | N/A |
| Software and Algorithms | ||
| ImageJ | National Institutes of Health (NIH) | https://imagej.nih.gov/ij/ |
| NIS-Elements | Nikon | https://www.microscope.healthcare.nikon.com/products/software/nis-elements |
| BellCurve for Excel | Social Survey Research Information Co. | https://bellcurve.jp/ex/ |
| BioRender | BioRender | https://biorender.com/ |
| Deposited Data | ||
| Source data for all figures, see Data S1 | This study | N/A |
Highlights:
Hepatic autophagy activation is required for exercise-induced metabolic benefits
Plasma or serum of exercised animals activates autophagy
FN1 is an exercise-elicited muscle-secreted autophagy-inducing circulating factor
FN1 induces autophagy and systemic metabolic benefits via liver α5β1 integrin pathway
Acknowledgements
We thank Dr. Noboru Mizushima for providing GFP-LC3 mice, and the following facilities at Northwestern University Feinberg School of Medicine for technical support: the Proteomics Core for proteomics studies, the Mouse Histology and Phenotyping Laboratory for mouse tissue sectioning, the Center for Advanced Microscopy/Nikon Imaging Center for confocal microscopy studies, and the Sanger Sequencing Facility for sequencing. The study is supported by NIH R01 DK113170, R01 DK123447, R01 DA056720, and BrightFocus Foundation Alzheimer’s Disease Research Award (to C. H.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing Interest Declaration
The authors declare no competing interests.
References
- 1.Ruegsegger GN, and Booth FW (2018). Health Benefits of Exercise. Cold Spring Harb Perspect Med 8. 10.1101/cshperspect.a029694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thyfault JP, and Bergouignan A (2020). Exercise and metabolic health: beyond skeletal muscle. Diabetologia 63, 1464–1474. 10.1007/s00125-020-05177-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Handschin C, and Spiegelman BM (2008). The role of exercise and PGC1alpha in inflammation and chronic disease. Nature 454, 463–469. 10.1038/nature07206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hu FB, Manson JE, Stampfer MJ, Colditz G, Liu S, Solomon CG, and Willett WC (2001). Diet, lifestyle, and the risk of type 2 diabetes mellitus in women. N Engl J Med 345, 790–797. 10.1056/NEJMoa010492. [DOI] [PubMed] [Google Scholar]
- 5.Kahn SE, Cooper ME, and Del Prato S (2014). Pathophysiology and treatment of type 2 diabetes: perspectives on the past, present, and future. Lancet 383, 1068–1083. 10.1016/S0140-6736(13)62154-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rocchi A, and He C (2017). Regulation of Exercise-Induced Autophagy in Skeletal Muscle. Curr Pathobiol Rep 5, 177–186. 10.1007/s40139-017-0135-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mizushima N, and Komatsu M (2011). Autophagy: renovation of cells and tissues. Cell 147, 728–741. 10.1016/j.cell.2011.10.026. [DOI] [PubMed] [Google Scholar]
- 8.Kaur J, and Debnath J (2015). Autophagy at the crossroads of catabolism and anabolism. Nat Rev Mol Cell Biol 16, 461–472. 10.1038/nrm4024. [DOI] [PubMed] [Google Scholar]
- 9.Kuma A, and Mizushima N (2010). Physiological role of autophagy as an intracellular recycling system: With an emphasis on nutrient metabolism. Semin Cell Dev Biol 21, 683–690. [DOI] [PubMed] [Google Scholar]
- 10.He C, and Klionsky DJ (2009). Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet 43, 67–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rocchi A, and He C (2015). Emerging roles of autophagy in metabolism and metabolic disorders. Front Biol (Beijing) 10, 154–164. 10.1007/s11515-015-1354-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lahiri V, Hawkins WD, and Klionsky DJ (2019). Watch What You (Self-) Eat: Autophagic Mechanisms that Modulate Metabolism. Cell Metab 29, 803–826. 10.1016/j.cmet.2019.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim KH, and Lee MS (2014). Autophagy--a key player in cellular and body metabolism. Nat Rev Endocrinol 10, 322–337. 10.1038/nrendo.2014.35. [DOI] [PubMed] [Google Scholar]
