

Abstract
Purpose of review:
Somatic mutations, described as non-inherited changes in DNA that arise and are passed on to descendant cells, are well known to cause cancers; however, it is increasingly appreciated that the propagation of somatic mutations within a tissue may have a role in causing non-neoplastic disorders and abnormalities in elderly individuals. The non-malignant clonal expansion of somatic mutations in the hematopoietic system is termed clonal hematopoiesis. This review will briefly discuss how this condition has been linked to various age-related diseases outside the hematopoietic system.
Recent findings:
Clonal hematopoiesis, resulting from leukemic driver gene mutations or mosaic loss of the Y chromosome in leukocytes, is associated with the development of various forms of cardiovascular disease, including atherosclerosis and heart failure, in a mutation-dependent manner.
Summary:
Accumulating evidence shows that clonal hematopoiesis represents a new mechanism for cardiovascular disease and a new risk factor that is as prevalent and consequential as the traditional risk factors that have been studied for decades.
Keywords: CHIP, ARCH, Inflammation, Fibrosis
Introduction:
Cardiologists and cardiovascular researchers are well aware of the role that inherited genetics play in cardiovascular disease; i.e. pathological conditions caused by a DNA variant in one or more of our genes passed on from parents to children. For example, the mutated genes that contribute to familial hypercholesterolemia (1) and hypertrophic cardiomyopathy (2, 3) have been identified, and genome-wide association studies have expanded our understanding of the heritable basis of cardiovascular disease (CVD) (4). Studies with induced pluripotent stem (iPS) cells and knockout mice studies have revealed the effects of inherited mutations on the development of disease processes in the cardiovascular system. However, heritable mutations can have relatively small predictive value for the development of CVD in the population and do little to explain the pathologies of advanced age, which is the greatest CVD risk factor. In contrast, relatively little progress has been made in understanding the role that somatic mutations, which linearly accumulate in cells over a lifetime, play in CVD. Previous studies focusing on post-zygotic mutations have been conducted primarily in the context of cancer, which rarely arises in cardiovascular organs. Recently, experimentalists and clinicians have come to appreciate that the accumulation of somatic mutations in the blood system plays an unexpected role in the progression of CVD (5, 6).
Clonal hematopoiesis (CH) is the process through which the proportion of mutant leukocytes increases following the acquisition of driver gene mutations that provide a growth or survival advantage to hematopoietic stem/progenitor cells (HSPCs). Mutations in HSPCs may occur early in life, and as people age, the advantages conferred by the acquired driver gene mutations allow mutant clones to grow to measurable levels. Most people are estimated to have 10-20 unique and detectable clones in their blood by age 70 (7). Expectedly, CH can lead to the development of hematologic malignancies; however, the incidence of progression to frank malignancy is low, and most CH patients display few or no hematologic manifestations. Rather, manifestations of CH-associated disease can often be seen across organ systems as mutant blood cells circulate throughout the body and enter tissues of the nervous, respiratory, musculoskeletal, and cardiovascular systems (8). There, they display disruptive behavior compared to cells derived from wild type HSPCs, leading to pathological responses to tissue damage or disease. The behavior of mutant leukocyte clones is dependent on several variables, including the specific mutation, cell type, or tissue microenvironment. Some may enhance inflammation in the wall of a blood vessel (9), while others may promote fibrosis in the heart (10). In this brief review, we discuss the molecular basis of CH and its variable impact on cardiovascular disease.
