

Abstract
Hybrid phospholipid/block copolymer membranes where polymers and lipids are molecularly mixed or phase-separated into polymer-rich and lipid-rich domains are promising drug delivery materials. Harnessing the chemical diversity of polymers and the biocompatability of lipids is a compelling approach to design the next generation of drug carriers. Here, we report on the development of a microfluidics-based strategy analogous to produce lipid nanoparticles (LNPs) for the nanomanufacturing of multilayered hybrid nanoparticles (HNPs). Using X-ray scattering, Cryo-electron, and polarized microscopy we show that phosphatidylcholine (PC) and PBD-b-PEO (poly(butadiene-block-ethylene oxide)) hybrid membranes can be nanomanufactured by microfluidics into HNPs with dense and multilayered cores which are ideal carriers of low-solubility drugs of the Biopharmaceutical Classification System (BCS) II and IV such as antimalarial DSM265 and Paclitaxel, respectively.
Graphical Abstract
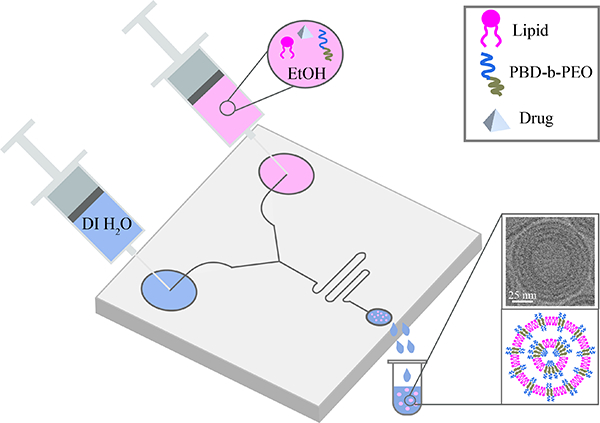
In this paper, we describe a microfluidics route to develop new structures of hybrid polymer and lipid nanoparticles (HNPs) which are beneficial to formulate poorly soluble drugs.
Introduction
A variety of nanostructures are formed via self-assembly of phospholipids (PLs) and amphiphilic block copolymers (BCPs)1,2, the most eminent phase being planar stacks of one or multiple bilayers, characteristic of the lamellar phase. This has led to the scientific discovery of polymersomes3–5 — liposome analogues that enabled many advances in drug delivery applications6,7. Polymerosomes are versatile nanocarriers with tunable membrane properties, such as size, permeability, and functionality7. Nevertheless, issues of biocompatability, biodegradability, and immunogenicity8 often arise for these fully synthetic systems. In addition, BCP vesicles have considerably less affinity to cell membranes, unmatched flexibility, and poor fusion ability compared to PL-based vesicles. Despite the inherent instability and limited shelf life of PL systems, a considerable number of lipid-based formulations have received great interest and are FDA-approved (e.g. Onpattro® (Patisiran) and Vyondys 53® (Golodirsen)) or undergoing clinical trials9. In the past few years there has been an explosion of advances in messenger RNA (mRNA) vaccines and currently two lipid-based mRNA vaccines for the SARS-CoV-2 virus have acquired emergency use authorization by the FDA and other international agencies10. The development of hybrid membrane systems combining the properties of PLs and BCPs could be a promising approach in drug carrier design. These controlled drug delivery systems would consist of stabilized nanoscaled hybrid membranes comprising PLs and BCPs11,11,12.
High-throughput screening (HTS) and combinatorial chemistry schemes for small-molecule drug discovery and almost always generate compounds of poor water solubility, high lipophilicity, and high molecular weight making them difficult to formulate with conventional drug delivery systems13–15. The Biopharmaceutics Classification System (BCS) classifies pharmaceutical commodities into one of the four categories according to their solubility and permeability and these four categories are: class I (high permeability/high solubility), class II (high permeability/low solubility), class III (low permeability/high solubility), and class IV (low permeability/low solubility) drugs16. Class II and class IV (low solubility) categories include many of the newly synthesized drug molecules16,17, denoting that the solubility or rate of dissolving in the gastrointestinal tract are the pronounced constrains for their absorption18. Various commonly used systems to increase bioavailability of these drug candidates include nanoparticle-based formulations19–22, lipid and polymer-based drug delivery systems23–28, and pro-drugs29–31.
Winzen et al. recently demonstrated that, PDMS-b-PMOXA/cholesterol hybrid vesicles show a drastically increased bending stiffness compared to pure PDMS-b-PMOXA polymersomes and PDMS-b-PMOXA/DMPC (1,2-dimyristoyl-sn-glycero-3-phosphocholine) vesicles which was attributed to a higher packing density due to the presence of cholesterol in hybrid vesicle membranes32. Another study demonstrated that the cell binding efficacy of nanovesicles can be significantly improved when prepared from PBD-b-PEO (poly(butadiene-block-ethylene oxide)) and HSPC (hydrogenated soy phosphatidylcholine) blends compared with pure block copolymer33. Compared to the extensive current approaches for manufacturing processes of pure lipid and pure polymer systems, only a few studies have exploited the preparation of hybrid membranes of PL and BCP as drug carriers. Here, we demonstrate a bench-scale setup for the synthesis of hybrid NPs (HNPs) and low solubility drug–loaded HNPs in a scale– independent manner using a microfluidics platform. Microfluidics nanoparticle synthesis are based on nanoprecipitation and provide a versatile approach to enable simple and consistent assembly of nanoparticles using a ‘bottom-up’ approach34–37. The NanoAssemblr™ is a recent microfluidics device development that exhibits high degree of control over size and polydispersity where staggered herringbone structures (SHM) induce rapid mixing by chaotic fluid flow (Precision NanoSystems Inc., Vancouver, Canada).
Our goal in this work is two-fold. First, we optimized the process of HNP assembly using a benchtop microfluidic device. Second, we explored the functionality of HNPs as a promising low solubility drug delivery platform candidate.
Experimental
Materials
1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), 1,2-dipalmitoyl-sn-glycero-3-phosphocholine (DPPC) suspended in chloroform were obtained from Avanti Polar Lipids (Alabama, USA). Amphiphilic diblock copolymer, poly(butadiene-b-ethylene oxide) (PBD-b-PEO), catalog number P19015-BdEO, was acquired from Polymer Source, Inc. (Quebec, Canada). The polydispersity is 1.06 and its average molecular weight (Mn) is 4000 with PBD block (rich in 1,4 microstructure) 2500 and PEO block 1500, respectively. In this study, we chose PBD-b-PEO since the previous work showed formation of phase-separated hybrid vesicles with domains38–40. Paclitaxel was purchased from Thermo Fisher Scientific (Waltham; MA, USA). DSM265 was purchased from MedChemExpress (MedChemExpress LLC; NJ, USA). All chemicals and materials were used as received.
