

Abstract
Over the last 2 decades or so, hypervalent iodine compounds, such as diaryliodonium salts and aryliodonium ylides, have emerged as useful precursors for labeling homoarenes and heteroarenes with no-carrier-added cyclotron-produced [18F]fluoride ion (t1/2 = 109.8 min). They permit rapid and effective radiofluorination at electron-rich as well as electron-deficient aryl rings, and often with unrestricted choice of ring position. Consequently, hypervalent aryliodine compounds have found special utility as precursors to various small-molecule 18F-labeling synthons and to many radiotracers for biomedical imaging with positron emission tomography. This review summarizes this advance in radiofluorination chemistry, with emphasis on precursor synthesis, radiofluorination mechanism, method scope, and method application.
Keywords: aryliodonium ylide, diaryliodonium salt, fluorine 18, PET, radiofluorination, radiotracer
1 ∣. INTRODUCTION
The molecular imaging modality of positron emission tomography (PET) relies on its use of radiotracers labeled with positron emitters for achieving biochemical specificity when they are applied in biomedical research programs or in drug development campaigns. For several reasons, 18F is a favored radionuclide for labeling such radiotracers. First, this radionuclide is readily produced from cyclotrons in very high activities and high molar activities as [18F] fluoride ion, according to the 18O(p,n)18F reaction on [18O]water.1 Moreover, the half-life of 18F (109.8 min) is long enough to allow radiotracer kinetics to be followed in vivo over a few hours and also usefully permits transport of 18F over considerable distances between radionuclide production and radiotracer imaging centers. The decay of 18F is almost purely (97%) by emission of a relatively low-energy positron (β+, 0.634 MeV), and this permits higher-resolution PET images to be obtained than with other useful positron emitters. Almost all PET radiotracers are metabolized in vivo.2,3 Defluorination of many 18F-labeled PET radiotracers gives [18F]fluoride ion, which binds avidly to bone. This bone uptake can become problematic for measuring radioactivity in nearby tissue and thereby can compromise the value of PET scans. This is particularly so for brain scans when radioactivity is taken up in the skull.2,3 Therefore, radiotracers need to be designed to avoid radiodefluorination in vivo. Labeling at an aryl or heteroaryl carbon is a favored strategy for this purpose. Consequently, the field of PET radiotracer development has sought to develop a battery of methods for unrestricted introduction of [18F]fluoride ion at aryl and heteroaryl groups in late-stage radiosynthesis.4-9 This review specifically covers the growing utility of hypervalent aryliodine compounds as precursors for use in such methods. The coverage is intended to be illustrative rather than exhaustive and builds on a previous review of this area.10
Historically, in the 1980s, classical aryl nucleophilic substitution reactions (SNAr)11 became the first useful methods for labeling arenes with [18F]fluoride ion, quickly supplanting the use of the earlier but very much less useful Wallach12 and Balz-Schiemann reactions13 (Figure 1). The introduction of the aminopolyether, Kryptofix 2.2.2 (K 2.2.2; 4,7,13,16,21,24-hexaoxa-1,10-diazabicyclo[8.8.8] hexacosane), as a phase transfer agent for [18F]fluoride ion by the Jülich group greatly facilitated this advance.14 Although classical SNAr reactions with [18F]fluoride ion have led to the development of many successful PET radiotracers, and continue to do so, there are limitations in the substrate scope of this method.4-10 Generally, homoarene precursors must contain a good leaving group that is activated by an electron-withdrawing group in the ortho or para position. Displacement of a leaving group from the meta position is only possible with nitro as the activating group, and then only under quite forcing conditions to give moderate yields.15 For heteroarenes, such as substituted pyridines, an activating group may not be required, but low yields may be obtained even under forcing conditions.16-18 Therefore, a need to expand the range of arenes that might be labeled with [18F]fluoride ion was long apparent during the early unfolding of the PET radiochemistry field. This was especially so, because many important radiotracers, such as [18F]l-6-fluoro-DOPA, have electron-rich rings, and traditionally they had been labeled with electrophilic agents such as cyclotron-produced [18F]fluorine or the derivative [18F]acetyl hypofluorite, each of which is difficult to produce and use, with yet further limitations of low yield, very low molar activity, and low reaction efficiency.19 Importantly, use of [18F]fluoride ion enables radiotracers to be labeled at high no-carrier-added (NCA) molar activity, as is essential for a potentially large range of radiotracers targeting low-density binding sites (eg, receptors and enzymes) in vivo.3
FIGURE 1.

Classical methods for the radiofluorination of arenes with [18F]fluoride ion and some of their limitations
In 1995, a quite long-standing paper by Van der Puy20 piqued my interest because this described the conversion of a class of hypervalent iodine compounds, namely, diaryliodonium salts (ArI+Ar1X−), into fluoroarenes by decomposition (X = BF4) or by treatment with KF (X = BF4, CF3COO, TsO, or Cl) in either the solid or solution phase. Moderate yields were reported from the thermal decomposition of symmetrical tetrafluoroborate salts (Ar = Ar1) in the presence of KF, not only for p-fluorochlorobenzene (39%) but also for fluorobenzene (85%), m-fluoronitrobenzene (38%), and p-fluoroanisole (44%) (Figure 2). These results for fluorination at electron-rich rings or in the meta position indicated the operation of a nonclassical SNAr mechanism with important potential for expanding chemistry with [18F]fluoride ion to the synthesis of electron-rich [18F]fluoroarenes and for the introduction of [18F]fluoride ion into meta positions. Trial reactions of [18F]fluoride ion with simple diaryliodonium salts in aprotic solvents using K 2.2.2 as the cryptand for potassium ion from added potassium carbonate base quickly affirmed this potential (Figure 3).21 Over the last 2 decades or so, this early report has been followed with extensive development of approaches to using diaryliodonium salts and other hypervalent iodine compounds for radiofluorination.10 Important motivations in this research have been to expand and improve the repertoire of methods for the syntheses of diverse hypervalent aryliodine precursors, to control reaction chemoselectivity between possible radiofluorination products, to increase the understanding of mechanism, and to extend the methodology to prepare useful labeling synthons and useful PET radiotracers. This review now proceeds to summarize these developments.
FIGURE 2.

Pyrolysis of diaryliodonium fluoroborates to give fluoroarenes20
FIGURE 3.

Yields for [18F]fluoroarenes from the radiofluorination of some simple diaryliodonium salts in acetonitrile21
2 ∣. DISCUSSION
2.1 ∣. Some structural features of hypervalent aryliodine compounds
As a basis for subsequent discussion on reactivity and mechanism, it is valuable to highlight important structural features of classes of hypervalent aryliodine compounds, such as the diaryliodonium salts and aryliodonium ylides that will be featuring prominently in this review. The term hypervalent is used to indicate a compound of a main group element that contains more than an octet of electrons in the valence shell. These compounds are commonly named following the N-X-L nomenclature, where N denotes the number of electrons formally assigned to the central atom, X its elemental symbol, and L the number of bonded ligands. Diaryliodonium salts (denoted ArI+Ar1X−) are 10-I-3 compounds and belong to a broader class of iodine(III) compounds, commonly known as λ3-iodanes. They show approximately T-shaped geometry with one aryl ring occupying an axial position and the other an equatorial position, but with fast and easy exchange of aryl ring position occurring through a process known as Berry pseudorotation (Figure 4). Highly electronegative ligands (eg, halide) tend to occupy an approximately apical position. Apical bonding through the central iodine atom is often referred to as “hypervalent” bonding, or as 3-center 4-electron (3c-4e) bonding.
FIGURE 4.

Exchange of axial and equatorial aryl ligands in diaryliodonium salts through pseudorotation
The crystal structures of some diaryliodonium salts have been published, including those of some chlorides,22-24 and recently those of 2 fluorides.25 In the solid state, many of these compounds, such as diphenyliodonium chloride, exist as anion-bridged dimers held together by secondary iodine-halogen bonds (Figure 5). Tetrameric structures have also been observed, as exemplified by the 2 reported examples of diaryliodonium fluorides.25 Whereas diaryliodonium salts appear to be fully ionized in polar protic solvents such as water, they may also exist as oligomers in organic solution. Such oligomers have been detected with mass spectrometry.24
FIGURE 5.

Examples of primary (black) and secondary (red) bonding patterns in single crystals of some aryliodine(III) compounds
Aryliodonium ylides are compounds in which the central iodine(III) atom carries a formal positive charge and is attached directly to an anionic site. In this review, aryliodonium ylides are denoted as ArI+—−CX2 where X is an electron-withdrawing substituent, such as an acyl substituent. Iodonium ylides are also found to be T-shaped molecules with C—I—C angles close to 90°, indicating the ylidic C─I bond to be zwitterionic (Figure 5).26 The CX2 moiety may be cyclic and if so may impart useful stability to the ylide. Cyclic ylides are of primary interest later in this review.
(Diacyloxyiodo)arenes (ArI(RCO2)2), such as the commercially available (diacetoxyiodo)arene [PhI(OAc)2, DIB, PID, or PIDA], show a T-shaped geometry with the phenyl ring occupying an equatorial position and the 2 acetoxy groups the axial positions in a trigonal-bipyramidal arrangement of bonds and lone pairs around the central iodine atom.27 The strong bonds between the central iodine atom to a carbon atom of the phenyl group and to one oxygen atom of each carboxylate group plus 2 secondary bonds to the 2 remaining oxygen atoms give an overall pentagonal-planar bonding arrangement (Figure 5). The simple trifluoroacetoxy analog [PhI(CF3CO2)2; PIFA] exists as a bridged dimer in the solid state.28
2.2 ∣. Synthesis of aryliodine(III) precursors for radiofluorination
For the development and expansion of the utility of any new radiofluorination methodology, precursors need to be readily accessible through effective and versatile synthetic methods. Ideally, these precursors should also be adequately stable for prolonged storage. Here, some of the main routes to aryliodine(III) precursors are surveyed.
2.2.1 ∣. (Diacyloxyiodo)arenes
(Diacyloxyiodo)arenes feature prominently as precursors or as nonisolated intermediates to other hypervalent iodine(III) compounds, particularly diaryliodonium salts29 and aryliodonium ylides.30 Recently, they have also been investigated as substrates for radiofluorination.31 Some (diacyloxyiodo)arenes are commercially available, such as PIDA and PIFA. Numerous methods are available for the synthesis of (diacyloxyiodo)arenes.32 These methods are of 2 main types: (1) oxidation of iodoarenes in the presence of the desired carboxylic acid or (2) ligand exchange in PIDA with the desired acid. So far, the oxidation route has been the most used for preparing (diacyloxyiodo)arenes as substrates for radiofluorination31 or as intermediates for other radiofluorination precursors (Figure 6).33 Peracetic acid,31,34,35 sodium periodate,36,37 or sodium perborate,31,37,38 each in acetic acid, have been favored as oxidants, and they have given numerous diversely substituted (diacyloxyiodo)arenes, including (diacyloxyiodo)heteroarenes, as stable crystalline solids in moderate to high yields. In a few cases, where the substituent is strongly electron withdrawing, a better yield may be obtained by making the trifluoroacetate with Oxone (KHSO5·0.5KHSO4·0.5K2SO4) in trifluoroacetic acid as oxidant followed by ligand exchange with acetic acid to access the diacetate.31
FIGURE 6.

Oxidative methods for preparing various substituted (diacetoxyiodo)arenes from iodoarenes
Recently, a method has been developed for synthesizing (diacetoxyiodo)arenes in the absence of protic acids.39 This method is based on treating a substituted iodoarene with Selectfluor (N-chloromethyl-N′-fluoro-triethylenediammonium bis(tetrafluoroborate); F-TEDA-BF4) and TMSOAc (Figure 6). This method is applicable to electron-rich and electron-poor iodoarenes and tolerates both acid- and base-sensitive functional groups.
2.2.2 ∣. Diaryliodonium salts
Diaryliodonium salts are generally stable to air and moisture and may be prepared on a multigram scale.39 Several diaryliodonium salts are now commercially available. Our laboratory's experience is that diaryliodonium salts can be stored for months, even years, in the cold and dark without decomposition. Another laboratory has similarly reported a highly functionalized iodonium salt to be stable for 18 months at room temperature under argon,38 and yet another reports a diaryliodonium salt precursor to be stable in a crimp-sealed amber vial at 5°C to 8°C under atmospheric conditions for at least 2 years.40 Salts with paired electron-rich rings may however show some storage instability. For example, 4-anisyl(2-thienyl) iodonium bromide decomposes within 2 weeks under ambient conditions.41
Methods for synthesizing diaryliodonium salts abound and have been extensively reviewed.29,42 Only some of the most popular and useful methods are mentioned here by way of illustration.
From the perspective of using diaryliodonium salts for radiotracer synthesis, unsymmetrical salts (Ar ≠ Ar1) are invariably preferred over symmetrical salts. One reason is that a simple cheap arene can serve as the source of one ring partner. Another reason is that the iodoarene that is extruded from a chemoselective radiofluorination reaction can usually be separated readily with high-performance liquid chromatography (HPLC) from the desired radiofluorination product. Unsymmetrical salts for radiofluorination are designed so that one aryl ring serves as the target for radiofluorination while the other ring serves as an electron-rich “spectator ring” that is relatively much less prone to radiofluorination (see below). As a formal anion, tosylate or another organic anion is often preferred over halide to confer good solubility of the salt in the organic solvent to be used for radiofluorination (eg, dimethylformamide [DMF] and acetonitrile [MeCN]). Simple metathesis reactions can be used to replace the anion with the one that is desired.37,43-47 Nonetheless, halide anions have sometimes been found preferable for high radiofluorination yields.
Unsymmetrical diaryliodonium salts are now relatively easy to prepare along various pathways (Figure 7). A typical synthesis of an unsymmetrical salt involves oxidation of an iodoarene to a λ3-iodane, either a (diacyloxyiodo)arene or a [hydroxy(tosyloxy)iodo]arene (HTIA), followed by ligand exchange with an electron-rich arene (eg, mesitylene,35 1,3,5-trimethoxybenzene,33 anisole,33 or thiophene),33,48 or a more nucleophilic arylating agent (eg, arylsilane,49,50 arylborate,51 or arylstannane52). The simple λ3-iodane, [hydroxy(tosyloxy)iodo]benzene (PhI(OH)OTs),53 commonly known as Koser's reagent or HTIB, is stable and commercially available. HTIAs, including Koser's reagent, may be converted into congeners through ligand exchange with a substituted iodoarene, ie, ArI(OH)OTs + Ar1I → Ar1I(OH)OTs + ArI .45,53 Direct mild syntheses of HTIAs from iodine and a substituted arene or from an iodoarene have also been reported.52-54 A (diacyloxyiodo)arene or an HTIA may be used as generated in situ, even in the presence of the Brønsted acid used for its synthesis.33,35 If isolated, these hypervalent reagents may be used in either the presence44 or absence of a Brønsted acid.55,56 The absence of a Brønsted acid allows a diaryliodonium salt having 1 or more acid-labile protecting groups, such as a Boc or ester group, to be prepared.55,56 Nevertheless, reactions of (diacyloxyiodo)arenes or HTIAs with electron-rich arenes, such as mesitylene, 1,3,5-trimethoxybenzene, anisole, or thiophene, do usually require acidic conditions. Clearly, regioselectivity cannot be an issue for reactions with the 1,3,5-tri-substituted arenes. The reactions with anisole and thiophene are found to be remarkably regioselective, for the 4-position in anisole and the 2-position in thiophene. The reactions with silanes, borates, and stannanes confer excellent regioselectivity under milder often acid-free conditions. Use of aryl borates is especially attractive because they are quite readily accessible, stable, and nontoxic. Arylstannanes are also often used as they may usually be prepared from the corresponding iodides. Yields of diaryliodonium salts from the metallated arenes are usually moderate but useful.
FIGURE 7.

