

Abstract
Measurable residual disease (MRD) is a powerful prognostic factor in acute myeloid leukemia (AML). However, pre-treatment molecular predictors of immunophenotypic MRD clearance remain unclear. We analyzed a dataset of 211 patients with pre-treatment next-generation sequencing who received induction chemotherapy and had MRD assessed by serial immunophenotypic monitoring after induction, subsequent therapy, and allogeneic stem cell transplant (allo-SCT). Induction chemotherapy led to MRD− remission, MRD+ remission, and persistent disease in 35%, 27%, and 38% of patients, respectively. With subsequent therapy, 34% of patients with MRD+ and 26% of patients with persistent disease converted to MRD−. Mutations in CEBPA, NRAS, KRAS, and NPM1 predicted high rates of MRD−, while mutations in TP53, SF3B1, ASXL1 and RUNX1 and karyotypic abnormalities including inv(3), monosomy 5 or 7 predicted low rates of MRD−. Patients with fewer individual clones were more likely to achieve MRD− remission. Among 132 patients who underwent allo-SCT, outcomes were favorable whether patients achieved early MRD− after induction or later MRD− after subsequent therapy prior to allo-SCT. As MRD conversion with chemotherapy prior to allo-SCT is rarely achieved in patients with specific baseline mutational patterns and high clone numbers, upfront inclusion of these patients into clinical trials should be considered.
Keywords: acute myeloid leukemia, measurable residual disease, minimal residual disease, MRD, flow cytometry, genetic, bone marrow transplant
Introduction:
Although intensive induction chemotherapy for acute myeloid leukemia (AML) leads to remission in the majority of patients, relapse rates remain high without subsequent consolidative chemotherapy or allogeneic stem cell transplant (allo-SCT).1, 2 Relapse after achieving a morphologic remission can be attributed to a failure to eradicate residual leukemic cells.3 Measurable residual disease (MRD) describes the presence of residual leukemic cells which are not detectable by means of conventional morphologic assessments.4 MRD assessment by flow cytometry and/or molecular genetic studies has been shown to be a powerful prognostic factor in AML, including in the prediction of relapse and survival following allo-SCT.5–11
While AML MRD is highly prognostic for future outcomes, the efficacy of current therapies in clearing immunophenotypic MRD in specific AML molecular subsets is not well characterized. One recent study explored the impact of diagnostic genetics in persistence of post-treatment molecular MRD.12 However, no studies have comprehensively examined the role of pre-treatment molecular mutations on post-treatment immunophenotypic MRD. Multiparametric flow cytometry (MFC)-MRD testing remains the most common MRD detection methodology in AML.11 Despite the increasing use of molecular testing across multiple AML treatment timepoints, many AML patients do not have mutations amenable to molecular MRD evaluation. It remains unknown whether pre-treatment mutational patterns, detected by either bulk or single cell sequencing, can predict the success of immunophenotypic MRD clearance.
The finding of persistent MRD following induction chemotherapy currently presents the clinician with a dilemma. It is unclear whether MRD can be eradicated by additional therapy prior to allo-SCT, or if further pre-transplant therapy would likely be futile with current approaches. Identification of such predictive biomarkers of MRD clearance would enable more individualized treatment selection and allow the upfront inclusion of patients with low likelihood of MRD clearance with standard induction therapy for in clinical trials. The objective of this study was to identify pre-induction genetic predictors of MRD clearance following 7+3 induction chemotherapy (7+3) and prior to allo-SCT. We retrospectively analyzed 211 patients who received 7+3 with serial high sensitivity flow cytometric immunophenotypic MRD monitoring. We also performed single cell sequencing in a subset of patients. Our data show that the presence of specific mutations, cytogenetic features, and low clone number at diagnosis predicts MRD clearance with chemotherapy.
Methods:
Data source and patient eligibility:
Data were retrospectively collected at Memorial Sloan Kettering Cancer Center (MSKCC). All patients with AML per 2016 World Health Organization classification,13 who received induction chemotherapy with a 7-day continuous infusion of cytarabine and 3 days of an anthracycline for newly diagnosed AML from 04/2014 to 03/2020, were included in the initial patient population (Supplemental Figure 1). Of 232 patients who received 7+3, 211 patients with pre-treatment next generation sequencing were included in the final analysis after exclusion of patients for the following reasons: death during induction therapy prior to response assessment (N=9), absence of MRD assessment by flow cytometry (N=8), receipt of Iomab-B as investigational pre-allo-SCT conditioning therapy (N=3), and no response assessment prior to allo-SCT (N=1). The study was approved by the MSKCC institutional review board.
Response criteria and MRD assessment:
Response to therapy was assessed using the 2017 European LeukemiaNet (ELN) response criteria modified by the MRD status.14 MRD was assessed by MFC analysis of bone marrow aspirate (BMA) samples as described previously (for details see Supplemental Methods).15 Any level of residual disease was considered MRD positive (MRD+). MRD− remission was defined as complete remission (CR), CR with incomplete count recovery (CRi), or morphologic leukemia-free state (MLFS) with no evidence of MRD by flow cytometry (CR/CRi/MLFS MRD−). Response was determined at three clinically relevant timepoints: (i) best response after completion of 7+3 prior to any other therapy, (ii) temporally closest response prior to allo-SCT and (iii) best response after allo-SCT before any further line of therapy (Supplemental Figure 1A). Overall survival (OS) was calculated from cycle 1 day 1 of 7+3 until death or time of last follow-up. OS after allo-SCT was calculated from the day of cell infusion until death or time of last follow-up.
Cytogenetic and molecular data:
Prior to 7+3 induction therapy, cytogenetic and mutational analyses were performed on BMA or peripheral blood (Supplemental Figure 1A). If both were available, data from BMA were used preferentially (for details see Supplemental Methods). 16, 17
Single-cell sequencing
Bone marrow (n=9 patients) or peripheral blood samples (n=2) from a selected subset of 11 patients with different rates and kinetics of MRD clearance over time were subjected to single-cell DNA sequencing using previously described processing and analysis methods (Supplemental Methods).18
Statistical analysis:
Categorical patient characteristics were summarized by frequency. Associations between patient characteristics, allo-SCT characteristics and responses after induction and prior to allo-SCT were evaluated by Fisher’s exact test. T-test with unequal variances was used to compare continuous single-cell features between groups of response. Associations between cytogenetic abnormalities, molecular predictors and the response after induction chemotherapy were tested by Fisher’s exact test and odds ratios with 95% confidence intervals (CIs) were estimated by fitting logistic regression.
To explore these associations in multivariable model, a forward stepwise logistic regression with Bayesian Information Criterion (BIC) selection was applied to the data.
Kaplan-Meier survival analysis was used to estimate time to event outcomes such as OS, time to MRD clearance, loss of MRD− and relapse.
Patients who died or received allo-SCT before achieving remission were censored at the date of earliest event. Among patients achieving CR/CRi/MLFS MRD− response, time to loss of MRD− was defined as time from disease assessment until morphologic relapse, MRD+ or death; patients without any of these events were censored on date of last follow-up. Similarly, among patients achieving a CR/CRi/MLFS response, time to loss of response was defined as time from disease assessment until morphologic relapse, or death; patients without any of these events were censored on date of last follow-up. Estimated median follow-up was calculated using the reverse Kaplan-Meier method. Associations with clinical predictors are evaluated by log-rank test and effects were estimated using univariate Cox proportional hazard model and presented as hazard ratios with 95% confidence intervals. Statistical significance was defined using a two-sided significance level at 0.05. All statistical analyses were using R version 3.6.1.
Results:
Patient and treatment characteristics:
Patient and treatment characteristics for all 211 patients are detailed in Supplemental Table 4. Transplant characteristics of patients who received a subsequent allo-SCT are shown in Supplemental Table 5. Patients were categorized by their MRD response after 7+3 and prior to allo-SCT, respectively. There were 136 (64%) patients with de novo AML and 75 (36%) patients with secondary AML (defined as therapy-related AML, AML with myelodysplasia-related changes [AML-MRC], and AML arising from a prior myeloproliferative neoplasm [MPN])13 (Supplemental Table 4). After completion of 7+3, a total of 69 (33%) patients received consolidation therapy (45 patients proceeded to allo-SCT after consolidation), whereas 95 (45%) patients required intensive re-induction for persistent disease or salvage therapy for relapsed disease (51 patients proceeded to allo-SCT); 36 (17%) patients proceeded directly to allo-SCT. Supplemental Figure 1B provides an overview of patient selection and treatment allocation.
A total of 132 patients (62%) underwent allo-SCT (Supplemental Table 5). Patients received allo-SCT from a matched related, matched unrelated, or alternative donor in 25%, 52%, and 23% of cases, respectively. A plurality of patients received an unmodified peripheral blood stem cell (PBSC) transplant (40%), whereas 34%, 11% and 15% received a CD34 selected PBSC, a bone marrow or a double umbilical cord blood stem cell transplant. Conditioning intensity was myeloablative, reduced intensity (RIC), and non-myeloablative in 56%, 43% and 1% of patients, respectively.
MRD is cleared both early after induction chemotherapy and later with additional antileukemic therapy prior to allo-SCT:
Induction chemotherapy resulted in MRD− remission, MRD+ remission, and persistent disease in 73 (35%), 58 (27%), and 80 (38%) of all patients, respectively (Supplemental Figure 1B+C). We first evaluated baseline disease and patient characteristics associated with MRD clearance (Supplementary Table 5). Compared to patients with MRD+ remission or persistent disease, patients who achieved an MRD− remission with 7+3 induction chemotherapy were more likely to be younger than 60 years old (MRD−: 68%; MRD+: 34%, p=0.001; persistent disease: 45%, p=0.03), to have de novo AML (MRD− 78%; MRD+: 59%, p=0.02; persistent disease: 56%, p=0.006) and to have ELN favorable disease (MRD−: 49%; MRD−: 21%, p=0.002; persistent disease: 4%, p<0.001) 14.
