

Abstract
Background:
The tyrosine kinase receptors Axl and MerTK are highly overexpressed in head and neck cancer (HNC) cells, where they are critical drivers of survival, proliferation, metastasis, and therapeutic resistance.
Methods:
We investigated the role of Axl and MerTK in creating an immunologically ‘cold’ tumor immune microenvironment (TIME) by targeting both receptors simultaneously with a small molecule inhibitor of Axl and MerTK (INCB081776). Effects of INCB081776 and/or anti-PDL1 on mouse oral cancer (MOC) cell growth and on the TIME were evaluated.
Results:
Targeting Axl and MerTK can reduce M2 and induce M1 macrophage polarization. In vivo, INCB081776 treatment alone or with anti-PDL1 appears to slow MOC tumor growth, increase pro-inflammatory immune infiltration, and decrease anti-inflammatory immune infiltration.
Conclusions:
This data indicates that simultaneous targeting of Axl and MerTK with INCB081776, either alone or in combination with anti-PDL1, slows tumor growth and creates a pro-inflammatory TIME in mouse models of HNC.
Keywords: Axl, MerTK, head and neck cancer, PDL1, tumor microenvironment
Introduction
In the United States, there are approximately 54,000 newly diagnosed head and neck cancer (HNC) patients each year, with over 11,000 associated deaths1. HNC is a highly complex and heterogeneous disease, arising from multiple sites in the mucosal lining of the aerodigestive tract including the oral cavity, tongue, salivary glands, pharynx, and larynx. These tumors are typically associated with alcohol and/or tobacco use, but human papillomavirus (HPV) has been discovered to cause a subset of HNC tumors2. Standard treatment approaches for HNC currently include surgery, radiation, and chemotherapy, with several immune checkpoint inhibitors (ICIs) being recently approved3,4. However, the prognosis for HNC has remained relatively unchanged for decades; the five-year relative survival rate between 2011–2017 was only 67%1.
Progression of solid-tumor cancers is determined not only by the oncogenic alterations within the tumor cells themselves but also by the makeup of the environment that surrounds the tumor cells5,6. The tumor immune microenvironment (TIME) typically includes fibroblasts, endothelial cells, and immune cells from both the innate and adaptive immune systems5. The typical TIME is considered to be immunosuppressive as most infiltrating immune cells, including regulatory T (Treg) cells, and M2 macrophages, are anti-inflammatory5–7. However, some cancers, including melanoma and non-small cell lung cancer (NSCLC), have relatively “hot” TIMEs with high levels of T cell and other pro-inflammatory cell infiltration5,8,9. These cancers also typically have higher response rates to ICIs, such as those targeting programmed death ligand 1 (PDL1)8,10. Unfortunately, response rates to ICIs even in these hot cancers are rarely greater than 40%, and most patients that initially respond will eventually develop resistance11,12. Cancers with “cold” TIMEs, characterized by a lack of T cell infiltration, have much lower ICI response rates. HNC patients, for example, initially respond to ICIs only about 15% of the time13, and face similar issues with acquired resistance. Therapeutic strategies to turn the TIME from cold to hot are therefore desperately needed.
Polarization of macrophages in the TIME plays a key role in setting the “temperature” of the environment. M2 macrophages are known to secrete, among other things, IL-10, an anti-inflammatory cytokine that can inhibit T cell proliferation14,15. M1 macrophages, in turn, secrete pro-inflammatory cytokines such as IL-6 and IL-1β14. Behavior of macrophages in non-cancer and cancer settings is at least partially regulated by Tyro3, Axl, and MerTK, the TAM family of receptor tyrosine kinases (RTKs)16. The TAM family is essential for macrophage-facilitated phagocytosis of apoptotic cells in normal tissue16. Growth arrest specific 6 (Gas6) and Protein S, the ligands for the TAMs, bind to phosphatidylserine residues on the surface of apoptotic cells; therefore, the TAMs act as a bridge between the phagocyte and the dying cell, facilitating efficient efferocytosis16.
The TAMs also play a critical role in macrophage polarization in solid tumors. Specifically, TAM receptor signaling inhibits M1 (anti-tumor) polarization and induces M2 (pro-tumor) polarization14. In addition to these roles in macrophages, previous work from our laboratory showed that Axl and MerTK cooperate to drive HNC via tumor intrinsic mechanisms17–20. Axl and MerTK are often overexpressed in HNC as compared to normal oral tissue17,21. In one study, Axl expression was found in 82% of HNC patient-derived xenografts (PDXs)17, while another study discovered MerTK expression in nearly 70% of HNSCC tumors21. Axl and MerTK signaling leads to cancer cell proliferation, survival, metastasis, and resistance to chemotherapeutics17–20. Additionally, tumor-bound Axl was recently implicated in creating a cold TIME22, suggesting that TAMs can impact tumors by both intrinsic and extrinsic mechanisms.
In this study, we investigate the effects of Axl and MerTK signaling on the tumor immune microenvironment in HNC. Using a novel kinase inhibitor targeting both Axl and MerTK (INCB081776)23, we demonstrate that simultaneous inhibition of Axl and MerTK leads to a pro-inflammatory, or ‘hot’ TIME in HNC, and that this inhibition of TAM signaling can enhance efficacy of ICI therapies.
Materials and Methods
Therapeutics:
INCB081776 was provided by Incyte Corporation (Wilmington, DE, USA) under an MTA. R428 and MRX2843 were obtained from MedChemExpress (Monmouth Junction, NJ, USA). Antibodies for in vivo use were purchased from BioXCell (Lebanon, NH, USA): rat IgG2b isotype control (#BE0090) and anti-PDL1 (#BE0101).
Cell Lines and Patient-Derived Xenografts (PDXs):
The mouse oral cancer MOC1, MOC2, and MOC22 cell lines were obtained from Dr. Ravindra Uppaluri (Dana-Farber Cancer Institute, Boston, MA, USA), and were maintained in DMEM/Ham’s F12 media (2:1) with 5% fetal bovine serum (FBS), 1% penicillin/streptomycin, 500ng/mL hydrocortisone, 0.5ng/mL murine epidermal growth factor (Thermo Fisher Scientific, Waltham, MA, USA), and 5ug/mL insulin (MilliporeSigma, St. Louis, MO, USA)24. The Chinese hamster ovary (CHO) parental, CHO-EGFR/Axl, and CHO-EGFR/MerTK cell lines were provided by Dr. Raymond B. Birge (Rutgers New Jersey Medical School, Newark, NJ, USA) and maintained in Ham’s F12 media with 10% FBS and 1% penicillin/streptomycin25. CHO-EGFR/Axl and CHO-EGFR/MerTK were also treated with 400ug/mL G418. The UWSCC1 and UWSCC64 PDXs were obtained from University of Wisconsin-Madison HNC SPORE resources. Cell Counting Kit 8 (Dojindo Molecular Technologies, Rockville, MD, USA) was used to assay cell viability according to manufacturer’s instructions.
Immunoblot Analysis:
Whole-cell lysate was obtained from cells using RIPA buffer (50mM HEPES, pH 7.4, 150mM NaCl, 0.1% Tween-20, 10% glycerol, 2.5mM EGTA, 1mM EDTA, 1mM DTT, 1mM Na3VO4, 1mM PMSF, 1mM beta-glycerophosphate (BGP), 10ug/mL leupeptin, and 10ug/mL aprotinin). Immunoblot analysis was performed as previously described20. Antibodies (Supplemental Table S1) were used according to the manufacturer’s instructions.
Bone Marrow Derived Macrophage (BMDM) Generation:
Animal procedures and maintenance were conducted in accordance with the University of Wisconsin-Madison School of Medicine and Public Health IACUC guidelines. Female C57BL/6N mice (Charles River Laboratories, Wilmington, MA, USA) were euthanized by CO2 asphyxiation, followed by cervical dislocation. The tibia and femur were removed and separated, and the epiphysis of each bone was removed. Both bones were placed, cut side down, in a 0.65mL tube that had a hole pierced in the bottom using a 25G needle. The 0.65mL tube was placed inside a 1.5mL tube, and both tubes and the bones were centrifuged at maximum speed for 30 seconds. The bone marrow pellet was resuspended in complete DMEM (4.5g/dL glucose, 10% FBS, 1% penicillin/streptomycin) and passed through a 70um strainer. Cells were centrifuged at 300xg for 10 minutes. Red blood cells were lysed using ACK Lysing Buffer (Thermo Fisher Scientific) for 5 minutes, and remaining cells were washed and plated in complete DMEM containing 20ng/mL murine macrophage colony stimulating factor (MCSF; PeproTech, Cranbury, NJ, USA). Cells were incubated at 37°C, 5%CO2 for seven days.
BMDM Assays:
BMDMs (M0 macrophages) were plated and maintained in complete DMEM (4.5g/dL glucose, 10% FBS, 1% penicillin/streptomycin) with 10ng/mL murine MCSF (PeproTech). For M1 macrophage polarization, BMDMs were treated with 10ng/mL lipopolysaccharide (LPS; MilliporeSigma) and 50ng/mL murine interferon gamma (IFNγ; PeproTech) for 24 hours. For M2 macrophage polarization, BMDMs were treated with 25ng/mL murine IL-4 (PeproTech) and 25ng/mL murine IL-13 (PeproTech) for 24 hours. Cytokines were removed and macrophages were treated with 6uM INCB081776 or with vehicle for 24 hours. At the end of the indicated times, macrophages were lysed using RLT Lysis Buffer from RNeasy Mini Kit (Qiagen, Hilden, Germany).
RNA Isolation, cDNA Synthesis, and qPCR:
Total RNA was isolated from cells using the RNeasy Mini Kit (Qiagen). The RNA was then quantified by a NanoDrop spectrophotometer (Thermo Fisher Scientific). The RNA was reverse transcribed to cDNA using qScript cDNA SuperMix (Quantabio, Beverly, MA, USA). Quantitative real time PCR (qPCR) was carried out using either the Bio-Rad CFX96 Real-Time PCR System (Bio-Rad, Hercules, CA, USA) or the QuantStudio 6 Pro (Thermo Fisher Scientific). TaqMan Fast Advanced Master Mix and TaqMan probes (Thermo Fisher Scientific) were used (Supplemental Table S2). The levels of gene expression were analyzed using the ΔΔCT method. Eef1a1 and Ubc were used as the normalization controls for macrophage polarization analysis, and GAPDH and Ubc were used for normalization of the cytokine production analysis.
