

Abstract
Introduction:
Nicotinamide adenine dinucleotide (NAD) is a coenzyme central to metabolism and energy production. NAD+-dependent deacetylase sirtuin 3 (SIRT3) regulates the acetylation levels of mitochondrial proteins that are involved in mitochondrial homeostasis. Fasting up-regulates hepatic SIRT3 activity, which requires mitochondrial NAD+. What is the mechanism, then, to transport more NAD+ into mitochondria to sustain enhanced SIRT3 activity during fasting?
Objective:
SLC25A51 is a recently discovered mitochondrial NAD+ transporter. We tested the hypothesis that, during fasting, increased expression of SLC25A51 is needed for enhanced mitochondrial NAD+ uptake to sustain SIRT3 activity. Because the fasting-fed cycle and circadian rhythm are closely linked, we further tested the hypothesis that SLC25A51 is a circadian regulated gene.
Methods:
We examined Slc25a51 expression in the liver of fasted mice, and examined its circadian rhythm in wild-type mice and those with liver-specific deletion of the clock gene BMAL1 (LKO). We suppressed Slc25a51 expression in hepatocytes and the mouse liver using shRNA-mediated knockdown, and then examined mitochondrial NAD+ levels, SIRT3 activities, and acetylation levels of SIRT3 target proteins (IDH2 and ACADL). We measured mitochondrial oxygen consumption rate using Seahorse analysis in hepatocytes with reduced Slc25a51 expression.
Results:
We found that fasting induced the hepatic expression of Slc25a51, and its expression showed a circadian rhythm-like pattern that was disrupted in LKO mice. Reduced expression of Slc25a51 in hepatocytes decreased mitochondrial NAD+ levels and SIRT3 activity, reflected by increased acetylation of SIRT3 targets. Slc25a51 knockdown reduced the oxygen consumption rate in intact hepatocytes. Mice with reduced Slc25a51 expression in the liver manifested reduced hepatic mitochondrial NAD+ levels, hepatic steatosis and hypertriglyceridemia.
Conclusions:
Slc25a51 is a fasting-induced gene that is needed for hepatic SIRT3 functions.
Keywords: Mitochondria, NAD, SLC25A51, SIRT3
1. Introduction
Nicotinamide adenine dinucleotide (NAD) is a coenzyme that is critical for metabolism and ATP production. NAD consists of two nucleotides, one containing an adenine nucleobase and the other nicotinamide. Oxidized NAD+ plays critical roles in oxidative metabolism and a range of cellular processes as it is required by multiple enzymes either as a cofactor or as a substrate [1–3].
The NAD+-dependent deacetylase sirtuin 3 (SIRT3) is localized to mitochondria [4–9]. SIRT3 regulates the acetylation levels of mitochondrial proteins that are involved in mitochondrial metabolism and homeostasis. SIRT3 is especially critical in the liver for regulating fatty acid oxidation through the deacetylation of mitochondrial enzymes during fasting [4,10,11].
Circadian cycles and the fed-fasting cycle are closely coupled, and the circadian clock is a molecular oscillator that coordinates nutrient storage and use throughout the daily periods. The circadian clock coordinates mitochondrial oxidative capacity through rhythmic regulation of NAD+ levels through the NAD+ salvage pathway [11,12]. Thus, circadian control of mitochondrial NAD+ availability, along with SIRT3 activity, modulates mitochondrial oxidative function during the fed-fasting cycle.
CLOCK (Clock Circadian Regulator) and BMAL1 (Brain and Muscle ARNT-Like 1) are the core transcription factors that drive the oscillation of genes in a tissue-specific manner throughout the circadian cycles [13]. The rate-limiting enzyme in NAD+ biosynthesis is nicotinamide phosphoribosyltransferase (NAMPT), and levels of intracellular NAD+ display circadian oscillations that are regulated by the core clock machinery CLOCK:BMAL1 [14]. NAMPT is generally considered a cytoplasm-localized enzyme that is upregulated during fasting [14]. During fasting, sustainment of mitochondrial NAD+ levels would be required for SIRT3 to facilitate fatty acid oxidation [11,12], but it is unknown how these levels are controlled.
SLC25A51 was recently identified as a mitochondrial NAD+ transporter in mammalian cells for importing cytosolic NAD+ into the mitochondrial matrix [15–20]. Indeed, SLC25A51-null cells have significant decreases in mitochondrial matrix NAD+ levels, oxidative tricarboxylic acid (TCA) cycle flux, and mitochondrial respiration. Therefore, we hypothesized that the mitochondrial NAD+ transporter SLC25A51 may be upregulated during fasting for sustainment of SIRT3 activity to meet the cell’s energy demand. Because the fasting-fed cycle and circadian rhythm are closely linked, we further hypothesized that SLC25A51 could be a circadian regulated gene.
To test these hypotheses, we examined expression levels of Slc25a51 in the liver of mice treated with fasting and through circadian periods. We also examined effects of depleting Slc25a51 on SIRT3 targets in hepatocytes and the mouse liver.
2. Materials and methods
2.1. Mice
Mice were housed at 22–24 °C with a 14-hour light, 10-hour dark cycle and provided with ad libitum water and a chow diet (6 % calories from fat, 8664; Harlan Teklad, Indianapolis, IN) unless otherwise indicated. Eight-week-old mice were treated with 20-hour fasting with fed mice as controls. To examine circadian rhythm, four-month-old male wild-type (WT) and BMAL1 liver specific KO mice [21] were housed in 12-hour light-dark cycles with free access to food and water for at least 2 weeks before switching to constant darkness for 24 h to allow endogenous clocks to free run. Male C57B6J mice on HFD for one month and control mice were ordered from the Jackson laboratory (strain #:380050 and 380,056, respectively). Mice were anesthetized with isofluorane before undergoing cervical dislocation. Liver samples from three to five mice per time period per genotype group were collected in constant darkness every 3 h for a 24-hour period. All animal protocols were approved by the Animal Care and Use Committee of Wayne State University.
2.2. RNA extraction, quantitative real-time PCR
Dissected tissues were immediately placed into RNAlater solution (Ambion, Austin, TX) for subsequent RNA extraction. Total RNA was isolated from tissues with RNeasy Tissue Mini Kit with deoxyribonuclease treatment (Qiagen, Valencia, CA). One microgram of RNA was reverse transcribed to cDNA using random hexamers (Superscript; Ambion). Relative expression levels were calculated, and β-actin was used as an internal control.