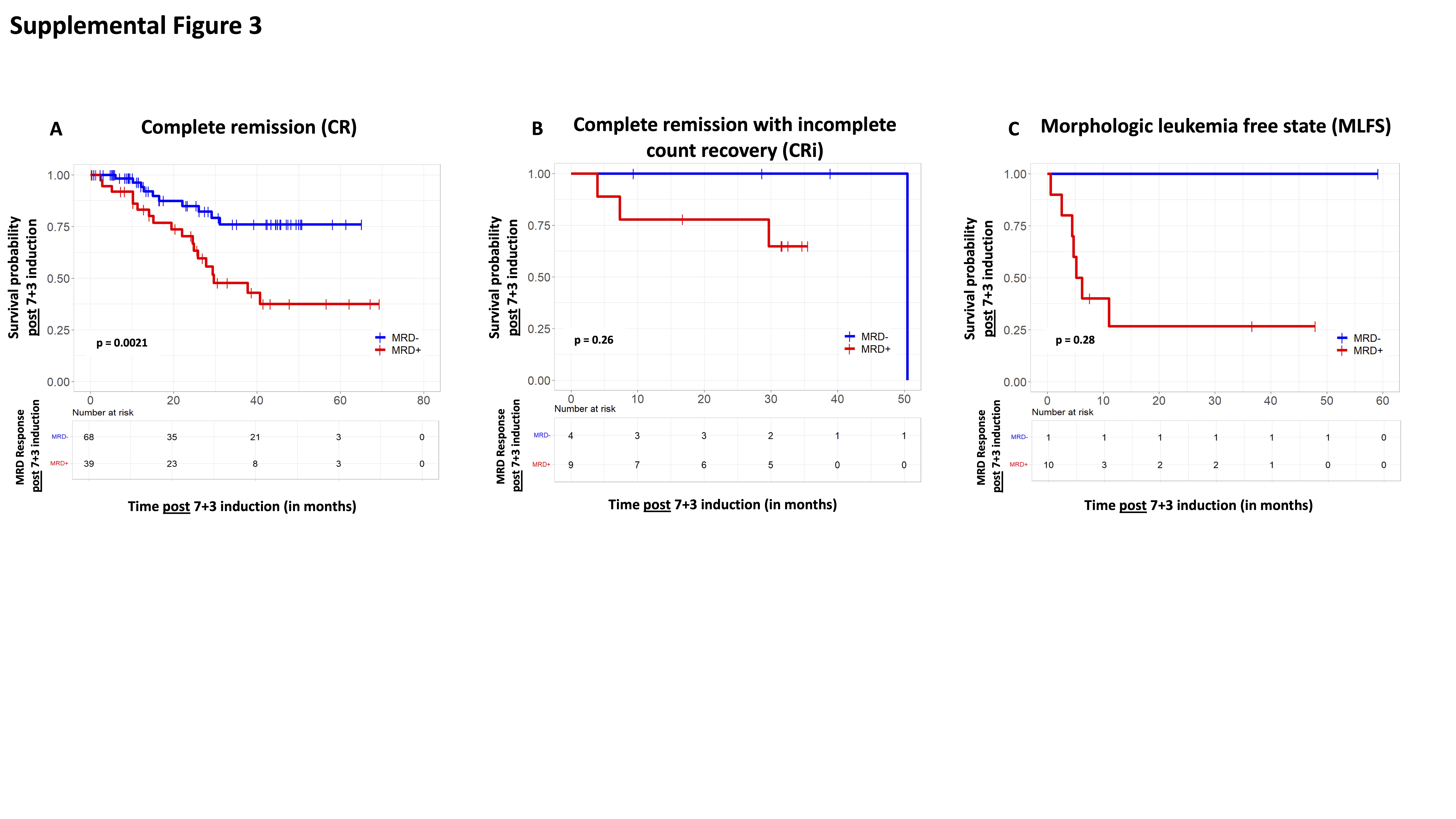
We analyzed whether MRD response after induction chemotherapy added prognostic significance beyond baseline ELN 2017 molecular risk on overall survival (OS) after induction chemotherapy (Supplemental Figure 2). We observed that MFC-MRD status after induction chemotherapy added prognostic significance beyond baseline ELN 2017 molecular risk profile for ELN favorable risk (p< 0.001; Supplemental Figure 2A). While there was a trend towards improved OS for patients with ELN adverse risk who achieved MRD− after induction chemotherapy, it was not statistically significant (p=0.16; Supplemental Figure 2C). In patients with ELN intermediate risk, achievement of MRD− with induction chemotherapy did not seem to improve survival (p=0.77; Supplemental Figure 2B). We also analyzed the prognostic impact of MRD for patients who have achieved a morphological response of CR, CRi and MLFS separately. We found a clear statistically significant prognostic impact of MRD for patients who achieved CR after induction chemotherapy (p=0.0021). While we observed the same trend for the prognostic impact of MRD in the setting of CRi and MLFS, it did not reach statistical significance, possibly due to smaller numbers of patients with these responses (p=0.26 and p=0.28) (Supplemental Figure 3).
The subsequent clinical course and clinical outcomes of all patients based on MRD response after 7+3 is summarized in Supplemental Figure 1A+B and Supplemental Figure 4. Of 73 patients with MRD− remission post induction chemotherapy, 21 patients proceeded to allo-SCT directly without further therapy, whereas 52 patients received further therapy with 27 of these patients ultimately receiving an allo-SCT (Supplemental Figure 1B). Of 58 patients with MRD+ remission after induction chemotherapy, 14 patients proceeded directly to allo-SCT, whereas 44 patients received further therapy and 28 of these subsequently proceeded to allo-SCT. Of 80 patients with persistent disease after induction chemotherapy, 2 patients directly underwent allo-SCT whereas 78 patients received further therapy and 40 of these subsequently proceeded to allo-SCT.
In the cohort of 132 patients who received allo-SCT, 45 (34%) patients achieved MRD− remission early on after completion of 7+3 induction chemotherapy, whereas 27 (21%) patients had either persistent disease or MRD+ remission post-induction but subsequently achieved MRD− remission prior to allo-SCT (Supplemental Figure 1B+D). Of the 27 patients who were converted to MRD− prior to allo-SCT, 14 (52%) and 13 (48%) were converted from MRD+ and persistent disease after induction chemotherapy. For patients converted from MRD+ to MRD− remission, conversion to MRD− was mainly achieved with consolidation therapy (75%) and less frequently with intensive reinduction (10%), non-intensive therapy (10%) and maintenance therapy (5%). In patients converted from persistent disease to MRD− remission, conversion to MRD− was achieved with intensive reinduction, consolidation, and non-intensive therapy in 60%, 25%, and 15% of patients, respectively. In contrast, 15 (11%) and 45 (34%) patients underwent allo-SCT with persistent AML or in MRD+ remission, respectively.
There were statistically significant differences in terms of graft and donor source between patients who underwent allo-SCT by MRD response status (Supplemental Table 5). Patients with persistent disease were more likely to have received an unmodified PBSC graft and to have had a matched unrelated donor than patients with MRD− or MRD+ remission. Conditioning regimen intensity was similar across disease status categories.
Rates of early MRD clearance after induction chemotherapy depend on pre-treatment cytogenetic and molecular characteristics:
We first asked which pre-treatment cytogenetic and genetic characteristics predict an early MRD− remission after 7+3. In the oncoprint shown in Figure 1 pre-treatment cytogenetic and molecular features are sorted based on the lowest to highest rate of MRD clearance after induction chemotherapy. Supplemental Table 6 shows MRD response rates and odds ratios for achieving MRD− remission after induction chemotherapy for each molecular feature.
Figure 1: Cytogenetic and genetic predictors of MRD− response after induction chemotherapy.