Some pathways for the synthesis of unsymmetrical diaryliodonium salts
Methods have been developed recently for converting an arene-iodoarene pair into an unsymmetrical iodonium salt under mild conditions, where either aryl partner may be electron rich or electron deficient. These methods typically use m-chloroperoxybenzoic acid (mCPBA) as oxidant, in the presence of triflic acid,57,58 toluenesulfonic acid,59 or BF3OEt2.60 A wide range of substituents in the aryl partners is tolerated. These substituents include Me,57-60 1,3,5-tri-Me,58,59 F,58,60 Cl,57,58 Br,58,60 I,57,60 CF3,58,60 NO2,57,58 MeO,57-60 CO2H,58 and CHO.60 This method has also been adapted for the single-pot preparation of N-heteroaryliodonium salts.61 The salts were isolated in protonated form but could then be deprotonated on basic alumina eluted with dichloromethane (DCM)/methanol. Recently, mCPBA in triflic acid has been found effective for the 1-pot synthesis of a wide range of substituted aryl(2,4,6-trimethoxyphenyl)iodonium triflates from iodoarenes.62 Trifluoroacetates have been prepared similarly by use of trifluoroacetic acid instead of triflic acid.63
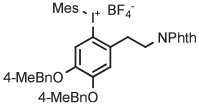
A recent method for the synthesis of acid-sensitive diaryliodonium salts uses a Lewis acid rather than a Brønsted acid (Figure 7).39 In this method, a (diacetoxyiodo)arene is generated from an iodoarene with Selectfluor in the presence of TMSOAc and then treated with a metal aryltrifluoroborate plus TMSOTf. This method has provided several highly functionalized diaryliodonium triflates in moderate to high yields. Functionalities in these salts have included N-succinimidyl, carboxylate esters, protected amino acid side chains, or polyethylene glycol chains.
Of further note is an efficient 1-pot pathway developed for the preparation of aryl(substituted-isoquinolinyl) iodonium triflates based on treating mesoionic carbene silver complexes with the hypervalent iodine reagent, PhI(Py′)2(OTf)2 where Py′ is a substituted pyridine (Figure 8).64 The method displayed tolerance for a wide variety of substituents (eg, alkyl, aryl, halogen, OMe, and ester) and gave 40% to 94%.yields.
FIGURE 8.

Synthesis of phenyl(substituted-isoquinolinyl) iodonium triflates
Symmetrical salts are occasionally useful precursors. An advantage conferred by a symmetrical salt precursor is that only 1 18F-labeled arene can be produced from the radiofluorination. This can be especially advantageous for the more difficult labeling of electron-rich arenes. For example [18F]4-fluoroanisole can be prepared in good yield from bis(4-anisyl)iodonium trifluoroacetate,21 and likewise [18F]2-fluoroanisole from bis(2-anisyl)iodonium chloride.65 Simple 18F-labeling synthons may be prepared from protected symmetrical diaryliodonium salts, as exemplified by a synthesis of [18F]4-fluorophenol.47
Symmetrical salts may be accessed by a variety of methods.29,42 Very early methods typically used harsh acidic conditions,43,66 such as treatment of an arene with NaIO4/I2 in concentrated sulfuric acid.43 Most of the currently useful methods for preparing symmetrical salts are special cases of the methods for preparing unsymmetrical salts, as already summarized. In addition, symmetrical diaryliodonium triflates have been prepared simply by treating arenes with iodine and mCPBA in triflic acid.58
2.2.3 ∣. Aryliodonium ylides
Several methods exist for preparing iodonium ylides.30 For the purpose of radiofluorination, stable crystalline spirocyclic aryliodonium ylides have been prepared readily from (diacetoxyiodo)arenes by treatment with spirocyclic derivatives of Meldrum's acid (2,2-dimethyl-1,3-dioxane-4,6-dione) in 10% sodium bicarbonate ethanol (Figure 9).65 This method is highly versatile with respect to both the aryl partner and the spirocyclic derivative. Open-ring analogs may also be prepared. A simple 1-pot 2-step procedure has been described for preparing aryliodonium ylides from the respective iodoarenes.68 Iodoarenes are first oxidized with mCPBA in DCM and then treated with Meldrum's acid and KOH. This method has also been applied to prepare various spirocyclic ylides.67 In general, low to moderate yields are obtained (Figure 9).
FIGURE 9.

Routes to aryliodonium ylide and spirocyclic iodonium ylides
In summary, methods for the synthesis of hypervalent aryliodine compounds for radiofluorination are relatively cheap, numerous, and versatile for the incorporation of desired functionality. Therefore, the syntheses of these compounds are no longer a major impediment to their use in radiofluorination. Considering their high reactivities, most of these compounds show remarkably adequate storage stability.
2.3 ∣. Practical approaches to radiofluorination
All approaches to radiofluorination begin with isolation of [18F]fluoride ion from proton-irradiated [18O]water (Figure 10). Typically, this is achieved by evaporation of the [18O]water in the presence of base (eg, K2CO3-K 2.2.2) or by entrapment of the [18F]fluoride ion on a quaternary ammonium ion exchange resin followed by elution with a solution of base or base containing a phase transfer agent (eg, K 2.2.2 or 18-crown-6). The [18F]fluoride ion can then be taken to dryness with 2 or more cycles of MeCN addition and evaporation of the generated azeotrope.
FIGURE 10.

Conventional and minimalist approaches to labeling substrates with [18F]fluoride ion
Conventionally, the radiofluorination of a hypervalent aryliodine precursor is based on a batch reaction of 18F−-K+-K 2.2.2 complex with a small amount of precursor (often 1-15 mg) in a small volume (0.5-1.0 mL) of a polar aprotic solvent within a septum-sealed glass or glassy carbon vessel. In some instances, other bulky cations are used for solubilization of the [18F]fluoride ion, such as K+-18-crown-6, Cs+, or R4N+ (R = Et or nBu). Typically, the cation is introduced as a carbonate or bicarbonate salt. This approach is readily automated for radiotracer production and has been performed in custom-built or commercial radiosynthesis devices.38,40,55,69-71
Other approaches have also been explored for performing and studying the radiofluorination of diaryliodonium salts. One approach is described as “minimalist” as it uses neither the base nor other additives that are introduced in the conventional batch approach (Figure 10).72 In this approach, the [18F]fluoride ion is first trapped on an anion exchange resin, which is then flushed with methanol, followed by the precursor iodonium salt in methanol. The obtained solution of substrate and [18F]fluoride ion is then evaporated, taken up in the reaction solvent, and heated. This type of procedure reduces the number of operational step, saves processing time, and is compatible with base-sensitive precursors and products.
The reactivity of diaryliodonium salts towards [18F] fluoride ion is such that reactions proceed to useful yields in solvents with low water content.56,65,73 Some salts are sufficiently reactive in solvent with high (28%) water content.74 As in the minimalist approach, this can avoid the lengthy and tedious need to dry the aqueous [18F]fluoride ion obtained from the cyclotron target.
Use of a commercial microfluidic apparatus (NanoTek, Advion), which incorporates a capillary silica tube as a microreactor, permits several radiofluorination reactions to be performed in sequence on a single day under highly controlled conditions of reagent concentration, time, and temperature (Figure 11). The microreactor is fed at constant rates from 2 reservoirs, one containing a precursor solution and the other containing the [18F]fluoride ion reagent solution. As a result, the study of reaction kinetics and even the derivation of Arrhenius activation energies become feasible.65 The microfluidic approach has more recently been extended to use hypervalent substrates for the synthesis of labeling synthons and for the routine production of a PET radiotracer, [18F]FPEB.75
FIGURE 11.

Layout of a microfluidic apparatus for the investigation of the radiofluorination of hypervalent aryliodine precursors
A pyrolytic approach has been developed specifically to the radiofluorination of diaryliodonium salts. The [18F]fluoride salt is generated from a nonfluoride precursor in a solvent, such as MeCN, followed by drying and then thermolysis in a predominantly nonpolar aprotic solvent such as benzene, 10% MeCN in toluene, or toluene (Figure 12).38,76 This approach has been incorporated successfully into commercially available automated radiosynthesis platforms such as the Synthera and TRACERlab modules.
FIGURE 12.

A pyrolytic method for the radiofluorination of diaryliodonium salts
The copper-mediated radiofluorination of aryl(mesityl)iodonium salts has recently been adapted to allow radiofluorination of C—H groups in electron-rich aryl and heteroarenes in situ.77 The arene is treated directly with [hydroxy(tosyloxy)iodo]mesitylene and TMSOTf in DCM to form the aryl(mesityl)iodonium salt in situ over several hours, which is then subjected to Cu-mediated radiofluorination with [18F]fluoride ion. This approach has allowed late-stage radiofluorination of toluenes, anisoles, anilines, pyrroles, and thiophenes in mostly good to high yields.
2.4 ∣. Mechanisms of radiofluorination
2.4.1 ∣. Diaryliodonium salts: “copper free”
The mechanism of the reactions of diaryliodonium salts with [18F]fluoride ion matches that of many other nucleophiles.78,79 Reactions proceed through ligand exchange with the nucleophile followed by “ligand coupling” and reductive elimination of iodoarene (Figure 13). For unsymmetrical iodonium salts, either iodoarene may be eliminated and either arylated nucleophile may be produced. The ratio of the arylated nucleophiles is termed the “chemoselectivity” of the process. For diaryliodonium salts that do not carry ortho alkyl substituents, radiofluorination of the most electron-deficient aryl ring is favored.36,80,81 For unsymmetrical salts that carry 1 or more ortho alkyl substituents, the ortho-substituted ring may be radiofluorinated in preference to a more electron-deficient ring.65,80 This reactivity behavior had been seen in analogous reactions of diaryliodonium salts with heavier halide ions and had been dubbed the “ortho effect.”82,83 The ortho effect is one feature that distinguishes these reactions from classical SNAr reactions. The abilities of ortho substitution patterns to impart an ortho effect in radiofluorination reactions have been ranked as 2,6-di-Me > 2,4,6-tri-Me > Br > Me > Et ≈ i-Pr > H > OMe.65
FIGURE 13.

Mechanism of the radiofluorination of unsymmetrical diaryliodonium salts
Computational studies have been performed with the aim of understanding both chemoselectivity and the ortho effect in the radiofluorination of diaryliodonium salts.25,80,84,85 An early computational and experimental study of the radiofluorination of heteroaryl(phenyl) iodonium salts came to the important conclusion that chemoselectivity was dictated by the difference in energy of the 2 possible transition states and not by the difference in the 2 ground state energies represented by axial-equatorial switching of the 2 aryl groups. Subsequent computational studies have reinforced this conclusion. Hill and Holland85 performed extensive density functional theory computations on para-substituted phenyl(aryl)iodonium salts as precursors for radiofluorination and described the 2 possible transition states in terms of bond distances for the central iodine and fluorine to each other and to aryl carbons, plus the distribution of charge between these 4 atoms. Linear correlations with positive slope were found between Hammett constants (σm,p) and calculated transition state energy differences, indicating that electron-withdrawing substituents promote radiofluorination at the ipso aryl ring. Similar findings were found for meta-substituted salts and thus accord with experimental findings.34 In this regard, Ross et al measured a linear correlation between the rate of radiofluorination of aryl(2-thienyl)iodonium bromides and Hammett constant (σ) with a positive slope.36
High-level computations have described geometries for the pair of transition states for the reaction of fluoride ion with a phenyl(2-tolyl)iodonium salt, each of which has [18F]fluoride ion loosely bonded to the hypervalent iodine and to the ipso carbon of an equatorial ring (Figure 14).25 The 2 transition states mainly differ by which aryl ring is in the equatorial position. A significant difference is that the fluorine-to-aryl carbon distance is slightly longer in the lower-energy transition state. The 2 transition states rapidly interconvert through a low-energy barrier.25 Therefore, in accord with the Curtin-Hammett principle,86 chemoselectivity is dictated by the size of this energy barrier and not by ground state or product state energy differences. In the phenyl(2-tolyl) iodonium salt example, the difference in transition state energies is computed to be 0.9 kcal/mol, which predicts an ortho effect and a chemoselectivity of about 2 in favor of the formation of 2-fluorotoluene over fluorobenzene. This agrees well with earlier experimental determination (0.63 kcal/mol).65
FIGURE 14.

Computed transition state geometries and energies (relative to ground state) for the fluorination of a phenyl(2-tolyl)iodonium salt.25 TSA leads to fluorobenzene and TSB to 2-fluorotoluene. TSB has somewhat lower energy (by 0.9 kcal/mol) such that 2-fluorotoluene is the preferred product. Bond distances are in Ångströms
Experimentally, it is found that chemoselectivities are fixed and highly repeatable under any set of reaction conditions. Therefore, it is possible to design unsymmetrical diaryliodonium salts that are highly chemoselective for undergoing radiofluorination to the desired [18F] fluoroarene. In these salts, one of the aryl rings is more electron rich and much less susceptible to radiofluorination than the other. As mentioned earlier, these aryl groups are termed spectator groups, and often in practice they are phenyl, 4-tolyl, 4-anisyl, 2,4,6-trimethoxyphenyl, or 2-thienyl, but not mesityl because of the ortho effect.
Various hypotheses have been put forward for the ortho effect, mostly based on steric and electronic considerations in ground state structures.82,83 Recently, computational studies suggest that the ortho effect may be ascribed to differences in electrostatic interactions between each of the 2 possible transition states.25 Substituents other than alkyl, such as methoxy, may not impart the ortho effect.25
Yields from the radiofluorinations of some diaryliodonium salts have shown dependence on salt anion.36,87 Such effects are not easily rationalized but may point to a role in the reaction pathway for oligomeric salts held together by anion bridges, where the anion must be displaced to allow radiofluorination to proceed.25 Experimental24,88 and computational studies25 support the existence of diaryliodonium salts oligomers in organic solution. Other factors giving rise to anion dependence are also possible, such as perhaps small differences in salt purities.
Generally, the radiofluorinations of diaryliodonium salts have been assumed to be regiospecific. However, the thermal decompositions of 5-methoxy-substituted [2.2]paracyclophane(4-anisyl)iodonium fluoride was found to give some 3-fluoroanisole in addition to the expected 4-fluoroanisole (Figure 15).89 The 7-methoxy analog also gave a mixture of 2 fluoro isomers. These results might be explained by the operation of an aryne mechanism due to electronic and steric effects.
FIGURE 15.

Pyrolysis of 5-methoxy-[2.2]paracyclophane(4-anisyl)iodonium fluoride gives 3-fluoroanisole in addition to 4-fluoroanisole
A free radical scavenger, TEMPO (2,2,6,6-tetramethyl-1-piperidinyloxyl), has been found beneficial in many but not all radiofluorinations of diaryliodonium salts.24,34,65,69,90 TEMPO may be acting to inhibit thermal or photochemical radical decomposition of diaryliodonium salt precursor, depending on salt structure.
2.4.2 ∣. Diaryliodonium salts: copper-mediated
The inclusion of a copper salt in the fluorination of a diaryliodonium salt has a profound effect on reaction mechanism and outcome.91,92 The ortho effect may be abolished, and the fluoride ion may tend to go to the more electron-deficient ring. In the radiofluorination of aryl(mesityl)iodonium salts (ArI+(Mes)X−), yield and chemoselectivity for [18F]fluoroarene (Ar18F) versus [18F]mesityl fluoride (Mes18F) have been found to depend strongly on the selected Cu precatalyst and solvent, under some conditions strongly favoring production of the [18F]fluoroarene and under others [18F] mesityl fluoride. The use of stable and commercially available (MeCN)4CuOTf in DMF at 85°C for 20 minutes gave high chemoselectivity for Ar18F versus Mes18F for a wide range of Ar groups, including electron-rich groups, such as 4-anisyl or 2,4,6-trimethoxyphenyl. Thus, under these conditions, a mesityl group serves as a very effective spectator group, giving generally very high chemoselectivity for Ar18F. Yields vary from low to high depending on the substitution pattern in the aryl ring. Tetrafluoroborate was the favored salt anion for optimal yield.
Detailed theoretical and experimental investigations41,92 suggest that the mechanism of these reactions involves a Cu(I/III) catalytic cycle (Figure 16). Experimental evidence suggests that where the catalyst is introduced as a Cu(II) species (eg, Cu(OTf)2), this is first reduced to a Cu(I) species by DMF. Oxidation of a Cu(I) species by the MesI+Ar species is then considered to be the rate-limiting step on the path to producing the [18F] fluoroarene.
FIGURE 16.

