

Abstract
Synapse dysfunction and loss are central features of neurodegenerative diseases, caused in part by the accumulation of protein oligomers. Amyloid-β, tau, prion, and α-synuclein oligomers bind to the cellular prion protein (PrPC), resulting in the activation of macromolecular complexes and signaling at the post-synapse, yet the early signaling events are unclear. Here we sought to determine the early transcript and protein alterations in the hippocampus during the pre-clinical stages of prion disease. We used a transcriptomic approach focused on the early-stage, prion-infected hippocampus of male wild-type mice, and identify immediate early genes, including the synaptic activity response gene, Arc/Arg3.1, as significantly upregulated. In a longitudinal study of male, prion-infected mice, Arc/Arg-3.1 protein was increased early (40% of the incubation period), and by mid-disease (pre-clinical), phosphorylated AMPA receptors (pGluA1-S845) were increased and metabotropic glutamate receptors (mGluR5 dimers) were markedly reduced in the hippocampus. Notably, sporadic Creutzfeldt-Jakob disease (sCJD) post-mortem cortical samples also showed low levels of mGluR5 dimers. Together, these findings suggest that prions trigger an early Arc response, followed by an increase in phosphorylated GluA1 and a reduction in mGluR5 receptors.
Keywords: Prion disease, Amyloid, Neurodegeneration, mGluR5, Immediate early genes
1. Introduction
Prion diseases are rapidly progressive neurodegenerative disorders caused by the accumulation of prion protein aggregates (PrPSc) in the central nervous system (CNS) (Caughey and Raymond, 1991; Pan et al., 1993; Prusiner, 1982; Prusiner, 1991). The initial clinical presentation commonly includes behavioral changes, cognitive decline, visual disturbances, ataxia, and / or motor symptoms, depending on the brain region(s) affected (Tee et al., 2018). Synapse loss has been extensively reported as an early neuropathological finding in experimental prion disease models (Belichenko et al., 2000; Brown et al., 2001; Cunningham et al., 2003; Ferrer, 2002; Gray et al., 2009; Jeffrey et al., 2000), and the onset of clinical signs correlates with a marked reduction in synaptic proteins (Cunningham et al., 2003; Siskova et al., 2013). In mouse models, this reduction in synaptic proteins is reportedly due to activation of the unfolded protein response and sustained repression of protein translation (Herrmann et al., 2015; Moreno et al., 2012), yet how PrPSc triggers endoplasmic reticulum stress is unclear. Recent studies indicate that the PrPC is a key mediator of synaptic toxicity at the cell surface, binding PrPSc as well as amyloid-β and tau oligomers, altering macromolecular synaptic complexes, and triggering intracellular kinases, including p38 MAP kinase and Fyn kinase in prion and Alzheimer’s disease, respectively (Fang et al., 2018; Haas et al., 2016; Um et al., 2013; Um et al., 2012). Nevertheless, our understanding of the earliest protein expression changes and signaling cascades initiated by PrPC at the synapse is limited.
PrPC is highly expressed in neurons, in the soma, axons, and pre-synaptic terminal and postsynaptic density (Herms et al., 1999; Moya et al., 2000; Um et al., 2012) and reportedly functions to promote neurite outgrowth (Santuccione et al., 2005), enhance neuroprotection (Bounhar et al., 2001; Roucou et al., 2005), and maintain myelin integrity in the peripheral nervous system (Bremer et al., 2010; Kuffer et al., 2016). PrPC is GPI-anchored in lipid rafts, thus signaling requires interaction with transmembrane protein partners, such as neural cell adhesion molecule (NCAM) (Schmitt-Ulms et al., 2001), metabotropic glutamate receptor 5 (mGluR5) (Haas et al., 2016), or a signaling complex that includes the NMDA receptor and low-density lipoprotein receptor-related protein-1 (LRP1) (Khosravani et al., 2008; Mantuano et al., 2020; You et al., 2012).
Dissecting the early events in prion disease has been challenging, due in part to the variation in results reported from different models, brain regions, prion strains, and timepoints. Temporal gene expression studies of bulk whole brain or specific brain regions have shown pre-clinical alterations in prion infection. However, while some reports indicate that astroglial and microglial transcripts are elevated in early disease and may be a driver of late-stage synaptic loss (Bourgognon et al., 2021; Carroll et al., 2020; Hwang et al., 2009; Sorce et al., 2020), others show neuronal transcript alterations consistent with heightened NMDA receptor activity (Majer et al., 2012). In agreement with the latter, prion exposure of cultured neurons reportedly activates NMDA receptors and causes spine collapse within 24 h (Fang et al., 2016). Collectively, these studies indicate a marked glial response and synaptic loss, yet differ in the sequence of events and in the earliest triggers of neuronal degeneration in the prion-affected brain.
Prion disease models are useful for determining the sequence of events in neurodegeneration, as the disease is initiated by prion exposure and the timing is predictable (Collinge, 2016). Given that synaptic protein alterations and glial activation are implicated in the loss of synapses, we performed an unbiased transcriptome-wide analysis of the hippocampus from prion- or mock-inoculated mice to gain insight into the early disease events, prior to synaptic degeneration. We then followed with a more detailed analysis of protein levels and their post-translational alterations in mice and sCJD patients. We found that immediate early genes (IEGs) were among the most abundant upregulated transcripts in the prion-infected hippocampus. We pursued a specific analysis of Arc/Arg3.1 (activity regulated cytoskeleton-associated protein), one of the most strongly induced genes in response to neuronal firing and a master regulator of long-term synaptic plasticity (Fu and Zuo, 2011; Shepherd and Bear, 2011; Shepherd et al., 2006). By quantitatively measuring changes in Arc, post-synaptic receptors, and synaptic signaling proteins, we begin to show how prions impact glutamate receptor-linked signaling in early disease.
2. Materials and methods
2.1. Prion inoculation of mice
C57BL/6J mice were maintained under specific pathogen-free conditions on a 12:12 light/dark cycle. Mice had access to standard laboratory chow and water ad libitum. Mice (6 – 8 weeks old) were anesthetized with ketamine and xylazine and inoculated into the left parietal cortex with 30 μl of 1% 22L prion-infected brain homogenate prepared from terminally ill mice. Strain 22L is a mouse-adapted prion originally derived from sheep scrapie and was a kind gift from Dr. Michael Oldstone. The 22L prion strain is characterized by diffuse prion aggregates, synaptic and neuronal loss, and microglial activation in the brain (Cunningham et al., 2005).
Mice (n = 3 – 5) were euthanized approximately monthly post-inoculation until clinical disease, and also two weeks after initial clinical symptoms (timepoints: 20, 40, 60, 80, 90, and 100% of the disease course). The timepoints were approximated based on previous studies using the 22L strain, however, the actual time to terminal disease may differ among cohorts. Mice were considered terminal according to clinical criteria including ataxia, kyphosis, stiff tail, hind leg clasp, and hind leg paresis. The brain was halved, and one hemibrain was immediately frozen on dry ice. The other hemibrain was formalin-fixed for 2 – 3 days, then immersed in 96% formic acid for 1 h, washed in water, and post-fixed in formalin for 2–4 days. Transverse brain sections (2 mm) were paraffin-embedded for histological analysis. The remaining brain sections were frozen for biochemical analyses. Three additional cohorts of male mice were inoculated for RNAseq (n = 1) and immunoblotting (n = 3) experiments. The hippocampus was collected and immediately stored in RNAlater™–ICE or frozen on dry ice. No mice were excluded from the analyses. Schematics of the experimental design are displayed in Figs. 1, 2, and 4 (schematics created with Biorender.com).
Fig. 1.