Hybrid Nanoparticles Synthesis Using a Bench-Scale Device
The self-assemble of NPs by microfluidics was accomplished by using the NanoAssemblr™ Benchtop platform (Precision Nanosystems Inc.). Pure PL (DOPC and DPPC:Chol (10:5 wt/wt ratio equivalent to 50 mol%), pure BCP and hybrid NPs with PL:BCP (4:1 mol ratio) were dissolved in ethanol at specific 8 mg/mL concentration. Syringe pumps introduce a lipid/ethanol solution trough one of the two channels on the microfluidics herringbone cartridge, meanwhile the aqueous phase is fed into the second channel. Microfluidics optimization can be performed by tuning the flow rate ratio (the ratio between the organic phase and the aqueous phase) and the total flow rate (the speed at which the two channels are administered through the microfluidic chip) using the NanoAssemblr™ software. Flow rate ratios of 1:1, 2:3, 1:2 and 1:1 were tested as well as total flow rates of 5–15 mL/min. For the synthesis of drug encapsulated NPs, PTX and DSM265 were added to the lipid/polymer phase at specific concentrations. All production was performed in open air, room RH and temperature.
Sample Purification via Dialysis
Samples were purified using the Maxi 6000 Pur-A-Lyzer kit with MWCO of 6kD. For removal of solvent (EtOH) and unencapsulated drug, samples were loaded into the Pur-A-Lyzer tube and then placed in a stirred beaker containing DI water. The dialysis buffer was change 3 times (every 3–4 hrs). All samples were retrieved at the initial volume.
Dynamic Light Scattering (DLS)
Polydispersity index (PDI) and particle size (Z-average) of the dispersions were determined using a dynamic light scattering equipment (DLS) with a Malvern Zetasizer Nano ZS with a 633 nm wavelength laser (Malvern Instruments, Worcestershire, U.K.). The scattering angle was set to 173° and data was processed using the Stokes-Einstein equation to calculate hydrodynamic radii, and cumulative analysis of the experimental correlation function. Zeta potential measurements were also performed on a Malvern Zetasizer Nano ZS equipment at RT. Electrophoretic calculations of zeta potential were made using the Smoluchowski approximation. All solutions were measured using disposable polystyrene cuvettes and 10x dilution with DI water. A total volume of 1 mL was needed for each size measurement.
Nanoparticle Tracking Analysis (NTA)
We measured the concentration of the dispersions by a nanoparticle tracking analyzer (NTA), NanoSight NS300 (Quantum Design, Tokyo, Japan). The measurements carried out in open air, room RH and temperature. Samples were diluted 1/1000 with DI water.
Small/Wide-Angle X-ray Scattering (SAXS/WAXS)
SAXS and WAXS experiments were performed to examine the nanoparticle structure and drug crystallization. Synchrotron SAXS and WAXS were conducted by using a 13.3 keV X-ray beam at beamline 12-ID-B of the Advanced Photon Source at Argonne National Laboratory. For simultaneous SAXS and WAXS data collection, the Pilatus2M (Dectris) and Pilatus300 (Dectris) detectors with pixel sizes of 0.172 mm were used, respectively. The acquired data was radially integrated on-site using MATLAB™ software developed by beamline 12-ID-B. Silver behenate powder standard was used to calibrate the sample to detector distance (SDD) and the SDDs for SAXS and WAXS were 2 and 0.46 m, respectively. All bulk samples at 100 mM concentration were hydrated with MilliQ water in quartz capillaries (Hilgenberg, Germany). Before the hydration step, samples were vacuum-dried in desiccator for days to remove all organic solvents. All 25 mM colloidal NPs were prepared using NanoAssemblr™ and transferred to quartz capillaries (Hilgenberg Glas, Germany). Before measurements quartz capillaries were flame sealed and kept hydrated for a couple of days. Experiments were all conducted in ambient conditions.
Cryo-EM sample preparation and data collection
Neat lipid, block copolymer and their hybrid samples for cryogenic transmission electron microscopy were prepared on a lacey carbon-coated 200 mesh grid (Electron Microscopy Sciences) using a Vitrobot Mark IV (Thermo Fisher Scientific). Briefly, 3.5 μL of sample was applied to the glow-discharged surface of the grid at a chamber temperature of 25 °C with a relative humidity level of 100 %, and then vitrified in liquid ethane. Before vitrification samples were blotted for 5 s with a blot force of 1. All Cryo-EM grids were imaged using a 200 keV Glacios (Thermo Fisher Scientific) TEM equipped with a Falcon4 direct electron detector. Images were collected at 120kX magnification (physical pixel size 1.3 Å) with a total dose of 6.9 e−/Å2 and a defocus of −5 μm using EPU automated acquisition software (Thermo Fisher).
Cross-polarization microscopy
Cross-polarized images were obtained in water at room temperature to check the presence of birefringence from multilayered structure using a Zeiss Axio Scope A1 microscope with 50×/0.75 EC Epiplan HD objective lens (Carl Zeiss Microscopy, NY, USA). Images were processed using the Zen software (Carl Zeiss AG).
Confocal Laser Scanning Microscopy
Hybrid MLVs were prepared following the gentle film hydration method. 80 mol% of DPPC PL with 0.1 mol% of 16:0 Liss Rhod PE, 18 mol% of PBD-b-PEO, and 2 mol% (PBD-b-PEO)-FITC were mixed in chloroform at a stock concentration of 100 mM. A volume of 15 mL of each stock solution was pipetted into 1.5 mL amber glass vial. The chloroform was evaporated under vacuum overnight protected from light exposure. For the hydration step, 1 mL of 100 mM sucrose/MilliQ water was introduced to the vial and incubated in the dark for 3 h at 45°C. Imaging was performed within 2 days. For Confocal Laser Scanning Microscopy (CLSM) imaging of Giant MLVs, we used a LSM 800 confocal microscope (Carl Zeiss Microimaging, Jena, Germany) with a Plan-Apochromat 63×/1.40 Oil M27 objective lens. Giant vesicles were transferred to a coverslip (No. 1.5) before imaging. Images were processed in Zen Blue software. Two sets of filters were used for detecting signals from Rhod B (excitation peak at 543 nm/emission peak at 565 nm) and FITC (excitation peak at 495 nm/emission peak at 519 nm).
