

Abstract
The lipocalin (LCN) family members, a group of small extracellular proteins with 160–180 amino acids in length, can be detected in all kingdoms of life from bacteria to human beings. They are characterized by low similarity of amino acid sequence but highly conserved tertiary structures with an eight-stranded antiparallel β-barrel which forms a cup-shaped ligand binding pocket. In addition to bind small hydrophobic ligands (i.e., fatty acids, odorants, retinoids, and steroids) and transport them to specific cells, lipocalins (LCNs) can interact with specific cell membrane receptors to activate their downstream signaling pathways, and with soluble macromolecules to form the complex. Consequently, lipocalins exhibit great functional diversity. Accumulating evidence has demonstrated that lipocalin family proteins exert multiple layers of function in the regulation of many physiological processes and human diseases (i.e., cancers, immune disorders, metabolic disease, neurological/psychiatric disorders, and cardiovascular disease). In this review, we firstly introduce the structural and sequence properties of lipocalins. Next, six lipocalins including apolipoprotein D (ApoD), ApoM, lipocalin 2 (LCN2), LCN10, retinol-binding protein 4 (RBP4), and Lipocalin-type prostaglandin D synthase (L-PGDS) which have been characterized so far are highlighted for their diagnostic/prognostic values and their potential effects on coronary artery disease and myocardial infarction injury. The roles of these 6 lipocalins in cardiac hypertrophy, heart failure, diabetes-induced cardiac disorder, and septic cardiomyopathy are also summarized. Finally, their therapeutic potential for cardiovascular disease is discussed in each section.
Keywords: Lipocalin, Biomarker, Coronary artery disease, Myocardial infarction, Cardiac hypertrophy, Cardiomyopathy
1. Introduction
Lipocalins (LCNs), a group of typically small extracellular proteins with 160–180 amino acids in length, belong to the calycin superfamily that also includes other 4 families: acid-binding proteins (ABPs), avidins, triabins, and metalloproteinase inhibitors (MPIs) (Flower, North, & Attwood, 1993). The term “lipocalin” was initially proposed by Pervaiz & Brew in 1987 for a group of proteins including α1-microglobulin (A1M, also referred to as protein HC), human α1-acid glycoprotein (AGP), bovine β-lactoglobulin, and serum retinol-binding protein (RBP), which was based on their sequence homologies, disulfide bond pattern, and functional similarities in their ability to bind lipophilic molecules inside a cup-shaped protein fold (Pervaiz & Brew, 1987). Over the past decades, there are >1000 LCN genes identified among bacteria, plants, fungi, animals, and at least 20 LCN-like genes exist in the human genome (Charkoftaki, et al., 2019; Du, et al., 2015; Urzua, et al., 2006). Interestingly, lipocalin genes have a conserved arrangement of exons/introns with a primary pattern including 7 exons and 6 introns (C. Guo, et al., 2010; Hamil, et al., 2003; Suzuki, et al., 2006). They are usually organized into clusters at the same chromosome, which may be generated by functional divergence or duplications with evolutionary time (Hamil, et al., 2003; Suzuki, et al., 2004; Suzuki, et al., 2006). For example, human LCN genes are mostly detected in the Chromosome 9, and mouse LCNs are mostly clustered in chromosome 2 (Fig. 1). Furthermore, 18 of 20 human LCN genes have mouse orthologs (Table 1) which share a high degree of similarity (Charkoftaki, et al., 2019; Du, et al., 2015; Urzua, et al., 2006). Among all 20 LCN genes, LCN7 shows the highest percent similarity (90%), whereas LCN15 and LCN13 (OBP2A) display the lowest percent identity (39%) to their mouse orthologs (Table 1). Please note that LCN1 (also known as tear per-albumin or von Ebner’s gland protein) (Glasgow, et al., 1993; Holzfeind, Merschak, Wojnar, & Redl, 1997; Lassagne, Ressot, Mattei, & Gachon, 1993) and PAEP [progestagen-associated endometrial protein, also referred to as glycodelin (GD), a secreted immunosuppressive glycoprotein] (Joshi, Smith, & Stokes, 1980) are found only in human, whereas Lcn3, 4, 5 11, 16, 17 and all the functional major urinary protein genes (MUPs) exist only in mouse (Charkoftaki, et al., 2019). Human LCN6 gene is the orthologous gene of mouse Lcn5 (Hamil, et al., 2003).
Figure 1: A lipocalin gene cluster located at the mouse chromosome 2 and the human chromosome 9.
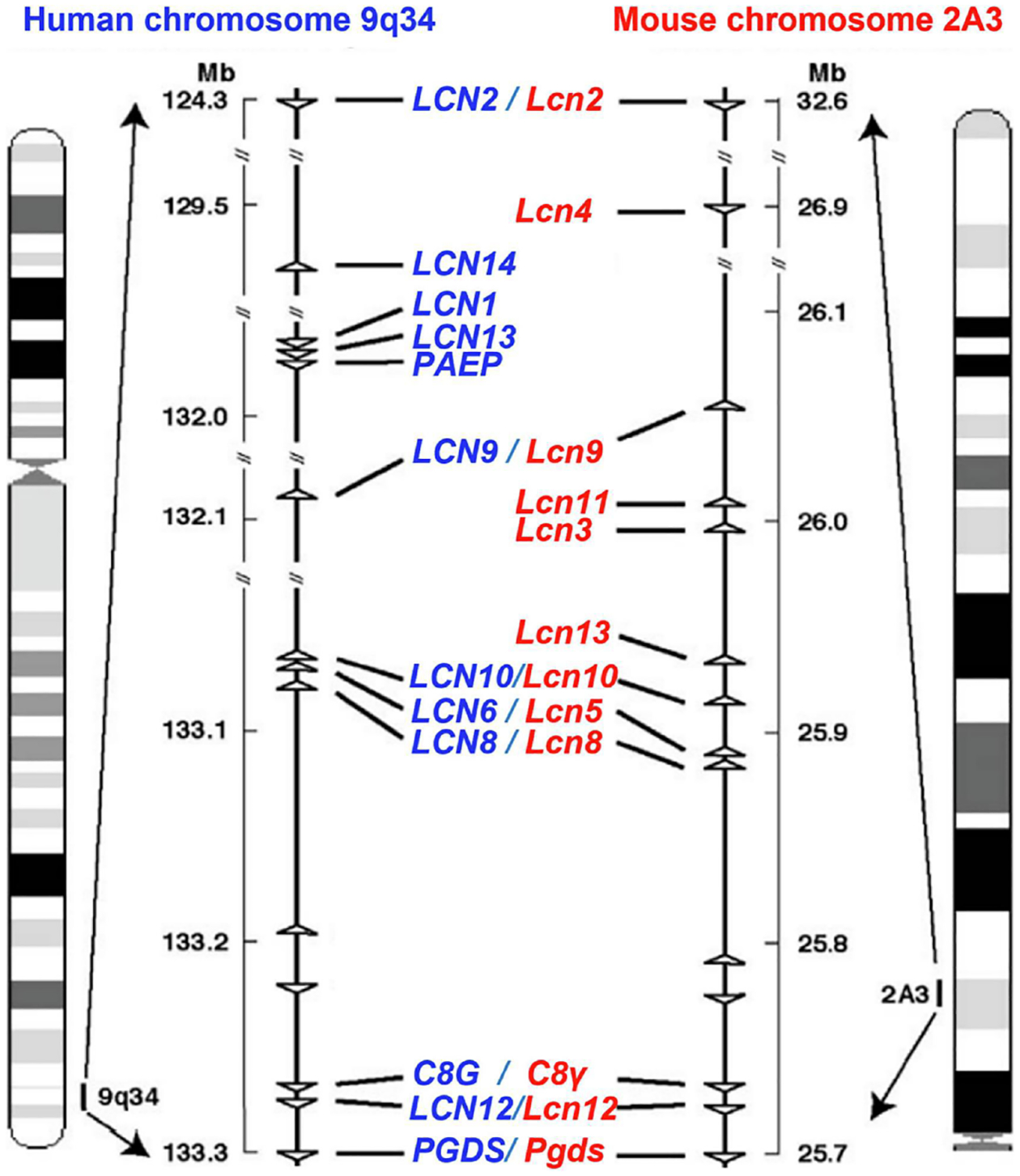
Syntonically homologous cluster of lipocalin genes at the chromosomes is conserved between human and mouse. Each triangle symbol represents gene locus and gene orientation. Adapted from Suzuki et al. (2004).
Table1.
List of the 24 LCN-like genes in the human and mouse—with gene symbols, chromosomal location, and their similarity, expressed as percent identity (%).
| Gene symbol (human) | Chromosome | Gene symbol (mouse) | Chromosome | Similarity (identity%) |
|---|---|---|---|---|
| LCN1 (TLC, VEGP) | 9q34 | NA | NA | NA |
| LCN2 (NGAL) | 9q34 | Lcn2 | 2B; 2 22.09 cM | 62 |
| NA | Lcn3–5 | 2; 2A3 | NA | |
| LCN6 (UNQ643) | 9q34.3 | Lcn6 | 2; 2A3 | 74 |
| LCN7 (TINAGL1) | 1p35.2 | Lcn7 | 4; 4D2.2 | 90 |
| LCN8 (EP17) | 9q34.3 | Lcn8 | 2; 2A3 | 70 |
| LCN9 (HEL 129) | 9q34.3 | Lcn9 | 2; 2A3 | 54 |
| LCN10 | 9q34.3 | Lcn10 | 2; 2A3 | 64 |
| NA | NA | Lcn11 | 2; 2A3 | NA |
| LCN12 | 9q34.3 | Lcn12 | 2; 2A3 | 56 |
| LCN13 (OBP2A) | 9q34 | Lcn13 | 2; 2A3 | 39 |
| LCN14 (OBP2B) | 9q34 | Lcn14 | 2; 2A3 | 50 |
| LCN15 (UNQ2541) | 9q34.3 | Lcn15 | 2; 2A3 | 39 |
| AMBP (A1M) | 9q32-q33 | Ambp | 3 B3; 433.96 cM | 77 |
| APOD (Apolipoprotein D) | 3q29 | Apod | 16 B2; 16 21.41 cM | 74 |
| APOM (Apolipoprotein M) | 6p21 | Apom | 17; 17 B1 | 81 |
| C8G | 9q34.3 | C8γ | 2 A3; 2 17.31 cM | 76 |
| ORM1 (AGP1) | 9q32 | Orm1 | 4 B3; 4 33.96 cM | 49 |
| ORM2 (AGP2) | 9q32 | Orm2 | 4 B3; 4 33.96 cM | 44 |
| PAEP (GD) | 9q34 | NA | NA | NA |
| PTGDS (L-PGDS) | 9q34.2-q34.3 | Ptgds | 2 A3; 2 17.28 cM | 72 |
| RBP4 (Retinol-bing protein-4) | 10q23.33 | Rbp4 | 19 C2; 19 32.75 cM | 86 |
LCNs were originally characterized to transport or store a range of small hydrophobic molecules such as odorants, retinol, steroid hormones, and various secondary metabolites (Flower, 1995, 1996; Flower, North, & Sansom, 2000). However, owing to their common molecular recognition patterns that include: 1) binding to specific cell membrane receptors, 2) high affinity to small hydrophobic molecules, and 3) the transient formation of complexes with soluble macromolecules, it is now becoming increasingly clear that lipocalins exert multiple layers of biological functions in the modulation of cell proliferation, differentiation, death, and ageing (Akerstrom, Flower, & Salier, 2000; Flower, 1995, 1996; Grzyb, Latowski, & Strzalka, 2006). They are also associated with the regulation of immune response/inflammation, smell reception, reproduction, cancer development, metabolic disorders, and cardiovascular remodeling (Ganfornina, Akerstrom, & Sanchez, 2022; Redl & Habeler, 2022; Virtanen, 2021). As carrier proteins, LCNs could act in the general clearance of intracellular and extracellular hydrophobic molecules (Flower, 1996). Clinically, LCNs have been extensively explored as biochemical biomarkers for the diagnosis of human disease (Bacci, Cavallari, de Rozier-Alves, Alves Bda, & Fonseca, 2015; Bergwik, Kristiansson, Allhorn, Gram, & Akerstrom, 2021; Krol-Grzymala, et al., 2022; Sawyer, 2021; Sivalingam, et al., 2017; Steinhoff, Lass, & Schupp, 2021; Wong & Tse, 2021; Zabetian-Targhi, Mahmoudi, Rezaei, & Mahmoudi, 2015). In this review, we firstly introduce the structural and sequence properties of LCNs, and then summarize their diverse roles in coronary artery disease, cardiac vessel integrity, myocardial infarction, cardiac hypertrophy, and their associated heart failure. We also highlight both beneficial and detrimental effects of LCNs in diabetic and septic cardiomyopathy. Finally, their therapeutic potentials to the treatment of cardiovascular disease are discussed.
2. Characterization of Lipocalin Family Proteins
It is well recognized that the similarity of amino acid sequences among lipocalin family members is very low falling below 20% (Flower, et al., 2000). However, their three-dimensional structure is highly preserved with eight antiparallel β-strands (labelled A-H) forming a fold or calyx in a cylindrical manner with a “closed end” on one side and an “open end” on the opposite side (Flower, et al., 2000) (Fig. 2). The “open end” provides an access into the central cavity for the internal ligand-binding with and transporting small hydrophobic compounds including lipids, odorants (e.g., pheromones), retinoids, and steroid hormones (Flower, et al., 2000; Grzyb, et al., 2006; Schlehuber & Skerra, 2005). The eight β-strands, connected by seven loops (L1-L7), coil in a right-handed manner around a central axis and interact through transversal hydrogen bonds (Grzyb, et al., 2006). The first loop L1 is a large and flexible omega-type loop which functions as a dynamic lid for the “open end” of the β -barrel, and the other six are short hairpin-type loops (Flower, et al., 1993) (Fig. 2). Previous work has performed analysis for the conservation of amino acid sequence and structure within lipocalin proteins and identified that there is a common lipocalin fold characterized by three large structurally conserved regions (SCRs): SCR1 (strand A and its preceding 310-like helix), SCR2 (strands F and G, and their linking loop L6), and SCR3 (strand H and adjoining residues) (Flower, et al., 1993; Flower, et al., 2000) (Fig. 2). Despite the fact that other SCRs of the common core are identified, they are small and can be neglected. For example, one recent study by Du et al showed that human lipocalin proteins share four SCRs (Du, et al., 2015).
Figure 2: Structure of the lipocalin fold.
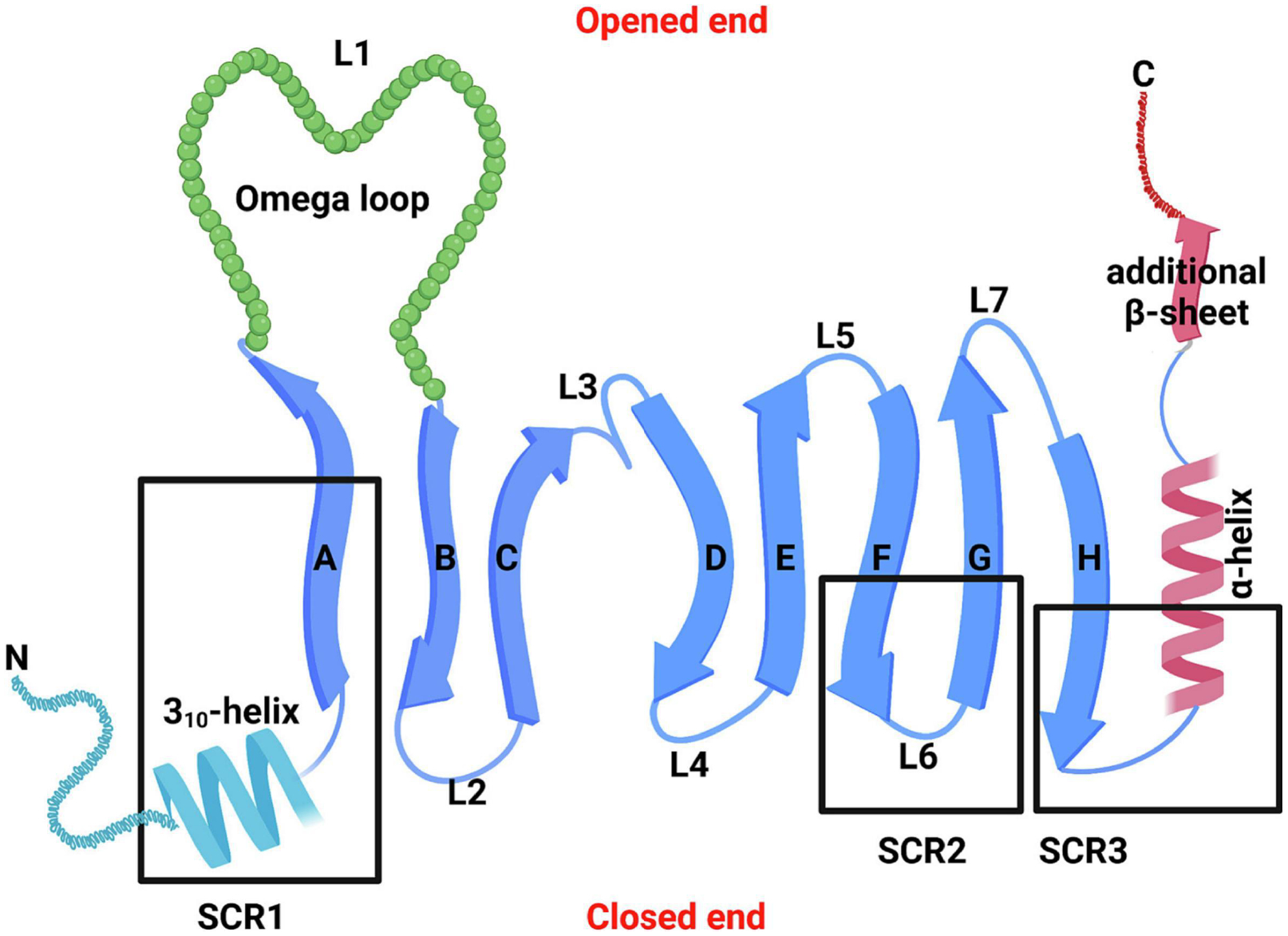
The eight β-strands of the antiparallel β-sheet plus one additional β-strand at the C-terminal are shown as arrows and marked A-H. The N-terminal 310 like helix and C-terminal α-helix are also marked. Connection loops are labelled L1-L7. There are four loops (L1, L3, L5, and L7) located at one end of the lipocalin L-barrel which open for the internal ligand-binding site. In parallel, there are three L-hairpin loops (L2, L4 and L6) at the other end and the N-terminal polypeptide chain crosses this end of the barrel to link strand A through a conserved 310 helix to close this end of the barrel. The three structurally conserved regions (SCRs) of the lipocalin fold (SCR1, SCR2, and SCR3) are labelled as heavy boxes. Adapted from Flower et al. (2000).