IsoPlexis:
MOC2 cells were treated with 6uM of INCB081776 or vehicle for 24 hours, after which they were treated with 18ng/mL of Gas6 (R&D Systems, Minneapolis, MN, USA) or vehicle for 6 hours. Supernatant from each treatment group was collected and analyzed with CodePlex Secretome chips (Mouse Inflammation panel) on the IsoSpark Duo (IsoPlexis, Branford, CT, USA). Results were analyzed using the IsoSpeak software (IsoPlexis).
In vivo Mouse Experiments:
Animal procedures and maintenance were conducted in accordance with the University of Wisconsin-Madison School of Medicine and Public Health IACUC guidelines. PDXs were propagated using R2G2 mice (Envigo, Indianapolis, IN, USA), then inoculated by subcutaneous injection into the dorsal flank of female athymic nude mice (Envigo). MOC cell lines were inoculated by subcutaneous injection into the dorsal flank of 7–8 weeks old female C57BL/6N (Charles River Laboratories) or female athymic nude mice (Envigo). All cells were resuspended in PBS containing Matrigel (50% v/v, Corning Inc., Corning, NY, USA). When tumors attained a volume of ~200mm3, as measured by a caliper, mice were randomized into groups and treatment was initiated. Inhibitor or equivalent volume of vehicle was administered by intraperitoneal injection (anti-PDL1 or IgG), oral gavage (R428, MRX2843, INCB081776), or chow (INCB081776).
Tumor Dissociation and Flow Cytometry:
Animal procedures and maintenance were conducted in accordance with the University of Wisconsin-Madison School of Medicine and Public Health IACUC guidelines. Tumors were extracted following euthanasia and placed into a 5mL tube containing 1mL of dissociation buffer (1X HBSS, 5% FBS). Each tumor was minced into fine pieces using sterile scissors, and an additional 4mL of dissociation buffer was added. Tumor pieces were transferred to a gentleMACS C Tube (Miltenyi Biotec, Bergisch Gladbach, Germany) and loaded into a gentleMACS Octo Dissociator with Heaters (Miltenyi Biotec). Cells were dissociated using the 37C_mTDK1 program. Cells were then passed through a 70um strainer and washed with PBS. Cells were stained with live/dead dye on ice for 30 minutes, washed, and resuspended in flow buffer (PBS without Ca/Mg, 2% FBS, 2mM EDTA, 25ug/mL DNase I) containing Fc Block (Biolegend, San Diego, CA, USA). Cells were then stained with fluorescent antibodies on ice for 30 minutes. Cells were washed and resuspended in flow buffer. If intracellular staining was needed, cells were then permeabilized overnight using the FoxP3/Transcription Factor Staining Buffer Kit (Tonbo Biosciences) according to manufacturer instructions. Cells were then stained with fluorescent intracellular antibodies at room temperature for 30 minutes. Cells were washed and resuspended in flow buffer. UltraComp beads (Thermo Fisher Scientific) were stained with fluorescent antibodies for 15–30 minutes on ice. Beads were washed and resuspended in flow buffer. Rainbow calibration particles (Spherotech, Lake Forest, IL, USA) were used to normalize across experiments. All samples were run on an Attune NxT flow cytometer (Thermo Fisher Scientific). Analysis was performed in FlowJo (BD, Franklin Lakes, NJ; RRID:SCR_008520). Supplemental Table S3 details the fluorescent antibodies used, and Supplemental Table S4 provides gating paths.
Immunohistochemistry (IHC):
Animal procedures and maintenance were conducted in accordance with the University of Wisconsin-Madison School of Medicine and Public Health IACUC guidelines. Tumor tissue samples were collected, fixed in 10% neutral buffered formalin, and paraffin embedded. Antigen retrieval was performed by heating the sections in 10mM citrate buffer (pH 6.0) in a decloaking chamber. Samples were incubated with rat anti-CD8a (Thermo Fisher Scientific #14080882, 1:500), rat anti-CD4 (Thermo Fisher Scientific #14976682, 1:500), or rat anti-FoxP3 (Thermo Fisher Scientific #14577382, 1:200). Sections were stained using the ImmPRESS HRP Goat Anti-Rat IgG Polymer Detection Kit (Vector Laboratories, Burlingame, CA, #MP-7404–50). Antibody binding was revealed by the addition of 3,3’-diaminobenzidine substrate (Vector Laboratories). Tissues were counterstained with Mayer’s hematoxylin (Thermo Fisher Scientific). Tissues were examined using an Olympus BX51 microscope. Quantitation of staining intensity was performed as previously described26 using FIJI.
Statistical Analysis:
Statistical analyses were performed using GraphPad Prism (GraphPad Software, Inc.; RRID:SCR_002798). Differences were considered significant when P < 0.05.
To analyze the longitudinal tumor growth data (volumes) for each study, log-transformed tumor growth over time was modeled and compared between treatment groups using linear mixed-effects models, in which individual samples were modeled as a random effect, while treatment groups, time and their interaction were modelled as fixed effects. Specifically, the fixed-effect model matrices were parameterized such that all main and interactive effects between respective treatment groups and time were estimated. Intra-tumor correlation was accommodated via random intercepts. Relative Ratios (RR) were reported for each treatment group relative to the vehicle group. For studies with two-drug experiments, the synergistic effect of the combination treatment was assessed through a three-way interaction between time, the first drug, and the second drug. R (v. 4.1.1; R Core Team 2021) and package ‘lme4’ (v. 1.1.27.1; Bates et. al. 2015) were used for analysis, and ‘lmerTest’ (v. 3.1.3; Kuznetsova et. al. 2017) was used to calculate p-values using Satterthwaite approximation.
Data Availability:
The data generated in this study are available upon request from the corresponding author.
Results
Axl and MerTK kinase activity in HNC can be inhibited by INCB081776
The ability of INCB081776 to inhibit signaling through Axl and MerTK was first tested using genetically modified Chinese hamster ovary (CHO) cell lines25. These cell lines stably expressed a chimeric receptor, where the extracellular domain of the epidermal growth factor receptor (EGFR) was fused to the intracellular kinase domain of either Axl or MerTK (Figure 1A&B). Stimulation of cells with EGF led to phosphorylation of the intracellular Axl/MerTK domains, but pre-incubation with INCB081776 efficiently inhibited activation of Axl/MerTK directly, as well as downstream survival pathway (Akt) activation (Figures 1C&D). Next, we confirmed that the MOC models24 being used expressed Axl and MerTK (Figure 1E). The three MOC lines have been extensively characterized; MOC2 exhibits a fast-growing, immune-evasive phenotype, while MOC1 and MOC22 exhibit a slow-growing, immunogenic phenotype27. We then tested the ability of INCB081776 to inhibit basal level Axl and MerTK phosphorylation in the MOC cell lines (Figure 1F). Phosphorylation of Axl was inhibited in both MOC1 and MOC2, and phosphorylation of MerTK and Akt was inhibited by INCB081776 in all three MOC cell lines. The LC50 of INCB081776 in each MOC cell line was also determined (Supplemental Figure S1), with an average LC50 of ~11uM. Collectively, these data indicate that INCB081776 is an efficient inhibitor of Axl/MerTK activation and signaling.
Figure 1: Axl and MerTK kinase activity in HNC can be inhibited by INCB081776.
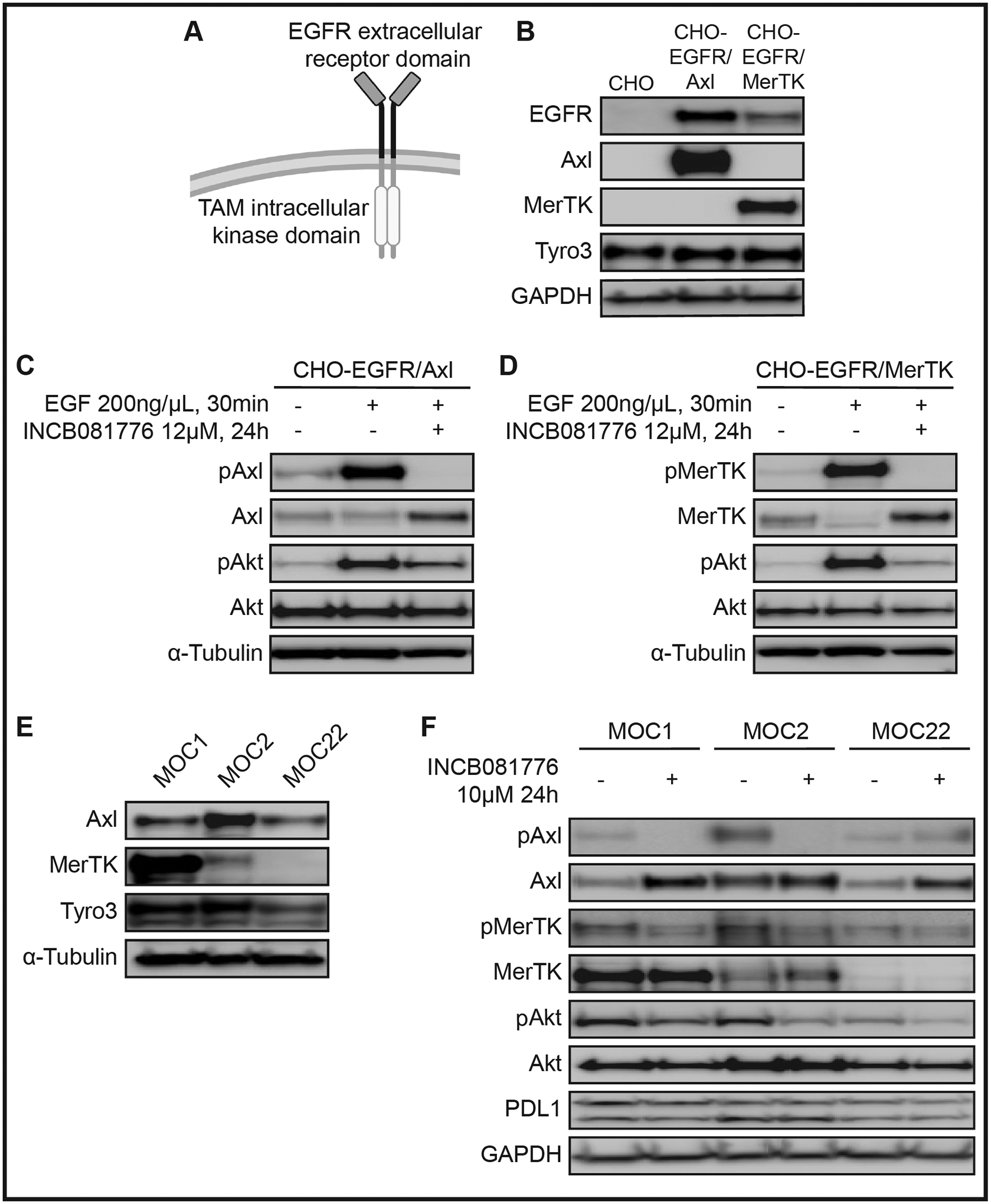
(A) Chinese hamster ovary (CHO) cell lines expressing chimeric receptors. The extracellular receptor domain of epidermal growth factor receptor (EGFR) is fused to the intracellular kinase domain of a TAM-family RTK, either Axl or MerTK. (B) Whole cell lysates from the parental (CHO) or chimeric (CHO-EGFR/Axl, CHO-EGFR/MerTK) cell lines were harvested and fractionated via SDS-PAGE, followed by immunoblotting for the indicated proteins. GAPDH was the loading control. CHO-EGFR/Axl (C) or CHO-EGFR/MerTK (D) cells were treated with either vehicle or 12μM INCB081776 for 24 hours, then treated with vehicle or 200ng/μL epidermal growth factor (EGF) for 30 minutes. Whole cell lysates were harvested and fractionated via SDS-PAGE, followed by immunoblotting for the indicated proteins. α-Tubulin was the loading control. (E) Whole cell lysates were harvested and fractionated via SDS-PAGE, followed by immunoblotting for the indicated proteins. Tubulin was the loading control. (F) Cells were treated with 10μM INCB081776 for 24 hours. Whole cell lysates were harvested and fractionated via SDS-PAGE, followed by immunoblotting for the indicated proteins. GAPDH was the loading control.