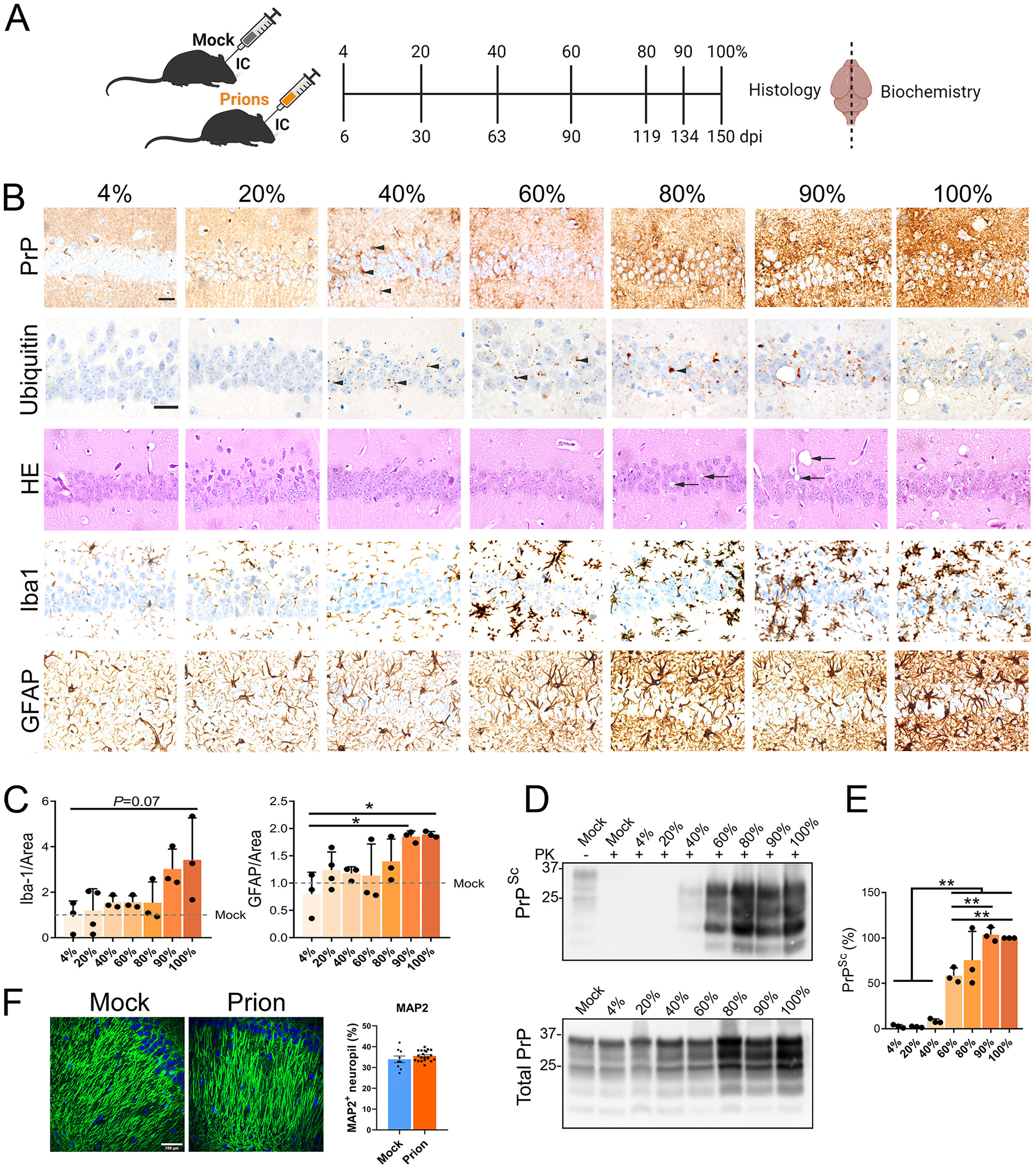
Prion-induced pathology and PrPSc were detected by the 40% timepoint in prion-exposed WT mice. (A) Schematic shows the seven brain collection timepoints inoculation. (B) Representative hippocampal sections from prion-inoculated mice immunolabeled for PrP, ubiquitin, microglia (Iba1), and astrocytes (GFAP), or stained with hematoxylin and eosin (HE). PrPSc and ubiquitin puncta are visible from the 40% timepoint (black arrowheads) and increase to terminal disease. Spongiform degeneration (black arrows) appears in later disease stages. Scale bar, 50 μm. (C) Quantification of Iba1 and GFAP stained area. (D) Immunoblot shows PK-resistant PrPSc (top) and total PrP (PrPC and PrPSc) (bottom) in hemibrain. (E) Quantification of PK-resistant PrPSc normalized to the average terminal PrPSc level (POM19 antibody). (F) Representative images of hippocampal CA1 from mock- or prion-inoculated mice immunolabeled for MAP2 and quantified for MAP2+ neuropil area. Scale bar, 100 μm. For panels C and E, one-way ANOVA and Tukey HSD post-hoc test; *P ≤ 0.05; **P ≤ 0.01; *** P ≤ 0.001, n = 3 – 4 mice per group. For panel F, Welch’s t-test; n = 3 mice (mock) and 7 mice (prion), 3 – 4 images per tissue section.
Fig. 2.
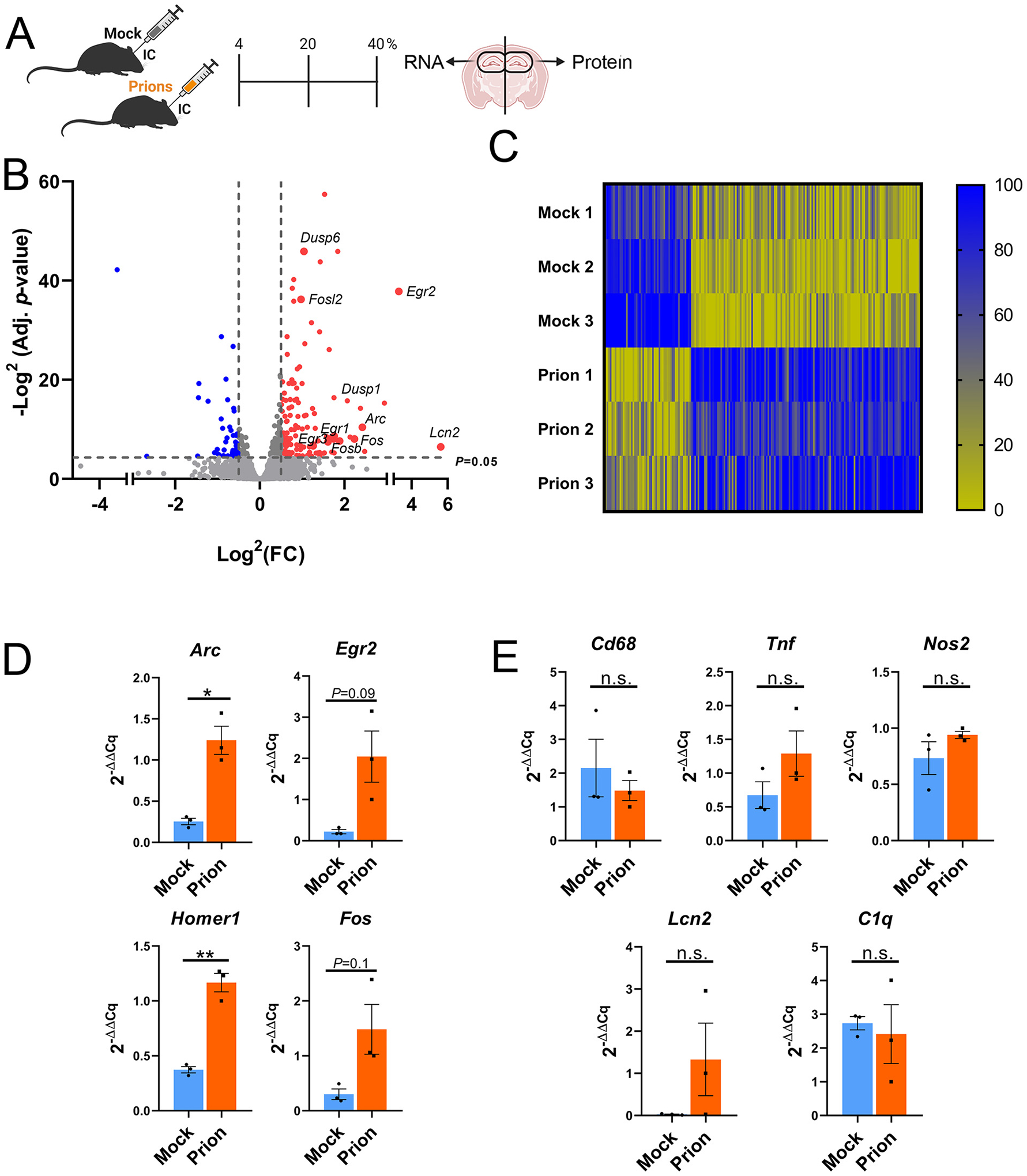
Profiling hippocampal transcripts in mock- and prion-inoculated mice reveals an early increase in immediate early genes. (A) Schematic shows the 40% time point when mock- or prion-inoculated hippocampi were isolated for RNA and protein analysis. (B) Volcano plot shows genes in prion-inoculated hippocampi that significantly differ from mock-inoculated hippocampus. (C) Heat map of genes significantly altered in prion-infected animals. Genes were lane normalized to improve visualization of changes. Within each lane, highest value = 100, lowest value = 0. (D, E) qRT-PCR for IEG genes (D), and inflammation-associated genes (E). n = 3 male mice per group. Welch’s t-test, *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001.
Fig. 4.