Primer sequences for mouse Slc25a51 were: forward, 5′-ATGATGGACTCCGAAGCACAT-3′; reverse, 5′-GGGTAAGTGATCGCCACGTT-3′. Primer sequences for mouse SIRT3 were: forward, 5′-GAGCGGCCTCTACAGCAAC-3′; reverse, 5′-GGAAGTAGTGAGTGACATTGGG-3′. Primer sequences for mouse β-actin were: forward, 5′-GTGACGTTGACATCCGTAAAGA-3′; reverse, 5′-GCCGGACTCATCGTACTCC-3. The shRNA Clone (TRCN0000123900, The Genetic Perturbation Platform) against Slc25a51 has the target sequence: GCTCGAATACAGTCTCAGATT, and the hairpin sequence: 5′-CCGG-GCTCGAATACAGTCTCAGATT-CTCGAG-AATCTGAGACTGTATTCGAGC-TTTTTG-3′. pLKO.1-puro sequence with the shRNA insert was used to transfect Hepa1-6 cells to generate a stable cell line. The adeno-associated virus serotype 8 (AAV8) to express shRNA targeting Slc25a51 was generated by VectorBuilder (Chicago, IL).
2.3. Co-immunoprecipitation, western blotting analysis, and mitochondrial isolation
Cells were cultured in 10-cm dishes, and grown cells were then transfected with culture medium only (non-treated), scramble shRNA, or Slc25a51 shRNA. Treated cells were harvested in 1 ml of lysis buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 1 mM NaF, 0.1 mM NaVa, 1 % NP-40, and 0.05 % SDS) and subjected to protease inhibitors. Next, cell lysates were collected via centrifugation at 13,000 rpm for 15 min.
For western blot analysis, 20 μg of total cell lysate were separated on SDS-polyacrylamide gel electrophoresis (PAGE) and transferred to a PVDF membrane, before incubation with either rabbit antibodies specific to ACADL (PA5-82450, Fisher) or IDH2 (PA5-82450, Fisher, Hampton, NH). The GAPDH levels were used for normalization. For the co-immunoprecipitation assay, 400 μg of cell lysates were incubated with each antibody ACADL or IDH2-conjugated agarose bead at 4 °C for overnight. The beads with bound proteins were washed for 3 times with a lysis buffer and were subsequently boiled in a 1× SDS-PAGE loading buffer for western blotting to detect the acetylation using anti-acetylated lysine-peroxidase antibody (SAB5200092, Sigma, St. Louis, MO). The band intensity was quantified using ImageJ, and the acetylation levels were normalized to total protein levels. Mitochondria were isolated using the Qproteome Mitochondria Isolation Kit (Qiagen, Hilden, Germany). SIRT activity was measured using the Universal SIRT Activity Assay Kit (Abcam, Cambridge, UK). Triglyceride levels were measured using triglyceride quantification kit (Sigma, St. Louis, MO).
2.4. In vitro circadian synchronization of mouse primary hepatocytes
Primary hepatocytes isolated from C57BL/6J mice were infected with recombinant adenovirus expressing BMAL1 short hairpin RNA (shRNA), or lacZ as a controls, for 24 h before being subjected to serum shock (50 % horse serum) for 2 h for circadian synchronization [22]. After serum shock synchronization, the shock medium was replaced with serum-free medium. Cell lysates were collected at 8-h intervals between 24 h (circadian 0 h) and 72 h (circadian 48 h) post–serum shock for western blot analysis.
2.5. Mitochondrial stress test
Stable cell lines were seeded at 10,000 cells/well onto a 0.1 % gelatin coated Seahorse XFe24 cell culture microplate (#100777-004, Agilent Technologies, Santa Clara, CA, USA) and cultured overnight in DMEM (#10-013-CV, Corning; Manassas, VA, USA) supplemented with 10 % FBS and 1 % penicillin/streptomycin. The next day, growth media was replaced with 675 μL/well XF DMEM media, pH 7.4 (#103575-100, Agilent Technologies; Santa Clara, CA, USA) supplemented with 10 mM glucose (#103577-100, Agilent Technologies; Santa Clara, CA, USA) and 10 mM sodium pyruvate (#103578-100, Agilent Technologies; Santa Clara, CA, USA) without phenol red or FBS. Cells were incubated in a CO2 free incubator for 1 h to degas the media. The oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were simultaneously measured in an XFe24 Seahorse Extracellular Flux Analyzer according to manufacturer’s protocol. Mitochondrial stress tests were performed via sequential injection of 1 μM oligomycin, 2 μM trifluoromethoxy carbonylcyanide phenylhydrazone (FCCP), and 1 μM rotenone/antimycin A using the Seahorse XF Cell Mito Stress Test Kit (#103015-100, Agilent Technologies, Santa Clara, CA, USA) following the manufacturer’s protocol. Calculations for the mitochondrial stress test readouts were performed as specified by the manufacturer.
2.6. Statistical analysis
Data are expressed as the mean ± s.e.m. Statistical significance was tested with unpaired two-tailed Student’s t-tests unless otherwise indicated. One-Way Analysis of Variance (ANOVA) was used to compare the means of three groups, e.g., hepatic expression levels of Slc25a51 in fed, fasting, and re-fed mice. The correlation between expression levels of Slc25a51 and SIRT3 was tested using linear regression analysis with the software OriginPro. The circadian pattern was tested using the Cosinor analysis with cosinor.online [23]. The differences were considered statistically significant if P < 0.05.
3. Results
3.1. Slc25a51 expression is induced by fasting in the mouse liver
We hypothesized that expression of Slc25a51 is induced by fasting in the liver because Slc25a51 may be needed to transport NAD+ into mitochondria when energy demand is high. To test the hypothesis, we examined Slc25a51 expression in the mouse liver. The experimental group contained both male and female mice, and the mice were fasted for 20 h. qPCR analysis was then performed to examine Slc25a51 expression.
Fasting increased liver Slc25a51 expression in both male (Fig. 1A) and female mice (Fig. 1B) at least 2-fold. We next examined liver Slc25a51 expression in male mice treated with 20 h of fasting followed by 4 h of refeeding. Fasting induced Slc25a51 expression in the liver, and Slc25a51 expression returned to its basal level 4 h after refeeding (Fig. 1C). In other words, expression of liver Slc25a51 was significantly reduced (>70 %) 4 h after refeeding, compared to fasted mice. Therefore, Slc25a51 is a fasting-regulated gene in the mouse liver.