Results and Discussion
We study the synthesis mechanism of HNPs by nanoprecipitation and microfluidics which are scalable approaches for use in nanomedicine37. Johnson and Prud’homme pioneered bulk methods to produce nanoparticles with hydrophobic cargo via flash nanoprecipitation41,42. Then later, with advancements in microfluidics technologies, microfluidics-based nanoprecipitation methods have emerged as other avenues to generate particles with narrow size distribution43. The goal of our paper is to develop the co-assembly of multilayers of polymers and lipids at the nanoscale into hybrid nanoparticles (HNPs) having narrow size distributions and small (ca 100 nm in diameter) average size. With this in mind, we employ a well-established bottom-up microfluidic approach having a staggered herringbone mixer (SHM) chip by Precision NanoSystems (Figure 1)44. We characterized the HNPs size and size distribution using light scattering and nanoparticle analysis systems, the HNPs structure with X-ray scattering, as well as microscopy (electron and polarized). Finally, we studied their potential as formulations of BCS II and IV drugs to optimize loading and suppress drug aggregation and crystallization.
Fig. 1.

Schematic drawing of hybrid nanoparticles microfluidic synthesis. Water and lipid/polymer/ethanol supplying fluids are concurrently administered in two separated channels. A herringbone pattern is used to stimulate the chaotic advection of water and lipid/polymer/ethanol laminar streams. Produced monodisperse ethanol-in-water emulsion droplets are stabilized by a lipid layer. Consecutive removal of ethanol from the emulsions by dialysis (at RT for 24 hrs) produces monodisperse and small nanoparticles. Flow conditions and composition enable the formation of multi-layered nanostructures.
Microfluidic Synthesis
We have previously implemented microfluidics synthesis to manufacture LNPs with intricate nanostructures such as cubosomes36. In this study, Kim et al. discovered that the structures obtained in the bulk phase diagram of judiciously chosen lipid mixtures in excess water could be stabilized in nanoparticle and colloidally stable forms. One important aspect was to control the physical-chemical properties of the emulsion droplets developed through the microfluidics process. These cubosomes, termed cuboplexes, were able to encapsulate siRNA in their aqueous domains36,45. In this work, we focus on developing an analogous manufacturing method for the production of hybrid nanoparticles (HNPs) with structures optimized to formulate low water-solubility drugs. Firstly, we set out to optimize the following working parameters: i) the choice of lipid and polymer molecular systems, ii) lipid/polymer compositions, iii) initial stock concentration, iv) flow rate ratio (FRR), and v) total flow rate (TFR). To achieve this, two phospholipid systems and two hybrid polymer/lipid formulations were prepared. We chose molecular systems that have been previously characterized for phase and structural behavior in our lab38,46–49. This includes DOPC (1,2-dioleoyl-sn-glycero-3-phosphocholine), DPPC:Chol (1,2-dipalmitoyl-snglycero-3-phosphocholine:Cholesterol), DOPC:PBD-b-PEO, and DPPC:Chol:PBD-b-PEO. Pure PL (DOPC and DPPC:Chol (10:5 wt/wt ratio equivalent to 50 mol%), pure BCP and hybrid NPs with PL:BCP (4:1 mol ratio) were dissolved in ethanol at specific 8 mg/mL concentration. NPs were produced at 1:1 FRR and 15 mL/min TFR and the effect of the hybridization on the resulting NP size, polydispersity index (PDI), and concentration was investigated post solvent removal (Figure 2). Vesicle size range was characterized by Dynamic Light Scattering (DLS) and concentration of the particles was validated by Nanoparticle Tracking Analysis (NTA).
Fig. 2.

Microfluidic parameters that affect the z-average particle size (blue circles), PDI (pink squares) and NTA nanoparticle flow (snapshots). (A) The effect of TFR for DOPC LNP formulations (2 mg/mL initial PL concentration, 1:1 FRR). (B) The effect of FRR for DOPC LNPs (2 mg/mL initial concentration, 5 mL/min TFR). The effect of adding PBD-b-PEO for (C) DPPC/Chol (1/1 mol ratio) PL (8 mg/mL initial PL concentration, 5 mL/min TFR, 1:1 FRR), and (D) DOPC PL formulations (2 mg/mL initial PL concentration, 5 mL/min TFR, 1:1 FRR). Results represent mean +/− standard deviation (SD), n = 3. Scale bar: 1 μm.
Figure 2 DLS and NTA data obtained for NPs prepared at different compositions and microfluidics flow conditions. We observe that as TFR increases (Figure 2A), DOPC at 2 mg/mL initial concentration and constant 1:1 FRR (blue circles), LNP size decreases while maintaining a reasonably low PDI (pink squares). The NTA data suggests (snapshot images) that increasing TFR results in a NP concentration increase from 0.5 × 1011 NPs/mL to 1.3 × 1011 NPs/mL. NP size and TFR negative correlation trend is in line with previously reported studies where our lab investigated cubosome LNPs36,45. To assess the effect of FRR on NP size and concentration (Figure 2B), we tested the same PL (DOPC) at 2 mg/mL and 5 mL/min TFR. DLS data revealed that NP size decreases from 315.9 nm to 39.7 nm as the FRR (lipid channel to buffer channel) decreases and NTA particle count also decreases from 3 × 1011 NPs/mL to 1.2 × 1011 NPs/mL. As with TFR, FRR variation does not seem to significantly affect the PDI. In summary, the flow ratio affects NP size and concentration much more effectively than the total flow rate. With this experiment, we were able to set optimal flow conditions to prepare PL NPs at a FRR of 1:1 and a TFR of 5 mL/min.
To consider how incorporation of BCP will affect the properties of HNPs, we compared samples using two lipids with different alkyl chain: DOPC having a double bond on the 9th carbon of each hydrocarbon tail (melting temperature Tm of −17 °C) and fully saturated DPPC (Tm of 41 °C). The ability to nanomanufacture LNPs at ambient conditions using high Tm lipids is important as it endows thermal stability to LNPs. To account for this, we incorporated cholesterol (Chol) to facilitate the production of LNPs. The incorporation of 50 mol% cholesterol has been displayed to unite the phase transition temperatures of PLs (lowering the effective Tm of saturated lipids and raising that of unsaturated analogues)50–52. Using the optimized microfluidics flow conditions (FRR of 1:1 and TFR of 5 mL/min), we prepared DPPC LNP formulations containing Chol (50 mol%) at ambient conditions of temperature and humidity.