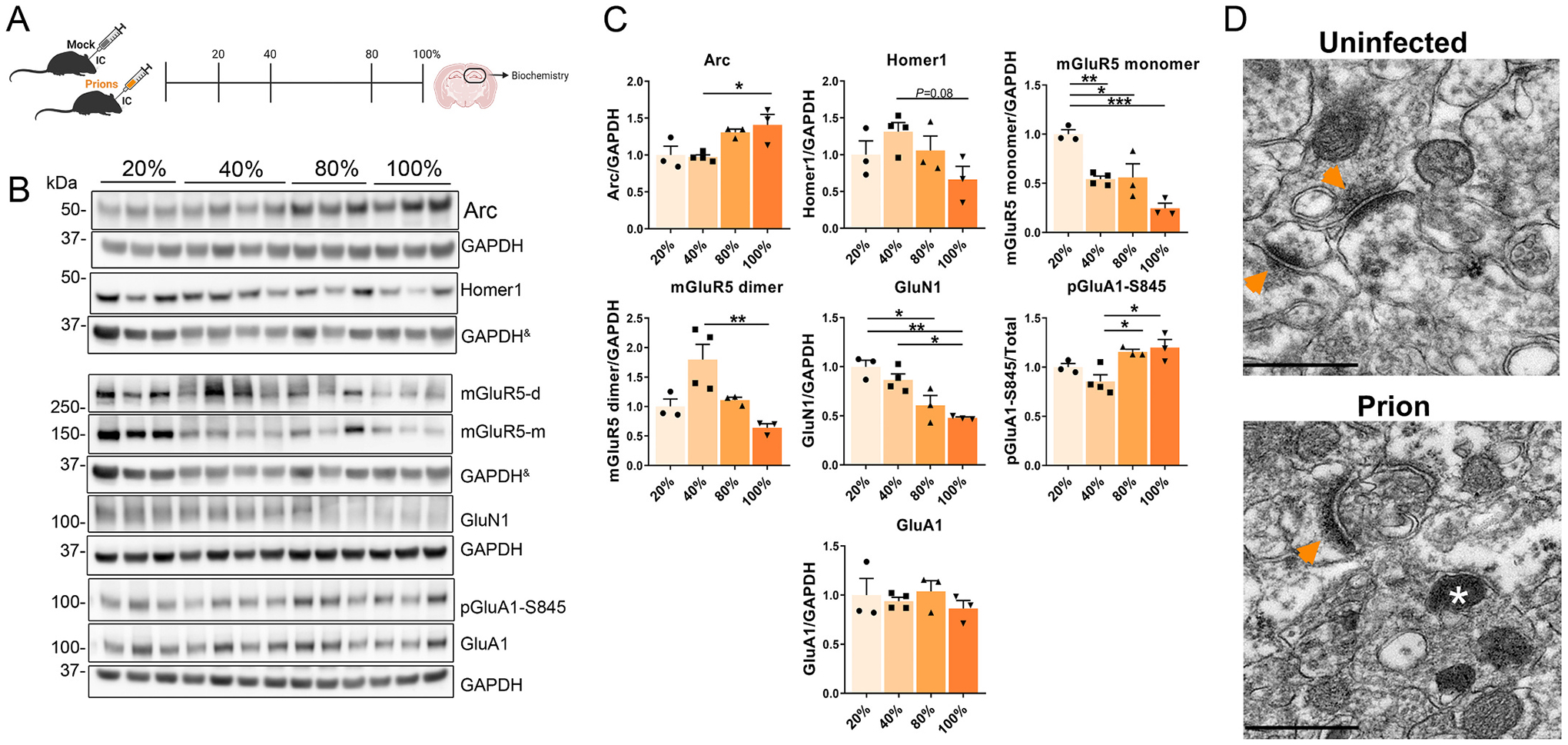
Longitudinal study of IEGs and synaptic proteins in the prion-infected hippocampus. (A) Schematic shows the four timepoints used to interrogate disease-associated protein alterations in the brain. (B, C) Western blot and quantification of IEGs and synaptic proteins, including receptors (mGluR5, GluN1, and GluA1). & indicates the same GAPDH loading controls for proteins immunoblotted on the same membrane. n = 3 – 4 male mice per group, One-way ANOVA and Tukey HSD post hoc test; *P ≤ 0.05; **P ≤ 0.01; ***P ≤ 0.001. (D) TEM images of the CA1 region of the hippocampus, arrowheads = post synapse density; * = lysosome, scale bar = 500 nm.
2.2. Western blots
Hemi-brains were homogenized in PBS (10% w/v) using a Beadbeater™ tissue homogenizer. Hippocampi were similarly homogenized in PBS using either a Beadbeater™ or by vortexing in 200 μl of 2% N-lauryl sarcosine in PBS with Phos-STOP™ (Pierce) and 1.0 mm zirconia-silicate beads (BioSpecific Products). Nucleases [Benzonase™ (Sigma)] and protease inhibitors (Complete TM) were added, and samples were lysed on ice for 15 min, then centrifuged at 2000g at 4 °C for 5 min. Proteins in the supernatant were quantified by bicinchoninic acid assay, and equivalent amounts of protein were electrophoresed through a 10% Bis-Tris gel (Invitrogen) and transferred to a nitrocellulose membrane by wet blotting. Membranes were incubated with primary antibodies overnight at 4 °C followed by incubation with an HRP-conjugated secondary antibody. The immunoblots were developed using a chemiluminescent substrate (ECL Supersignal West Dura or Femto, ThermoFisher Scientific) and visualized on a Fuji LAS 4000 imager. Densitometry analysis was performed using MultiGauge software (Fujifilm).
2.3. PrPSc concentration by sodium phosphotungstic acid (NaPTA) precipitation
PrPSc was concentrated from mouse brains by sodium phosphotungstic acid (NaPTA) precipitation prior to western blotting (Wadsworth et al., 2001). Briefly, 100 μl aliquots of 10% brain homogenate in an equal volume of 4% N-lauryl sarcosine in PBS were incubated for 30 min, then digested with an endonuclease [Benzonase™ (Sigma)] followed by treatment with 100 μg/ml proteinase K (PK) at 37 °C for 30 min. After the addition of NaPTA, MgCl2, and protease inhibitors (Complete TM, Roche), extracts were incubated at 37 °C for 30 min and centrifuged at 18,000 g for 30 min at 37 °C. The pellets were resuspended in 0.1% N-lauryl sarcosine prior to electrophoresis and blotting as described above.
2.4. Histological analyses
Tissue sections were cut from blocks of formalin-fixed, paraffin embedded mouse brains. Tissue sections (4 – 5 μm) were stained using hematoxylin and eosin or immunolabeled using a Ventana Discovery Ultra (Ventana Medical Systems, Tucson, AZ, USA) and antibodies targeting glial fibrillary acidic protein (GFAP) (DAKO), ionized calcium binding adaptor molecule 1 (Iba1) (Wako), PrP (SAF84; Cayman Chemical), and ubiquitin (DAKO). Antigen retrieval was independently optimized for each epitope to yield the maximal signal-to-noise ratio. For PrP, slides were incubated in protease 2 (P2) for 20 min followed by antigen retrieval in CC1 (Tris-based; pH 8.5; Ventana) for 64 min at 95 °C. For GFAP, P2 was used for 16 min. For Iba1 and ubiquitin, CC1 was used for 40 min. Sections were incubated in primary antibody for 32 min at 37 °C, followed by secondary antibody (HRP-coupled goat anti-rabbit; OmniMap system; Ventana) for 12 min at 37 °C, and then 3,3′-diaminobenzidine (DAB) substrate.
For MAP2 immunolabeling, sections were deparaffinized, heated in citrate buffer (Sigma) with 0.05% Tween20 in a pressure cooker for 30 min, quenched in 3% H2O2 in methanol for 15 min, and blocked in TNB buffer [0.5% TSA Blocking Reagent (PerkinElmer) in 100 mM Tris-HCl buffer (pH 7.5) with 150 mM NaCl] containing 5% goat serum. All primary and secondary antibodies were diluted in TNB buffer with 5% goat serum. Slides were then incubated in the primary antibodies, MAP2 (mouse) (Cell Signaling Technologies, #4542; 1:100) overnight at 4 °C. The MAP2 antibody was visualized using biotin-conjugated anti-mouse IgG (Jackson ImmunoResearch) (30 min), then streptavidin-HRP (Jackson ImmunoResearch) (30 min), followed by tyramide-Alexa488 (Invitrogen). All slides were incubated in DAPI and mounted using ProLong Gold (ThermoFisher Scientific).
2.5. Confocal microscopy and microtubule associated protein 2 (MAP2) analysis
Z-stack images (0.5 μm step size) of MAP2 immunolabeled hippocampus (CA1) were acquired using the Eclipse Ti2-E (Nikon) microscope. Images were acquired using the laser scanning confocal mode (A1R HD, Nikon) and an S Fluor 40× NA 1.30 oil objective. All imaging functions were integrated into the NIS elements software (High Content Analysis package).
To determine the MAP2 immunolabeled area, the images were deconvolved, the best focal field selected, and the ImageJ software was utilized to determine the percentage of the area of neuropil covered by MAP2 (% MAP2+ neuropil).
2.6. Transmission electron microscopy of mouse brain
Two mice (uninfected = 1 and terminal 22L = 1) were prepared by immersion fixation of a 2 mm thick coronal section at the level of the hippocampus in modified Karnovsky’s fixative for 2 days. Brain samples were treated for 1 h in 96% formic acid, washed in 0.15 M sodium cacodylate buffer 3 times, and post-fixed in modified Karnovsky’s fixative for 3 days. CA1 hippocampal sections were then immersed in 1% osmium tetroxide in 0.15 M cacodylate buffer for 1 h and stained in 2% uranyl acetate for 1 h. Samples were dehydrated in ethanol, embedded in Durcupan epoxy resin (Sigma-Aldrich), sectioned at 50 to 60 nm on a Leica UCT ultramicrotome, and placed on Formvar and carbon-coated copper grids. Sections were stained with 2% uranyl acetate for 5 min and Sato’s lead stain for 1 min. Grids were viewed using a JEOL 1200EX II (JEOL) transmission electron microscope and photographed using a Gatan digital camera (Gatan).
2.7. qRT-PCR
Brain samples in RNAlater–ICE™ were thawed, lysed, and immersed in 5.3 M guanidine isothiocyanate for 24 h, prior to RNA purification using the PureLink RNA isolation kit (ThermoFisher Scientific). qRT-PCR was performed as previously published with minor modifications (Ojeda-Juarez et al., 2020). Briefly, 500 ng of bulk RNA was reverse transcribed into cDNA using the iScript cDNA Synthesis kit (Bio-Rad). Amplification reactions contained reverse transcriptase, Power PCR SYBR Green Master Mix (Applied Biosystems), and specific primers (designed using Primer-BLAST (https://www.ncbi.nlm.nih.gov/tools/primer-blast/); Supplementary Table 1). PCR amplification was performed on a CFX96 system (Bio-Rad) using the following conditions: 10 min at 95 °C and 40 cycles of 30 s at 95 °C, 1 min at 59 °C, and 1 min at 72 °C. A denaturation step was added at the end of the amplification reaction for Tm analysis. The results obtained were analyzed using Bio-Rad CFX Manager 3.1 (Bio-Rad). The relative amount of mRNA of every gene versus the internal control (GAPDH) was calculated following the 2−(ΔΔCq) method.