Mechanism proposed for the radiofluorination of a phenyl(mesityl)iodonium salt in the presence of (MeCN)4CuOTf in dimethylformamide
2.4.3 ∣. Aryliodonium ylides
The radiofluorination of aryliodonium ylides probably proceeds through a mechanism like that of the radiofluorination of diaryliodonium salts, except that the auxiliary ligand is not considered to be susceptible to radiofluorination (Figure 17).85,93 Therefore, there is no chemoselectivity consideration. Labeling reactions have been performed with low amounts of ylide (eg, 2 mg) in DMF with Et4NHCO3 as base at 120°C for 10 minutes and have given generally low to moderate yields on a wide range of aryl and heteroaryliodonium ylides.67 Simple arenes with electron-rich rings have been obtained with this method in moderate yield, such as [18F] fluoromesitylene (45%). Labeling in the meta position was also moderately successful to produce, for example, [18F]3-fluoropyridine (65%) and [18F]3-fluorotrifluoromethyl benzene (71%).
FIGURE 17.

Mechanism proposed for the radiofluorination of aryliodonium ylides
One study has observed the formation of regioisomers in the radiofluorination of electron-rich ylides, such as ylides of 4-iodoanisole and 4-benzyloxyiodobenzene derived from Meldrum's acid.92 The [18F]4-fluoroarene and [18F]3-fluoroarene products were obtained in a ratio of 3:1 from the 4-anisyliodonium ylide and 20:11 from the 4-benzyloxyphenyliodonium ylide. Lack of regioselectivity has also been observed for the radiofluorination of more elaborate but structurally related ylides designed as precursors to radiotracers.94 These findings indicate involvement of an aryne mechanism for electron-rich aryliodonium ylides, as has already been alluded to for the radiofluorinations of highly electron-rich diaryliodonium salts.
2.4.4 ∣. Other classes of hypervalent aryliodine compound
(Diacetoxyiodo)arenes undergo radiofluorination with [18F]fluoride ion.31 One mechanism for this reaction likely involves ligand exchange. Thus, quantum chemical calculations predict that formation of [(acetoxyfluoro) iodo]benzene from (diacetoxyiodo)benzene is thermodynamically favored with a low-energy barrier of 16.4 kcal/mol and that onward conversion of this intermediate into fluorobenzene has a free energy of activation (ΔG) of 33.8 kcal/mol.
Although iodylarenes (ArIO2) are hypervalent aryliodine compounds, they only undergo radiofluorination with [18F]fluoride ion in high yields when the aryl ring is strongly activated with an ortho- or para-electron-withdrawing group,95 in a manner suggestive of a classical SNAr mechanism.
2.5 ∣. Radiosynthesis of labeling synthons
Tracer molecules that are not readily amenable to late-stage labeling with [18F]fluoride ion may be amenable to labeling with synthons derived from [18F]fluoride ion. Popular synthons are electrophilic agents, such as 18F-labeled aryl halides, benzaldehydes, benzyl halides, benzyl azides, and aryl carboxylic acid esters.96,97 Major applications of such synthons are in the labeling of peptides, proteins, and other macromolecules.98 They may also be used for labeling small molecules. Many of these synthons may be accessed through classical SNAr reactions with [18F]fluoride ion, but improved methods are always sought, as well as access to all regioisomers.
2.5.1 ∣. From diaryliodonium salts
Table 1 lists many of the labeling synthons that have been prepared from diaryliodonium salts. Overall, moderate to good yields of the listed synthons have been obtained from rapid reactions (1-45 min) at low to moderately high temperatures (80°C-200°C). Not all the listed examples have been isolated for application.
TABLE 1.
Labeling synthons prepared from diaryliodonium salts
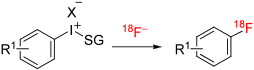
| ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Entry | Labeling Synthon | Precursor |
Labeling conditions |
Yield,a % | Reference | |||||
| R1 | SG | X− | Solvent | Mode | °C/W | min | ||||
| 1 | 18F]1-Chloro-4-fluorobenzene | 4-Cl | 4-An | TsO | DMSO | Batch | 80 | 20 | 80 | Way et al99 |
| 2 | [18F]1-Chloro-4-fluorobenzene | 4-Cl | 4-An | TsO | DMSO | MW | ns | 5 | 70 | Way et al99 |
| 3 | [18F]1-Bromo-2-fluorobenzene | 2-Br | Ph | Cl− | DMF | MR | 190 | 4 | 68 | Chun et al65 |
| 4 | [18F]1-Bromo-4-fluorobenzene | 4-Br | 2-Thi | Br− | DMF | Batch | 130 | 45 | 70 | Way et al99 |
| 5 | [18F]1-Bromo-4-fluorobenzene | 4-Br | 4-BrC6H4 | Br− | DMF | Batch | 130 | 10 | 65 ± 10 | Ermert et al102 |
| 6 | [18F]1-Fluoro-4-iodobenzene | 4-I | 4-IC6H4 | TsO− | DMF | Batch | 80-160 | 40 | 21-46 | Wuest and Kniess100 |
| 7 | [18F]1-Fluoro-4-iodobenzene | 4-I | 4-IC6H4 | Cl− | DMF | MW | 120 | 5 | 15-70 | Wuest and Kniess100 |
| 8 | [18F]1-Fluoro-4-iodobenzene | 4-I | 4-An | TfO− | DMF | Batch | 130 | 10 | 40 ± 7 | Kügler et al101 |
| 9 | [18F]1-Fluoro-4-iodobenzene | 4-I | 2-Thi | Br− | DMF | Batch | 130 | 45 | 60 ± 9 | Way et al99 |
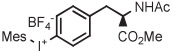
| 10 | [18F]1-Fluoro-4-iodobenzene | 4-I | Mes | BF4− | DMF | Batch (Cu) | 85 | 20 | 35 ± 8 | Ichiishi et al41 |
| 11 | [18F]3-Fluorobenzaldehyde | 3-CHO | Ph | Br− | DMF | MW | 110 | 5 | 80 | Basuli et al87 |
| 12 | [18F]3-Fluorobenzaldehyde | 3-CHO | Mes | BF4− | DMF | Batch (Cu) | 85 | 20 | 35 ± 1 | Ichiishi et al41 |
| 13 | [18F]3-Fluorobenzaldehyde | 3-CHO | Mes | BF4− | DMF | Batch (Cu) | 85 | 20 | 85 | Zlatopolskiy et al105 |
| 14 | [18F]3-Fluorobenzaldehyde | 3-CHO | 4-An | I− | DMSO | Batch | 130 | 10 | 20-25 | Richarz et al72 |
| 15 | [18F]3-Fluorobenzaldehyde | 3-CHO | 2-Thi | Cl− | DMF | MR | 180 | ~5 | 46 | Chun and Pike46 |
| 16 | [18F]4-Fluorobenzaldehyde | 4-CHO | 4-An | Cl− | DMF | MR | 180 | ~5 | 73 | Chun and Pike46 |
| 17 | [18F]4-Fluorobenzaldehyde | 4-CHO | Ph | CF3CO2− | DMF | MR | 180 | ns | 83 | Charlton and Carroll106 |
| 18 | [18F]2-Fluorobenzyl chloride | 2-CH2Cl | 4-An | TsO− | DMF | MR | 180 | ~4 | 55 | Chun and Pike46 |
| 19 | [18F]3-Fluorobenzyl chloride | 3-CH2Cl | 5-Me-2-Thi | Cl− | DMF | MR | 200 | ~4 | 33 | Chun and Pike46 |
| 20 | [18F]4-Fluorobenzyl chloride | 4-CH2Cl | TMP | TsO− | DMF | MR | 180 | ~3 | 52 | Chun and Pike46 |
| 21 | [18F]2-Fluorobenzyl bromide | 2-CH2Br | TMP | TsO− | DMF | MR | 160 | ~3 | 50 | Chun and Pike46 |
| 22 | [18F]3-Fluorobenzyl bromide | 3-CH2Br | 2-Thi | Cl− | DMF | MR | 200 | ~4 | 31 | Chun and Pike46 |
| 23 | [18F]4-Fluorobenzyl bromide | 4-CH2Br | TMP | TsO− | DMF | MR | 140 | ~4 | 20 | Chun and Pike46 |
| 24 | [18F]2-Fluorobenzyl azide | 2-CH2N3 | 4-An | TsO− | DMF | MR | 180 | ~3 | 41 | Chun and Pike44 |
| 25 | [18F]3-Fluorobenzyl azide | 3-CH2N3 | 4-An | TsO− | DMF | MR | 180 | ~3 | 39 | Chun and Pike44 |
| 26 | [18F]4-Fluorobenzyl azide | 4-CH2N3 | 4-An | Br− | DMF | MR | 200 | ~1.5 | 40 | Chun and Pike44 |
| 27 | [18F]4-Fluorophenolb | 4-OH | 2-Thi | Br− | DMF | Batch | 130 | 15-20 | (34-36) | Ross et al37 |
| 28 | [18F]4-Fluorophenolb | 4-OH | 4-BnO-C6H4 | TsO− | DMF | MW | 1 | 52 ± 3 | Heifer et al47 | |
| 29 | N-Succinimidyl 4-[18F]fluorobenzoate |
|
2-Thi | CF3CO2− | DMF/TEMPO | Batch | 130 | 5 | 4-23 | Yan et al112 Carrol and Yan113 |
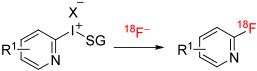
| ||||||||||
| 30 | [18F]2-Fluoro-6-chloropyridine | 6-Cl | 4-An | TsO− | DMF | MR | 100 | ~4 | 60 | Chun and Pike114 |
| 31 | [18F]2-Fluoro-5-bromopyridine | 5-Br | 4-An | TsO− | DMF | MR | 120 | ~4 | 64 | Chun and Pike114 |
| 32 | [18F]2-Fluoro-6-bromopyridine | 6-Br | 4-An | TsO− | DMF | MR | 180 | ~4 | 53 | Chun and Pike114 |
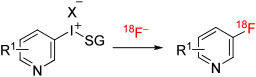
| ||||||||||
| 33 | [18F]3-Fluoro-2-chloropyridine | 2-Cl | 4-An | TsO− | DMF | MR | 180 | ~3 | 36 | Chun and Pike114 |
| 34 | [18F]3-Fluoro-2-bromopyridine | 2-Br | 4-An | TsO− | DMF | MR | 160 | ~4 | 28 | Chun and Pike114 |
| 35 | [18F]3-Fluoro-6-chloropyridine | 6-Cl | 4-An | TsO− | DMF | MR | 180 | ~4 | 28 | Chun and Pike114 |
| 36 | [18F]3-Fluoro-6-bromopyridine | 6-Br | 4-An | TsO− | DMF | MR | 160 | ~4 | 30 | Chun and Pike114 |
Abbreviations: (Cu), Cu-mediated reaction; DMF, dimethylformamide; DMSO, dimethyl sulfoxide; MR, microreactor (microfluidic); ns, not specified; MW, microwave (batch); TEMPO, 2,2,6,6-tetramethyl-1-piperidinyloxyl.
Values in plain type are yields before isolation. Values in bold and parentheses are yields after isolation.
Process is 2 steps from BnO-protected salt. Data are for the radiofluorination step.
[18F]Fluorohalobenzenes have been accessed rapidly in moderate to good yields (Table 1, entries 1-10).41,65,99-102 Notably, among these labeling synthons, [18F]4-fluoroiodobenzene (entries 6-10)100 can be prepared efficiently and has been applied to the radiosynthesis of several candidate radiotracers through efficient transition metal–mediated coupling reactions. These radiotracers include 18F-labeled nucleosides,103 a D4 receptor ligand,101 and a cyclooxygenase 2 inhibitor104 (Figure 18).
FIGURE 18.

