

Abstract
SHP2 is a phosphatase/adaptor protein that plays an important role in various signaling pathways. Its mutations are associated with cancers and developmental diseases. SHP2 contains a protein tyrosine phosphatase (PTP) and two SH2 domains. Selective inhibition of these domains has been challenging due to the multitude of homologous proteins in the proteome. Here, we developed a monobody, synthetic binding protein, that bound to and inhibited the SHP2 PTP domain. It was selective to SHP2 PTP over close homologs. A crystal structure of the monobody-PTP complex revealed that the monobody bound both highly conserved residues in the active site and less conserved residues in the periphery, rationalizing its high selectivity. Its epitope overlapped with the interface between the PTP and N-terminal SH2 domains that is formed in auto-inhibited SHP2. By using the monobody as a probe for the accessibility of the PTP active site, we developed a simple, nonenzymatic assay for the allosteric regulation of SHP2. The assay showed that, in the absence of an activating phospho-Tyr ligand, wild-type SHP2 and the “PTP-dead” C459E mutant were predominantly in the closed state in which the PTP active site is inaccessible, whereas the E76K and C459S mutants were in the open, active state. It also revealed that previously developed monobodies to the SH2 domains, ligands lacking a phospho-Tyr, weakly favored the open state. These results provide corroboration for a conformational equilibrium underlying allosteric regulation of SHP2, provide powerful tools for characterizing and controlling SHP2 functions, and inform drug discovery against SHP2.
Keywords: synthetic binding protein, drug discovery, allostery, protein engineering
The Src-homology 2 (SH2) domain-containing phosphatase 2 (SHP2) is important for a variety of cellular processes, including cell growth, differentiation, and migration.1,2 In particular, SHP2 is required for full activation and sustained signaling of extra-cellular-regulated kinases (ERKs) via receptor tyrosine kinase pathways.3–6 Furthermore, mutations in SHP2 are strongly associated with childhood hematological malignancies such as juvenile myelomonocytic leukemia, and developmental disorders such as Noonan syndrome7 and LEOPARD (Lentigines, Electrocardiogram abnormalities, Ocular hypertelorism, Pulmonic stenosis, Abnormalities of genitalia, Retardation of growth, and Deafness) syndrome.8,9 Consequently, SHP2 inhibitors are being developed as anti-cancer therapeutics both for monotherapy and combination therapies.10–17 The precise mechanisms of how SHP2 contributes to signaling and human disease are still incompletely understood. To better understand these mechanisms, generating specific inhibitors and activators towards SHP2 would greatly aid investigation of SHP2 function.
SHP2 comprises two SH2 domains, termed N-SH2 and C-SH2, respectively, a protein tyrosine phosphatase (PTP) domain, and a C-terminal disordered region containing multiple phosphorylation sites (Figure 1(a)). Selectively targeting these domains, as well as other modular domain families, with small molecule inhibitors has been challenging due to the presence of highly homologous members in the SH2 and PTP families. The SH2 family contains 121 members,18 and the classical PTP family to which SHP2 belongs contains 41 members.19
Figure 1.
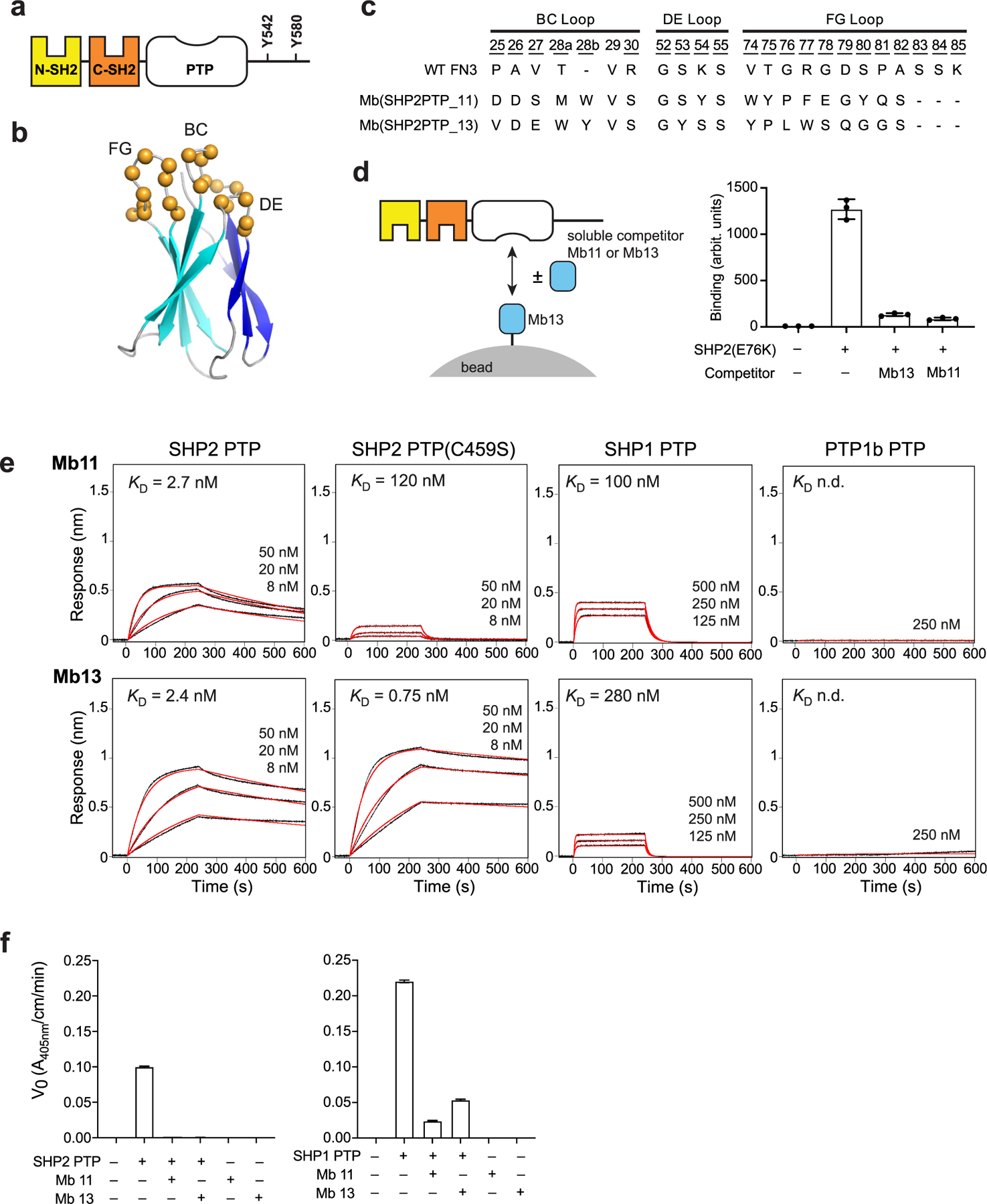
Monobodies binding to SHP2 PTP. (a) Domain organization of SHP2. (b) Schematic of the monobody library design. The b-strands of the FN3 scaffold are labeled A-G, and diversified residues are shown as colored spheres. Figure modified from Figure 1(a) of Hantschel et al. 2020. (c) Amino acid sequences of the diversified loops of the selected monobodies and wild-type FN3. Residue numbers for diversified positions are underlined. (d) Competition between SHP2(E76K) binding of Mb11 and Mb13, as tested using yeast surface display. (e) BLI sensorgrams of the monobodies binding to the wild-type PTP domain of SHP2, SHP1 and PTP1B, as well as SHP2 PTP(C459S). The analyte concentrations are shown over the sensorgrams. The KD values were determined using global fitting of the 1:1 binding model. The red curves show the best fit. (f) Inhibition of the PTP catalytic activity of SHP2 and SHP1 by Mb11 and Mb13 at 500 nM.
The catalytic activity of the SHP2 PTP domain is important for ERK activation. Mutations that impair catalytic activity in the PTP domain of SHP2 block ERK activation in response to insulin,20,21 and are associated with LEOPARD syndrome.22 However, what critical substrates SHP2 needs to dephosphorylate in order to elicit these effects remain a mystery. To our knowledge, no small molecule inhibitors that target exclusively the PTP domain discriminate SHP2 and its close homolog, SHP1, by more than 10-fold.23–26 Developing inhibitors that selectively target the PTP domain will help identify critical substrates of SHP2 and delineate the role of the PTP domain for SHP2 signaling versus its SH2 domains.
Because of extreme challenges in developing small molecule inhibitors against members of modular domain families, we and others have utilized synthetic binding proteins as “tool biologics”.27,28 We previously developed monobodies, synthetic binding proteins based on a human fibronectin type III (FN3) domain,27,29,30 that selectively recognized either the N-SH2 or C-SH2 domain of SHP2 in the context of the human proteome.31 Unlike conventional antibodies, which rely on disulfide formation for proper folding and function, the monobody scaffold contains no cysteines, making monobodies ideally suited as genetically encoded intracellular reagents. In addition to SHP2 SH2 domains, we have successfully developed monobodies that are exquisitely selective, including those selective to oncogenic RAS mutants.32 The need for a specific inhibitor for the PTP domain of SHP2 motivated us to generate highly specific monobodies directed towards the SHP2 PTP domain.
Structural and biochemical studies have defined the framework of the allosteric regulation of SHP2 functions in which the PTP domain is auto-inhibited by the N-SH2 domain in the basal state.33,34 Binding of phosphotyrosine (pY)-containing peptides to the N-SH2 domain relieves this auto-inhibitory mechanism and shifts the enzyme to the open, active conformation.35 Many disease-associated mutations of SHP2 are known or expected to alter the open-closed equilibrium. The most successful inhibitors of SHP2 have exploited this regulatory mechanism. They bind to the interface between the SH2 domains and PTP, which is not highly conserved among SHP2 homologs, and stabilize the closed conformation.10 Although the open-closed equilibrium of SHP2 can be monitored by measuring its PTP activity or directly monitoring the conformation by NMR spectroscopy, a simpler assay can accelerate mechanistic studies and drug discovery.
Here we describe new monobodies highly specific to SHP2 versus the closely related PTP domains of SHP1 and PTP1B. A crystal structure of a monobody–PTP complex defines the mechanism of inhibition and provides rationale for its high selectivity. We developed a simple, nonenzymatic assay using these monobodies to monitor the conformational equilibrium of SHP2, and characterized effects of mutations and ligands.