Fig. 1.
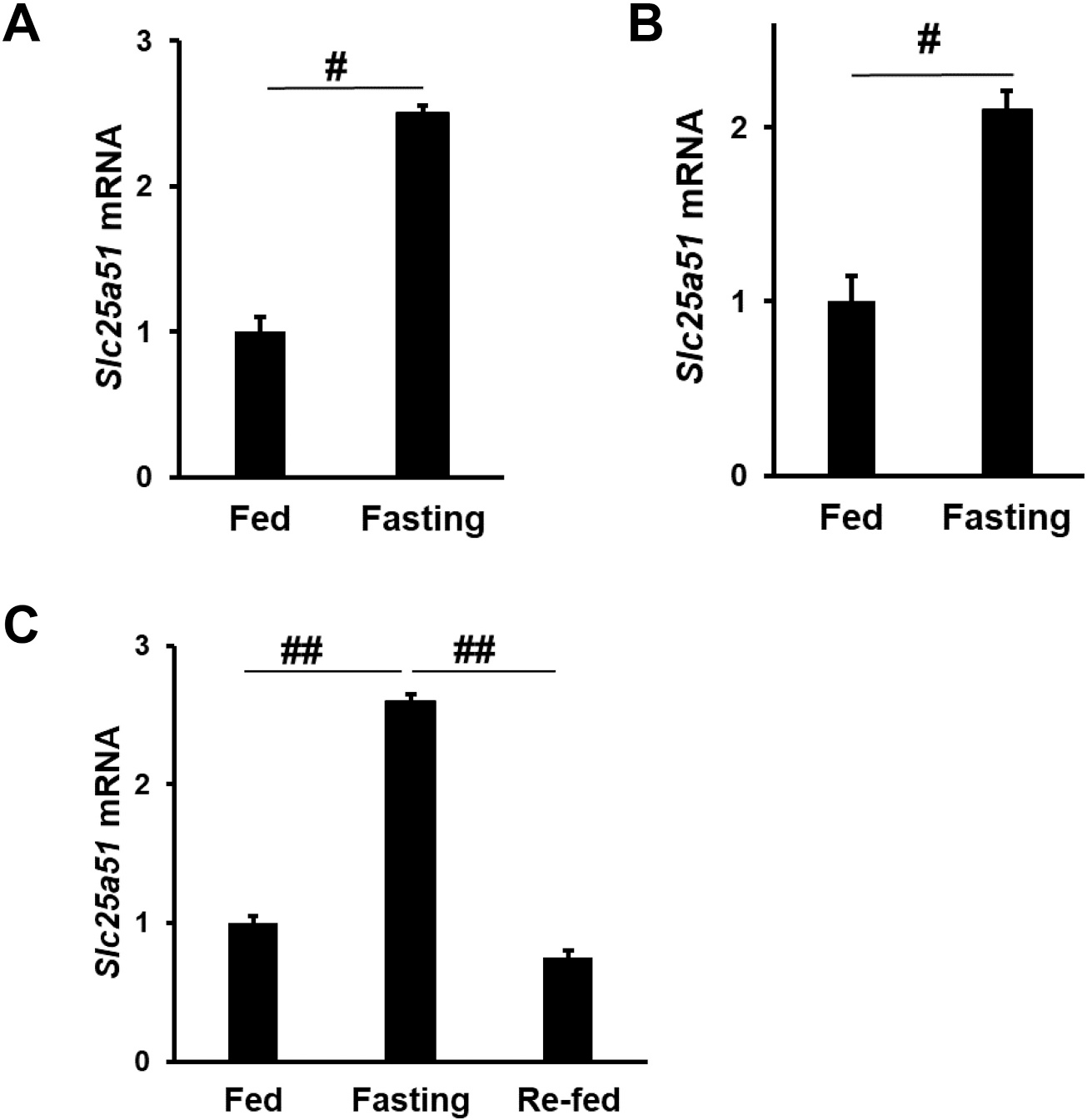
Slc25a51 is induced by fasting in the mouse liver. Hepatic mRNA levels of Slc25a51 in male (A) and female mice (B) with or without 20-hour fasting. Male mice, N = 3 per group. Female mice N = 5 per group. (C) Hepatic mRNA levels of Slc25a51 in male mice with 20-hour fasting, followed by 4-hour re-feeding. N = 4, 5, 3, for the fed, fasting, and re-fed groups, respectively. #, p < 0.01. ##, p ≪ 0.01. Data are represented as mean ± SEM.
3.2. Expression of Slc25a51 correlates with that of SIRT3
It has been established that liver SIRT3 is induced by fasting [4]. To examine the relationship between SIRT3 and Slc25a51, we examined liver SIRT3 expression levels in the same samples used for analyzing Slc25a51 expression. Fasting significantly induced the expression of SIRT3 in the liver of both male (Fig. 2A) and female mice (Fig. 2B), consistent with previous reports [4]. Four hours after refeeding, expression levels of liver SIRT3 returned to basal levels (Fig. 2C).
Fig. 2.

Slc25a51 expression is correlated with that of SIRT3 in the mouse liver. Hepatic mRNA levels of SIRT3 in male (A) and female mice (B) with or without 20-hour fasting. Male mice, N = 3 per group. Female mice N = 5 per group. (C) Hepatic mRNA levels of SIRT3 in male mice with 20-hour fasting, followed by 4-hour re-feeding. N = 4, 5, 3, for the fed, fasting, and re-fed groups, respectively. #, p < 0.01. ##, p ≪ 0.01. Data are represented as mean ± SEM. (D) Correlation between the expression levels of Slc25a51 and SIRT3 in mice subjected to fasting and refeeding.
Slc25a51 and SIRT3 showed similar patterns of expression in response to fasting and refeeding, so we analyzed the correlation of their expression levels. Linear regression showed that expression of Slc25a51 significantly correlated with that of SIRT3 (R = 0.77, P ≪ 0.01) (Fig. 2D). Therefore, hepatic expression of SIRT3 and Slc25a51 correlated in vivo in response to fasting and re-feeding.