The results (Figure 2C) suggests that inclusion of cholesterol yields DPPC LNPs of small size (around 200 nm) and confined size distribution (PDI = 0.1). These results confirm that microfluidics can be used in ambient environment to assemble NPs comprising lipids with transition temperatures above room temperature. Figure 2C shows that incorporation of 20 mol% PBD-b-PEO reduces the average HNP size to around 100 nm and concentration (from 6.5 × 1011 to 3.8 × 1011 NPs/mL, presented in Table 1). Analogous results are obtained for DOPC-based HNPs (Figure 2D). We have previously shown Figure 3H that a composition of 20 mol% PBD-b-PEO BCP and 80 mol% DPPC (or DOPC) PL results in well-mixed hybrid membranes in micron-scaled vesicles. An increase in PDI of HNPs is noted which can arise from non-hybridized BCP aggregates circling through the microfluidics set-up. It is noteworthy that the size of the DOPC-based HNPs is around 268 nm versus 100 nm for DPPC-based HNPs. Particle size in the range of 50–250 nm is required for the use of nanomedicine in nano-scaled therapeutics53. This could be attributed to the fact that DPPC-Chol based particles are more stable against NP-NP fusion leads to bigger particles and wider size distribution. Indeed we observe that HNPs are more polydisperse having considerably higher PDI (PDI = 0.6) compared to DPPC HNPs (PDI = 0.2). Table 1 shows how the concentration of nanoparticles is affected for LNPs prepared with DOPC versus DPPC, as well as the effect of hybridization with DPPC/PBD-b-PEO (4:1 mol ratio). In addition to the concentration of particles, size and PDI (Figure 2) blue circles and pink squares, respectively) we also measured the Zeta potential of the NPs. The zeta potential values are within the range of what is recommended for LNPs54,55 but we note that as NPs are hybridized with PBD-b-PEO, the zeta potential decreases from around −29 to −32 mV (DOPC) and −12 to −17 mV (DPPC) wich is consistent with what has been observed previously on the effect of adding polyethylene glycyol (PEG) to NPs55. This confirms that the PBD-b-PEO is indeed hybridized in the phospholipid (DOPC and DPPC/chol) LNP membrane.
Table 1.
Effect of mixing block copolymer with different types of PL (DOPC vs DPPC) on zeta potential and concentration of NPs.
| Sample Information | ζ Potential [mV] | Conc. [# of NPs/mL] |
|---|---|---|
|
| ||
| DOPC LNPs | −29.4 ± 0.7 | (0.5 ± 0.3) × 1011 |
| DPPC LNPs | −12.3 ± 0.2 | (6.5 ± 0.1) × 1011 |
| DOPC HNPs | −31.6 ± 0.5 | (0.4 ± 0.1) × 1011 |
| DPPC HNPs | −16.7 ± 0.3 | (3.8 ± 0.1) × 1011 |
DOPC, hybrid DOPC/PBD-b-PEO (4:1 mol ratio), DPPC, and hybrid DPPC/PBD-b-PEO (4:1 mol ratio) were manufactured using microfluidics at optimal flow rates and ratios. Results represent mean +/− SD, n = 3.
Fig. 3.

Structural characterization of polymer–lipid HNPs (1:4 molar ratio of polymer to lipid). (A) SAXS profiles obtained for hydrated neat PBD-b-PEO (denoted as polymer, blue line), DOPC (pink line), and their mixture (denoted as hybrid, green line). (B) SAXS profiles obtained for corresponding microfluidically synthesized PBD-b-PEO, DOPC, and their mixture NPs. POM image of DOPC NPs in included in the inset. (C,D) Cryo-EM images of (C) DOPC and (D) hybrid DPPC/Chol/PBD-b-PEO NPs. (E) SAXS profiles obtained for hydrated neat PBD-b-PEO (blue line), DPPC/Chol (pink line), and their mixture (green line). (F) SAXS profiles obtained for corresponding microfluidically synthesized PBD-b-PEO, DPPC/Chol, and their mixture NPs. A POM image of DPPC/Chol NPs is included in the inset. (G) Cryo-EM image of DPPC/Chol (50/50, mole ratio) NPs. (H) Cross-sectional CLSM image of a DPPC/PBD-b-PEO (80 mol% DPPC and 20 mol% PBD-b-PEO) hybrid giant multilamellar vesicle (HGMV). (PBD-b-PEO)-FITC and 16:0 Liss Rhod PE were used to label polymer-rich and lipid-rich domains, respectively. Rhod B fluorescence signal in magenta (excitation wavelength at 561 nm and detection wavelength 558–700 nm) and FITC fluorescence signal in green (excitation wavelength at 488 nm and detection wavelength 400–544 nm) are in the merged image of two channels. Scale bar: (C,D,G) 50 nm, (B) 3 μm, (F) 1 μm, and (H) 5 μm.
Structural Characterization of Lipid–Polymer Hybrid Nanoparticles
To elucidate structural properties of PBD-b-PEO and DPPC/DOPC molecules within the hybrid membrane of nanoparticles, we implemented Small Angle X-ray Scattering (SAXS). Figures 3A and 3E show an I vs. q 1D SAXS profiles for the equilibrium highly concentrated (100 mM) bulk lamellar phase in excess water as controls. The SAXS data for pure BCP and pure DOPC (Figure 3A) demonstrates a sequence of peaks at equivalent interpeak distances noted as L00n, where n indicates the diffraction order. The expected SAXS motif emerges from multilamellar arrangement of repeated self-assembled bilayer stacks inside the capillary. The equation d =2πn/qn, where qn corresponds to the location of the nth order diffraction peak, was used to determine the interlayer spacing of the lamellar structure. According to this, the d-spacings of the multilamellar structure in BCP (blue line) and PL (magenta line) are 174 and 65 Å, respectively. In hybrid membrane (green line) lamellar phases arising from both domains are still apparent. Our lab has previously discovered this in the same PL–BCP system and noticed that it emerges as the consequence of correlation between the BCP-rich and the PL-rich domains across many layers38,46,47. Analogous structures can be obtained for other BCPs such as methoxy-poly(ethylene glycol)-b-poly(ε-caprolactone) (mPEG-b-PCL) hybridized with DPPC48. The lamellar d-spacing of DOPC in the hybrid system increased to 78 Å and that of PBD-b-PEO increased to 232 Å. The adjustment of the d spacing is a good indication of the fact that BCPs and lipids are hybridized within the bilayer. Specifically, stretching of hydrophobic domains arise to minimize the interfacial tension between BCP-rich and PL-rich domains and expansion of hydration layers are expected due to the fact that PL and BCP hydrophilic moieties have preferential hydrogen bonding networks56,57.