INCB081776 affects tumor growth in human and mouse models of HNC
Previously, we reported that MerTK mediates resistance to Axl targeting strategies, and we proposed that targeting both Axl and MerTK simultaneously could improve treatment outcomes19. Therapeutics targeting either Axl (R42828) or MerTK (MRX284329) alone have previously been developed, and we directly compared the efficacy of those two single-target therapies to the dual-target inhibitor INCB081776. We implanted tumors from a PDX model of HNC that expresses Axl and MerTK (UWSCC1) into nude mice and treated with both single-target therapies (R428 and MRX2843), alone and in combination, and with the dual-target therapy INCB081776 (Figure 2A). Neither R428 nor MRX2843, when given alone, had any impact on tumor growth. However, when R428 and MRX2843 were given together, they significantly slowed tumor growth, just as INCB081776 treatment did. To strengthen these findings, we tested INCB081776 on tumors from a second Axl/MerTK-expressing PDX model of HNC (UWSCC64, Figure 2B) in nude mice. As in UWSCC1, treatment with INCB081776 significantly slowed tumor growth in UWSCC64. Together, this data suggests that single-target agents against either Axl or MerTK were insufficient to slow tumor growth. INCB081776, a dual-target therapy, was able to significantly slow tumor growth, suggesting that tumor-bound Axl/MerTK play a critical role in driving HNC tumor growth. Finally, to determine if INCB081776 could impair tumor growth in syngeneic model systems, we treated MOC1 and MOC2 tumors implanted in C57BL/6N mice with INCB081776. As in the PDX models, INCB081776 treatment significantly slowed tumor growth in syngeneic models of HNC (Figures 2C–D). Collectively, this data suggests that both human and mouse HNC tumor growth is slowed by INCB081776 treatment.
Figure 2: INCB081776 affects tumor growth in human and mouse models of HNC.
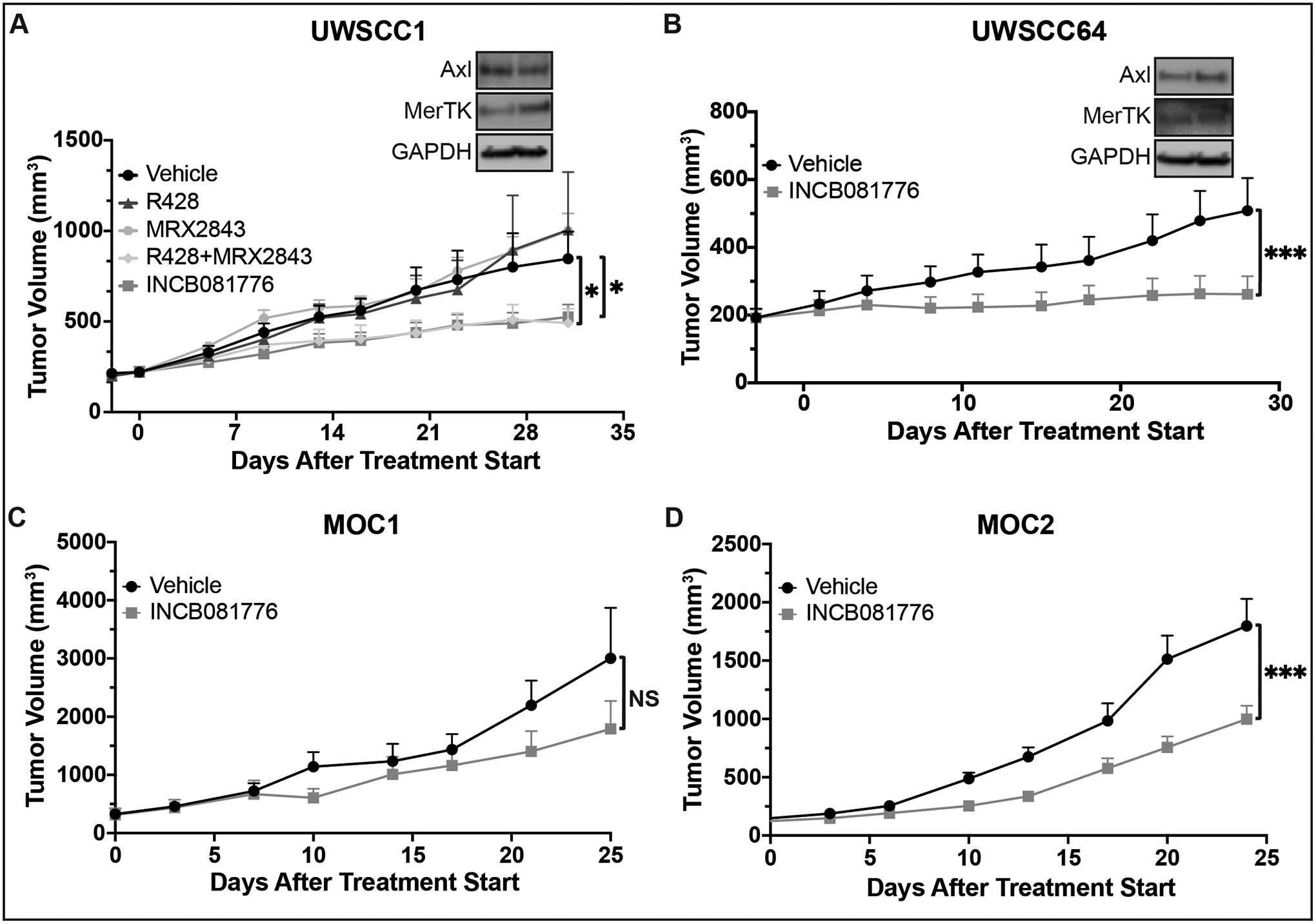
(A-B) Subcutaneous xenografts were generated in nude mice using the UWSCC1 (A) and UWSCC64 (B) HNC cell lines. Whole cell lysates were harvested and fractionated via SDS-PAGE, followed by immunoblotting for the indicated proteins. GAPDH was the loading control. (A) Tumor-bearing mice were treated with vehicle, 25mg/kg R428, 12.5–20mg/kg MRX2843, the combination of R428 and MRX2843, or 30mg/kg INCB081776, administered twice daily via oral gavage (BID). (B) Tumor-bearing mice were treated with vehicle or 30mg/kg BID INCB081776. (C-D) Subcutaneous tumors were generated in syngeneic (C57BL/6N) mice using the MOC1 (C) and MOC2 (D) HNC cell lines. (C) Tumor-bearing mice were treated with vehicle or 30mg/kg BID INCB081776. (D) Tumor-bearing mice were treated with vehicle- or INCB081776-containing chow (equivalent to 200mg/kg/day). There was no difference in efficacy when INCB081776 was dosed at 30mg/kg BID or in chow (data not shown). (A-D) Tumor volume was measured (n = 6–12 per group). Mean values and SEMs are shown. *, P < 0.05; ***, P < 0.001; NS, not significant.
Targeting Axl/MerTK signaling leads to an anti-tumor cytokine signature
Given that inhibition of Axl and MerTK signaling led to decreased tumor growth, we next sought to understand how Axl/MerTK signaling on the tumor cell was leading to a pro-tumor environment. The secretion of cytokines such as TNFα30,31, GMCSF32,33, IL-1β34–36, and MCP-137 have long been implicated in the progression of HNC, so we considered that Axl and MerTK signaling could be contributing to the production of these cytokines. We measured expression of cytokines in MOC1 and MOC2 following inhibition of Axl/MerTK signaling with INCB081776 (Figure 3A–B), finding that treatment with INCB081776 decreased expression of all four cytokines analyzed. When implanted in syngeneic mice, MOC2 cells produce faster-growing and more aggressive tumors than MOC1. To examine the effects of INCB081776 on HNC tumor progression more closely, we treated MOC2 cells with the natural Axl/MerTK ligand, Gas6, alone and in combination with INCB081776 and measured the effects on secretion of GMCSF and MCP-1 via IsoPlexis (Figure 3C–D). Surprisingly, secretion of GMCSF was only significantly decreased following treatment with the combination of Gas6 and INCB081776 (Figure 3C). Levels of MCP-1 were significantly decreased following treatment with INCB081776, whether it was given alone or in combination with Gas6 (Figure 3D). Collectively, these data show that inhibition of Axl/MerTK signaling with INCB081776 leads to a decrease in the expression and secretion of pro-tumor cytokines.
Figure 3: INCB081776 decreases levels of pro-tumor cytokines in MOC cells.