Notably, some LCNs contains three SRCs, whereas other LCNs have only one or two SCRs (Flower, et al., 2000). Accordingly, the LCN family can be divided in two groups: kernel LCNs and outlier LCNs. The Kernel lipocalins, representing a core set of LCNs, share three conserved sequence motifs corresponding to the three main SCRs of the lipocalin fold (Flower, et al., 2000; Grzyb, et al., 2006). The outlier LCNs typically share no more than two SCRs and are more diverse. Based on this categorization, A1M, ApoD, complement C8 gamma chain (C8γ), LCN2, lipocalin-type prostaglandin D2 synthase (L-PGDS), PEAP, RBP4, and all mouse MUPs belong to kernel lipocalins; while AGP1, AGP2, ApoM, LCN1, LCN13, and LCN14 are included in the outlier category (Charkoftaki, et al., 2019; Du, et al., 2015; Flower, et al., 2000; Grzyb, et al., 2006). It is important to note here, among all apolipoproteins (ApoA, ApoB, ApoC, ApoD, ApoE, ApoF, ApoH ApoL, ApoM), both ApoD and ApoM share no amino acid sequence similarities with other apolipoproteins but < 20% homology with lipocalins and both contain distinct SCR motifs which ApoD protein has all three SCRs of LCN (kernel lipocalin), whereas ApoM protein contains one SCR only (outlier lipocalin) (Charkoftaki, et al., 2019; Du, et al., 2015).
Although lipocalin proteins are structurally highly conserved and similar, their binding ligands and receptors are divergent, leading to their functional different (Akerstrom, et al., 2000; Ganfornina, et al., 2022; Grzyb, et al., 2006; Redl & Habeler, 2022; Virtanen, 2021). For example, LCN1, one of four major proteins in human tears functions as a lipid sponge on the ocular surface (Glasgow, et al., 1993; Holzfeind, et al., 1997; Lassagne, et al., 1993). LCN2 can bind metalloproteinase 9 (MMP9), lipoprotein receptor-related protein 2 (LRP2), toll-like receptor 4 (TLR4), and 24p3R to mediate various inflammatory response (Guardado, Ojeda-Juarez, Kaul, & Nordgren, 2021; Jaberi, et al., 2021). LCN7 (also named tubulointerstitial nephritis antigen-like 1, TINAGL1) acts as a matricellular regulator of angiogenesis through activation of the TGF-β signaling pathway and increased secretion of VEGF (Brown, et al., 2010; L. Sun, Dong, Gu, Guo, & Yu, 2019). LCN13 and LCN14 may regulate glucose homeostasis (Buhler, et al., 2021; Cho, Zhou, Sheng, & Rui, 2011; Lee, et al., 2016; Zhou & Rui, 2013). In addition, LCNs could be internalized into cells, and transferred to lysosomes either for degradation [i.e., L-PGDS] or performing their function there (Mohri, et al., 2006; Nagata, et al., 2009). They can also interact with mitochondria in damaged cells (Kristiansson, et al., 2020; Olsson, et al., 2013). For example, Olsson et al previously showed that A1M could bind with high affinity to apoptotic cells and is localized to mitochondria through specifically binding to a subunit of Complex I (Olsson, et al., 2013). Furthermore, extracellular LCNs exist in diverse formats: bound to protein partners, lipoprotein particles, or membrane vesicles, which may yield varied effects (Redl & Habeler, 2022; Virtanen, 2021). As lipid-associated proteins, LCNs have been well studied in the regulation of metabolic diseases (see reviews elsewhere (Bacci, et al., 2015; Christoffersen, 2021; Frances, Tavernier, & Viguerie, 2021; Jaberi, et al., 2021; Nono Nankam & Bluher, 2021; Zabetian-Targhi, et al., 2015)). However, recent years have also witnessed the great progress made on the investigation of major LCNs (i.e., ApoD, ApoM, LCN2, LCN10, L-PGDS, and RBP4) in the field of cardiovascular diseases, as what we focus on below.
3. Lipocalins in Coronary Artery Disease and Myocardial Infarction
Coronary artery disease (CAD) is recognized to be the leading cause of death worldwide (Frak, et al., 2022; Hetherington & Totary-Jain, 2022; Schuett, Lehrke, Marx, & Burgmaier, 2015). The critical pathological change observed in CAD patients is atherosclerosis with the cholesterol and lipid-based blockage that usually causes chronic inflammation and myocardial infarction (Frak, et al., 2022; Hetherington & Totary-Jain, 2022; Schuett, et al., 2015). LCNs are secreted proteins and can be detected in the blood, urine, and other body fluids (Akerstrom, et al., 2000; Flower, 1995, 1996; Ganfornina, et al., 2022; Grzyb, et al., 2006; Redl & Habeler, 2022; Virtanen, 2021). Over past decades, therefore, tremendous effort has been made to define lipocalins as diagnostic and prognostic biomarkers for atherosclerosis and ischemic heart disease. Currently, several LCN proteins (i.e., LCN2 and RBP4) have been identified as risk factors to promote atherosclerosis and myocardial injury (Cheng, et al., 2014; Dong, Li, & Tang, 2015; Hemdahl, et al., 2006; G. Huang, et al., 2012; F. Li, Xia, Li, & Yang, 2014; Lindberg, et al., 2012; Y. Liu, Wang, Chen, & Xia, 2015; Sahinarslan, et al., 2011; Shibata, et al., 2020; Sivalingam, et al., 2018; Soylu, et al., 2015; Wan, et al., 2014). By contrast, multiple LCNs (i.e., ApoD, ApoM, and L-PGDS) are found to have anti-atherogenic properties and protective effects against myocardial infarction injury (Ahnstrom, et al., 2008; Annema, et al., 2022; Hosbond, et al., 2014; Inoue, et al., 2008; Kurano & Yatomi, 2018; Sarjeant, et al., 2003; Su, Jiao, Yang, & Ye, 2009; H. Sun, et al., 2019; Tsukamoto, et al., 2013; X. Zhang, et al., 2021). Therefore, the following contents focus on the roles of LCN2, RBP4, ApoD, ApoM, and L-PGDS in CAD and ischemia heart disease.
3.1. Lipocalins serve as biomarkers for coronary artery disease and ischemic heart disease:
3.1.1. Apolipoprotein D (ApoD)
Apolipoprotein D (ApoD) is an atypical apolipoprotein with a structure like lipocalins and belongs to LCN family (Weech, et al., 1991). While it is a component of the high-density lipoprotein (HDL), its relative abundance in HDL particles is low (1–2%) (Rassart, Desmarais, Najyb, Bergeron, & Mounier, 2020). Nonetheless, the majority of circulating ApoD (~ 83%) is carried by HDL particles, while a small fraction is bound to low-density lipoproteins (LDLs) and very low-density lipoproteins (VLDLs) (Blanco-Vaca, Via, Yang, Massey, & Pownall, 1992; Rassart, et al., 2020; Soiland, et al., 2007; Weech, et al., 1991). Recently, increasing evidence has indicated that aberrant expression of ApoD is linked to the altered lipid metabolism and risk of CAD (Annema, et al., 2022; Sarjeant, et al., 2003; H. Sun, et al., 2019; Tsukamoto, et al., 2013; X. Zhang, et al., 2021). For example, Sun et al (2019) utilized iTRAQ labeling followed by 2D LC-MS/MS to compare the urinary proteome of CAD cohorts (n=22) to healthy controls (n=22) (H. Sun, et al., 2019). Their results revealed that urine ApoD levels were significantly lower in CAD patients than healthy cohorts. Further analysis showed that ApoD together with TFF1 (Trefoil Factor 1) could diagnose CAD with 85% sensitivity and 99% specificity. However, Tsukamoto et al (2013) (Tsukamoto, et al., 2013) reported that increased ApoD deposition is detectable in the atherosclerotic lesions of humans, and the plasma ApoD levels were markedly increased in an atherosclerotic mouse model (ApoE-KO). To clarify such a difference, a recent study by Zhang et al (2021) conducted a deep RNA-sequencing on whole blood collected from a cohort of CAD patients with early myocardial infarction (MI, n=55), high coronary artery calcification (CAC) without prior MI (n=72), and controls (n=71) (X. Zhang, et al., 2021). They observed that ApoD was significantly downregulated in all CAD patients with either early MI or high CAC, compared with controls. Controversially, Annema et al (2022) measured serum levels of ApoD in 531 Caucasian individuals who underwent coronary angiography (356 males and 175 females) and follow-up period over a median of 5.8 years (Annema, et al., 2022). These authors found that the higher cumulative incidence rates of mortality and adverse cardiovascular events is positively associated with the increased serum levels of ApoD. These data suggest that high levels of circulating ApoD could be a biomarker of poor prognosis in patients with suspected or established coronary artery disease. Collectively, it will be warranted to investigate whether the elevation of serum ApoD levels in human patients may either contribute to CAD development or alternatively, implicate a compensatory mechanism or is an innocent bystander.
3.1.2. Apolipoprotein M (ApoM)
Apolipoprotein M (ApoM) was initially identified by Xu et al (1999) (N. Xu & Dahlback, 1999) as an apolipoprotein. However, later studies found that unlike most apolipoproteins, ApoM protein structure has a hydrophobic binding pocket resembling lipocalin proteins (Christoffersen, et al., 2006). ApoM is mainly synthesized in the liver and kidney, and to a minor extent, in adipose tissue (Christoffersen, et al., 2008; Sramkova, et al., 2019). While ApoM in the circulation is primarily carried by HDLs, it can bind to other lipoprotein subtypes including LDL, VLDLs, and chylomicrons (Christoffersen, et al., 2006; N. Xu & Dahlback, 1999). At present, numerous clinical investigations have focused on the role of ApoM in the prognosis of patients with coronary heart disease and myocardial infarction (Ahnstrom, et al., 2008; Kurano & Yatomi, 2018; Su, et al., 2009). Disappointingly, two earlier studies by Ahnström et al (2008) and Su et al (2009) suggest that plasma levels of apolipoprotein M are not associated with the risk of coronary artery disease (Ahnstrom, et al., 2008; Su, et al., 2009). However, Wolfrum et al (2005) found that silencing of ApoM expression in mice with small interfering RNA (siRNA) resulted in the accumulation of large (Wolfrum, Poy, & Stoffel, 2005), cholesterol-rich HDL particles, due to impaired conversion of HDL to pre-β-HDL, a subclass of lipid-poor apolipoproteins that serves as a key acceptor of peripheral cellular cholesterol (Wroblewska, 2011). This study suggests that reduced expression of ApoM may influence HDL and cholesterol metabolism and further affect the susceptibility to CAD. Along this line, the subsequent studies were shifted to explore single nucleotide polymorphisms (SNPs) of the proximal promoter region of ApoM gene in the whole blood of CAD patients (H. Guo, Zhao, Zhang, Chen, & Zhang, 2015; Jiao, et al., 2007; W. W. Xu, et al., 2008; P. H. Zhang, et al., 2016; Z. Zhang, Chu, & Yin, 2013; L. Zheng, et al., 2014; L. Zheng, et al., 2009). These SNPs may affect the transcription activity and expression levels of ApoM (H. Guo, et al., 2015; Jiao, et al., 2007; W. W. Xu, et al., 2008; P. H. Zhang, et al., 2016; Z. Zhang, et al., 2013; L. Zheng, et al., 2014; L. Zheng, et al., 2009). So far, at least 5 SNPs have been identified including T-1628G, C-1065A, T-855C, and T-778C, and C-724del that are linked to CAD and myocardial infarction (H. Guo, et al., 2015; Jiao, et al., 2007; W. W. Xu, et al., 2008; P. H. Zhang, et al., 2016; Z. Zhang, et al., 2013; L. Zheng, et al., 2014; L. Zheng, et al., 2009). For example, both Jiao et al (2007) and Zheng et al (2009) reported that there is a prominent association between the T-778C polymorphism and the risk of CAD in the Chinese population (Jiao, et al., 2007; L. Zheng, et al., 2009). Xu et al (2008) analyzed T-855C mutant allele in 418 CAD patients and 372 controls and showed that T-855C polymorphism carriers had an increased risk for CAD (W. W. Xu, et al., 2008). Accordingly, Zheng et al (2014) reported that occurrences of alleles T-1628G, T-855C and C-724del were significantly higher in CAD patients compared to non-CAD patients (L. Zheng, et al., 2014). They further confirmed that −855C and −724del could decrease ApoM expressions. These results were also validated by Zhang et al (2016) who showed that T-855C in the ApoM promoter could be a biomarker and provides binding sites for AP-2α, leading to a reduction of ApoM transcription activity (P. H. Zhang, et al., 2016).
Furthermore, Guo et al (2015) investigated the relationship between −724del in the ApoM promoter and myocardial infarction. They observed that the −724Del frequency in the MI group was significantly higher than that in the controls (H. Guo, et al., 2015), Accordingly, plasma ApoM levels were remarkably decreased but the total cholesterol (TC) levels were dramatically increased in MI patients with −724Del allele, in comparison to control samples. These results indicate that −724del polymorphism of ApoM promoter suppresses its transcription activity, leading to the downregulation of ApoM protein expression, and increased the risk of MI. Nonetheless, it remains unclear and needs to further determine whether other ApoM SNPs in addition to −724del could serve as a biomarker for ischemic heart disease.
3.1.3. Lipocalin 2 (LCN2)
LCN2 was initially identified from simian virus 40 (SV-40)–infected mouse kidney cells in 1989 by Hraba-Renevey et al (Hraba-Renevey, Turler, Kress, Salomon, & Weil, 1989) and later, the LCN2 protein was isolated from human neutrophil granules, and named neutrophil gelatinase-associated lipocalin (NGAL) (Kjeldsen, Johnsen, Sengelov, & Borregaard, 1993). At the present, LCN2 is defined as a pleiotropic glycoprotein that belongs to the lipocalin family (Charkoftaki, et al., 2019). In addition to secretion by activated neutrophils, LCN2 can be also released by multiple cell types including endothelial cells, macrophages, epithelial cells, kidney tubular cells and hepatocytes (Flo, et al., 2004). The expression of NGAL is dynamically altered in several acute and chronic pathological processes such as cell differentiation and proliferation, fibrosis, inflammation, iron trafficking, and metabolic disorders (Helanova, Spinar, & Parenica, 2014). NGAL has gained increasing attention as a potent early biomarker of renal injury, inflammation and more recently as a valuable biomarker of atherosclerosis and ischemic heart disease (Cheng, et al., 2014; Hemdahl, et al., 2006; Lindberg, et al., 2012; Sahinarslan, et al., 2011; Shibata, et al., 2020; Sivalingam, et al., 2018; Soylu, et al., 2015).
In the case of atherosclerotic arteries, numerous groups have reported that increased levels of serum or urinal LCN2 correlate with atherosclerosis risk factors, disease severity burden and mortality. For example, Ni et al (2013) measured serum LCN2 levels in a Chinese cohort of 261 in-patients who underwent coronary angiography (169 men and 92 postmenopausal women; CAD: 188 and non-CAD: 73). They observed that serum LCN2 levels were significantly higher in male patients than females and interestingly, significant differences or correlations were seen only in men with CAD, compared to non-CAD (Ni, et al., 2013).
Similarly, Lahiri et al (2018) reported that plasma LCN2 in patients admitted with acute coronary syndromes (ACS) can be considered as a possible new risk marker in ACS because it is able to predict hospital mortality (Lahiri, Alex, & George, 2018). Likewise, Soylu et al (2015) collected peripheral blood samples from 45 patients with normal coronary arteries (NCA) who underwent coronary angiography and 47 patients with non-ST elevation ACS (NSTE-ACS) (Soylu, et al., 2015). They observed that serum levels of LCN2 were remarkably higher in the NSTE-ACS group, in comparison to with the NCA control samples. Further analysis showed that higher levels of LCN2 were correlated well with the complexity of lesion and severity of CAD patients with NSTE-ACS. These results suggest that serum levels of LCN2 on admission are positively associated with the burden of atherosclerosis in ACS patients with NSTE.
In the case of patients with STEMI (ST segment elevation myocardial infarction), Kirbis et al (2015) prospectively measured admission and in-hospital urine LCN2 by investigating 61 consecutive STEMI patients after primary percutaneous coronary intervention. They found that urine LCN2 was elevated very early after STEMI, suggesting it might be a marker of high risk for acute heart failure in an individual patient (Kirbis, Gorenjak, & Sinkovic, 2015). Similarly, Lindberg et al (2012), Helanova et al (2015), Barbarash et al (2017), Li et al (2019) consistently demonstrated that higher LCN2 level in the blood of patients with STEMI is an independent predictor of mortality (Barbarash, et al., 2017; Helanova, et al., 2015; C. Li, et al., 2019; Lindberg, et al., 2012). Together, these clinical observations strongly implicate that LCN2 is a risk biomarker for CAD and IHD (ischemic heart disease).