2.8. RNAseq
For RNAseq, tissues were homogenized TRIzol (1 mL per 100 mg of tissue) and incubated for 24 h. Samples were then incubated in chloroform for 5 min and centrifuged at 12,000 g for 15 min at 4 °C. The aqueous upper phase containing RNA was transferred to a tube containing an equal volume of 70% ethanol and vortexed. RNA was treated with DNases and isolated using the PureLink RNA isolation kit (ThermoFisher Scientific). Total RNA was assessed for quality using an Agilent Tapestation 4200, and 1 μg of RNA from samples with an RNA Integrity Number (RIN) greater than 7.0 were used to generate RNA sequencing libraries using the TruSeq Stranded mRNA Sample Prep Kit (Illumina, San Diego, CA). Samples were processed following the manufacturer’s instructions, modifying RNA shear time to five min. Resulting libraries were multiplexed and sequenced with 75 basepair (bp) single reads (SR75) to a depth of approximately 25 million reads per sample on an Illumina HiSeq400. Samples were demultiplexed using bcl2fastq Conversion Software (Illumina, San Diego, CA). QC and RNAseq were conducted at the IGM Genomics Center, University of California, San Diego, La Jolla, CA.
2.9. RNA-seq data processing
RNA-sequencing reads were trimmed of adaptor sequences using cutadapt (v1.4.0) and mapped to repetitive elements (RepBase v18.04) using the STAR (v2.4.0i). Reads that did not map to repetitive elements were then mapped to the mouse genome (mm9). GENCODE (v19) gene annotations and featureCounts (v.1.5.0) were used to create read count matrices. Differential expression analysis was performed with DESeq2 v1.32.0 on the raw counts matrix.
2.10. Bioinformatics analysis
Ingenuity Pathway Analysis (IPA; version 70,750,971) was used to analyze 197 hDEG to determine top diseases and disorders, as well as top networks altered. Settings included direct and indirect relationships with the following filters: (tissues = Astrocytes OR Hypothalamus OR Olfactory Bulb OR Pyramidal neurons OR Granule cells OR Substantia Nigra OR Microglia OR Ventricular Zone OR Nucleus Accumbens OR Other Neurons OR Choroid Plexus OR Caudate Nucleus OR Spinal Cord OR Amygdala OR Cerebellum OR Subventricular Zone OR Putamen OR Medulla Oblongata OR Granule Cell Layer OR Dorsal Root Ganglion OR Gray Matter OR White Matter OR Sciatic Nerve OR Cerebral Ventricles OR Parietal Lobe OR Corpus Callosum OR Cerebral Cortex OR Cortical neurons OR Thalamus OR Brain OR Hippocampus OR Purkinje cells OR Striatum OR Trigeminal Ganglion OR Neurons not otherwise specified OR Other Nervous System OR Pituitary Gland OR Nervous System not otherwise specified OR Brainstem).
2.11. Primary antibodies for western blots
The following antibodies were used for western blotting: mouse anti-PrP [1:10,000, POM19, amino acids 201–225; POM1 (Polymenidou et al., 2008)]; anti-beta actin (1:5000, Genetex); anti-GAPDH (1:5000, Novus Biologicals #NB300–221SS); anti-αtubulin (1:1000, Cell Signaling Technologies #3873); anti-synapsin I (1:10,000, Fisher Scientific #AB1543MI); anti-synaptophysin (1:10,000, Invitrogen #MA1213); anti-phosphorylated synapsin I (1:1000; Cell Signaling Technology; #2311); anti-PSD-95 (1:5000, Cell Signaling Technology #3450); anti-SNAP25 (1:10,000, Cell Signaling Technology #5308); anti-mGluR5 (1:5000, Cell Signaling Technology #55920); anti-phosphorylated GluA1-S845 (1:1000, Cell Signaling Technology #8084); anti-GluA1 (1:1000, Cell Signaling Technology #13185); anti-Arc (1:2000, Proteintech #16290–1-AP); anti-EGR1 (1:1000, Cell Signaling Technology #4154); anti-EGR2 (1:1000, Novus Biologicals #NB110–59723SS); anti-Homer1 (1:1000, Cell Signaling Technology #8231); anti-GluN1 (1:1000, Cell Signaling Technology #5704).
2.12. Statistical analysis
Analysis of histopathological data, qRT-PCR analysis for mRNA expression, western blotting data, and Pearson correlation were performed using Prism software (GraphPad Software, Inc.). Comparisons between two groups were made by Welch’s t-test, whereas multiple groups were compared by one-way analysis of variance (ANOVA) followed by Tukey HSD post hoc test. P-values ≤0.05 were considered statistically significant.
3. Results
3.1. Prion infection induces the upregulation of immediate early gene transcripts
The hippocampus is commonly affected in prion disease (Cunningham et al., 2003; Godsave et al., 2008; Hilton et al., 2013), yet the signaling pathways that contribute to early synapse loss and eventual neuronal death are unclear. To determine the early transcriptional changes in the prion-infected hippocampus, we first defined an early disease timepoint with evidence of dysregulated proteostasis. We performed a longitudinal analysis of prion- (strain 22L) or mock-inoculated wild type (WT) mice (C57BL/6), and assessed the hippocampus for PrPSc levels, spongiform degeneration, gliosis, and ubiquitinated inclusions at seven different time points from 6 days post-inoculation (dpi) to terminal disease (approximately 150 dpi; 100% timepoint) (Fig. 1A–D). In the prion-infected hippocampus, PrPSc and ubiquitin inclusions appeared early (63 dpi, 40% of the incubation period) (Fig. 1B). In contrast, spongiform change, astrogliosis, and microgliosis (trending; P = 0.07) were observed in later disease (90–119 dpi; 60–80%), coincident with the onset of clinical signs (Fig. 1B–C). Consistent with the histology, immunoblotting revealed PK-resistant PrPSc at the 40% time point (Fig. 1D–E). At this time, there was no detectable dendritic injury as determined by MAP2 immunolabeling (Fig. 1F).
We therefore performed RNA-seq analysis from prion- and mock-infected hippocampus from male mice at the 40% timepoint (Fig. 2A). Comparison between the hippocampal transcripts of prion- and mock-inoculated mice revealed 334 significantly altered genes (adjusted P ≤ 0.05; Supplementary Table 2). We further focused on 197 highly differentially expressed genes (hDEGs; log2(FC) ≥ 0.5 and ≤ −0.5; Supplementary Table 3), in which 142 genes showed a higher expression and 55 genes showed a lower expression (Fig. 2B–C, Supplementary Table 3). Interestingly, four of the top 10 upregulated genes were the IEGs, Arc, Fos, Fosb, and Egr2. Further analysis revealed that several other IEGs were significantly upregulated, namely Homer1, Dusp1, Nr4a1, Fosl2, Egr1, Egr3, and Dusp6 (Fig. 2C), suggesting an early increase in neuronal activity and synaptic plasticity (Korb and Finkbeiner, 2011; Peebles et al., 2010). Fewer genes were significantly decreased; the top 10 downregulated genes included Ccl28, Calml4, Tmem267, Tnc, Tbx3os1, Acr, and Krt2.
Endolysosomal and autophagy-related transcripts were also increased, including Arl4d, Arl5b, Adam8, Adrb1, Osbpl3, and Lrrk2. Notably, there were limited changes in activated microglia- or astrocyte-associated transcripts, for example, there were no increases in Gfap, Tgfb1, Ptges2, Cd14, Cd68, C1qb, consistent with our histologic findings and reports indicating hippocampal gliosis occurs in mid to late disease (Supplementary Table 2) (Hwang et al., 2009; Vincenti et al., 2015). Sorce and colleagues recently reported gene expression changes in mouse hippocampus at 8 weeks post prion-inoculation (RML strain) (Sorce et al., 2020), finding Homer1 significantly upregulated, Egr1 trending higher, and microglia- or astrocyte-associated transcripts (i.e., Gfap, Tgfb1, Ptges2, Cd14, Cd68, C1qb) unchanged or significantly decreased, which was congruent with our results and suggests IEGs may be increased in early prion disease induced by two distinct strains.
To corroborate the elevated IEG transcripts, we conducted quantitative real-time PCR (qRT-PCR) on hippocampal RNA from the same mice. Congruent with the RNA-seq findings, Arc and Homer1 transcripts were significantly increased and Egr2 was trending high (P = 0.09) (Fig. 2D). Transcripts associated with activated microglia, Tnf, Cd68, and Nos2, were unchanged when compared to mock-inoculated mice (Fig. 2E). Thus, the increase in IEG transcripts suggests that alterations in synaptic plasticity are among the earliest transcriptional responses to prions.
To identify biological functions associated with the hDEG signature, we utilized Ingenuity Pathway Analysis software (IPA). IPA revealed that, of the significantly enriched pathways, ‘Neurological Disease’ had the most DEGs, (111)(P-value: 4.95 × 10−2–1.01 × 10−5). Genes altered in the module included Arc, Egr1, Fos, Dusp6, and other IEGs. Additionally, the top network algorithmically generated based on hDEG, was significantly associated with ‘Neurological Disease’ and ‘Lipid Metabolism’ corroborating the pathway analysis (Fig. S1).
3.2. Prions induce an early increase in Arc, yet a reduction of mGluR5
To determine whether the IEG proteins were also elevated, we measured Arc/Arg-3.1 (referred to hereafter as Arc), EGR1, Homer-1, and c-Fos in the contralateral hippocampus (same brains used for RNA-seq). Consistent with the RNAseq data, soluble Arc and c-Fos were increased by 1.9- and 1.7-fold, respectively, whereas EGR1 and Homer-1 levels were trending higher than in mock brain (2- and 1.4-fold, respectively; P-values = 0.08, and 0.09) (Fig. 3A–B). To verify the increase in Arc, we inoculated an additional cohort (male mice) and again found Arc protein increased in the prion-infected hippocampus at the 40% timepoint (Fig. S2).
Fig. 3.