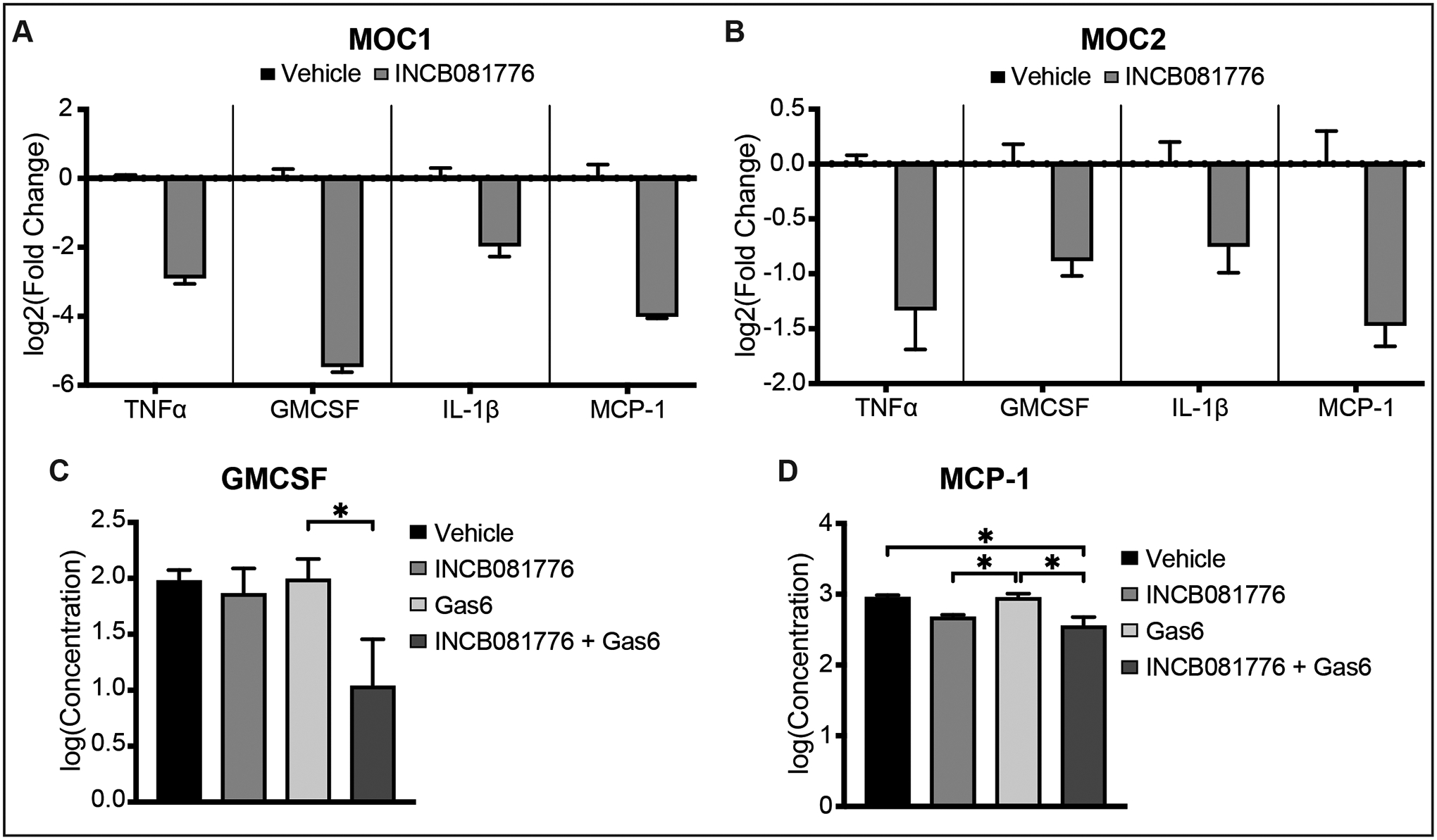
(A-B) Cells were treated with vehicle or 10μM INCB081776 for 24 hours. The expression levels of the indicated genes were determined by qPCR. Mean values and SEMs are shown (n = 3 per group) and are representative of three independent experiments. (C-D) MOC2 cells were treated with vehicle or 10μM INCB081776 for 24 hours, then treated with vehicle or 18ng/mL Gas6 for 6 hours. Supernatant was collected and levels of GMCSF (C) or MCP-1 (D) were determined by IsoPlexis. Levels of TNFα and IL-10 were below the limit of detection for the assay. Mean values and SEMs are shown (n = 2 per group) and are representative of two independent experiments. *, P < 0.05.
INCB081776 treatment can prevent M2 polarization and induce M1 polarization
In the non-cancer setting, Axl and MerTK are found primarily on macrophages, where they regulate polarization status38. We hypothesized that targeting Axl and MerTK signaling using INCB081776 could affect macrophage polarization. We first tested naïve macrophages (M0s), adding INCB081776 for 30 hours and measuring the expression of signature M1 (anti-tumor) and M2 (pro-tumor) genes via qPCR (Figures 4A–C). Treatment with INCB081776 increased expression of the M1-associated genes (Figure 4B) and decreased expression of M2-associated genes (Figure 4C). Next, using either M0s or macrophages polarized to an M2 state (M2s), we repeated the treatment with INCB081776 (Figure 4D). Again, we showed that naïve macrophages treated with INCB081776 had increased expression of the M1-associated genes (Figure 4E) and decreased expression of the M2-associated genes (Figure 4F). Interestingly, macrophages already polarized to the M2 state had increased expression of half of the M1 genes (Figure 4G) and decreased expression of M2 genes (Figure 4H) following treatment with INCB081776. Collectively, these data suggest that targeting Axl/MerTK signaling with INCB081776 can shift macrophage polarization to an anti-tumor, pro-inflammatory state.
Figure 4: INCB081776 prevents M2 polarization and induces M1 polarization.
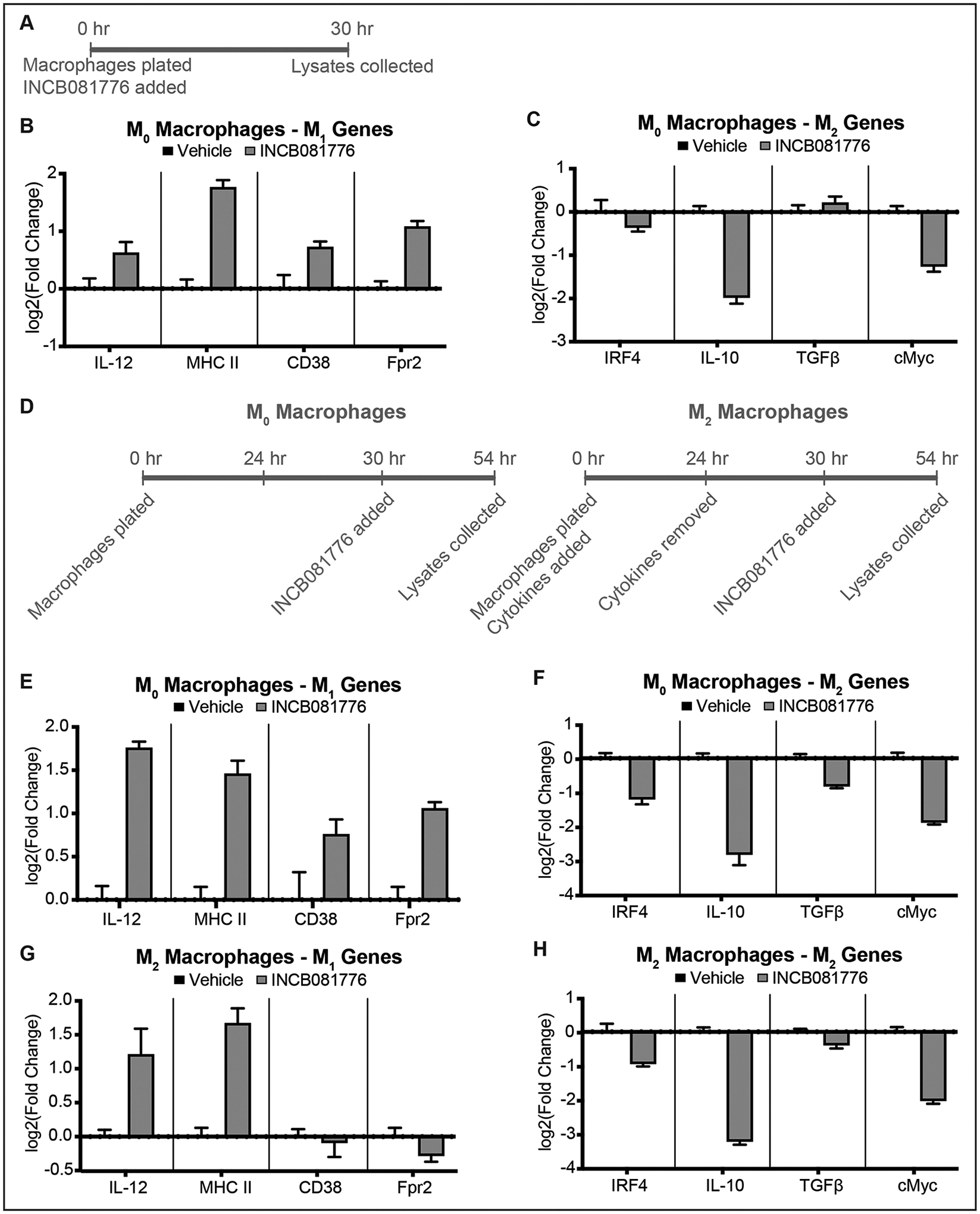
(A) Experimental timeline for (B) and (C). (B-C) Bone marrow derived macrophages (BMDMs) were treated with vehicle or 6μM INCB081776 for 24 hours. The expression levels of M1-associated genes (B) or M2-associated genes (C) were determined by qPCR. Mean values and SEMs are shown (n = 3 per group) and are representative of three independent experiments. (D) Experimental timeline for (E-H). (E-H) BMDMs were treated with vehicle (E-F) or 25ng/mL each of IL-4 and IL-13 (G-H) for 24 hours, then treated with 6μM INCB081776 for 24 hours. The expression levels of M1-associated genes (E, G) or M2-associated genes (F, H) were determined by qPCR. Mean values and SEMs are shown (n = 3 per group) and are representative of three independent experiments.
INCB081776 creates a hotter TIME in mouse HNC tumors
To understand if inhibition of Axl/MerTK signaling leads to a more pro-inflammatory, or “hotter” TIME in vivo, we utilized the MOC1 and MOC2 cell lines in syngeneic mice. MOC1 tumors have been characterized as “hot”, with relatively more pro-inflammatory immune infiltrate than their “cold” counterparts, MOC2 tumors27. Despite these baseline differences, both MOC1 and MOC2 tumors were more highly infiltrated with pro-inflammatory immune cells following treatment with INCB081776 (Figures 5–6).