3.1.4. Lipocalin-type prostaglandin D synthase (L-PGDS):
L-PGDS was originally discovered by Clausen (1961) as a β-trace in human cerebrospinal fluid (Clausen, 1961), and later was found to be identical to human L-PGDS (Hoffmann, et al., 1993; Watanabe, Urade, Mader, Murphy, & Hayaishi, 1994). L-PGDS is unique in its bifunctional character as both an enzyme synthesizing prostaglandin D2 (PGD2) (Urade, Fujimoto, & Hayaishi, 1985) and a lipophilic ligand-carrier protein of the lipocalin family (Urade & Hayaishi, 2000b). The amino acid sequence of L-PGDS has the homology with the lipocalin family members and possesses a typical lipocalin fold of β-barrel (Urade & Hayaishi, 2000a). While L-PGDS was initially detected in the central nervous system and male genital organs of various mammals (Clausen, 1961), it is also distributed in myocardial cells, atrial endocardial cells, and smooth muscle cells of the arteriosclerotic intima (Urade & Hayaishi, 2000a, 2000b). Eguchi et al (1997) reported that L-PGDS is accumulated in the atherosclerotic plaque of coronary arteries with severe stenosis. They further measured the plasma levels of L-PGDS in human patients with stable angina and showed that the L-PGDS levels were significantly higher in the cardiac vein than in the coronary artery (Eguchi, et al., 1997). These results indicate that L-PGDS is present in the stenotic site of patients with stable angina and can be secreted from the myocardium into the coronary circulation.
Along this line, a multicenter cooperative study by Inoue et al (2008) measured serum levels in a large cohort of 1013 consecutive patients with stable CAD and 241 controls (Inoue, et al., 2008). Their results indicated that the L-PGDS levels in CAD patients were significantly lower than controls. Multiple regression analysis further revealed that the L-PGDS level was the most powerful independent predictor of the coronary severity score. This study suggests that the reduced L-PGDS levels are related to the severity of CAD, and the measurement of serum L-PGDS levels can be helpful for screening of stable CAD prior to coronary angiography. Hosbond et al (2014) reported that L-PGDS is not a biomarker of atherosclerotic manifestations (Hosbond, et al., 2014), however, this investigation is based on a single blood test from a small cohort of 30 patients with ACS which appears to be less valuable.
3.1.5. Retinol binding protein 4 (RBP4)
The RBP4 was initially discovered by Quadro et al (1999) as an adipokine that binds specifically to vitamin A (retinol) and delivering it from the liver to target tissues and is therefore regulating circulating levels of vitamin A (Quadro, et al., 1999). RBP4 is produced mainly by the liver and adipose tissue (Christou, Tselepis, & Kiortsis, 2012). Accordingly, RBP4 has been well characterized to involve the development of obesity and insulin resistance (Christou, et al., 2012). RBP4 has been proposed to involve in the pathophysiology of CAD, but accumulated findings on the association of RBP4 levels with CAD are inconsistent and even opposite, as reviewed below.
An earlier clinical investigation by Mallat et al (2009) demonstrated that serum level of RBP4 does not provide added value for predicting CAD risk beyond traditional risk factors (Mallat, et al., 2009). Subsequently, Sun et al (2013) determined plasma levels of full-length and several C-terminally truncated subfractions of RBP4 among 468 women who developed CHD and 472 matched controls in the Nurses’ Health Study cohort (Q. Sun, et al., 2013). They observed that higher circulating full-length and total RBP4 levels but not truncated RBP4 are significantly associated with increased risk of woman patients with CHD in a time-dependent fashion. Consistently, a later study by Liu et al (2015) who also measured the plasma levels of RBP4 in 447 women (246 with ACS and 201 with stable CAD), and plasma levels of RBP4 were remarkably higher in stable CAD patients with complex coronary lesions than in those with simple lesions (Y. Liu, et al., 2015). Further multiple logistic regression analysis indicated that higher levels of RBP4 independently correlated with a 23% higher risk for complex lesions, and total plasma levels of RBP4 were predictors of cardiac death in female patients.
Regardless gender, Calo et al (2014) investigated the relationship between RBP4 levels and the presence and severity of CAD (Calo, Maiolino, Pagnin, Vertolli, & Davis, 2014), Lambadiari et al (2014), Li et al (2014), Perumalsamy et al (2021), Qian et al (2022), and Nar et al (2021) determined the relationship between RBP4 levels and the presence/severity of CAD. All these studies consistently observed that serum levels of RBP4 were greatly elevated in CAD patients compared to non-CAD cohorts, and independently correlated with CAD severity and adverse cardiovascular outcomes (Lambadiari, et al., 2014; F. Li, Xia, Li, et al., 2014; Nar, Sanlialp, & Nar, 2021; Perumalsamy, Ahmad, & Huri, 2021; Qian, Yan, Xu, Fang, & Ma, 2022).
Controversially, Wang et al (2018) investigated the role of RBP4 and the possible correlation between RBP4 levels and sex hormones, especially testosterone and estradiol, in CAD patients (n = 440) and healthy controls (n = 218) (H. Wang, et al., 2018). They observed that serum levels of RBP4 were significantly lower in male CAD patients, especially those with acute MI (myocardial infarction), than in control samples. However, there was no significant difference in serum RBP4 levels in female CAD patients between both groups. In addition, these authors found that RBP4 levels positively correlated with testosterone in male CAD patients (r = 0.124, p< 0.05). Logistic regression analysis revealed that RBP4 is a protective factor for CAD. Finally, they conclude that serum levels of RBP4 are significantly decreased and positively related with testosterone in men with CAD. Higher levels of RBP4 correlate well with lower risk of CAD. These results indicate that RBP4 could play a potential protective role for male but not for female CAD patients, which is totally opposite with other groups’ findings, mentioned above.
Taken together, numerous epidemiological studies have investigated the correlation of serum RBP4 levels with the risk of CAD over the past years. Most studies have revealed similar findings that elevated serum RBP4 levels are positively related to CAD (Calo, et al., 2014; Lambadiari, et al., 2014; F. Li, Xia, Li, et al., 2014; Y. Liu, et al., 2015; Mallat, et al., 2009; Nar, et al., 2021; Perumalsamy, et al., 2021; Qian, et al., 2022; Q. Sun, et al., 2013), whereas a nonsignificant or even negative correlations between RBP4 levels and CAD have also documented in other studies (Cubedo, et al., 2014; T. Liu, et al., 2019; Pan, et al., 2020; H. Wang, et al., 2018). Therefore, further studies will be performed to discern whether RBP4 levels really correlate with the risk of CAD, using a large cohort of CAD patients from multiple center and different races/populations.
3.2. Protective effects of ApoD, ApoM and L-PGDS against CAD and MI
As mentioned above, ApoD, a member of lipocalin family, plays a critical role in transporting lipids and other small hydrophobic molecules for metabolism (Sanchez & Ganfornina, 2021). Given that majority of circulating ApoD is bound with HDL, and dyslipidemia with increased LDL/ triglyceride and decreased HDL levels often predisposes an individual at risk to CAD (Sanchez & Ganfornina, 2021), ApoD therefore should have a great impact on the pathogenesis of CAD and its related myocardial infarction injury. In this regard, an earlier study by Sarjeant et al (2003) investigated whether ApoD could affect vascular smooth muscle cell (VSMC) proliferation, a critical culprit for atherosclerosis and arterial restenosis after angioplasty (Sarjeant, et al., 2003). They firstly observed that ApoD is accumulated in human atheromatous plaque but not in normal coronary arteries, suggesting the regulatory effect of ApoD on VSMCs during atherosclerosis. Further experimental results revealed that intracellular accumulation of ApoD can selectively inhibit PDGF-BB–induced VSMC proliferation by preventing translocation of phospho-ERK1/2 to the nucleus. These data suggest that an increased ApoD deposition in atherosclerotic lesions could be ascribed to a compensatory response of ApoD to help cholesterol removal from peripheral cells or derive from the consequence of defects in ApoD-mediated cholesterol trafficking (Sarjeant, et al., 2003). Indeed, Tsukamoto et al (2013) utilized a double-knockout mouse model (dKO) with homozygous null mutations in the HDL receptor (SR-BI) and apolipoprotein E (ApoE) genes which spontaneously develops occlusive, atherosclerotic CAD and ischemic heart failure (Tsukamoto, et al., 2013). They observed that the expression of ApoD in the dKO hearts was dramatically upregulated by 80-fold at the age of 43 days, compared with control mouse hearts. Interestingly, in mouse ischemic hearts, the expression of ApoD was substantially upregulated in non-infarcted but not in infarcted tissue at 48 h post-MI (Tsukamoto, et al., 2013). Considering that ApoD was previously shown to protect against hypertoxic stress in flies and mice and to be activated by neuronal injury (Sanchez & Ganfornina, 2021), therefore, Tsukamoto et al (2013) hypothesized that elevation of ApoD may be a protective mechanism of cardiac cells (cardiomyocytes, endothelia cells, etc.) in response to vessel occlusion and consequent ischemic stress (Tsukamoto, et al., 2013). Using ApoD-overexpressing and ApoD-KO mice, these authors clearly identified ApoD as a powerful protector against myocardial infarction injury (Tsukamoto, et al., 2013). Collectively, the mechanism underlying ApoD-mediated cardio-protection against ischemia injury is associated with: 1) a reduced atherosclerosis by disrupting p-ERK nuclear translocation; 2) an increased cell viability in cardiomyocytes and vessel endothelia cells (ECs), possibly due to the properly folded conformation of ApoD and its antioxidant activity, 3) increased internalization of plasma-derived ApoD, and/or (4) a regulation of cell differentiation related to the angiogenic response (Fig. 3).
Figure 3: ApoD-mediated protection against atherosclerosis and myocardial infarction (MI) injury.
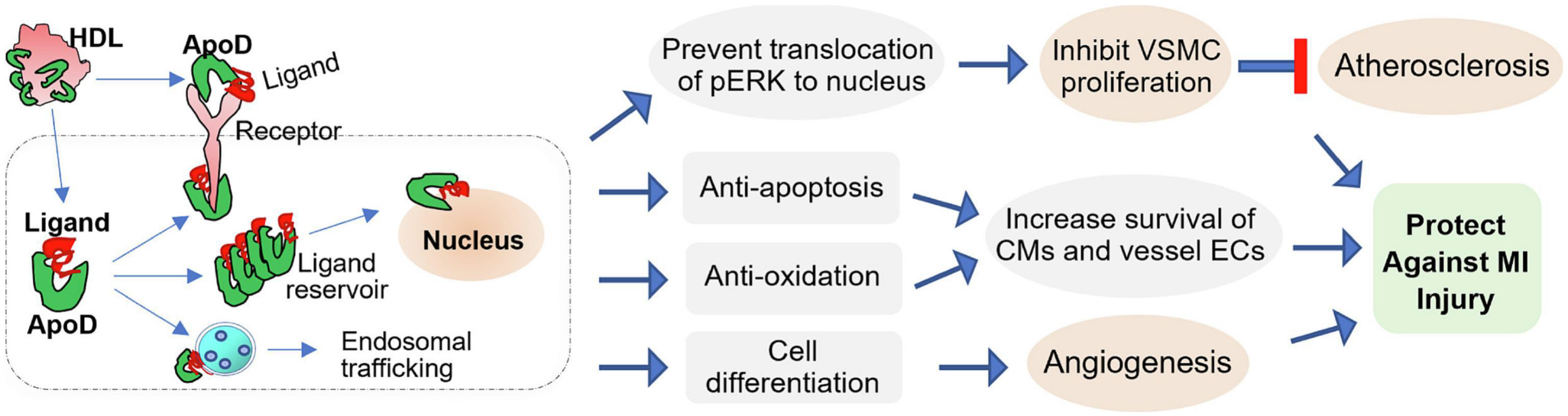
Circulating ApoD can be internalized into the cell in which ApoD binds hydrophobic ligands, while may pile up as an intracellular reservoir to stimulate the effect on gene transcription of nuclear receptor ligands or prevent ligand translocation to the nucleus. ApoD is also able to exert endosomal trafficking of its bound ligands/proteins. In addition, ApoD can interact with transmembrane receptors and complex with other proteins either inside or outside the cell. Accordingly, ApoD has been identified to prevent pERK translocation to the nucleus, leading to the inhibition of aberrant VSMC proliferation and thereby, attenuation of atherosclerosis. Moreover, increased ApoD levels can protect cardiomyocytes (CMs) and vessel endothelial cells (ECs) against apoptosis and oxidation as well as promote cell differentiation-related angiogenesis, leading to reduced myocardial infarction (MI) injury.
Like ApoD, circulating ApoM also binds to HDL particles. Earlier studies showed that ApoM-bound HDL had atheroprotective effects against LDL-oxidation via stimulating cholesterol efflux from macrophages and regulating pre-beta HDL formation (Christoffersen, et al., 2008; Christoffersen, et al., 2006; Sramkova, et al., 2019; Wolfrum, et al., 2005). However, a later study by Christoffersen et al (2011) discovered that ApoM could function as a chaperon to the bioactive sphingolipid, sphingosine-1-phosphate (S1P), with ~70% of S1P bound to ApoM (Christoffersen, et al., 2011). Further experimental results from other groups indicate that the complex of HDL-ApoM/S1P is able to connect HDL with endothelial cells to maintain an intact endothelial barrier and limit endothelial inflammation and apoptosis (Y. Liu & Tie, 2019; Ruiz, Frej, et al., 2017; Ruiz, Okada, & Dahlback, 2017). So far, several studies have showed that ApoM and S1P individually provide anti-atherogenic potential (Elsoe, et al., 2012; X. S. Huang, Zhao, Hu, & Luo, 2007; Wolfrum, et al., 2005). However, high levels of either ApoM or S1P could be associated with dyslipidemia (Hajny, Borup, Elsoe, & Christoffersen, 2021). In this regard, there are some discrepancies about the understanding of the ApoM/S1P-mediated protection against atherogenesis. For example, overexpression of ApoM in LDL-R-deficiency mice demonstrated protective effects against atherosclerosis (Christoffersen, et al., 2008; Sramkova, et al., 2019; Wolfrum, et al., 2005). However, such ApoM-mediated favorable consequence on atherosclerosis was only displayed in mouse models with unchanged LDL-cholesterol in comparsion to controls (Christoffersen, et al., 2008). Otherwise, elevation of ApoM levels caused even accelerated atherosclerosis (Christoffersen, 2021). The mechanism underlying such opposite results is unclear, but the impact of ApoM on the circulating LDL/VLDL particles and the lipoprotein lipase activity may play a role. Regarding S1P, overexpression of S1P showed beneficial to suppress or retard the progression of atherosclerosis in animal models through reduced macrophage apoptosis (Feuerborn, et al., 2017), but super-physiological concentrations of S1P displayed in SGLT2-knockout mice could induce dyslipidemia (Bektas, et al., 2010) which would have a potential harmful consequence on the cardiovascular system. Using ApoM-transgenic mice in which plasma levels of S1P are elevated by 3-fold, Morel et al (2016) showed that elevation of ApoM/S1P significantly reduced myocardial infarction injury by phosphorylating the gap junction protein connexin 43, and short-term treatment with S1P has a therapeutic potential in the fight against myocardial infarction damage (Morel, et al., 2016). Collectively, the complex of ApoM/S1P may have either beneficial or harmful consequence on the cardiovascular system. Therefore, it will be greatly needed for more investigations to understand the interplay between these molecules.
Unlike ApoD and ApoM as HDL-binding proteins, L-PGDS is the only member of LCN family characterized as an enzyme involved in the synthesis of Prostaglandin D2 (PGD2), a molecule that involves in the regulation of many patho-physiological functions (Urade, 2021). In addition, L-PGDS binds retinoids, thyroids, amyloid β (Aβ) peptides, and diverse types of lipophilic ligands (Hoffmann, et al., 1993; Urade, et al., 1985; Urade & Hayaishi, 2000a, 2000b; Watanabe, et al., 1994). Currently, it is well documented that L-PGDS has neuro-protective effects against damage following either acute or chronic neurological disorders (Hoffmann, et al., 1993; Urade, et al., 1985; Urade & Hayaishi, 2000a, 2000b; Watanabe, et al., 1994). Nonetheless, accumulating evidence has implicated that L-PGDS is highly expressed in the heart including in cardiomyocytes (Eguchi, et al., 1997; Han, et al., 2009; Katsumata, et al., 2014; Otsuki, et al., 2003; Tokudome, et al., 2009). Importantly, Tokudome et al (2009) reported that glucocorticoid-mediated protection during myocardial ischemia/reperfusion (I/R) is largely ascribed to the activation of L-PGDS-derived PGD2 biosynthesis in cardiomyocytes (Tokudome, et al., 2009). Consistently, Katsumata et al (2014) showed that PGD2 protects heart against I/R injury by activating Nrf2 predominantly via PGF(2α) receptor (Katsumata, et al., 2014). While reduced L-PGDS levels may contribute to the severity of CAD, it remains unclear whether elevation of L-PGDS is beneficial for the treatment of CAD. In addition, future studies will be needed to explore whether L-PGDS has a therapeutic value in ischemic heart disease.