Results
Potent and selective monobody inhibitors of the PTP domain of SHP2
Using established methods that combine phage display and yeast surface display, we developed two monobodies that bound to the SHP2 PTP domain (Figure 1(b) and (c)). In order to enrich highly specific monobodies, we combined positive and negative library sorting in the yeast display phase. In the first and third rounds of library sorting, we enriched clones that bound to the SHP2 PTP target, whereas in the second round we enriched clones that marginally bound to the homologous PTP domains of SHP1 and PTP1B. Although we used two monobody libraries of distinct designs, one in which amino acid diversities were localized in three loops and the other in which a beta-sheet surface and two loops were diversified,36 both monobody clones originated from the “loop” library. Mb(SHP2PTP_11) and Mb(SHP2PTP_13) have distinct sequences in the diversified loops. Hereafter, we will refer to these monobodies as Mb11 and Mb13 for brevity. Mb11 and Mb13 competed against each other for binding SHP2(E76K), indicating that they bind to overlapping epitopes in SHP2 PTP (Figure 1(d)).
Binding measurements using biolayer interferometry (BLI) showed that Mb11 and Mb13 bound to SHP2 PTP with KD values of 2.7 and 2.4 nM, respectively (Figure 1(e)). They also respectively bound to the PTP domains of SHP1 40 and 120-fold more weakly, and exhibited no detectable binding to PTP1B PTP at 250 nM (Figure 1(e)). These differences in KD are mainly manifested in differences in the dissociation rate constant, kd (Supplementary Table 1), consistent with common observations that kd dictates affinity in protein–protein interactions, particularly among closely related proteins.37,38 Together, these monobodies have high affinity and high specificity toward the SHP2 PTP domain.
Mb11 exhibited much weaker binding to PTP (C459S), a mutation of an active-site residue, whereas Mb13 bound more tightly. These results suggest that, although these two monobodies bound to an overlapping epitope in SHP2-PTP, they interact with different parts of the PTP active site.
We next tested whether these monobodies inhibited the activity of the PTP domain, using purified monobody protein samples and paranitrophenyl phosphate (pNPP) as a substrate. At SHP2 PTP and monobody concentrations of 0.5 and 1.0 µM, conditions in which we can achieve sufficient assay sensitivity and near saturation of PTP with a monobody estimated from their KD values, both monobodies completely inhibited the PTP activity (Figure 1(f)). The monobodies showed partial inhibition of SHP1 PTP, consistent with their lower affinity to the PTP domain of SHP1. In addition, the small molecule inhibitor of SHP2, cryptotanshinone, that binds to the active site pocket of the PTP domain25 inhibited binding of Mb13 to SHP2 (Supplementary Figure 1), further supporting that Mb13 binds the PTP catalytic site. These results indicate Mb11 and Mb13 to be potent and specific inhibitors of SHP2 PTP.
Crystal structure of the SHP2 PTP–Mb13 complex
To elucidate the mechanism of inhibition and the structural basis for the high specificity, we determined the crystal structure of the SHP2 PTP–Mb13 complex at 2.4 Å resolution (Table 1). There are two SHP2 PTP/Mb13 complexes in the asymmetric unit (Supplementary Figure 2(a)). The two copies of the monobody form extensive crystal contacts via an extended intermolecular β-sheet involving β-strand A and the N-terminal tail. This mode of crystal contact has been seen in β-rich proteins such as monobodies and nanobodies.27,39,40 The overall folds of the PTP domain of SHP2 and Mb13 are also similar to previously determined structures (Cα rmsd < 0.5 Å for both the PTP domain and the monobody, excluding the diversified loops for the monobody).31,33 The interface between the two complexes within the asymmetric unit are essentially equivalent, involving essentially the same set of residues, although the buried surface area values are substantially different due to conformational differences at the interface periphery (Table 1; Supplementary Table 2; Supplementary Figure 2(b)). We chose complex 1 consisting of chains A and B for the following analysis, because complex 1 contained fewer disordered residues than complex 2.
Table 1.
Crystallographic information, refinement statistics and interface properties of the monobody Mb13/SHP2 PTP complex (PDB ID: 7TVJ).
| Data Collection* | |
|---|---|
| Space group | P1 |
| Cell dimensions | |
| a, b, c (Å) | 48.46, 48.49, 102.07 |
| α, β, γ (°) | 80.14, 77.53, 61.63 |
| Resolution (Å) | 2.39 (2.48–2.39) |
| Rmerge† | 0.084 (0.245) |
| I / σI | 6.7 (2.3) |
| Completeness (%) | 93.3 (92.1) |
| Redundancy | 1.8 (1.7) |
| Refinement | |
| Resolution (Å) | 2.39 |
| No. reflections | 28,944 |
| R‡ / Rfree§ | 0.23/0.26 |
| No. atoms | 5801 |
| Protein | 5551 |
| Ligand | 26 |
| Water | 224 |
| B-factors | |
| Protein | 34.33 |
| Ligand | 32.15 |
| Water | 30.03 |
| R.m.s. deviations1 | |
| Bond lengths (Å) | 0.0022 |
| Bond angles (°) | 0.54 |
| Ramachandran analysis1 | |
| Favored | 97.35% |
| Outlier | 0.59% |
| Interface analysis | |
| Sc2 | 0.744 (complex 1), 0.738 (complex 2) |
| Buried surface area (Å2)3 | |
| SHP2 PTP | 752.6 (chain A), 660 (chain D, Mb13 Y57 rotamer A), 684 (chain D, Mb13 Y57 rotamer B) |
| Mb13 | 814.2 (chain B), 730.5 (chain E, Mb13 Y57 rotamer A), 756.9 (chain E, Mb13 Y57 rotamer B) |
Values for highest resolution shell shown in parentheses.
over i observations of a reflection hkl.
R = Σ | |F(obs)|-|F(calc)| | /Σ IF(obs)I.
Rfree is R with 5% of reflections sequestered before refinement.
R.m.s. deviations and Ramachandran statistics are determined by MolProbity analysis61 in the PHENIX software suite.59
Sc values were calculated using the Sc program62 in the CCP4 suite.63 Complexes 1 and 2 refer to chains A and B, and chains D and E, respectively.
Buried surface area calculation was determined using the Protein interfaces, surfaces and assemblies service PISA at the European Bioinformatics Institute (https://www.ebi.ac.uk/pdbe/prot_int/pistart.html).64
The monobody is situated directly over the active site of the PTP domain (Figure 2(a)), strongly suggesting that Mb13 primarily acts as a competitive inhibitor that blocks the access of substrates to the active site. The epitope of SHP2 PTP for Mb13 is not centered over the active site. Rather, Mb13 primarily interacts with a shallow pocket adjacent to the deep active site pocket (Figure 2(b)). The location of the Mb13 epitope is similar to the interface between the PTP and N-SH2 domains in the full-length SHP2 structure (Figure 2(c)).33 For brevity, we will refer to the N-SH2-binding surface of PTP as the epitope for N-SH2. The epitopes of SHP2 PTP for Mb13 and N-SH2 are similar in size (750 Å and 900 Å2 buried surface area, respectively) (Figure 2(b) and (c)). These observations suggest that binding of Mb13 and N-SH2 to SHP2 PTP is mutually exclusive (see the next section for evidence supporting this view). Although they bind to similar surfaces, Mb13 and N-SH2 show little similarity in the use of secondary structure elements or amino acid chemistries (Figure 2(b) and (c)). Therefore, Mb13 shows no signs of mimicry of the PTP-binding interface of N-SH2, and rather it defines a distinct mode of recognizing this surface, as we have seen in other monobody-target complexes.30
Figure 2.
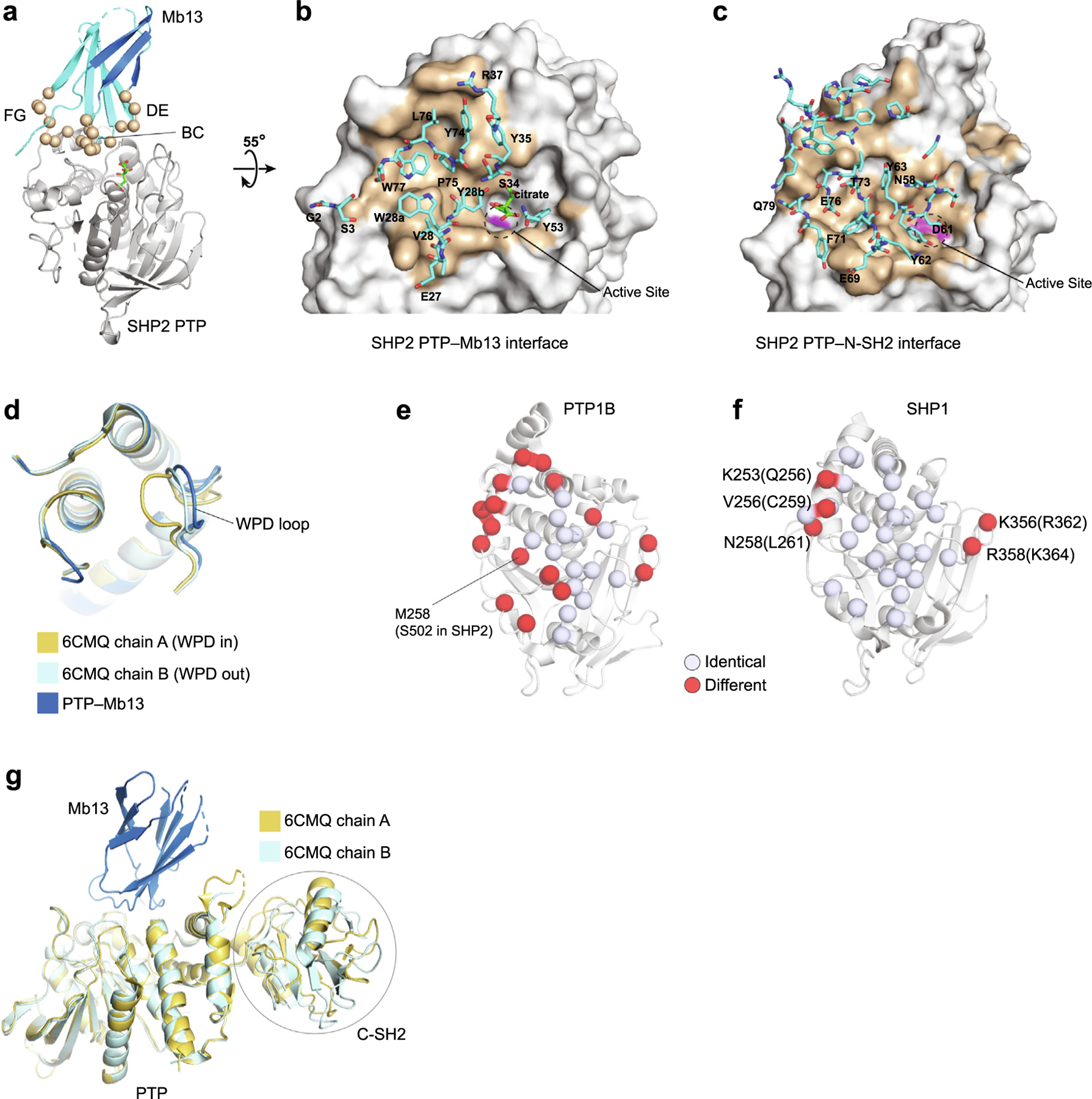
Crystal structure of the SHP2 PTP–Mb13 complex. (a) The PTP–Mb13 complex. Residues diversified in the library are shown as spheres. (b) Close up of the PTP–Mb13 interface, in which PTP is represented as a surface model with the epitope in brown and residues within the paratope of Mb13, as defined as those within 5 Å of PTP atoms, are represented as sticks and labeled. The active-site is marked with the dashed circle, with the C459 surface in magenta. (c) Close up of the SHP2 PTP–N-SH2 interface in the autoinhibited SHP2 structure (PDB ID 2SHP) as viewed in the same orientation as in panel b. Residues of N-SH2 within 5 Å of PTP atoms are represented as sticks. A subset of residues is labeled for brevity. (d) Comparison of the PTP conformation in the Mb13 complex with representative, WPD-in and WPD-out conformations reported previously (PDB ID 6CMQ). Only the vicinity of the active site is shown for clarity. (e, f). Conservation of Mb13 epitope residues, defined as those residues that are located within 5 Å of Mb13, between SHP2 and PTP1B (e) and between SHP2 and SHP1 (f). The identical residues in the epitope are shown as white spheres and different residues in red. The different residues in SHP1 are labeled and their corresponding SHP2 residues are shown in the parentheses. (g) Superposition of the SHP2 PTP–Mb13 complex with the SHP2ΔN-SH2 structure (PDB ID 6CMQ) using the PTP, showing that there are no steric clashes between Mb13 and the C-SH2 domains in the SHP2ΔN -SH2 structure.