Text of review:
From a cardiologist’s perspective, CH can be a foreign concept due to the complexity of classifying CH and the fact that its association with the cardiovascular system involves concepts from the fields of hematology, oncology, and immunology. CH is typically defined simply as the mutation-induced aberrant growth of a single HSPC, regardless of the size of the mutant clones (the proportion of mutated cells). The sensitivity of the DNA sequencing methods determines the size of the clone that can be detected. Greater DNA sequencing depths and error-correction methodologies allow for more sensitive detection of mutant clones, but this also increases the costs of CH analysis (11). Targeting a panel of fewer, yet “higher-yield”, regions of the genome instead of the whole genome or exome allows for a more cost-effective approach to detecting smaller clones. Variant allele frequency (VAF) is a technical measure of the percentage of sequencing reads with mutations, and the mutant allele frequency in blood is considered a proxy for clone size in the HSPC compartment. Along with clone size, the type of driver mutation and its locus can provide further nuance for understanding the physiological effects of a CH mutation. A single gene can be affected by single-nucleotide variants (SNVs) or small insertions/deletions (Indels). Among these, CH caused by mutations in a defined set of genes with a predisposition for the development of leukemia and having a VAF of more than 2% is known as CHIP (clonal hematopoiesis of indeterminate potential) (12). There are more than 100 CHIP driver gene candidates. Mutations most frequently occur in the epigenetic regulators DNMT3A, TET2, and ASXL1, and clones arising from these variants have been linked to CVD (13). The term “mosaic chromosomal alternation” (mCA), is used to characterize chromosomal changes such as chromosomal gain or loss, loss of heterozygosity (LOH), and other chromosomal modifications of autosomal and sex-linked chromosomes. Hematopoietic loss of the sex-linked Y chromosome in males is reported to be associated with CVD (10, 14, 15), whereas chromosomal alternations of autosomes do not appear to be associated with CVD (16).
CH with leukemia driver mutations and CVD
Two articles published in the New England Journal of Medicine in 2014 are commonly referenced to highlight the clinical importance of CHIP (17, 18). In these longitudinal studies, whole exome sequences of peripheral blood cells from a total of about twenty-nine thousand individuals were examined. Both of these studies reported that CHIP increases the risk for all-cause mortality by 40-50%. In a secondary analysis, Jaiswal et al. (18) found that the risk for coronary artery disease (CAD) and stroke was higher in individuals with CHIP (HR=2.0; 95% CI: 1.2 – 3.4 for CAD and HR= 2.6; 95% CI: 1.4 – 4.8 for stroke). This seminal finding led to experimental studies of CHIP and CVD (9, 13, 19). Promising results then spurred investigations, both epidemiological and experimental, into the role of CH in other age-related diseases, particularly those of the cardiovascular system (8).
A notable study established an epidemiological link between CHIP and heart failure prognosis. Dorsheimer et al. (20) examined 200 patients from three clinical trials that evaluated intracoronary autologous bone marrow cell transplantation to treat ischemic heart failure. Bone marrow cells were sequenced to identify CHIP, and mutations in the DNMT3A and TET2 genes were present in 38 of the 200 cases. Combined DNMT3A- and TET2-mediated CHIP was associated with increased risk for heart failure mortality and rehospitalization in patients with ischemic heart failure (HR=2.1; 95% CI: 1.1-4.0). This work was subsequently corroborated and extended by additional analyses (21–25). More recently, the association between CHIP and the incidence of heart failure was examined by Yu et al. in a meta-analysis of four TOPMed cohorts (ARIC, CHS, JHS, and WHI) and the UK Biobank, totaling 56,597 individuals (26). They found that CHIP increased the risk of heart failure onset by 25% (HR=1.25; 95% CI: 1.13-1.35) and was individually associated with increased risk of heart failure either with (HR=1.26; 95% CI: 1.07-1.36) or without previous coronary artery disease (HR=1.21; 95% CI: 0.97-1.64). Additionally, they found mutations in TET2, JAK2, and ASXL1 were each individually associated with increased risk for heart failure in a dose-dependent manner (HR=1.29; 95% CI: 1.18-2.14 for TET2, HR=2.50; 95% CI: 1.35-4.69 for JAK2, HR=1.58; 95% CI: 1.20-2.08 for ASXL1). Surprisingly, mutations in the commonly mutated driver gene, DNMT3A, were not associated with heart failure (HR=1.09; 95% CI: 0.95-1.24).
Experimental studies have provided evidence for a mechanistic link between hematopoietic mutations in CHIP driver genes and CVD. While most studies have focused on models of TET2 CH, other CHIP genes examined include DNMT3A, ASXL1, JAK2, TP53, and PPM1D, and the experimental models have included atherosclerosis, ischemic and non-ischemic heart failure, doxorubicin-induced cardiac toxicity and others (8, 27–33). While functional differences have been observed between the different CHIP driver genes, all appear to promote inflammatory processes.