3.3. Detrimental effects of LCN2 and RBP4 in CAD and MI.
LCN2 was initially identified as a protein isolated from specific neutrophil granules released at sites of infection and inflammation in human and was validated to be covalently bound with neutrophil gelatinase (Kjeldsen, et al., 1993). LCN2 has bacteriostatic properties through its sequestering iron from bacterial siderophores (Holmes, Paulsene, Jide, Ratledge, & Strong, 2005; Nairz, et al., 2009). In the case of atherosclerotic arteries, LCN2 may play a key role in vascular remodeling, because LCN2 can form a complex with MMP-9 and thereby, prevent MMP-9 degradation and reinforces its proteolytic activity contributing to the formation of an unstable atherosclerotic plaque (Hemdahl, et al., 2006). Indeed, the complex of LCN2/MM9 has been detected in atherosclerotic plaques and its concentration is increased in plaques with intramural hematoma and central necrosis (Cruz, Gaiao, Maisel, Ronco, & Devarajan, 2012; Hemdahl, et al., 2006; Leclercq, et al., 2007; te Boekhorst, et al., 2011). Recently, to evaluate the functional role of Lcn2 in different stages of atherosclerosis, Amersfoort et al (2018) generated a double knockout mouse model in which the expression of low-density lipoprotein receptor (LDLR) and Lcn2 are deficient (Amersfoort, et al., 2018). They showed that the contribution of Lcn2 to experimental atherosclerosis is stage-dependent as it appears to limit lesion development, whereas it potentially involves plaque instability at more advanced stages of atherosclerosis. In the case of ischemic heart disease, Sung et al (2017) observed that myocardial Lcn2 was significantly upregulated in mouse ischemic hearts, compared to sham-group (Sung, et al., 2017). Accordingly, both Lcn2-KO mice displayed less cardiac ischemic injury than WT mice. Furthermore, treatment with recombinant Lcn2 protein promoted ischemia-induced cardiac cell death and myocardial dysfunction through inhibiting the protective autophagic response to ischemia. Consistently, Xu et al (2012) showed that LCN2 could induce cardiomyocyte apoptosis by increasing intracellular iron accumulation (G. Xu, et al., 2012). In addition, Song et al (2018) showed that holo-Lcn2 (Lcn2-siderophore-iron, 1:3:1) promotes mitochondrial ROS generation and suppresses mitochondrial oxidative phosphorylation in the cultured adult rat cardiomyocytes, which may contribute to ischemia-triggered cardiac damage (Song, et al., 2018). Put all together, these data suggest that Lcn2 may play a detrimental role in the pathogenesis of coronary artery disease and ischemic heart disease.
Regarding RBP4, while it is well appreciated as a plasma transport protein for delivering retinol from liver to the tissues, it may have harmful effects in the pathophysiology of CAD and myocardial infarction injury, as summarized in Figure 4. For example, Li et al (2014) treated rat aortic smooth muscle cells (RASMCs) with different doses of RBP4 (1 and 4 μg/mL) and observed that RBP4 stimulated aberrant VSMC proliferation and migration via activation of the MAPK pathway, which may contribute to the formation of atherosclerotic plaques in vivo (F. Li, Xia, Sheikh, et al., 2014). Moreover, Liu et al (2017) reported that increased levels of RBP4 in aortic atherosclerotic lesions of both human and mice could promote the formation of macrophage-derived foam cells through the activation of scavenger-receptor CD36-mediated cholesterol uptake via the Toll-like receptor 4, but not retinol, thus accelerating the progression of atherosclerosis (Y. Liu, et al., 2017). Lastly, RBP4 can play important role in the regulation of endothelial inflammatory response and mitochondrial dysfunction, both of which greatly accelerate or influence atherogenesis in the coronary artery (J. Wang, et al., 2015). As for the detrimental role of RBP4 in ischemic heart disease, Zhang et al (2021) recently showed that RBP4 was significantly increased both in vivo ischemic mouse hearts and in vitro hypoxic cardiomyocytes (K. Z. Zhang, et al., 2021). These authors further found that the elevation of RBP4 may activate cardiomyocyte pyroptosis upon acute myocardial infarction, evidenced by that overexpression of RBP4 greatly stimulated LRP3 inflammasome and Caspase-1 activities and thereby, promoting gasdermin-D (GSDMD)-mediated pyroptosis. Using co-immunoprecipitation assay, the same authors identified that RBP4 bound directly with NLRP3 in cardiomyocytes. What’s more, silencing of RBP4 in mouse hearts significantly reduced infarct size, limited AMI-induced pyroptosis and myocardial depression, in comparison to control samples. Taken together, these observations clearly indicate that RBP4 is a harmful factor by promoting cardiomyocyte pyroptosis via interaction with NLRP3 in the ischemic heart.
Figure 4: RBP4-mediated harmful effects on atherosclerosis and myocardial infarction (MI) injury.
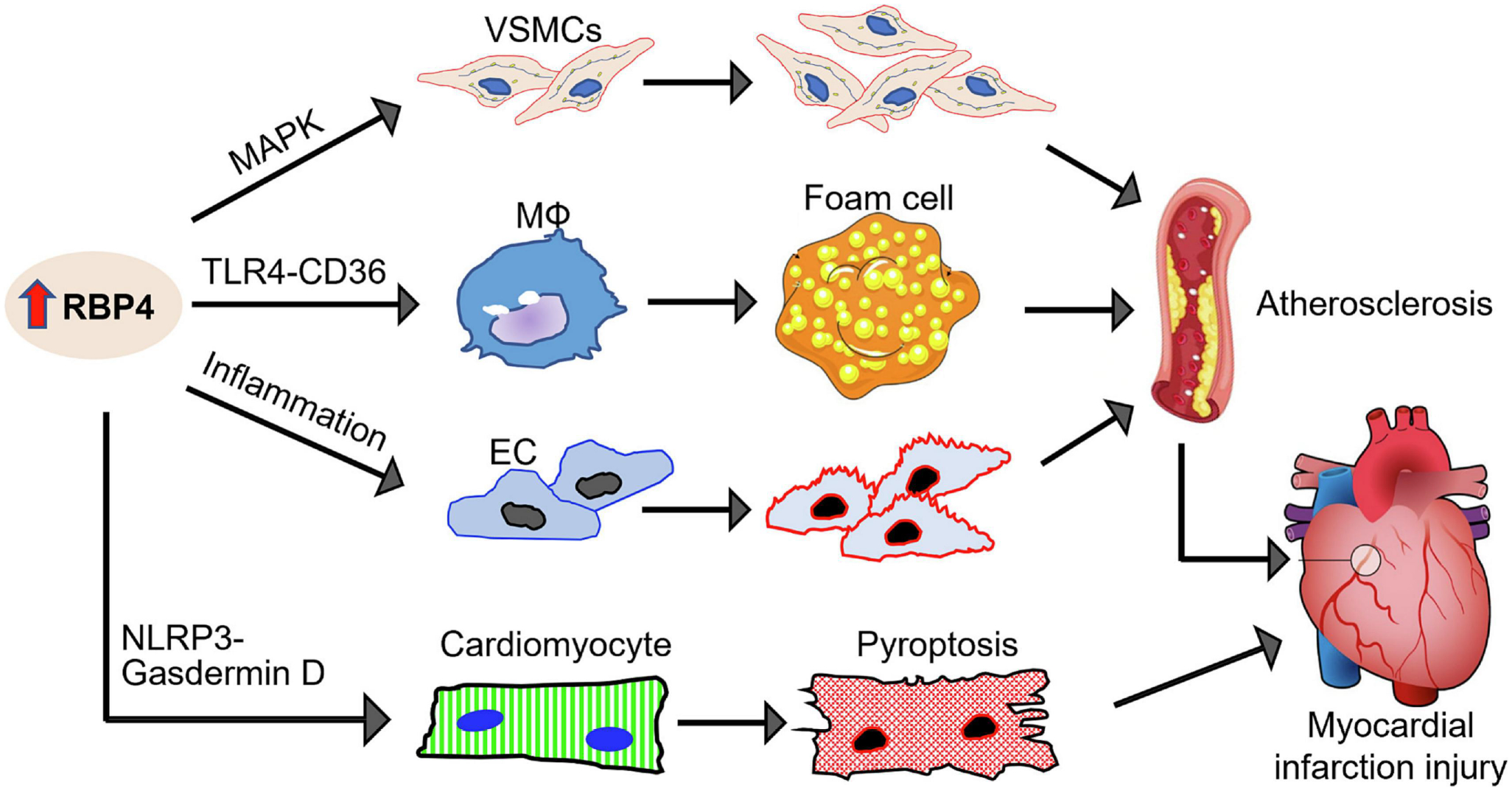
Elevation of RBP4 can: 1) activate MAPK pathway and promote aberrant VSMC proliferation; 2) interact with TLR4 and activate CD36-mediated cholesterol uptake in macrophages (MФs) which form foam cells; 3) increase mitochondrial dysfunction and inflammation in endothelial cells (ECs). All these significantly accelerate atherogenesis. In addition, elevation of RBP4 can interact directly with NLRP3 in cardiomyocytes, leading to GSDMD (gasdermin-D)-dependent pyroptosis and thereby, aggravate myocardial infarction injury.
4. Lipocalins in Cardiac Hypertrophy and Heart Failure
Pathological cardiac hypertrophy occurs usually in response to hypertension, increased workload, or chronic stress, which finally leads to heart failure and is the single most important risk factor for cardiovascular death (Frieler & Mortensen, 2015). Among human lipocalin proteins, two members including RBP4 and LCN2 have been well characterized to positively regulate pathological cardiac remodeling and heart failure, as summarized below.
Solini et al (2009) observed that serum RBP4 levels are elevated in patients with hypertension without metabolic syndrome (Solini, Santini, Madec, Rossi, & Muscelli, 2009). Consistently, Kraus et al (2015) utilized animal model and showed that elevation of serum RBP4 levels raises blood pressure (BP), whereas Lowering RBP4 reduces BP through enhanced eNOS-mediated vasodilatation (Kraus, et al., 2015). These studies suggest that RBP4 may play a direct role in the regulation of BP and involve pressure overload-induced cardiac hypertrophy. Moreover, Gao et al (2016) also observed that circulating levels of RBP4 and adipose expression of RBP4 are significantly increased in cardiac hypertrophic mice induced by transverse aortic constriction and angiotensin-II (Ang-II) infusion, compared to controls (Gao, et al., 2016). Importantly, treatment of neonatal cardiomyocyte with RBP4 greatly increased myocyte size and enhanced protein synthesis together with elevated expression of hypertrophic markers by activating TLR4/MyD88-mediated inflammatory pathways. Supportively, Rbp4-knockout mice are protected from Ang-II-induced cardiac hypertrophy (Kraus, et al., 2015).
Recently, Marques et al (2017) utilized genetic and environmental experimental mouse models to assess the association of LCN2 with concentric cardiac hypertrophy and its relation to the normal variation of human heart size and cardiac hypertrophy in diabetes mellitus (Marques, et al., 2017). They clearly demonstrated that higher levels of LCN2 are well correlated with cardiac hypertrophy, whereas adult Lcn2-knockout mice display significantly smaller hearts than wild-type controls. In human studies, higher LCN2 expression was also found to correlate well with cardiac hypertrophy in healthy individuals and in patients with type 2 diabetes mellitus. The further in vitro studies by Marques et al revealed that overexpression of Lcn2 in neonatal cardiomyocytes activates hypertrophic pathways, causing an increase in cardiomyocyte size but a decrease in their proliferation (Marques, et al., 2017). Collectively, this study suggests that Lcn2 is able to positively regulate cardiac hypertrophy. In support of these findings, Kim et al (2018) investigated the association between plasma levels of LCN2 and cardiac hypertrophy in patients with chronic kidney disease (CKD). They showed that higher plasma levels of LCN2 were independent predictors of cardiac hypertrophy and heart failure in patients with pre-dialysis CKD (Kim, et al., 2018).
More recently, Del Gaudio et al (2021) reported that loss of ApoM and loss of S1P transporter, Spns2, both can exaggerate cardiac hypertrophy in hypertension (Del Gaudio, et al., 2021), suggesting a protective role of ApoM/S1P signaling in hypertension and pathological cardiac hypertrophy. As for other lipocalin family members, while the expression levels of cardiac LCN6 and LCN10 in human patients with end-stage heart failure have been detected to be significantly lower than healthy controls (di Salvo, et al., 2015), their possible roles in cardiac hypertrophy are very limited.
5. Lipocalins in Diabetes-Induced Cardiac Dysfunction
Patients with diabetes have an increased risk of developing cardiovascular remodeling (Gustafsson, et al., 2004). Diabetes-induced heart failure (also termed as diabetic cardiomyopathy, DCM) occurs usually in the absence of other cardiac risk factors e.g. coronary artery disease, hypertension, or congenital heart diseases (Y. Zheng, Ley, & Hu, 2018). Over the past decades, a variety of mediators/factors including insufficient myocardial angiogenesis, cardiac fibrosis, cell death, lipid accumulation, inflammation, and oxidative stress have been defined as contributors to DCM (Jia, Hill, & Sowers, 2018). In this regard, lipocalin family members are well recognized to regulate lipid metabolism, insulin sensitivity, inflammatory response, oxidative stress, and angiogenesis, which are expected to play fundamental role in diabetic cardiomyopathy. However, the effects of lipocalins on the heart during diabetes have relatively less been studied. For example, LCN2 is a proinflammatory adipokine which is significantly elevated in the patients with obesity and diabetes (Y. Wang, et al., 2007) and clinical studies have also observed that there is a strong positive correlation between circulating LCN2 and heart failure (Chan, Sung, & Sweeney, 2015). Nonetheless, it remains unclear whether elevation of LCN2 directly contribute to diabetic cardiomyopathy.
Recently, we observed that the expression of Lcn2 is remarkably increased, whereas the Lcn10 expression is significantly downregulated in macrophages under metabolic stress conditions (Q. Li, et al., 2022). Accordingly, using a global knockout mouse model, we showed that ablation of Lcn10 could exacerbate diabetes-induced cardiomyopathy by skewing macrophages towards a pro-inflammatory phenotype and increased cardiac inflammatory response (Q. Li, et al., 2022). Mechanistically, we identified that Lcn10 deficiency in macrophages largely prevented Nr4a1 translocation to nuclei and disrupted its anti-inflammatory signaling pathway (Q. Li, et al., 2022). Our study suggests that reduction of Lcn10 may contribute to diabetes-induced cardiac dysfunction. Future investigations would be needed to elucidate whether elevation of Lcn10 expression/activity in macrophages or directly administration of recombinant Lcn10 protein has therapeutic potential for the treatment of diabetes-induced low-grade inflammation and concomitant cardiomyopathy. In addition, it will be warranted to explore whether other lipocalin family members are involved in diabetes cardiomyopathy.
6. Lipocalins in Sepsis-Induced Cardiomyopathy
Sepsis is a life-threatening syndrome due to multi-organ dysfunction induced by a dysregulated immune response to infection (Rudd, et al., 2020). Importantly, myocardial depression has been elucidated to be a major culprit to the increased risk of mortality and morbidity in patients with severe sepsis (Lin, et al., 2022). Although decades of intensive study, the precise mechanisms underlying sepsis-triggered immune dysregulation and cardiac dysfunction remain obscure (Beesley, et al., 2018; Carbone, Liberale, Preda, Schindler, & Montecucco, 2022; Hollenberg & Singer, 2021). As mentioned above, lipocalin family proteins have demonstrated diverse properties in the regulation of oxidative stress, immune response, vascular permeability, and cell survival, all of which are associated with sepsis. Therefore, in this regard, lipocalins may play good, bad, or ugly roles in the modulation of sepsis-triggered cardiomyopathy, which are discussed below.
Among human lipocalin family members, LCN2 is well characterized to serves as the potential biomarker in the assessment of severity and prediction of prognosis of patients with sepsis (Bagshaw, et al., 2010; Lentini, de Cal, Clementi, D’Angelo, & Ronco, 2012; Martensson, et al., 2010). In addition, Wang et al (2017) and Liu et al (2022) both observed that high plasma LCN2 correlates with high mortality and myocardial dysfunction in severe sepsis and septic shock (W. Liu, et al., 2022; B. Wang, et al., 2017). These clinical investigations suggest that increased levels of Lcn2 may have harmful effects and contribute to the pathogenesis of sepsis. However, an earlier study by Flo et al. (2004) demonstrated that exogenous Lcn2 directly inhibited the growth of Escherichia coli (E. coli) via its binding specificity for siderophores (Flo, et al., 2004). Furthermore, the presence or administration of Lcn2 protected against oxidative stress through upregulation of antioxidant enzymes (e.g., superoxide dismutase, heme oxygenase 1) in vivo during sepsis (Srinivasan, et al., 2012; Zhao, Wei, & Xu, 2015). Indeed, numerous studies have suggested that Lcn2 protects from organ failure in sepsis models (H. Li, et al., 2018; Srinivasan, et al., 2012; Wu, et al., 2010; Zhao, et al., 2015). Accordingly, Lcn2-null mice exhibited an increase in the bacterial burden and exacerbated sepsis (Bellmann-Weiler, et al., 2013; Z. Liu, Petersen, & Devireddy, 2013; Nairz, et al., 2015; Toyonaga, et al., 2016; Warszawska, et al., 2013). Therefore, in view of all these data, it could be hypothesized that Lcn2 offers therapeutic promise against sepsis-induced myocardial cardiomyopathy. Controversially, a recent study by Huang et al (2022) showed that LCN2 deficiency led to the improvement of cardiac function in bacterial endotoxin (LPS)-induced septic mice through the inhibition of ferroptosis in cardiomyocytes (Y. Huang, et al., 2022). The in vitro studies further revealed that Lcn2 induced cardiomyocyte ferroptosis by increasing intracellular labile iron pool (LIP) via interacting with its receptor 24p3R on the surface of myocytes. Another study by Liu et al (2022) indicated that LCN-2 may involve in the process of septic cardiomyopathy through stimulating lipid accumulation and mitochondrial dysfunction (W. Liu, et al., 2022). Therefore, future studies with directly injection of recombinant Lcn2 protein into bacterial sepsis model rather than endotoxin-induced sepsis model are warranted to discern whether Lcn2 may harness host immune system to protect against sepsis-induced heart failure or Lcn2 may cause cardiomyocyte ferroptosis and lipid toxicity, leading to septic myocardial injury (Fig. 5).