Figure 5: INCB081776 creates a pro-inflammatory TIME in MOC1 tumors.
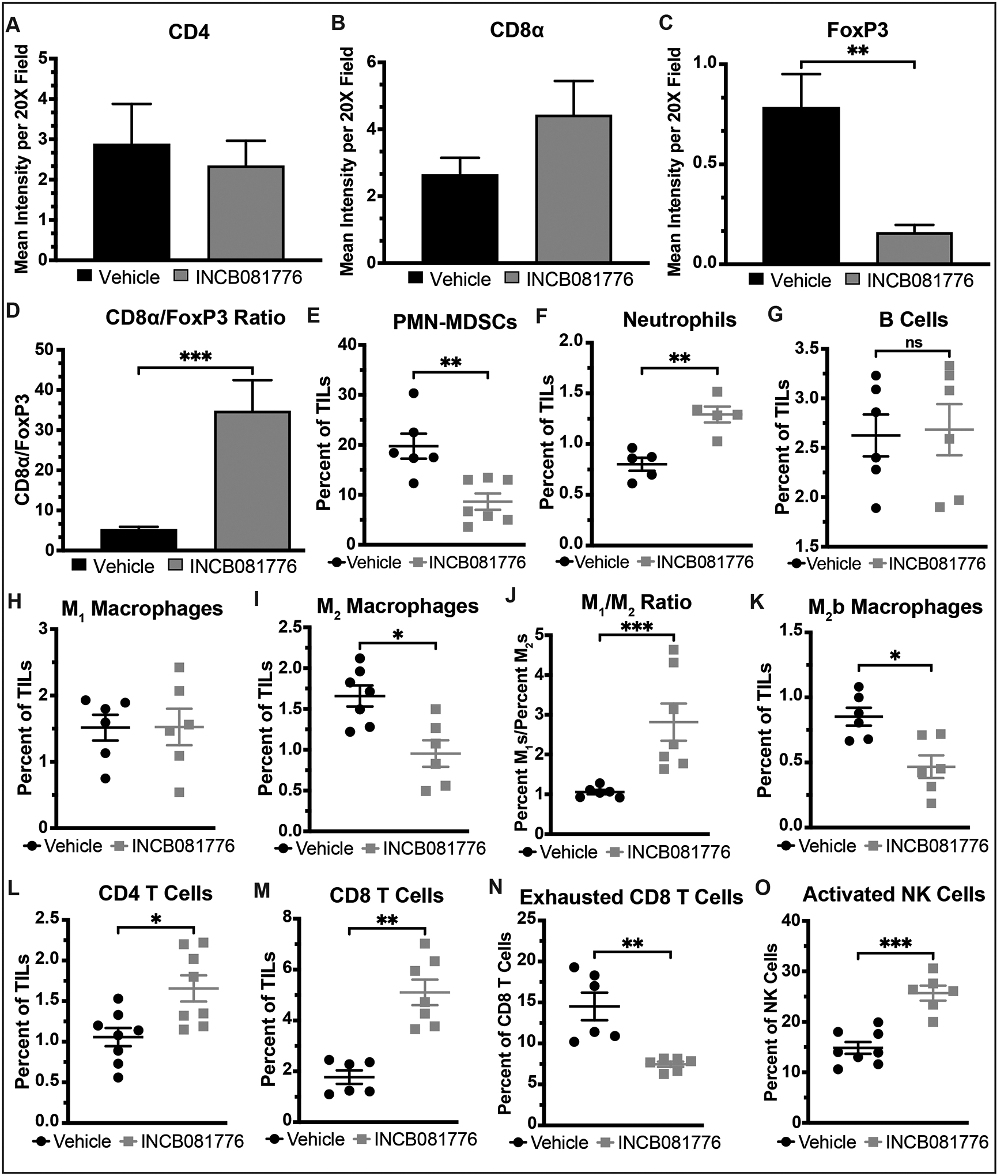
(A-O) Subcutaneous tumors were generated in syngeneic (C57BL/6N) mice using the MOC1 cell line. Tumor-bearing mice were treated with vehicle or INCB081776 via chow (~200mg/kg/day) for 7 days. (A-D) Tumors were stained using IHC. Image quantification was performed, and the CD8α/FoxP3 ratio was calculated. Mean values and SEMs are shown (n = 5–10 per group). **, P < 0.01; ***, P < 0.001. Representative images are shown in Supplemental Figure S2. (E-O) Tumor-infiltrating lymphocytes (TILs) were analyzed via flow cytometry. Mean values and SEMs are shown (n = 5–8 per group). *, P < 0.05; **, P < 0.01; ***, P < 0.001; NS, not significant.
Figure 6: INCB081776 creates a pro-inflammatory TIME in MOC2 tumors.
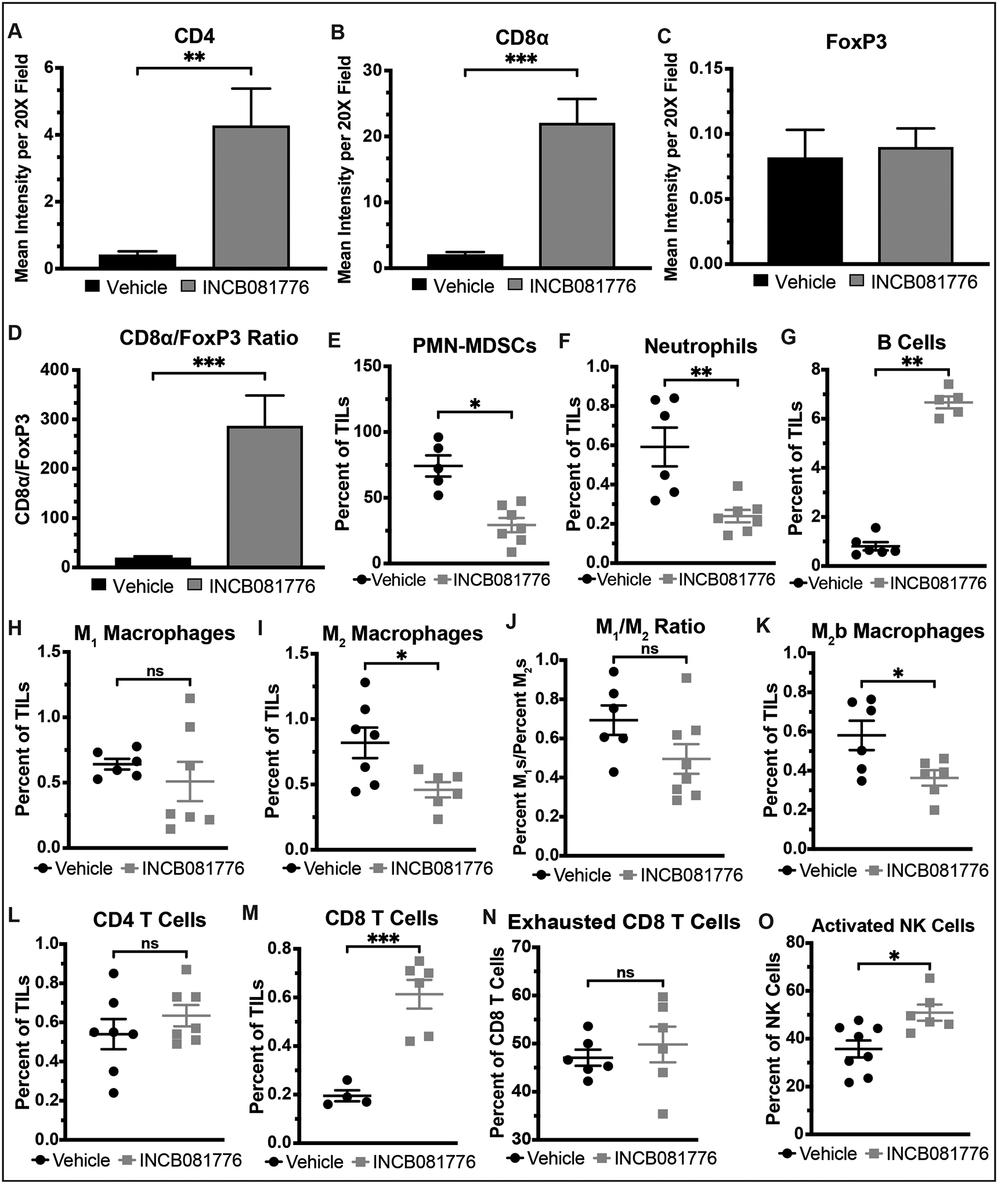
(A-O) Subcutaneous tumors were generated in syngeneic (C57BL/6N) mice using the MOC2 cell line. Tumor-bearing mice were treated with vehicle or INCB081776 via chow (~200mg/kg/day) for 12 days. (A-D) Tumors were stained using IHC. Image quantification was performed, and the CD8α/FoxP3 ratio was calculated. Mean values and SEMs are shown (n = 12 per group). **, P < 0.01; ***, P < 0.001. Representative images are shown in Supplemental Figure S2. (E-O) Tumor-infiltrating lymphocytes (TILs) were analyzed via flow cytometry. Mean values and SEMs are shown (n = 4–8 per group). *, P < 0.05; **, P < 0.01; ***, P < 0.001; NS, not significant.
Immunohistochemical analysis of MOC1 tumors showed that, while treatment with INCB081776 did not significantly change the levels of CD4 or CD8 T cells present in MOC1 tumors (Figure 5A–B), the level of FoxP3+ cells was significantly decreased in treated tumors (Figure 5C), resulting in a large increase in the CD8/FoxP3 ratio (Figure 5D). Flow cytometric analysis further confirmed that Axl/MerTK inhibition creates a hot TIME, with INCB081776 treatment resulting in decreased levels of pro-tumor immune cells, such as PMN-MDSCs and M2 macrophages, and increased levels of anti-tumor immune cells like CD8 T cells and activated NK cells (Figure 5E–O). Of note, the M1/M2 macrophage ratio was significantly increased (Figure 5J) and the level of exhausted CD8 T cells was significantly decreased (Figure 5N). Overall, these data indicate that treatment with INCB081776 results in a significantly hotter TIME in MOC1 tumors.