LOY in leukocytes and CVD
The X and Y sex chromosomes determine the biological sex of an individual. At birth, men have one X and one Y chromosome in every somatic cell; however, a portion of somatic cells may lose their Y chromosome with age. This phenomenon is commonly observed in blood cells, and it is referred to as hematopoietic mosaic loss of Y chromosome (mLOY). The absence of the Y chromosome in an individual blood cell does not result in the transformation of a male cell into a female cell; instead, it can be conceptualized as a transition of genotype from XY to XØ. Similar to CHIP, aging is the greatest risk factor for developing mLOY. A recent study showed that mLOY is detectable in more than 40% of 70-year-old men, making mLOY the most prevalent somatic mutation in humans (34, 35). A history of smoking can also be a significant risk factor for mLOY, yet population studies have shown these associations appear to be transient, as it is reversible after smoking cessation (36).
Epidemiological studies have revealed the associations between mLOY and various age-related diseases (Figure 1). For example, it has been found that males who display mLOY are more likely to develop Alzheimer’s disease and certain forms of cancer and have shorter lifespans by an average of 5.5 years (37). Recently, we investigated the statistical association between mLOY and cardiovascular (CV) disease using data from the UK Biobank and found that mLOY is associated with an increased risk of CVD mortality. Specifically, mLOY in greater than 40% of leukocytes confers a 1.8-fold greater risk of heart failure death and a 2.4-fold greater risk of congestive heart failure death (10).
Figure 1. Associations between mosaic loss of Y chromosome (LOY) in leukocytes and various age-associated diseases.
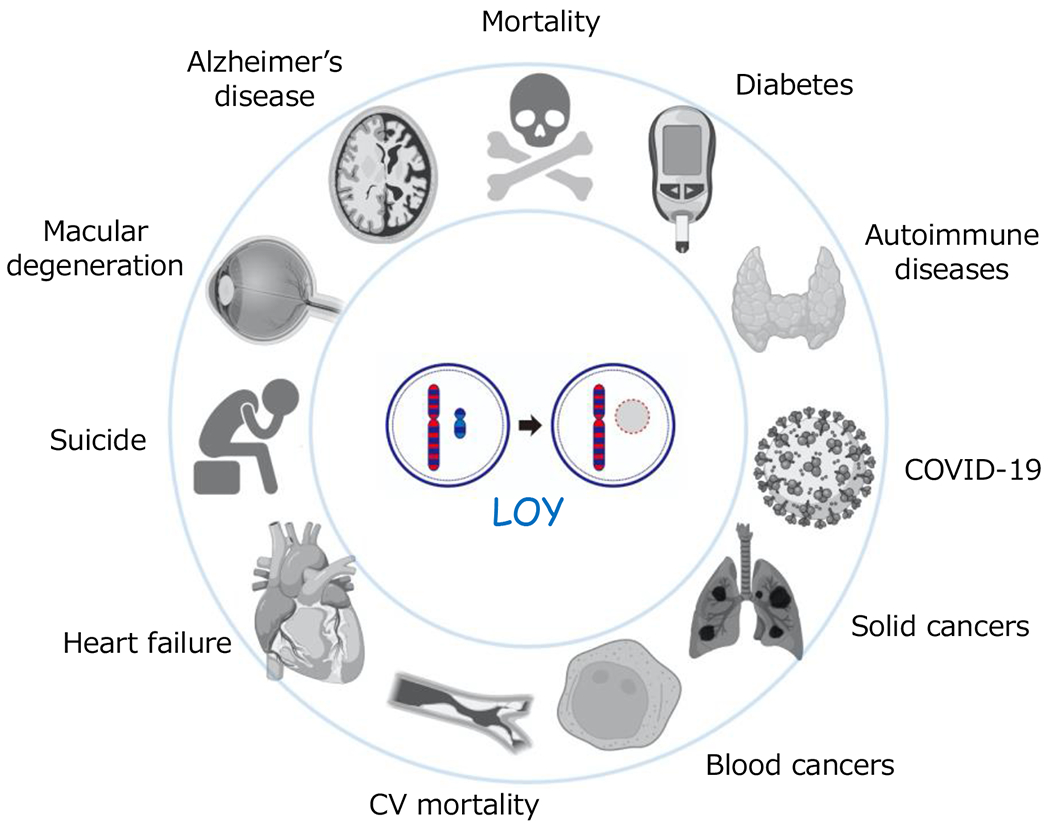
LOY has been epidemiologically associated with many diseases and mortality, yet mechanistic mechanisms linking LOY to specific diseases have been understudied.