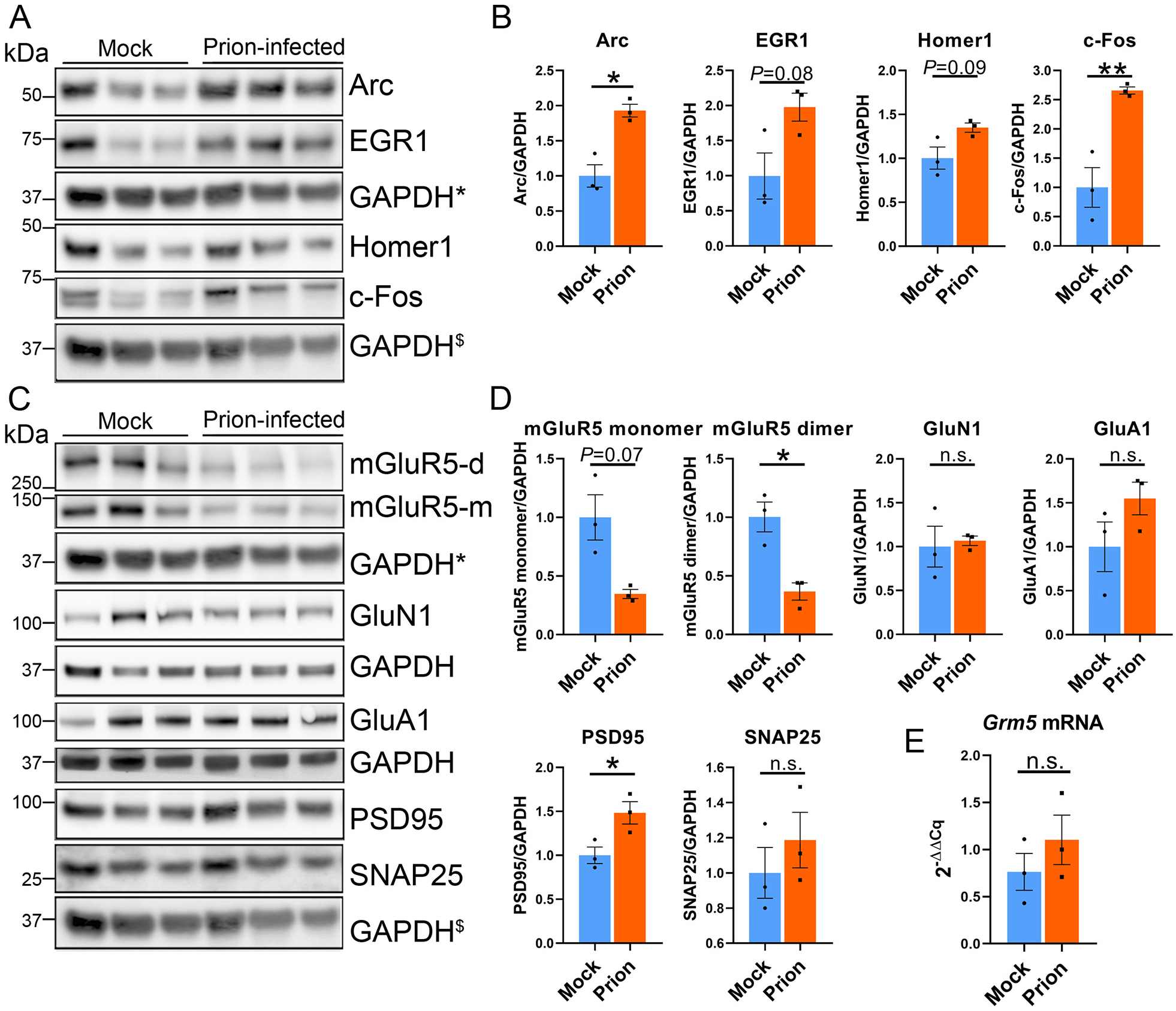
Increased Arc protein correlates with a decrease in mGluR5, but not GluN1 or GluA1. (A, B) Immunoblots and quantification of hippocampal proteins show increases in Arc and c-Fos at the 40% timepoint post-prion exposure. (C, D) Immunoblots and quantification of synapse-associated proteins, including receptors (mGluR5, GluN1, and GluA1), a presynaptic protein (SNAP25), and a postsynaptic protein (PSD-95) reveal a decrease in mGluR5 dimers, and a modest increase in PSD-95. (E) qRT-PCR reveals no change in Grm5 transcripts. * and $ indicate the same GAPDH loading controls for proteins immunoblotted on the same membrane. n = 3 male mice per group. Welch’s t-test, *P ≤ 0.05; **P ≤ 0.01.
Prions reportedly initiate a signaling cascade that includes activation of NMDA receptors (Fang et al., 2018), thus the early elevation in Arc may indicate prion-linked synaptic activity. Arc is required in synaptic plasticity and mediates the endocytosis of post-synaptic receptors, including AMPA receptors, to facilitate long-term depression (LTD) following synaptic stimulation (Waung et al., 2008; Wilkerson et al., 2018). Signaling through mGluR5 and NMDA receptors results in Arc transcription, and primes neurons for homeostatic downscaling. To determine whether mGluR5, NMDA, or AMPA receptors may be altered congruent with elevated Arc, we measured hippocampal levels of mGluR5, GluN1, and GluA1 at the 40% timepoint. While GluN1 and GluA1 levels were unchanged, strikingly, mGluR5 monomer and dimer (membrane-bound) levels were reduced by more than 60% [monomers: 65 ± 4% reduction (trending low); dimers: 63 ± 7% reduction (mean ± standard error (SE)] (Fig. 3C–D). There was no evidence for structural loss to explain the reduced mGluR5, as there was no loss of dendrites at the 40% timepoint (Fig. 1F), or loss in the pre-synaptic vesicle protein, SNAP25, or post-synaptic density proteins; PSD-95 was modestly increased (Fig. 3C–D). Additionally, hippocampal transcripts of Grm5, which codes for mGluR5, were unchanged, indicating that the reduction in mGluR5 protein was not due to changes in gene expression (Fig. 3E).
3.3. Longitudinal study: Increased phosphorylated GluA1 following prion exposure
To define how Arc and synaptic proteins change from early to late disease progression in the prion-infected hippocampus, we performed a longitudinal study of prion and mock-inoculated mice and analyzed hippocampal proteins at approximately 20%, 40%, 80% of the incubation period, and at terminal prion disease (Fig. 4A). There was no change in any of the assessed proteins at the 20% timepoint, yet by the 40% timepoint, Arc was increased as compared to mock controls (Fig. S3A–B), consistent with our previous cohorts. Arc levels peaked at terminal disease (Fig. 4B–C). We then evaluated changes in synaptic proteins and receptors in the prion-inoculated mice. The pre-synaptic proteins, synaptophysin and VAMP2, were reduced over time, whereas phosphorylated synapsin I (pSyn1-S9) was increased at terminal disease (Fig. S4B–C). Notably, mGluR5 monomers had decreased by almost 50% at the 40% timepoint and decreased by approximately 75% at terminal disease (Fig. 4B–C). By comparison, GluN1 levels were also reduced, but not until late disease (80%) (Fig. 4C). Changes in hippocampal levels of Arc, synaptophysin, VAMP2, pSyn1-S9, and mGluR5 were likely due to prions, as protein levels were unaltered in mock-inoculated control mice (Fig. S5).
To probe for synaptic proteins potentially altered by Arc, we next assessed the phosphorylation status of GluA1 at S845, as Arc promotes AMPA receptor internalization. pGluA1-S845 promotes the synaptic retention of GluA1 and facilitates long term potentiation (LTP), and thus can be used as a proxy for surface GluA1 (Banke et al., 2000; Diering et al., 2014; Ehlers, 2000; Esteban et al., 2003; Kim and Ziff, 2014; Lee et al., 2000; Lee et al., 2003; Man et al., 2007; Oh et al., 2006). While total GluA1 levels were unchanged, pGluA1-S845 levels were increased by the 80% time point [also observed when compared to mock (Fig. S3A–B)] and remained elevated at terminal disease (Fig. 4B–C), suggesting GluA1 is at the cell surface. While mGluR5 or NMDA activation drive an increase in synaptic Arc to facilitate AMPA receptor endocytosis (Chowdhury et al., 2006; Wilkerson et al., 2018; Wilkerson et al., 2014), here the persistently high level of pGluA1-S845 suggests that the Arc response was ineffective.
To determine whether there were alterations in synapse structure, we performed transmission electron microscopy (TEM) on terminal prion-inoculated (22 L) mouse brain and uninfected control brain. At terminal disease, the hippocampus showed markedly curved PSDs not observed in the uninfected control (Fig. 4D). Collectively, these data show that prions induce elevated levels of pGluA1-S845 and markedly alter PSD structure.
3.4. Sporadic CJD patients show a marked reduction in mGluR5
Finally, to determine whether IEG and synaptic receptor levels differ in the brain of patients with sCJD, we performed immunoblotting on occipital cortex from sCJD [n = 8, subtypes MM1 and MV1 (n = 2), MM1–2 (n = 2) and MV1–2 (n = 2), MV2 (n = 2)] and control (n = 6) post-mortem samples (Table 1 & Table 2). sCJD patient disease duration ranged from 4 to 27 months and cortical PrPSc levels negatively correlated with disease duration (Pearson r = −0.79; P = 0.02) (Fig. 5A – B). There were no significant alterations in Arc or Homer1 (Fig. 5C), however, most sCJD brains showed a massive reduction in mGluR5 dimers [reduction of 76 ± 8% of control values (mean ± SE)], suggesting low levels of membrane-bound mGluR5 receptors [samples 2 and 5 (MV1–2 and MM1–2) had a less pronounced decrease in dimers] (Fig. 5D–E). The reduction in mGluR5 dimers was specific, as GluN1, GluA1, and mGluR5 monomers were unaltered. Additionally, the levels of pre- and post-synaptic proteins, synaptophysin, VAMP2, and PSD-95, were similar to controls (Fig. 5D–E), despite the control patient population being significantly older (Table 3).