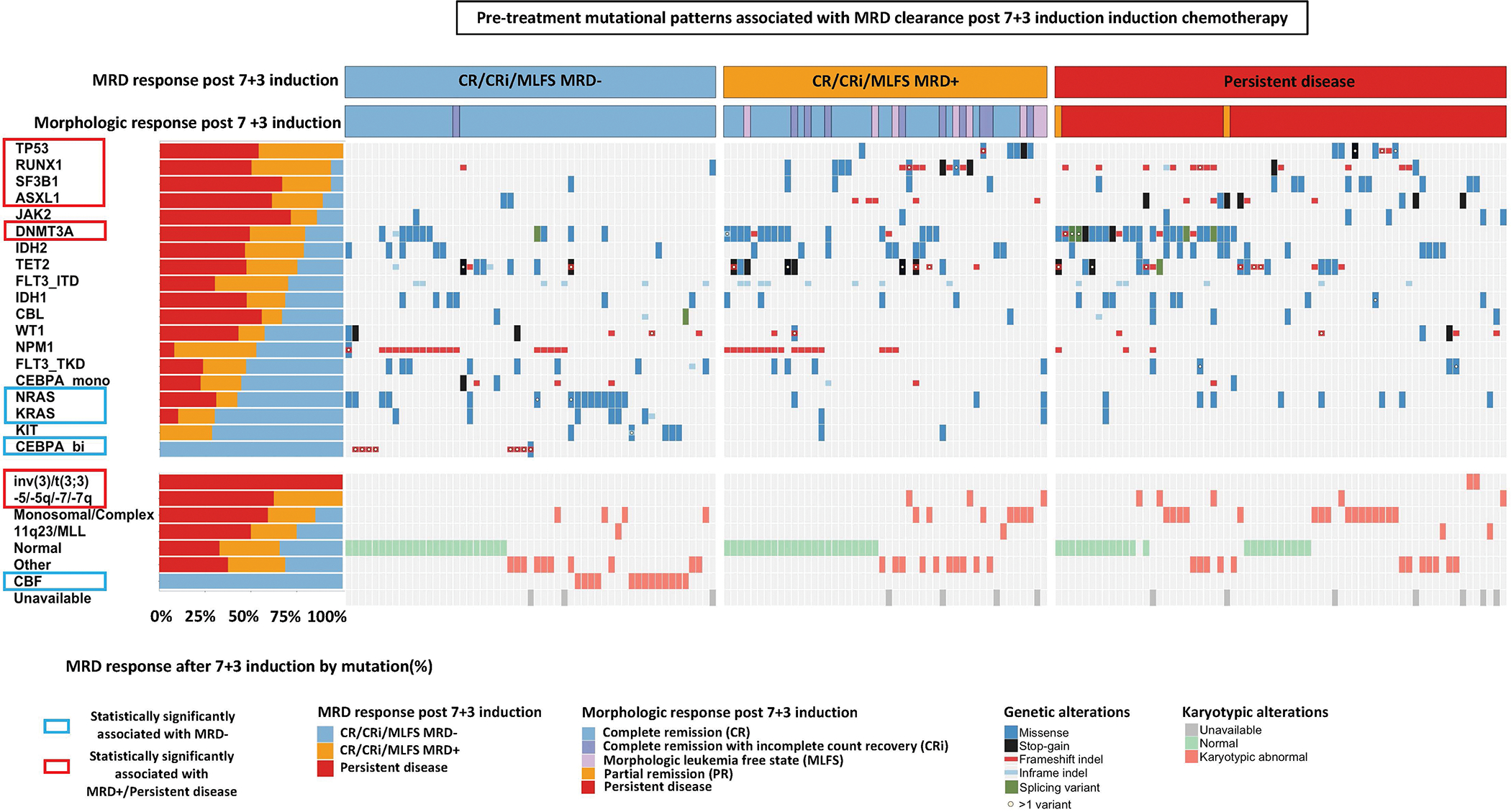
Univariate analyses of pre-treatment genetic predictors of achieving MRD− CR/CRi/MLFS after 7+3 induction chemotherapy. Each column and row denote an individual patient and gene/cytogenetic abnormality, respectively. MRD response are shown in the top rows. Genes and cytogenetic abnormalities are each are ordered by odds ratio (OR) of achieving an MRD− CR/CRi/MLFS from lowest to highest. Patients who had at least one mutation in the 19 most commonly mutated genes covered in all sequencing platforms are displayed.
Among cytogenetic abnormalities, we observed that all patients with core-binding factor (CBF) leukemia (i.e., t(8;21)(q22;q22) or inv(16)(p13q22)/t(16;16)(p13;q22)) achieved an MRD− remission with 7+3. Conversely, none of the patients with inv(3)/t(3:3), monosomy 5/del(5q), or monosomy 7/del(7q) achieved MRD− remission.
All patients with biallelic CEBPA mutations achieved MRD− remission (MRD− remission: 100%, OR for MRD− remission: NA; p=0.0001). In addition, mutations in NRAS (MRD− remission: 58%, OR for MRD− remission: 3.01 [95% CI: 1.29–6.97]; p=0.014) and KRAS (70%; OR: 4.78 [95% CI: 1.2–19.12]; p=0.034) predicted high rates of MRD− remission after 7+3. In contrast, none of the patients (0/13) with TP53 mutations achieved MRD− remission after induction chemotherapy (p=0.005). Furthermore, mutations in RUNX1 (13%, OR: 0.11 [95% CI: 0.03–0.47], p<0.001), SF3B1 (7%, OR: 0.12 [95% CI: 0.02–0.95], p=0.021), ASXL1 (11%; OR: 0.21 [95% CI: 0.05–0.95]; p=0.036) and DNMT3A (20%, OR 0.38 [95% CI: 0.18–0.80]; p=0.011) predicted low rates of MRD clearance after 7+3.
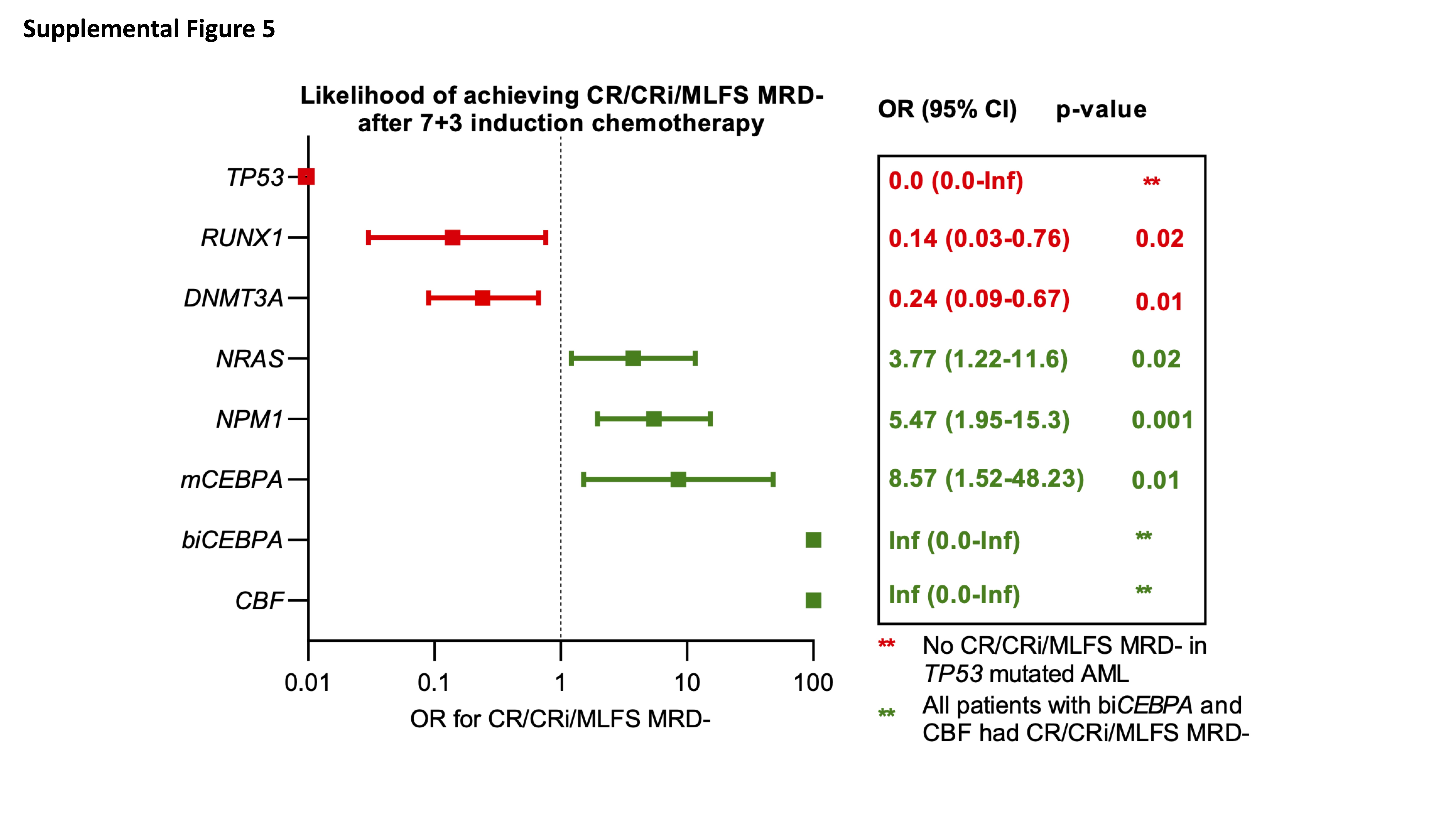
Stepwise logistic regression on a set of genes with a frequency of >5% and cytogenetic abnormalities as predictors of MRD clearance showed that CBF as well as mutations in CEBPA, NRAS, NPM1 were statistically significant predictors of MRD clearance with 7+3. Conversely, mutations in TP53, RUNX1 and DNMT3A remained statistically significant predictors of failure to clear MRD with 7+3 induction (Supplemental Figure 5).
Presence of mutations in TP53 and SF3B1 pre-treatment predict failure of MRD conversion prior to allo-SCT:
We next asked whether baseline molecular predictors were also able to predict a failure of MRD conversion prior to allo-SCT even if additional therapy was administered. In the oncoprint shown in Figure 2 cytogenetic and molecular features are sorted based on the lowest to highest MRD clearance rate prior to allo-SCT. Supplemental Table 7 shows MRD response rates and odds ratios for achieving MRD− remission prior to allo-SCT for each molecular feature.
Figure 2: Cytogenetic and genetic predictors of MRD− response pre allo-SCT.