How can the post-natal loss of a sex-determination chromosome in blood cells affect illnesses in elderly men? One explanation is that hematopoietic mLOY is a biomarker of an individual’s genomic instability, affecting their susceptibility to diseases throughout the body. In fact, it has been reported that germline variants associated with the risk of mLOY involve genes implicated in cell proliferation, cell cycle regulation, DNA damage response, and apoptosis, and that these variants overlap with cancer susceptibility (34, 38). Another possibility is that blood cells lacking the Y chromosome behave differently from euploid blood cells, and they directly impact the organs they infiltrate. The latter possibility is supported by transcriptomic analyses of human leukocytes, which reveal that genes expressed by the Y chromosome, or autosomes, are dysregulated in mLOY leukocytes (39). Thus, we carried out mouse experiments to test whether a causal link potentially exists between mLOY and CV disease (10). By employing CRISPR/Cas9 technology to target the repetitive regions in the centromere of the Y chromosome, it was possible to produce hematopoietic stem/progenitor cells (HSPCs) deficient for the entire Y chromosome. Mice reconstituted with HSPCs without the Y chromosome (mLOY mice) were prone to fibrosis in the lungs, kidneys, and heart upon unperturbed aging, indicating that mLOY plays a direct role in the age-related decline of organ function in males (Figure 2). Additionally, mLOY mice developed more severe heart failure following cardiac stress induced by surgical constriction of the aorta. Histologically, the hearts of mLOY mice showed increased fibrosis and a greater number of cardiac fibroblasts. Importantly, unlike the hematopoietic cells, the cardiac fibroblasts in the mLOY mice retain their Y chromosome, and theoretically, their genotype should be identical to that of wild-type cells. Therefore, we hypothesized that the over-activation of fibroblasts observed in mLOY mice would be a response to the presence of blood cells lacking the Y chromosome. Single-cell RNA sequencing analysis and immunohistochemical analysis revealed that Y-chromosome deficient macrophages recruited to the heart showed aberrant profibrotic differentiation and greater production of profibrotic molecules such as TGF-β1. In turn, the cardiac fibroblasts in the hematopoietic mLOY mice displayed elevated TGF-β/SMAD pathway activation. These findings imply that cardiac macrophages lacking the Y chromosome stimulate cardiac fibroblasts to produce extracellular matrix. Furthermore, post-surgical treatment with a neutralizing antibody that blocks TGF-β ameliorated the difference in experimental heart failure severity between mLOY and control mice. Thus, understanding a patient’s mLOY status is expected to aid in the identification of high-risk patient groups for diseases associated with excessive fibrosis, and this could aid in treatment decisions.
Figure 2. Loss of Y chromosome in leukocytes is causally linked to heart failure.
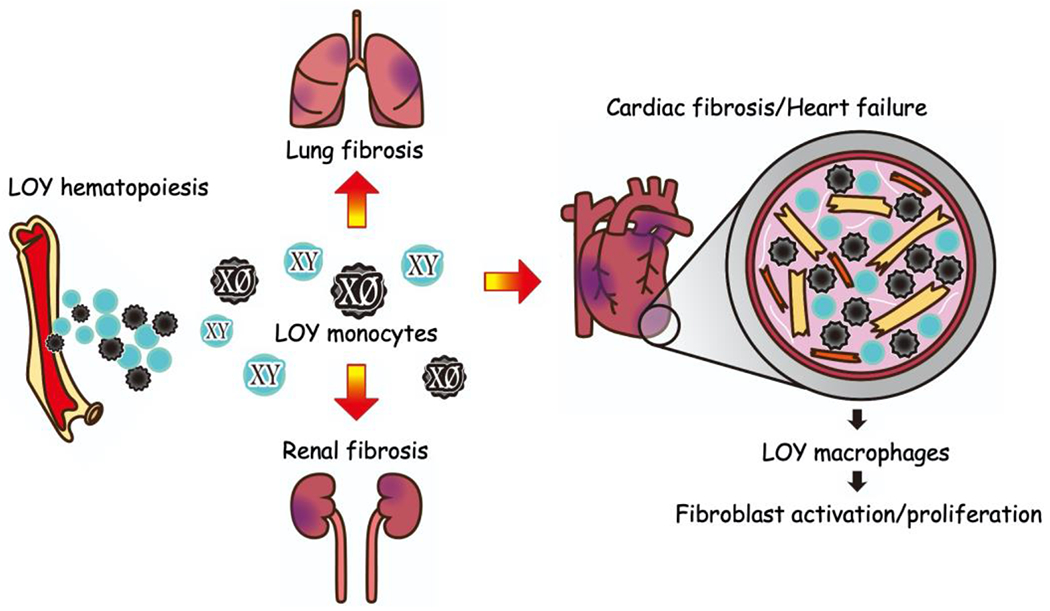
A male is considered to have hematopoietic mosaic loss of Y chromosome (mLOY) when the fraction of leukocytes in blood rises over the detection threshold. Compared to controls, aged mice with hematopoietic mLOY developed more fibrosis in their lungs, kidneys. Circulating mLOY monocytes recruited to the heart are more likely to differentiate into pro-fibrotic macrophages, resulting in TGFβ-dependent activation of cardiac fibroblasts, fibroblast proliferation, and extracellular matrix tissue deposition.