- 14.Klionsky DJ, Petroni G, Amaravadi RK, Baehrecke EH, Ballabio A, Boya P, Bravo-San Pedro JM, Cadwell K, Cecconi F, Choi AMK, et al. (2021). Autophagy in major human diseases. EMBO J 40, e108863. 10.15252/embj.2021108863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lira VA, Okutsu M, Zhang M, Greene NP, Laker RC, Breen DS, Hoehn KL, and Yan Z (2013). Autophagy is required for exercise training-induced skeletal muscle adaptation and improvement of physical performance. FASEB J 27, 4184–4193. 10.1096/fj.13-228486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.He C, Bassik MC, Moresi V, Sun K, Wei Y, Zou Z, An Z, Loh J, Fisher J, Sun Q, et al. (2012). Exercise-induced BCL2-regulated autophagy is required for muscle glucose homeostasis. Nature 481, 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Grumati P, Coletto L, Schiavinato A, Castagnaro S, Bertaggia E, Sandri M, and Bonaldo P (2011). Physical exercise stimulates autophagy in normal skeletal muscles but is detrimental for collagen VI-deficient muscles. Autophagy 7, 1415–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Liu X, Niu Y, Yuan H, Huang J, and Fu L (2015). AMPK binds to Sestrins and mediates the effect of exercise to increase insulin-sensitivity through autophagy. Metabolism 64, 658–665. 10.1016/j.metabol.2015.01.015. [DOI] [PubMed] [Google Scholar]
- 19.Drake JC, Wilson RJ, Laker RC, Guan Y, Spaulding HR, Nichenko AS, Shen W, Shang H, Dorn MV, Huang K, et al. (2021). Mitochondria-localized AMPK responds to local energetics and contributes to exercise and energetic stress-induced mitophagy. Proc Natl Acad Sci U S A 118. 10.1073/pnas.2025932118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lo Verso F, Carnio S, Vainshtein A, and Sandri M (2014). Autophagy is not required to sustain exercise and PRKAA1/AMPK activity but is important to prevent mitochondrial damage during physical activity. Autophagy 10, 1883–1894. 10.4161/auto.32154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Medina DL, Di Paola S, Peluso I, Armani A, De Stefani D, Venditti R, Montefusco S, Scotto-Rosato A, Prezioso C, Forrester A, et al. (2015). Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat Cell Biol 17, 288–299. 10.1038/ncb3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vainshtein A, and Hood DA (2016). The regulation of autophagy during exercise in skeletal muscle. J Appl Physiol (1985) 120, 664–673. 10.1152/japplphysiol.00550.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Sanchez AM, Bernardi H, Py G, and Candau RB (2014). Autophagy is essential to support skeletal muscle plasticity in response to endurance exercise. Am J Physiol Regul Integr Comp Physiol 307, R956–969. 10.1152/ajpregu.00187.2014. [DOI] [PubMed] [Google Scholar]
- 24.Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Overvatn A, Bjorkoy G, and Johansen T (2007). p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 282, 24131–24145. 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- 25.Nicklin P, Bergman P, Zhang B, Triantafellow E, Wang H, Nyfeler B, Yang H, Hild M, Kung C, Wilson C, et al. (2009). Bidirectional transport of amino acids regulates mTOR and autophagy. Cell 136, 521–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Vega ME, and Schwarzbauer JE (2016). Collaboration of fibronectin matrix with other extracellular signals in morphogenesis and differentiation. Curr Opin Cell Biol 42, 1–6. 10.1016/j.ceb.2016.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Espelage L, Al-Hasani H, and Chadt A (2020). RabGAPs in skeletal muscle function and exercise. J Mol Endocrinol 64, R1–R19. 10.1530/JME-19-0143. [DOI] [PubMed] [Google Scholar]