3.3. Slc25a51 expression showed a circadian rhythm-like pattern in primary hepatocytes and was controlled by BMAL1
A key metabolic aspect of circadian oscillations is a natural fast-fed cycle that corresponds in rodents with day and night activity. Bass’ group found that NAD+ levels are regulated by the circadian transcription factor CLOCK, and that a NAD+ cycle drives mitochondrial oxidative metabolism [11]. Because Slc25a51 transports NAD+ into mitochondria, Slc25a51 expression may also be regulated by circadian rhythm.
To test this hypothesis, we first examined the circadian rhythm of Slc25a51 in primary hepatocytes using serum shock, which is a method to study circadian rhythm in cells [22]. We examined Slc25a51 expression in primary hepatocytes subjected to serum shock for circadian synchronization.
Although the Cosinor analysis, a statistical method that tests rhythms, did not show a significant 24-h rhythm (P = 0.15), over a period of 48 h, Slc25a51 showed a circadian rhythm-like pattern by having a statistically significant higher expression at hour C8 than at hour C24. Consistently, Slc25a51 expression is higher at hour C32 than at hour C48 (Fig. 3A). Therefore, Slc25a51 expression in hepatocytes exhibits a circadian rhythm-like pattern, consistent with a previous report [24].
Fig. 3.
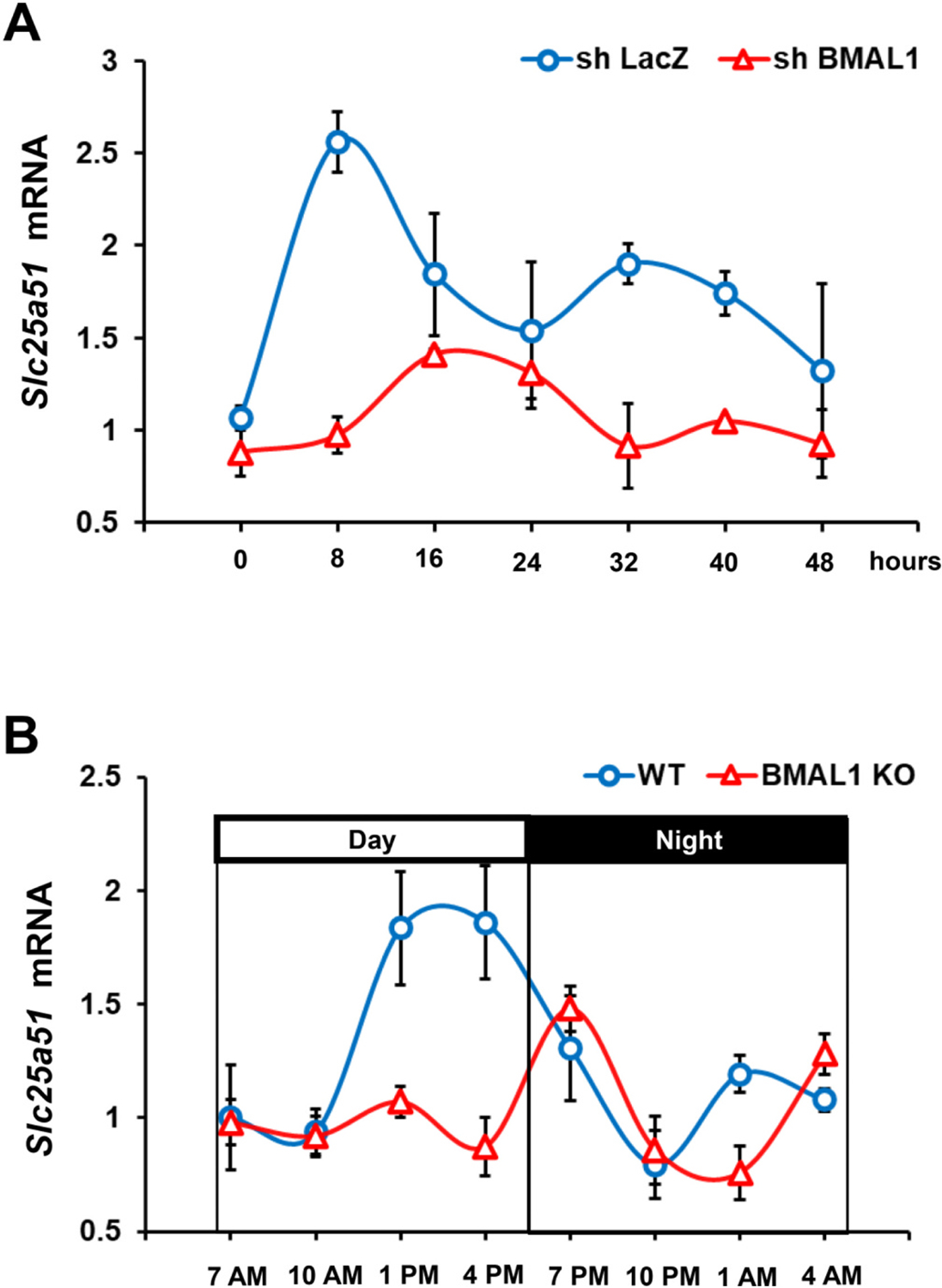
Liver Slc25a51 expression showed a circadian rhythm-like pattern and is a downstream target of BMAL1. A) Slc25a51 expression in primary hepatocytes with or without BMAL1 knockdown. Primary hepatocytes isolated from C57BL/6J mice were infected with recombinant adenovirus expressing BMAL1 short hairpin RNA (shRNA) for 24 h before being subjected to serum shock (50 % horse serum) for 2 h for circadian synchronization. After serum shock synchronization, the shock medium was replaced with serum-free medium. Cell lysates were collected at 8-h intervals between 24 h (circadian 0 h) and 72 h (circadian 48 h) post–serum shock. B) Slc25a51 expression in the liver collected from WT mice and mice with liver-specific KO of BMAL1 every 3 h during the 24 h circadian period. Data are represented as mean ± SEM.
BMAL1 is the core circadian oscillator that is critical in controlling the expressions of circadian related genes. To test whether BMAL1 regulates Slc25a51 expression, we knocked down the expression of BMAL1 using the adenoviral expression of BMAL1 shRNA. Cells were then subjected to serum shock for circadian synchronization. Interestingly, the circadian pattern of Slc25a51 expression was disrupted in hepatocytes with reduced expression of BMAL1 (Fig. 3A), indicating that Slc25a51 expression is under the control of BMAL1.