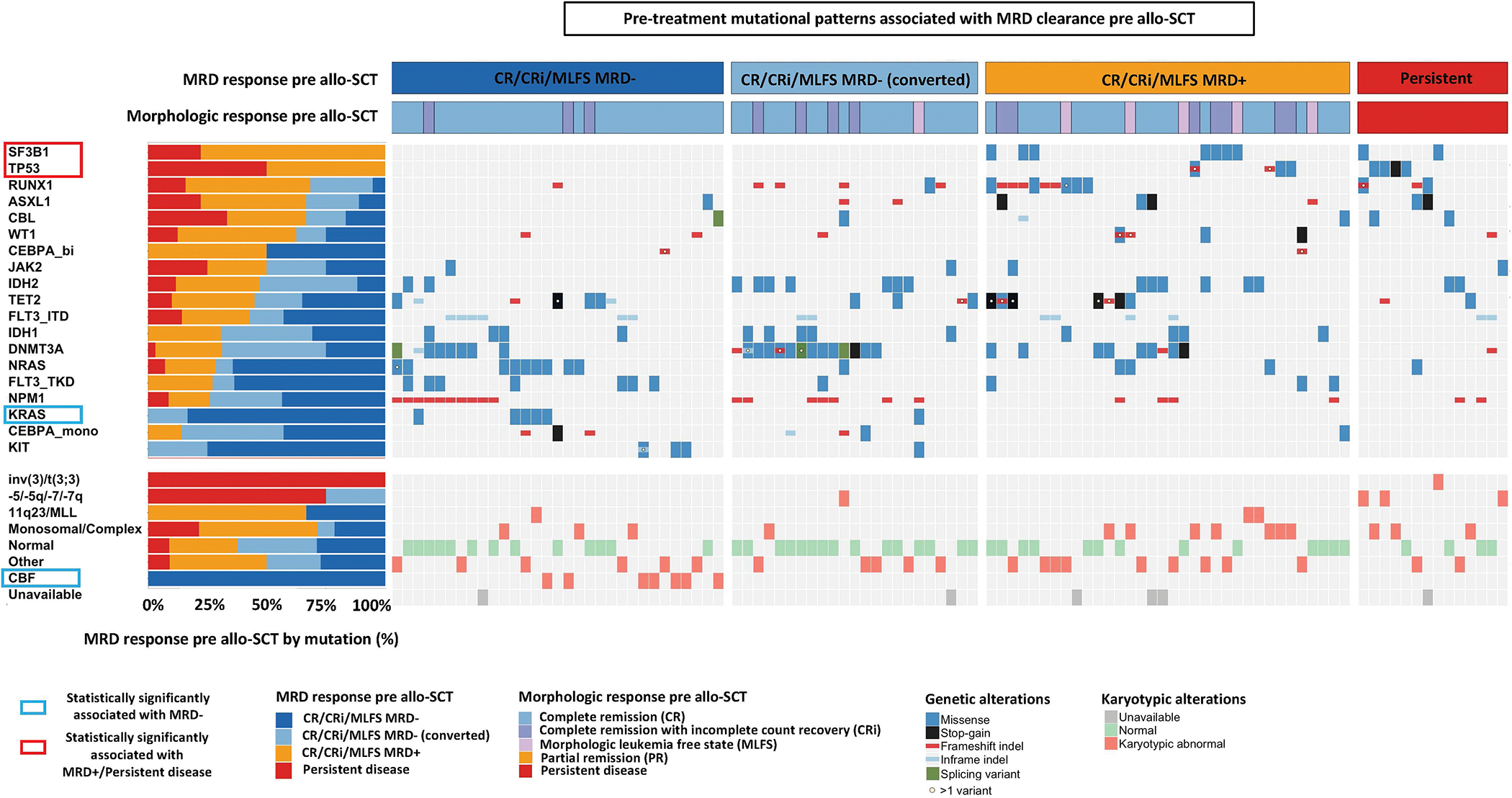
Univariate analyses of pre-treatment genetic predictors of achieving MRD− CR/CRi/MLFS prior to allo-SCT. Each column and row denote an individual patient and gene/cytogenetic abnormality, respectively. MRD response are shown in the top rows. Genes and cytogenetic abnormalities are each are ordered by odds ratio (OR) of achieving an MRD− CR/CRi/MLFS from lowest to highest. Patients who had at least one mutation in the 19 most commonly mutated genes covered in all sequencing platforms are displayed.
While all patients with CBF leukemia achieved MRD− remission prior to transplant, no patients with mutations in TP53 or SF3B1 achieved an MRD− remission prior to allo-SCT despite induction and post-induction therapy including consolidation and salvage therapy. In a multivariable model with stepwise logistic regression, mutations in KRAS remained statistically significantly associated with higher rates of MRD clearance rates prior to allo-SCT, while mutations in TP53 and SF3B1 remained statistically significant predictors of lower rates of MRD clearance rates prior to allo-SCT.
Clonal diversity prior to treatment impacts MRD clearance with induction chemotherapy:
Lastly, we studied whether clonal diversity as assessed on a single cell level could provide additional predictive information regarding MRD clearance after 7+3 and prior to allo-SCT. We selected a group of 11 patients based on differences in MRD clearance over time including patients with early and durable MRD clearance as well as patients who failed to clear MRD or initially cleared MRD but then quickly relapsed. We then used single cell sequencing to assess clonal diversity as estimated by the number of individual clones prior to starting 7+3 and tracked the patients MRD clearance pattern over time. Clones were defined as cells with identical protein-encoding single-nucleotide variants and a bootstrapping approach was applied to identify clones that included at least ten cells. Figure 3A shows a swimmer plot of these 11 patients.
Figure 3: Impact of clonal diversity on MRD response on the single cell level.
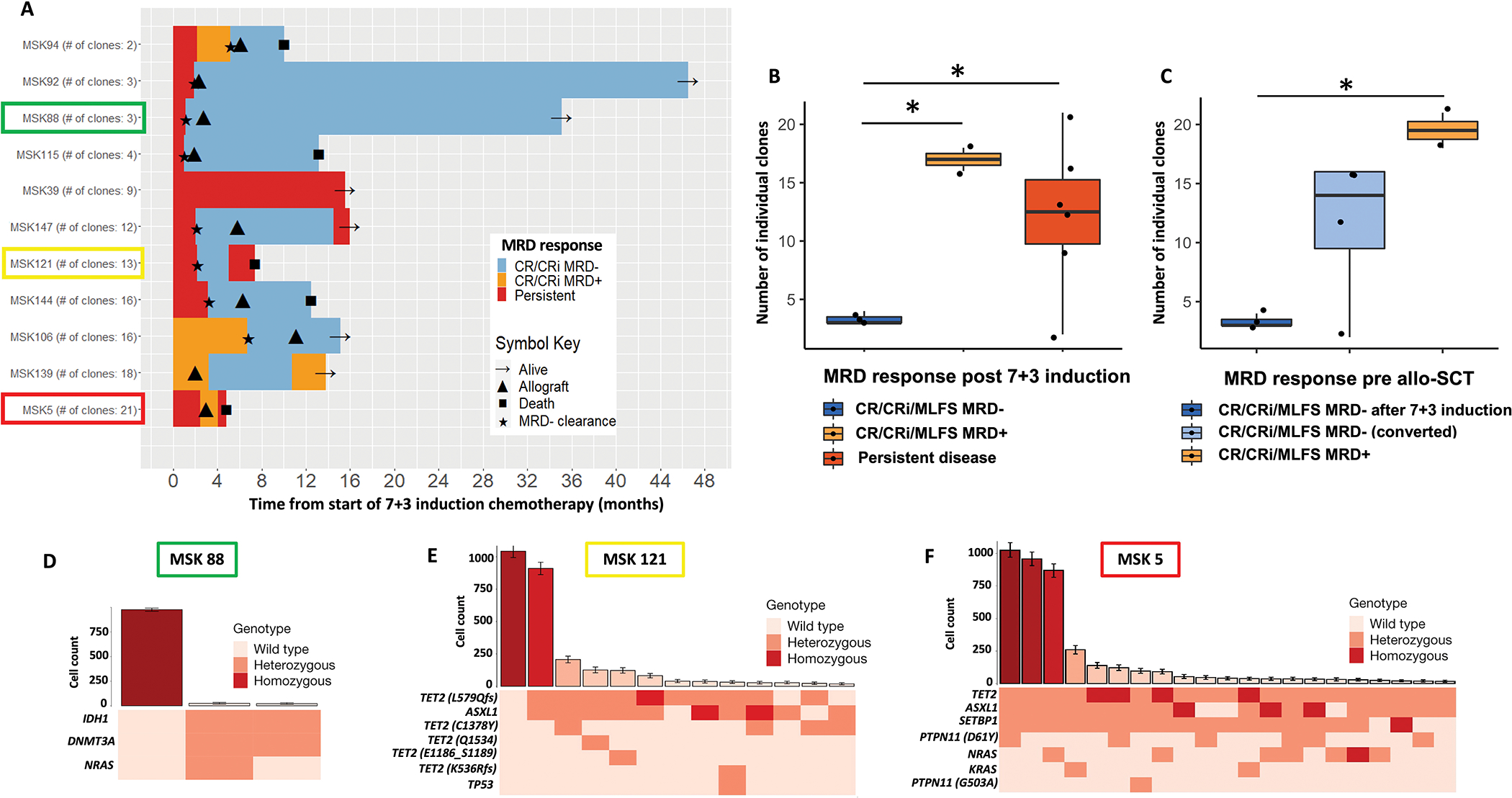
(A) Swimmer’s plot showing clonal diversity as estimated by the number of individual clones prior to starting 7+3 (as assessed by single cell analysis) and tracking MRD clearance with treatment over time. Patients are ordered by number of individual clones lowest to highest.
(B-C) Impact of clonal diversity as assessed by the number of individual clones at time of diagnosis on MRD clearance. (B) MRD clearance rates (based on pre-treatment number of individual clones) with induction chemotherapy including patients with MRD−, MRD+ and persistent disease after induction chemo. (C) MRD clearance rates (based on pre-treatment number of individual clones) prior to allo-SCT including patients with early MRD− after induction chemo, converted MRD− after additional therapy and MRD+ disease pre allo-SCT.