The SHP2 PTP/Mb13 interface buries approximately 750 Å2, which is comparable to other monobody/target complexes.30,31,39,41 The BC, DE, and FG loops of Mb13 provide almost all of the buried surface area, burying approximately 350, 50, and 350 Å2, respectively. Out of the 20 residues diversified in the monobody library, 12 are located in the interface, indicating a good match between the observed interface and the library design (Figure 2(a) and (b)). The structure also supports the general trend that monobodies bind functional sites of proteins even though they are selected in an unbiased manner regarding their epitopes, or where within the target protein they should bind.30
The PTP active site contains clear electron density, which we assigned as a citrate anion (Supplementary Figure 2(c)), a component of the crystallization solution. An addition of citrate did not substantially change the affinity of Mb13 to SHP2 PTP (Supplementary Figure 2(d)), suggesting that the citrate ion does not play a crucial role in the interaction between Mb13 and the PTP domain.
We also observed electron density that suggested the presence of a disulfide bond between the side chain of active site C459 and that of nearby C367 (Supplementary Figure 2(c)). The electron density is consistent with partial occupancies as a mixture of reduced and disulfide forms. Another nearby cysteine residue, C333, was predominately reduced. This structure is similar to the oxidized form of a mutant associated with Noonan syndrome, SHP2(N308D),42 and further supports that C367 is the “backdoor” cysteine that forms disulfide bonding with the active site cysteine in WT SHP2.43 Our crystal structure therefore provides direct structural-based evidence for one state anticipated from this mechanism. It is unlikely that the disulfide bond between C459 and C367 is necessary for Mb13 binding, because binding measurements were performed using buffers with fresh DTT, which should minimize disulfide formation and because the monobody bound SHP2 PTP(C459S) (Figure 1(e)). Interestingly, oxidation of SHP2 PTP with H2O2 reduced Mb13 binding and subsequent reduction with DTT, following established procedures,42,44 restored the binding (Supplementary Figure 2(e)), with an accompanying change primarily in ka (Supplementary Table 2). This change in ka suggests a model in which oxidation stabilizes the PTP conformation that is less favorable for Mb13 binding. However, as the PTP domain contains a total of five Cys residues that could be oxidized, the mechanism of this oxidation-mediated inhibition will be elucidated in future studies.
We speculated that because Mb13 is distinctly different from a substrate of SHP2 PTP, i.e., a linear peptide harboring a pY moiety, the Mb13 complex might capture a novel conformation of the SHP2 PTP active site. However, the PTP active site showed minimal differences from previously reported, “WPD-out” conformation, in which the WPD loop is situated distant from the catalytic site (Figure 2(d)).33,34,45
The SHP2 PTP–Mb13 interface provides a rationale for the high specificity of Mb13 for SHP2 and against PTP1B. Seventeen of the 32 residues within the Mb13 epitope, defined as those within 5 Å of Mb13 atoms, are different in PTP1B PTP (Figure 2(e)). In particular, S502 in SHP2 corresponds to M258 in PTP1B. This difference would cause a steric clash with W77 of Mb13, rationalizing why Mb13 exhibits no detectable binding to PTP1B. In contrast, a similar comparison with SHP1 revealed that the epitope for Mb13 is highly conserved between SHP2 and SHP1. Out of the 32 residues in the Mb13 epitope, only five are different between SHP2 and SHP1, and these differences lie along the periphery of the PTP–-Mb13 interface (Figure 2 (f)). Unlike the PTP1B case, we did not find a SHP1 residue that would clearly disrupt the Mb13-SHP1 interaction. Three of the five residues that differ between SHP2 and SHP1 and are located in the Mb13 epitope, Q256, C259 and L261, contribute a total of ~ 100 A2 of buries surface area (Supplementary Table 2), suggesting that these differences are likely to be important for the discrimination of SHP2 and SHP1 by Mb13. We speculate that Mb13 discriminates SHP2 from SHP1 by recognizing subtle conformational differences caused collectively by these residues and/or by residues in the so-called second shell, i.e., those that do not directly contact Mb13. Future research utilizing systematic mutagenesis will define the basis for the high specificity of Mb13 that can discriminate the nearly identical surfaces between SHP1 and SHP2 PTPs.
Probing the PTP active-site accessibility using a monobody
The overlap between the Mb13 epitope and the N-SH2 epitope (Figure 2(b) and (c)) suggests mutually exclusive binding of N-SH2 and Mb13 to the PTP domain. This in turn suggests that we can use Mb13 as a probe for assessing the accessibility of the PTP active site in the full-length SHP2 protein (Figure 3(a)). Indeed, the KD of Mb13 to full-length SHP2 was 130 nM, much weaker than to the isolated PTP domain, as measured using BLI (Figure 3(b)). Also, the association rate constant, ka, was much reduced by 40-fold relative to the ka for the Mb13–PTP interaction, whereas the dissociation rate constant, kd, was similar (Supplementary Table 1). The crystal structure of the SHP2 PTP–Mb13 complex suggests that the presence of the C-SH2 domain in SHP2ΔN -SH2 minimally affects the PTP-Mb13 interaction (Figure 2(g)). Mb13 bound to SHP2 that lacked the N-SH2, SHP2ΔN -SH2, with affinity similar to PTP alone (KD = 7 nM; Figure 3(b)) and also with similar kinetic parameters. These results strongly support the need to displace the N-SH2 domain in full-length SHP2 for Mb13 to bind to SHP2 PTP, which in turn supports the capability of the assay with Mb13 to probe the accessibility of SHP2 PTP. In a simple, two-state model in which full-length SHP2 is assumed to exist in either the open or closed state, and the closed state has no affinity to Mb13, the equilibrium constant for the open-closed transition, Keq, is expressed as:
where is the KD of Mb13 to the full-length SHP2 protein, and is the KD to the isolated PTP. Accordingly, the Keq for wild-type SHP2 was 54. This number is consistent with that estimated using a PTP activity assay and NMR spectroscopy (see the next section).34,46
Figure 3.
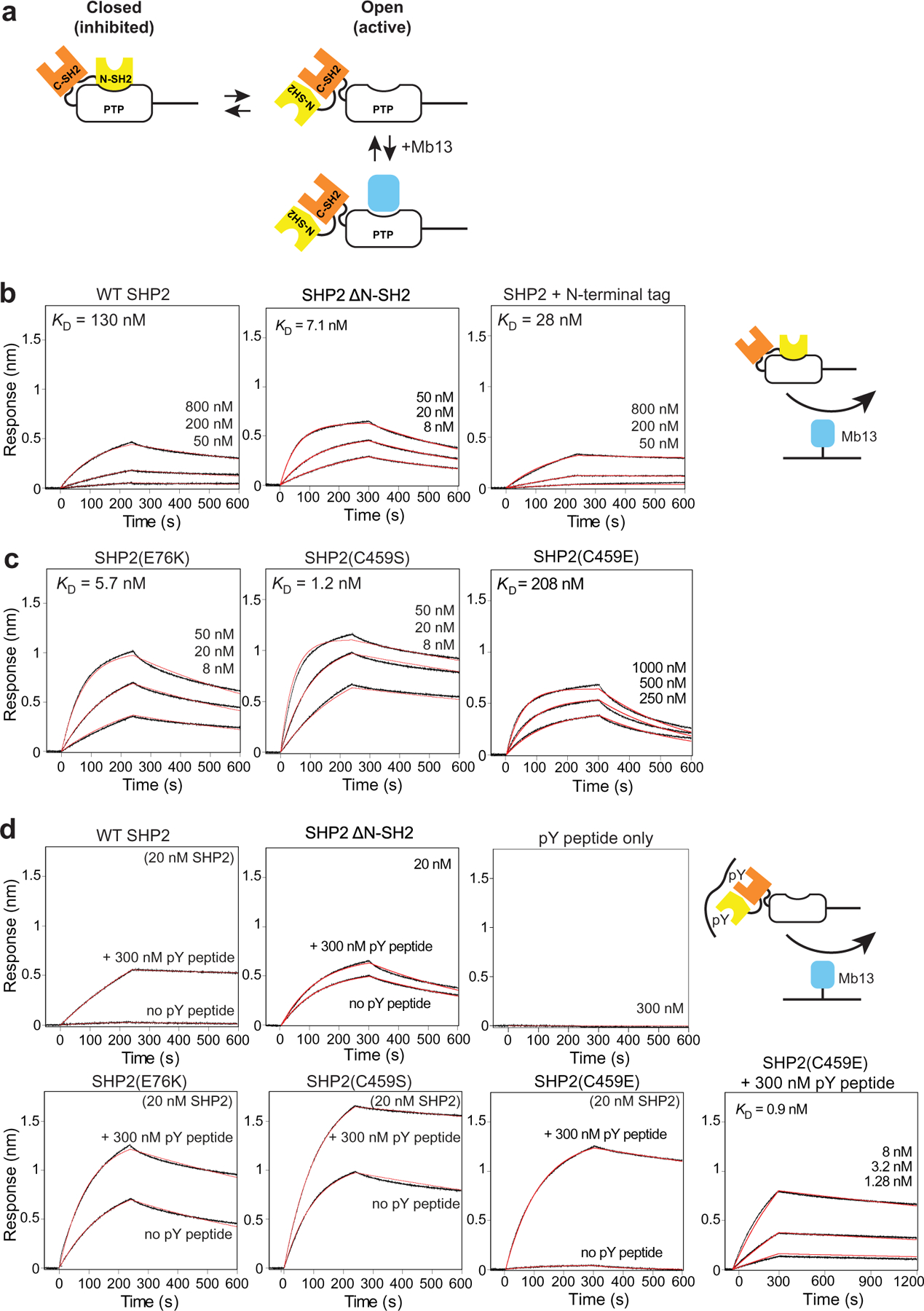
Characterization of SHP2 allostery using Mb13 as a probe for the accessibility of the PTP active site. (a) Cartoon depicting the open-closed equilibrium of SHP2 and binding of Mb13 to the open state but not to the closed state. (b) BLI sensorgrams for the interaction of the full-length, ΔN-SH2 and N-terminally tagged constructs of WT SHP2 with Mb13 immobilized on a sensor tip. The tag consists of His6, Avi-tag and a TEV cleavage sequence. (c) BLI sensorgrams for the interaction of full-length SHP2 mutants with Mb13 immobilized on a sensor tip. (d) BLI sensorgrams for the interaction of the indicated SHP2 constructs with Mb13 in the absence and presence of the tandem pY peptide derived from GAB2. The right bottom panel shows the interaction of SHP2(C459E) in the presence of 300 nM pY peptide.