- 28.Salva MZ, Himeda CL, Tai PW, Nishiuchi E, Gregorevic P, Allen JM, Finn EE, Nguyen QG, Blankinship MJ, Meuse L, et al. (2007). Design of tissue-specific regulatory cassettes for high-level rAAV-mediated expression in skeletal and cardiac muscle. Mol Ther 15, 320–329. 10.1038/sj.mt.6300027. [DOI] [PubMed] [Google Scholar]
- 29.Koornneef A, Maczuga P, van Logtenstein R, Borel F, Blits B, Ritsema T, van Deventer S, Petry H, and Konstantinova P (2011). Apolipoprotein B knockdown by AAV-delivered shRNA lowers plasma cholesterol in mice. Mol Ther 19, 731–740. 10.1038/mt.2011.6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tasfaout H, Lionello VM, Kretz C, Koebel P, Messaddeq N, Bitz D, Laporte J, and Cowling BS (2018). Single Intramuscular Injection of AAV-shRNA Reduces DNM2 and Prevents Myotubular Myopathy in Mice. Mol Ther 26, 1082–1092. 10.1016/j.ymthe.2018.02.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang Q, Tang Y, Imbrogno K, Lu A, Proto JD, Chen A, Guo F, Fu FH, Huard J, and Wang B (2012). AAV-based shRNA silencing of NF-kappaB ameliorates muscle pathologies in mdx mice. Gene Ther 19, 1196–1204. 10.1038/gt.2011.207. [DOI] [PubMed] [Google Scholar]
- 32.Zincarelli C, Soltys S, Rengo G, and Rabinowitz JE (2008). Analysis of AAV serotypes 1–9 mediated gene expression and tropism in mice after systemic injection. Mol Ther 16, 1073–1080. 10.1038/mt.2008.76. [DOI] [PubMed] [Google Scholar]
- 33.Bachmann M, Kukkurainen S, Hytonen VP, and Wehrle-Haller B (2019). Cell Adhesion by Integrins. Physiol Rev 99, 1655–1699. 10.1152/physrev.00036.2018. [DOI] [PubMed] [Google Scholar]
- 34.Yang JT, Bader BL, Kreidberg JA, Ullman-Cullere M, Trevithick JE, and Hynes RO (1999). Overlapping and independent functions of fibronectin receptor integrins in early mesodermal development. Dev Biol 215, 264–277. 10.1006/dbio.1999.9451. [DOI] [PubMed] [Google Scholar]
- 35.Hamidi H, and Ivaska J (2018). Every step of the way: integrins in cancer progression and metastasis. Nat Rev Cancer 18, 533–548. 10.1038/s41568-018-0038-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bhullar IS, Li YS, Miao H, Zandi E, Kim M, Shyy JY, and Chien S (1998). Fluid shear stress activation of IkappaB kinase is integrin-dependent. J Biol Chem 273, 30544–30549. 10.1074/jbc.273.46.30544. [DOI] [PubMed] [Google Scholar]
- 37.Chen N, and Debnath J (2013). IkappaB kinase complex (IKK) triggers detachment-induced autophagy in mammary epithelial cells independently of the PI3K-AKT-MTORC1 pathway. Autophagy 9, 1214–1227. 10.4161/auto.24870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Criollo A, Senovilla L, Authier H, Maiuri MC, Morselli E, Vitale I, Kepp O, Tasdemir E, Galluzzi L, Shen S, et al. (2010). The IKK complex contributes to the induction of autophagy. EMBO J 29, 619–631. 10.1038/emboj.2009.364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Comb WC, Cogswell P, Sitcheran R, and Baldwin AS (2011). IKK-dependent, NF-kappaB-independent control of autophagic gene expression. Oncogene 30, 1727–1732. 10.1038/onc.2010.553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Wei Y, Pattingre S, Sinha S, Bassik M, and Levine B (2008). JNK1-mediated phosphorylation of Bcl-2 regulates starvation-induced autophagy. Mol Cell 30, 678–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pattingre S, Bauvy C, Carpentier S, Levade T, Levine B, and Codogno P (2009). Role of JNK1-dependent Bcl-2 Phosphorylation in Ceramide-induced Macroautophagy. J Biol Chem 284, 2719–2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pattingre S, Tassa A, Qu X, Garuti R, Liang XH, Mizushima N, Packer M, Schneider MD, and Levine B (2005). Bcl-2 antiapoptotic proteins inhibit Beclin 1-dependent autophagy. Cell 122, 927–939. [DOI] [PubMed] [Google Scholar]