Figure 3B shows the 1D SAXS profiles obtained for the equilibrium colloidal DOPC PL NPs, BCP NPs, and their hybrid HNPs (20 mM concentration). This consists of various diffraction peaks located at q positions with good correspondence with what was obtained for more concentrated bulk systems (as shown in Figure 3A). Sharp diffraction patterns from lipid-only NPs resulted in lamellar spacing d = 64 Å. Even though optical imaging does not have the resolution to image individual NPs, by applying cross-polarized optical microscopy (POM) we could see the characteristic birefringence signal for a multilayered lipid NP. Suspended BCP NPs showed a dominant form factor signal and a broad L001, L002 and L003 peaks with 146 Å d-spacing. The calculated lattice spacing of NPs decreased compared to that of the bulk BCP. HNPs yield sharp diffraction peaks from the lipid lamellar phase and the first small diffraction peak from the BCP lamellar phase. In HNPs the d-spacing for lipid domains increased to 79 Å (dDOPC =64 Å), while the BCP domain increased to 224 Å (dPBD–b–PEO =174 Å). Colloidal NPs are expected to have higher water contents compared to highly concentrated bulk samples.
SAXS experimental results obtained for DPPC-based NPs are shown in Figure 3E and F. From the 1D SAXS profiles (Figure 3E), the d-spacing of the multilamellar structure in concentrated DPPC bulk samples (magenta line) was calculated to be the same as for DOPC (d = 64 Å). Also in this case the POM image indicates the presence of a multilamellar NP. When mixed in a hybrid membrane, the lamellar d-spacing of DPPC and BCP increased to d = 77 Å and d = 209 Å respectively. From Figure 3F, we were able to extract a lamellar spacing for colloidal samples lower concentration. DPPC-based NPs maintained the same d = 64 Å as in the bulk sample, while the spacing for BCP NPs decreased to d=141 Å ( = 174 Å). The scattering intensity of peaks obtained for low concentration HNPs was not sufficient to determine d-spacing but it should be expected that the values are similar to those obtained for more concentrated samples as seen for all other systems discussed here.
The SAXS and POM data clearly indicates that pure polymer, lipid, and hybrid NPs can be synthesized to adopt a multilamellar structure at low and high concentration. However, SAXS is an average method so we can not extract the structure of individual particles. For this we need to resort to higher resolution electron microscopy methods. Figure 3C and D show Cryo-EM data obtained for NPs synthesized by microfluidics. Cryo-EM images indicate that microfluidic synthesis yields monodisperse NPs with average diameters of 100–200 nm (depending on the molecular system) which is consistent with DLS measurements as well as multilamellarity indirectly observed by SAXS and POM. DOPC NPs (Figure 3C) in general larger (200 nm) than DPPC NPs (100 nm) (Figure 3G) but DOPC NPs are denser comprising more layered bilayers. Average d-spacing values calculated from SAXS of DOPC NPs (d = 64 Å) and DPPC NPs (d = 64 Å) agree well with the distances measured from Cryo-EM images of individual DOPC NPs (d = 63 Å) and DPPC NPs (d = 60 Å). This indicates that NPs are rather uniform structurally throughout the solution. The d-spacing estimated by Cryo-EM image of hybrid DPPC/Chol/PBD-b-PEO NP is 78 Å. In SAXS, the intensity of the peaks for hybrid NPs is not strong enough to resolve d-spacing but the values agree well with d-spacing of the hybrid DPPC/PBD-b-PEO bulk composition (textitd = 77 Å for PL and textitd = 209 Å for BCP). In bulk SAXS, two d-spacings can be resolved but not in Cryo-EM. Further in-depth analysis of d-spacing variation from Cryo-EM images could convey nano-scale domain formation information that is beyond the scope of this paper. The differences in size and number of bilayers in a membrane stack is likely to be determined by membrane elasticity and viscosity properties which are expected to be different for lipid systems in the fluid state (DOPC) or liquid order-state (DPPC with cholesterol)50–52. Significantly, HNPs comprising DPPC PL and PBD-b-PEO BCP are also smaller (around 80 nm in diameter) and we observe that a large portion of the HNPs readily developed multilamellar bilayer stacks (Figure 3D) with a number of layers comparable to DPPC-based LNPs. BCPs are expected to indeed have membrane bending rigidity and viscosity that are higher than fluid lipid membranes as recently shown by Faizi et al.58. To the best of our knowledge this is the first direct imaging proof of multilamellarity in HNPs. These results are important because it indicates that the equilibrium structure of hybrid polymer/lipid membranes derived by SAXS and POM is conserved from the bulk to the nanoscale. Interestingly, preparing a micron-sized hybrid giant multilamellar vesicle (HGMV) comprising DPPC and PBD-b-PEO also results stacking of hybrid bilayers that are denser at the rim. Figure 3H shows confocal laser scanning microscopy (CLSM) images of a HGMV comprising RL-FITC labeled BCP domains (green) and RL-Rhod B labeled lipid domains (magenta). We observe the co-localization of PBD-b-PEO and DPPC fluorescence across layers indicating indeed the presence of a system of hybrid bilayer stacks. Fluorescence intensity variations due to the inherent susceptibility of the dyes to undergo bleaching, dye polarizability, and phase separation into PL-rich and BCP-rich domains can result in higher intensity of the green or magenta signal in certain vesicle regions. Nevertheless, it is clear that the fluorescence co-localization is uniform across layers and the HGMV is indeed hybrid and not comprised of BCP-only or PL-only individual membranes.
In previous work38,46–49 we have shown that in bulk systems, as well as in micro-scale hybrid vesicles using a variety of PLs and BCPs, in-membrane phase separation into PL-rich and BCP-rich domains is common. The phase-separated domain sizes can be sub-nm making it difficult to observe optically but often we can resolve X-ray diffraction patterns characteristic of registry of like-domains across multiple layers. At the nanoscale, HNPs X-ray data at low concentration seem to indicate the presence of BCP and PL diffraction peaks which is consistent with the presence of PL-rich and BCP-rich domains but no clear signs of phase separation, such as differing membrane thickness59, could be clearly detected by Cryo-EM. The hydrophobic discrepancy between the BCP and the PL chains leads to the existence of domains but PEO is anticipated to have more freedom at the water–membrane interface once it is inserted with the small headgroups of PL. Mixing PL with BCP at the molecular scale would weaken some of the PEO steric repulsion. This combined with hydrophobic mismatch would lead to BCP and PL hybridization at the nano or sub-nano scale. Hence, we speculate that HNPs have very small, nano- or sub-nanoscale domains.