We next evaluated the active-site accessibility of SHP2 mutants using an assay with the monobody probe. SHP2(E76K) is a mutation within N-SH2 that is associated with Noonan syndrome. It disrupts the interaction between N-SH2 and PTP domains and thus opens and activates SHP2.46 The KD value of the SHP2(E76K)-Mb13 interaction was 5.7 nM (Figure 3(c)), corresponding to Keq of 2.4. The difference in Keq between WT and SHP2 (E76K), 23, is in an excellent agreement with the value estimated from an activity assay and NMR spectroscopy, 2334 but smaller than an earlier estimate from a PTP activity assay, 555.46
Next, we examined a “PTP-dead” C459S mutant that is commonly used to test the role of the PTP activity in SHP2-dependent cellular functions. Clearly, a PTP activity assay cannot be applied to this mutant. C459S exhibited high affinity to Mb13, with a KD value of 1.2 nM (Figure 3(c)), comparable to E76K. With the KD value of Mb13-PTP(C459S), 0.75 nM, Keq for SHP2(C459S) was 1.6, indicating that it is nearly fully open, consistent with previous NMR studies.34 The kinetic parameters for SHP2(E76K) and SHP2(C459S) were also similar to those for the Mb13–SHP2 WT PTP interaction (Supplementary Table 1), further supporting this interpretation.
Another PTP-dead mutant, C459E, was reported to be in the closed state, unlike the C459S mutant, by Padua et al.34 Indeed, the affinity of Mb13 to the C459E mutant was low, with the KD value of 210 nM and a reduced ka value (Figure 3(c) and Supplementary Table 1). We excluded the possibility that the C459E mutation disrupted the binding of Mb13 (see the next section). Thus, our results provide independent support for the proposal by Padua et al. that the C459E mutation abrogates PTP activity with minimal perturbation of the conformational equilibrium of SHP2.
In addition, we examined the effect of adding an N-terminal tag to SHP2. N-terminal tagging is a common approach used for biochemical and cellular studies of SHP2. Interestingly, we saw a small but substantial change in the affinity of N-terminally tagged SHP2 to Mb13 (Figure 3(b)). This reduction of KD is primarily due to the reduction of kd (Supplementary Table 1), suggesting that this tag affected the dissociation of the complex. Because the N-terminus can be located near the interface between the monobody and PTP domains (Figure 3(a)), perhaps it is not surprising that an N-terminal tag can influence the monobodySHP2 interaction. Nevertheless, these results offer a cautionary note as to the use of tagged SHP2 molecules.
Assessment of SHP2 regulation with ligands
Binding of pY ligands to the N-SH2 domains weakens the interaction between the N-SH2 and PTP domains and thereby shifts the open-closed equilibrium toward the open state.47,48 We characterized this activation reaction using our assay with the Mb13 probe. An addition of a nearly saturating concentration (300 nM with respect to KD = 24 nM)31 of a tandem pY peptide, a peptide corresponding to residues of GAB2 containing two pY residues, dramatically increased the affinity of Mb13 to SHP2 (Figure 3(d)). Consistent with our expectation that the pY peptide can be hydrolyzed by the PTP activity of SHP2, we observed a loss of the activation effect in a second set of the measurements, except for the PTP-dead, C459S mutant (Supplementary Figure 3). These results confirm the allosteric activation of SHP2 by pY peptides. The changes in the kinetic parameters upon addition of the pY peptide occurred primarily in ka (Supplementary Table 1), which is consistent with a model that the peptide addition changes the accessibility of the Mb13-binding site and has no effects on the dissociation of Mb13 from the complex once formed.
The addition of the tandem pY peptide only marginally changed Mb13 binding to SHP2ΔN - SH2, but it enhanced Mb13 binding to SHP2 (E76K) and SHP2(C459S), although slightly (Figure 3(d)). In contrast to SHP2(C459S), SHP2 (C459E) exhibited strong enhancement with the addition of the pY peptide, further supporting that SHP2(C459E) is in the closed conformation in the absence of a pY peptide.
Next, we tested the effects of SHP099, a small molecule inhibitor that binds to the interface between the SH2 domains and the PTP domain and thereby stabilizes the closed form of SHP2.10 SHP099 reduced the affinity of E76K and C459S to Mb13 but not to the level of WT SHP2 in the absence of the inhibitor (Figure 4(a)), confirming the challenge of inhibiting activating mutations with this class of allosteric inhibitors.34,49
Figure 4.
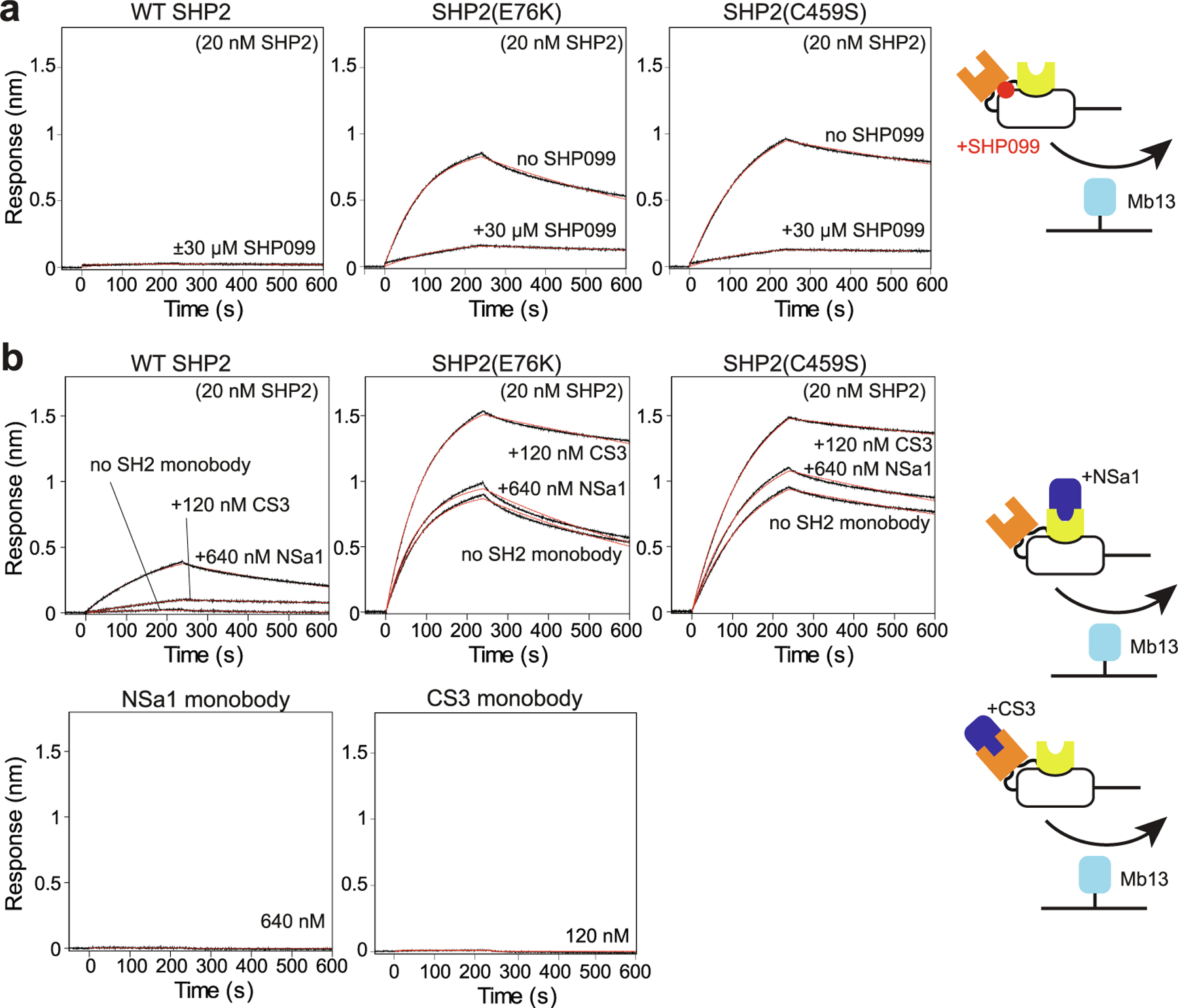
Effects of synthetic ligands on SHP2 allosteric regulation. (a) Effects of SHP099 on the interaction of the indicated SHP2 constructs with Mb13. (b) Effects of SH2-binding monobodies NSa1 and CS3. Data are shown in the same manner as in Figure 3.