- 43.Gonzalez-Franquesa A, Stocks B, Borg ML, Kuefner M, Dalbram E, Nielsen TS, Agrawal A, Pankratova S, Chibalin AV, Karlsson HKR, et al. (2021). Discovery of Thymosin Beta-4 as a Human Exerkine and Growth Factor. Am J Physiol Cell Physiol. 10.1152/ajpcell.00263.2021. [DOI] [PubMed] [Google Scholar]
- 44.Hartwig S, Raschke S, Knebel B, Scheler M, Irmler M, Passlack W, Muller S, Hanisch FG, Franz T, Li X, et al. (2014). Secretome profiling of primary human skeletal muscle cells. Biochim Biophys Acta 1844, 1011–1017. 10.1016/j.bbapap.2013.08.004. [DOI] [PubMed] [Google Scholar]
- 45.Norheim F, Raastad T, Thiede B, Rustan AC, Drevon CA, and Haugen F (2011). Proteomic identification of secreted proteins from human skeletal muscle cells and expression in response to strength training. Am J Physiol Endocrinol Metab 301, E1013–1021. 10.1152/ajpendo.00326.2011. [DOI] [PubMed] [Google Scholar]
- 46.Yoon JH, Yea K, Kim J, Choi YS, Park S, Lee H, Lee CS, Suh PG, and Ryu SH (2009). Comparative proteomic analysis of the insulin-induced L6 myotube secretome. Proteomics 9, 51–60. 10.1002/pmic.200800187. [DOI] [PubMed] [Google Scholar]
- 47.Pedersen BK, and Febbraio MA (2008). Muscle as an endocrine organ: focus on muscle-derived interleukin-6. Physiol Rev 88, 1379–1406. 10.1152/physrev.90100.2007. [DOI] [PubMed] [Google Scholar]
- 48.Qin B, Zhou Z, He J, Yan C, and Ding S (2015). IL-6 Inhibits Starvation-induced Autophagy via the STAT3/Bcl-2 Signaling Pathway. Sci Rep 5, 15701. 10.1038/srep15701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dutta RK, Kathania M, Raje M, and Majumdar S (2012). IL-6 inhibits IFN-gamma induced autophagy in Mycobacterium tuberculosis H37Rv infected macrophages. Int J Biochem Cell Biol 44, 942–954. 10.1016/j.biocel.2012.02.021. [DOI] [PubMed] [Google Scholar]
- 50.Pinto AP, da Rocha AL, Cabrera EMB, Marafon BB, Kohama EB, Rovina RL, Simabuco FM, Bueno Junior CR, de Moura LP, Pauli JR, et al. (2020). Role of interleukin-6 in inhibiting hepatic autophagy markers in exercised mice. Cytokine 130, 155085. 10.1016/j.cyto.2020.155085. [DOI] [PubMed] [Google Scholar]
- 51.Marasco MR, Conteh AM, Reissaus CA, Cupit J.E.t., Appleman EM, Mirmira RG, and Linnemann AK (2018). Interleukin-6 Reduces beta-Cell Oxidative Stress by Linking Autophagy With the Antioxidant Response. Diabetes 67, 1576–1588. 10.2337/db17-1280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Linnemann AK, Blumer J, Marasco MR, Battiola TJ, Umhoefer HM, Han JY, Lamming DW, and Davis DB (2017). Interleukin 6 protects pancreatic beta cells from apoptosis by stimulation of autophagy. FASEB J 31, 4140–4152. 10.1096/fj.201700061RR. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Delk NA, and Farach-Carson MC (2012). Interleukin-6: a bone marrow stromal cell paracrine signal that induces neuroendocrine differentiation and modulates autophagy in bone metastatic PCa cells. Autophagy 8, 650–663. 10.4161/auto.19226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Li XZ, Sui CY, Chen Q, Chen XP, Zhang H, and Zhou XP (2013). Promotion of autophagy at the maturation step by IL-6 is associated with the sustained mitogen-activated protein kinase/extracellular signal-regulated kinase activity. Mol Cell Biochem 380, 219–227. 10.1007/s11010-013-1676-9. [DOI] [PubMed] [Google Scholar]