Loading of Low-solubility Drugs into Hybrid Nanoparticles
So far, we have shown that size-controlled and rapid synthesis of polymer and lipid NPs as well as hybrid NPs having multiple layers in the NP cores can be achieved with the microfluidics method. These layers consist of alternating and periodic arrays of hydrophobic-hydrophilic environments that we hypothesize could have preferential interactions with drugs of varied levels of lipophilicity. To evaluate the applicability of high-throughput HNPs production as formulation agents of low-solubility drugs we tested the incorporation of DSM265 (an antiparasitic drug) and Paclitaxel (cancer and restenosis drug) (PTX) that categorize as class II and IV drugs of the Biopharmaceutics Classification System (BCS), respectively. The BCS systems defines two dominant factors affecting drug bioavailability: water solubility and permeability. BCS II drugs have high permeability but low solubility and BCS IV compounds have very low solubility and permeability making them very hard to formulate effectively. A critical issue with low solubility drugs is that once in aqueous environments drug-drug interactions are much more favorable than drug-solvent, leading to drug aggregation followed by crystallization that severely hinders drug activity. A suitable NP system would be able to not only encapsulate a tunable amount of drug as well as conserve it in a dispersed state. Drug crystallization is readily detected by X-ray diffraction or Wide Angle X-ray Scattering (WAXS) experiments. We have previously shown that PTX crystallization is significantly hindered when a hybrid DPPC and PBD-b-PEO bulk slab is used to encapsulate PTX38,46,47. In this work we want to evaluate if these properties can be translated into a nanoparticle drug delivery system.
Based on the optimisation results shown in Figure 2, both drugs were formulated within NPs prepared at a FRR of 1:1 and a TFR of 5 mL/min. Drugs were mixed with stock lipid/polymer feeding solution and injected in the anti-solvent channel at nominal concentrations of 4 mol% (PTX) and 5 mol% (DSM265). These formulations contained drugs and stock in the molar ratios of 1:25 (PTX:stock) and 1:20 (DSM265:stock). We evaluated drug crystallization by WAXS for NP systems (Figure 4C and D) and compared the results to corresponding bulk slabs (Figure 4A and B). The results obtained for DOPC, DPPC, PBD-b-PEO and their hybrids is shown in Figure 4. We could confirm our previous results obtained for PTX in bulk (Figure 4B). Several diffraction peaks from PTX crystals can be seen (black line). DPPC is in a gel-state and a peak arising from hexagonally packed alkyl chains can be−1 detected at around q = 1.5 Å−1. As PTX is loaded into a hybrid PBD-b-PEO system (light green line) there are no signs of significant drug crystallization and the DPPC chain packing characteristic peak broadens and shifts q position. This indicates the presence of favorable drug-polymer-lipid interactions over drug-drug and the onset of crystallization. For DPPC-only (light pink line) system we observe several diffraction peaks due to the presence of drug crystals. For the polymer-only (light blue line) system there are some indications of drug diffraction but not extensive. These results fully align with what was observed for NP systems (Figure 4D). However, unlike observed here, our previous studies in bulk systems showed that pure polymer systems had poor PTX dispersion performance.38,46–49. We attribute these differences to sample format. The time scale for clustering, nucleation, and aggregation of drug molecules will depend on the number of membranes (thickness), concentration of the drug, among other factors. Previously, PTX crystallization was evaluated in lipid, polymer, and hybrid films of well-controlled thickness. Here, both NPs and bulk samples are prepared in quartz capillaries to keep sample preparation consistency. We speculate that the difference in results have to do with bulk sample thickness. For example, thicker slabs of polymer-only film samples in our previous study show more drug crystallization than thin slabs of polymer-only quartz capillary samples. However, in uniform NP systems, this is not an issue. Importantly, we could confirm that the trend of lower drug crystallization in hybrid systems is applicable to all bulk and NP systems (in film and quartz capillary formats) as well as other drugs or lipids as it will be discussed below.
Fig. 4.

Simultaneous WAXS profiles of PL, BCP and hybrid membranes (4:1 lipid to polymer molar ratio) with drug integrated, and free drug in DI water. (A) WAXS profiles obtained for hydrated PBD-b-PEO (light blue line), DPPC (light pink line), their mixture in bulk (light green line) with 5 mol% DSM265 integrated, and free DSM265 in DI water (black line). (B) WAXS profiles obtained for hydrated PBD-b-PEO (light blue line), DPPC (light pink line), their mixture in bulk (light green line) with 3 mol% PTX integrated, and free PTX in DI water (black line). (C) WAXS profiles obtained for microfluidically synthesized PBD-b-PEO (blue line), DOPC (pink line), and their mixture NPs (green line) with 5 mol% DSM265. (D) WAXS profiles obtained for microfluidically synthesized PBD-b-PEO (blue line), DPPC/Chol (50/50, mole ratio) (pink line), and their mixture (green line) with 4 mol% PTX integrated.
To evaluate the formulation ability of DSM265, a potent antimalarial currently on clinical trials60–62, we loaded different amounts of drug into bulk slab and NP systems and the results are very promising. Figure 4A shows that that DSM265 in an aqueous environment readily crystallizes and yields several characteristic diffraction peaks (black line). At 5 mol% DSM265 loading into HNPs (faint green line) no diffraction peaks characteristic of DSM265 drug crystals could be detected. The diffraction data for hybrid systems in bulk or NP format are comparable as the only detected diffraction peaks arise from altered DPPC alkyl chain packing which is consistent with the fact that drug is dispersed in the membrane hydrophobic environment. For polymer-only (faint blue line) and DPPC-only (faint pink line) systems several DSM265 crystallization peaks in addition to the characteristic DPPC alkyl chain packing peak are clearly visible.
In general, increasing the encapsulated amount of hydrophobic drug destabilizes traditional liposomal (single bilayer vesicles) drug formulation due to the drug molecule crystallization63,64. Liposomes with 3–4 mol% PTX is the highest reported content with stability for weeks to months while 4–5 mol% PTX is stable in a span of hours to one day, and 8 mol% PTX liposomes remain stable for only 15 minutes65. In this study, samples were prepared days in advance before inspection by WAXS. On the basis of our previous studies of PTX in hybrid lipid-polymer systems38,46,47, we fixed DSM265 loading to 5 mol%, which displayed no signs of drug crystallization.