Conclusion:
CH due to somatic mutations in leukemic driver genes or mosaic loss of Y chromosome in leukocytes is epidemiologically associated with age-associated diseases, including worse prognosis in coronary artery disease and heart failure outcomes. Experimental studies in mice have shown that CH mutations are causally linked to CVD via enhancing inflammatory and fibrotic processes in the tissues they infiltrate. Future studies will need to further extend these studies to humans to assess whether mutated human leukocytes display pro-inflammatory and pro-fibrotic characteristics similar to murine cells (21), and whether anti-inflammation therapy in the case of CHIP or anti-fibrosis therapy in the case of mLOY ameliorates CH-driven CVD (40).
Key points:
There are several genetic patterns of CH including mutations in leukemic driver genes, mosaic chromosomal alternation in autosomes, and the loss of sex chromosomes.
CHIP and mLOY in leukocytes are common in elderly individuals and are associated with inferior survival and age-related CVDs such as atherosclerosis and heart failure.
Murine macrophages with leukemic driver mutations enhance inflammation in a gene-dependent manner.
mLOY in leukocytes accelerates tissue aging, including lung, renal and cardiac fibrosis in mice.
Cardiac macrophages deficient in the Y chromosome promote fibrotic responses in the heart.
Disclosure of funding:
This work was supported by the National Institutes of Health (NIH) grants AG073249, AG072095, HL142650, and HL152174 and NASA grant 80NSSC21K0549 to K.W.; Grant-in-Aid for Research Activity Start-up 21K20879 to S.S.; Grant-in-Aid for Scientific Research C 22K08162 to S.S.; and the MSD Life Science Foundation (S.S.); the Cardiovascular Research Fund (S.S.); The Japanese Heart Failure Society (S.S.); Kondou Kinen Medical Foundation (S.S.).
Footnotes
Conflicts of Interest: None
References
- 1.Goldstein JL, Brown MS. The LDL receptor. Arterioscler Thromb Vasc Biol. 2009;29(4):431–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Geisterfer-Lowrance AA, Kass S, Tanigawa G, et al. A molecular basis for familial hypertrophic cardiomyopathy: a beta cardiac myosin heavy chain gene missense mutation. Cell. 1990;62(5):999–1006. [DOI] [PubMed] [Google Scholar]
- 3.Jarcho JA, McKenna W, Pare JA, et al. Mapping a gene for familial hypertrophic cardiomyopathy to chromosome 14q1. The New England journal of medicine. 1989;321(20):1372–8. [DOI] [PubMed] [Google Scholar]
- 4.Walsh R, Jurgens SJ, Erdmann J, Bezzina CR. Genome-wide association studies of cardiovascular disease. Physiol Rev. 2023. [DOI] [PubMed] [Google Scholar]
- 5.Heimlich JB, Bick AG. Somatic Mutations in Cardiovascular Disease. Circ Res. 2022;130(1):149–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Libby P, Sidlow R, Lin AE, et al. Clonal Hematopoiesis: Crossroads of Aging, Cardiovascular Disease, and Cancer: JACC Review Topic of the Week. Journal of the American College of Cardiology. 2019;74(4):567–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mitchell E, Spencer Chapman M, Williams N, et al. Clonal dynamics of haematopoiesis across the human lifespan. Nature. 2022;606(7913):343–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Evans MA, Walsh K. Clonal hematopoiesis, somatic mosaicism, and age-associated disease. Physiol Rev. 2023;103(1):649–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Fuster JJ, MacLauchlan S, Zuriaga MA, et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science (New York, NY). 2017;355(6327):842–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.*.Sano S, Horitani K, Ogawa H, et al. Hematopoietic loss of Y chromosome leads to cardiac fibrosis and heart failure mortality. Science (New York, NY). 2022;377(6603):292–7. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study provided evidence of an epidemiological association between hematopoietic mosaic loss of Y and cardiovascular-related mortality. Additionally, this study was the first to report functional evidence of the loss of Y mutation confers a profibrotic phenotype to leukocytes harboring the mutation in mice, leading to increased fibrosis across organ systems and a decline in cardiac function.