Table 1.
Patient demographic data and clinical features.
| Patient number | Age of onset (years) | Disease duration (months) | PRNP genotype at codon 129 | sCJD subtype | Clinical signs at onset | Western blot: relative occipital PrPSc levels* | MRI: occipital cortex abnormalities |
|---|---|---|---|---|---|---|---|
| 1 | 60 | 20 | MV | 1–2 | Visuospatial | 0.09 | yes |
| 2 | 55 | 24 | MV | 1–2 | Behavior | 0.6 | no |
| 3 | 69 | 6 | MV | 1 | Behavior/Memory | 3.9 | no |
| 4 | 57 | 4 | MM | 1 | Motor | 3.2 | no |
| 5 | 69 | 10 | MM | 1–2 | Cognitive/Apraxia | 2.3 | yes |
| 6 | 69 | 15 | MV | 2 | Behavior/Memory | 0.9 | no |
| 7 | 56 | 10 | MM | 1–2 | Language | 1.2 | no |
| 8 | 79 | 27 | MV | 2 | Cognitive | 1.1 | yes |
Levels relative to a control case (PrP codon 129 MM1).
Table 2.
Demographic data for control cases.
| Patient number | Age at death (years) | Gender | Mini Mental State Examination (MMSE) | BRAAK combo |
|---|---|---|---|---|
| 1 | 80 | F | 29 | 1 |
| 2 | 93 | F | 30 | 1 |
| 3 | 87 | M | 29 | 1.1 |
| 4 | 102 | F | 27 | 1 |
| 5 | 71 | M | – | 1 |
| 6 | 83 | F | 29 | 1.1 |
Fig. 5.
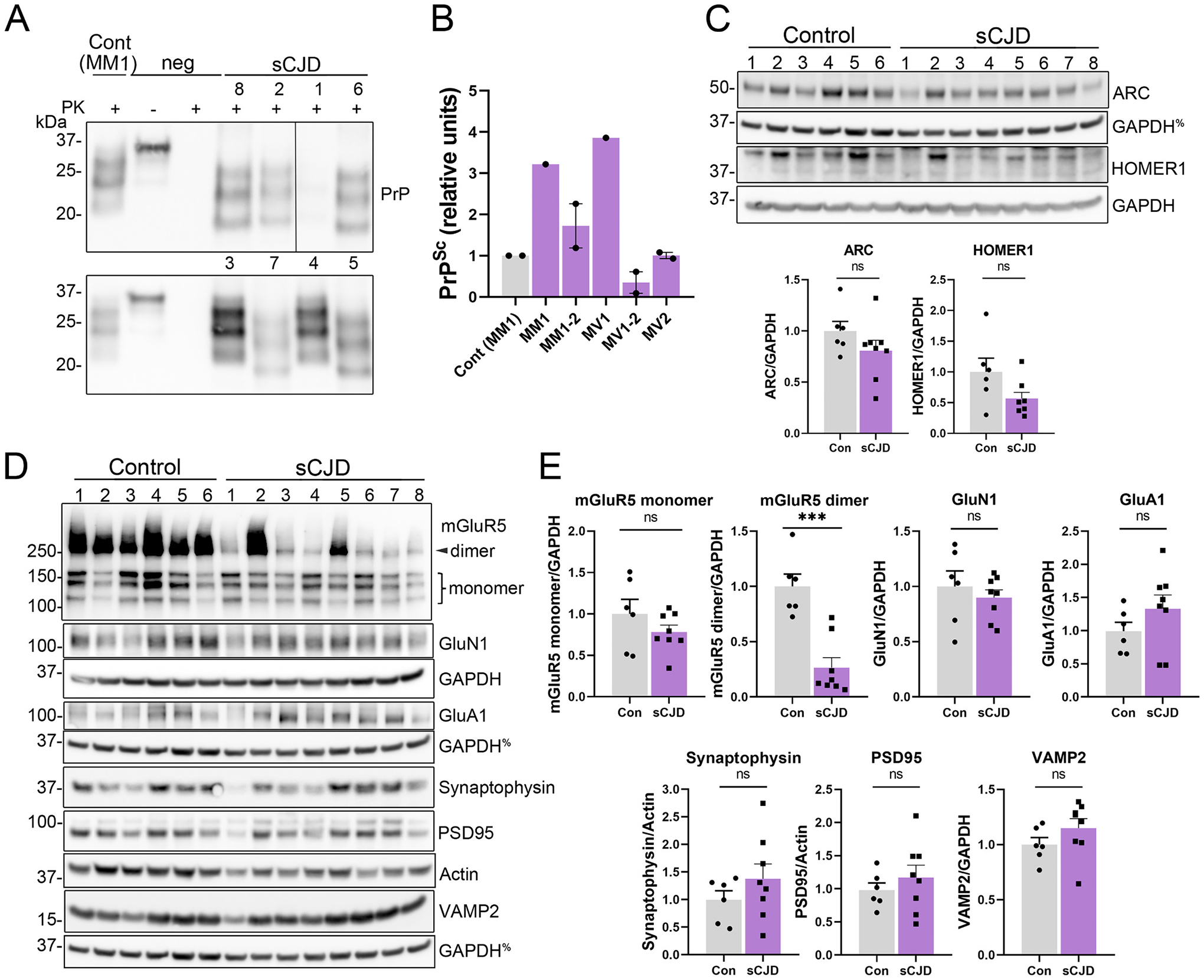
Occipital cortices from sCJD patients show a marked decrease in mGluR5 dimers. (A, B) Western blot and quantification of PK-resistant PrPSc in the occipital cortex of sCJD patients^. (C-E) Western blots and quantification of IEGs and synaptic proteins from control and sCJD patient cortical samples. %indicates the same GAPDH loading controls for proteins immunoblotted on the same membrane. n = 6 controls and n = 8 sCJD patients. Welch’s t-test. **P ≤ 0.01; ***P ≤ 0.001. ^Western blots previously published in (Orru et al., 2018).
Table 3.
Number of subjects, sex, and age at the time of death for control and sCJD subjects for samples used in Fig. 5. Age is shown as mean ± standard deviation (SD). Statistical analysis of age was performed using a Welch’s t-test.
| Table 3 | Occipital cortex | ||
|---|---|---|---|
| Characteristics | Control | sCJD | P value* |
| Number of subjects | 6 | 8 | – |
| Age at death (years) | 86 ± 11 | 66 ± 9 | 0.004 |
| Sex [male (%)] | 2 (33%) | 3 (38%) |
Taken together, these results suggest that prions induce neuronal synaptic activity in vivo, as Arc was upregulated and there was an increase in phosphorylated AMPA receptors. Additionally, mGluR5 dimers were markedly reduced, potentially as a compensatory mechanism for high neuronal activity.
4. Discussion
Synapse loss is thought to be an early response to prion aggregates in the brain, yet the initial molecular events triggered by prions in vivo are unclear. Here we use an unbiased transcriptomic analysis to identify immediate early genes (IEGs) as among the most significant, highly upregulated genes in the hippocampus in early prion disease, preceding the upregulation of inflammatory genes in male mice. Although Arc is required for mGluR5-LTD (Park et al., 2008; Waung et al., 2008), these data suggest that elevated Arc in the prion-infected hippocampus was associated with chronic synaptic activity, with pGluA1 (S845) increasing to terminal disease. Given reports that Arc induction is superseded by high NMDA receptor activity (Steward and Worley, 2001), here we define Arc as an early indicator of a disruption in glutamatergic synaptic homeostasis. A caveat is that the biochemical analysis performed here provides an average of hippocampal synaptic proteins, obscuring the heterogeneity of synaptic responses to prions. Nevertheless, this work reveals post-translational alterations and thereby opens new avenues for selectively evaluating how prions chronically deregulate synaptic homeostasis.
Arc plays a key role in synaptic plasticity by modulating AMPA receptor internalization and promoting LTD (Chowdhury et al., 2006; Zhang et al., 2019; Zhang et al., 2015). Following prion exposure, we observed an increase in Arc, yet with a notable lack of subsequent synaptic downscaling; GluA1 was phosphorylated at S845, suggesting membrane insertion at the post-synaptic density. Further investigation to reveal the cause of the persistent increase in pGluA1-S845 is necessary. Active CaMKII reportedly reduces Arc-driven internalization of AMPA receptors (Okuno et al., 2018; Zhang et al., 2019), thus it is possible that in prion disease, active CaMKII limits the access of Arc to the post-synaptic density, which could explain the seemingly persistent elevation in pGluA1-S845. Alternatively, downstream signaling events required for AMPA receptor down-regulation may be impaired in prion disease.
Previous in vitro work has demonstrated that PrPC and PrPSc interact with NMDA receptors, with one study reporting a direct interaction between PrPC and NMDA receptor subunits (Khosravani et al., 2008). Soluble, full length PrPC was shown to activate cell signaling events in an NMDA receptor- and LRP1-dependent manner, including Src family kinase transactivation of TrkA leading to downstream activation of ERK1/2 (Mantuano et al., 2020). Consistent with PrPC functioning as a ligand, PrPC was reported to reduce NMDA receptor activity by lowering the magnitude of non-desensitizing currents (Huang et al., 2021). In contrast to PrPC, Harris and colleagues showed that PrPSc rapidly activates NMDA receptors, stimulates p38 mitogen-activated protein kinase (MAPK) and other kinases, and leads to spine collapse (Fang et al., 2018). This spine retraction phenotype was prevented by the noncompetitive NMDA channel blocker, memantine, but not the mGluR5 receptor antagonist, 2-methyl-6-(phenylethynyl)-pyridine hydrochloride (MPEP). The current study reveals the early synaptic activity response to prions in vivo, including the early Arc response followed by an increase in pGluA1-S845, which has not been previously described. Collectively these findings suggest chronic synaptic activity and a failure to restore synaptic homeostasis, which could contribute to synapse loss. There was some variability observed in the degree of protein alteration between the cohorts, which may be caused by inter-cohort differences in the kinetics of disease progression. In future studies, it would be important to identify the molecular mechanisms driving the prion-induced increase in Arc as well as the apparent failure of Arc to regulate AMPA receptors.