To evaluate the effect of swapping DPPC for DOPC for the formulation of DSM265 in NPs (Figure 4C) we loaded 5 mol% DSM265 into pure DOPC LNPs (pink line), pure PBD-b-PEO NPs (blue line) and DOPC-PBD-b-PEO HNPs (green line). The results are rather striking. In this case, lipid alkyl chains are disordered and only a very broad peak should be obtained in the q range of 1.3 to 1.5 Å−1. It is perfectly clear that HNPs are far superior systems to hinder DSM265 crystallization and improve its solubility. It is noteworthy that HNP formulations (using any phospholipid) are likely to be more effective at improving drug solubility than lipid-only LNPs. Polymeric materials have vast chemical richness compared to lipids but used in neat form display much higher toxicity. The combination of bio-compatible lipids with polymeric molecular systems that we can be tuned to optimize drug-hybrid membrane interactions is likely to result in enhanced drug dispersion power of hybrid bulk and NP systems.
Conclusions
In this study we utilized benchtop microfluidic devices to manufacture hybrid lipid and polymer nanoparticles of different molecular composition intended for drug formulation. We found that flow conditions in the microfluidic reactor can be tuned to yield monodisperse HNPs in the size range of 100–200 nm in diameter. A comprehensive characterization approach based on X-ray diffraction, scattering, electron and optical microscopy is employed to show that HNPs are dense and highly structured. HNPs comprise periodic concentrically arranged hybrid lipid and polymer bilayers. Depending on the elastic and viscosity properties of the membranes, HNPs adjust the number of bilayers in the stack.
Hybrid systems both in bulk and NP form were almost always more beneficial than pure systems at encapsulating low solubility drugs. The most significant finding was the ability to formulate a potent low-solubility antimalarial (DSM265) under clinical trials that have not yet been successfully formulated. We observe that HNPs can disperse up to 5 mol% impeding DSM265 aggregation, nucleation, and crystallization.
These results show that the nanostructure and the incorporation of block copolymers into LNPs are important factors in designing drug delivery systems, and more research is required to fully resolve how these HNPs will interact with cells and their bio-distribution behavior.
Acknowledgements
This work was supported by the National Science Foundation under Grant No. DMR-1554435 (hybrid membranes) and the National Institutes of Health under Grant No. R01GM143723 (nanoparticles). The x-ray scattering experiments were performed using resources of the Advanced Photon Source, beamline 12-ID-C, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. This research was partially conducted in the Materials Research Laboratory Central Research Facilities, University of Illinois at Urbana-Champaign.
Footnotes
Conflicts of interest
Authors declare no conflict of interests.
Notes and references
- 1.Israelachvili JN, Mitchell DJ and Ninham BW, 1976, 72, 1525–1568. [Google Scholar]
- 2.Bates CM and Bates FS, Macromolecules, 2017, 50, 3–22. [Google Scholar]
- 3.Discher BM, Won Y-Y, Ege DS, Lee JC, Bates FS, Discher DE and Hammer DA, Science, 1999, 284, 1143–1146. [DOI] [PubMed] [Google Scholar]
- 4.Discher DE and Eisenberg A, Science, 2002, 297, 967–973. [DOI] [PubMed] [Google Scholar]
- 5.Kamat NP, Katz JS and Hammer DA, The journal of physical chemistry letters, 2011, 2, 1612–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lee JS and Feijen J, Journal of controlled release, 2012, 161, 473–483. [DOI] [PubMed] [Google Scholar]
- 7.Rideau E, Dimova R, Schwille P, Wurm FR and Landfester K, Chemical society reviews, 2018, 47, 8572–8610. [DOI] [PubMed] [Google Scholar]
- 8.Levine DH, Ghoroghchian PP, Freudenberg J, Zhang G, Therien MJ, Greene MI, Hammer DA and Murali R, Methods, 2008, 46, 25–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Adams D, Gonzalez-Duarte A, O’Riordan WD, Yang C-C, Ueda M, Kristen AV, Tournev I, Schmidt HH, Coelho T, Berk JL et al. , New England Journal of Medicine, 2018, 379, 11–21. [DOI] [PubMed] [Google Scholar]
- 10.Jackson LA, Anderson EJ, Rouphael NG, Roberts PC, Makhene M, Coler RN, McCullough MP, Chappell JD, Denison MR, Stevens LJ et al. , New England journal of medicine, 2020. [Google Scholar]
- 11.Chemin M, Brun P-M, Lecommandoux S, Sandre O and Le Meins J-F, Soft Matter, 2012, 8, 2867–2874. [Google Scholar]
- 12.Lim SK, De Hoog H-P, Parikh AN, Nallani M and Lied-berg B, Polymers, 2013, 5, 1102–1114. [Google Scholar]
- 13.Lipinski CA, Journal of Pharmacological and Toxicological Methods, 2000, 44, 235–249. [DOI] [PubMed] [Google Scholar]
- 14.Lipinski CA, Lombardo F, Dominy BW and Feeney PJ, Advanced drug delivery reviews, 2001, 23, 3–25. [DOI] [PubMed] [Google Scholar]
- 15.Keserü GM and Makara GM, nature reviews Drug Discovery, 2009, 8, 203–212. [DOI] [PubMed] [Google Scholar]
- 16.Amidon GL, Lennernäs H, Shah VP and Crison JR, Pharmaceutical research, 1995, 12, 413–420. [DOI] [PubMed] [Google Scholar]
- 17.Yu LX, Amidon GL, Polli JE, Zhao H, Mehta MU, Conner DP, Shah VP, Lesko LJ, Chen M-L, Lee VH et al. , Pharmaceutical research, 2002, 19, 921–925. [DOI] [PubMed] [Google Scholar]
- 18.Müllertz A, Ogbonna A, Ren S and Rades T, Journal of pharmacy and pharmacology, 2010, 62, 1622–1636. [DOI] [PubMed] [Google Scholar]
- 19.Niwa T and Danjo K, European Journal of Pharmaceutical Sciences, 2013, 50, 272–281. [DOI] [PubMed] [Google Scholar]
- 20.Bhakay A, Davé R and Bilgili E, Powder technology, 2013, 236, 221–234. [Google Scholar]
- 21.Vasconcelos T, Sarmento B and Costa P, Drug discovery today, 2007, 12, 1068–1075. [DOI] [PubMed] [Google Scholar]
- 22.Anselmo AC and Mitragotri S, Advanced drug delivery reviews, 2017, 108, 51–67. [DOI] [PubMed] [Google Scholar]
- 23.Humberstone AJ and Charman WN, Advanced drug delivery reviews, 1997, 25, 103–128. [Google Scholar]