- 11.Young AL, Challen GA, Birmann BM, Druley TE. Clonal haematopoiesis harbouring AML-associated mutations is ubiquitous in healthy adults. Nat Commun. 2016;7:12484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Steensma DP, Bejar R, Jaiswal S, et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood. 2015;126(1):9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Jaiswal S, Natarajan P, Silver AJ, et al. Clonal Hematopoiesis and Risk of Atherosclerotic Cardiovascular Disease. The New England journal of medicine. 2017;377(2):111–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haitjema S, Kofink D, van Setten J, et al. Loss of Y Chromosome in Blood Is Associated With Major Cardiovascular Events During Follow-Up in Men After Carotid Endarterectomy. Circ Cardiovasc Genet. 2017;10(4):e001544. [DOI] [PubMed] [Google Scholar]
- 15.Loftfield E, Zhou W, Graubard BI, et al. Predictors of mosaic chromosome Y loss and associations with mortality in the UK Biobank. Sci Rep. 2018;8(1):12316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Terao C, Suzuki A, Momozawa Y, et al. Chromosomal alterations among age-related haematopoietic clones in Japan. Nature. 2020;584(7819):130–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Genovese G, Kahler AK, Handsaker RE, et al. Clonal hematopoiesis and blood-cancer risk inferred from blood DNA sequence. The New England journal of medicine. 2014;371(26):2477–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jaiswal S, Fontanillas P, Flannick J, et al. Age-related clonal hematopoiesis associated with adverse outcomes. The New England journal of medicine. 2014;371(26):2488–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sano S, Oshima K, Wang Y, et al. Tet2-Mediated Clonal Hematopoiesis Accelerates Heart Failure Through a Mechanism Involving the IL-1beta/NLRP3 Inflammasome. Journal of the American College of Cardiology. 2018;71(8):875–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dorsheimer L, Assmus B, Rasper T, et al. Association of Mutations Contributing to Clonal Hematopoiesis With Prognosis in Chronic Ischemic Heart Failure. JAMA Cardiol. 2019;4(1):25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.*.Abplanalp WT, Cremer S, John D, et al. Clonal Hematopoiesis-Driver DNMT3A Mutations Alter Immune Cells in Heart Failure. Circ Res. 2021;128(2):216–28. [DOI] [PubMed] [Google Scholar]; This investigation using single-cell RNA-Seq data from patient-derived heart samples demonstrated increased expression of inflammatory signaling molecules among leukocytes with DNMT3A mutations, supporting several experimental studies that suggest CHIP contributes to disease processes via pro-inflammatory innate immune cells.