Figure 5: Paradoxical role of LCN2 in sepsis-induced myocardial depression.
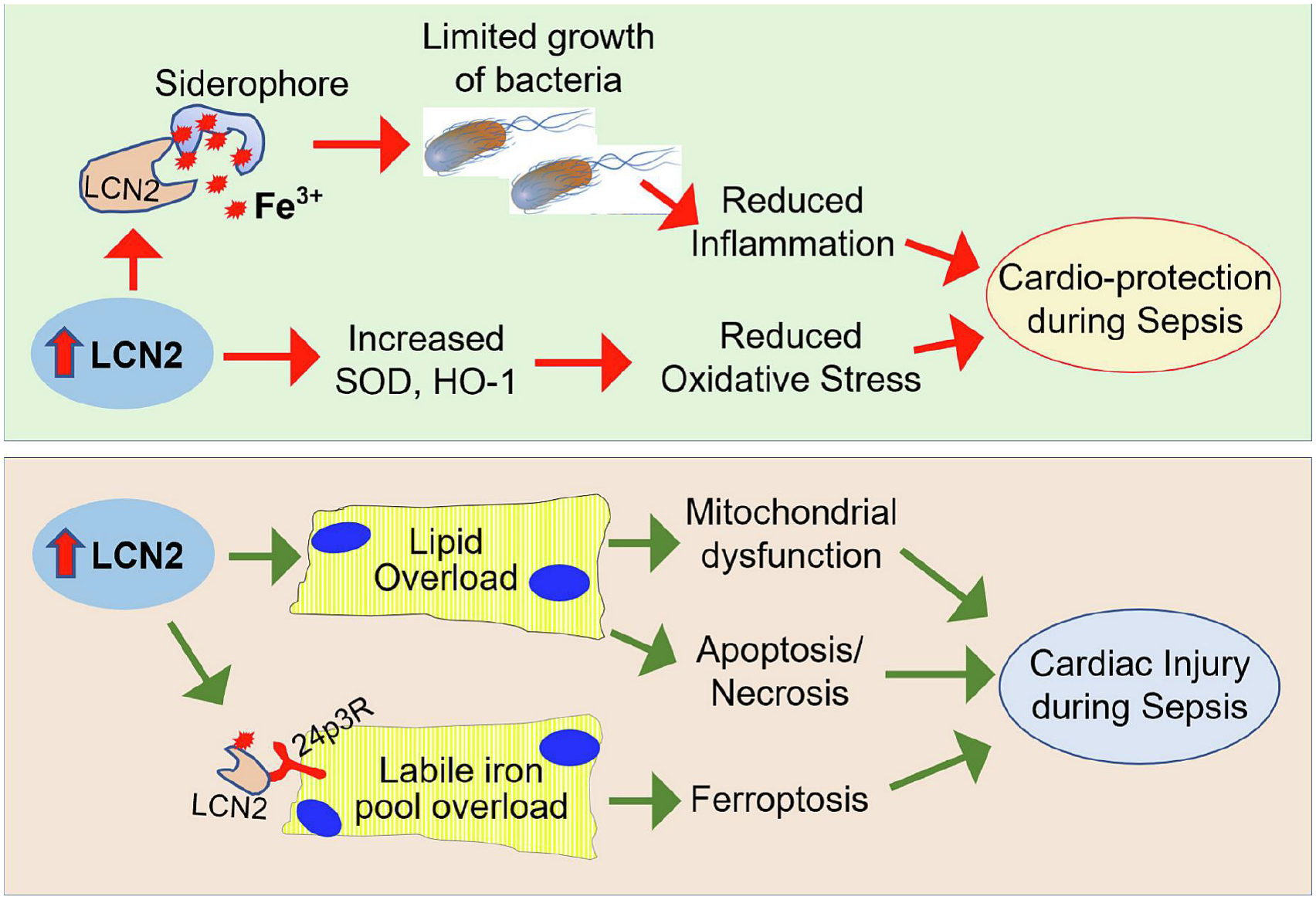
Increased levels of LCN2 could bind siderophore and limit bacteria to use of iron for growth, leading to reduced inflammation. In addition, Lcn2 can upregulate the expression of superoxide dismutase (SOD) and heme oxygenase-1 (HO-1), leading to reduced oxidative stress. Therefore, LCN2-mediated anti-inflammation and anti-oxidation should provide protection against sepsis-induced cardiac dysfunction. However, LCN2 is able to promote lipid accumulation and labile iron pool overload in cardiomyocytes, leading to apoptosis, necrosis, and ferroptosis, which exaggerate sepsis-induced cardiac injury.
Recently, Wang et al (2021) explored whether circulating Lcn10 serves as a prognostic tool in septic patients with myocardial depression (L. Wang, et al., 2021). They demonstrated that higher levels of serum LCN10 in septic patients are well correlated with the incidence of cardiac depression triggered by sepsis. Importantly, LCN10 appears a more reliable for the prediction of 28-day mortality than other biomarkers used clinically (L. Wang, et al., 2021). Using Lcn10-KO mouse model, we elucidated that loss of Lcn10 significantly promoted sepsis-induced cardiac dysfunction through increasing vascular leakage (Zhao H, 2023). By contrast, both endogenous and exogenous elevation of Lcn10 significantly reduced endothelial permeability and improved cardiac function during sepsis (Zhao H, 2023). Our results suggest that injection of recombinant Lcn10 protein could have therapeutic effects against sepsis-caused cardiomyopathy and mortality.
Unlike increased circulating LCN2 and LCN10 in septic patients, Kumaraswamy et al (2012) and Frej et al (2016) observed that the plasma levels of ApoM were significantly reduced in patients with severe sepsis (Kumaraswamy, Linder, Akesson, & Dahlback, 2012) (Frej, et al., 2016). As a carrier of S1P in HDL for endothelial barrier protection (Christoffersen, et al., 2011; Y. Li, et al., 2020), the decreased ApoM could contribute to the increased vascular leakage observed in sepsis. Indeed, Fan et al (2020) reported that the plasma levels of HDL/S1P were significantly decreased and negatively correlated with endothelial damage in sepsis, both in the animal model and in the septic patients (Fan, et al., 2020). The in vivo HDL/S1P injection significantly reduced lung oedema and endothelial leakage in septic rats (Fan, et al., 2020). In addition, Kurano et al (2018) showed that ApoM protected mice from LPS-induced organ injury and death (Kurano, et al., 2018). Therefore, it will be very interesting to examine whether elevation of either ApoM or S1P has therapeutic potential for the improvement of sepsis-induced cardiac dysfunction and survival. As for the role of other lipocalin family members in sepsis-induced myocardial depression, there is very limited information in the literature.
7. Summary and Future Perspectives
Lipocalin family proteins, albeit the very low similarity of their amino acid sequences (< 20%), share a common three-dimensional structure (named lipocalin-fold) consisting of an eight-stranded antiparallel β -barrel (Charkoftaki, et al., 2019; Du, et al., 2015; Flower, et al., 1993; Pervaiz & Brew, 1987). They are not only important carriers of preferentially hydrophobic molecules, but also bind other ligands (i.e., siderophores, intact proteins) and membrane receptors (Akerstrom, et al., 2000; Flower, 1995, 1996; Flower, et al., 2000; Ganfornina, et al., 2022; Grzyb, et al., 2006; Redl & Habeler, 2022; Virtanen, 2021). Furthermore, they can internalize in cells and traffic to intracellular organelles (i.e., mitochondria, lysosomes) and nuclei (Asimakopoulou, et al., 2017; Kurozumi, et al., 2020; Yammine, Zablocki, Baron, Terzi, & Gallazzini, 2019). Consequently, they exert diverse biological and physiological effects (i.e., behavior, chemo-sensation, development, and reproduction) and are involved in a variety of pathological processes in many human diseases such as cancers, immune disorders, metabolic disease, neurological/psychiatric disorders, and cardiovascular disease. This review presents a detailed summary of major lipocalins in cardiovascular diseases which have been investigated so far. Since lipocalins are mainly extracellular proteins that can be detected in the blood, urine, and other body fluids, therefore, numerous studies have focused on the identification of lipocalins as biomarkers for the diagnosis and prognosis of cardiovascular diseases. In this review, we center on 6 major lipocalins including ApoD, ApoM, LCN2, LCN10, RBP4, and L-PGDS, and heavily summarize their diagnostic/prognostic values and their potential effects on coronary artery disease and myocardial infarction injury. We also highlight the roles of these 6 lipocalins in cardiac hypertrophy and heart failure as well as diabetic cardiomyopathy and sepsis-induced myocardial depression. Their possible therapeutic strategies for the treatment of cardiovascular diseases are also discussed in each section.
Currently, the large number of lipocalins have been identified to date. However, the individual pathophysiological function and its underlying mechanisms remains largely unclear. In particular, the knowledge about lipocalin receptors, cellular uptake of lipocalins, and the related signaling pathways is still limited. In this regard, future studies will be greatly needed to elucidate whether lipocalins interact with the distinctive or common receptor on cardiovascular cells, and which lipocalin could be used to develop pharmaceutical drug for cardiovascular disease. It is important to note here that human lipocalins have been chosen as appropriate scaffolds for protein engineering to generate anticalin drugs, because lipocalin-fold has highly structural plasticity of their binding sites which can be modified to specifically recognize and tightly bind disease-related molecular targets (Achatz, Jarasch, & Skerra, 2022; Schiefner, Gebauer, Richter, & Skerra, 2018; Schiefner & Skerra, 2015; Skerra, 2008). Anticalin proteins are usually generated by combinatorial design based on natural lipocalins which contain a densely central β-barrel supporting four structurally variable loops that form a binding pocket (Rothe & Skerra, 2018). Accordingly, reshaping of this variable loop region could yield a different anticalin protein (lipocalin isotype) with distinct ligand specificities or physiological functions. Initial application of these anticalin proteins was driven by the discovery of some structural similarity between their binding sites and those of immunoglobulins (antibodies) (Schlehuber & Skerra, 2005). The first LCN1-derived anticalin protein was developed for therapeutically targeting human VEGF-A, a key factor of tumor angiogenesis and ocular disease (Ferrara & Kerbel, 2005). The human LCN2 has been proved particularly successful for the design of binding proteins with novel specificities and so far, more than 20 Lcn2-based anticalins have been generated towards different medically relevant target proteins, mainly in the areas of cancer and inflammatory diseases [see reviews elsewhere (Achatz, et al., 2022; Deuschle, Ilyukhina, & Skerra, 2021)]. Indeed, anticalin proteins can be produced by modified human lipocalins with specificities for prescribed targets of interest, ranging from small molecules to proteins/peptides. The affinities of the resulting anticalin proteins can be competitive for or even better than those accessible by antibody technology. Furthermore, anticalin proteins comprise a single polypeptide chain that folds into a stable eight-stranded β-barrel with exposed N- and C-terminals (neither of which is part of the binding pocket) which makes them ideal building blocks to generate multi-valent, multi-paratopic and multi-specific proteins, thus offering novel therapeutic modalities for human diseases. Most importantly, anticalin proteins have low immunogenic potential, as they are modified from soluble human lipocalins that are enriched in plasma or other body fluids and only a limited number of amino acids are altered from their natural counterparts. All these advantages of anticalin proteins mentioned above make them attractive as therapeutics for cardiovascular disease. Therefore, it will be worthy to spend effort on developing such anticalin drugs aiming at cardiovascular disorders by selecting suitable targets, and then designing and modifying human lipocalins.
Acknowledgements
The research in Dr. Guo-Chang Fan’s lab is supported by NIH grants R01 GM-132149, and HL-160811, and R35 GM-149538.
List of Abbreviations
- A1M
α1-microglobulin
- Aβ
amyloid β
- ABPs
acid-binding proteins
- ACS
acute coronary syndromes
- AGP
α1-acid glycoprotein
- ApoD
apolipoprotein D
- ApoM
apolipoprotein M
- BP
blood pressure
- C8γ
complement C8 gamma chain
- CAC
coronary artery calcification
- CAD
Coronary artery disease
- CKD
chronic kidney disease
- dKO
double-knockout mouse model
- ECs
endothelia cells
- GD
glycodelin
- GSDMD
gasdermin-D
- HDL
high-density lipoprotein
- LCNs
Lipocalins
- LCN2
Lipocalin 2
- LDLs
low-density lipoproteins
- LDLR
low-density lipoprotein receptor
- LIP
labile iron pool
- L-PGDS
lipocalin-type prostaglandin D2 synthase
- LPS
lipopolysaccharide
- LRP2
lipoprotein receptor-related protein 2
- MMP9
metalloproteinase 9
- MUPs
major urinary protein genes
- MPIs
metalloproteinase inhibitors
- NCA
normal coronary arteries
- NGAL
neutrophil gelatinase-associated lipocalin
- NSTE-ACS
non-ST elevation ACS
- PAEP
progestagen-associated endometrial protein
- PGD2
prostaglandin D2
- RASMCs
rat aortic smooth muscle cells
- RBP4
retinol binding protein 4
- ROS
reactive oxygen species
- SCRs
structurally conserved regions
- SNPs
single nucleotide polymorphisms
- STEMI
ST segment elevation myocardial infarction
- TC
total cholesterol
- TINAGL1
tubulointerstitial nephritis antigen-like 1
- TLR4
toll-like receptor 4
- VLDLs
very low-density lipoproteins
- VSMC
vascular smooth muscle cell
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest
All authors including H.-H. Yang, X. Wang, S. Li, Y. Liu, R. Akbar, and G.-C. Fan confirm that this article content has no conflict of interest.