Examples of compounds labeled with synthons prepared from hypervalent aryliodine precursors. Blue indicates the partial structure derived from the labeling synthon
[18F]Fluorobenzaldehydes are well known to be useful labeling synthons, especially for multistep syntheses leading to labeled amino acids.97 Only the 2- and 4-regioisomers are readily accessible through classical SNAr reactions, whereas not only the 4-regioisomer but also the 3-regioisomer can be obtained through the radiofluorination of diaryliodonium salts in good yields (Table 1, entries 11-17).46,72,105,106 It is notable that the formyl group resists possible oxidation during the syntheses of the salts and their radiofluorination reactions.
All regioisomers of the [18F]fluorobenzyl chlorides,46 bromides,46 and azides44 have been obtained in moderate yields from single-step radiofluorination reactions under microfluidic conditions using short residence times (≤4 min) at quite high temperatures (160°C-200°C) (Table 1, entries 18-26). In the syntheses of the [18F] fluorobenzyl halides, potential competing displacement of the labile halogen was not a confounding issue. These single-step radiosyntheses are attractive compared with conventional multistep syntheses based on SNAr. [18F]3-Fluorobenzyl bromide has also been prepared from [18F]3-fluorobenzaldehyde, itself prepared from a diaryliodonium salt, and applied to label an anticancer agent, lapatinib, with 18F (Figure 18).107 The [18F] fluorobenzyl azides have utility for labeling macromolecules through “click reactions” with alkyne derivatives,108 as exemplified by the labeling of proteins109 and oligonucleotides110 (Figure 18).
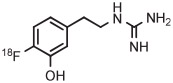
[18F]Fluorophenoxy compounds are usually inaccessible through simple single-step SNAr reactions because of unfavorable aryl ring electronics. [18F]4-Fluorophenol is a useful labeling synthon for such compounds.97 [18F]4-Fluorophenol has been prepared in 2 steps in 34% to 36% overall yield through the radiofluorination of 4-benzyloxyphenyl(2-thienyl)iodonium bromide in DMF at 130°C for 5 minutes, followed by almost quantitative removal of the benzyl group with ammonium formate in methanol in the presence of palladium black (Table 1, entry 27).37 Use of a bis(benzyloxyphenyl)iodonium tosylate precursor and microwave heating improved the yield of [18F]4-fluorophenol to 52%. TEMPO did not improve the yield of this reaction (entry 28).47
[18F]Fluorobenzoic acid esters can be useful labeling synthons, as exemplified by [18F]N-succinimidyl 4-fluorobenzoate, which is widely used for labeling peptides and proteins.97 The conventional method for synthesizing [18F]N-succinimidyl 4-fluorobenzoate, based on initial SNAr on the triflate salt of ethyl 4-(trimethylammonium) benzoate, is quite high yielding (38%) but takes 3 steps over about an hour.111 [18F]N-Succinimidyl 4-fluorobenzoate has been prepared in a single step from an aryl(2-thienyl)iodonium trifluoroacetate in 13% to 23% yields in shorter synthesis times (Table 1, entry 29).112,113
[18F]Fluorohalopyridines are potentially useful labeling agents as they should, for example, participate in coupling chemistry like that of the [18F]fluorohalobenzenes. Several [18F]fluorohalopyridines have been synthesized in a microfluidic apparatus (NanoTek, Advion) from 4-anisyl(halopyridyl)iodonium tosylates in DMF at temperatures in the range of 100°C to 180°C with microreactor residence times of about 3 to 4 minutes.112 Labeling yields ranged from 53% to 60% for [18F]2-fluorohalopyridines (Table 1, entries 30-32) and from 28% to 36% for [18F]3-fluorohalopyridines (entries 33-36).
2.5.2 ∣. From aryliodonium ylides
The use of aryliodonium ylides to prepare labeling synthons is quite recent, and there are consequently relatively few examples. Nonetheless, the known examples generally show moderate to good yields from rapid reactions at quite low temperatures (Table 2).
TABLE 2.
Some labeling synthons prepared from aryliodonium ylides
| |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Entry | Labeling Synthon | Precursor |
Labeling Conditions |
Yield,b % | References | ||||
| R1 | Aux−a | Mode | Solvent | °C | min | ||||
| 1 | [18F]3-Fluorobromobenzene | 3-Br | C | Batch | DMF | 130 | 10 | 72 ± 3 | Yuan et al115 |
| 2 | [18F]4-Fluoroiodobenzene | 4-I | A | Batch | DMSO | 110 | 15 | 70 ± 10 | Kügler et al101 |
| 3 | [18F]3-Fluorobenzaldehyde | 3-CHO | B | Batch | DMF | 120 | 10 | 7-52 | Petersen et al116 |
| 4 | [18F]3-Fluorobenzyl azide | 3-CH2N3 | B | Batch | DMF | 120 | ns | 69 ± 8 | Wang et al117 |
| 5 | [18F]4-Fluorobenzyl azide | 4-CH2N3 | B | Batch | DMF | 120 | 10 | 70 ± 4 (52 ± 2) | |
| 6 | [18F]4-Fluorobenzyl azide | 4-CH2N3 | B | Batch | DMF | 120 | 10 | (25 ± 10) c | Rotstein et al67 |
| 7 | [18F]4-Fluorobenzyl azide | 4-CH2N3 | B | MR | DMF | 210 | ~1 | 68 ± 5 (24 ± 0) | Calderwood et al75 |
| 8 | [18F]3-Fluorobenzoic acid methyl ester | 3-CO2Me | B | Batch | DMF | 120 | 10 | 77 ± 7 | Rotstein et al67 |
| 9 | [18F]2-(3-Fluorophenyl)ethylamined | 3-(CH2)2NHBoc | A | Batch | DMF | 110 | 10 | ~70 | Drerup et al118 |
| |||||||||
| 10 | [18F]1-(2-Azidoethoxy)-4-bromo-2-fluorobenzene | 2-(O(CH2)2N3) | B | Batch | DMF | 120 | 10 | 69 ± 2 | Wang et al117 |
| 11 | [18F]1-((2-Azidoethoxy)methyl)-4-bromo-2-fluorobenzene | 2-(CH2 O(CH2)2N3) | B | Batch | DMF | 120 | 10 | 90 ± 2 | Wang et al117 |
Abbreviations: DMF, dimethylformamide; DMSO, dimethyl sulfoxide; MR, microreactor (microfluidic); ns, not specified.
Values in plain type are yields before isolation. Values in bold and parentheses are yields after isolation.
Uncorrected for decay.
Process is 2 steps. Data are for the radiofluorination step.
Thus, [18F]3-fluorobromobenzene has been prepared in 72% yield through radiofluorination of a spiroadamantyl aryliodonium ylide (Table 2, entry 1).115 This labeling synthon, prepared in this manner, has been coupled to a cyclic amide in the presence of copper(I) iodide to give a candidate AMPA (α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid) receptor radiotracer in 72% yield (Figure 18).115 After work-up and purification, the final overall yield of the radiotracer was about 15%. [18F]4-Fluoroiodobenzene has been obtained from the radiofluorination of an ylide prepared from Meldrum's acid, namely, (2,2-dimethyl-5,7-dioxo-1,3-dioxocan-6-yl) (4-iodophenyl)iodonium, in dimethyl sulfoxide (DMSO) at 110°C in the presence of t-butylammonium bicarbonate in 70% yield (entry 2). This yield is quite similar to that obtained from a symmetrical diaryliodonium salt (60%) (Table 1, entry 9)99 and is appreciably higher than that from an unsymmetrical diaryliodonium salt (40%) (Table 1, entry 8).101 [18F]4-Fluoroiodobenzene prepared from the ylide precursor has been used in a multistep radiosynthesis of a candidate D4 receptor radiotracer, known as [18F]FAUC 316 (Figure 18).101 This radiotracer was obtained for clinical use in 10% yield after 80 minutes of preparation time.
Spirocyclopentyl aryliodonium ylides have been used to prepare several useful labeling synthons. [18F]3-Fluorobenzaldehyde has been prepared in variable yield (7%–52%) yield (Table 2, entry 3).116 [18F]3-Fluorobenzyl azide (entry 4)117 and [18F]4-fluorobenzyl azide67,75,117 (entries 5-7) have been prepared in good nonisolated yields. Isolated yields for the 4-fluoro isomers ranged from low to moderate (entries 5-7). Other labeling synthons that carry reactive substituents at a benzyl position have also been prepared, including [18F]3-fluorobenzoic acid methyl ester (77%) (entry 8).67 [18F]2-(3-Fluorophenyl) ethylamine has also been prepared in good yield (~70%) for labeling enzyme inhibitors (entry 9).118
Long-chain alkyl azido labeling synthons have been prepared through the radiofluorinations of “ortho-oxygen-stabilized iodonium derivatives” (Figure 19) in impressively high yields (Table 2, entries 10 and 11). In ortho-oxygen-stabilized iodonium derivatives, secondary bonding from the oxygen of a pendant ortho aliphatic substituent to the iodonium center provides extra thermal stability, which may account for the high radiofluorination yields. These labeling synthons were successfully applied to labeling single-stranded DNA aptamers (Figure 18).117,119
FIGURE 19.