We previously reported monobodies NSa1 and CS3 that respectively bind to the N-SH2 and C-SH2 domains of SHP2.31 They have high specificity to their respective target SH2 domains among human SH2 domains, and also have high affinity in spite of not containing a pY moiety. Importantly, unlike pY ligands that show cross-reactivity to both SH2 domains, NSa1 does not bind to C-SH2, and CS3 does not bind to N-SH2, allowing us to perturb the two SH2 domains independently. We tested the effects of these monobodies using our assay at saturating concentrations (Figure 4(b)). NSa1 increased the binding of Mb13 to WT SHP2 but had no effects on the two activated mutants. By contrast, CS3 enhanced the binding of the SHP2 mutants to Mb13, to an extent similar to those observed with the GAB2 tandem pY peptide. CS3 also enhanced the binding of WT SHP2 somewhat, but the binding was still much weaker than the mutants or WT SHP2 in the presence of the pY ligand.
Assessment of SHP2 PTP accessibility in cells
Finally, we examined whether the monobody probes can be used to assess the conformation of endogenous SHP2 in HEK293 cells. Upon EGF stimulation, the level of phosphorylated SHP2 was increased as expected, with a concomitant increase in the phosphorylated ERK level, a marker for the activation of the RAS-MAPK signaling pathway (Supplementary Figure 4). We captured SHP2 from cell lysates using Mb11 and Mb13, and detected using western blotting. Based on the characterization using a series of SHP2 mutants and fragments described above, it is highly likely that these monobodies preferentially capture SHP2 in the open state. Both Mb11 and Mb13 captured SHP2 (Figure 5(a)), indicating that these monobodies are sufficiently selective to recognize endogenous SHP2 in the context of cell lysates. EGF stimulation increased the amount of captured SHP2 by 3-fold with either monobody, consistent with a model that the activation of the EGF pathway increased the concentration of the open state of SHP2 that can be captured with the monobodies.
Figure 5.
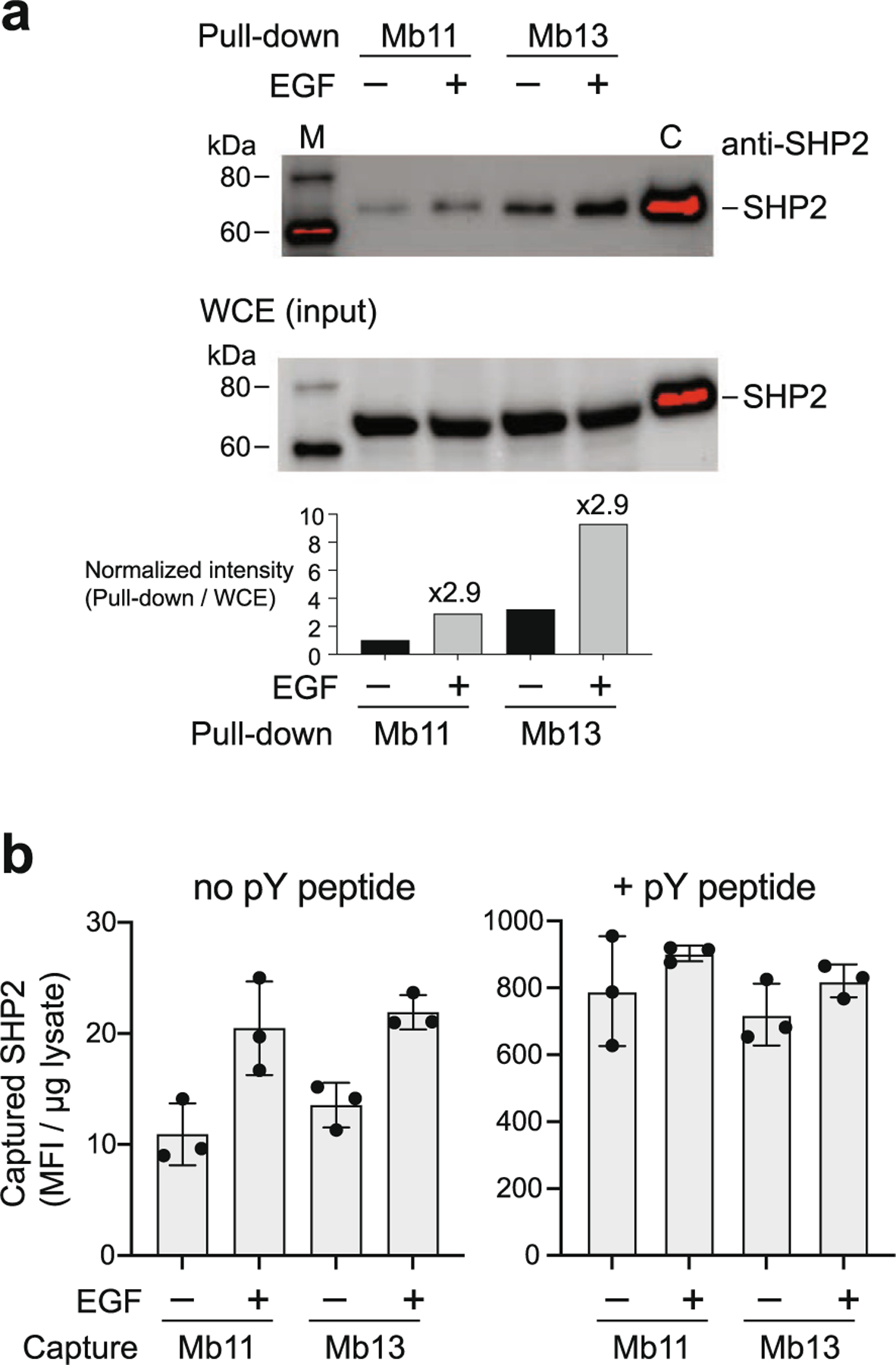
Assessing the level of activated SHP2 in cells using Mb11 and Mb13. (a) The amounts of SHP2 in HEK293 cells that were captured using Mb11 or Mb13 with and without EGF stimulation (5 min with 50 ng/ml). The captured SHP2 protein was detected using an anti-SHP2 antibody. Purified SHP2 was included as a control in lane C. (b) Quantification of captured SHP2 using Mb11 or MB13 as a capture reagent in the absence or presence of GAB2 pY peptide using flow cytometry. The raw data are provided in Supplementary Figure 5.
We also implemented a more rapid method for profiling the accessibility of SHP2 PTP by the monobodies using bead-based capture and detection using flow cytometry.50,51 In this method we captured SHP2 from lysates using Mb11 or Mb13 immobilized on magnetic beads. After washing the beads, captured SHP2 was detected using an anti-SHP2 antibody followed by a fluorescently labeled secondary antibody. We found that, although the flow cytometry signals were linear with respect to the amount of lysates added to the beads, there was substantial background signal (Supplementary Figure 5). Therefore, we used the slope of the flow cytometry signal determined with 2, 5 and 10 mg lysates as the assay readout, rather than the raw signal intensities at a single lysate amount, thereby eliminating the contribution of the background signals. Using this assay, we determined that the level of captured SHP2 increases by 2-fold upon EGF stimulation (Figure 5(b)), in agreement with the results with monobody capture and Western blotting detection described above. In addition, to determine the amount of total SHP2 in the lysates, we treated the lysates with the GAB2 pY peptide prior to capturing with a monobody. This treatment dramatically increased the signals by ~40-fold, and eliminated differences between samples with and without EGF stimulation (Figure 5(b)). The enhancement in the captured SHP2 level after EGF treatment but without the pY peptide addition (approximately 8–10; Figure 5 (b), left panel) corresponds to ~1% of total SHP2 captured after the pY peptide treatment (750–900; Figure 5(b), right panel), suggesting that only a small fraction of cellular SHP2 is in the state that can be captured with the monobodies, even when the cells are strongly stimulated with a growth factor.
Discussion
The monobody inhibitors described in this work achieve high levels of discrimination between the PTP domains of SHP2 and SHP1 (Figure 1), despite targeting highly conserved regions near the active site. These molecules represent additional examples of highly selective monobodies directed to challenging intracellular targets.28 Such high selectivity has thus far been unachievable with small molecules. Also, the KD and IC50 values of these monobodies are similar, if not more potent, than those reported for small molecules against the isolated PTP domain (NSC-87877, 320 nM; PHPS4, 630 nM; 14a, 800 nM; and cryptotanshinone, 22.5 µM).23–26 Whereas this study was focused on biochemical and structural aspects, we envision that these monobodies, used as genetically encoded intracellular biologics,28 will be uniquely powerful tools for dissecting the roles of SHP2 in the cellular context.
PTP domains have been particularly challenging targets for small molecules due to the chemical nature of the active site. The active site is highly charged and sensitive to redox, which consequently gives rise to highly charged compounds and “redox cycling compounds”.52 By contrast, the paratope of Mb13 contains mostly neutral, aromatic amino acid residues (Figure 2 (b)). This mode of inhibiting a PTP might be recapitulated using macrocycles and peptidomimetics, modalities that are more amenable for making cell permeable molecules than proteins.
The assay for the SHP2 conformational equilibrium that we developed here (Figure 3) complements previously reported ones.10,34 It is sensitive, uses small quantities of samples, and does not rely on enzyme activity. Because Mb13 does not cross-react with the SH2 domains, unlike pY-containing molecules, we envision that this assay has fewer interferences. It should be useful for characterizing effects of SHP2 mutants on the conformational equilibrium and screening for SHP2 inhibitors and activators.
Using this assay, we discovered that the two SH2-binding monobodies weakly favor the open form of SHP2. In our original work,31 we were unable to determine whether these monobodies activate SHP2, because of the low sensitivity of the PTP activity assay that we employed. To our knowledge, there are no reported activators of SHP2 that do not contain a pY moiety. This finding demonstrates the feasibility of developing a new class of SHP2 regulators. The superposition of the N-SH2 domain in the auto-inhibited form of full-length SHP2 and in the complex with NSa1 mono-body show conformational differences in the regions of the N-SH2 domain that interact with the PTP domain (Supplementary Figure 6). Because these regions in the NSa1 complex are involved in crystal contact, we cannot exclude the possibility that the observed conformational differences do not exist in solution. However, this observation suggests that NSa1 stabilizes a conformation of the N-SH2 domain that reduces its affinity to the PTP domain, which leads to favoring the open state. Based on the crystal structure of a monobody closely related to CS3, CS1, in complex with the C-SH2 domain, CS3 should bind to an epitope of the C-SH2 domain that does not present steric clashes. As such, it is unclear how CS3 preferentially stabilizes the open state.
The assay also showed that only a small fraction of WT SHP2 is in the open conformation even when cells are strongly activated with a growth factor. In contrast, a pathological mutant, SHP2(E76K), is uniformly in the open conformation. We envision that this difference in the conformational equilibrium could be exploited to selectively degrade disease-associated SHP2 mutants. Monobodies, including those targeting SHP2 SH2 domains, have been used as building blocks for selective “biodegraders”.53–56 Although Mb11 or Mb13 is not selective to a specific mutation, they are selective to the open conformation. As such, we speculate that one can build degraders selective to activating mutants of SHP2. Clearly, selectivity to SHP2 PTP among all PTPs is important for such applications, and these monobodies may already satisfy this requirement.