It is worth noting that mGluR5 dimer levels were reduced in early-to-mid (pre-clinical) experimental prion disease and continued to progressively decrease. Moreover, mGluR5 dimer levels were markedly reduced in the terminal sCJD patient brain as compared to aged control brains. mGluR5 dimers are expressed on the cell surface of neurons and glial cells, and are constitutively endocytosed, with most recycled mGluR5 returning to the cell surface, and the remainder ubiquitinated by the E3 ligase, Siah-1A (Moriyoshi et al., 2004), and degraded in the lysosome (Ko et al., 2012). Reports indicate that PrPC interacts with mGluR5, triggering intracellular signaling (Haas et al., 2014; Um et al., 2013). Interestingly, prion-infected mice treated with an mGluR5 antagonist showed a modestly prolonged survival (Goniotaki et al., 2017). Given that phosphorylated AMPA receptors may be indicators of high synaptic activity, the reduced mGluR5 level is intriguing and raises the question of a possible compensatory downregulation of mGluR5. It remains unresolved whether the reduction in mGluR5 is beneficial or detrimental to synaptic homeostasis, and whether the reduction is strain specific. Also, since astrocytes and microglia also express mGluR5, it is possible that glial mGluR5 levels are altered in prion infection. Future studies to define the mechanism underlying the reduction in mGluR5 dimers, as well as the cell type(s) driving the reduction, would be important to understanding whether the loss is due to degradation of mGluR5 at the synapse. Regardless of the mechanism of mGluR5 loss, PET-ligands that target mGluR5 receptors are currently available (Mecca et al., 2021) and may be immediately useful to explore for diagnosing prion disease and for tracking therapeutic efficacy.
5. Conclusion
We have shown evidence suggesting that prions induce synaptic activity in the hippocampus, with important consequences including AMPA receptor activation in the pre-clinical stage of disease. Future studies to further investigate the cross-talk between glutamatergic receptors in the early response to prions as well as studies of how alterations in synaptic activity cause synapse loss would be warranted.
Supplementary Material
Acknowledgments
We thank the National Prion Disease Pathology Surveillance Center (NPDPSC) for prion typing and all patients and their families for participating in research. We also thank Dr. Nicole Bata for advice on ImageJ image analysis, Karina Barbosa for technical support with the IPA software, Timothy Meerloo and Ying Jones at the UCSD Electron Microscopy Facility for TEM sample preparation, Eric Griffis at the UCSD Nikon Imaging Center for technical assistance, the animal care staff at UC San Diego for excellent animal care, Jeff Metcalf and the laboratory of Dr. Robert Rissman for providing patient samples, and Dr. Julia A. Callender for critical review of the manuscript.
Funding
This study was supported by the National Institutes of Health grants NS069566 (CJS), NS076896 (CJS), NS047101 (UCSD Microscopy Core), 1S10OD023527 (EM Facility), and AG062429 (UCSD Shiley-Marcos Alzheimer’s Disease Research Center). MDG was funded by the National Institutes of Health grants (AG031189; AG062562; AG055619) and the Michael J. Homer Family Fund. JAL was supported by a Ruth L. Kirschstein Institutional National Research Award from the National Institutes of Health (NS103588).
Declaration of Competing Interest
Dr. Geschwind has consulted for Adept Field Consulting (Backbay consulting), Ascel Health LLC, Anderson Boutwell Traylor, Best Doctors Inc., Biohaven Pharma Inc., Bioscience Pharma Partners, LLC (BPP), Clarion Consulting, First Thought Consulting, Grand Rounds Inc./UCSF Second Opinion Inc., Maupin Cox Legoy, Quest Diagnostics, Smith & Hennessey LLC and Trinity Partners LLC. He has received speaking honoraria for various medical center lectures, Oakstone Publishing and Wolters Kluwer. He has received past research support from Alliance Biosecure, CurePSP, the Tau Consortium, Quest Diagnostics, and NIH. Dr. Geschwind serves on the board of directors for San Francisco Bay Area Physicians for Social Responsibility and on the editorial board of Dementia & Neuropsychologia.
Abbreviations:
- MPEP
2-methyl-6-(phenylethynyl)-pyridine hydrochloride
- AMPA
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- Arc
activity regulated cytoskeleton-associated protein
- PrPSc
aggregated prion protein
- PrPC
cellular prion protein
- CNS
central nervous system
- hDEGs
highly differentially expressed genes
- IEGs
immediate early genes
- IPA
Ingenuity Pathway Analysis
- LTD
long-term depression
- LTP
long-term potentiation
- LRP1
low-density lipoprotein receptor-related protein-1
- mGluR5
metabotropic glutamate receptor 5
- MAP2
microtubule associated protein 2
- MAPK
mitogen-activated protein kinase
- NMDA
N-methyl-D-aspartate
- NCAM
neural cell adhesion molecule
- PSD
post-synaptic density
- PK
proteinase K
- qRT-PCR
quantitative real-time polymerase chain reaction
- NaPTA
sodium phosphotungstic acid
- sCJD
sporadic Creutzfeldt-Jakob disease
- TEM
transmission electron microscopy
Footnotes
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.nbd.2022.105834.
Availability of data and materials
The datasets used for the present study are available from the GEO depository (accession number GSE207251). Additionally, a list of hDEGs is available as supplementary material, and all data is available from the corresponding author.
Data availability
Data will be made available on request.
References
- Banke TG, et al. , 2000. Control of GluR1 AMPA receptor function by cAMP-dependent protein kinase. J. Neurosci 20, 89–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belichenko PV, et al. , 2000. Dendritic and synaptic alterations of hippocampal pyramidal neurones in scrapie-infected mice. Neuropathol. Appl. Neurobiol 26, 143–149. [DOI] [PubMed] [Google Scholar]
- Bounhar Y, et al. , 2001. Prion protein protects human neurons against Bax-mediated apoptosis. J. Biol. Chem 276, 39145–39149. [DOI] [PubMed] [Google Scholar]
- Bourgognon JM, et al. , 2021. Inhibition of neuroinflammatory nitric oxide signaling suppresses glycation and prevents neuronal dysfunction in mouse prion disease. Proc. Natl. Acad. Sci. U. S. A 118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bremer J, et al. , 2010. Axonal prion protein is required for peripheral myelin maintenance. Nat. Neurosci 13, 310–318. [DOI] [PubMed] [Google Scholar]
- Brown D, et al. , 2001. Early loss of dendritic spines in murine scrapie revealed by confocal analysis. Neuroreport 12, 179–183. [DOI] [PubMed] [Google Scholar]
- Carroll JA, et al. , 2020. RNA-seq and network analysis reveal unique glial gene expression signatures during prion infection. Mol. Brain 13, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caughey B, Raymond GJ, 1991. The scrapie-associated form of Prp is made from a cell-surface precursor that is both protease-sensitive and phospholipase-sensitive. J. Biol. Chem 266, 18217–18223. [PubMed] [Google Scholar]
- Chowdhury S, et al. , 2006. Arc/Arg3.1 interacts with the endocytic machinery to regulate AMPA receptor trafficking. Neuron 52, 445–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collinge J, 2016. Mammalian prions and their wider relevance in neurodegenerative diseases. Nature 539, 217–226. [DOI] [PubMed] [Google Scholar]
- Cunningham C, et al. , 2003. Synaptic changes characterize early behavioural signs in the ME7 model of murine prion disease. Eur. J. Neurosci 17, 2147–2155. [DOI] [PubMed] [Google Scholar]
- Cunningham C, et al. , 2005. Neuropathologically distinct prion strains give rise to similar temporal profiles of behavioral deficits. Neurobiol. Dis 18, 258–269. [DOI] [PubMed] [Google Scholar]
- Diering GH, et al. , 2014. PKA-GluA1 coupling via AKAP5 controls AMPA receptor phosphorylation and cell-surface targeting during bidirectional homeostatic plasticity. Neuron 84, 790–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehlers MD, 2000. Reinsertion or degradation of AMPA receptors determined by activity-dependent endocytic sorting. Neuron 28, 511–525. [DOI] [PubMed] [Google Scholar]
- Esteban JA, et al. , 2003. PKA phosphorylation of AMPA receptor subunits controls synaptic trafficking underlying plasticity. Nat. Neurosci 6, 136–143. [DOI] [PubMed] [Google Scholar]
- Fang C, et al. , 2016. A neuronal culture system to detect prion Synaptotoxicity. PLoS Pathog 12, e1005623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang C, et al. , 2018. Prions activate a p38 MAPK synaptotoxic signaling pathway. PLoS Pathog 14, e1007283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrer I, 2002. Synaptic pathology and cell death in the cerebellum in Creutzfeldt-Jakob disease. Cerebellum 1, 213–222. [DOI] [PubMed] [Google Scholar]
- Fu M, Zuo Y, 2011. Experience-dependent structural plasticity in the cortex. Trends Neurosci 34, 177–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godsave SF, et al. , 2008. Cryo-immunogold electron microscopy for prions: toward identification of a conversion site. J. Neurosci 28, 12489–12499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goniotaki D, et al. , 2017. Inhibition of group-I metabotropic glutamate receptors protects against prion toxicity. PLoS Pathog 13, e1006733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray BC, et al. , 2009. Selective presynaptic degeneration in the synaptopathy associated with ME7-induced hippocampal pathology. Neurobiol. Dis 35, 63–74. [DOI] [PubMed] [Google Scholar]