In MOC2 tumors, treatment with INCB081776 was similarly demonstrated to create a hotter TIME in vivo (Figure 6). Levels of both CD4 and CD8 T cells measured by IHC were significantly increased following INCB081776 treatment (Figure 6A–B), but levels of FoxP3+ cells were not increased (Figure 6C) in treated tumors, resulting in a large increase in the CD8/FoxP3 ratio (Figure 6D). Flow cytometric analysis further confirmed that Axl/MerTK inhibition creates a hot TIME in MOC2 tumors, with INCB081776 resulting in decreased levels of pro-tumor immune cells, like PMN-MDSCs and M2 macrophages, and increased levels of anti-tumor immune cells like CD8 T cells and activated NK cells (Figure 6E–O). Interestingly, the level of B cells was significantly increased following Axl/MerTK inhibition (Figure 6G). Collectively, these data indicate that Axl/MerTK inhibition with INCB081776 results in a significantly hotter TIME in MOC2 tumors.
INCB081776 and αPDL1 cooperate to slow tumor growth and create a hot TIME in HNC tumors
We next investigated the potential impact of INCB081776 treatment creating a hotter TIME. Given that tumors with hot TIMEs typically have higher response rates to ICIs8,10, we hypothesized that the combination of INCB081776 with an antibody targeting PDL1 (αPDL1) would result in a treatment response superior to either therapeutic alone.
MOC1 tumors exhibited an attenuated response to INCB081776 alone, but treatment with αPDL1 alone and in combination with INCB081776 significantly slowed tumor growth (Figure 7A). After 7 days, treatment with INCB081776 alone resulted in significant changes in the levels of immune infiltrate, as did treatment with αPDL1 alone and in combination with INCB081776 (Figure 7B–M). Notably, the level of M1 macrophages did not increase significantly except when INCB081776 and αPDL1 were given together, and the level of total Tregs did not decrease significantly except under that same combination treatment (Figure 7D, 7I). Immunohistochemical analysis confirmed the effectiveness of the combination of INCB081776 and αPDL1 in creating and maintaining a hot TIME (Supplemental Figures S2 and S3). Collectively, these data indicate that INCB081776 cooperates with αPDL1 treatment to significantly slow tumor growth and create a hotter TIME in MOC1 tumors.
Figure 7: Combined INCB081776 and αPDL1 treatment slows growth and creates a pro-inflammatory TIME in MOC1 tumors.
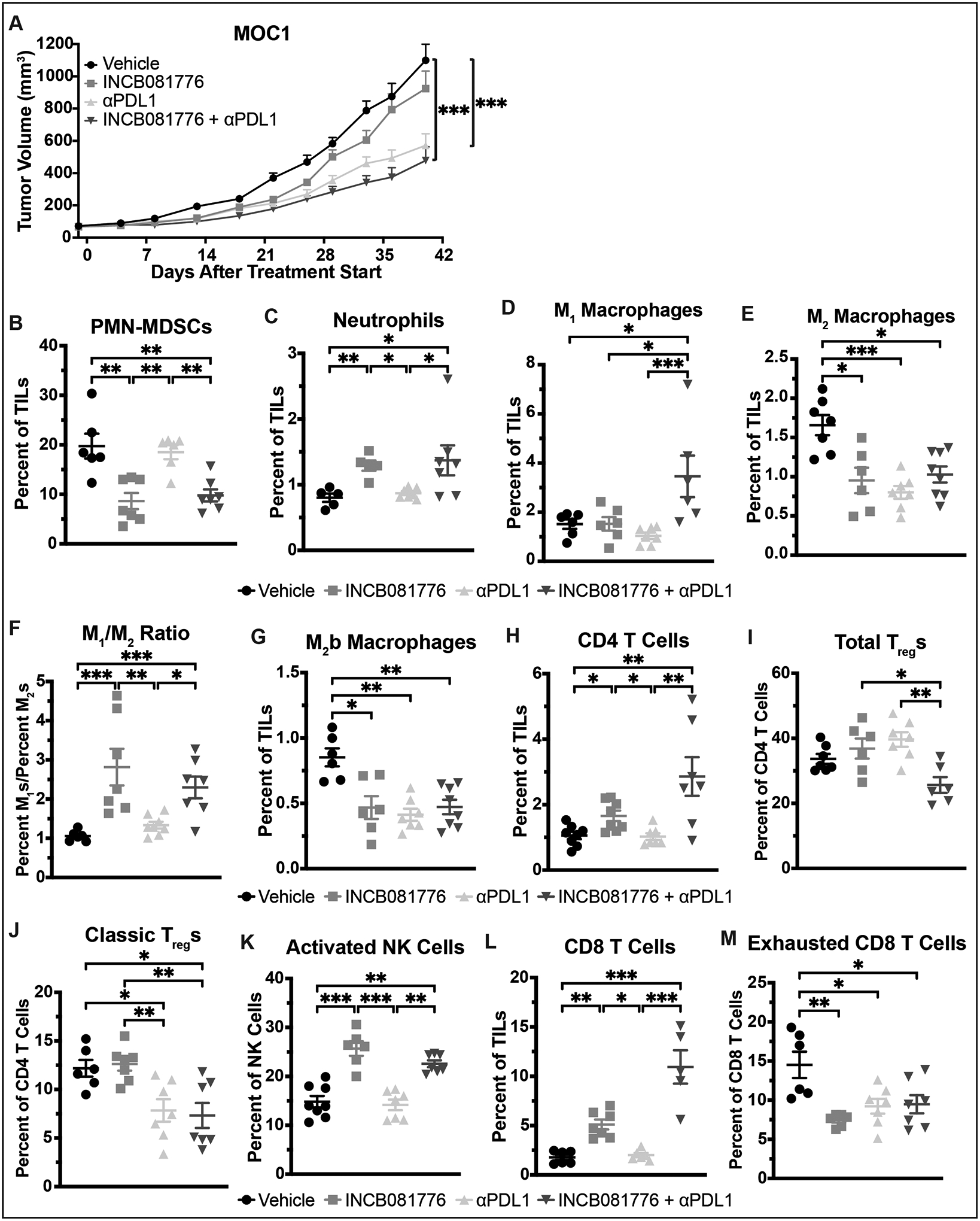
(A-M) Subcutaneous tumors were generated in syngeneic (C57BL/6N) mice using the MOC1 cell line. (A) Tumor-bearing mice were treated with vehicle, INCB081776 (via chow, ~200mg/kg/day), 2mg/kg αPDL1, or the combination of INCB081776 and αPDL1. Tumor volume was measured (n = 12–14 per group). Mean values and SEMs are shown. ***, P < 0.001. (B-M) Tumor-bearing mice were treated with vehicle, INCB081776 (via chow, ~200mg/kg/day), 2mg/kg αPDL1, or the combination of INCB081776 and αPDL1 for 7 days. Tumor-infiltrating lymphocytes (TILs) were analyzed via flow cytometry. Mean and SEMs are shown (n = 5–8 per group). *, P < 0.05; **, P < 0.01; ***, P < 0.001. Data for vehicle and INCB081776 groups were previously shown in Figure 5.
In MOC2 tumors, the combination of INCB081776 and αPDL1 similarly slowed tumor growth and created a hotter TIME in vivo (Figure 8). Tumor growth delay was observed following treatment with INCB081776 or αPDL1 alone, as well as with INCB081776 and αPDL1 in combination (Figure 8A). All three treatment groups significantly altered the levels of immune infiltrate, with each treatment having a slightly different effect (Figure 8B–M). Of note, the level of classic Tregs did not decrease significantly except following treatment with the combination of INCB081776 and αPDL1 (Figure 8J). The combination treatment also increased levels of anti-tumor cells like CD8 T cells and activated NK cells (Figure 8K–L) and decreased levels of pro-tumor cells like exhausted CD8 T cells (Figure 8M). Immunohistochemical analysis confirmed the effectiveness of the combination of INCB081776 and αPDL1 in creating and maintaining a hot TIME (Supplemental Figures S4 and S5). Overall, these data indicate that inhibition of Axl/MerTK with INCB081776 cooperates with αPDL1 therapy to significantly slow tumor growth and create a hotter TIME in both MOC1 and MOC2 tumors.
Figure 8: Combined INCB081776 and αPDL1 treatment slows growth and creates a pro-inflammatory TIME in MOC2 tumors.