Methods
Protein expression and purification
Genes for SHP1 and PTP1B were amplified from plasmids obtained from the PlasmID Repository at Harvard Medical School (catalog numbers, HsCD00376930 and HsCD00002513, respectively). The gene for SHP2 was kindly provided by Dr. Oliver Hantschel. Fragments containing the protein tyrosine phosphatase (PTP) domains of SHP2 (residues 224–529, UniProt entry Q06124), SHP1 (residues 241–530, UniProt entry P29350), and PTP1B (residues 2–283, UniProt entry P18031) were produced by sticky-end PCR and cloned in the pHBT vector.31 Monobody proteins and full-length SHP2 proteins were also produced using the pHBT vector.
Proteins were produced in BL21(DE3) cells containing the pBirAcm plasmid (Avidity) in the presence of 50 µM biotin for in vivo biotinylation. All proteins were purified using Ni-Sepharose columns (Cytiva), and their apparent monodispersity was confirmed using a Superdex 75 size-exclusion column (Cytiva). All PTPs were further purified by size-exclusion on Superdex 75 before dialysis in TBS (50 mM Tris, 150 mM NaCl, pH 7.5) and 1 mM DTT. For crystallization, phosphatase assay and BLI experiments, the affinity tag for each protein was removed using the tobacco etch virus (TEV) protease and the cleaved proteins were purified by passing through a Ni-Sepharose column. Full-length SHP2 and monobody proteins for BLI measurements were further purified using a Superdex 75 column. A GAB2 fragment (residues 605–650 according to UniProt Q9UQC2) containing Y614 and Y643 was produced as a fusion with yeast SUMO and phosphorylated using the kinase domain of EphA3 as described previously.31
Phage display and yeast surface display
The monobody libraries and selection methods have been previously described.39,36,57 The target concentrations used for rounds 1, 2, 3, and 4 of phage display were 200 nM, 100 nM, 50 nM, and 20 nM, respectively. Phages were captured onto biotinylated targets immobilized to streptavidin-coated magnetic beads (Streptavidin MagneSphere Para-magnetic Particles; Promega, Z5481/2) and then eluted with 0.1 M Gly-HCl, pH 2.1. After gene shuffling among phage clones within each enriched population,36 we performed three rounds of library selection by yeast surface-display using a target concentration of 200 nM SHP2 PTP in the first positive sorting round, 100 nM of SHP1 and 100 nM PTP1B in the second negative sorting round, and 100 nM SHP2 PTP in the final positive sorting round. Yeast surface displayed experiments were performed as described previously36 except that mouse anti-V5 antibody (Thermo) and Dylight 650-conjugated goat anti-mouse IgG (Thermo) were used in place of equivalent reagents. Affinity maturation in yeast display format was performed similarly to initial sorting experiments except binding reaction was done at 30 °C in the presence of 200 µM DTT for 30 min before quenching with ice-cold BSS.
Competition binding assay for testing the specificity of monobodies was performed using yeast surface display as follows. Approximately 105 yeast cells for each monobody clone were incubated with a mixture of 100 nM biotinylated, cognate target and 3 µM competitor, from which the biotinylation tag had been removed, in 20 μl BSS buffer (50 mM Tris–HCl, 150 mM NaCl, pH 8, 1 mg/ml bovine serum albumin (BSA) and 1 mM EDTA). After 30 min incubation on ice with shaking, the cell suspensions were transferred to the wells of a 96-well filter plate (MultiScreenHTS HV, 0.45 μm pore size; Millipore), drained by vacuum, and washed twice by 100 µl of BSST (BSS buffer containing 0.1% Tween 20). Next, 20 µl of 10 µg/ml Dylight 650-conjugated to streptavidin (Thermo) in BSS was added to each of the wells. After incubation on ice with shaking for 30 min, the cells were washed with BSST twice in the same manner described as above. The cells were finally suspended in 400 µl of BSS and five thousand events were analyzed using a Guava EasyCyte 6/L flow cytometer (Millipore). The experiment was performed in triplicate.
Cryptotanshinone inhibition
Cryptotanshinone was acquired from Sigma (C5624–5MG). 5 nM Mb13 monobody was incubated with 20 µg/ml streptavidin-coupled Dynabeads M280 (Invitrogen) for 30 min on ice in BSS buffer (50 mM Tris–HCl, 150 mM NaCl, pH 8, 1 mg/ml BSA, 1 mM DTT, and 1 mM EDTA). The beads were then washed and blocked with 1 mM biotin for 30 min, followed by another wash and resuspension in BSS buffer. 200 nM SHP2 PTP was pre-incubated with or without 100 µM cryptotanshinone for 30 min at 30 °C in BSS buffer. Ten microliters of the Mb13-bead solution were then transferred to a well of a 96-well filter plate (MultiScreenHTS HV, 0.45 µm pore size; Millipore) and drained by vacuum. Twenty µl of the SHP2 PTP mixture were then transferred to the wells of the filter plate containing the drained beads and the plate was incubated at 30 °C with shaking for 30 min. The wells were drained and washed twice with ice-cold BSST (BSS buffer containing 0.1% Tween 20). After draining, 20 µl of 10 µg/ml of Dylight 650-conjugated to streptavidin (Thermo) in BSS was added to each of the wells. After incubation on ice with shaking for 30 min, the beads were washed with BSST twice in the same manner described as above. The beads were resuspended in 200 µl BSST and the fluorescence emission in the Red2 channel was analyzed for 5000 events on a Guava EasyCyte 6/L flow cytometer (Millipore).
PTP activity assay
The activities of SHP2 and SHP1 PTP domains were measured using chromogenic substrate pNPP. Reactions were performed in a 96-well clear, flat-bottom polystyrene plate in 60 mM HEPES-NaOH buffer, pH 7.2 containing 75 mM NaCl, 75 mM KCl, 1 mM EDTA, 0.05% Tween 20 and 5 mM DTT, in a final volume of 100 µl at 25 °C. PTPs (final concentration 0.5 µM) and monobodies (final concentration 1 µM) were added to the solution and incubated ~1 min at 25 °C, then reactions were started by addition of final 1 mg/ml pNPP (P4744, MilliporeSigma). Absorbance at 405 nm was monitored every 1 min for 60 min in a microplate reader (BioTek Epoch 2, Agilent). Measurements were performed in triplicate.
X-ray crystallography
Mb13 and SHP2 PTP were mixed and purified as a complex from a Superdex 75 column (Cytiva) in 10 mM Tris, 50 mM NaCl, pH 8.0, concentrated to a total protein concentration of 9.3 mg/ml, and crystallized in 0.1 M sodium citrate tribasic dihydrate pH 5.0, 2% Tascimate, and 16% PEG 3350 at 19 °C by the hanging drop vapor diffusion method. Crystals were frozen in a mixture of 80% mother liquor and 20% ethylene glycol.
X-ray diffraction data were collected at the Advanced Photon Source (Argonne National Laboratory) beamline 24-ID-C. Data were processed and scaled with XDS.58 The Mb13/SHP2 PTP structure was determined by molecular replacement in PHENIX59 from a multicopy search using the catalytic PTP domain structure of SHP2 (PDB ID 3ZM1) and a monobody structure excluding loop regions (PDB ID 3K2M). Manual model building, solvent addition, and refinement of the structures were conducted iteratively using Coot60 and phenix.refine. Molecular graphics were generated using PyMOL (https://www.pymol.org).
Biolayer interferometry
The interactions were monitored at 22 °C using an Octet R8 instrument (Sartorius). A biotinylated monobody was immobilized on a streptavidin-coated tip at the level of 0.5 nm and its interaction with analytes was monitored in 60 mM HEPES buffer, pH 7.2, containing 75 mM NaCl, 75 mM KCl, 1 mM EDTA, 0.05% P-20, 1 mM DTT, 1 µM biotin and 0.5 % (w/v) BSA (BLI buffer). Tips were regenerated using 0.1 M glycine HCl buffer, pH 2.1. The data were analyzed using the software provided by the vendor.
For measurement with oxidized SHP2 PTP, a SHP2 PTP sample diluted to 0.5 µM in BLI buffer without DTT was incubated with 2 mM H2O2 for 15 min at room temperature, following published procedures.42,44 This oxidized SHP2 PTP sample was further diluted in BLI buffer (without H O or DTT) to the final concentration for measurement. As a re-reduced control, oxidized PTP was treated with final 5 mM DTT, incubated 15 min on ice, then diluted in BLI buffer for measurement.
Cell culture
HEK293 cells were cultured in Dulbecco’s Modified Eagle’s Medium supplemented with 10% fetal bovine serum and antibiotic–antimycotic mixture (Gibco) at 37 °C in a humidified incubator with 5% CO2. Where applicable, cells were cultured for 48 hours after medium change and stimulated with 50 ng/ml of EGF for 5 min.
Preparation of whole cell extract (WCE)
Cells (approximately 3.6 × 107 cells) were washed in phosphate-buffered saline (PBS), suspended in 10 mM Tris HCl buffer pH 7.9 containing 420 mM NaCl, 0.1% NP-40, protease inhibitor cocktail (Roche) and 1 mM sodium orthovanadate, and lysed by a total of five freeze–thaw cycles. To eliminate RNA and DNA, the lysates were incubated with 25 units/ml of benzonase nuclease at 4 °C for 30 min and centrifuged at 10,000 xg at 4 °C for 30 min. After the centrifugation, the supernatant (approximately 2 ml) was collected and filtered with 0.45 µm syringe filter.
Immunoprecipitation
M-280 Dynabeads (Thermo, 11206D) were washed two times with PBS containing 0.1% BSA and 0.02% Tween 20 with the beads incubated for 10 minutes in each washing step. Five hundred pmol of a biotinylated monobody were immobilized onto 50 µl of the pre-washed Dynabeads by incubation at 4 °C with rotation for one hour. Excess biotin-binding sites of the beads were blocked with 5 µM of biotin, and the monobody-immobilized Dynabeads were washed with PBS containing 0.1% BSA and 0.02% Tween 20 two times. One milliliter of whole cell lysates was incubated with 30 µl of washed M-280 Dynabeads to eliminate proteins that bind nonspecifically to the beads. After removing the beads, the precleared lysates were incubated with 50 µl of monobody-immobilized Dynabeads for 1.5 hours at 4 °C with rotation, and washed three times with 20 mM Tris-HCl buffer pH 8.0 containing 10% glycerol, 5 mM MgCl2, 480 mM NaCl, 0.5% NP-40 and 0.1% Sodium deoxycholate with 10 minutes of incubation at 4 °C for each washing step. The M280 beads with captured proteins in 50 ml of 10 mM Tris HCl buffer pH 8.0 containing 150 mM NaCl and 0.1% SDS were heated in boiling water for 5 minutes. The eluted proteins were characterized by Western blot analysis.