- 55.Bostrom P, Wu J, Jedrychowski MP, Korde A, Ye L, Lo JC, Rasbach KA, Bostrom EA, Choi JH, Long JZ, et al. (2012). A PGC1-alpha-dependent myokine that drives brown-fat-like development of white fat and thermogenesis. Nature 481, 463–468. 10.1038/nature10777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jedrychowski MP, Wrann CD, Paulo JA, Gerber KK, Szpyt J, Robinson MM, Nair KS, Gygi SP, and Spiegelman BM (2015). Detection and Quantitation of Circulating Human Irisin by Tandem Mass Spectrometry. Cell Metab 22, 734–740. 10.1016/j.cmet.2015.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wang J, Meng M, Li M, Guan X, Liu J, Gao X, Sun Q, Li J, Ma C, and Wei L (2020). Integrin alpha5beta1, as a Receptor of Fibronectin, Binds the FbaA Protein of Group A Streptococcus To Initiate Autophagy during Infection. mBio 11. 10.1128/mBio.00771-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Moffat J, Grueneberg DA, Yang X, Kim SY, Kloepfer AM, Hinkle G, Piqani B, Eisenhaure TM, Luo B, Grenier JK, et al. (2006). A lentiviral RNAi library for human and mouse genes applied to an arrayed viral high-content screen. Cell 124, 1283–1298. 10.1016/j.cell.2006.01.040. [DOI] [PubMed] [Google Scholar]
- 59.Wiederschain D, Wee S, Chen L, Loo A, Yang G, Huang A, Chen Y, Caponigro G, Yao YM, Lengauer C, et al. (2009). Single-vector inducible lentiviral RNAi system for oncology target validation. Cell Cycle 8, 498–504. 10.4161/cc.8.3.7701. [DOI] [PubMed] [Google Scholar]
- 60.Sarbassov DD, Guertin DA, Ali SM, and Sabatini DM (2005). Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307, 1098–1101. [DOI] [PubMed] [Google Scholar]
- 61.Rudin CM, Durinck S, Stawiski EW, Poirier JT, Modrusan Z, Shames DS, Bergbower EA, Guan Y, Shin J, Guillory J, et al. (2012). Comprehensive genomic analysis identifies SOX2 as a frequently amplified gene in small-cell lung cancer. Nat Genet 44, 1111–1116. 10.1038/ng.2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Stewart SA, Dykxhoorn DM, Palliser D, Mizuno H, Yu EY, An DS, Sabatini DM, Chen IS, Hahn WC, Sharp PA, et al. (2003). Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA 9, 493–501. 10.1261/rna.2192803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mizushima N, Yamamoto A, Matsui M, Yoshimori T, and Ohsumi Y (2004). In vivo analysis of autophagy in response to nutrient starvation using transgenic mice expressing a fluorescent autophagosome marker. Mol Biol Cell 15, 1101–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kuramoto K, Wang N, Fan Y, Zhang W, Schoenen FJ, Frankowski KJ, Marugan J, Zhou Y, Huang S, and He C (2016). Autophagy activation by novel inducers prevents BECN2-mediated drug tolerance to cannabinoids. Autophagy 12, 1460–1471. 10.1080/15548627.2016.1187367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kuramoto K, Kim YJ, Hong JH, and He C (2021). The autophagy protein Becn1 improves insulin sensitivity by promoting adiponectin secretion via exocyst binding. Cell Rep 35, 109184. 10.1016/j.celrep.2021.109184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yamamoto S, Kuramoto K, Wang N, Situ X, Priyadarshini M, Zhang W, Cordoba-Chacon J, Layden BT, and He C (2018). Autophagy Differentially Regulates Insulin Production and Insulin Sensitivity. Cell Rep 23, 3286–3299. 10.1016/j.celrep.2018.05.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All original data for creating all graphs in the paper are provided in Data S1.
This study did not generate/analyze datasets/code.
Any additional information required to reanalyze the data reported in this paper is available from the Lead Contact upon request.