- 22.Abplanalp WT, Mas-Peiro S, Cremer S, et al. Association of Clonal Hematopoiesis of Indeterminate Potential With Inflammatory Gene Expression in Patients With Severe Degenerative Aortic Valve Stenosis or Chronic Postischemic Heart Failure. JAMA Cardiol. 2020;5(10):1170–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Assmus B, Cremer S, Kirschbaum K, et al. Clonal haematopoiesis in chronic ischaemic heart failure: prognostic role of clone size for DNMT3A- and TET2-driver gene mutations. Eur Heart J. 2021;42(3):257–65. [DOI] [PubMed] [Google Scholar]
- 24.Cremer S, Kirschbaum K, Berkowitsch A, et al. Multiple Somatic Mutations for Clonal Hematopoiesis Are Associated With Increased Mortality in Patients With Chronic Heart Failure. Circ Genom Precis Med. 2020;13(4):e003003. [DOI] [PubMed] [Google Scholar]
- 25.Kiefer KC, Cremer S, Pardali E, et al. Full spectrum of clonal haematopoiesis-driver mutations in chronic heart failure and their associations with mortality. ESC Heart Fail. 2021;8(3):1873–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.**.Yu B, Roberts MB, Raffield LM, et al. Supplemental Association of Clonal Hematopoiesis With Incident Heart Failure. Journal of the American College of Cardiology. 2021;78(1):42–52. [DOI] [PMC free article] [PubMed] [Google Scholar]; This study leveraged the TOPMed cohort to provide epidemiologic evidence that clonal hematopoiesis of indeterminant potential is associated with the onset of heart failure. Interestingly, this study demonstrated that some but not all driver genes were individually associated with an increased risk of developing heart failure, suggesting CHIP may be associated with disease pathogenesis in a mutation-specific manner.
- 27.Yura Y, Miura-Yura E, Katanasaka Y, et al. The Cancer Therapy-Related Clonal Hematopoiesis Driver Gene Ppm1d Promotes Inflammation and Non-Ischemic Heart Failure in Mice. Circ Res. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sano S, Oshima K, Wang Y, et al. CRISPR-Mediated Gene Editing to Assess the Roles of Tet2 and Dnmt3a in Clonal Hematopoiesis and Cardiovascular Disease. Circ Res. 2018;123(3):335–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sano S, Wang Y, Ogawa H, et al. TP53-mediated therapy-related clonal hematopoiesis contributes to doxorubicin-induced cardiomyopathy by augmenting a neutrophil-mediated cytotoxic response. JCI Insight. 2021;6(13). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sano S, Wang Y, Yura Y, et al. JAK2 (V617F) -Mediated Clonal Hematopoiesis Accelerates Pathological Remodeling in Murine Heart Failure. JACC Basic Transl Sci. 2019;4(6):684–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Min KD, Polizio AH, Kour A, et al. Experimental ASXL1-Mediated Clonal Hematopoiesis Promotes Inflammation and Accelerates Heart Failure. J Am Heart Assoc. 2022;11(19):e026154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zekavat SM, Viana-Huete V, Matesanz N, et al. TP53-mediated clonal hematopoiesis confers increased risk for incident atherosclerotic disease. Nat Cardiovasc Res. 2023: 10.1038/s44161-022-00206-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Fidler TP, Xue C, Yalcinkaya M, et al. The AIM2 inflammasome exacerbates atherosclerosis in clonal haematopoiesis. Nature. 2021;592(7853):296–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Thompson DJ, Genovese G, Halvardson J, et al. Genetic predisposition to mosaic Y chromosome loss in blood. Nature. 2019;575(7784):652–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Forsberg LA, Halvardson J, Rychlicka-Buniowska E, et al. Mosaic loss of chromosome Y in leukocytes matters. Nat Genet. 2019;51(1):4–7. [DOI] [PubMed] [Google Scholar]
- 36.Dumanski JP, Rasi C, Lönn M, et al. Mutagenesis. Smoking is associated with mosaic loss of chromosome Y. Science (New York, NY). 2015;347(6217):81–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Jobling MA, Tyler-Smith C. Human Y-chromosome variation in the genome-sequencing era. Nat Rev Genet. 2017;18(8):485–97. [DOI] [PubMed] [Google Scholar]
- 38.Wright DJ, Day FR, Kerrison ND, et al. Genetic variants associated with mosaic Y chromosome loss highlight cell cycle genes and overlap with cancer susceptibility. Nat Genet. 2017;49(5):674–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dumanski JP, Halvardson J, Davies H, et al. Immune cells lacking Y chromosome show dysregulation of autosomal gene expression. Cell Mol Life Sci. 2021;78(8):4019–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Svensson EC, Madar A, Campbell CD, et al. TET2-Driven Clonal Hematopoiesis and Response to Canakinumab: An Exploratory Analysis of the CANTOS Randomized Clinical Trial. JAMA Cardiol. 2022;7(5):521–8. [DOI] [PMC free article] [PubMed] [Google Scholar]