3.4. Slc25a51 expression showed a circadian rhythm-like pattern and was controlled by BMAL1 in the mouse liver
We next examined Slc25a51 expression in livers collected from mice every 3 h during the 24-hour circadian period. Expression levels of Slc25a51 during the daytime were higher than at night (Fig. 3B). However, the Cosinor analysis revealed a circadian rhythm-like pattern (P = 0.07). The reason for not reaching statistical significance was likely due to the limited data point (samples were collected every 3 h), because in CircaDB, a database of mammalian circadian gene expression profiles [25], Slc25a51 (samples were collected every hour [26]) showed a statistically significant 24-hour pattern (Supplemental Fig. 1).
To test whether BMAL1 regulates Slc25a51 expression in vivo, we examined the expression of liver Slc25a51 in mice in which BMAL1 was specifically deleted in the liver. Liver samples were collected from liver-specific BMAL1 knockout (LKO) mice every 6 h during a 24-hour circadian period, and the same regimen was performed in control mice. The circadian regulation of Slc25a51 seen in the WT mice was disrupted in the LKO mice. During the daytime, expression of Slc25a51 in WT mice was higher than that in the LKO mice (Fig. 3B). These results show that Slc25a51 is a downstream target of BMAL1 in vivo.
3.5. Reduced Slc25a51 expression suppressed mitochondrial NAD+ levels and SIRT3 activity in hepatocytes
To determine the mitochondrial enzymatic activities supported by Slc25a51 in liver, we used a previously published and validated shRNA [15] to deplete Slc25a51 in Hepa 1–6 cells, a mouse hepatocyte cell line. Expression of the shRNA reduced Slc25a51 mRNA expression about 80 % relative to the scramble shRNA in Hepa 1–6 cells (Fig. 4A). We examined NAD+ levels in mitochondria isolated with the Qproteome Mitochondria Isolation Kit. Total mitochondrial NAD+ levels were decreased approximately 70 % in cells with the Slc25a51 knockdown (Fig. 4B).
Fig. 4.

Reduced Slc25a51 expression suppressed mitochondrial NAD+ levels and SIRT3 activity in hepatocytes. A) shRNA mediated knockdown of Slc25a51 in Hepa 1–6 cells, with scramble RNA as controls. B) Mitochondrial NAD+ levels in Hepa 1–6 cells with reduced Slc25a51 expression. C) Mitochondrial SIRT activity in Hepa1-6 cells with Slc25a51 knockdown. (D) Western blotting analysis using acetylated lysine specific antibody following immunoprecipitation using IDH2 and ACADL antibodies. Band intensity quantification for acetylation of IDH2 (E) and ACADL (F) was performed using ImageJ, normalized to the total levels of IDH2 and ACADL, respectively. G) TG level in Hepa1-6 cells with Slc25a51 knockdown. Data are represented as mean ± SEM. #, P < 0.01.
The enzymatic activity of SIRT3 requires NAD+ and it has a KM(NAD+) of ~100 μM [27]. Thus, SIRT3 activity is poised to be impaired by diminishment of mitochondrial NAD+, and SIRT3 is also a predominant deacetylase in hepatic mitochondria. To test whether loss of Slc25a51 impairs mitochondrial SIRT deacetylase activity, we examined deacetylase activities in isolated mitochondria from cells depleted of Slc25a51. Deacetylase activity was reduced in mitochondria of cells with Slc25a51 knockdown (Fig. 4C). We further examined the acetylation levels of two established SIRT3 mitochondrial targets, IDH2 and ACADL [28]. Western blot analyses using pan acetyl-lysine antibodies with immunoprecipitation of either IDH2 or ACADL showed that both of these targets had increased acetylation levels when Slc25a51 was depleted (Fig. 4D–F). Consistent with a link between reduced SIRT3 activity and increased triglyceride (TG) accumulation [29], the TG content was increased in cells with Slc25a51 knockdown (Fig. 4G).
Overall the data indicated that depletion of Slc25a51 reduced SIRT3 activity through the limitation of mitochondrial NAD+ concentrations.
3.6. Reduced Slc25a51 expression suppressed oxygen consumption in hepatocytes
We examined mitochondrial oxygen consumption rate (OCR) using a Seahorse Extracellular Flux Analyzer to do real-time and live cell analysis in hepatocytes. Glycolytic activity of these cells was also indirectly determined by the extracellular acidification rate (ECAR), which measured lactic acid production as an indicator of glycolytic activity [30]. Both OCR and ECAR were suppressed in the cells with reduced Slc25a51 expression (Fig. 5A, B). Basal respiration of the knockdown cell line was reduced 18 % compared to the control (Fig. 5C). During the mitochondrial stress test, the knockdown cells showed a 20 % reduction in ATP-coupled respiration after the injection of oligomycin (Fig. 5D) and a 26 % reduction in maximal respiration after the injection of FCCP compared to scramble controls (Fig. 5E). Therefore, reduced Slc25a51 expression suppresses oxygen consumption in hepatocytes.
Fig. 5.

Reduced Slc25a51 expression suppressed oxygen consumption in hepatocytes. A Seahorse Extracellular Flux Analyzer was used to determine (A) oxygen consumption rate (OCR), (B) extracellular acidification rate (ECAR), (C) Basal respiration, (D) ATP-coupled respiration, and (E) maximal respiration in Hepa 1–6 cells with shRNA mediated Slc25a51 knockdown, with scramble RNA as controls. Data are represented as mean ± SEM. #, P < 0.01.
3.7. Reduced Slc25a51 expression in the mouse liver caused hepatic steatosis and hypertriglyceridemia
In hepatocytes, reduced Slc25a51 expression suppressed mitochondrial NAD+ levels and increased acetylation levels of SIRT3 targets, and functionally, caused reduced oxygen consumption and TG accumulation. We therefore hypothesized that in mice reduced Slc25a51 expression in the liver affects hepatic mitochondrial NAD+ levels, acetylation levels of SIRT3 targets, and TG metabolism. To test the hypothesis, we performed tail-vein injection of adeno-associated virus serotype 8 (AAV8) to express shRNA targeting Slc25a51, with scramble AAV8-shRNA as controls. Two weeks following virus injection, livers were dissected from 20-hour fasted mice for analysis.