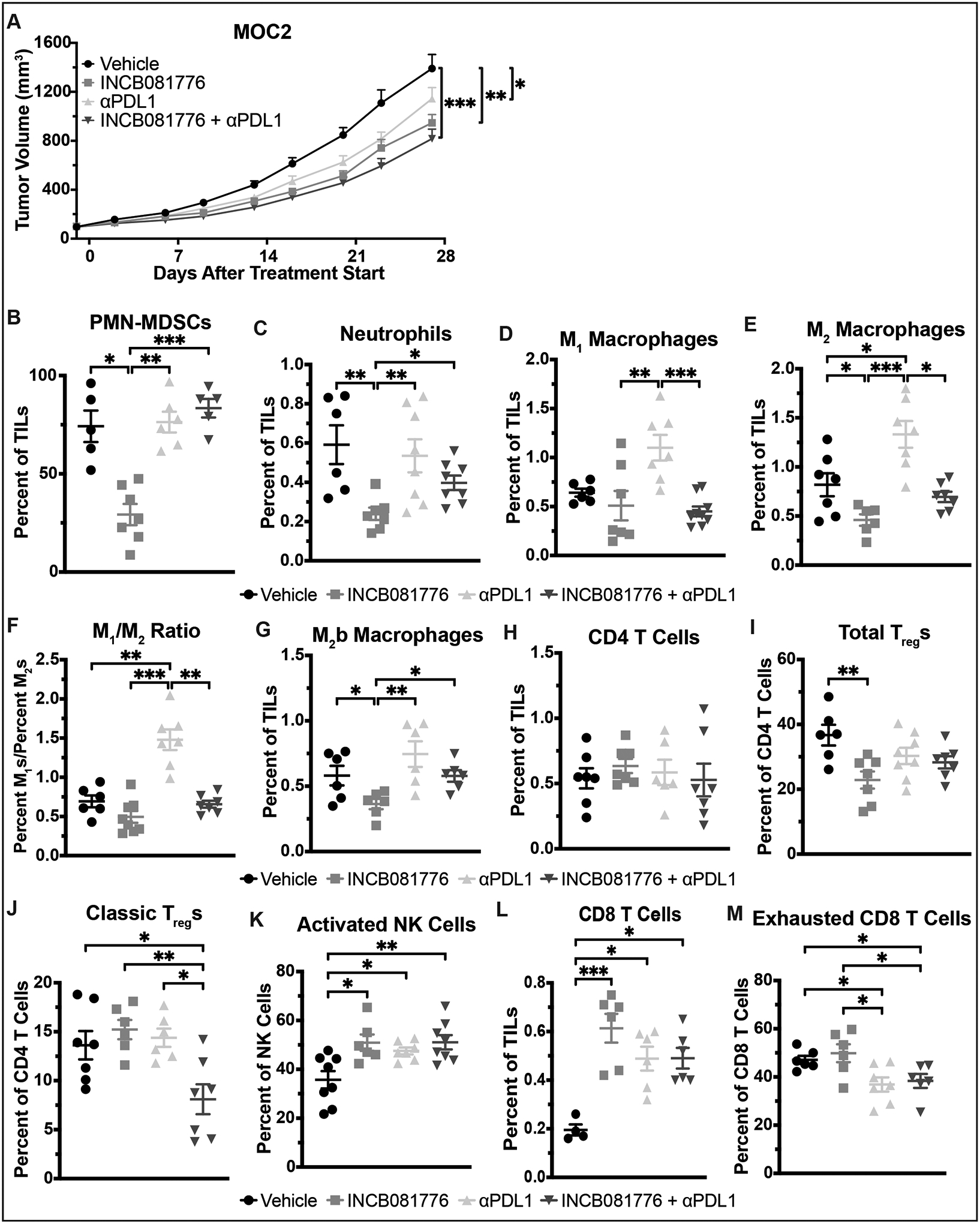
(A-M) Subcutaneous tumors were generated in syngeneic (C57BL/6N) mice using the MOC2 cell line. (A) Tumor-bearing mice were treated with vehicle, INCB081776 (via chow, ~200mg/kg/day), 10mg/kg αPDL1, or the combination of INCB081776 and αPDL1. Tumor volume was measured (n = 12–14 per group). Mean values and SEMs are shown. *, P < 0.05; **, P < 0.01; ***, P < 0.001. (B-M) Tumor-bearing mice were treated with vehicle, INCB081776 (via chow, ~200mg/kg/day), 1mg/kg αPDL1, or the combination of INCB081776 and αPDL1 for 12 days. Tumor-infiltrating lymphocytes (TILs) were analyzed via flow cytometry. Mean and SEMs are shown (n = 5–8 per group). *, P < 0.05; **, P < 0.01; ***, P < 0.001. Data for vehicle and INCB081776 groups were previously shown in Figure 6.
Discussion
In the decades since their discovery, the TAM receptor tyrosine kinases have been increasingly implicated in solid-tumor growth and progression, for both their oncogenic effects and their immunomodulatory roles39,40. This report presents data suggesting that 1) Axl and MerTK signaling leads to an anti-inflammatory, pro-tumor TIME in HNC, 2) inhibition of this signaling using INCB081776 can slow tumor growth and alter the levels of immune infiltrate to create a more pro-inflammatory, anti-tumor TIME, and 3) combining INCB081776 treatment with standard-of-care immune checkpoint inhibitor αPDL1 can enhance its effects on tumor growth and the TIME. Notably, we showed that INCB081776 has the largest impact on immunologically cold tumors, suggesting a high potential clinical benefit of Axl/MerTK inhibition in this patient population.
The impact of Axl/MerTK signaling on solid-tumor growth is well-established, as studies both by our laboratory and others have shown that inhibition of Axl and/or MerTK can significantly slow tumor progression in many different cancer types, including head and neck17,19, pancreatic41, lung41,42, breast23,41–45, bladder23, colon42,44, ovarian42, and skin cancer46. The current study confirms these findings, showing that dual inhibition of Axl and MerTK with INCB081776 slows tumor growth in both mouse and human models of HNC (Figure 2).
Axl and MerTK are most significantly expressed in the non-cancer setting on macrophages, where they play a critical role in mediating the efferocytosis function14,16. The two receptors also regulate the expression of various cytokines that can affect macrophage polarization14. Previous studies have primarily focused on the effects of Axl signaling alone, reporting that Axl inhibition or genetic loss results in decreases in cytokines related to immunosuppressive cell recruitment (MCP-142,47, MIP-1α22,42, MIP-1β22,42, RANTES22,42, and eotaxin48), myeloid-supportive cytokines (MCSF22,47,48, GMCSF22, and GCSF22,48), NF-κB target cytokines (TNFα22,48, IL-622,48, and IL-1α22), and others (IL-1β48, IL-748). We were able to corroborate some of these findings, showing that inhibition of Axl/MerTK signaling with INCB081776 decreases the expression/secretion of TNFα, IL-1β, GMCSF, and MCP-1 (Figure 3). This study represents one of the first evaluations of cytokines regulated by Axl and MerTK in HNC.
Interestingly, the four cytokines identified in our analysis as being upregulated by Axl/MerTK signaling in HNC cells (TNFα, IL-1β, GMCSF, and MCP-1) have all been previously implicated in the progression of HNC. Through its interactions with the PI3K-Akt pathway30, the c-REL/ΔNp63α complex31, and TAp7331, TNFα promotes cell survival and proliferation in HNC. Likewise, MCP-1 activates Akt, ERK, and STAT3 to promote survival of HNC cells; high MCP-1 expression has been associated with poor overall survival in HNC patients37. The interleukin IL-1β has been identified as a key driver of HNC, playing a role in every stage of the malignancy. Elevated expression of IL-1β in the local microenvironment is associated with an increased risk of carcinogenesis35, and IL-1β stimulation of HNC cells upregulates the expression of fascin34 and Snail36, which promote invasiveness and the epithelial-mesenchymal transition, respectively. Similarly, GMCSF has been associated with angiogenesis32 and increased invasion potential/metastasis33 in HNC, through its role as a growth factor for endothelial cells32 and its upregulation of matrix metalloproteinases and related proteins33. Each of these cytokines have been individually identified as potential therapeutic targets in HNC, but our data indicates that they are regulated, at least in part, by Axl and MerTK signaling (Figure 3). This suggests that a single therapeutic against Axl and MerTK – in place of therapeutics targeting each cytokine individually – could reverse the negative effects of these cytokines on HNC progression.
Axl and MerTK also play key roles in regulating macrophage polarization in both cancer and non-cancer settings14. Specifically, signaling through Axl and MerTK lead to M2 macrophage polarization, which is commonly associated with an anti-inflammatory, pro-tumor phenotype14. Thus, we hypothesized that inhibition of Axl/MerTK signaling would halt and/or reverse M2 polarization and potentially lead to M1 macrophage polarization, a pro-inflammatory, anti-tumor phenotype. This hypothesis was supported by our data, which showed that inhibition of Axl/MerTK with INCB081776 decreased expression of M2-associated genes and increased expression of M1-associated genes in primary BMDMs, including those that had been polarized to the M2 state before INCB081776 treatment (Figure 4). The ability of Axl and MerTK targeting agents to affect polarization in such a manner has been described previously; both the Axl inhibitor BGB32448 and the MerTK inhibitor sitravatinib41 were shown to decrease expression of M2-associated genes in BMDMs. However, neither compound was associated with increased expression of M1-associated genes as in the current study, suggesting that dual inhibition of Axl and MerTK may be necessary to increase M1 polarization levels.
One of the most significant findings of the current study was the shift in the so-called ‘temperature’ of the TIME from cold to hot following Axl/MerTK dual inhibition with INCB081776 (Figures 5–6). The current study is one of the first to suggest Axl/MerTK regulation of the TIME in HNC. Other studies have previously suggested a link between the TAM family of RTKs and the immune system in melanoma46 or in breast22,25,45,47, ovarian42, lung41, or colon23,44 cancer. Interestingly, the overall immuno-oncological effects of targeting the TAMs appear to be consistent regardless of which specific TAM receptor is being targeted. Inhibition of either Axl22,42,47 or MerTK41,45 alone, by either genetic or pharmacological means, has been shown to increase pro-inflammatory immune infiltration in solid tumors. Likewise, previous studies targeting two or more TAMs simultaneously have demonstrated a shift in the TIME from cold to hot following TAM inhibition23,25,44,46. The specific cell types involved and the magnitude of the shift in the TIME vary between each study, but most describe an increase in CD8 T cell infiltration following treatment with a TAM-targeting agent; this increase in CD8 T cells was also observed in the current study (Figures 5B, 5L, 6B, and 6L).
Finally, we combined the dual Axl/MerTK inhibitor INCB081776 with an ICI against PDL1 (αPDL1). ICIs show increased efficacy in tumors with hot TIMEs8,10, and we showed that INCB081776 can shift the TIME from cold to hot, so we hypothesized that the combination of INCB081776 and αPDL1 could result in a better tumor response than either treatment alone. Tumor growth was significantly slowed by the combination treatment (Figures 7A, 8A); these data are supported by other studies showing a similar delay in tumor growth or an increase in survival time following treatment with either a single-41,42,47 or multi-TAM23,25,43,44,46 agent. Moreover, the addition of αPDL1 to INCB081776 treatment resulted in a further shift of the TIME; overall, the combination treatment group had a hotter TIME than either therapy alone (Figures 7–8). This trend has been previously observed in other studies23,25,41,43,44,46,47, though the current study represents some of the first evidence for this combinatorial effect in HNC.
The current study demonstrated that the Axl/MerTK targeting agent INCB081776 can significantly alter the growth patterns of HNC tumors, either alone or in combination with αPDL1, through its effects on immunomodulatory molecules, macrophage polarization, and the tumor immune microenvironment as a whole. Besides INCB081776, over 30 therapeutics that target one or more of the TAMs have been made; the specific agents, their characteristics, and their clinical status have been extensively reviewed39,49–51. Dual inhibition of Axl and MerTK could drastically improve clinical efficacy of ICI therapeutics in HNC, potentially leading to more positive patient outcomes.
Supplementary Material
Acknowledgements
The authors thank the University of Wisconsin Carbone Cancer Center (UWCCC) Flow Cytometry Laboratory, the UWCCC Experimental Animal Pathology Laboratory, and the UWCCC Biostatistics Shared Resource, all supported by P30 CA014520, for use of their facilities and services. The authors also thank Dr. Ravindra Uppaluri for the use of the MOC cell lines, and Incyte Corporation for supplying the quantities of INCB081776 necessary to complete these experiments.