(E-F) Representative clonal repertoire for a patient with low clonal diversity (E: MSK 88) in contrast to two patients with high clonal diversity (F: MSK 5 and G: MSK121), respectively. The top panel shows the number of cells (mean ± 95% confidence intervals (CI)) identified with a given genotype and ranked by decreasing frequency. The bottom panel shows a heat map indicates mutation zygosity for each clone.
Patients who achieved MRD− remission after 7+3 had a lower number of clones (mean: 3.33; SD: 0.58) compared to patients with MRD+ remission (mean: 17.0; SD: 1.41; p = 0.03) or persistent disease (mean: 12.2; SD: 6.43; p = 0.02) (Figure 3B).
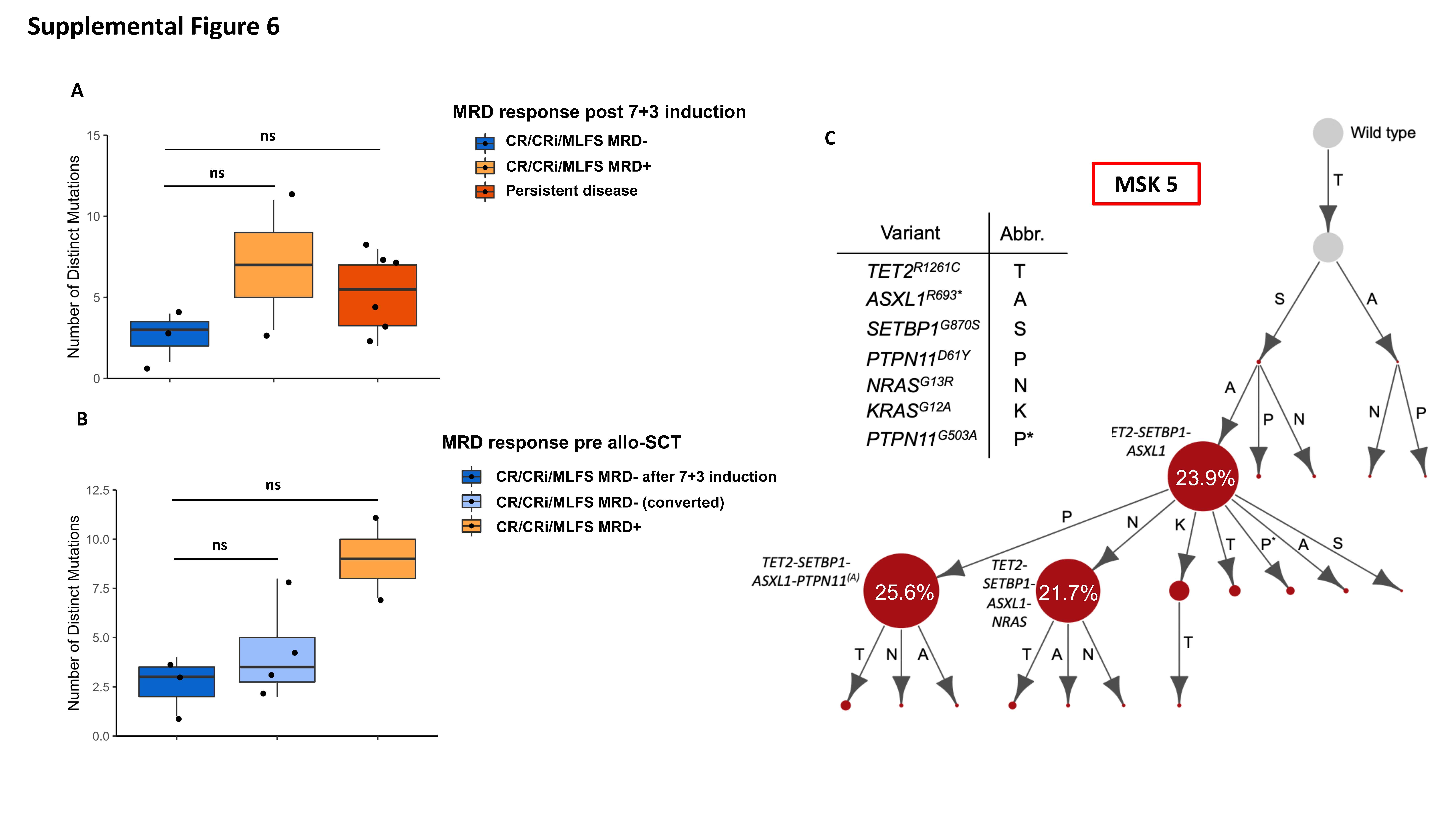
Similar associations were found between clonal diversity as assessed by number of clones at time of diagnosis and MRD clearance prior to allo-SCT. Patients who achieved MRD− remission early after 7+3 had a significantly lower number of clones (mean: 3.33; SD: 0.58 vs. mean: 19.5; SD: 2.12; p = 0.05) compared to patients with residual MRD prior to allo-SCT (Figure 3C). Importantly, the number of mutations at diagnosis was not statistically significantly different between patients who cleared MRD with 7+3 compared to patients with MRD+ or persistent disease (Supplemental Figure 6A). Similarly, the number of mutations was not statistically significantly different between patients with MRD− and MRD+/persistent disease prior to allo-SCT (Supplemental Figure 6B).
The clonal repertoires of three representative cases including one patient with lower clonal diversity (MSK 88) who cleared MRD, another patient (MSK 121) who initially cleared MRD but eventually relapsed, and one patient with higher clonal diversity (MSK 5) who never cleared MRD are shown in Figure 3D–F. MSK 88 who achieved MRD− immediately after 7+3 had fewer and mutationally less complex clones (Figure 3D) compared to MSK 121 (Figure 3E) and MSK 5 (Figure 3F).
Lastly, we used a Markov decision process with reinforcement learning to generate evolutionary trajectories to delineate the sequence of somatic genetic events during myeloid transformation. Patient MSK 5 demonstrated complex evolutionary trajectories, with progressive clonal dominance and subsequent subclonal propagation (Supplemental Figure 6C).
Allo-SCT leads to high rates of MRD clearance, however the durability of MRD− remission after allo-SCT is dependent on MRD status prior to allo-SCT:
We observed high rates of MRD− remission following allo-SCT (111 [84%] patients) with only 2% and 10% of patients showing persistent disease or MRD+ remission at time of first disease status assessment after allo-SCT, respectively. By the first post-transplant BMA at a median of 33 days, 35 out of 46 (76%) and 8 out of 15 patients (53%) who entered transplant with MRD+ remission or persistent AML, respectively, had cleared MRD (Figure 4A). The median duration of MRD− remission after allo-SCT was not reached for patients with MRD− remission after 7+3 or who converted to MRD− remission prior to allo-SCT (not statistically different from MRD− remission after 7+3 induction, HR: 1.40; 95% CI: 0.44–4.42; p=0.57). Despite high rates of initial MRD clearance with allo-SCT, the durability of MRD− after allo-SCT was significantly shorter for patients with MRD+ (median duration of MRD−: 35.5 months; HR: 2.70; 95% CI:1.09–6.63; p=0.03) and persistent disease (median duration of MRD−: 8.1 months; HR: 7.88; 95% CI: 2.67–23.06; p<0.001) compared to patients with MRD− remission after 7+3 induction (Figure 4B).
Figure 4: Outcomes post allo-SCT based on the MRD response pre allo-SCT:
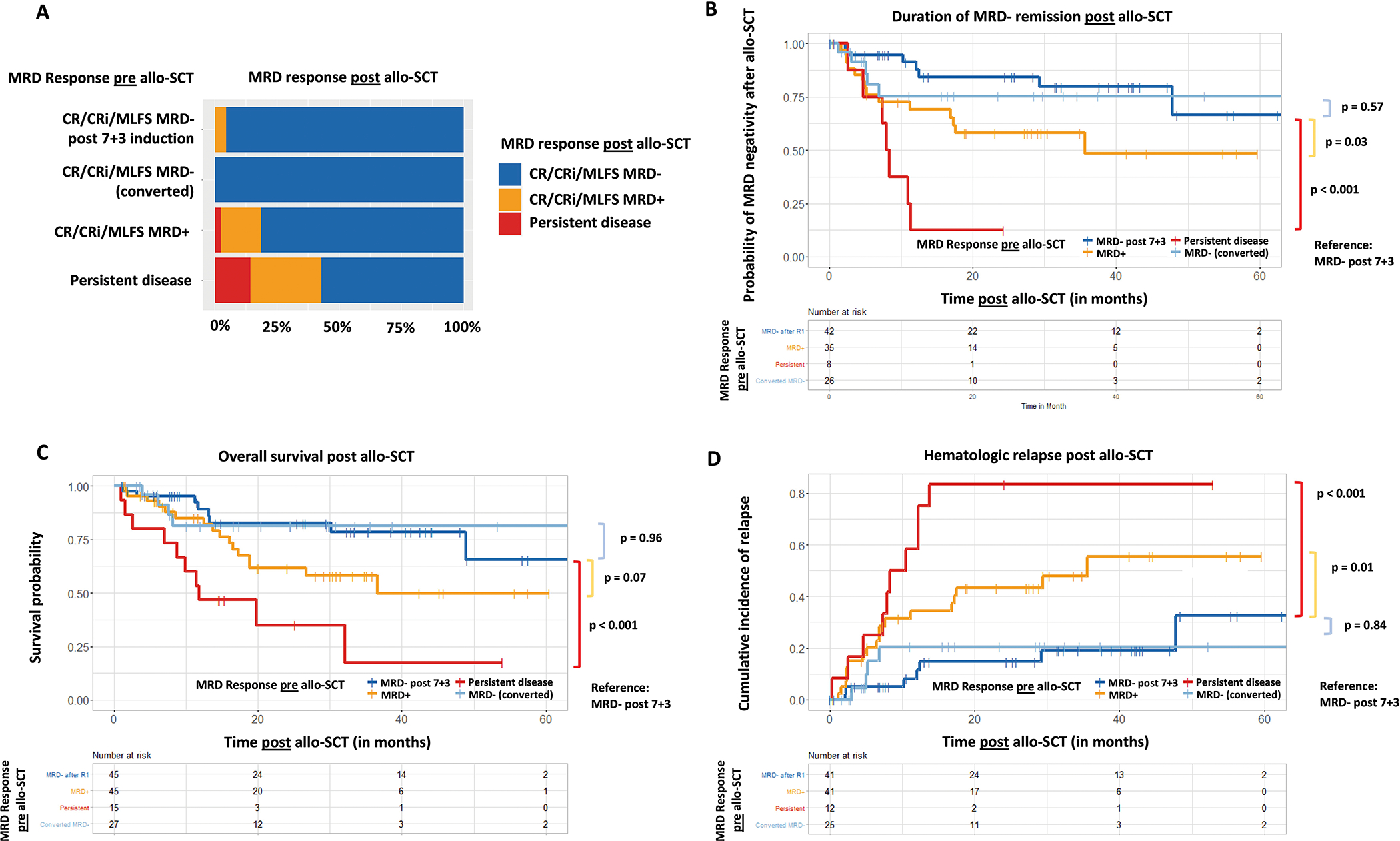
(A) MRD response post allo-SCT based on MRD response achieved pre allo-SCT. (B) Duration of MRD−negativity, overall survival (C) and cumulative relapse rates (D) post allo-SCT by pre allo-SCT MRD response.