Examples of ortho oxygen-stabilized aryliodonium ylides
2.6 ∣. Radiosynthesis of radiotracers
Although hypervalent aryliodine precursors have shown utility for preparing a wide variety of labeling synthons and these synthons have been used to meet challenging radiosynthesis requirements, the ideal is for radiotracers to be labeled at a late stage, preferably in a single step or a single labeling step followed by a single deprotection step. The utility of hypervalent aryliodine precursors for meeting this ideal is now summarized.
2.6.1 ∣. From diaryliodonium salts (copper free)
Translocator protein (TSPO) 18 kDa is a recognized biomarker for neuroinflammation. Extensive efforts have been made to produce high-performing radiotracers for imaging TSPO 18 kDa. [18F]FDAA1106, a candidate radiotracer for imaging this target, was the first radiotracer to be produced from a diaryliodonium salt.120 Radiofluorination of crude 4-anisyl(aryl)iodonium tosylate in DMSO at 80°C for 20 minutes gave [18F]FDAA1106 in moderate yield (46%) (Table 3, entry 1). In this early study, the iodonium salt was used in crude form because of concern about its stability. [18F]4-Fluoroanisole was a significant by-product (19%). In this case, a classical SNAr reaction would not have been feasible for the radiosynthesis of [18F]FDAA1106 because there is no strong electron-withdrawing substituent in the ortho or para position to the fluoro substituent. The other radiotracer examples shown in Table 3 are also structures for which classical SNAr would be either inapplicable or, at best, poorly applicable for 18F labeling.
TABLE 3.
Examples of radiotracers prepared from diaryliodonium salts under copper-free conditions
| Entry | Radiotracer |
Precursor (ArI+SGX−) |
Labeling Technique |
Labeling Conditions |
Yield,a % | Reference | |||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Name | Structure | ArI+ | SG | X− | Solvent | °C/W | min | ||||
| 1 | [18F]FDAA1106 |
|
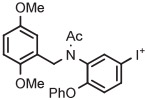
|
4-An | TsO− | Batch (1 step) | DMSO | 80 | 20 | 46 | Zhang et al120 |
| 2 | [18F]MTEB |
|
|
4-An | TsO− | MW (1 step) | DMF with TEMPO | 100 | 2 × 2 | 20 ± 1 | Telu et al69 |
| 3 | [18F]SP203 |
|
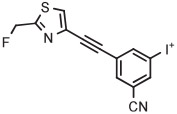
|
4-An | TsO− | MW (1 step) | DMF with TEMPO | 100 | 2 × 2 | 33 ± 8 | Telu et al69 |
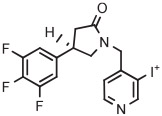
| 4 | [18F]UCB-H |
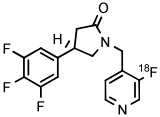
|
|
4-An | TfO− | Batch (1 step) | MeCN with TEMPO | 125 | 10 | 50 (34 ± 2) | Warnier et al40 |
| 5 | [18F]6-FBT |
|
|
3-Thi | TsO− | Batch (1 step) | MeCN with TEMPO | 130 | 10 | 60 ± 6a (19-40) | Lee et al56 |
| 6 | [18F]Flumazenil |
|
|
4-Tol | TsO− | Batch (1 step) | DMF with TEMPO | 150 | 5 | 67 ± 3 (54 ± 9) | Moon et al70,126 |
| 7 | [18F]Fluoro-palonosetron |
|
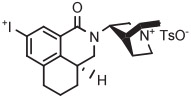
|
4-An | TsO− | Batch (1 step) | DMF with TEMPO | 140 | 12 | 14 (6) | Mu et al127 |
| 8 | [18F]FIMX |
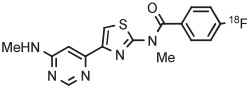
|
|
Ph | TsO− | MW (1 step) | DMSO with TEMPO | 90 | 2.5 | 38 (20) | Xu et al55 |
| 9 | Matrix metalloproteinase radioligand |
|
|
4-An | ClO4− | Batch (2 steps) | DMF-trace H2O | 130 | 3 | 70 (10-20) | Selivanova et al128 |
| 10 | [18F]CB91 |
|
|
2-Thi | TFA− | Microreactor (1 step) | DMSO | 190 | 0.25 | 42 ± 7 | Saccomanni et al130 |
| 11 | 5-HTc receptor radioligand |
|
|
4-Tol | TsO− | Batch with TEMPO (2 steps) | DMF | 130 | 10 | (7.8 ± 2.7) | Kim et al131 |
| 12 | [18F]MFBG |
|
|
4-An | TfO− | Pyrolysis of 18F− salt (2 steps) | Toluene-MeCN | 120 | 8 | (31) | Hu et al71 |
| 13 | [18F]FMHPG |
|
|
2-Thi | Br− | Batch with TEMPO (3 steps) | DMF-H2O | 150 | 25 | 45 (7) | Jang et al133 |
| 14 | [18F]6-FDA |
|
|
4-An | TfO− | Pyrolysis of 18F− salt (2 steps) | Toluene | 150 | 4 | 46 ± 7 (36 ± 4) or (72 ± 10) | Neumann et al38 |
| 15 | [18F]FDOPA |
|
|
4-An | TfO− | Pyrolysis of 18F− salt (2 steps) | Diglyme | 140 | 5 | 40 (14 ± 4) | Kuik et al137 |
Abbreviations: DMF, dimethylformamide; DMSO, dimethyl sulfoxide; [18F]FIMX, [18F]4-fluoro-N-methyl-N-(4-(6-(methylamino)pyrimidin-4-yl)thiazol-2-yl)benzamide; [18F]FMHPG, [18F]4-fluoro-3-hydroxyphenethylguanidine; [18F]MFBG, [18F]1-(3-fluorobenzyl)guanidine; [18F]6-FBT, [18F]4-(6-fluorobenzo[d]thiazol-2-yl)-N-methylaniline; [18F]6-FDA, [18F]6-fluorodopamine; [18F]SP203, 3-fluoro-5-(2-(2-[18F] (fluoromethyl)-thiazol-4-yl)ethynyl)benzonitrile; MW, microwave (batch); TEMPO, 2,2,6,6-tetramethyl-1-piperidinyloxyl.
Values in plain type are yields before isolation. Values in bold and parentheses are yields after isolation.
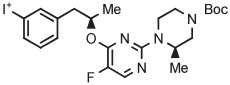
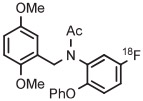
[18F]MTEB and a diarylalkyne congener 3-fluoro-5-(2-(2-[18F](fluoromethyl)-thiazol-4-yl)ethynyl)benzonitrile ([18F]SP203) are high-performing radiotracers for imaging brain metabotropic glutamate receptor subtype 5 (mGlu5). [18F]MTEB has been obtained in 20% yield through the radiofluorination of a 4-anisyl(aryl)iodonium tosylate precursor in DMF at 160°C under microwave irradiation for 4 minutes in the presence of TEMPO (Table 3, entry 2).69 [18F]SP203 has been obtained similarly from the corresponding 4-anisyl(aryl)iodonium tosylate in 33% yield (entry 3).69 On the basis of experiments in a microfluidic apparatus, the chemoselectivities of the radiofluorination reactions for the desired radiotracers over [18F]4-fluoroanisole are exceptionally high (>44), and much higher than from precursors having a phenyl spectator group. TEMPO was beneficial for improved yield in both microreactor syntheses and batch syntheses. The precursors were found to be stable for several months when stored in a refrigerator.
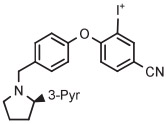
Synaptic vesicle glycoprotein 2A is a biomarker for synaptic loss in epilepsy and Alzheimer's disease. [18F] UCB-H has been developed as the first radiotracer for imaging brain synaptic vesicle glycoprotein 2A in vivo.40 The original production of this radiotracer had involved several steps performed over 2 hours, starting with [18F]3-fluoroisonicotinaldehyde as labeling synthon, giving less than 15% yield.40 Subsequently, a much improved single-step method has been devised for the preparation of [18F]UCB-H based on radiofluorination of a 4-anisyl(3-pyridyl)iodonium tosylate in MeCN at 125°C for 10 minutes in the presence of TEMPO.40 [18F]UCB-H was produced regularly for clinical use according to this method in 34 ± 2% yield and with very high molar activity (815 GBq/μmol) (Table 3, entry 4). The difficulty of radiofluorinations in the 3-position of pyridyl compounds with classical SNAr reactions is well known.16 This radiosynthesis exemplifies the utility of aryl(3-pyridyl) iodonium salts for introducing [18F]fluoride ion into the 3-position of pyridyl substrates. In this work, the authors stressed the need to obtain the iodonium salt in fully deprotonated form (through the use of a basic alumina), as otherwise radiochemical yields were substantially decreased. Crystallization of the salt was important for purification and for ensuring good solubility.
The secondary arylamine, [18F]4-(6-fluorobenzo[d] thiazol-2-yl)-N-methylaniline ([18F]6-FBT), is a candidate radiotracer for imaging brain β-amyloid plaques in Alzheimer's disease. Two-step syntheses of [18F]6-FBT from 5 N-Boc-protected diaryliodonium tosylates with different spectator groups have been compared.56 Each salt was radiofluorinated in MeCN in the presence of TEMPO at 130°C for 10 minutes. [18F]N-Boc-protected-6-FBT was produced from the salts with 2-Thi, 3-Thi, 4-Tol, Ph, or 4-An as spectator group in 34%, 60%, 27%, 24%, and 19% yield, respectively. Deprotection with 3M HCl in ethyl acetate gave [18F]6-FBT in 30%, 41%, and 19% overall yield from radiosyntheses that started with the salts having 2-Thi, 3-Thi, and Ph spectator groups, respectively (Table 3, entry 5).
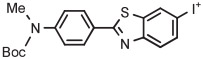
11C-labeled flumazenil is a PET radiotracer long used for studying brain benzodiazepine receptors.121 [18F] Flumazenil has now been found to be an equally effective radiotracer.122 The longer half-life of 18F allows [18F] flumazenil to be used at imaging sites remote from cyclotrons. The original method for producing [18F]flumazenil, based on radiofluorination of a nitro precursor,123 has a history of low and unreliable production yield.124,125 [18F] Flumazenil has since been obtained in over 67% yield from an aryl(4-tolyl)iodonium tosylate by radiofluorination in DMF in the presence of TEMPO at 150°C for 5 minutes (Table 3, entry 6).70 In the development of this method, several key parameters were carefully evaluated and optimized, including spectator group (Ph, 2-Thi, 3-Thi, 3-An, 4-An, and 4-Tol), precursor amount (2, 4, and 8 mg), solvent (MeCN, DMF, and DMSO), base (n-Bu4NHCO3 and K 2.2.2-K2CO3), temperature (100°C, 125°C, and 150°C), time (5 and 15 min), presence or absence of TEMPO radical scavenger, and stability of salt during reaction.126 The optimal conditions have been operated for the automated routine production of [18F]flumazenil for imaging experiments over a 2.5-year period, and they have achieved impressive performance.70 Thus, the average yield was 53% (n = 94), and the mean molar activity was 572 GBq/μmol (entry 6).
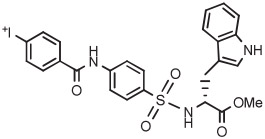
Another compound, resembling flumazenil in having a fluoro substituent in meta orientation to a carboxamido group, is fluoro-palonosetron. Radiofluorination of an anisyl(aryl)iodonium tosylate precursor under conditions similar to those used for the labeling of flumazenil gave [18F]fluoropalonosetron in a nonoptimized yield of 14% (Table 3, entry 7).125 By contrast, a matrix metalloproteinase radioligand having a carboxamido group in para orientation to fluorine was radiofluorinated in 2 steps from a protected precursor, in which the initial radiofluorination yield was 70% (Table 3, entry 8).128
In some cases, spectator groups other than 4-anisyl or 2-thienyl have been used successfully. Although the phenyl group is not the most effective spectator group for achieving high chemoselectivity in radiofluorination, aryl(phenyl)iodonium salts can still be quite attractive as precursors to radiotracers because of their relative ease of synthesis from commercially available Koser's reagent. Moreover, any [18F]fluorobenzene by-product from their radiofluorinations is easily separated off by evaporation or chromatography. An example of the use of phenyl as a spectator group, in addition to that already mentioned ([18F]FDAA1106; Table 3, entry 1), is the radiosynthesis of the mGlu1 receptor radiotracer, [18F]4-fluoro-N-methyl-N-(4-(6-(methylamino)pyrimidin-4-yl)thiazol-2-yl)benzamide ([18F]FIMX), which contains a fluoro substituent in para orientation to a carboxamido group. Microwave-promoted (90-W) radiofluorination of an N-Boc-protected aryl(phenyl)iodonium tosylate in DMSO for 2.5 minutes gave [18F]FIMX in 38% yield (entry 9).55 Fortuitously, complete deprotection occurred during the radiofluorination reaction. The precursor showed excellent stability during cold storage in the dark. [18F]FIMX could therefore be prepared regularly by this method for clinical use.129 No [18F]fluorobenzene was observed in this radiosynthesis. Prior attempts to prepare this radiotracer through SNAr on a protected nitro precursor had failed.
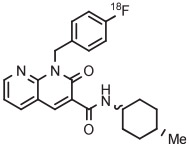
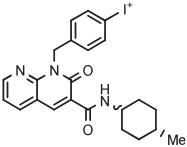
The synthesis of [18F]CB91, a CB2 receptor radiotracer, provides an example of the effective use of a 2-thienyl spectator group. In this case, radiochemistry optimization and later high-activity radiotracer production were performed in a commercial microfluidic apparatus (NanoTek, Advion). [18F]CB91 was obtained as a mixture of cis and trans isomers in 42% yield from reactions in DMSO at 190°C using about 25 seconds of microreactor residence time (Table 3, entry 10).130 CB91 contains a fluorine substituent on a deactivated phenyl ring and therefore would have been a poor candidate for direct labeling through a classical SNAr reaction.
A candidate 5-HT2C receptor radioligand having a 4-(3-fluorophenethoxy)pyrimidyl substructure has been labeled in 7.8% isolated yield for imaging use in 2 steps from an N-Boc-protected aryl(4-tolyl)iodonium tosylate precursor in DMF in the presence of TEMPO.131 The radiofluorination was conducted for 10 minutes at 130°C, followed by rapid removal of the Boc group with 2M hydrochloric acid (Table 3, entry 11).
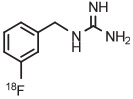
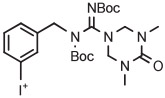
[18F]1-(3-Fluorobenzyl)guanidine ([18F]MFBG) is a promising radiotracer for imaging neuroblastoma. Earlier syntheses of this radiotracer had required 3 steps from [18F]fluoride ion that were overall inefficient.132 An efficient 2-step synthesis of [18F]MFBG has been developed from 4-anisyl(aryl)iodonium triflate having a fully protected guanidinyl group, based on generation of the [18F]fluoride salt followed by thermolysis and acid deprotection (Table 3, entry 12).71 The synthesis has been fully automated on a commercial radiosynthesis instrument (Synthera, IBA). This procedure gives [18F]MFBG for clinical use in 56 minutes and in almost 3-fold higher yield (31%) than previous methods.
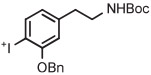
[18F]4-Fluoro-3-hydroxyphenethylguanidine ([18F] FMHPG) is a useful radiotracer for quantifying regional cardiac sympathetic nerve density. An attempt to produce this radiotracer through radiofluorination on an iodonium salt of a fully protected MHPG followed by single-step deprotection had proven unsuccessful.133 Nevertheless, multistage synthesis starting with the radiofluorination of O-benzyl-N-Boc-protected aryl(2-thieny)iodonium bromide in the presence of TEMPO to give [18F]t-butyl 3-(benzyloxy)-4-fluorophenethylcarbamate in 45% yield (Table 3, entry 13) followed by 2 more steps to install the guanadinyl group and deprotect gave [18F]FMHPG for clinical use in 7% overall yield.133
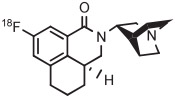
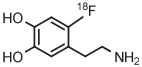
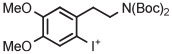
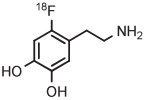
[18F]6-Fluorodopamine ([18F]6-FDA) is an established radiotracer for imaging the cardiac norepinephrine transporter. Historically, [18F]6-FDA has been produced through inefficient electrophilic methods starting from cyclotron-produced [18F]fluorine gas. Although [18F]6-FDA can be synthesized in 4 steps starting with radiofluorination on a nitro precursor, overall yield is low (9%).134 More recently, a protected 4-anisyl(aryl) iodonium triflate has been developed as a precursor for [18F]6-FDA.38 Generation of the [18F]fluoride salt from this triflate precursor in MeCN, followed by evaporation of solvent, thermolysis in toluene, and deprotection, gives [18F]6-FDA. [18F]6-Fluorodopamine for clinical use has been produced with this precursor on one automated radiochemistry platform (Synthera, IBA) in 36% yield after 65 minutes and on another automated platform (TRACERlab FX-FN, GE) in 72% yield after 130 minutes (Table 3, entry 14).
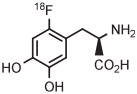
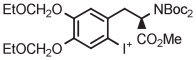
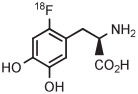
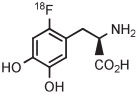
For several decades, [18F]FDOPA has been a key PET radiotracer, mainly for studies of Parkinson disease. However, the widespread use of [18F]FDOPA has been hampered because of the long-standing absence of a simple method for production in high yield from [18F]fluoride ion.135 Therefore, efforts have been made to synthesize [18F]FDOPA through radiofluorination of a protected diaryliodonium salt followed by deprotection. According to a commercial report,136 [18F]FDOPA can be prepared in 30% to 40% yield by conversion of a 4-anisyl(aryl) iodonium triflate into the [18F]fluoride salt in MeCN followed by solvent evaporation, thermolysis at 140°C for 5 minutes in diglyme, and then deprotection. The method has since been adapted to produce [18F]FDOPA for clinical use in 14 ± 4% overall yield in a synthesis time of 117 minutes (Table 3, entry 15).137 The protected precursor is commercially available.