AAV8-mediated hepatic expression of shRNA-Slc25a51 effectively reduced Slc25a51 expression by approximately 30 %, compared to the mice with control scramble shRNA (Fig. 6A). Reduced Slc25a51 expression consistently reduced the level of the mitochondrial NAD+ levels (Fig. 6B). Because SIRT3 activity depends on NAD+, we then investigated the SIRT3 activity by examining the acetylation levels of SIRT3 target proteins IDH2 and ACADL. Western blot analyses using pan acetyl-lysine antibodies with immunoprecipitation of either IDH2 or ACADL showed that both of these targets had increased acetylation in the livers with reduced Slc25a51 expression (Fig. 6C–E). Because reduced SIRT3 activity is associated with hepatic steatosis, we did Oil Red O staining, which is a method to stain TG. We observed an obvious increase in Oil Red O staining in mice with reduced Slc25a51 expression (Fig. 6F), and we then examined liver TG level, which was consistently significantly increased (Fig. 6G). We further examined circulating TG levels, and the mice with reduced Slc25a51 expression showed hypertriglyceridemia (Fig. 6H).
Fig. 6.

Reduced Slc25a51 expression in the mouse liver suppressed mitochondrial NAD+ levels and caused hepatic steatosis and hypertriglyceridemia. (A) AAV8-shRNA mediated knockdown of Slc25a51 expression in the mouse liver, with scramble AAV8-shRNA as controls. (B) Mitochondrial NAD+ levels in the liver with reduced Slc25a51 expression. (C) Western blotting analysis using acetylated lysine specific antibody following immunoprecipitation using IDH2 and ACADL antibodies. Band intensity quantification for acetylation of IDH2 (D) and ACADL (E) was performed using ImageJ, normalized to the total levels of IDH2 and ACADL, respectively. (F) Oil Red O staining for liver sections. G) Liver TG levels and H) serum TG levels. Mice were fasted for 20 h. N = 5 in each group. Data are represented as mean ± SEM. *, P < 0.05; #, P < 0.01.
4. Discussion
SIRT3 is critical for maintaining mitochondrial integrity and function, which can impact cell survival, death and metabolic pathways. With its central role in mitochondrial biology, SIRT3 mediates the adaptation of increased energy demands during fasting by deacetylating and restoring activity to mitochondrial proteins when acetyl-CoA levels are elevated due to increased fatty acid oxidation in mitochondria [4–9]. SIRT3 activity involves the irreversible cleavage of the glycosidic bond between nicotinamide and the ADP-ribose, and thus SIRT3 activity results in consumption of oxidized NAD+ [4,5,7]. With high SIRT3 activity, oxidized NAD+ would need to be replenished to sustain an oxidizing mitochondrial environment. [28,31–36]. An unanswered question, therefore, was whether mitochondrial concentrations of NAD+ were sufficient to sustain SIRT3 activity or whether import through the recently discovered Slc25a51 is required in these scenarios.
We found that during fasting Slc25a51 was induced in the liver and that the loss of the transporter resulted in diminished mitochondrial NAD+ levels and SIRT3 activity. During the periods needing enhanced fatty acid oxidation, fasting and daytime, expression of Slc25a51 was elevated. The diurnal rhythmicity of Slc25a51 expression can be driven by light/dark or fed/fasting cycles. Lazar’s lab did time-restricted feeding, in which food was available only during the light phase to uncouple these cycles, and found that diurnal expression pattern of Slc25a51 was disrupted by the time-restricted feeding, suggesting that the diurnal rhythmicity is primarily driven by the feeding/fasting cycle [24].
The rate-limiting enzyme in NAD biosynthesis, NAMPT, is also a fasting-induced gene that is under the regulation of circadian rhythm [11,14]. During fasting, NAMPT is induced and synthesizes more NAD. Our data indicate that Slc25a51 is expressed to transport NAD+ into mitochondria to sustain SIRT3 activity. Prolonged SIRT3 activity would eventually require replenishment of mitochondrial NAD+ concentrations due to the intrinsic turnover of the dinucleotide as part of its enzymatic mechanism. Additionally, the imported NAD+ can directly sustain fatty acid β-oxidation in the mitochondria, as it is required to drive oxidation of the β-carbon on hydroylacyl co-A molecules through 3-hydroxyacyl co-A dehydrogenase (ScHAD or HADH). Although both enzymes are poised to be impaired by insufficient mitochondrial NAD+ concentrations, it may be that dysregulation initially impairs SIRT3 activity and then fatty-acid oxidation is further exacerbated through loss of HADH oxidative activity. The enzyme kinetics data showed KM(NAD+) of SIRT3 at ~100 μM [27] and that of HADH I and II at <5 μM [37].
Overall the data suggest that SLC25A51-mediated NAD+ uptake is necessary to sustain mitochondrial NAD+ levels to coordinate the upregulation of SIRT3 activity and β-oxidation in response to fasting. We propose that these coordinated actions enable cells to effectively perform fatty acid oxidation to cope with energy demand during fasting (Fig. 7). These findings also raise the following questions for future studies.
Fig. 7.

A model of how fasting may coordinate NAMPT and SLC25A51 to transport NAD+ into mitochondria for SIRT3 activity. During fasting, SIRT3 and NAMPT are upregulated, and the expression of the mitochondrial NAD+ transporter SLC25A51 is coordinately increased to sustain SIRT3 activity. Increased fatty acid oxidation may involve SIRT3 independent enzymes, such as hydroxyl-acyl CoA dehydrogenase.
We examined liver Slc25a51 expression, but what are expression levels in other tissues and cell types in response to fasting and circadian rhythm? SIRT3 is abundantly expressed, especially enriched in mitochondria-rich tissues, such as WAT, brown fat, muscle, kidney, and brain. At least in WAT, another metabolism-active tissue, we found that Slc25a51 showed a similar pattern of change. That is, WAT Slc25a51 and SIRT3 expression were induced by fasting and significantly correlated (Supplemental Fig. 2). Future research will need to address the expression changes in other tissues, as well as the functional implications in terms of SIRT3 functions in the absence of Slc25a51.
NAMPT, Slc25a51, and SIRT3 were induced by fasting. However, when re-feeding occurs, what mechanisms do cells use to degrade the proteins or return their activities back to basal levels?