Post-transplant outcomes are similar for patients who clear MRD early after induction chemotherapy or later with additional therapy prior to allo-SCT:
The clinical course following allo-SCT and outcomes for all patients based on their MRD response prior to allo-SCT is summarized in Supplemental Figure 4B. At a median follow up of 29.9 months, median OS was not reached for patients who achieved early MRD− after 7+3 induction and patients who converted from MRD+ remission to MRD− remission later (with additional therapy but prior to allo-SCT). Both early MRD clearance and MRD conversion later were associated with statistically significantly (p<0.001) improved OS compared with patients who continued to have MRD+ (median OS: 36.6 months [95% CI: 18.8 months – not reached]) or persistent disease (median OS: 11.8 months [95% CI: 8.8 months – not reached]) prior to allo-SCT (Figure 4C). Similar associations were observed for cumulative relapse rates, which were significantly higher for patients with MRD+ remission (HR: 3.15 [95% CI: 1.3–7.63]; p=0.01) or persistent disease (HR: 7.07 [95% CI: 2.66–18.9]; p<0.001) compared to patients with MRD− remission (Figure 4D).
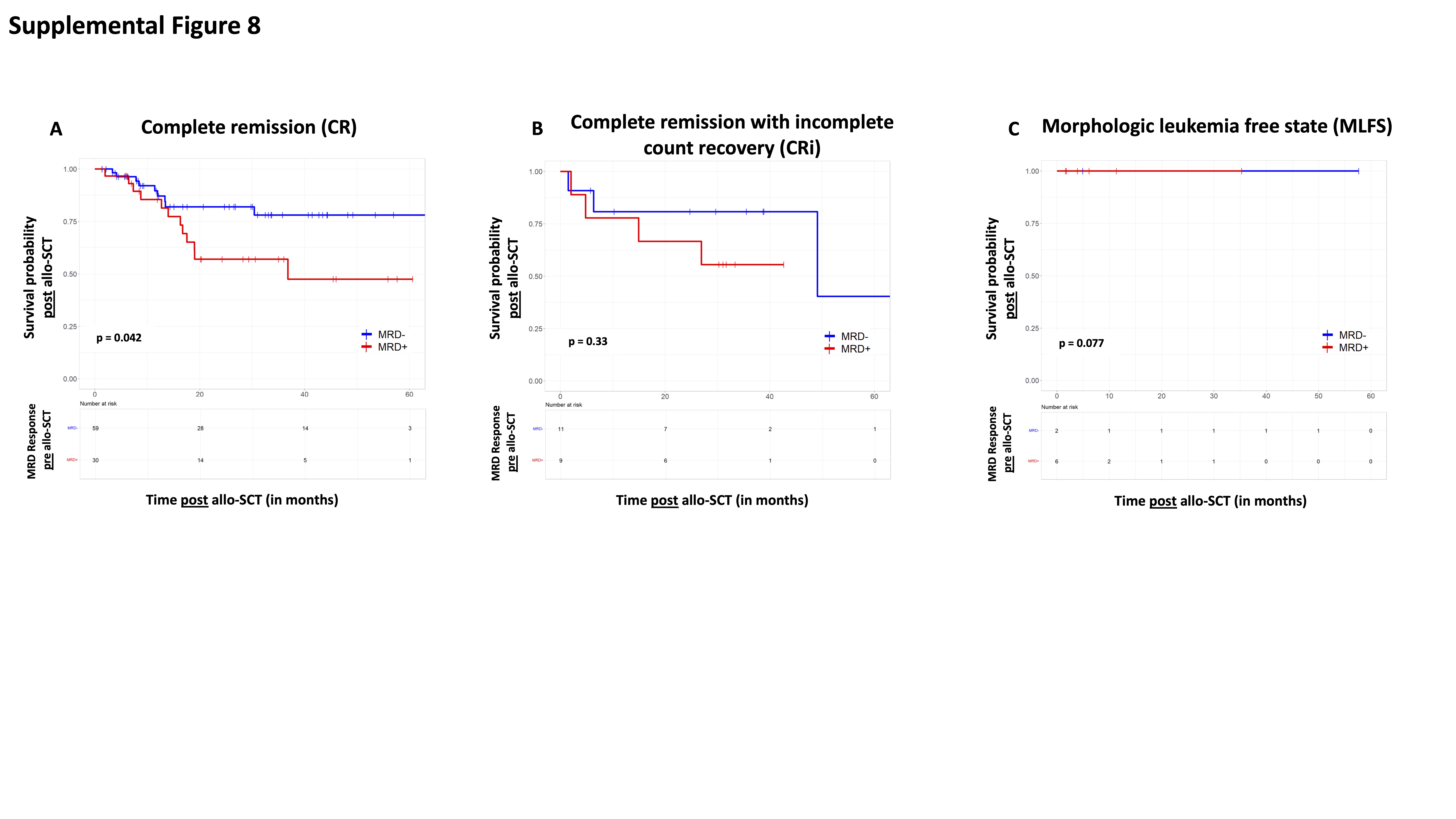
We also analyzed whether MRD response pre-allo-SCT added prognostic significance beyond baseline ELN 2017 molecular risk on overall survival (OS) after allo-SCT (Supplemental Figure 7). Similar to MRD status after induction chemotherapy, we observed that MRD status pre-allo-SCT added prognostic significance beyond baseline ELN 2017 molecular risk profile for ELN favorable risk (p = 0.02; Supplemental Figure 7A). While there was a trend towards improved OS for patients with ELN adverse risk who achieved MRD− pre-allo-SCT, it was not statistically significant (p=0.077; Supplemental Figure 7C). In patients with ELN intermediate risk, achievement of MRD− with induction chemotherapy did not seem to improve survival (p=0.46; Supplemental Figure 7B). Lastly, we found that MRD had a statistically significant prognostic impact in patients who achieved CR pre allo-SCT (p = 0.042), whereas in patients who achieved a CRi or MLFS pre allo-SCT, MRD status did not result in statistically significant differences in post allo-SCT OS (p = 0.33 and p = 0.077) with the caveat that only a small number of patients was examined for each of the morphologic responses (Supplemental Figure 8).
Discussion:
Using a robustly annotated dataset of 211 AML patients who all received intensive induction chemotherapy in the frontline setting and were serially monitored for MRD by highly sensitive MFC, we not only confirm the prognostic importance of MRD in AML but show several novel findings: (i) specific cytogenetic and mutational patterns as well as clonal diversity assessed prior to treatment start can predict the likelihood of MRD clearance with chemotherapy (ii) early and late conversion to an MRD− response prior to allo-SCT lead to similar post-allo-SCT outcomes. Notably, MRD conversion with chemotherapy prior to allo-SCT is rarely achieved in specific molecularly defined subsets of AML.
Given the well-documented prognostic impact of MRD status at various timepoints during the treatment course,5, 8, 11, 19–21 understanding pre-treatment genetic predictors of MRD clearance at the time of diagnosis can potentially guide treatment selection. While patients who are likely to achieve MRD clearance early on with chemotherapy alone should be treated with standard of care chemotherapy, patients who are highly unlikely to achieve MRD clearance should be prioritized for clinical trials. In our cohort, a combination of pre-treatment cytogenetic abnormalities, genetic mutations, and measures of clonal diversity identified patients who are very likely vs. highly unlikely to achieve MRD clearance after induction chemotherapy and prior to allo-SCT. As expected, CBF chromosomal translocations and mutations in CEBPA and NPM1 predicted high MRD clearance with 7+3 induction chemotherapy. Interestingly, RAS mutations predicted high MRD clearance rates with induction chemotherapy which is in contrast to what has been observed with venetoclax based therapy, arguing for the use of chemotherapy in AML patients with RAS pathway mutations.22 Conversely, no AML patients with inv(3)/t(3:3), monosomy 5/del(5q), monosomy 7/del(7q) and with mutations in TP53 (0/13) and only 1/15 patients with a mutation in SF3B1 cleared MRD following induction chemotherapy. Strikingly, no AML patients with mutations in TP53 or SF3B1 were able to clear MRD prior to allo-SCT even when further consolidation and/or salvage chemotherapy was administered.