General observations from Table 3 are that 4-anisyl has been selected most frequently as the spectator group, and weakly nucleophilic tosylate as the counterion. Functional group tolerance in the radiofluorinations is quite broad, but free amino, phenol, guanidinyl, and carboxyl groups have generally been protected. Amounts of precursor used in the radiosyntheses are generally low and very compatible with the use of a small volume of a polar aprotic solvent (eg, DMSO, DMF, and MeCN) in batch reactions. Different large cations have been used for solubilization of the [18F] fluoride ion, including Cs+, Et4N+, Bu4N+, and K+-K 2.2.2, and these have not always given similar results in direct comparisons. The addition of the radical scavenger TEMPO has been found beneficial in many of these reactions. Reactions operate in a quite low temperature range (80°C-190°C) and are generally brief, proceeding to useful yields in just a few minutes. The radioactive fluoro derivatives of spectator groups have never been reported as being problematic in radiotracer separations. All the reactions may be regarded as NCA, and where reported, the molar activities are in the usual range for NCA radiofluorinations by other methods.
2.6.2 ∣. From diaryliodonium salts (copper mediated)
The [(MeCN)4CuOTf]-mediated radiofluorinations of protected aryl(mesityl)iodonium tetrafluoroborates for the radiosyntheses of [18F]FDAA1106, protected [18F]l-4-fluorophenylalanine ([18F]4-FPhe), and protected [18F]6-FDA, under minimalist conditions in DMF at 85°C for 20 minutes, have been reported to proceed in exceptionally high yields of 93%, 81% to 92%, and 71% to 94%, respectively (Table 4, entries 1-3).105 A minimalist approach has since been used to produce [18F]FDAA1106, [18F]4-FPhe, and [18F]6-FDA for clinical use in 60%, 53% to 66%, and 46% overall yields, respectively.105 Zischler et al have subsequently reported yields of 41% and 56% for the production of [18F]FDAA1106 and [18F]4-FPhe, respectively (entries 1 and 3).136
TABLE 4.
Radiotracers prepared from aryl(mesityl)iodonium salts: copper mediated
| Entry | Target radiotracer |
Precursor Structure |
Labeling Technique |
Labeling Conditions |
Yield,a % | Reference | |||
|---|---|---|---|---|---|---|---|---|---|
| Name | Structure | Solvent | °C | min | |||||
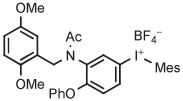
| 1 | [18F]FDAA1106 |
|
|
Minimalist (1 step) | dmf | 85 | 20 | 93 (60) (41 ± 3) | Zlatopolskiy et al,105 Zischler et al138 |
| 2 | [18F]4-FPhe |
|
|
Minimalist (2 steps) | DMF | 85 | 20 | 81-92 (53-66) (56 ± 5) | Zlatopolskiy et al,105 Zischler et al138 |
| 3 | [18F]6-FDA |
|
|
Minimalist (2 steps) | DMF | 85 | 20 | 71-94 (46) | Zlatopolskiy et al105 |
| 4 | [18F]FDOPA |
|
|
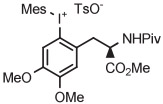
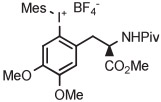
Batch | DMF | 85 | 20 | 17 ± 5 | Ichiishi et al41 |
| 5 | [18F]FDOPA |
|
|
Batch | DMF | 85 | 20 | 31 ± 3 | Ichiishi et al41 |
| 6 | [18F]4-FPhe |
|
|
Batch | DMF | 85 | 20 | 23 ± 5 | Ichiishi et al41 |
Abbreviations: DMF, dimethylformamide; [18F]4-FPhe, [18F]l-4-fluorophenylalanine; [18F]6-FDA, [18F]6-fluorodopamine.
Values in plain type are yields before isolation. Values in bold and parentheses are yields after isolation.
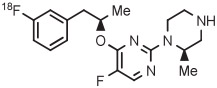
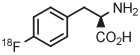
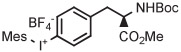
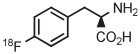
Copper-mediated radiofluorination of a fully protected aryl(mesityl)iodonium tosylate (6 μmol) with 18F−-18-crown-6-K2CO3 in DMF (0.25 mL) for 20 minutes at 85°C provided a protected [18F]FDOPA in 17% yield (Table 4, entry 4).41 Radiofluorination yield almost doubled to 31% when an analogous tetrafluoroborate salt was used (entry 5). Exchange of [18F]fluoride ion with the tetrafluoroborate anion occurred to some extent, and therefore, molar activity was only moderately high at 11 GBq/μmol. Radiofluorinations of appropriate aryl(mesityl)iodonium tetrafluoroborates under the same conditions gave protected [18F]4-FPhe in 23% yield (entry 6).41
Overall, copper-mediated radiofluorinations of aryl(mesityl)iodonium salts are highly attractive because they share many of the beneficial features of the copper-free radiofluorinations of diaryliodonium salts, such as a need to use only small amounts of precursor and a requirement for only moderate reaction temperatures and short reaction times. The precursor salts are usefully stable for months in the dark. Moreover, radiofluorination reactions proceed cleanly to single desired products, which is attractive for reducing separation challenges. The moderate molar activities obtained from the use of high-yielding tetrafluoroborate salts are acceptable for some radiotracers, such as [18F]FDOPA, [18F]4-FPhe, and [18F]6-FDA but unacceptable for radiotracers intended to bind to low-density protein targets, such as [18F]FDAA1106. A further drawback is a need to test that final radiotracer products for human administration are not unacceptably contaminated with copper. Normally, however, only very low and acceptable levels of copper are found in chromatographically purified products.
2.6.3 ∣. From aryliodonium ylides
Aryliodonium ylides having the auxiliary group derived from Meldrum's acid have been used to prepare [18F]4-FPPMP and[18F]3-FPPMP as candidate radiotracers for norepinephrine transporter and serotonin transporter, respectively (Table 5, entries 1 and 2).94 Low but useful yields were obtained from N-Boc-protected precursors over 2 steps. These radiotracers have 18F in highly electron-rich aryl rings. The radiofluorination of the precursor for [18F]4-FPPMP in MeCN at 130°C for 20 minutes also gave the Boc-protected [18F]3-fluoroisomer (~10%) in addition to the expected Boc-protected [18F]4-fluoroisomer (20%), thereby implicating the operation of an aryne-type mechanism.
TABLE 5.
Radiotracers prepared from aryliodonium ylides
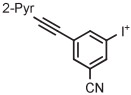
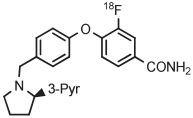
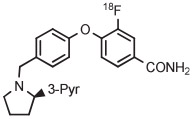
| Entry | Radiotracer Target |
Precursor (ArI+Aux−) |
Labeling conditions |
Yield,b % | Reference | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Name | Structure | ArI+ | Aux−a | Mode | Solvent | °C | min | |||
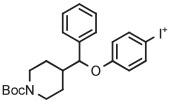
| 1 | [18F]4-FPPMP |
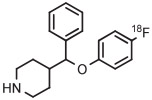
|
|
A | Batch (2 steps) | MeCN | 130 | 20 | 20 | Cardinale et al94 |
| 2 | [18F]3-FPPMP |
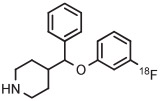
|
|
A | Batch (2 steps) | MeCN | 120 | 20 | (20 ± 5) | Cardinale et al94 |
| 3 | [18F]FPEB |
|
|
B | Batch (1 step) | DMF | 80 | 5 | 49 ± 6 | Stephenson et al139 |
| 4 | [18F]FPEB |
|
|
B | MR (1 step) | DMF | 200 | ~1 | (20 ± 5) | Stephenson et al139 |
| 5 | [18F]LY2459989 |
|
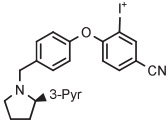
|
B | MR (2 steps) | DMF | 120 | 5 | 76 | Cai et al140 |
| 6 | [18F]LY2459989 |
|
|
B | Batch (2 steps) | DMF | 80 | 5 | 49 (30-43) | Cai et al140 |
| 7 | [18F]PFH |
|
|
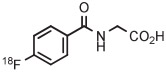
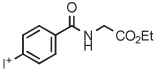
B | Batch (2 steps) | DMF | 130 | 10 | (46 ± 3) | Nkepang et al142 |
| 8 | [18F]4-FPhe |
|
|
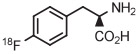
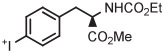
B | Batch (2 steps) | DMF | 120 | 10 | 55 ± 4 | Rotstein et al67 |
| 9 | [18F]CIMBI-198 |
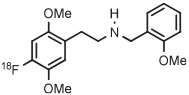
|
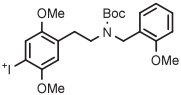
|
B | Batch with TEMPO (2 step) | DMF | 135 | 20 | 12 ± 3 (2) | Petersen et al143 |
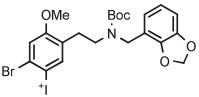
| 10 | 18F-Labeled 5-HT2A receptor radiotracer |
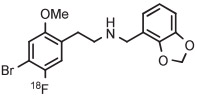
|
|
B | Batch (2 steps) with TEMPO | DMF | 135 | 20 | 76-77 (10-12) | Petersen et al143 |
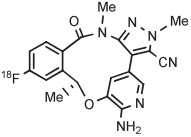
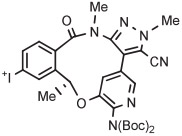
| 11 | [18F]Lorlatinib analog |
|
|
B | Batch (2 steps) | DMF | 80 | 10 | (14) c | Collier et al144 |
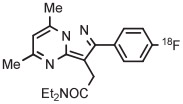
| 12 | [18F]FDPA |
|
|
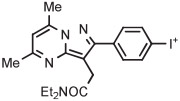
C | Batch (1 step) | DMF | 120 | 15 | 40 ± 3 | Wang et al145 |
| 13 | [18F]FDPA |
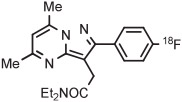
|
|
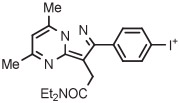
C | Batch (1 step) | MeCN | 120 | 12 | 45 ± 8 | Wang et al145 |
| 14 | [18F]Safinamide |
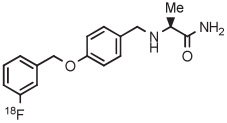
|
|
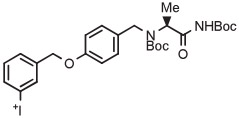
C | Batch (2 steps) | DMF | 120 | 10 | 15 | Rotstein et al93 |
| 15 | [18F]FMT |
|
|
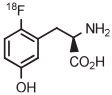
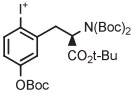
C | Batch (2 steps) | DMF | 120 | 20 | 60 (12)cb | Rotstein et al93 |
| 16 | [18F]MFBG |
|
|
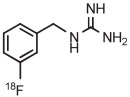
C | Batch (2 steps) | DMF | 120 | 10 | 70 (14)c | Rotstein et al93 |
Abbreviations: DMF, dimethylformamide; [18F]FMT, [18F]l-2-fluoro-meta-tyrosine; MeCN, acetonitrile; [18F]MFBG, [18F]1-(3-Fluorobenzyl)guanidine; TEMPO, 2,2,6,6-tetramethyl-1-piperidinyloxyl.
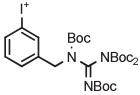
Values in bold and parentheses are yields after isolation.
Not decay corrected.
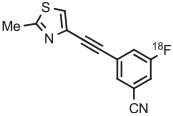
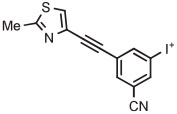
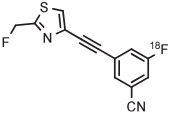
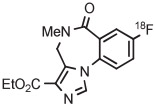
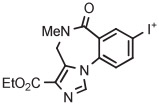
Stable spirocyclopentyl aryliodonium ylides are gaining considerable attention for PET radiotracer synthesis and production (Table 5, entries 3-11). [18F]FPEB is a preferred radiotracer for imaging brain mGlu5 receptors. However, synthesis by radiofluorination through classical SNAr is low yielding because of lack of activation by ring substituents. [18F]FPEB has been synthesized through radiofluorination of a spirocyclopentyl aryliodonium ylide in DMF at 80°C for 5 minutes in 49% yield (Table 5, entry 3).139 This reaction has been used to produce [18F]FPEB for clinical use in moderately high yield (20%) in an automated microfluidic apparatus (entry 4).139 Each production required less than 1 hour. The precursor was found to be stable for 2 months at room temperature. [18F]FPEB shares the same [18F]fluoro-3-cyano,5-alkynylaryl structural motif as the earlier mentioned mGlu5 receptor radiotracers, [18F]MTEB and [18F]SP203. The yield of [18F]FPEB from iodonium ylide precursor is quite comparable with those of [18F]MTEB and [18F]SP203 from 4-anisyl(aryl)iodonium tosylates (Table 3, entries 2 and 3).
A spirocyclopentyl aryliodonium ylide has been used in a 2-step synthesis of [18F]LY2459989, a κ-opioid receptor radiotracer.140 After the initial radiofluorination, the aryl nitrile group is oxidized to a carboxamido group with hydrogen peroxide. Optimal radiofluorination reactions in a microfluidic apparatus used DMF as solvent at 120°C for 1 minute and gave high incorporation of radioactivity (76%) into the nitrile intermediate (Table 5, entry 5). For high-activity production, the precursor was treated with [18F]fluoride ion in a vial at 80°C for 5 minutes followed by nitrile hydrolysis to give [18F]LY2459989 in 30% to 43% yield (entry 6). These yields were 30-fold higher than those obtained when starting with an SNAr reaction in which [18F]fluoride ion displaced an iodo or nitro group. In these SNAr reactions, the meta nitrile electron-withdrawing group provides only weak leaving group activation, and this activation is also countermanded by an ortho aryloxy group.
[18F]p-Fluorohippurate ([18F]PFH) shows promise for imaging renal function. The original radiosynthesis of [18F]PFH consisted of a 2-pot 4-step procedure that provided the radiotracer in 95 minutes in an estimated yield of 60% to 65%.141 By contrast, treatment of a protected spirocyclopentyl aryliodonium ylide with [18F]fluoride ion in DMF for 10 minutes at 130°C followed by basic hydrolysis for 5 minutes at 130°C gave [18F]PFH in 46% yield after 45 minutes (Table 5, entry 7).142 A spirocyclopentyliodonium ylide has also been used under similar radiofluorination conditions to produce N,O-protected [18F]4-FPhe (entry 8).67
Petersen et al prepared [18F]CIMBI-198 in 2 steps from an N-Boc-protected spirocyclopentyl iodonium ylide (Table 5, entry 9).143 The radiofluorination step proceeded in only low yield (12%), allowing the final product to be isolated in only 2% yield (entry 9). In [18F]CIMBI-198, the 18F is present in a highly substituted electron-rich aryl ring. A congeneric structure and candidate 5-HT2A receptor radioligand was labeled in much higher isolated yield (10%-12%), with the radiofluorination step proceeding in high yield (76%-77%) (entry 10).116
A di-Boc-protected spirocyclopentyl aryliodonium ylide precursor was successfully radiofluorinated in DMF under quite mild conditions (80°C; 10 min) to give [18F]lorlatinib, a candidate ROS1/ALK radiotracer, in useful isolated yield (14% non-decay-corrected) (Table 5, entry 11).144 By comparison, classical SNAr on a nitro precursor gave only 1% radiochemical yield.
A spiroadamantyl aryliodonium ylide has also been used to prepare another radiotracer for TSPO 18 kDa, namely, [18F]FDPA.145 The choice of base for use in the radiofluorination reaction was found to be critical. An experimental yield of 40% was obtained for reactions conducted in DMF (1.0 mL) at 120°C for 15 minutes with 2 mg of precursor in the presence of 12 mg of t-butyl ammonium mesylate (Table 5, entry 12). This method was modified to produce the radiotracer for clinical use in 45% yield (entry 13).
[18F]Safinamide, [18F]l-2-fluoro-meta-tyrosine ([18F] FMT), and [18F]MFBG have also been prepared from protected spiroadamantyl aryliodonium ylides.93 [18F] Safinamide was obtained in 15% yield through radiofluorination of precursor in DMF at 120°C for 10 minutes followed by deprotection (Table 5, entry 13). Treatment of the [18F]FMT precursor gave 60% radioactivity incorporation after 20 minutes at 120°C in DMF. After deprotection and HPLC purification, [18F]FMT was obtained in 12% non-decay-corrected yield (entry 14). The radiosynthesis time was about 1 hour. Treatment of the [18F]MFBG precursor with [18F]fluoride ion gave protected [18F]MFBG intermediate in 70% yield with great reproducibility.93 Deprotection and HPLC purification provided [18F]MFBG for clinical use in 14% non-decay-corrected yield in less than 75 minutes (entry 16). This yield is somewhat lower than the production yield from an automated synthesis of [18F]MFBG from a 4-anisyl(aryl)iodonium triflate (31%) (Table 3, entry 12).
3 ∣. CONCLUSION
The radiofluorination of hypervalent aryliodine precursors is now established as a useful methodology for the radiosynthesis of [18F]fluoroarenes as labeling synthons and PET radiotracers from NCA cyclotron-produced [18F]fluoride ion. The methodology is broadly applicable to electron-rich arenes and heteroarenes in addition to electron-deficient arenes, and often without restriction on position of labeling. The hypervalent aryliodine precursors are synthetically accessible from inexpensive materials and are generally adequately stable for storage. Only small amounts are required in labeling procedures that are free of heavy transition metals and readily adaptable to automation under current good manufacturing practice conditions.
ACKNOWLEDGEMENTS
V.W.P. is supported by the Intramural Research Program of the National Institutes of Health (NIMH: ZIA MH002793). The author is thankful to colleagues (Drs Lu, Simeon, and Telu) for review of the manuscript and helpful comments.
Biography