References
- Achatz S, Jarasch A, & Skerra A (2022). Structural plasticity in the loop region of engineered lipocalins with novel ligand specificities, so-called Anticalins. J Struct Biol X, 6, 100054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahnstrom J, Axler O, Jauhiainen M, Salomaa V, Havulinna AS, Ehnholm C, Frikke-Schmidt R, Tybjaerg-Hansen A, & Dahlback B (2008). Levels of apolipoprotein M are not associated with the risk of coronary heart disease in two independent case-control studies. J Lipid Res, 49, 1912–1917. [DOI] [PubMed] [Google Scholar]
- Akerstrom B, Flower DR, & Salier JP (2000). Lipocalins: unity in diversity. Biochim Biophys Acta, 1482, 1–8. [DOI] [PubMed] [Google Scholar]
- Amersfoort J, Schaftenaar FH, Douna H, van Santbrink PJ, Kroner MJ, van Puijvelde GHM, Quax PHA, Kuiper J, & Bot I (2018). Lipocalin-2 contributes to experimental atherosclerosis in a stage-dependent manner. Atherosclerosis, 275, 214–224. [DOI] [PubMed] [Google Scholar]
- Annema W, Gawinecka J, Muendlein A, Saely CH, Drexel H, & von Eckardstein A (2022). Elevated levels of apolipoprotein D predict poor outcome in patients with suspected or established coronary artery disease. Atherosclerosis, 341, 27–33. [DOI] [PubMed] [Google Scholar]
- Asimakopoulou A, Fulop A, Borkham-Kamphorst E, de Leur EV, Gassler N, Berger T, Beine B, Meyer HE, Mak TW, Hopf C, Henkel C, & Weiskirchen R (2017). Altered mitochondrial and peroxisomal integrity in lipocalin-2-deficient mice with hepatic steatosis. Biochim Biophys Acta Mol Basis Dis, 1863, 2093–2110. [DOI] [PubMed] [Google Scholar]
- Bacci MR, Cavallari MR, de Rozier-Alves RM, Alves Bda C, & Fonseca FL (2015). The impact of lipocalin-type-prostaglandin-D-synthase as a predictor of kidney disease in patients with type 2 diabetes. Drug Des Devel Ther, 9, 3179–3182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagshaw SM, Bennett M, Haase M, Haase-Fielitz A, Egi M, Morimatsu H, D’Amico G, Goldsmith D, Devarajan P, & Bellomo R (2010). Plasma and urine neutrophil gelatinase-associated lipocalin in septic versus non-septic acute kidney injury in critical illness. Intensive Care Med, 36, 452–461. [DOI] [PubMed] [Google Scholar]
- Barbarash OL, Bykova IS, Kashtalap VV, Zykov MV, Hryachkova ON, Kalaeva VV, Shafranskaya KS, Karetnikova VN, & Kutikhin AG (2017). Serum neutrophil gelatinase-associated lipocalin has an advantage over serum cystatin C and glomerular filtration rate in prediction of adverse cardiovascular outcome in patients with ST-segment elevation myocardial infarction. BMC Cardiovasc Disord, 17, 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beesley SJ, Weber G, Sarge T, Nikravan S, Grissom CK, Lanspa MJ, Shahul S, & Brown SM (2018). Septic Cardiomyopathy. Crit Care Med, 46, 625–634. [DOI] [PubMed] [Google Scholar]
- Bektas M, Allende ML, Lee BG, Chen W, Amar MJ, Remaley AT, Saba JD, & Proia RL (2010). Sphingosine 1-phosphate lyase deficiency disrupts lipid homeostasis in liver. J Biol Chem, 285, 10880–10889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellmann-Weiler R, Schroll A, Engl S, Nairz M, Talasz H, Seifert M, & Weiss G (2013). Neutrophil gelatinase-associated lipocalin and interleukin-10 regulate intramacrophage Chlamydia pneumoniae replication by modulating intracellular iron homeostasis. Immunobiology, 218, 969–978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergwik J, Kristiansson A, Allhorn M, Gram M, & Akerstrom B (2021). Structure, Functions, and Physiological Roles of the Lipocalin alpha(1)-Microglobulin (A1M). Front Physiol, 12, 645650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco-Vaca F, Via DP, Yang CY, Massey JB, & Pownall HJ (1992). Characterization of disulfide-linked heterodimers containing apolipoprotein D in human plasma lipoproteins. J Lipid Res, 33, 1785–1796. [PubMed] [Google Scholar]
- Brown LJ, Alawoki M, Crawford ME, Reida T, Sears A, Torma T, & Albig AR (2010). Lipocalin-7 is a matricellular regulator of angiogenesis. PLoS One, 5, e13905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buhler L, Maida A, Vogl ES, Georgiadi A, Takacs A, Kluth O, Schurmann A, Feuchtinger A, von Toerne C, Tsokanos FF, Klepac K, Wolff G, Sakurai M, Ekim Ustunel B, Nawroth P, & Herzig S (2021). Lipocalin 13 enhances insulin secretion but is dispensable for systemic metabolic control. Life Sci Alliance, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calo LA, Maiolino G, Pagnin E, Vertolli U, & Davis PA (2014). Increased RBP4 in a human model of activated anti-atherosclerotic and antiremodelling defences. Eur J Clin Invest, 44, 567–572. [DOI] [PubMed] [Google Scholar]
- Carbone F, Liberale L, Preda A, Schindler TH, & Montecucco F (2022). Septic Cardiomyopathy: From Pathophysiology to the Clinical Setting. Cells, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan YK, Sung HK, & Sweeney G (2015). Iron metabolism and regulation by neutrophil gelatinase-associated lipocalin in cardiomyopathy. Clin Sci (Lond), 129, 851–862. [DOI] [PubMed] [Google Scholar]
- Charkoftaki G, Wang Y, McAndrews M, Bruford EA, Thompson DC, Vasiliou V, & Nebert DW (2019). Update on the human and mouse lipocalin (LCN) gene family, including evidence the mouse Mup cluster is result of an “evolutionary bloom”. Hum Genomics, 13, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng JM, Akkerhuis KM, Meilhac O, Oemrawsingh RM, Garcia-Garcia HM, van Geuns RJ, Piquer D, Merle D, du Paty E, Galea P, Jaisser F, Rossignol P, Serruys PW, Boersma E, Fareh J, & Kardys I (2014). Circulating osteoglycin and NGAL/MMP9 complex concentrations predict 1-year major adverse cardiovascular events after coronary angiography. Arterioscler Thromb Vasc Biol, 34, 1078–1084. [DOI] [PubMed] [Google Scholar]
- Cho KW, Zhou Y, Sheng L, & Rui L (2011). Lipocalin-13 regulates glucose metabolism by both insulin-dependent and insulin-independent mechanisms. Mol Cell Biol, 31, 450–457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christoffersen C (2021). Apolipoprotein M-A Marker or an Active Player in Type II Diabetes? Front Endocrinol (Lausanne), 12, 665393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christoffersen C, Jauhiainen M, Moser M, Porse B, Ehnholm C, Boesl M, Dahlback B, & Nielsen LB (2008). Effect of apolipoprotein M on high density lipoprotein metabolism and atherosclerosis in low density lipoprotein receptor knock-out mice. J Biol Chem, 283, 1839–1847. [DOI] [PubMed] [Google Scholar]
- Christoffersen C, Nielsen LB, Axler O, Andersson A, Johnsen AH, & Dahlback B (2006). Isolation and characterization of human apolipoprotein M-containing lipoproteins. J Lipid Res, 47, 1833–1843. [DOI] [PubMed] [Google Scholar]
- Christoffersen C, Obinata H, Kumaraswamy SB, Galvani S, Ahnstrom J, Sevvana M, Egerer-Sieber C, Muller YA, Hla T, Nielsen LB, & Dahlback B (2011). Endothelium-protective sphingosine-1-phosphate provided by HDL-associated apolipoprotein M. Proc Natl Acad Sci U S A, 108, 9613–9618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christou GA, Tselepis AD, & Kiortsis DN (2012). The metabolic role of retinol binding protein 4: an update. Horm Metab Res, 44, 6–14. [DOI] [PubMed] [Google Scholar]
- Clausen J (1961). Proteins in normal cerebrospinal fluid not found in serum. Proc Soc Exp Biol Med, 107, 170–172. [DOI] [PubMed] [Google Scholar]
- Cruz DN, Gaiao S, Maisel A, Ronco C, & Devarajan P (2012). Neutrophil gelatinase-associated lipocalin as a biomarker of cardiovascular disease: a systematic review. Clin Chem Lab Med, 50, 1533–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cubedo J, Padro T, Cinca J, Mata P, Alonso R, & Badimon L (2014). Retinol-binding protein 4 levels and susceptibility to ischaemic events in men. Eur J Clin Invest, 44, 266–275. [DOI] [PubMed] [Google Scholar]
- Del Gaudio I, Rubinelli L, Sasset L, Wadsack C, Hla T, & Di Lorenzo A (2021). Endothelial Spns2 and ApoM Regulation of Vascular Tone and Hypertension Via Sphingosine-1-Phosphate. J Am Heart Assoc, 10, e021261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deuschle FC, Ilyukhina E, & Skerra A (2021). Anticalin(R) proteins: from bench to bedside. Expert Opin Biol Ther, 21, 509–518. [DOI] [PubMed] [Google Scholar]
- di Salvo TG, Yang KC, Brittain E, Absi T, Maltais S, & Hemnes A (2015). Right ventricular myocardial biomarkers in human heart failure. J Card Fail, 21, 398–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong H, Li X, & Tang Y (2015). Serum Retinol-Binding Protein-4 Level is a High Risk Factor for Coronary Heart Disease in Chinese. Clin Lab, 61, 1675–1678. [DOI] [PubMed] [Google Scholar]
- Du ZP, Wu BL, Wu X, Lin XH, Qiu XY, Zhan XF, Wang SH, Shen JH, Zheng CP, Wu ZY, Xu LY, Wang D, & Li EM (2015). A systematic analysis of human lipocalin family and its expression in esophageal carcinoma. Sci Rep, 5, 12010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eguchi Y, Eguchi N, Oda H, Seiki K, Kijima Y, Matsu-ura Y, Urade Y, & Hayaishi O (1997). Expression of lipocalin-type prostaglandin D synthase (beta-trace) in human heart and its accumulation in the coronary circulation of angina patients. Proc Natl Acad Sci U S A, 94, 14689–14694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elsoe S, Ahnstrom J, Christoffersen C, Hoofnagle AN, Plomgaard P, Heinecke JW, Binder CJ, Bjorkbacka H, Dahlback B, & Nielsen LB (2012). Apolipoprotein M binds oxidized phospholipids and increases the antioxidant effect of HDL. Atherosclerosis, 221, 91–97. [DOI] [PubMed] [Google Scholar]
- Fan Y, Chen J, Liu D, Li W, Wang H, Huang Y, & Gao C (2020). HDL-S1P protects endothelial function and reduces lung injury during sepsis in vivo and in vitro. Int J Biochem Cell Biol, 126, 105819. [DOI] [PubMed] [Google Scholar]
- Ferrara N, & Kerbel RS (2005). Angiogenesis as a therapeutic target. Nature, 438, 967–974. [DOI] [PubMed] [Google Scholar]
- Feuerborn R, Becker S, Poti F, Nagel P, Brodde M, Schmidt H, Christoffersen C, Ceglarek U, Burkhardt R, & Nofer JR (2017). High density lipoprotein (HDL)-associated sphingosine 1-phosphate (S1P) inhibits macrophage apoptosis by stimulating STAT3 activity and survivin expression. Atherosclerosis, 257, 29–37. [DOI] [PubMed] [Google Scholar]
- Flo TH, Smith KD, Sato S, Rodriguez DJ, Holmes MA, Strong RK, Akira S, & Aderem A (2004). Lipocalin 2 mediates an innate immune response to bacterial infection by sequestrating iron. Nature, 432, 917–921. [DOI] [PubMed] [Google Scholar]
- Flower DR (1995). Multiple molecular recognition properties of the lipocalin protein family. J Mol Recognit, 8, 185–195. [DOI] [PubMed] [Google Scholar]
- Flower DR (1996). The lipocalin protein family: structure and function. Biochem J, 318 (Pt 1), 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flower DR, North AC, & Attwood TK (1993). Structure and sequence relationships in the lipocalins and related proteins. Protein Sci, 2, 753–761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flower DR, North AC, & Sansom CE (2000). The lipocalin protein family: structural and sequence overview. Biochim Biophys Acta, 1482, 9–24. [DOI] [PubMed] [Google Scholar]
- Frak W, Wojtasinska A, Lisinska W, Mlynarska E, Franczyk B, & Rysz J (2022). Pathophysiology of Cardiovascular Diseases: New Insights into Molecular Mechanisms of Atherosclerosis, Arterial Hypertension, and Coronary Artery Disease. Biomedicines, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frances L, Tavernier G, & Viguerie N (2021). Adipose-Derived Lipid-Binding Proteins: The Good, the Bad and the Metabolic Diseases. Int J Mol Sci, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frej C, Linder A, Happonen KE, Taylor FB, Lupu F, & Dahlback B (2016). Sphingosine 1-phosphate and its carrier apolipoprotein M in human sepsis and in Escherichia coli sepsis in baboons. J Cell Mol Med, 20, 1170–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frieler RA, & Mortensen RM (2015). Immune cell and other noncardiomyocyte regulation of cardiac hypertrophy and remodeling. Circulation, 131, 1019–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganfornina MD, Akerstrom B, & Sanchez D (2022). Editorial: Functional Profile of the Lipocalin Protein Family. Front Physiol, 13, 904702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao W, Wang H, Zhang L, Cao Y, Bao JZ, Liu ZX, Wang LS, Yang Q, & Lu X (2016). Retinol-Binding Protein 4 Induces Cardiomyocyte Hypertrophy by Activating TLR4/MyD88 Pathway. Endocrinology, 157, 2282–2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glasgow BJ, Heinzmann C, Kojis T, Sparkes RS, Mohandas T, & Bateman JB (1993). Assignment of tear lipocalin gene to human chromosome 9q34–9qter. Curr Eye Res, 12, 1019–1023. [DOI] [PubMed] [Google Scholar]
- Grzyb J, Latowski D, & Strzalka K (2006). Lipocalins - a family portrait. J Plant Physiol, 163, 895–915. [DOI] [PubMed] [Google Scholar]
- Guardado S, Ojeda-Juarez D, Kaul M, & Nordgren TM (2021). Comprehensive review of lipocalin 2-mediated effects in lung inflammation. Am J Physiol Lung Cell Mol Physiol, 321, L726–L733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo C, Lian Y, Liu Q, Liu J, Zhang Y, & Lin D (2010). Soluble expression and characterization of a mouse epididymis-specific protein lipocalin6. Protein Expr Purif, 69, 64–67. [DOI] [PubMed] [Google Scholar]
- Guo H, Zhao XX, Zhang XJ, Chen W, & Zhang J (2015). Functional study of −724I/D polymorphism in apolipoprotein M (apoM) gene promoter region and its association with myocardial infarction. Med Sci Monit, 21, 371–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gustafsson I, Brendorp B, Seibaek M, Burchardt H, Hildebrandt P, Kober L, Torp-Pedersen C, Danish Investigatord of A, & Mortality on Dofetilde Study, G. (2004). Influence of diabetes and diabetes-gender interaction on the risk of death in patients hospitalized with congestive heart failure. J Am Coll Cardiol, 43, 771–777. [DOI] [PubMed] [Google Scholar]
- Hajny S, Borup A, Elsoe S, & Christoffersen C (2021). Increased plasma apoM levels impair triglyceride turnover in mice. Biochim Biophys Acta Mol Cell Biol Lipids, 1866, 158969. [DOI] [PubMed] [Google Scholar]
- Hamil KG, Liu Q, Sivashanmugam P, Anbalagan M, Yenugu S, Soundararajan R, Grossman G, Rao AJ, Birse CE, Ruben SM, Richardson RT, Zhang YL, O’Rand MG, Petrusz P, French FS, & Hall SH (2003). LCN6, a novel human epididymal lipocalin. Reprod Biol Endocrinol, 1, 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han F, Takeda K, Ishikawa K, Ono M, Date F, Yokoyama S, Furuyama K, Shinozawa Y, Urade Y, & Shibahara S (2009). Induction of lipocalin-type prostaglandin D synthase in mouse heart under hypoxemia. Biochem Biophys Res Commun, 385, 449–453. [DOI] [PubMed] [Google Scholar]
- Helanova K, Littnerova S, Kubena P, Ganovska E, Pavlusova M, Kubkova L, Jarkovsky J, Pavkova Goldbergova M, Lipkova J, Gottwaldova J, Kala P, Toman O, Dastych M, Spinar J, & Parenica J (2015). Prognostic impact of neutrophil gelatinase-associated lipocalin and B-type natriuretic in patients with ST-elevation myocardial infarction treated by primary PCI: a prospective observational cohort study. BMJ Open, 5, e006872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helanova K, Spinar J, & Parenica J (2014). Diagnostic and prognostic utility of neutrophil gelatinase-associated lipocalin (NGAL) in patients with cardiovascular diseases--review. Kidney Blood Press Res, 39, 623–629. [DOI] [PubMed] [Google Scholar]
- Hemdahl AL, Gabrielsen A, Zhu C, Eriksson P, Hedin U, Kastrup J, Thoren P, & Hansson GK (2006). Expression of neutrophil gelatinase-associated lipocalin in atherosclerosis and myocardial infarction. Arterioscler Thromb Vasc Biol, 26, 136–142. [DOI] [PubMed] [Google Scholar]
- Hetherington I, & Totary-Jain H (2022). Anti-atherosclerotic therapies: Milestones, challenges, and emerging innovations. Mol Ther, 30, 3106–3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann A, Conradt HS, Gross G, Nimtz M, Lottspeich F, & Wurster U (1993). Purification and chemical characterization of beta-trace protein from human cerebrospinal fluid: its identification as prostaglandin D synthase. J Neurochem, 61, 451–456. [DOI] [PubMed] [Google Scholar]
- Hollenberg SM, & Singer M (2021). Pathophysiology of sepsis-induced cardiomyopathy. Nat Rev Cardiol, 18, 424–434. [DOI] [PubMed] [Google Scholar]
- Holmes MA, Paulsene W, Jide X, Ratledge C, & Strong RK (2005). Siderocalin (Lcn 2) also binds carboxymycobactins, potentially defending against mycobacterial infections through iron sequestration. Structure, 13, 29–41. [DOI] [PubMed] [Google Scholar]
- Holzfeind P, Merschak P, Wojnar P, & Redl B (1997). Structure and organization of the porcine LCN1 gene encoding Tear lipocalin/von Ebner’s gland protein. Gene, 202, 61–67. [DOI] [PubMed] [Google Scholar]
- Hosbond SE, Diederichsen AC, Pedersen L, Rasmussen LM, Munkholm H, Gerke O, Poulsen TS, & Mickley H (2014). Lipocalin-type prostaglandin D synthase is not a biomarker of atherosclerotic manifestations. Scand J Clin Lab Invest, 74, 219–227. [DOI] [PubMed] [Google Scholar]