Our data also show that patients with higher clonal diversity pre-treatment as assessed by the number of individual clones had lower MRD− response rates with chemotherapy. Importantly, clonal diversity was defined at single cell resolution. In contrast, simple estimation of clonal diversity by number of mutations, which can easily be done by bulk sequencing, failed to predict MRD clearance with induction chemotherapy. Clonal diversity has not been previously associated with the likelihood of AML MRD clearance, and our data show that clonal complexity may serve as a novel predictor of clinical response to chemotherapy at the level of MRD.
As shown in prior studies, MRD− remission prior to allo-SCT was associated with reduced rates of relapse and improved OS post allo-SCT5, 23. Our study adds the critical and novel information that outcomes following allo-SCT were comparable for patients who achieved an early MRD− remission following induction therapy and patients who converted to MRD−negativity after additional post-induction therapy. Thus, the finding of MRD positivity immediately following induction chemotherapy does not necessarily portend a poor prognosis if MRD−negativity can be achieved following additional therapy prior to allo-SCT. Patients who achieved MRD−negativity following additional therapy prior to allo-SCT had no difference in duration of post-transplant MRD−negativity, relapse rate or survival after allo-SCT compared to patients who achieved MRD− directly after 7+3.
Given the favorable post-transplant outcomes we observed in patients with later achievement of MRD− prior to allo-SCT, our results emphasize that eradication of MRD prior to allo-SCT should be an important therapeutic goal. However, our findings also suggest that patients with specific pre-treatment highly adverse molecular features (inv(3), TP53, SF3B1) are unlikely to benefit from standard chemotherapy to convert MRD+ to MRD−. While some of these patients might achieve a morphologic remission, an MRD− remission is rarely achieved. Early inclusion in clinical trials may be the best option for MRD+ patients with these adverse baseline molecular features. Whether an alternative induction strategy with venetoclax-based combinations in patients with high molecular risk AML could be more successful in clearing MRD requires further study. One recent report found high rates of MRD clearance with combination fludarabine, cytarabine, granulocyte colony-stimulating factor, and idarubicin + venetoclax.24 However, current venetoclax combinations may still not clear AML MRD with specific adverse molecular profiles such as mutations in splicing factors and TP53.22, 25, 26 Encouraging preliminary results in patients with MDS and oligoblastic AML including those with TP53 mutations have been reported for the combination of the anti-CD47 antibody magrolimab in combination with azacitidine.27 Targeting splicing factor mutations such as SF3B1 has remained challenging in AML and MDS with limited efficacy seen in clinical trials with splicing inhibitors such as H3B-8800.28
A major challenge to the routine use of MRD in treatment selection in AML is the variability of MRD assessment techniques and detection thresholds.29 Efforts to standardize and improve technologies for MRD assessment by both flow cytometry and NGS are ongoing.4, 11, 29, 30 Our study includes the use of highly sensitive and standardized MFC to assess for MRD assessment. While a study by Murdock et al has presented data on molecularly defined MRD12, MFC remains the standard method for MRD assessment for most AML patients in current clinical practice in the United States.
Interestingly, we observed that MRD status as defined by MFC after induction chemotherapy and pre-allo-SCT added prognostic significance beyond baseline ELN 2017 molecular risk profile for patients with ELN favorable risk disease. While there seemed to be a trend towards improved survival for patients with ELN adverse risk AML who achieved MRD− after induction chemotherapy and prior to allo-SCT, it did not reach statistical significance. For patients with ELN intermediate risk disease, we did not observe a significant difference in survival outcomes between patients achieving MRD− vs. those who remained MRD+. We note that the lack of statistically significant association between MRD status and survival in ELN intermediate and adverse risk groups may be due to smaller sample sizes which limit statistical power.
Our finding that MFC MRD status adds prognostic information in AML ELN favorable risk patients is partly in contrast to the findings of Murdock et al, who recently showed that molecular MRD status by NGS did not add prognostic significance beyond baseline molecular risk profile.12 Notably, 51% of the AML patient population in our study were younger adults <60 years old, while all patients in Murdock et al were older adults age 60 and above. Apart from differences in patient and transplant characteristics between those two studies, there are also inherent differences in the methods of MRD assessment in both studies (MFC in our study vs. NGS mutational analysis in the study by Murdock et al). Larger studies are needed to determine how to best incorporate baseline molecular risk and post treatment MRD by both MFC and NGS mutational analysis into a comprehensive risk assessment for patients with AML.
Strengths of our manuscript include the large, well-annotated patient cohort uniformly treated initially with intensive induction chemotherapy, required availability of baseline NGS, and comprehensive clinical and MFC MRD data collected longitudinally at various time points across the treatment course. We note that the retrospective and single-center design of this study could limit the generalizability of our results. Single cell sequencing analysis was also only available for a relatively small subset of patients. Future single-cell sequencing studies with larger sample sizes are necessary to validate the association of clonal diversity with resistance to MRD eradication.
In conclusion, we found that specific molecular features including cytogenetic abnormalities, genetic mutations, and clone numbers prior to induction chemotherapy are powerful predictors of AML MRD clearance. We also observed that early or late conversion to MRD− prior to allo-SCT is associated with favorable post-allo-SCT outcomes. Our data argue for future strategies tailored to patients with adverse molecular features and high clonal diversity who are currently unlikely to achieve AML MRD clearance.
Supplementary Material
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Acknowledgments:
M.S. received funding from the MSKCC Clinical Scholars T32 Program under award number 2T32 CA009512-31. This work was funded by a Conquer Cancer Foundation Young Investigator Award (award number GC241610). A.D.G. received funding from an American Society of Hematology (ASH) Fellow Scholar Award in Clinical Research and a Conquer Cancer Foundation Young Investigator Award. Any opinions, findings, and conclusions expressed in this material are those of the author(s) and do not necessarily reflect those of the American Society of Clinical Oncology® or Conquer Cancer®. Research reported in this publication was supported by the NCI of the National Institutes of Health under Award Number P30 CA016359 and P01 CA23766, and Cancer Center Support Grant/Core Grant to Memorial Sloan Kettering Cancer Center (P30 CA008748) The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
The remaining authors declare no competing financial interests.
Author disclosures:
M.S. has consulted for Curis Oncology, Boston Consulting and is member of the advisory board for Novartis and Kymera. S.F.C. has consulted for and holds equity interest in Imago Biosciences. M.B.G. reports research support from Amgen, Sanofi, and Actinium Pharmaceuticals and has served in an advisory capacity for Sanofi and Allogene. B.C.S. received research funding from Amgen, Janssen, Celgene, Quintiles, Pfizer, CSL Behring, Sanofi, Adienne, Kite, Jazz, and Actinuum. B.G. received research funding from Actinium. W.X. received research funding from Stemline Therapeutics. J.G. served as a consultant for the Gerson Lehman Group. S.G. received research funding from Amgen, Actinuum, Celgene, Jansen, Quintiles, Pfizer, CSL Behring, Sanofi, Adienne, Kite, JAZZ, Johnson&Johnson. S.G. served on the advisory committees or Board of Director’s of Amgen, Actinuum, Johnson& Johnson, Jensen, Jazz Pharmaceuticals, Takeda, Novartis, KITE, Spectrum Pharma, BMS, Sanofi, Pfizer, GSK. M.A.P reports honoraria from Abbvie, Astellas, Bristol-Myers Squibb, Celgene, Equilium, Incyte, Karyopharm, Kite/Gilead, Merck, Miltenyi Biotec, MorphoSys, Novartis, Nektar Therapeutics, Omeros, OrcaBio, Takeda, and VectivBio AG, Vor Biopharma. M.A.P. serves on DSMBs for Cidara Therapeutics, Medigene, Sellas Life Sciences, and Servier, and the scientific advisory board of NexImmune. M.A.P. has ownership interests in NexImmune and Omeros. M.A.P. has received research support for clinical trials from Incyte, Kite/Gilead, Miltenyi Biotec, and Novartis. E.M.S. received research funding from Bayer; was a consultant for Amgen, AbbVie, Seattle Genetics, and Biotheryx; served as a consultant and received research funding from Syndax; was a member of the Board of Directors or advisory committee for PTC Therapeutics and Syros; served as a consultant and was member of the Board of Directors or advisory committee for Astellas Pharmaceutical, Agios Pharmaceuticals, and Genentech; served as a consultant, received research funding, and was a member of the Board of Directors or advisory committee for Daiichi-Sankyo, Celgene Pharmaceuticals, and Novartis; and is a current equity holder in privately held Auron Therapeutics. M.A. reports honoraria from Biocartis, Invivoscribe, physician educational resources, Peerview institute for medical education, clinical care options, RMEI medical education. M.A. served as consultant in advisory boards for Janssen Global Services, Bristol-Myers Squibb, AstraZeneca, Roche and Merck. M.S.T. received research funding from AbbVie, Cellerant, Orsenix, ADC Therapeutics, Glycomimetics, Rafael, and Amgen; was a member of the Board of Directors or advisory committee for Bioline rx, Daiichi-Sankyo, KAHR, Rigel, Delta Fly Pharma, Oncolyze, Jazz Pharma, Roche, and Novartis; received research funding from and was a member of the Board of Directors or advisory committee for BioSight; has served on advisory boards for Innate Pharaceuticals, Kura Oncology, and Syros Pharmaceuticals; and has a patent and received royalties from UpToDate. A.D.G. received research funding from Celularity, ADC Therapeutics, Aprea, AROG, Pfizer, Prelude, and Trillium; received research funding from and served as a consultant for Aptose and Daiichi Sankyo; served as a consultant and member of advisory committees for Astellas, Celgene, and Genentech; received research funding from, served as a consultant for, and was a member of advisory committees for AbbVie; and received honoraria from Dava Oncology. None of these relationships were related to the development of this manuscript.