Victor W. Pike is Chief of the PET Radiopharmaceutical Sciences Section of the Molecular Imaging Branch of the National Institute of Mental Health. His main research interest is in developing 11C- or 18F-labeled PET radiotracers for imaging specific proteins (enzymes, transporters, and receptors) that have relevance to the study and treatment of neuropsychiatric disorders. He has coauthored well over 300 peer-reviewed publications stemming from his research over nearly 4 decades.
Footnotes
DISCLAIMER
Although V.W.P. is employed by the US Government, views expressed in this article do not represent those of the US government.
This review is dedicated to friend and colleague, Professor Heinz H. Coenen, in recognition of his productive scientific research, service and leadership over his career within the field of PET radiotracer development, including the area covered by this review.
REFERENCES
- 1.Guillaume M, Luxen A, Nebeling B, Argentini M, Clark JC, Pike VW. Recommendations for fluorine-18 production. Appl Radiat Isot. 1991;42:749–762. [Google Scholar]
- 2.Pike VW. PET radiotracers: crossing the blood-brain barrier and surviving metabolism. TiPs. 2009;30:431–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pike VW. Considerations in the development of reversibly binding PET radioligands for brain imaging. Curr Med Chem. 2016;23:1818–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Coenen HH. Fluorine-18 labeling methods: features and possibilities of basic reactions. In: Schubiger PA, Lehmann L, Friebe M, eds. Ernst Schering Research Foundation, Workshop 62PET Chemistry—The Driving Force in Molecular Imaging. Berlin: Springer-Verlag; 2007:15–50. [DOI] [PubMed] [Google Scholar]
- 5.Cai L, Lu S, Pike VW. Chemistry with [18F]fluoride ion. Eur J Org Chem. 2008;2853–2873. [Google Scholar]
- 6.Kim DW, Jeong H-J, Lim ST, Sohn M-H. Recent trends in the nucleophilic [18F]-radiolabeling method with no-carrier-added [18F]fluoride. Nucl Med Mol Imaging. 2010;44:25–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Tredwell M, Gouverneur V. 18F labeling of arenes. Angew Chem Int Ed. 2012;51:11426–11437. [DOI] [PubMed] [Google Scholar]
- 8.Cole EL, Stewart MN, Littich R, Hoareau R, Scott PJH. Radiosyntheses using fluorine-18: the art and science of late stage fluorination. Curr Top Med Chem. 2014;14:875–900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Preshlock S, Tredwell M, Gouverneur V. F-18-Labeling of arenes and heteroarenes for applications in positron emission tomography. Chem Rev. 2016;116:719–766. [DOI] [PubMed] [Google Scholar]
- 10.Yusubov MS, Svitich DY, Larkina MS, Zhdankin VV. Applications of iodonium salts and iodonium ylides as precursors for nucleophilic fluorination in positron emission tomography. ARKIVOC. 2013. (i);364–395. [Google Scholar]
- 11.Terrier F. The SNAr reactions; mechanistic aspects. In: Modern Nucleophilic Aromatic Substitution. Weinheim, Germany: Wiley-VCH; 2011, Ch. 1:1–94. [Google Scholar]
- 12.Wallach O. Ueber das Verhalten einiger Diazo- und Diazoamidoverbindungen. Justus Liebigs Ann Chem. 1886;235:233–255. [Google Scholar]
- 13.Balz G, Schiemann G. Über aromatische Fluorverbindungen, I.: Ein neues Verfahren zu ihrer Darstellung. Ber dtsch Chem Ges A/B. 1927;60:1186–1190. [Google Scholar]
- 14.Hamacher K, Coenen HH, Stöcklin G. Efficient stereospecific synthesis of no-carrier-added 2-[18F]-fluoro-2-deoxy-d-glucose using aminopolyether supported nucleophilic substitution. J Nucl Med. 1986;27:235–238. [PubMed] [Google Scholar]
- 15.Constantinou M, Shah F, Pike VW. Radiofluoridations of m-substituted nitrobenzenes. J Label Compd Radiopharm. 2001;44(Suppl. 1):S889–S891. [Google Scholar]
- 16.Dollé F. Fluorine-18-labelled fluoropyridines: advances in radiopharmaceutical design. Curr Pharm Des. 2005;11:3221–3235. [DOI] [PubMed] [Google Scholar]
- 17.Dolci L, Dollé F, Jubeau S, Vaufrey F, Crouzel C. 2-[18F] Fluoropyridine by no-carrier-added nucleophilic aromatic substitution with K[18F]F-K222. J Label Compd Radiopharm. 1999;42:975–985. [Google Scholar]
- 18.Naumiec GR, Cai L, Lu Y, Pike VW. Quinuclidine and DABCO enhance the radiofluorinations of 5-substituted 2-halopyridines. Eur J Org Chem. 2017; In press [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Luxen A, Guillaume M, Melega WP, Pike VW, Solin O, Wagner R. Production of 6-[18F]fluoro-L-DOPA—a critical review. Nucl Med Biol. 1992;19:149–158. [DOI] [PubMed] [Google Scholar]
- 20.Van der Puy M. Conversion of diaryliodonium salts to fluoroarenes. J Fluor Chem. 1982;21:385–392. [Google Scholar]
- 21.Pike VW, Aigbirhio FI. Reactions of cyclotron-produced [18F] fluoride with diaryliodonium salts—a novel single-step route to no-carrier-added [18F]fluoroarenes. J Chem Soc Chem Commun. 1995;21:2215–2216. [Google Scholar]
- 22.Alcock NW, Countryman RM. Secondary bonding. Part I. Crystal and molecular structures of (C6H5)2IX X = (CI, Br, or I). J Chem Soc Dalton Trans. 1977;217–219. [Google Scholar]
- 23.Alcock NW, Countryman RM. Secondary bonding. Part 14. Structural isomerism in diaryliodonium halides and the structure of di(p-toly1)iodonium bromide. J Chem Soc Dalton Trans. 1987;193–196. [Google Scholar]
- 24.Lee Y-S, Hodošček M, Chun J-H, Pike VW. Conformational structure and energetics of 2-methylphenyl(2′-methoxyphenyl) iodonium chloride: evidence for solution clusters. Chem-A Eur J. 2010;16:10418–10423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lee Y-S, Chun J-H, Hodošček M, Pike VW. Crystal structures of diaryliodonium fluorides and their implications for fluorination mechanisms. Chem A Eur J. 2017;23:4353–4363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhu C, Yoshimura A, Solntsev P, et al. New highly soluble dimedone-derived iodonium ylides: preparation, X-ray structure, and reaction with carbodiimide leading to oxazole derivatives. Chem Commun. 2012;48:10108–10110. [DOI] [PubMed] [Google Scholar]
- 27.Lee C-K, Max TCW, Li W-K, Kirner JF. Iodobenzene diacetate. Acta Crystallogr. 1977, B33;1620–1622. [Google Scholar]
- 28.Alcock NW, Harrison WD, Howes C. Secondary bonding. Part 13. Aryl-tellurium(IV) and -iodine(III)acetates and trifluoroacetates. The crystal and molecular structures of bis-(p-methoxyphenyl)tellurium diacetate, μ-oxo-bis[diphenyltrifluoroacetoxytellurium] hydrate, and [bis(trifluoroacetoxy)iodo]benzene. J Chem Soc Dalton Trans. 1984;1709–1716. [Google Scholar]
- 29.Zhdankin VV. Iodonium salts. In: Hypervalent Iodine Chemistry: Preparation, Structure and Properties of Polyvalent Iodine Compounds. Chichester, UK: John Wiley & Sons; 2014:76–84. [Google Scholar]
- 30.Zhdankin VV. Iodonium ylides. In: Hypervalent Iodine Chemistry: Preparation, Structure and Properties of Polyvalent Iodine Compounds. John Wiley & Sons; 2014:99–105. [Google Scholar]
- 31.Haskali MB, Telu S, Lee Y-S, Morse CL, Lu S, Pike VW. An investigation of (diacetoxyiodo)arenes as precursors for preparing no-carrier-added [18F]fluoroarenes from cyclotron-produced [18F]fluoride ion. J Org Chem. 2016;81:297–302. [DOI] [PMC free article] [PubMed] [Google Scholar]; See in addition: Haskali MB, Telu S, Lee Y-S, Morse CL, Lu S, Pike VW. Correction to “An investigation of (diacetoxyiodo)arenes as precursors for preparing no-carrier-added [18F]fluoroarenes from cyclotron-produced [18F]fluoride ion.” J Org Chem. 2016;81:2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhdankin VV. Organoiodine(III) carboxylates. In: Hypervalent Iodine Chemistry: Preparation, Structure and Properties of Polyvalent Iodine Compounds. John Wiley & Sons; 2014:35–41. [Google Scholar]
- 33.Chun J-H, Pike VW. Regiospecific syntheses of functionalized diaryliodonium tosylates via [hydroxy(tosyloxy)iodo]arenes generated in situ from (diacetoxyiodo)arenes. J Org Chem. 2012;77:1931–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chun J-H, Lu S, Pike VW. Radiosyntheses of meta-substituted [18F]fluoroarenes from [18F]fluoride ion and diaryliodonium tosylates within a microreactor. Eur J Org Chem. 2011;4439–4447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shah A, Pike VW, Widdowson DA. Synthesis of functionalised unsymmetrical diaryliodonium salts. J Chem Soc Perkin Trans. 1997;1:2463–2465. [Google Scholar]
- 36.Ross L, Ermert J, Hocke C, Coenen HH. Nucleophilic 18F-fluorination of heteroaromatic iodonium salts with no-carrier-added [18F]fluoride ion. J Am Chem Soc. 2007;129:8018–8025. [DOI] [PubMed] [Google Scholar]
- 37.Ross TL, Ermert J, Coenen HH. Synthesis of no-carrier-added 4-[18F]fluorophenol from 4-benzyloxyphenyl-(2-thienyl) iodonium bromide. Molecules. 2011;16:7621–7626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Neumann KD, Qin L, Vāvere AL, et al. Efficient automated syntheses of high specific activity 6-[18F]fluorodopamine using a diaryliodonium salt precursor. J Label Compd Radiopharm. 2016;59:30–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Qin L, Hu B, Neumann KD, et al. A mild and general one-pot synthesis of densely functionalized diaryliodonium salts. Eur J Org Chem. 2015;5919–5924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Warnier C, Lemaire C, Becker G, et al. Enabling efficient positron emission tomography (PET) imaging of synaptic vesicle glycoprotein 2A (SV2A) with a robust and one-step radiosynthesis of a highly potent 18F-labeled ligand ([18F] UCB-H). J Med Chem. 2016;59:8955–8966. [DOI] [PubMed] [Google Scholar]
- 41.Ichiishi N, Brooks AF, Topczewski JJ, Rodnick ME, Sanford MS, Scott PJH. Copper-catalyzed [18F]fluorination of (mesityl) (aryl)iodonium salts. Org Lett. 2014;16:3224–3227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Merritt EA, Olofsson B. Diaryliodonium salts: a journey from obscurity to fame. Angew Chem Int Ed. 2009;48:9052–9070. [DOI] [PubMed] [Google Scholar]
- 43.Beringer FM, Falk RA, Karniol M, et al. Diaryliodonium salts. IX. The synthesis of substituted diphenyliodonium salts. J Am Chem Soc. 1959;81:342–351. [Google Scholar]
- 44.Chun J-H, Pike VW. Single-step radiosyntheses of “18F-labeled click synthons” from azide-functionalized diaryliodonium salts. Eur J Org Chem. 2012;4541–4547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cardinale J, Ermert J, Coenen HH. Convenient preparation of (4-iodophenyl)aryliodonium salts. Tetrahedron. 2012;68:4112–4116. [Google Scholar]
- 46.Chun J-H, Pike VW. Single-step syntheses of no-carrier-added functionalized [18F]fluoroarenes as labeling synthons from diaryliodonium salts. Org Biomol Chem. 2013;11:6300–6306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Helfer A, Meléan JC, Ermert J, Infantino A, Coenen HH. Bis(4-benzyloxyphenyl) iodonium salts as effective precursors for the no-carrier-added radiosynthesis of 4-[18F]fluorophenol. Appl Radiat Isot. 2013;82:264–267. [DOI] [PubMed] [Google Scholar]
- 48.Margida AJ, Koser GF. Direct condensation of [hydroxy(tosyloxy)iodo]arenes with thiophenes. A convenient, mild synthesis of aryl(2-thienyl)iodonium tosylates. J Org Chem. 1984;49:3643–3646. [Google Scholar]
- 49.Koser GF, Wettach RH, Smith CS. New methodology in iodonium salt synthesis, reactions of [hydroxy(tosyloxy) iodo]arenes with aryltrimethylsilanes. J Org Chem. 1980;45:1543–1544. [Google Scholar]
- 50.Carman CS, Koser GF. Regiospecific synthesis of aryl(2-furyl) iodonium tosylates, a new class of iodonium salts, from [hydroxy(tosyloxy)iodo]arenes and 2-(trimethylsilyl)furans in organic solvents. J Org Chem. 1983;48:2534–2539. [Google Scholar]
- 51.Carroll MA, Pike VW, Widdowson DA. New synthesis of diaryliodonium sulfonates from arylboronic acids. Tetrahedron Lett. 2000;41:5393–5396. [Google Scholar]
- 52.Pike VW, Butt F, Shah A, Widdowson DA. Facile synthesis of substituted diaryliodonium tosylates by treatment of aryltributylstannanes with Koser's reagent. J Chem Soc Perkin Trans. 1999;1:245–248. [Google Scholar]
- 53.Koser GF, Wettach RH. [Hydroxy tosyliodo]benzene, a versatile reagent for the mild oxidation of iodoarenes at the iodine atom by ligand transfer. J Org Chem. 1980;45:1542–1543. [Google Scholar]
- 54.Merritt EA, Carneiro VMT, Silva LF Jr, Olofsson B. Facile synthesis of Koser's reagent and derivatives from iodine or iodoarenes. J Org Chem. 2010;75:7416–7419. [DOI] [PubMed] [Google Scholar]
- 55.Xu R, Zanotti-Fregonara P, Zoghbi SS, et al. Synthesis and evaluation in monkey of [18F]4-fluoro-N-methyl-N-(4-(6-(methylamino)pyrimidin-4-yl)thiazol-2-yl)benzamide ([18F] FIMX): a promising radioligand for PET imaging of brain metabotropic glutamate receptor 1 (mGluR1). J Med Chem. 2013;56:9146–9155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lee BC, Kim JS, Kim BS, et al. Aromatic radiofluorination and biological evaluation of 2-aryl-6-[18F]fluorobenzothiazoles as a potential positron emission tomography imaging probe for β-amyloid plaques. Bioorg Med Chem. 2011;19:2980–2990. [DOI] [PubMed] [Google Scholar]
- 57.Bielawski M, Olofsson B. High-yielding one-pot synthesis of diaryliodonium triflates from arenes and iodine or iodoarenes. Chem Commun. 2007;2521–2523. [DOI] [PubMed] [Google Scholar]
- 58.Bielawski M, Zhu M, Olofsson B. Efficient and general one-pot synthesis of diaryliodonium triflates: optimization, scope and limitations. Adv Synth Catal. 2007;349:2610–2618. [Google Scholar]
- 59.Zhu M, Jalalian N, Olofsson B. One-pot synthesis of diaryliodonium salts using toluenesulfonic acid: a fast entry to electron-rich diaryliodonium tosylates and triflates. Synlett. 2008;592–596. [Google Scholar]
- 60.Bielawski M, Aili D, Olofsson B. Regiospecific one-pot synthesis of diaryliodonium tetrafluoroborates from aryl boronic acids and iodoarenes. J Org Chem. 2008;73:4602–4607. [DOI] [PubMed] [Google Scholar]
- 61.Bielawski M, Malmgren J, Pardo LM, Wikmark Y, Olofsson B. One-pot synthesis and applications of N-heteroaryl iodonium salts. ChemOpen. 2014;3:19–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Seidl TL, Sundalam SK, McCullough B, Stuart DR. Unsymmetrical aryl(2,4,6-trimethoxyphenyl)iodonium salts: one-pot synthesis, scope, stability, and synthetic studies. J Org Chem. 2016;81:1998–2009. [DOI] [PubMed] [Google Scholar]
- 63.Carreras V, Sandtorv AH, Stuart DR. Synthesis of aryl(2,4,6-trimethoxyphenyl)iodonium trifluoroacetate salts. J Org Chem. 2017;82:1279–1284. [DOI] [PubMed] [Google Scholar]
- 64.Yuan Z, Cheng R, Chen P, Liu G, Liang SH. Efficient pathway for the preparation of aryl(isoquinoline)iodonium-(III) salts and synthesis of radiofluorinated isoquinolines. Angew Chem Int Ed. 2016;55:11882–11886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chun J-H, Lu S, Lee Y-S, Pike VW. Fast and high-yield microreactor syntheses of ortho-substituted [18F]fluoroarenes from reactions of [18F]fluoride ion with diaryliodonium salts. J Org Chem. 2010;75:3332–3338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Beringer FM, Drexler M, Grindler EM, Lumpkin CC. Diaryliodonium salts. I. Synthesis. J Am Chem Soc. 1953;75:2705–2708. [Google Scholar]
- 67.Rotstein BH, Stephenson NA, Vasdev N, Liang SH. Spirocyclic hypervalent iodine(III)-mediated radiofluorination of non-activated and hindered aromatics. Nat Commun. 2014;5:4365–4371. [DOI] [PubMed] [Google Scholar]
- 68.Cardinale J, Ermert J. Simplified synthesis of aryliodonium ylides by a one-pot procedure. Tetrahedron Lett. 2013;54:2067–2069. [Google Scholar]
- 69.Telu S, Chun J-H, Siméon FG, Lu S, Pike VW. Effective syntheses of mGluR5 PET radioligands through the radiofluorination of diaryliodonium salts. Org Biomol Chem. 2011;9:6629–6638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Moon BS, Park JH, Lee HJ, Lee BC, Kim SE. Routine production of [18F]flumazenil from iodonium tosylate using a sample pretreatment method: a 2.5-year production report. Mol Imaging Biol. 2014;16:619–625. [DOI] [PubMed] [Google Scholar]
- 71.Hu B, Vavere AL, Neumann KD, Shulkin BL, DiMagno SG, Snyder SE. A practical, automated synthesis of meta-[18F] fluorobenzylguanidine for clinical use. ACS Chem Neurosci. 2015;6:1870–1879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Richarz R, Krapf P, Zarrad F, Urusova E, Neumaier AB, Zlatopolskiy BD. Neither azeotropic drying, nor base nor other additives: a minimalist approach to 18F-labeling. Org Biomol Chem. 2014;12:8094–8099. [DOI] [PubMed] [Google Scholar]
- 73.Pascali G, De Simone M, Matesic L, Greguric I, Salvadori PA. Tolerance of water in microfluidic radiofluorinations: a potential methodological shift? J Flow Chem. 2014;4:86–91. [Google Scholar]