- Haas LT, et al. , 2014. Therapeutic molecules and endogenous ligands regulate the interaction between brain cellular prion protein (PrPC) and metabotropic glutamate receptor 5 (mGluR5). J. Biol. Chem 289, 28460–28477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas LT, et al. , 2016. Metabotropic glutamate receptor 5 couples cellular prion protein to intracellular signalling in Alzheimer’s disease. Brain 139, 526–546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herms J, et al. , 1999. Evidence of presynaptic location and function of the prion protein. J. Neurosci 19, 8866–8875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann US, et al. , 2015. Prion infections and anti-PrP antibodies trigger converging neurotoxic pathways. PLoS Pathog 11, e1004662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hilton KJ, et al. , 2013. Early hippocampal synaptic loss precedes neuronal loss and associates with early behavioural deficits in three distinct strains of prion disease. PLoS One 8, e68062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S, et al. , 2021. Mutation of copper binding sites on cellular prion protein abolishes its inhibitory action on NMDA receptors in mouse hippocampal neurons. Mol. Brain 14, 117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang D, et al. , 2009. A systems approach to prion disease. Mol. Syst. Biol 5, 252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffrey M, et al. , 2000. Synapse loss associated with abnormal PrP precedes neuronal degeneration in the scrapie-infected murine hippocampus. Neuropathol. Appl. Neurobiol 26, 41–54. [DOI] [PubMed] [Google Scholar]
- Khosravani H, et al. , 2008. Prion protein attenuates excitotoxicity by inhibiting NMDA receptors. J. Cell Biol 181, 551–565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S, Ziff EB, 2014. Calcineurin mediates synaptic scaling via synaptic trafficking of Ca2+ permeable AMPA receptors. PLoS Biol 12, e1001900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko SJ, et al. , 2012. PKC phosphorylation regulates mGluR5 trafficking by enhancing binding of Siah-1A. J. Neurosci 32, 16391–16401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korb E, Finkbeiner S, 2011. Arc in synaptic plasticity: from gene to behavior. Trends Neurosci 34, 591–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuffer A, et al. , 2016. The prion protein is an agonistic ligand of the G protein-coupled receptor Adgrg6. Nature 536, 464–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HK, et al. , 2000. Regulation of distinct AMPA receptor phosphorylation sites during bidirectional synaptic plasticity. Nature 405, 955–959. [DOI] [PubMed] [Google Scholar]
- Lee HK, et al. , 2003. Phosphorylation of the AMPA receptor GluR1 subunit is required for synaptic plasticity and retention of spatial memory. Cell 112, 631–643. [DOI] [PubMed] [Google Scholar]
- Majer A, et al. , 2012. Early mechanisms of pathobiology are revealed by transcriptional temporal dynamics in hippocampal CA1 neurons of prion infected mice. PLoS Pathog 8, e1003002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Man HY, et al. , 2007. Regulation of {alpha}-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptor trafficking through PKA phosphorylation of the Glu receptor 1 subunit. Proc. Natl. Acad. Sci. U. S. A 104, 3579–3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mantuano E, et al. , 2020. A soluble derivative of PrP(C) activates cell-signaling and regulates cell physiology through LRP1 and the NMDA receptor. J. Biol. Chem 295, 14178–14188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mecca AP, et al. , 2021. Effect of age on brain metabotropic glutamate receptor subtype 5 measured with [(18)F]FPEB PET. Neuroimage 238, 118217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno JA, et al. , 2012. Sustained translational repression by eIF2alpha-P mediates prion neurodegeneration. Nature 485, 507–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moriyoshi K, et al. , 2004. Seven in absentia homolog 1A mediates ubiquitination and degradation of group 1 metabotropic glutamate receptors. Proc. Natl. Acad. Sci. U. S. A 101, 8614–8619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moya KL, et al. , 2000. Immunolocalization of the cellular prion protein in normal brain. Microsc. Res. Tech 50, 58–65. [DOI] [PubMed] [Google Scholar]
- Oh MC, et al. , 2006. Extrasynaptic membrane trafficking regulated by GluR1 serine 845 phosphorylation primes AMPA receptors for long-term potentiation. J. Biol. Chem 281, 752–758. [DOI] [PubMed] [Google Scholar]
- Ojeda-Juarez D, et al. , 2020. Lipocalin-2 mediates HIV-1 induced neuronal injury and behavioral deficits by overriding CCR5-dependent protection. Brain Behav. Immun 89, 184–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuno H, et al. , 2018. Inverse synaptic tagging: an inactive synapse-specific mechanism to capture activity-induced Arc/arg3.1 and to locally regulate spatial distribution of synaptic weights. Semin. Cell Dev. Biol 77, 43–50. [DOI] [PubMed] [Google Scholar]
- Orru CD, et al. , 2018. Prion seeds distribute throughout the eyes of sporadic creutzfeldt-jakob disease patients. mBio 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan KM, et al. , 1993. Conversion of alpha-helices into beta-sheets features in the formation of the scrapie prion proteins. Proc. Natl. Acad. Sci. U. S. A 90, 10962–10966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S, et al. , 2008. Elongation factor 2 and fragile X mental retardation protein control the dynamic translation of arc/Arg3.1 essential for mGluR-LTD. Neuron 59, 70–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peebles CL, et al. , 2010. Arc regulates spine morphology and maintains network stability in vivo. Proc. Natl. Acad. Sci. U. S. A 107, 18173–18178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polymenidou M, et al. , 2008. The POM monoclonals: a comprehensive set of antibodies to non-overlapping prion protein epitopes. PLoS One 3, e3872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prusiner SB, 1982. Novel proteinaceous infectious particles cause scrapie. Science 216, 136–144. [DOI] [PubMed] [Google Scholar]
- Prusiner SB, 1991. Molecular biology of prion diseases. Science 252, 1515–1522. [DOI] [PubMed] [Google Scholar]
- Roucou X, et al. , 2005. Cellular prion protein inhibits proapoptotic Bax conformational change in human neurons and in breast carcinoma MCF-7 cells. Cell Death Differ 12, 783–795. [DOI] [PubMed] [Google Scholar]
- Santuccione A, et al. , 2005. Prion protein recruits its neuronal receptor NCAM to lipid rafts to activate p59fyn and to enhance neurite outgrowth. J. Cell Biol 169, 341–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt-Ulms G, et al. , 2001. Binding of neural cell adhesion molecules (N-CAMs) to the cellular prion protein. J. Mol. Biol 314, 1209–1225. [DOI] [PubMed] [Google Scholar]
- Shepherd JD, Bear MF, 2011. New views of Arc, a master regulator of synaptic plasticity. Nat. Neurosci 14, 279–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepherd JD, et al. , 2006. Arc/Arg3.1 mediates homeostatic synaptic scaling of AMPA receptors. Neuron 52, 475–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siskova Z, et al. , 2013. Brain region specific pre-synaptic and post-synaptic degeneration are early components of neuropathology in prion disease. PLoS One 8, e55004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorce S, et al. , 2020. Genome-wide transcriptomics identifies an early preclinical signature of prion infection. PLoS Pathog 16, e1008653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steward O, Worley PF, 2001. Selective targeting of newly synthesized Arc mRNA to active synapses requires NMDA receptor activation. Neuron 30, 227–240. [DOI] [PubMed] [Google Scholar]
- Tee BL, et al. , 2018. Prion diseases. Neurol. Clin 36, 865–897. [DOI] [PubMed] [Google Scholar]
- Um JW, et al. , 2012. Alzheimer amyloid-beta oligomer bound to postsynaptic prion protein activates Fyn to impair neurons. Nat. Neurosci 15, 1227–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Um JW, et al. , 2013. Metabotropic glutamate receptor 5 is a coreceptor for Alzheimer abeta oligomer bound to cellular prion protein. Neuron 79, 887–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vincenti JE, et al. , 2015. Defining the microglia response during the time course of chronic neurodegeneration. J. Virol 90, 3003–3017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wadsworth JDF, Joiner S, Hill AF, Campbell TA, Desbruslais M, Luthert PJ, Collinge J, 2001. Tissue distribution of protease resistant prion protein in variant CJD using a highly sensitive immuno-blotting assay. Lancet 358, 171–180. [DOI] [PubMed] [Google Scholar]
- Waung MW, et al. , 2008. Rapid translation of Arc/Arg3.1 selectively mediates mGluR-dependent LTD through persistent increases in AMPAR endocytosis rate. Neuron 59, 84–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkerson JR, et al. , 2014. A role for dendritic mGluR5-mediated local translation of Arc/Arg3.1 in MEF2-dependent synapse elimination. Cell Rep 7, 1589–1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkerson JR, et al. , 2018. Roles for arc in metabotropic glutamate receptor-dependent LTD and synapse elimination: implications in health and disease. Semin. Cell Dev. Biol 77, 51–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You H, et al. , 2012. Abeta neurotoxicity depends on interactions between copper ions, prion protein, and N-methyl-D-aspartate receptors. Proc. Natl. Acad. Sci. U. S. A 109, 1737–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, et al. , 2015. Structural basis of arc binding to synaptic proteins: implications for cognitive disease. Neuron 86, 490–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, et al. , 2019. Arc oligomerization is regulated by CaMKII phosphorylation of the GAG domain: an essential mechanism for plasticity and memory formation. Mol. Cell 75 (13–25), e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets used for the present study are available from the GEO depository (accession number GSE207251). Additionally, a list of hDEGs is available as supplementary material, and all data is available from the corresponding author.
Data will be made available on request.