Western blot analysis
WCE and immunoprecipitated proteins were separated in SDS-PAGE and transferred to PVDF membrane. The membrane was blocked with blocking buffer (50 mM Tris HCl pH 7.5 containing 150 mM NaCl, 5% BSA and 0.1% Tween20). The membrane was probed with anti-SHP2 antibody (Santa Cruz sc-7384, mouse monoclonal IgG, dilution 1:1,000), anti-phosphorylated SHP2 (pY580) antibody (Cell Signaling, 3703, rabbit IgG, dilution 1:1,000), anti-Erk antibody (Cell Signaling, 9102S, lot 23, rabbit IgG, dilution 1:5,000) or anti-phosphorylated Erk antibody (Cell Signaling, 9101S, lot 28, rabbit IgG, dilution 1:5,000) in blocking buffer and rinsed with TBS buffer (50 mM Tris HCl pH 7.5, 150 mM NaCl) containing 0.1% Tween20. The detection was performed with anti-mouse IgG-HRP (Thermo, 31432, goat IgG, dilution 1:10,000) or anti-rabbit IgG-HRP (Thermo, 31462, goat IgG, dilution 1:10,000) in TBS buffer (50 mM Tris HCl pH 7.5, 150 mM NaCl) containing 0.25% BSA and 0.1% Tween20 and Pierce ECL 2 Western Blotting Substrate (Pierce, 80196).
Bead-based binding assays
Five microliters of M-280 Dynabeads (Thermo, 11206D) was suspended in 100 ml of 50 mM Tris HCl pH 8.0 containing 150 mM NaCl, 0.1% BSA, 1 mM EDTA pH 8.0, 1 mM DTT and 40 pmol of biotinylated monobody, and incubated for an hour at 4 °C with rotation. Excess biotin-binding sites of the beads were blocked with 200 µl of 5 µM biotin. The monobody-immobilized Dynabeads were resuspended in 10 mM Tris HCl pH 7.9 containing 420 mM NaCl and 0.1 % NP-40, and incubated with 25, 50 and 100 µg of WCE proteins for 30 min at room temperature with gentle shaking. GAB2 peptide was mixed with WCE at a final concentration of 1 µM immediately before the incubation with the beads. After the incubation, the beads were rinsed with 50 mM Tris HCl pH 8.0 containing 150 mM NaCl, 0.1 % BSA, 1 mM EDTA pH 8.0, 1 mM DTT and 0.1 % Tween20 three times using a 96-well filter plate (Millipore, MSHVN4550), and incubated with anti-SHP2 antibody (Santa Cruz, sc-7384, lot H2916; 1:50 dilution) in 50 mM Tris HCl pH 8.0 containing 150 mM NaCl, 0.1 % BSA, 1 mM EDTA pH 8.0, 1 mM DTT and 1 mM sodium orthovanadate. After 30 min incubation with gentle shaking at room temperature, the beads were washed three times and incubated with Dylight 650 anti-mouse IgG antibody (Thermo, 84545, lot PB195113; 1:100 dilution) in 50 mM Tris HCl pH 8.0 containing 150 mM NaCl, 0.1 % BSA, 1 mM EDTA pH 8.0, 1 mM DTT and 1 mM sodium orthovanadate. After 30 min incubation with gentle shaking at 4 °C in dark, the beads were washed three times and resuspended in 50 mM Tris HCl pH 8.0 containing 150 mM NaCl, 0.1 % BSA, 1 mM EDTA pH 8.0 and 1 mM DTT. The binding of SHP2 to monobodies were analyzed on a HyperCyt screener (Sartorius). Signals reported are median fluorescence intensities.
Supplementary Material
Acknowledgments
We thank B. G. Neel for advice and discussion. This work was supported by the National Institutes of Health grants R01 CA194864 and R01 CA212608 (S.K.). The core facilities of NYU School of Medicine were partially supported by the Cancer Center Support Grant P30CA016087. This work is based upon research conducted at the Northeastern Collaborative Access Team beamlines, which are funded by the National Institute of General Medical Sciences from the National Institutes of Health (P30 GM124165). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02–06CH11357.
Footnotes
CRediT authorship contribution statement. Fern Sha: Conceptualization, Methodology, Investigation, Writing – original draft. Kohei Kurosawa: Methodology, Investigation, Writing – review & editing. Eliezra Glasser: Methodology, Investigation, Writing – review & editing. Gayatri Ketavarapu: Investigation, Writing – review & editing. Samara Albazzaz: Investigation, Writing – review & editing. Akiko Koide: Methodology, Investigation, Writing – review & editing. Shohei Koide: Conceptualization, Investigation, Writing – original draft, Writing – review & editing, Supervision.
Declaration of Competing Interest
The authors declare the following financial interests/personal relationships which may be considered as potential competing interests: S.K. is a co-founder and holds equity in Revalia Bio and Aethon Therapeutics; has received consultation fees from Black Diamond Therapeutics and Aethon Therapeutics; has received research funding from Puretech Health, Argenx BVBA, Black Diamond Therapeutics and Aethon Therapeutics. A.K. and S.K. are listed as inventors on issued and pending patents on the monobody technology filed by The University of Chicago (US Patent 9512199 B2 and related pending applications). The other authors declare no competing interests.
Appendix A. Supplementary Data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jmb.2023.168010.
References
- 1.Chan G, Kalaitzidis D, Neel BG, (2008). The tyrosine phosphatase Shp2 (PTPN11) in cancer. Cancer Metastasis Rev 27, 179–192. [DOI] [PubMed] [Google Scholar]
- 2.Dance M, Montagner A, Salles JP, Yart A, Raynal P, (2008). The molecular functions of Shp2 in the Ras/Mitogen-activated protein kinase (ERK1/2) pathway. Cell. Signal 20, 453–459. [DOI] [PubMed] [Google Scholar]
- 3.Shi ZQ, Lu W, Feng GS, (1998). The Shp-2 tyrosine phosphatase has opposite effects in mediating the activation of extracellular signal-regulated and c-Jun NH2-terminal mitogen-activated protein kinases. J. Biol. Chem 273, 4904–4908. [DOI] [PubMed] [Google Scholar]
- 4.Saxton TM, Henkemeyer M, Gasca S, Shen R, Rossi DJ, Shalaby F, Feng GS, Pawson T, (1997). Abnormal mesoderm patterning in mouse embryos mutant for the SH2 tyrosine phosphatase Shp-2. EMBO J 16, 2352–2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Shi ZQ, Yu DH, Park M, Marshall M, Feng GS, (2000). Molecular mechanism for the Shp-2 tyrosine phosphatase function in promoting growth factor stimulation of Erk activity. Mol. Cell Biol 20, 1526–1536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang SQ, Tsiaras WG, Araki T, Wen G, Minichiello L, Klein R, Neel BG, (2002). Receptor-specific regulation of phosphatidylinositol 3’-kinase activation by the protein tyrosine phosphatase Shp2. Mol. Cell Biol 22, 4062–4072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tartaglia M, Mehler EL, Goldberg R, Zampino G, Brunner HG, Kremer H, van der Burgt I, Crosby AH, et al. , (2001). Mutations in PTPN11, encoding the protein tyrosine phosphatase SHP-2, cause Noonan syndrome. Nat. Genet 29, 465–468. [DOI] [PubMed] [Google Scholar]
- 8.Digilio MC, Conti E, Sarkozy A, Mingarelli R, Dottorini T, Marino B, Pizzuti A, Dallapiccola B, (2002). Grouping of multiple-lentigines/LEOPARD and Noonan syndromes on the PTPN11 gene. Am. J. Hum. Genet 71, 389–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Legius E, Schrander-Stumpel C, Schollen E, Pulles-Heintzberger C, Gewillig M, Fryns JP, (2002). PTPN11 mutations in LEOPARD syndrome. J. Med. Genet 39, 571–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen YN, LaMarche MJ, Chan HM, Fekkes P, Garcia-Fortanet J, Acker MG, Antonakos B, Chen CH, et al. , (2016). Allosteric inhibition of SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nature 535, 148–152. [DOI] [PubMed] [Google Scholar]
- 11.Fedele C, Li S, Teng KW, Foster CJR, Peng D, Ran H, Mita P, Geer MJ, et al. , (2021). SHP2 inhibition diminishes KRASG12C cycling and promotes tumor microenvironment remodeling. J. Exp. Med 218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang Y, Mohseni M, Grauel A, Diez JE, Guan W, Liang S, Choi JE, Pu M, et al. , (2021). SHP2 blockade enhances anti-tumor immunity via tumor cell intrinsic and extrinsic mechanisms. Sci. Rep 11, 1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bagdanoff JT, Chen Z, Acker M, Chen YN, Chan H, Dore M, Firestone B, Fodor M, et al. , (2019). Optimization of Fused Bicyclic Allosteric SHP2 Inhibitors. J. Med. Chem 62, 1781–1792. [DOI] [PubMed] [Google Scholar]
- 14.Sarver P, Acker M, Bagdanoff JT, Chen Z, Chen YN, Chan H, Firestone B, Fodor M, et al. , (2019). 6-Amino-3-methylpyrimidinones as Potent, Selective, and Orally Efficacious SHP2 Inhibitors. J. Med. Chem 62, 1793–1802. [DOI] [PubMed] [Google Scholar]
- 15.Nichols RJ, Haderk F, Stahlhut C, Schulze CJ, Hemmati G, Wildes D, Tzitzilonis C, Mordec K, et al. , (2018). RAS nucleotide cycling underlies the SHP2 phosphatase dependence of mutant BRAF-, NF1- and RAS-driven cancers. Nat. Cell Biol 20, 1064–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ruess DA, Heynen GJ, Ciecielski KJ, Ai J, Berninger A, Kabacaoglu D, Gorgulu K, Dantes Z, et al. , (2018). Mutant KRAS-driven cancers depend on PTPN11/SHP2 phosphatase. Nat. Med 24, 954–960. [DOI] [PubMed] [Google Scholar]
- 17.Mainardi S, Mulero-Sanchez A, Prahallad A, Germano G, Bosma A, Krimpenfort P, Lieftink C, Steinberg JD, et al. , (2018). SHP2 is required for growth of KRAS-mutant non-small-cell lung cancer in vivo. Nat. Med 24, 961–967. [DOI] [PubMed] [Google Scholar]
- 18.Machida K, Thompson CM, Dierck K, Jablonowski K, Karkkainen S, Liu B, Zhang H, Nash PD, et al. , (2007). High-throughput phosphotyrosine profiling using SH2 domains. Mol. Cell 26, 899–915. [DOI] [PubMed] [Google Scholar]
- 19.Sacco F, Perfetto L, Castagnoli L, Cesareni G, (2012). The human phosphatase interactome: An intricate family portrait. FEBS Lett 586, 2732–2739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Milarski KL, Saltiel AR, (1994). Expression of catalytically inactive Syp phosphatase in 3T3 cells blocks stimulation of mitogen-activated protein kinase by insulin. J. Biol. Chem 269, 21239–21243. [PubMed] [Google Scholar]