- Hraba-Renevey S, Turler H, Kress M, Salomon C, & Weil R (1989). SV40-induced expression of mouse gene 24p3 involves a post-transcriptional mechanism. Oncogene, 4, 601–608. [PubMed] [Google Scholar]
- Huang G, Wang D, Khan UI, Zeb I, Manson JE, Miller V, Hodis HN, Budoff MJ, Merriam GR, Harman MS, Brinton EA, Cedars MI, Su Y, Lobo RA, Naftolin F, Santoro N, Taylor HS, & Wildman RP (2012). Associations between retinol-binding protein 4 and cardiometabolic risk factors and subclinical atherosclerosis in recently postmenopausal women: cross-sectional analyses from the KEEPS study. Cardiovasc Diabetol, 11, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang XS, Zhao SP, Hu M, & Luo YP (2007). Apolipoprotein M likely extends its anti-atherogenesis via anti-inflammation. Med Hypotheses, 69, 136–140. [DOI] [PubMed] [Google Scholar]
- Huang Y, Zhang N, Xie C, You Y, Guo L, Ye F, Xie X, & Wang J (2022). Lipocalin-2 in neutrophils induces ferroptosis in septic cardiac dysfunction via increasing labile iron pool of cardiomyocytes. Front Cardiovasc Med, 9, 922534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inoue T, Eguchi Y, Matsumoto T, Kijima Y, Kato Y, Ozaki Y, Waseda K, Oda H, Seiki K, Node K, & Urade Y (2008). Lipocalin-type prostaglandin D synthase is a powerful biomarker for severity of stable coronary artery disease. Atherosclerosis, 201, 385–391. [DOI] [PubMed] [Google Scholar]
- Jaberi SA, Cohen A, D’Souza C, Abdulrazzaq YM, Ojha S, Bastaki S, & Adeghate EA (2021). Lipocalin-2: Structure, function, distribution and role in metabolic disorders. Biomed Pharmacother, 142, 112002. [DOI] [PubMed] [Google Scholar]
- Jia G, Hill MA, & Sowers JR (2018). Diabetic Cardiomyopathy: An Update of Mechanisms Contributing to This Clinical Entity. Circ Res, 122, 624–638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiao GQ, Yuan ZX, Xue YS, Yang CJ, Lu CB, Lu ZQ, & Xiao MD (2007). A prospective evaluation of apolipoprotein M gene T-778C polymorphism in relation to coronary artery disease in Han Chinese. Clin Biochem, 40, 1108–1112. [DOI] [PubMed] [Google Scholar]
- Joshi SG, Smith RA, & Stokes DK (1980). A progestagen-dependent endometrial protein in human amniotic fluid. J Reprod Fertil, 60, 317–321. [DOI] [PubMed] [Google Scholar]
- Katsumata Y, Shinmura K, Sugiura Y, Tohyama S, Matsuhashi T, Ito H, Yan X, Ito K, Yuasa S, Ieda M, Urade Y, Suematsu M, Fukuda K, & Sano M (2014). Endogenous prostaglandin D2 and its metabolites protect the heart against ischemia-reperfusion injury by activating Nrf2. Hypertension, 63, 80–87. [DOI] [PubMed] [Google Scholar]
- Kim IY, Kim JH, Kim MJ, Lee DW, Hwang CG, Han M, Rhee H, Song SH, Seong EY, & Lee SB (2018). Plasma neutrophil gelatinase-associated lipocalin is independently associated with left ventricular hypertrophy and diastolic dysfunction in patients with chronic kidney disease. PLoS One, 13, e0205848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirbis S, Gorenjak M, & Sinkovic A (2015). The role of urine neutrophil gelatinase--associated lipocalin (NGAL) in acute heart failure in patients with ST--elevation myocardial infarction. BMC Cardiovasc Disord, 15, 49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kjeldsen L, Johnsen AH, Sengelov H, & Borregaard N (1993). Isolation and primary structure of NGAL, a novel protein associated with human neutrophil gelatinase. J Biol Chem, 268, 10425–10432. [PubMed] [Google Scholar]
- Kraus BJ, Sartoretto JL, Polak P, Hosooka T, Shiroto T, Eskurza I, Lee SA, Jiang H, Michel T, & Kahn BB (2015). Novel role for retinol-binding protein 4 in the regulation of blood pressure. FASEB J, 29, 3133–3140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kristiansson A, Davidsson S, Johansson ME, Piel S, Elmer E, Hansson MJ, Akerstrom B, & Gram M (2020). alpha(1)-Microglobulin (A1M) Protects Human Proximal Tubule Epithelial Cells from Heme-Induced Damage In Vitro. Int J Mol Sci, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krol-Grzymala A, Sienkiewicz-Szlapka E, Fiedorowicz E, Rozmus D, Cieslinska A, & Grzybowski A (2022). Tear Biomarkers in Alzheimer’s and Parkinson’s Diseases, and Multiple Sclerosis: Implications for Diagnosis (Systematic Review). Int J Mol Sci, 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumaraswamy SB, Linder A, Akesson P, & Dahlback B (2012). Decreased plasma concentrations of apolipoprotein M in sepsis and systemic inflammatory response syndromes. Crit Care, 16, R60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurano M, Tsuneyama K, Morimoto Y, Shimizu T, Jona M, Kassai H, Nakao K, Aiba A, & Yatomi Y (2018). Apolipoprotein M Protects Lipopolysaccharide-Treated Mice from Death and Organ Injury. Thromb Haemost, 118, 1021–1035. [DOI] [PubMed] [Google Scholar]
- Kurano M, & Yatomi Y (2018). Sphingosine 1-Phosphate and Atherosclerosis. J Atheroscler Thromb, 25, 16–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurozumi S, Alsaeed S, Orah N, Miligy IM, Joseph C, Aljohani A, Toss MS, Fujii T, Shirabe K, Green AR, Aleskandarany MA, & Rakha EA (2020). Clinicopathological significance of lipocalin 2 nuclear expression in invasive breast cancer. Breast Cancer Res Treat, 179, 557–564. [DOI] [PubMed] [Google Scholar]
- Lahiri A, Alex AG, & George PV (2018). Estimating the prevalence of elevated plasma neutrophil gelatinase associated lipocalin level in patients with acute coronary syndromes and its association with outcomes. Indian Heart J, 70, 220–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambadiari V, Kadoglou NP, Stasinos V, Maratou E, Antoniadis A, Kolokathis F, Parissis J, Hatziagelaki E, Iliodromitis EK, & Dimitriadis G (2014). Serum levels of retinol-binding protein-4 are associated with the presence and severity of coronary artery disease. Cardiovasc Diabetol, 13, 121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lassagne H, Ressot C, Mattei MG, & Gachon AM (1993). Assignment of the human tear lipocalin gene (LCN1) to 9q34 by in situ hybridization. Genomics, 18, 160–161. [DOI] [PubMed] [Google Scholar]
- Leclercq A, Houard X, Philippe M, Ollivier V, Sebbag U, Meilhac O, & Michel JB (2007). Involvement of intraplaque hemorrhage in atherothrombosis evolution via neutrophil protease enrichment. J Leukoc Biol, 82, 1420–1429. [DOI] [PubMed] [Google Scholar]
- Lee JT, Huang Z, Pan K, Zhang HJ, Woo CW, Xu A, & Wong CM (2016). Adipose-derived lipocalin 14 alleviates hyperglycaemia by suppressing both adipocyte glycerol efflux and hepatic gluconeogenesis in mice. Diabetologia, 59, 604–613. [DOI] [PubMed] [Google Scholar]
- Lentini P, de Cal M, Clementi A, D’Angelo A, & Ronco C (2012). Sepsis and AKI in ICU Patients: The Role of Plasma Biomarkers. Crit Care Res Pract, 2012, 856401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li C, Zhang Z, Peng Y, Gao H, Wang Y, Zhao J, & Pan C (2019). Plasma neutrophil gelatinase-associated lipocalin levels are associated with the presence and severity of coronary heart disease. PLoS One, 14, e0220841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Xia K, Li C, & Yang T (2014). Retinol-binding protein 4 as a novel risk factor for cardiovascular disease in patients with coronary artery disease and hyperinsulinemia. Am J Med Sci, 348, 474–479. [DOI] [PubMed] [Google Scholar]
- Li F, Xia K, Sheikh MS, Cheng J, Li C, & Yang T (2014). Retinol binding protein 4 promotes hyperinsulinism-induced proliferation of rat aortic smooth muscle cells. Mol Med Rep, 9, 1634–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Feng D, Cai Y, Liu Y, Xu M, Xiang X, Zhou Z, Xia Q, Kaplan MJ, Kong X, & Gao B (2018). Hepatocytes and neutrophils cooperatively suppress bacterial infection by differentially regulating lipocalin-2 and neutrophil extracellular traps. Hepatology, 68, 1604–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Li Y, Huang W, Wang X, Liu Z, Chen J, Fan Y, Peng T, Sadayappan S, Wang Y, & Fan GC (2022). Loss of Lipocalin 10 Exacerbates Diabetes-Induced Cardiomyopathy via Disruption of Nr4a1-Mediated Anti-Inflammatory Response in Macrophages. Front Immunol, 13, 930397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Zhou J, Qiu J, Huang Z, Wang W, Wu P, & Feng A (2020). Berberine reduces gut-vascular barrier permeability via modulation of ApoM/S1P pathway in a model of polymicrobial sepsis. Life Sci, 261, 118460. [DOI] [PubMed] [Google Scholar]
- Lin YM, Lee MC, Toh HS, Chang WT, Chen SY, Kuo FH, Tang HJ, Hua YM, Wei D, Melgarejo J, Zhang ZY, & Liao CT (2022). Association of sepsis-induced cardiomyopathy and mortality: a systematic review and meta-analysis. Ann Intensive Care, 12, 112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindberg S, Pedersen SH, Mogelvang R, Jensen JS, Flyvbjerg A, Galatius S, & Magnusson NE (2012). Prognostic utility of neutrophil gelatinase-associated lipocalin in predicting mortality and cardiovascular events in patients with ST-segment elevation myocardial infarction treated with primary percutaneous coronary intervention. J Am Coll Cardiol, 60, 339–345. [DOI] [PubMed] [Google Scholar]
- Liu T, Han C, Sun L, Ding Z, Shi F, Wang R, Wang W, Shan W, Zhang Y, Hu N, Liu J, & Bu H (2019). Association between new circulating proinflammatory and anti-inflammatory adipocytokines with coronary artery disease. Coron Artery Dis, 30, 528–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu W, Guo X, Jin L, Hong T, Zhang Q, Su F, Shen Y, Li S, & He B (2022). Lipocalin-2 participates in sepsis-induced myocardial injury by mediating lipid accumulation and mitochondrial dysfunction. Front Cardiovasc Med, 9, 1009726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, & Tie L (2019). Apolipoprotein M and sphingosine-1-phosphate complex alleviates TNF-alpha-induced endothelial cell injury and inflammation through PI3K/AKT signaling pathway. BMC Cardiovasc Disord, 19, 279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Wang D, Chen H, & Xia M (2015). Circulating retinol binding protein 4 is associated with coronary lesion severity of patients with coronary artery disease. Atherosclerosis, 238, 45–51. [DOI] [PubMed] [Google Scholar]
- Liu Y, Zhong Y, Chen H, Wang D, Wang M, Ou JS, & Xia M (2017). Retinol-Binding Protein-Dependent Cholesterol Uptake Regulates Macrophage Foam Cell Formation and Promotes Atherosclerosis. Circulation, 135, 1339–1354. [DOI] [PubMed] [Google Scholar]
- Liu Z, Petersen R, & Devireddy L (2013). Impaired neutrophil function in 24p3 null mice contributes to enhanced susceptibility to bacterial infections. J Immunol, 190, 4692–4706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallat Z, Simon T, Benessiano J, Clement K, Taleb S, Wareham NJ, Luben R, Khaw KT, Tedgui A, & Boekholdt SM (2009). Retinol-binding protein 4 and prediction of incident coronary events in healthy men and women. J Clin Endocrinol Metab, 94, 255–260. [DOI] [PubMed] [Google Scholar]
- Marques FZ, Prestes PR, Byars SG, Ritchie SC, Wurtz P, Patel SK, Booth SA, Rana I, Minoda Y, Berzins SP, Curl CL, Bell JR, Wai B, Srivastava PM, Kangas AJ, Soininen P, Ruohonen S, Kahonen M, Lehtimaki T, Raitoharju E, Havulinna A, Perola M, Raitakari O, Salomaa V, Ala-Korpela M, Kettunen J, McGlynn M, Kelly J, Wlodek ME, Lewandowski PA, Delbridge LM, Burrell LM, Inouye M, Harrap SB, & Charchar FJ (2017). Experimental and Human Evidence for Lipocalin-2 (Neutrophil Gelatinase-Associated Lipocalin [NGAL]) in the Development of Cardiac Hypertrophy and heart failure. J Am Heart Assoc, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martensson J, Bell M, Oldner A, Xu S, Venge P, & Martling CR (2010). Neutrophil gelatinase-associated lipocalin in adult septic patients with and without acute kidney injury. Intensive Care Med, 36, 1333–1340. [DOI] [PubMed] [Google Scholar]
- Mohri I, Taniike M, Okazaki I, Kagitani-Shimono K, Aritake K, Kanekiyo T, Yagi T, Takikita S, Kim HS, Urade Y, & Suzuki K (2006). Lipocalin-type prostaglandin D synthase is upregulated in oligodendrocytes in lysosomal storage diseases and binds gangliosides. J Neurochem, 97, 641–651. [DOI] [PubMed] [Google Scholar]
- Morel S, Christoffersen C, Axelsen LN, Montecucco F, Rochemont V, Frias MA, Mach F, James RW, Naus CC, Chanson M, Lampe PD, Nielsen MS, Nielsen LB, & Kwak BR (2016). Sphingosine-1-phosphate reduces ischaemia-reperfusion injury by phosphorylating the gap junction protein Connexin43. Cardiovasc Res, 109, 385–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagata N, Fujimori K, Okazaki I, Oda H, Eguchi N, Uehara Y, & Urade Y (2009). De novo synthesis, uptake and proteolytic processing of lipocalin-type prostaglandin D synthase, beta-trace, in the kidneys. FEBS J, 276, 7146–7158. [DOI] [PubMed] [Google Scholar]
- Nairz M, Schroll A, Haschka D, Dichtl S, Sonnweber T, Theurl I, Theurl M, Lindner E, Demetz E, Asshoff M, Bellmann-Weiler R, Muller R, Gerner RR, Moschen AR, Baumgartner N, Moser PL, Talasz H, Tilg H, Fang FC, & Weiss G (2015). Lipocalin-2 ensures host defense against Salmonella Typhimurium by controlling macrophage iron homeostasis and immune response. Eur J Immunol, 45, 3073–3086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nairz M, Theurl I, Schroll A, Theurl M, Fritsche G, Lindner E, Seifert M, Crouch ML, Hantke K, Akira S, Fang FC, & Weiss G (2009). Absence of functional Hfe protects mice from invasive Salmonella enterica serovar Typhimurium infection via induction of lipocalin-2. Blood, 114, 3642–3651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nar G, Sanlialp SC, & Nar R (2021). Retinol binding protein 4 levels relate to the presence and severity of coronary artery disease. J Med Biochem, 40, 384–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni J, Ma X, Zhou M, Pan X, Tang J, Hao Y, Lu Z, Gao M, Bao Y, & Jia W (2013). Serum lipocalin-2 levels positively correlate with coronary artery disease and metabolic syndrome. Cardiovasc Diabetol, 12, 176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nono Nankam PA, & Bluher M (2021). Retinol-binding protein 4 in obesity and metabolic dysfunctions. Mol Cell Endocrinol, 531, 111312. [DOI] [PubMed] [Google Scholar]
- Olsson MG, Rosenlof LW, Kotarsky H, Olofsson T, Leanderson T, Morgelin M, Fellman V, & Akerstrom B (2013). The radical-binding lipocalin A1M binds to a Complex I subunit and protects mitochondrial structure and function. Antioxid Redox Signal, 18, 2017–2028. [DOI] [PubMed] [Google Scholar]
- Otsuki M, Gao H, Dahlman-Wright K, Ohlsson C, Eguchi N, Urade Y, & Gustafsson JA (2003). Specific regulation of lipocalin-type prostaglandin D synthase in mouse heart by estrogen receptor beta. Mol Endocrinol, 17, 1844–1855. [DOI] [PubMed] [Google Scholar]
- Pan Y, Wang L, Xie Y, Tan Y, Chang C, Qiu X, & Li X (2020). Characterization of differentially expressed plasma proteins in patients with acute myocardial infarction. J Proteomics, 227, 103923. [DOI] [PubMed] [Google Scholar]
- Perumalsamy S, Ahmad WAW, & Huri HZ (2021). Retinol-Binding Protein-4-A Predictor of Insulin Resistance and the Severity of Coronary Artery Disease in Type 2 Diabetes Patients with Coronary Artery Disease. Biology (Basel), 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pervaiz S, & Brew K (1987). Homology and structure-function correlations between alpha 1-acid glycoprotein and serum retinol-binding protein and its relatives. FASEB J, 1, 209–214. [DOI] [PubMed] [Google Scholar]
- Qian K, Yan X, Xu C, Fang Y, & Ma M (2022). Association Between Circulating Retinol-Binding Protein 4 and Adverse Cardiovascular Events in Stable Coronary Artery Disease. Front Cardiovasc Med, 9, 829347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quadro L, Blaner WS, Salchow DJ, Vogel S, Piantedosi R, Gouras P, Freeman S, Cosma MP, Colantuoni V, & Gottesman ME (1999). Impaired retinal function and vitamin A availability in mice lacking retinol-binding protein. EMBO J, 18, 4633–4644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rassart E, Desmarais F, Najyb O, Bergeron KF, & Mounier C (2020). Apolipoprotein D. Gene, 756, 144874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Redl B, & Habeler M (2022). The diversity of lipocalin receptors. Biochimie, 192, 22–29. [DOI] [PubMed] [Google Scholar]
- Rothe C, & Skerra A (2018). Anticalin((R)) Proteins as Therapeutic Agents in Human Diseases. BioDrugs, 32, 233–243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudd KE, Johnson SC, Agesa KM, Shackelford KA, Tsoi D, Kievlan DR, Colombara DV, Ikuta KS, Kissoon N, Finfer S, Fleischmann-Struzek C, Machado FR, Reinhart KK, Rowan K, Seymour CW, Watson RS, West TE, Marinho F, Hay SI, Lozano R, Lopez AD, Angus DC, Murray CJL, & Naghavi M (2020). Global, regional, and national sepsis incidence and mortality, 1990–2017: analysis for the Global Burden of Disease Study. Lancet, 395, 200–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz M, Frej C, Holmer A, Guo LJ, Tran S, & Dahlback B (2017). High-Density Lipoprotein-Associated Apolipoprotein M Limits Endothelial Inflammation by Delivering Sphingosine-1-Phosphate to the Sphingosine-1-Phosphate Receptor 1. Arterioscler Thromb Vasc Biol, 37, 118–129. [DOI] [PubMed] [Google Scholar]