Research reported in this publication was supported by a pilot grant from the University of Wisconsin Institute for Clinical and Translational Research (DLW, ICTR-UWCH RD12), a UWCCC Support Grant from the National Cancer Institute (P30CA014520), and a Wisconsin Head and Neck Cancer SPORE grant from the National Institute of Dental and Craniofacial Research (DLW, JYB, PMH, P50DE026787). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
References
- 1.Siegel RL, Miller KD, Fuchs HE & Jemal A Cancer statistics, 2022. CA Cancer J Clin 72, 7–33 (2022). [DOI] [PubMed] [Google Scholar]
- 2.Gillison ML, et al. Evidence for a Causal Association Between Human Papillomavirus and a Subset of Head and Neck Cancers. JNCI 92, 709–720 (2000). [DOI] [PubMed] [Google Scholar]
- 3.Ferris R & Gillison ML Nivolumab for Squamous-Cell Cancer of Head and Neck. N Engl J Med 376, 596 (2017). [DOI] [PubMed] [Google Scholar]
- 4.Burtness B, et al. Pembrolizumab alone or with chemotherapy versus cetuximab with chemotherapy for recurrent or metastatic squamous cell carcinoma of the head and neck (KEYNOTE-048): a randomised, open-label, phase 3 study. Lancet 394, 1915–1928 (2019). [DOI] [PubMed] [Google Scholar]
- 5.Duan Q, Zhang H, Zheng J & Zhang L Turning Cold into Hot: Firing up the Tumor Microenvironment. Trends in Cancer 6, 605–618 (2020). [DOI] [PubMed] [Google Scholar]
- 6.Binnewies M, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nature Medicine 24, 541–550 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Scharping NE & Delgoffe GM Tumor microenvironment metabolism: a new checkpoint for anti-tumor immunity. Vaccines (Basel) 4, 46 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vareki SM High and low mutational burden tumors versus immunologically hot and cold tumors and response to immune checkpoint inhibitors. Journal for ImmunoTherapy of Cancer 6, 157 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gajewski TF The next hurdle in cancer immunotherapy: overcoming the non-T-cell-inflamed tumor microenvironment. Seminars in Oncology 42, 663–671 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zemek RM, et al. Sensitization to immune checkpoint blockade through activation of a STAT1/NK axis in the tumor microenvironment. Science Translational Medicine 11, eaav7816 (2017). [DOI] [PubMed] [Google Scholar]
- 11.Rausch MP & Hastings KT Chapter 9 - Immune Checkpoint Inhibitors in the Treatment of Melanoma: From Basic Science to Clinical Application. in Cutaneous Melanoma: Etiology and Therapy (eds. Ward WH & Farma JM) (Codon Publications, Brisbane (AU), 2017). [PubMed] [Google Scholar]
- 12.Xia L, Liu Y & Wang Y PD-1/PD-L1 Blockade Therapy in Advanced Non-Small-Cell Lung Cancer: Current Status and Future Directions. The Oncologist 24, S31–S41 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ran X & Yang K Inhibitors of the PD-1/PD-L1 axis for the treatment of head and neck cancer: current status and future perspectives. Drug Des Devel Ther 11, 2007–2014 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Myers KV, Amend SR & Pienta KJ Targeting Tyro3, Axl and MerTK (TAM receptors): implications for macrophages in the tumor microenvironment. Mol Cancer 18, 94 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Couper KN, Blount DG & Riley EM IL-10: The Master Regulator of Immunity to Infection. The Journal of Immunology 180, 5771–5777 (2008). [DOI] [PubMed] [Google Scholar]
- 16.Lemke G Biology of the TAM Receptors. Cold Spring Harb Perspect Biol 5(2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Brand TM, et al. AXL Is a Logical Molecular Target in Head and Neck Squamous Cell Carcinoma. Clin Cancer Res 21, 2601–2612 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brand TM, et al. AXL mediates resistance to cetuximab therapy. Cancer Res 74, 5152–5164 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar] [Research Misconduct Found]
- 19.McDaniel NK, et al. MERTK Mediates Intrinsic and Adaptive Resistance to AXL-targeting Agents. Mol Cancer Ther 17, 2297–2308 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McDaniel NK, et al. AXL Mediates Cetuximab and Radiation Resistance Through Tyrosine 821 and the c-ABL Kinase Pathway in Head and Neck Cancer. Clin Cancer Res (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.von Massenhausen A, et al. MERTK as a novel therapeutic target in head and neck cancer. Oncotarget 7, 32678–32694 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Aguilera TA, et al. Reprogramming the immunological microenvironment through radiation and targeting Axl. Nat Commun 7, 13898 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rios-Doria J, et al. A Potent and Selective Dual Inhibitor of AXL and MERTK Possesses Both Immunomodulatory and Tumor-Targeted Activity. Front Oncol 10, 598477 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Judd NP, et al. ERK1/2 Regulation of CD44 Modulates Oral Cancer Aggressiveness. Cancer Research 72, 365–374 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kasikara C, et al. Pan-TAM Tyrosine Kinase Inhibitor BMS-777607 Enhances Anti-PD-1 mAb Efficacy in a Murine Model of Triple-Negative Breast Cancer. Cancer Res 79, 2669–2683 (2019). [DOI] [PubMed] [Google Scholar]
- 26.Crowe AR & Yue W Semi-quantitative Determination of Protein Expression using Immunohistochemistry Staining and Analysis: An Integrated Protocol. Bio Protoc 9(2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cash H, et al. mTOR and MEK1/2 inhibition differentially modulate tumor growth and the immune microenvironment in syngeneic models of oral cavity cancer. Oncotarget 6, 36400–36417 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Holland SJ, et al. R428, a selective small molecule inhibitor of Axl kinase, blocks tumor spread and prolongs survival in models of metastatic breast cancer. Cancer Res 70, 1544–1554 (2010). [DOI] [PubMed] [Google Scholar]
- 29.Minson KA, et al. The MERTK/FLT3 inhibitor MRX-2843 overcomes resistance-conferring FLT3 mutations in acute myeloid leukemia. JCI Insight 1, e85630 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sandra F, et al. TNF inhibited the apoptosis by activation of Akt serin/threonine kinase in the human head and neck squamous cell carcinoma. Cellular Signaling 14, 771–778 (2002). [DOI] [PubMed] [Google Scholar]
- 31.Lu H, et al. TNF-alpha promotes c-REL/DeltaNp63alpha interaction and TAp73 dissociation from key genes that mediate growth arrest and apoptosis in head and neck cancer. Cancer Res 71, 6867–6877 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ninck S, et al. Expression profiles of angiogenic growth factors in squamous cell carcinomas of the head and neck. Int J Cancer 106, 34–44 (2003). [DOI] [PubMed] [Google Scholar]
- 33.Tomita T, et al. Granulocyte-macrophage colony-stimulating factor upregulates matrix metalloproteinase-2 (MMP-2) and membrane type-1 MMP (MT1-MMP) in human head and neck cancer cells. Cancer Letters 156, 83–91 (2000). [DOI] [PubMed] [Google Scholar]
- 34.Lee MK, Park JH, Gi SH & Hwang YS IL-1beta Induces Fascin Expression and Increases Cancer Invasion. Anticancer Res 38, 6127–6132 (2018). [DOI] [PubMed] [Google Scholar]
- 35.Wu T, et al. Modulation of IL-1beta reprogrammes the tumor microenvironment to interrupt oral carcinogenesis. Sci Rep 6, 20208 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.St John MA, et al. Proinflammatory mediators upregulate snail in head and neck squamous cell carcinoma. Clin Cancer Res 15, 6018–6027 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ji WT, Chen HR, Lin CH, Lee JW & Lee CC Monocyte chemotactic protein 1 (MCP-1) modulates pro-survival signaling to promote progression of head and neck squamous cell carcinoma. PLoS One 9, e88952 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Myers KV, Amend SR & Pienta KJ Targeting Tyro3, Axl and MerTK (TAM receptors): implications for macrophages in the tumor microenvironment. Molecular Cancer 18 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mikolajczyk A, et al. Two-Front War on Cancer-Targeting TAM Receptors in Solid Tumour Therapy. Cancers (Basel) 14(2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhou L & Matsushima GK Tyro3, Axl, Mertk receptor-mediated efferocytosis and immune regulation in the tumor environment. Int Rev Cell Mol Biol 361, 165–210 (2021). [DOI] [PubMed] [Google Scholar]
- 41.Du W, Huang H, Sorrelle N & Brekken RA Sitravatinib potentiates immune checkpoint blockade in refractory cancer models. JCI Insight 3(2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Guo Z, Li Y, Zhang D & Ma J Axl inhibition induces the antitumor immune response which can be further potentiated by PD-1 blockade in the mouse cancer models. Oncotarget 8, 89761–89774 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Davra V, et al. Axl and Mertk Receptors Cooperate to Promote Breast Cancer Progression by Combined Oncogenic Signaling and Evasion of Host Antitumor Immunity. Cancer Res 81, 698–712 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yokoyama Y, et al. Immuno-oncological Efficacy of RXDX-106, a Novel TAM (TYRO3, AXL, MER) Family Small-Molecule Kinase Inhibitor. Cancer Res 79, 1996–2008 (2019). [DOI] [PubMed] [Google Scholar]
- 45.Cook RS, et al. MerTK inhibition in tumor leukocytes decreases tumor growth and metastasis. J Clin Invest 123, 3231–3242 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Holtzhausen A, et al. TAM Family Receptor Kinase Inhibition Reverses MDSC-Mediated Suppression and Augments Anti-PD-1 Therapy in Melanoma. Cancer Immunol Res 7, 1672–1686 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Goyette MA, et al. Targeting Axl favors an antitumorigenic microenvironment that enhances immunotherapy responses by decreasing Hif-1alpha levels. Proc Natl Acad Sci U S A 118(2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ludwig KF, et al. Small-Molecule Inhibition of Axl Targets Tumor Immune Suppression and Enhances Chemotherapy in Pancreatic Cancer. Cancer Res 78, 246–255 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Zhu C, Wei Y & Wei X AXL receptor tyrosine kinase as a promising anti-cancer approach: functions, molecular mechanisms and clinical applications. Mol Cancer 18, 153 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Engelsen AST, et al. Dissecting the Role of AXL in Cancer Immune Escape and Resistance to Immune Checkpoint Inhibition. Front Immunol 13, 869676 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Di Stasi R, De Rosa L & D’Andrea LD Therapeutic aspects of the Axl/Gas6 molecular system. Drug Discov Today 25, 2130–2148 (2020). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data generated in this study are available upon request from the corresponding author.