Hepatic Slc25a51 is highly induced by fasting, and it was recently reported that it is also induced by protein intake [38]. Then, what are the transcription factors that directly mediate Slc25a51 expression in response to nutritional cues? CREBH and PPARα are liver-enriched fasting-activated transcription factors that mediate lipid metabolism. We examined the hepatic Slc25a51 expression in CREBH KO and PPARα KO mice under fed vs. fasting conditions [39], and there were no significant differences between WT and KO mice (Supplemental Fig. 3A, B). Therefore, at least, CREBH and PPARα are not likely to be the transcription factors that mediate Slc25a51 expression.
Is SLC25A51 involved in disease states? SIRT3 dysfunction is involved in many disease states, such as obesity, diabetes, and aging [29,40,41]. In humans, fatty liver is associated with reduced SIRT3 activity and mitochondrial protein hyperacetylation [29]. If SLC25A51 expression is reduced in these disease states, then that would result in less mitochondrial NAD+, providing a direct mechanism for these diseases. One-month HFD treatment did not change hepatic Slc25a51 expression in mice (Supplemental Fig. 3C); however, we cannot rule out the possibility that longer HFD treatment alters its expression.
One limitation of the current study is that we examined the expression levels of Slc25a51 mRNA, but do not have data showing its protein levels. We tested 2 commercially available antibodies against mouse Slc25a51, but did not observe positive signals using mouse liver samples. Thus, future studies need to examine Slc25a51 protein levels in response to fasting. Another limitation is that we used AAV8 to knock down the hepatic expression of Slc25a51, and the mice showed phenotypes, such as fatty liver, 2 weeks following virus injection. It would be ideal to study the relatively long-term effects of reduced Slc25a51 expression, in terms of whole-body metabolism, such as glucose homeostasis, insulin resistance, oxygen consumption, and respiration ratio. Rather than AAV8, Slc25a51 liver-specific knockout mice would be suitable for these studies.
In summary, we found that Slc25a51 expression was induced by fasting, showed a circadian rhythm-like pattern, and that Slc25a51 was needed for SIRT3 function in hepatocytes. These results suggest a mechanism by which the cell transports NAD+ into mitochondria during energy demand to sustain oxidation and SIRT3 activity.
Supplementary Material
Funding
This work was partially funded by National Institutes of Health (NIH) grants DK090313 and DK126908 (to KZ); HL134787 and DK132065 (to RZ); a Pilot and Feasibility Grant (to HK) from the Michigan Diabetes Research Center (NIH Grant P30-DK020572).
Abbreviations:
- ACADL
Acyl-CoA dehydrogenase-long chain
- BMAL1
Brain and Muscle ARNT-Like 1
- CLOCK
Clock Circadian Regulator
- IDH2
isocitrate dehydrogenase-mitochondrial
- NAD
nicotinamide adenine dinucleotide
- NAMPT
nicotinamide phosphoribosyltransferase
- shRNA
short hairpin RNA
- SIRT3
sirtuin 3
- TG
triglyceride
- WAT
white adipose tissue
Footnotes
Declaration of competing interest
The authors declare that they have no conflict of interest.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.metabol.2022.155275.
CRediT authorship contribution statement
R.Z. conceptualized the study, analyzed data, and wrote the manuscript. K.Z. conceptualized the study, analyzed data, and revised the manuscript. H.K. and M.J.L. conducted experiments and analyzed data. P.T.M. conducted the Seahorse experiments, analyzed data, and wrote sections related to Seahorse analysis. M.H. and X.A.C. analyzed data and revised the manuscript. R.Z. and K.Z. are the guarantors of this work, and as such, had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis.
References
- [1].Imai S, Armstrong CM, Kaeberlein M, Guarente L. Transcriptional silencing and longevity protein Sir2 is an NAD-dependent histone deacetylase. Nature 2000;403:795–800. [DOI] [PubMed] [Google Scholar]
- [2].Lin SJ, Defossez PA, Guarente L. Requirement of NAD and SIR2 for life-span extension by calorie restriction in Saccharomyces cerevisiae. Science 2000;289:2126–8. [DOI] [PubMed] [Google Scholar]
- [3].Revollo JR, Grimm AA, Imai S. The NAD biosynthesis pathway mediated by nicotinamide phosphoribosyltransferase regulates Sir2 activity in mammalian cells. J Biol Chem 2004;279:50754–63. [DOI] [PubMed] [Google Scholar]
- [4].Hirschey MD, Shimazu T, Goetzman E, Jing E, Schwer B, Lombard DB, et al. SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 2010;464:121–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Ahn BH, Kim HS, Song S, Lee IH, Liu J, Vassilopoulos A, et al. A role for the mitochondrial deacetylase Sirt3 in regulating energy homeostasis. Proc Natl Acad Sci U S A 2008;105:14447–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Onyango P, Celic I, McCaffery JM, Boeke JD, Feinberg AP. SIRT3, a human SIR2 homologue, is an NAD-dependent deacetylase localized to mitochondria. Proc Natl Acad Sci U S A 2002;99:13653–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Lombard DB, Alt FW, Cheng HL, Bunkenborg J, Streeper RS, Mostoslavsky R, et al. Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol Cell Biol 2007;27:8807–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Schwer B, Bunkenborg J, Verdin RO, Andersen JS, Verdin E. Reversible lysine acetylation controls the activity of the mitochondrial enzyme acetyl-CoA synthetase 2. Proc Natl Acad Sci U S A 2006;103:10224–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Huang JY, Hirschey MD, Shimazu T, Ho L, Verdin E. Mitochondrial sirtuins. Biochim Biophys Acta 1804;2010:1645–51. [DOI] [PubMed] [Google Scholar]