- 24.Chakraborty S, Shukla D, Mishra B and Singh S, European journal of pharmaceutics and biopharmaceutics, 2009, 73, 1–15. [DOI] [PubMed] [Google Scholar]
- 25.Guo S and Huang L, Biotechnology advances, 2014, 32, 778–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Guo X, Wang L, Wei X and Zhou S, Journal of Polymer Science Part A: Polymer Chemistry, 2016, 54, 3525–3550. [Google Scholar]
- 27.Alexis F, Pridgen E, Molnar LK and Farokhzad OC, Molecular pharmaceutics, 2008, 5, 505–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Paris JL, Gaspar R, Coelho F, De Beule P and Silva BF, bioRxiv, 2022. [Google Scholar]
- 29.Rautio J, Kumpulainen H, Heimbach T, Oliyai R, Oh D, Järvinen T and Savolainen J, Nature reviews Drug discovery, 2008, 7, 255–270. [DOI] [PubMed] [Google Scholar]
- 30.Rumondor AC, Dhareshwar SS and Kesisoglou F, Journal of pharmaceutical sciences, 2016, 105, 2498–2508. [DOI] [PubMed] [Google Scholar]
- 31.Huang L, Yang J, Wang T, Gao J and Xu D, Journal of Nanobiotechnology, 2022, 20, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Winzen S, Bernhardt M, Schaeffel D, Koch A, Kappl M, Koynov K, Landfester K and Kroeger A, Soft Matter, 2013, 9, 5883–5890. [Google Scholar]
- 33.Cheng Z, Elias DR, Kamat NP, Johnston ED, Poloukhtine A, Popik V, Hammer DA and Tsourkas A, Bioconjugate chemistry, 2011, 22, 2021–2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Belliveau NM, Huft J, Lin PJ, Chen S, Leung AK, Leaver TJ, Wild AW, Lee JB, Taylor RJ, Tam YK et al. , Molecular Therapy-Nucleic Acids, 2012, 1, e37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Akbarzadeh A, Rezaei-Sadabady R, Davaran S, Joo SW, Zarghami N, Hanifehpour Y, Samiei M, Kouhi M and Nejati-Koshki K, Nanoscale research letters, 2013, 8, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim H, Sung J, Chang Y, Alfeche A and Leal C, ACS nano, 2018, 12, 9196–9205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wan J, Shi L, Benson B, Bruzek MJ, Anthony JE, Sinko PJ, Prudhomme RK and Stone HA, Langmuir, 2012, 28, 13143–13148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kang M, Lee B and Leal C, Chemistry of Materials, 2017, 29, 9120–9132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nam J, Beales PA and Vanderlick TK, Langmuir, 2011, 27, 1–6. [DOI] [PubMed] [Google Scholar]
- 40.Le Meins J-F, Schatz C, Lecommandoux S and Sandre O, Materials today, 2013, 16, 397–402. [Google Scholar]
- 41.Johnson BK and Prud’homme RK, AIChE Journal, 2003, 49, 2264–2282. [Google Scholar]
- 42.Johnson BK and Prud’homme RK, Process and apparatuses for preparing nanoparticle compositions with amphiphilic copolymers and their use, 2012, US Patent 8,137,699. [Google Scholar]
- 43.Markwalter CE, Pagels RF, Wilson BK, Ristroph KD and Prud’homme RK, JoVE (Journal of Visualized Experiments), 2019, e58757. [DOI] [PubMed] [Google Scholar]
- 44.Whitesides GM, Science, 2002, 295, 647651. [DOI] [PubMed] [Google Scholar]
- 45.Kim H and Leal C, ACS nano, 2015, 9, 10214–10226. [DOI] [PubMed] [Google Scholar]
- 46.Kang M, Tuteja M, Centrone A, Topgaard D and Leal C, Advanced functional materials, 2018, 28, 1704356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tuteja M, Kang M, Leal C and Centrone A, Analyst, 2018, 143, 3808–3813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Go YK, Kambar N and Leal C, Polymers, 2020, 12, 1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Go YK and Leal C, Chemical reviews, 2021, 121, 13996–14030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Moghaddam B, Ali MH, Wilkhu J, Kirby DJ, Mohammed AR, Zheng Q and Perrie Y, International journal of pharmaceutics, 2011, 417, 235–244. [DOI] [PubMed] [Google Scholar]
- 51.Pan J, Tristram-Nagle S and Nagle JF, Physical Review E, 2009, 80, 021931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gracia RS, Bezlyepkina N, Knorr RL, Lipowsky R and Dimova R, Soft Matter, 2010, 6, 1472–1482. [Google Scholar]
- 53.Tang L, Yang X, Yin Q, Cai K, Wang H, Chaudhury I, Yao C, Zhou Q, Kwon M, Hartman JA et al. , Proceedings of the National Academy of Sciences, 2014, 111, 15344–15349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Patel VR and Agrawal Y, Journal of advanced pharmaceutical technology & research, 2011, 2, 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Bhattacharjee S, Journal of controlled release, 2016, 235, 337–351. [DOI] [PubMed] [Google Scholar]
- 56.Karathanou K and Bondar A-N, The Journal of Membrane Biology, 2018, 251, 461–473. [DOI] [PubMed] [Google Scholar]
- 57.Dahal UR and Dormidontova EE, Physical Chemistry Chemical Physics, 2017, 19, 9823–9832. [DOI] [PubMed] [Google Scholar]
- 58.Faizi HA, Dimova R and Vlahovska PM, Biophysical Journal, 2022, 121, 910–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Heberle FA, Doktorova M, Scott HL, Skinkle AD, Waxham MN and Levental I, Proceedings of the National Academy of Sciences, 2020, 117, 19943–19952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Phillips MA, Lotharius J, Marsh K, White J, Dayan A, White KL, Njoroge JW, El Mazouni F, Lao Y, Kokkonda S et al. , Science translational medicine, 2015, 7, 296ra111–296ra111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bélard S and Ramharter M, The Lancet Infectious Diseases, 2018, 18, 819–820. [DOI] [PubMed] [Google Scholar]
- 62.Llanos-Cuentas A, Casapia M, Chuquiyauri R, Hinojosa J-C, Kerr N, Rosario M, Toovey S, Arch RH, Phillips MA, Rozenberg FD et al. , The Lancet Infectious Diseases, 2018, 18, 874–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sharma A, Mayhew E, Bolcsak L, Cavanaugh C, Harmon P, Janoff A and Bernacki RJ, International journal of cancer, 1997, 71, 103–107. [DOI] [PubMed] [Google Scholar]
- 64.Çağdaş M, Sezer AD and Bucak S, Application of nanotechnology in drug delivery, 2014, 1, 1–50. [Google Scholar]
- 65.Straubinger RM and Balasubramanian SV, Methods in enzymology, 2005, vol. 391, pp. 97–117. [DOI] [PubMed] [Google Scholar]