- 74.Chun J-H, Telu S, Lu S, Pike VW. Radiofluorination of diaryliodonium tosylates under aqueous–organic and cryptand-free conditions. Org Biomol Chem. 2013;11:5094–5099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Calderwood S, Collier TL, Gouverneur V, Liang SH, Vasdev N. Synthesis of 18F-arenes from spirocyclic iodonium(III) ylides via continuous-flow microfluidics. J Fluor Chem. 2015;178:249–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang B, Qin L, Neumann KD, Uppaluri S, Cerny RL, Di Magno SG. Improved arene fluorination methodology for I(III) salts. Org Lett. 2010;12:3352–3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.McCammant MS, Thompson S, Brooks AF, Krska SW, Scott PJH, Sanford MS. Cu-mediated C─H 18F-fluorination of electron-rich (hetero)arenes. Org Lett. 2017;19:3939–3942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ochiai M, Kitagawa Y, Toyonari M. On the mechanism of α-phenylation of β-keto esters with diaryl-λ3-iodanes: evidence for a non-radical pathway. ARKIVOC. 2003, (vi);43–48. [Google Scholar]
- 79.Ochiai M In: Wirth T, ed. Hypervalent Iodine Chemistry: Modern Developments in Organic Synthesis. Vol.224 Berlin, Top Curr Chem: Springer Verlag; 2003:5–67. [Google Scholar]
- 80.Martín-Santamaría S, Carroll MA, Carroll CM, et al. Fluoridation of heteroaromatic iodonium salts—experimental evidence supporting theoretical prediction of the selectivity of the process. Chem Commun. 2000;649–650. [Google Scholar]
- 81.Shah A, Pike VW, Widdowson DA. The synthesis of [18F] fluoroarenes from the reaction of cyclotron-produced [18F] fluoride ion with diaryliodonium salts. J Chem Soc Perkin. 1998;1:2043–2046. [Google Scholar]
- 82.Yamada Y, Okawara M. Steric effect in the nucleophilic attack of bromide anion on diaryl- and aryl-2-thienyliodonium ions. Bull Chem Soc Jap. 1972;45:1860–1863. [Google Scholar]
- 83.Lancer KM, Wiegand GH. The ortho effect in the pyrolysis of iodonium halides. A case for a sterically controlled nucleophilic aromatic (SN) substitution reaction. J Org Chem. 1976;41:3360–3336. [Google Scholar]
- 84.Carroll MA, Martín-Santamaría S, Pike VW, Rzepa HS, Widdowson DA. An ab initio and MNDO-d SCF-MO computational study of stereoelectronic control in extrusion reactions of R2I-F iodine(III) intermediates. J Chem Soc Perkin Trans. 1999;2:2707–2714. [Google Scholar]
- 85.Hill DE, Holland JP. Computational studies on hypervalent iodonium(III) compounds as activated precursors for 18F radiofluorination of electron-rich arenes. Comput Theor Chem. 2015;1066:34–46. [Google Scholar]
- 86.Seeman JI. Effect of conformational change on reactivity in organic chemistry. Evaluations, applications, and extensions of Curtin-Hammett Winstein-Holness kinetics. Chem Rev. 1983;83:83–134. [Google Scholar]
- 87.Basuli F, Wu H, Griffiths GL. Syntheses of meta-[18F] fluorobenzaldehyde and meta-[18F]fluorobenzyl bromide from phenyl(3-formylphenyl)iodonium salt precursors. J Label Compd Radiopharm. 2011;54:224–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Ochiai M, Kida M, Sato K, et al. Association and dissociation of (Z)-(β-bromoalkenyl)-(phenyl)iodonium bromide in chloroform solution: detection of vinyl-λ3-iodane dimer in solution. Tetrahedron Lett. 1999;40:1559–1562. [Google Scholar]
- 89.Graskemper JW, Wang B, Qin L, Neumann KD, DiMagno SG.Unprecedented directing group ability of cyclophanes in arene fluorinations with diaryliodonium salts. Org Lett. 2011;13:3158–3161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Carroll MA, Nairne J, Smith G, Widdowson DA. Radical scavengers: a practical solution to the reproducibility issue in the fluoridation of diaryliodonium salts. J Fluor Chem. 2007;128:127–132. [Google Scholar]
- 91.Ichiishi N, Canty AJ, Yates B, Sanford MS. Cu-catalyzed fluorination of diaryliodonium salts with KF. Org Lett. 2013;15:5134–5137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ichiishi N, Canty AJ, Yates BF, Sanford MS. Mechanistic investigations of Cu-catalyzed fluorination of diaryliodonium salts: elaborating the CuI/CuIII manifold in copper catalysis. Organometallics. 2014;33:5525–5534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Rotstein BH, Wang L, Liu RY, et al. Mechanistic studies and radiofluorination of structurally diverse pharmaceuticals with spirocyclic iodonium(III) ylides. Chem Sci. 2016;7:4407–4417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Cardinale J, Ermert J, Humpert S, Coenen HH. Iodonium ylides for one-step, no-carrier-added radiofluorination of electron rich arenes, exemplified with 4-(([18F]fluoro-phenoxy)-phenylmethyl)piperidine NET and SERT ligands. RSC Adv. 2014;4:17293–17299. [Google Scholar]
- 95.Satyamurthy N, Barrio JR. Nucleophilic fluorination of aromatic compounds. 2010, WO 2010/008522 A2.
- 96.Ermert J. 18F-Labelled intermediates for radiosynthesis by modular build-up reactions: newer developments. Biomed Res Int. 2014; Article ID 812973. 10.1155/2014/812973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Wuest F. Fluorine-18 labeling of small molecules: the use of 18F-labeled fluoroarenes derived from no-carrier added [18F] fluoride as labeling precursors. In: Schubiger PA, Lehmann L, Friebe M, eds. Ernst Schering Research Foundation, Workshop 62PET Chemistry—The Driving Force in Molecular Imaging. Berlin: Springer-Verlag; 2007:51–78. [DOI] [PubMed] [Google Scholar]
- 98.Wester HJ, Schottelius M. Fluorine-18 labeling of peptides and proteins. In: Schubiger PA, Lehmann L, Friebe M, eds. Ernst Schering Research Foundation, Workshop 62PET Chemistry—The Driving Force in Molecular Imaging. Berlin: Springer-Verlag; 2007:79–111. [DOI] [PubMed] [Google Scholar]
- 99.Way J, Bouvet VC, Wuest F. Synthesis of 4-[18F] fluorohalobenzenes and palladium-mediated cross-coupling reactions for the synthesis of 18F-labeled radiotracers. Curr Org Chem. 2013;17:2138–2152. [Google Scholar]
- 100.Wuest FR, Kniess T. Synthesis of 4-[18F]fluoroiodobenzene and its application in Sonogashira cross-coupling reactions. J Label Compd Radiopharm. 2003;46:699–713. [Google Scholar]
- 101.Kügler F, Ermert J, Kaufholz P, Coenen HH. 4−[18F] Fluorophenylpiperazines by improved Hartwig-Buchwald N-arylation of 4-[18F]fluoroiodobenzene, formed via hypervalent λ3-iodane precursors: application to build-up of the dopamine D4 ligand [18F]FAUC 316. Molecules. 2015;20:470–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ermert J, Hocke C, Ludwig T, Gail R, Coenen HH. Comparison of pathways to the versatile synthon of no-carrier-added 1-bromo-4-[18F]fluorobenzene. J Label Compd Radiopharm. 2004;47:429–441. [Google Scholar]
- 103.Wuest FR, Kniess T. No-carrier added synthesis of 18F-labelled nucleosides using Stille cross-coupling reactions with 4-[18F]fluoroiodobenzene. J Label Compd Radiopharm. 2004;47:457–468. [Google Scholar]
- 104.Wuest FR, Hoehne A, Metz P. Synthesis of 18F-labelled cyclooxygenase-2 (COX-2) inhibitors via Stille reaction with 4-[18F] fluoroiodobenzene as radiotracers for positron emission tomography (PET). Org Biomol Chem. 2005;3:503–507. [DOI] [PubMed] [Google Scholar]
- 105.Zlatopolskiy BD, Zischler J, Krapf P, et al. Copper-mediated aromatic radiofluorination revisited: efficient production of PET tracers on a preparative scale. Chem A Eur J. 2015;21:5972–5979. [DOI] [PubMed] [Google Scholar]
- 106.Charlton M,Carroll MA. Synthesis of 4-[18F] fluorobenzaldehyde using diaryliodonium salt precursors in a microreactor. J Label Compd Radiopharm. 2013;56:50–63. [Google Scholar]
- 107.Basuli F, Wu H, Li C, Shi Z-D, Sulima A, Griffiths GL. A first synthesis of 18F-radiolabeled lapatinib: a potential tracer for positron emission tomographic imaging of ErbB1/ErbB2 tyrosine kinase activity. J Label Compd Radiopharm. 2011;54:633–636. [Google Scholar]
- 108.Pretze M, Pietzsch D, Mamat C. Recent trends in bioorthogonal click-radiolabeling reactions using fluorine-18. Molecules. 2013;18:8618–8665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Thonon D, Kech C, Paris J, Lemaire C, Luxen A. New strategy for the preparation of clickable peptides and labeling with 1-(azidomethyl)-4-[18F]-fluorobenzene for PET. Bioconjug Chem. 2009;20:817–823. [DOI] [PubMed] [Google Scholar]
- 110.Mercier F, Paris J, Kaisin G, et al. General method for labeling siRNA by click chemistry with fluorine-18 for the purpose of PET imaging. Bioconjug Chem. 2011;22:108–114. [DOI] [PubMed] [Google Scholar]
- 111.Mading P, Fuchtner F, Wust F. Module assisted synthesis of the bifunctional labeling agent N-succinimidyl 4-[18F] fluorobenzoate ([18F]SFB). Appl Radiat Isot. 2005;63:329–332. [DOI] [PubMed] [Google Scholar]
- 112.Yan R, Brichard L, Soloviev D, Aigbirhio FI, Carroll MA. The first single-step-single-pot synthesis of 4-[18F]SFB. J Label Compd Radiopharm. 2009;52:216 [Google Scholar]
- 113.Carrol MA, Yan R. Formation of 18F and 19F fluoroarenes bearing reactive functionalities. WO 2009/138763 A1.
- 114.Chun J-H, Pike VW. Selective syntheses of no-carrier-added 2- and 3-[18F]fluorohalopyridines through the radiofluorination of halopyridinyl(4'-methoxyphenyl)iodonium tosylates. Chem Commun. 2012;48:9921–9923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Yuan G, Jones GB, Vasdev N, Liang SH. Radiosynthesis and preliminary evaluation of 18F-labeled 2-(1-(3-fluorophenyl)-2-oxo-5-(pyrimidin-2-yl)-1,2-dihydropyridin-3-yl)benzonitrile for imaging AMPA receptors. Bioorg Med Chem Lett. 2016;26:4857–4860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Petersen IN, Kristensen JL, Herth MM. Nucleophilic 18F-labeling of spirocyclic iodonium ylide or boronic pinacol ester precursors: advantages and disadvantages. Eur J Org Chem. 2017;453–458. [Google Scholar]
- 117.Wang L, Jacobson O, Avdic D, et al. Ortho-stabilized 18F-azido click agents and their application in PET imaging with single-stranded DNA aptamers. Angew Chem Int Ed. 2015;54:12777–12781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Drerup C, Ermert J, Coenen HH. Synthesis of a potent aminopyridine-based nNOS-inhibitor by two recent no-carrier-added 18F-labelling methods. Molecules. 2016;21:1160. 10.3390/molecules21091160 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Jacobson O, Weiss ID, Wang L, et al. 18F-Labeled single-stranded DNA aptamer for PET imaging of protein tyrosine kinase-7 expression. J Nucl Med. 2015;56:1780–1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Zhang MR, Kumata K, Suzuki K. A practical route for synthesizing a PET ligand containing [18F]fluorobenzene using reaction of diphenyliodonium salt with [18F]F−. Tetrahedron Lett. 2007;48:8632–8635. [Google Scholar]
- 121.Pike VW, Halldin C, Crouzel C, et al. Radioligands for PET studies of central benzodiazepine receptors and PK (peripheral benzodiazepine) binding sites—current status. Nucl Med Biol. 1993;20:503–525. [DOI] [PubMed] [Google Scholar]
- 122.Odano I, Halldin C, Karlsson P, et al. [18F]Flumazenil binding to central benzodiazepine receptor studies by PET—quantitative analysis and comparisons with [11C]flumazenil. Neuroimage. 2009;45:891–902. [DOI] [PubMed] [Google Scholar]
- 123.Ryzhikov NN, Seneca N, Krasikova RN, et al. Preparation of highly specific radioactivity [18F]flumazenil and its evaluation in cynomolgus monkey by positron emission tomography. Nucl Med Biol. 2005;32:109–116. [DOI] [PubMed] [Google Scholar]
- 124.Massaweh G, Schirrmacher E, La Fougere C, et al. Improved work-up procedure for the production of [18F]flumazenil and first results of its use with a high-resolution research tomograph in human stroke. Nucl Med Biol. 2009;36:721–727. [DOI] [PubMed] [Google Scholar]
- 125.Mandap KS, Ido T, Kiyono Y, et al. Development of microwave-based automated nucleophilic [18F]fluorination system and its application to the production of [18F]flumazenil. Nucl Med Biol. 2009;36:403–409. [DOI] [PubMed] [Google Scholar]
- 126.Moon BS, Kil HS, Park JH, et al. Facile aromatic radiofluorination of [18F]flumazenil from diaryliodonium salts with evaluation of their stability and selectivity. Org Biomol Chem. 2011;9:8346–8355. [DOI] [PubMed] [Google Scholar]
- 127.Mu L, Herde AM, Rűefli PM, et al. Synthesis and pharmacological evaluation of [11C]granisetron, and [18F] fluoropalonosetron as PET probes for 5-HT3 receptor imaging. ACS Chem Neurosci. 2016;7:1552–1564. [DOI] [PubMed] [Google Scholar]
- 128.Selivanova SV, Stellfeld T, Heinrich TK, et al. Design, synthesis, and initial evaluation of a high affinity positron emission tomography probe for imaging matrix metalloproteinases 2 and 9. J Med Chem. 2013;56:4912–4920. [DOI] [PubMed] [Google Scholar]
- 129.Zanotti-Fregonara P, Xu R, Zoghbi SS, et al. The PET radioligand 18F-FIMX images and quantifies metabotropic glutamate receptor 1 in proportion to the regional density of its gene transcript in human brain. J Nucl Med. 2016;57:242–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Saccomanni G, Pascali G, Del Carlo S, et al. Design, synthesis and preliminary evaluation of 18F-labelled 1,8-naphthyridin- and quinolin-2-one-3-carboxamide derivatives for PET imaging of CB2 cannabinoid receptor. Bioorg Med Chem Lett. 2015;25:2532–2535. [DOI] [PubMed] [Google Scholar]
- 131.Kim J, Moon BS, Lee BC, et al. A potential PET radiotracer for the 5-HT2C receptor: synthesis and in vivo evaluation of 4(3-[18F]fluorophenethoxy)pyrimidine.ACS Chem Neurosci. 2017;8:996–1003. [DOI] [PubMed] [Google Scholar]
- 132.Zhang H, Huang R, Pillarsetty N, et al. Synthesis and evaluation of 18F-labeled benzylguanidine analogs for targeting the human norepinephrine transporter. Eur J Nucl Med Mol Imaging. 2014;41:322–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Jang KS, Jung Y-W, Gu G, et al. 4-[18F]Fluoro-m-hydroxyphenethylguanidine: a radiopharmaceutical for quantifying regional cardiac sympathetic nerve density with positron emission tomography. J Med Chem. 2013;56:7312–7323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Ding YS, Fowler JS, Gatley SJ, Dewey SL, Wolf AP, Schlyer DJ. Synthesis of high specific activity 6-[18F]fluorodopamine for positron emission tomography studies of sympathetic nervous tissue. J Med Chem. 1991;34:861–863. [DOI] [PubMed] [Google Scholar]
- 135.Edwards R, Wirth T. [18F]6-Fluoro-3,4-dihydroxy-l-phenylalanine—recent modern syntheses for an elusive radiotracer. J Label Compd Radiopharm. 2015;58:183–187. [DOI] [PubMed] [Google Scholar]
- 136.Ground Fluor Pharmaceuticals, Inc, 2014. Technical specification sheet (PDF) [Online]. Lincoln, Nebraska, USA. Available from: http://www.gfpharma.com/pubs/FDOPA_CutSheet.pdf [Google Scholar]
- 137.Kuik W-J, Kema IP, Brouwers AH, et al. In vivo biodistribution of no-carrier-added 6-18F-fluoro-3,4-dihydroxy-l-phenylalanine (18F-DOPA), produced by a new nucleophilic substitution approach, compared with carrier-added 18F-DOPA, prepared by conventional electrophilic substitution. J Nucl Med. 2015;56:106–112. [DOI] [PubMed] [Google Scholar]
- 138.Zischler J, Krapf P, Richarz R, Zlatopolskiy BD, Neumaier B. Automated synthesis of 4-[18F]fluoroanisole, [18F]DAA1106 and 4-[18F]FPhe using Cu-mediated radiofluorination under “minimalist” conditions. Appl Radiat Isot. 2016;115:133–137. [DOI] [PubMed] [Google Scholar]
- 139.Stephenson NA, Holland JP, Kassenbrock A, et al. Iodonium ylide–mediated radiofluorination of 18F-FPEB and validation for human use. J Nucl Med. 2015;56:489–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Cai Z, Li S, Pracitto R, et al. Fluorine-18-labeled antagonist for PET imaging of kappa opioid receptors. ACS Chem Neurosci. 2017;8:12–16. [DOI] [PubMed] [Google Scholar]
- 141.Awasthi V, Pathuri G, Agashe HB, Gali H. Synthesis and in vivo evaluation of p-18F-fluorohippurate as a new radiopharmaceutical for assessment of renal function by PET. J Nucl Med. 2011;52:147–153. [DOI] [PubMed] [Google Scholar]
- 142.Nkepang GN, Hedrick AF, Awasthi V, Gali H. Facile synthesis of para-[18F]fluorohippurate via iodonium ylide–mediated radiofluorination for PET renography. Bioorg Med Chem Lett. 2016;26:479–483. [DOI] [PubMed] [Google Scholar]
- 143.Petersen IN, Villadsen J, Hansen HD, et al. 18F-Labelling of electron-rich iodonium ylides: application to the radiosynthesis of potential 5-HT2A receptor PET ligands. Org Biomol Chem. 2017;15:4351–4358. [DOI] [PubMed] [Google Scholar]
- 144.Collier TL, Normandin MD, Stephenson NA, et al. Synthesis and preliminary PET imaging of 11C and 18F isotopologues of the ROS1/ALK inhibitor lorlatinib. Nat Commun. 2017; 8, Article number: 15761 (2017) doi: 10.1038/ncomms15761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Wang L, Cheng R, Fujinaga M, et al. A facile radiolabeling of [18F]FDPA via spirocyclic iodonium ylides: preliminary PET imaging studies in preclinical models of neuroinflammation. J Med Chem. 2017;60:5222–5227. [DOI] [PMC free article] [PubMed] [Google Scholar]