- [10].Yang H, Yang T, Baur JA, Perez E, Matsui T, Carmona JJ, et al. Nutrient-sensitive mitochondrial NAD+ levels dictate cell survival. Cell 2007;130:1095–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Peek CB, Affinati AH, Ramsey KM, Kuo HY, Yu W, Sena LA, et al. Circadian clock NAD+ cycle drives mitochondrial oxidative metabolism in mice. Science 2013;342:1243417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Levine DC, Hong H, Weidemann BJ, Ramsey KM, Affinati AH, Schmidt MS, et al. NAD(+) controls circadian reprogramming through PER2 nuclear translocation to counter aging. Mol Cell 2020;78(835–49):e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Nakahata Y, Sahar S, Astarita G, Kaluzova M, Sassone-Corsi P. Circadian control of the NAD+ salvage pathway by CLOCK-SIRT1. Science 2009;324:654–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Ramsey KM, Yoshino J, Brace CS, Abrassart D, Kobayashi Y, Marcheva B, et al. Circadian clock feedback cycle through NAMPT-mediated NAD+ biosynthesis. Science 2009;324:651–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Luongo TS, Eller JM, Lu MJ, Niere M, Raith F, Perry C, et al. SLC25A51 is a mammalian mitochondrial NAD(+) transporter. Nature 2020;588:174–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Kory N, Uit de Bos J, van der Rijt S, Jankovic N, Gura M, Arp N, et al. MCART1/SLC25A51 is required for mitochondrial NAD transport. Sci Adv 2020;6:eabe5310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Girardi E, Agrimi G, Goldmann U, Fiume G, Lindinger S, Sedlyarov V, et al. Epistasis-driven identification of SLC25A51 as a regulator of human mitochondrial NAD import. Nat Commun 2020;11:6145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Davila A, Liu L, Chellappa K, Redpath P, Nakamaru-Ogiso E, Paolella LM, et al. Nicotinamide adenine dinucleotide is transported into mammalian mitochondria. Elife 2018;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Ouyang Y, Bott AJ, Rutter J. Maestro of the SereNADe: SLC25A51 orchestrates mitochondrial NAD. Trends Biochem Sci 2021;46:348–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Ziegler M, Monne M, Nikiforov A, Agrimi G, Heiland I, Palmieri F. Welcome to the family: identification of the NAD(+) transporter of animal mitochondria as member of the solute carrier family SLC25. Biomolecules 2021;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Molusky MM, Ma D, Buelow K, Yin L, Lin JD. Peroxisomal localization and circadian regulation of ubiquitin-specific protease 2. PLoS One 2012;7:e47970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Balsalobre A, Damiola F, Schibler U. A serum shock induces circadian gene expression in mammalian tissue culture cells. Cell 1998;93:929–37. [DOI] [PubMed] [Google Scholar]
- [23].Molcan L Time distributed data analysis by Cosinor. 2019. 10.1101/805960. Online application. BioRxiv. [DOI] [Google Scholar]
- [24].Guan D, Xiong Y, Trinh TM, Xiao Y, Hu W, Jiang C, et al. The hepatocyte clock and feeding control chronophysiology of multiple liver cell types. Science 2020;369:1388–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Pizarro A, Hayer K, Lahens NF, Hogenesch JB. CircaDB: a database of mammalian circadian gene expression profiles. Nucleic Acids Res 2013;41:D1009–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Hughes ME, DiTacchio L, Hayes KR, Vollmers C, Pulivarthy S, Baggs JE, et al. Harmonics of circadian gene transcription in mammals. PLoS Genet 2009;5:e1000442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Feldman JL, Dittenhafer-Reed KE, Kudo N, Thelen JN, Ito A, Yoshida M, et al. Kinetic and structural basis for acyl-group selectivity and NAD(+) dependence in sirtuin-catalyzed deacylation. Biochemistry 2015;54:3037–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Yang W, Nagasawa K, Munch C, Xu Y, Satterstrom K, Jeong S, et al. Mitochondrial sirtuin network reveals dynamic SIRT3-dependent deacetylation in response to membrane depolarization. Cell 2016;167(985–1000):e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Kendrick AA, Choudhury M, Rahman SM, McCurdy CE, Friederich M, Van Hove JL, et al. Fatty liver is associated with reduced SIRT3 activity and mitochondrial protein hyperacetylation. Biochem J 2011;433:505–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].TeSlaa T, Teitell MA. Techniques to monitor glycolysis. Methods Enzymol 2014;542:91–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Howitz KT, Bitterman KJ, Cohen HY, Lamming DW, Lavu S, Wood JG, et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature 2003;425:191–6. [DOI] [PubMed] [Google Scholar]
- [32].Haigis MC, Sinclair DA. Mammalian sirtuins: biological insights and disease relevance. Annu Rev Pathol 2010;5:253–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Houtkooper RH, Pirinen E, Auwerx J. Sirtuins as regulators of metabolism and healthspan. Nat Rev Mol Cell Biol 2012;13:225–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Finkel T, Deng CX, Mostoslavsky R. Recent progress in the biology and physiology of sirtuins. Nature 2009;460:587–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Kim A, Koo JH, Lee JM, Joo MS, Kim TH, Kim H, et al. NRF2-mediated SIRT3 induction protects hepatocytes from ER stress-induced liver injury. FASEB J 2022;36:e22170. [DOI] [PubMed] [Google Scholar]
- [36].Gao P, You M, Li L, Zhang Q, Fang X, Wei X, et al. Salt-induced hepatic inflammatory memory contributes to cardiovascular damage through epigenetic modulation of SIRT3. Circulation 2022;145:375–91. [DOI] [PubMed] [Google Scholar]
- [37].Filling C, Keller B, Hirschberg D, Marschall HU, Jornvall H, Bennett MJ, et al. Role of short-chain hydroxyacyl CoA dehydrogenases in SCHAD deficiency. Biochem Biophys Res Commun 2008;368:6–11. [DOI] [PubMed] [Google Scholar]
- [38].Le Couteur DG, Solon-Biet SM, Parker BL, Pulpitel T, Brandon AE, Hunt NJ, et al. Nutritional reprogramming of mouse liver proteome is dampened by metformin, resveratrol, and rapamycin. Cell Metab 2021;33(2367–79):e4. [DOI] [PubMed] [Google Scholar]
- [39].Kim H, Wei J, Song Z, Mottillo E, Samavati L, Zhang R, et al. Regulation of hepatic circadian metabolism by the E3 ubiquitin ligase HRD1-controlled CREBH/PPARalpha transcriptional program. Mol Metab 2021;49:101192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Hirschey MD, Shimazu T, Jing E, Grueter CA, Collins AM, Aouizerat B, et al. SIRT3 deficiency and mitochondrial protein hyperacetylation accelerate the development of the metabolic syndrome. Mol Cell 2011;44:177–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Nogueiras R, Habegger KM, Chaudhary N, Finan B, Banks AS, Dietrich MO, et al. Sirtuin 1 and sirtuin 3: physiological modulators of metabolism. Physiol Rev 2012;92:1479–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.