- Ruiz M, Okada H, & Dahlback B (2017). HDL-associated ApoM is anti-apoptotic by delivering sphingosine 1-phosphate to S1P1 & S1P3 receptors on vascular endothelium. Lipids Health Dis, 16, 36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sahinarslan A, Kocaman SA, Bas D, Akyel A, Ercin U, Zengin O, & Timurkaynak T (2011). Plasma neutrophil gelatinase-associated lipocalin levels in acute myocardial infarction and stable coronary artery disease. Coron Artery Dis, 22, 333–338. [DOI] [PubMed] [Google Scholar]
- Sanchez D, & Ganfornina MD (2021). The Lipocalin Apolipoprotein D Functional Portrait: A Systematic Review. Front Physiol, 12, 738991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarjeant JM, Lawrie A, Kinnear C, Yablonsky S, Leung W, Massaeli H, Prichett W, Veinot JP, Rassart E, & Rabinovitch M (2003). Apolipoprotein D inhibits platelet-derived growth factor-BB-induced vascular smooth muscle cell proliferated by preventing translocation of phosphorylated extracellular signal regulated kinase 1/2 to the nucleus. Arterioscler Thromb Vasc Biol, 23, 2172–2177. [DOI] [PubMed] [Google Scholar]
- Sawyer L (2021). beta-Lactoglobulin and Glycodelin: Two Sides of the Same Coin? Front Physiol, 12, 678080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiefner A, Gebauer M, Richter A, & Skerra A (2018). Anticalins Reveal High Plasticity in the Mode of Complex Formation with a Common Tumor Antigen. Structure, 26, 649–656 e643. [DOI] [PubMed] [Google Scholar]
- Schiefner A, & Skerra A (2015). The menagerie of human lipocalins: a natural protein scaffold for molecular recognition of physiological compounds. Acc Chem Res, 48, 976–985. [DOI] [PubMed] [Google Scholar]
- Schlehuber S, & Skerra A (2005). Lipocalins in drug discovery: from natural ligand-binding proteins to “anticalins”. Drug Discov Today, 10, 23–33. [DOI] [PubMed] [Google Scholar]
- Schuett KA, Lehrke M, Marx N, & Burgmaier M (2015). High-Risk Cardiovascular Patients: Clinical Features, Comorbidities, and Interconnecting Mechanisms. Front Immunol, 6, 591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shibata K, Sato K, Shirai R, Seki T, Okano T, Yamashita T, Koide A, Mitsuboshi M, Mori Y, Hirano T, & Watanabe T (2020). Lipocalin-2 exerts pro-atherosclerotic effects as evidenced by in vitro and in vivo experiments. Heart Vessels, 35, 1012–1024. [DOI] [PubMed] [Google Scholar]
- Sivalingam Z, Erik Magnusson N, Grove EL, Hvas AM, Dalby Kristensen S, & Bojet Larsen S (2018). Neutrophil gelatinase-associated lipocalin (NGAL) and cardiovascular events in patients with stable coronary artery disease. Scand J Clin Lab Invest, 78, 470–476. [DOI] [PubMed] [Google Scholar]
- Sivalingam Z, Larsen SB, Grove EL, Hvas AM, Kristensen SD, & Magnusson NE (2017). Neutrophil gelatinase-associated lipocalin as a risk marker in cardiovascular disease. Clin Chem Lab Med, 56, 5–18. [DOI] [PubMed] [Google Scholar]
- Skerra A (2008). Alternative binding proteins: anticalins - harnessing the structural plasticity of the lipocalin ligand pocket to engineer novel binding activities. FEBS J, 275, 2677–2683. [DOI] [PubMed] [Google Scholar]
- Soiland H, Soreide K, Janssen EA, Korner H, Baak JP, & Soreide JA (2007). Emerging concepts of apolipoprotein D with possible implications for breast cancer. Cell Oncol, 29, 195–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solini A, Santini E, Madec S, Rossi C, & Muscelli E (2009). Retinol-binding protein-4 in women with untreated essential hypertension. Am J Hypertens, 22, 1001–1006. [DOI] [PubMed] [Google Scholar]
- Song E, Ramos SV, Huang X, Liu Y, Botta A, Sung HK, Turnbull PC, Wheeler MB, Berger T, Wilson DJ, Perry CGR, Mak TW, & Sweeney G (2018). Holo-lipocalin-2-derived siderophores increase mitochondrial ROS and impair oxidative phosphorylation in rat cardiomyocytes. Proc Natl Acad Sci U S A, 115, 1576–1581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soylu K, Aksan G, Nar G, Ozdemir M, Gulel O, Inci S, Aksakal A, Soylu AI, & Yilmaz O (2015). Serum neutrophil gelatinase-associated lipocalin levels are correlated with the complexity and the severity of atherosclerosis in acute coronary syndrome. Anatol J Cardiol, 15, 450–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sramkova V, Berend S, Siklova M, Caspar-Bauguil S, Carayol J, Bonnel S, Marques M, Decaunes P, Kolditz CI, Dahlman I, Arner P, Stich V, Saris WHM, Astrup A, Valsesia A, Rossmeislova L, Langin D, & Viguerie N (2019). Apolipoprotein M: a novel adipokine decreasing with obesity and upregulated by calorie restriction. Am J Clin Nutr, 109, 1499–1510. [DOI] [PubMed] [Google Scholar]
- Srinivasan G, Aitken JD, Zhang B, Carvalho FA, Chassaing B, Shashidharamurthy R, Borregaard N, Jones DP, Gewirtz AT, & Vijay-Kumar M (2012). Lipocalin 2 deficiency dysregulates iron homeostasis and exacerbates endotoxin-induced sepsis. J Immunol, 189, 1911–1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinhoff JS, Lass A, & Schupp M (2021). Biological Functions of RBP4 and Its Relevance for Human Diseases. Front Physiol, 12, 659977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su W, Jiao G, Yang C, & Ye Y (2009). Evaluation of apolipoprotein M as a biomarker of coronary artery disease. Clin Biochem, 42, 365–370. [DOI] [PubMed] [Google Scholar]
- Sun H, Wang D, Liu D, Guo Z, Shao C, Sun W, & Zeng Y (2019). Differential urinary proteins to diagnose coronary heart disease based on iTRAQ quantitative proteomics. Anal Bioanal Chem, 411, 2273–2282. [DOI] [PubMed] [Google Scholar]
- Sun L, Dong Z, Gu H, Guo Z, & Yu Z (2019). TINAGL1 promotes hepatocellular carcinogenesis through the activation of TGF-beta signaling-medicated VEGF expression. Cancer Manag Res, 11, 767–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Q, Kiernan UA, Shi L, Phillips DA, Kahn BB, Hu FB, Manson JE, Albert CM, & Rexrode KM (2013). Plasma retinol-binding protein 4 (RBP4) levels and risk of coronary heart disease: a prospective analysis among women in the nurses’ health study. Circulation, 127, 1938–1947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung HK, Chan YK, Han M, Jahng JWS, Song E, Danielson E, Berger T, Mak TW, & Sweeney G (2017). Lipocalin-2 (NGAL) Attenuates Autophagy to Exacerbate Cardiac Apoptosis Induced by Myocardial Ischemia. J Cell Physiol, 232, 2125–2134. [DOI] [PubMed] [Google Scholar]
- Suzuki K, Lareyre JJ, Sanchez D, Gutierrez G, Araki Y, Matusik RJ, & Orgebin-Crist MC (2004). Molecular evolution of epididymal lipocalin genes localized on mouse chromosome 2. Gene, 339, 49–59. [DOI] [PubMed] [Google Scholar]
- Suzuki K, Yu X, Chaurand P, Araki Y, Lareyre JJ, Caprioli RM, Matusik RJ, & Orgebin-Crist MC (2006). Epididymis-specific promoter-driven gene targeting: a transcription factor which regulates epididymis-specific gene expression. Mol Cell Endocrinol, 250, 184–189. [DOI] [PubMed] [Google Scholar]
- te Boekhorst BC, Bovens SM, Hellings WE, van der Kraak PH, van de Kolk KW, Vink A, Moll FL, van Oosterhout MF, de Vries JP, Doevendans PA, Goumans MJ, de Kleijn DP, van Echteld CJ, Pasterkamp G, & Sluijter JP (2011). Molecular MRI of murine atherosclerotic plaque targeting NGAL: a protein associated with unstable human plaque characteristics. Cardiovasc Res, 89, 680–688. [DOI] [PubMed] [Google Scholar]
- Tokudome S, Sano M, Shinmura K, Matsuhashi T, Morizane S, Moriyama H, Tamaki K, Hayashida K, Nakanishi H, Yoshikawa N, Shimizu N, Endo J, Katayama T, Murata M, Yuasa S, Kaneda R, Tomita K, Eguchi N, Urade Y, Asano K, Utsunomiya Y, Suzuki T, Taguchi R, Tanaka H, & Fukuda K (2009). Glucocorticoid protects rodent hearts from ischemia/reperfusion injury by activating lipocalin-type prostaglandin D synthase-derived PGD2 biosynthesis. J Clin Invest, 119, 1477–1488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toyonaga T, Matsuura M, Mori K, Honzawa Y, Minami N, Yamada S, Kobayashi T, Hibi T, & Nakase H (2016). Lipocalin 2 prevents intestinal inflammation by enhancing phagocytic bacterial clearance in macrophages. Sci Rep, 6, 35014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsukamoto K, Mani DR, Shi J, Zhang S, Haagensen DE, Otsuka F, Guan J, Smith JD, Weng W, Liao R, Kolodgie FD, Virmani R, & Krieger M (2013). Identification of apolipoprotein D as a cardioprotective gene using a mouse model of lethal atherosclerotic coronary artery disease. Proc Natl Acad Sci U S A, 110, 17023–17028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urade Y (2021). Biochemical and Structural Characteristics, Gene Regulation, Physiological, Pathological and Clinical Features of Lipocalin-Type Prostaglandin D(2) Synthase as a Multifunctional Lipocalin. Front Physiol, 12, 718002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urade Y, Fujimoto N, & Hayaishi O (1985). Purification and characterization of rat brain prostaglandin D synthetase. J Biol Chem, 260, 12410–12415. [PubMed] [Google Scholar]
- Urade Y, & Hayaishi O (2000a). Biochemical, structural, genetic, physiological, and pathophysiological features of lipocalin-type prostaglandin D synthase. Biochim Biophys Acta, 1482, 259–271. [DOI] [PubMed] [Google Scholar]
- Urade Y, & Hayaishi O (2000b). Prostaglandin D synthase: structure and function. Vitam Horm, 58, 89–120. [DOI] [PubMed] [Google Scholar]
- Urzua U, Roby KF, Gangi LM, Cherry JM, Powell JI, & Munroe DJ (2006). Transcriptomic analysis of an in vitro murine model of ovarian carcinoma: functional similarity to the human disease and identification of prospective tumoral markers and targets. J Cell Physiol, 206, 594–602. [DOI] [PubMed] [Google Scholar]
- Virtanen T (2021). Inhalant Mammal-Derived Lipocalin Allergens and the Innate Immunity. Front Allergy, 2, 824736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wan K, Zhao J, Deng Y, Chen X, Zhang Q, Zeng Z, Zhang L, & Chen Y (2014). A genetic polymorphism in RBP4 is associated with coronary artery disease. Int J Mol Sci, 15, 22309–22319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Chen G, Li J, Zeng Y, Wu Y, & Yan X (2017). Neutrophil gelatinase-associated lipocalin predicts myocardial dysfunction and mortality in severe sepsis and septic shock. Int J Cardiol, 227, 589–594. [DOI] [PubMed] [Google Scholar]
- Wang H, Zhou P, Zou D, Liu Y, Lu X, & Liu Z (2018). The role of retinol-binding protein 4 and its relationship with sex hormones in coronary artery disease. Biochem Biophys Res Commun, 506, 204–210. [DOI] [PubMed] [Google Scholar]
- Wang J, Chen H, Liu Y, Zhou W, Sun R, & Xia M (2015). Retinol binding protein 4 induces mitochondrial dysfunction and vascular oxidative damage. Atherosclerosis, 240, 335–344. [DOI] [PubMed] [Google Scholar]
- Wang L, Xie W, Li G, Hu B, Wu W, Zhan L, & Zou H (2021). Lipocalin 10 as a New Prognostic Biomarker in Sepsis-Induced Myocardial Dysfunction and Mortality: A Pilot Study. Mediators Inflamm, 2021, 6616270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Lam KS, Kraegen EW, Sweeney G, Zhang J, Tso AW, Chow WS, Wat NM, Xu JY, Hoo RL, & Xu A (2007). Lipocalin-2 is an inflammatory marker closely associated with obesity, insulin resistance, and hyperglycemia in humans. Clin Chem, 53, 34–41. [DOI] [PubMed] [Google Scholar]
- Warszawska JM, Gawish R, Sharif O, Sigel S, Doninger B, Lakovits K, Mesteri I, Nairz M, Boon L, Spiel A, Fuhrmann V, Strobl B, Muller M, Schenk P, Weiss G, & Knapp S (2013). Lipocalin 2 deactivates macrophages and worsens pneumococcal pneumonia outcomes. J Clin Invest, 123, 3363–3372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe K, Urade Y, Mader M, Murphy C, & Hayaishi O (1994). Identification of beta-trace as prostaglandin D synthase. Biochem Biophys Res Commun, 203, 1110–1116. [DOI] [PubMed] [Google Scholar]
- Weech PK, Provost P, Tremblay NM, Camato RN, Milne RW, Marcel YL, & Rassart E (1991). Apolipoprotein D--an atypical apolipoprotein. Prog Lipid Res, 30, 259–266. [DOI] [PubMed] [Google Scholar]
- Wolfrum C, Poy MN, & Stoffel M (2005). Apolipoprotein M is required for prebeta-HDL formation and cholesterol efflux to HDL and protects against atherosclerosis. Nat Med, 11, 418–422. [DOI] [PubMed] [Google Scholar]
- Wong YK, & Tse HF (2021). Circulating Biomarkers for Cardiovascular Disease Risk Prediction in Patients With Cardiovascular Disease. Front Cardiovasc Med, 8, 713191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wroblewska M (2011). The origin and metabolism of a nascent pre-beta high density lipoprotein involved in cellular cholesterol efflux. Acta Biochim Pol, 58, 275–285. [PubMed] [Google Scholar]
- Wu H, Santoni-Rugiu E, Ralfkiaer E, Porse BT, Moser C, Hoiby N, Borregaard N, & Cowland JB (2010). Lipocalin 2 is protective against E. coli pneumonia. Respir Res, 11, 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu G, Ahn J, Chang S, Eguchi M, Ogier A, Han S, Park Y, Shim C, Jang Y, Yang B, Xu A, Wang Y, & Sweeney G (2012). Lipocalin-2 induces cardiomyocyte apoptosis by increasing intracellular iron accumulation. J Biol Chem, 287, 4808–4817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu N, & Dahlback B (1999). A novel human apolipoprotein (apoM). J Biol Chem, 274, 31286–31290. [DOI] [PubMed] [Google Scholar]
- Xu WW, Zhang Y, Tang YB, Xu YL, Zhu HZ, Ferro A, Ji Y, Chen Q, & Fan LM (2008). A genetic variant of apolipoprotein M increases susceptibility to coronary artery disease in a Chinese population. Clin Exp Pharmacol Physiol, 35, 546–551. [DOI] [PubMed] [Google Scholar]
- Yammine L, Zablocki A, Baron W, Terzi F, & Gallazzini M (2019). Lipocalin-2 Regulates Epidermal Growth Factor Receptor Intracellular Trafficking. Cell Rep, 29, 2067–2077 e2066. [DOI] [PubMed] [Google Scholar]
- Zabetian-Targhi F, Mahmoudi MJ, Rezaei N, & Mahmoudi M (2015). Retinol binding protein 4 in relation to diet, inflammation, immunity, and cardiovascular diseases. Adv Nutr, 6, 748–762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang KZ, Shen XY, Wang M, Wang L, Sun HX, Li XZ, Huang JJ, Li XQ, Wu C, Zhao C, Liu JL, Lu X, & Gao W (2021). Retinol-Binding Protein 4 Promotes Cardiac Injury After Myocardial Infarction Via Inducing Cardiomyocyte Pyroptosis Through an Interaction With NLRP3. J Am Heart Assoc, 10, e022011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang PH, Gao JL, Pu C, Feng G, Wang LZ, Huang LZ, & Zhang Y (2016). A single-nucleotide polymorphism C-724 /del in the proter region of the apolipoprotein M gene is associated with type 2 diabetes mellitus. Lipids Health Dis, 15, 142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, van Rooij JGJ, Wakabayashi Y, Hwang SJ, Yang Y, Ghanbari M, Bos D, Consortium B, Levy D, Johnson AD, van Meurs JBJ, Kavousi M, Zhu J, & O’Donnell CJ (2021). Genome-wide transcriptome study using deep RNA sequencing for myocardial infarction and coronary artery calcification. BMC Med Genomics, 14, 45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Chu G, & Yin RX (2013). Apolipoprotein M T-778C polymorphism is associated with serum lipid levels and the risk of coronary artery disease in the Chinese population: a meta-analysis. Lipids Health Dis, 12, 135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, W. P, Wang X, Du W, Yang H-H, Liu YY, Cui SH, Huang W, Peng T, Chen J, Gao C, Wang Y, Sadayappan S, Ma C, Fan Y, Wang C, Fan GC. (2023). Lipocalin 10 is essential for protection against inflammation-triggered vascular leakage by activating LRP2-Ssh1 signaling pathway. Cardiovasc Res. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S, Wei Y, & Xu D (2015). Neutrophil gelatinase-associated lipocalin attenuates injury in the rat cecal ligation and puncture model of sepsis via apoptosis inhibition. Nephrology (Carlton), 20, 646–653. [DOI] [PubMed] [Google Scholar]
- Zheng L, Luo G, Zhang J, Mu Q, Shi Y, Berggren-Soderlund M, Nilsson-Ehle P, Zhang X, & Xu N (2014). Decreased activities of apolipoprotein m promoter are associated with the susceptibility to coronary artery diseases. Int J Med Sci, 11, 365–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng L, Luo G, Zhang X, Zhang J, Zhu J, Wei J, Mu Q, Chen L, Nilsson-Ehle P, & Xu N (2009). Determination of single-nucleotide polymorphism in the proximal promoter region of apolipoprotein M gene in coronary artery diseases. Int J Gen Med, 2, 177–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Ley SH, & Hu FB (2018). Global aetiology and epidemiology of type 2 diabetes mellitus and its complications. Nat Rev Endocrinol, 14, 88–98. [DOI] [PubMed] [Google Scholar]
- Zhou Y, & Rui L (2013). Lipocalin 13 regulation of glucose and lipid metabolism in obesity. Vitam Horm, 91, 369–383. [DOI] [PMC free article] [PubMed] [Google Scholar]