- 21.Noguchi T, Matozaki T, Horita K, Fujioka Y, Kasuga M, (1994). Role of SH-PTP2, a protein-tyrosine phosphatase with Src homology 2 domains, in insulin-stimulated Ras activation. Mol. Cell Biol 14, 6674–6682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tartaglia M, Martinelli S, Stella L, Bocchinfuso G, Flex E, Cordeddu V, Zampino G, Burgt I, et al. , (2006). Diversity and functional consequences of germline and somatic PTPN11 mutations in human disease. Am. J. Hum. Genet 78, 279–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chen L, Sung SS, Yip ML, Lawrence HR, Ren Y, Guida WC, Sebti SM, Lawrence NJ, et al. , (2006). Discovery of a novel shp2 protein tyrosine phosphatase inhibitor. Mol. Pharmacol 70, 562–570. [DOI] [PubMed] [Google Scholar]
- 24.Hellmuth K, Grosskopf S, Lum CT, Wurtele M, Roder N, von Kries JP, Rosario M, Rademann J, et al. , (2008). Specific inhibitors of the protein tyrosine phosphatase Shp2 identified by high-throughput docking. PNAS 105, 7275–7280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu W, Yu B, Xu G, Xu WR, Loh ML, Tang LD, Qu CK, (2013). Identification of cryptotanshinone as an inhibitor of oncogenic protein tyrosine phosphatase SHP2 (PTPN11). J. Med. Chem 56, 7212–7221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lawrence HR, Pireddu R, Chen L, Luo Y, Sung SS, Szymanski AM, Yip ML, Guida WC, et al. , (2008). Inhibitors of Src homology-2 domain containing protein tyrosine phosphatase-2 (Shp2) based on oxindole scaffolds. J. Med. Chem 51, 4948–4956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hantschel O, Biancalana M, Koide S, (2020). Monobodies as enabling tools for structural and mechanistic biology. Curr. Opin. Struct. Biol 60, 167–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Akkapeddi P, Teng KW, Koide S, (2021). Monobodies as tool biologics for accelerating target validation and druggable site discovery. RSC Med. Chem 12, 1839–1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koide A, Bailey CW, Huang X, Koide S, (1998). The fibronectin type III domain as a scaffold for novel binding proteins. J. Mol. Biol 284, 1141–1151. [DOI] [PubMed] [Google Scholar]
- 30.Sha F, Salzman G, Gupta A, Koide S, (2017). Monobodies and other synthetic binding proteins for expanding protein science. Protein Sci 26, 910–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sha F, Gencer EB, Georgeon S, Koide A, Yasui N, Koide S, Hantschel O, (2013). Dissection of the BCR-ABL signaling network using highly specific monobody inhibitors to the SHP2 SH2 domains. PNAS 110, 14924–14929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Teng KW, Tsai ST, Hattori T, Fedele C, Koide A, Yang C, Hou X, Zhang Y, et al. , (2021). Selective and noncovalent targeting of RAS mutants for inhibition and degradation. Nat. Commun 12, 2656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hof P, Pluskey S, Dhe-Paganon S, Eck MJ, Shoelson SE, (1998). Crystal structure of the tyrosine phosphatase SHP-2. Cell 92, 441–450. [DOI] [PubMed] [Google Scholar]
- 34.Padua RAP, Sun Y, Marko I, Pitsawong W, Stiller JB, Otten R, Kern D, (2018). Mechanism of activating mutations and allosteric drug inhibition of the phosphatase SHP2. Nat. Commun 9, 4507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vogel W, Lammers R, Huang J, Ullrich A, (1993). Activation of a phosphotyrosine phosphatase by tyrosine phosphorylation. Science 259, 1611–1614. [DOI] [PubMed] [Google Scholar]
- 36.Koide A, Wojcik J, Gilbreth RN, Hoey RJ, Koide S, (2012). Teaching an Old Scaffold New Tricks: Monobodies Constructed Using Alternative Surfaces of the FN3 Scaffold. J. Mol. Biol 415, 393–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Clackson T, Ultsch MH, Wells JA, de Vos AM, (1998). Structural and functional analysis of the 1:1 growth hormone:receptor complex reveals the molecular basis for receptor affinity. J. Mol. Biol 277, 1111–1128. [DOI] [PubMed] [Google Scholar]
- 38.Horn JR, Sosnick TR, Kossiakoff AA, (2009). Principal determinants leading to transition state formation of a protein-protein complex, orientation trumps side-chain interactions. PNAS 106, 2559–2564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wojcik J, Hantschel O, Grebien F, Kaupe I, Bennett KL, Barkinge J, Jones RB, Koide A, et al. , (2010). A potent and highly specific FN3 monobody inhibitor of the Abl SH2 domain. Nat. Struct. Mol. Biol 17, 519–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Tereshko V, Uysal S, Koide A, Margalef K, Koide S, Kossiakoff AA, (2008). Toward chaperone-assisted crystallography: protein engineering enhancement of crystal packing and X-ray phasing capabilities of a camelid single-domain antibody (VHH) scaffold. Protein Sci 17, 1175–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gilbreth RN, Esaki K, Koide A, Sidhu SS, Koide S, (2008). A dominant conformational role for amino acid diversity in minimalist protein-protein interfaces. J. Mol. Biol 381, 407–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Machado L, Critton DA, Page R, Peti W, (2017). Redox Regulation of a Gain-of-Function Mutation (N308D) in SHP2 Noonan Syndrome. ACS Omega 2, 8313–8318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen CY, Willard D, Rudolph J, (2009). Redox regulation of SH2-domain-containing protein tyrosine phosphatases by two backdoor cysteines. Biochemistry 48, 1399–1409. [DOI] [PubMed] [Google Scholar]
- 44.Haque A, Andersen JN, Salmeen A, Barford D, Tonks NK, (2011). Conformation-sensing antibodies stabilize the oxidized form of PTP1B and inhibit its phosphatase activity. Cell 147, 185–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yu ZH, Xu J, Walls CD, Chen L, Zhang S, Zhang R, Wu L, Wang L, et al. , (2013). Structural and mechanistic insights into LEOPARD syndrome-associated SHP2 mutations. J. Biol. Chem 288, 10472–10482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Keilhack H, David FS, McGregor M, Cantley LC, Neel BG, (2005). Diverse biochemical properties of Shp2 mutants. Implications for disease phenotypes. J. Biol. Chem 280, 30984–30993. [DOI] [PubMed] [Google Scholar]
- 47.Sugimoto S, Wandless TJ, Shoelson SE, Neel BG, Walsh CT, (1994). Activation of the SH2-containing protein tyrosine phosphatase, SH-PTP2, by phosphotyrosine-containing peptides derived from insulin receptor substrate-1. J. Biol. Chem 269, 13614–13622. [PubMed] [Google Scholar]
- 48.Darian E, Guvench O, Yu B, Qu CK, MacKerell AD Jr., (2011). Structural mechanism associated with domain opening in gain-of-function mutations in SHP2 phosphatase. Proteins 79, 1573–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.LaRochelle JR, Fodor M, Vemulapalli V, Mohseni M, Wang P, Stams T, LaMarche MJ, Chopra R, et al. , (2018). Structural reorganization of SHP2 by oncogenic mutations and implications for oncoprotein resistance to allosteric inhibition. Nat. Commun 9, 4508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hattori T, Koide A, Panchenko T, Romero LA, Teng KW, Corrado AD, Koide S, (2020). Multiplex bead binding assays using off-the-shelf components and common flow cytometers. J. Immunol. Methods 490, 112952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Nishikori S, Hattori T, Fuchs SM, Yasui N, Wojcik J, Koide A, Strahl BD, Koide S, (2012). Broad ranges of affinity and specificity of anti-histone antibodies revealed by a quantitative Peptide immunoprecipitation assay. J. Mol. Biol 424, 391–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Johnston PA, (2011). Redox cycling compounds generate H2O2 in HTS buffers containing strong reducing reagents–real hits or promiscuous artifacts? Curr. Opin. Chem. Biol 15, 174–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Fulcher LJ, Hutchinson LD, Macartney TJ, Turnbull C, Sapkota GP, (2017). Targeting endogenous proteins for degradation through the affinity-directed protein missile system. Open Biol 7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Roth S, Macartney TJ, Konopacka A, Chan KH, Zhou H, Queisser MA, Sapkota GP, (2020). Targeting Endogenous K-RAS for Degradation through the Affinity-Directed Protein Missile System. Cell Chem. Biol 27 1151–1163 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lim S, Khoo R, Juang YC, Gopal P, Zhang H, Yeo C, Peh KM, Teo J, et al. , (2021). Exquisitely Specific anti-KRAS Biodegraders Inform on the Cellular Prevalence of Nucleotide-Loaded States. ACS Cent. Sci 7, 274–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ludwicki MB, Li J, Stephens EA, Roberts RW, Koide S, Hammond PT, DeLisa MP, (2019). Broad-Spectrum Proteome Editing with an Engineered Bacterial Ubiquitin Ligase Mimic. ACS Cent. Sci 5, 852–866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Teng KW, Koide A, Koide S, (2022). Engineering Binders with Exceptional Selectivity. Methods Mol. Biol 2491, 143–154. [DOI] [PubMed] [Google Scholar]
- 58.Kabsch W, (2010). Xds. Acta Crystallogr. D Biol. Crystallogr 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Adams PD, Afonine PV, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, et al. , (2010). PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Emsley P, Cowtan K, (2004). Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr 60, 2126–2132. [DOI] [PubMed] [Google Scholar]
- 61.Chen VB, Arendall WB 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS, et al. , (2010). MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr 66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lawrence MC, Colman PM, (1993). Shape complementarity at protein/protein interfaces. J. Mol. Biol 234, 946–950. [DOI] [PubMed] [Google Scholar]
- 63.Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, et al. , (2011). Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr 67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Krissinel E, Henrick K, (2007). Inference of macromolecular assemblies from crystalline state. J. Mol. Biol 372, 774–797. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.