References:
- 1.Döhner H, Weisdorf DJ, Bloomfield CD. Acute Myeloid Leukemia. N Engl J Med 2015. Sep 17; 373(12): 1136–1152. [DOI] [PubMed] [Google Scholar]
- 2.Rucker FG, Schlenk RF, Bullinger L, Kayser S, Teleanu V, Kett H, et al. TP53 alterations in acute myeloid leukemia with complex karyotype correlate with specific copy number alterations, monosomal karyotype, and dismal outcome. Blood 2012. Mar 1; 119(9): 2114–2121. [DOI] [PubMed] [Google Scholar]
- 3.Schuurhuis GJ, Heuser M, Freeman S, Bene MC, Buccisano F, Cloos J, et al. Minimal/measurable residual disease in AML: a consensus document from the European LeukemiaNet MRD Working Party. Blood 2018. Mar 22; 131(12): 1275–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schuurhuis GJ, Heuser M, Freeman S, Béné M-C, Buccisano F, Cloos J, et al. Minimal/measurable residual disease in AML: a consensus document from the European LeukemiaNet MRD Working Party. Blood 2018; 131(12): 1275–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Araki D, Wood BL, Othus M, Radich JP, Halpern AB, Zhou Y, et al. Allogeneic Hematopoietic Cell Transplantation for Acute Myeloid Leukemia: Time to Move Toward a Minimal Residual Disease-Based Definition of Complete Remission? J Clin Oncol 2016. Feb 1; 34(4): 329–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ivey A, Hills RK, Simpson MA, Jovanovic JV, Gilkes A, Grech A, et al. Assessment of Minimal Residual Disease in Standard-Risk AML. N Engl J Med 2016. Feb 4; 374(5): 422–433. [DOI] [PubMed] [Google Scholar]
- 7.Jongen-Lavrencic M, Grob T, Hanekamp D, Kavelaars FG, Al Hinai A, Zeilemaker A, et al. Molecular Minimal Residual Disease in Acute Myeloid Leukemia. N Engl J Med 2018. Mar 29; 378(13): 1189–1199. [DOI] [PubMed] [Google Scholar]
- 8.Freeman SD, Virgo P, Couzens S, Grimwade D, Russell N, Hills RK, et al. Prognostic relevance of treatment response measured by flow cytometric residual disease detection in older patients with acute myeloid leukemia. J Clin Oncol 2013. Nov 10; 31(32): 4123–4131. [DOI] [PubMed] [Google Scholar]
- 9.Short NJ, Zhou S, Fu C, Berry DA, Walter RB, Freeman SD, et al. Association of measurable residual disease with survival outcomes in patients with acute myeloid leukemia: a systematic review and meta-analysis. JAMA oncology 2020; 6(12): 1890–1899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Short NJ, Rafei H, Daver N, Hwang H, Ning J, Jorgensen JL, et al. Prognostic impact of complete remission with MRD negativity in patients with relapsed or refractory AML. Blood Advances 2020; 4(24): 6117–6126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heuser M, Freeman SD, Ossenkoppele GJ, Buccisano F, Hourigan CS, Ngai LL, et al. 2021 Update Measurable Residual Disease in Acute Myeloid Leukemia: European LeukemiaNet Working Party Consensus Document. Blood 2021. Nov 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Murdock HM, Kim HT, Denlinger N, Vachhani P, Hambley B, Manning BS, et al. Impact of diagnostic genetics on remission MRD and transplantation outcomes in older patients with AML. Blood 2022. Jun 16; 139(24): 3546–3557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Arber DA, Orazi A, Hasserjian R, Thiele J, Borowitz MJ, Le Beau MM, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood 2016. May 19; 127(20): 2391–2405. [DOI] [PubMed] [Google Scholar]
- 14.Dohner H, Estey E, Grimwade D, Amadori S, Appelbaum FR, Buchner T, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood 2017. Jan 26; 129(4): 424–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Getta BM, Devlin SM, Levine RL, Arcila ME, Mohanty AS, Zehir A, et al. Multicolor Flow Cytometry and Multigene Next-Generation Sequencing Are Complementary and Highly Predictive for Relapse in Acute Myeloid Leukemia after Allogeneic Transplantation. Biol Blood Marrow Transplant 2017. 2017/07/01/; 23(7): 1064–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cheng DT, Mitchell TN, Zehir A, Shah RH, Benayed R, Syed A, et al. Memorial Sloan Kettering-Integrated Mutation Profiling of Actionable Cancer Targets (MSK-IMPACT): A Hybridization Capture-Based Next-Generation Sequencing Clinical Assay for Solid Tumor Molecular Oncology. J Mol Diagn 2015. May; 17(3): 251–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ptashkin RN, Benayed R, Ziegler J, Rema AB, Sadowska J, Kiecka I, et al. Abstract 3409: MSK-IMPACT Heme: Validation and clinical experience of a comprehensive molecular profiling platform for hematologic malignancies. Cancer Res 2019; 79(13 Supplement): 3409–3409. [Google Scholar]
- 18.Miles LA, Bowman RL, Merlinsky TR, Csete IS, Ooi AT, Durruthy-Durruthy R, et al. Single-cell mutation analysis of clonal evolution in myeloid malignancies. Nature 2020. Nov; 587(7834): 477–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Balsat M, Renneville A, Thomas X, Botton Sd, Caillot D, Marceau A, et al. Postinduction Minimal Residual Disease Predicts Outcome and Benefit From Allogeneic Stem Cell Transplantation in Acute Myeloid Leukemia With NPM1 Mutation: A Study by the Acute Leukemia French Association Group. J Clin Oncol 2017; 35(2): 185–193. [DOI] [PubMed] [Google Scholar]
- 20.Terwijn M, van Putten WL, Kelder A, van der Velden VH, Brooimans RA, Pabst T, et al. High prognostic impact of flow cytometric minimal residual disease detection in acute myeloid leukemia: data from the HOVON/SAKK AML 42A study. J Clin Oncol 2013. Nov 1; 31(31): 3889–3897. [DOI] [PubMed] [Google Scholar]
- 21.Thol F, Gabdoulline R, Liebich A, Klement P, Schiller J, Kandziora C, et al. Measurable residual disease monitoring by NGS before allogeneic hematopoietic cell transplantation in AML. Blood 2018. Oct 18; 132(16): 1703–1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.DiNardo CD, Tiong IS, Quaglieri A, MacRaild S, Loghavi S, Brown FC, et al. Molecular patterns of response and treatment failure after frontline venetoclax combinations in older patients with AML. Blood 2020. Mar 12; 135(11): 791–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Walter RB, Gooley TA, Wood BL, Milano F, Fang M, Sorror ML, et al. Impact of pretransplantation minimal residual disease, as detected by multiparametric flow cytometry, on outcome of myeloablative hematopoietic cell transplantation for acute myeloid leukemia. J Clin Oncol 2011. Mar 20; 29(9): 1190–1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.DiNardo CD, Lachowiez CA, Takahashi K, Loghavi S, Xiao L, Kadia T, et al. Venetoclax Combined With FLAG-IDA Induction and Consolidation in Newly Diagnosed and Relapsed or Refractory Acute Myeloid Leukemia. J Clin Oncol 2021; 39(25): 2768–2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Stahl M, Menghrajani K, Derkach A, Chan A, Xiao W, Glass J, et al. Clinical and molecular predictors of response and survival following venetoclax therapy in relapsed/refractory AML. Blood Adv 2021. Mar 9; 5(5): 1552–1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tiong IS, Dillon R, Ivey A, Teh T-C, Nguyen P, Cummings N, et al. Venetoclax induces rapid elimination of NPM1 mutant measurable residual disease in combination with low-intensity chemotherapy in acute myeloid leukaemia. Br J Haematol 2021; 192(6): 1026–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sallman DA. Tolerability and efficacy of the first-in-class anti-CD47 antibody magrolimab combined with azacitidine in MDS and AML patients: Phase Ib results. In: David Andrew Sallman MAMASADJLSKWBDTJBPVDJGMRSKJV, Moffitt Cancer Center TFL, City of Hope Medical Center DCA, University of Oklahoma Health Sciences Center OCOK, Columbia Univ Medcl Ctr NYNY, University of Kansas Medical Center SCRIKCKS, et al. , editors.; 2020; ASCO Virtual Scientific Program: American Society of Clinical Oncology; 2020. [Google Scholar]
- 28.Steensma DP, Wermke M, Klimek VM, Greenberg PL, Font P, Komrokji RS, et al. Results of a Clinical Trial of H3B-8800, a Splicing Modulator, in Patients with Myelodysplastic Syndromes (MDS), Acute Myeloid Leukemia (AML) or Chronic Myelomonocytic Leukemia (CMML). Blood 2019; 134(Supplement_1): 673–673. [Google Scholar]
- 29.Bewersdorf JP, Shallis RM, Boddu PC, Wood B, Radich J, Halene S, et al. The minimal that kills: Why defining and targeting measurable residual disease is the “Sine Qua Non” for further progress in management of acute myeloid leukemia. Blood Rev 2019. Dec 20: 100650. [DOI] [PubMed] [Google Scholar]
- 30.Hourigan CS, Gale RP, Gormley NJ, Ossenkoppele GJ, Walter RB. Measurable residual disease testing in acute myeloid leukaemia. Leukemia 2017. Jul; 31(7): 1482–1490. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.