

Abstract

The recent emergence of SARS-CoV-2 in the human population has caused a global pandemic. The virus encodes two proteases, Mpro and PLpro, that are thought to play key roles in the suppression of host protein synthesis and immune response evasion during infection. To identify the specific host cell substrates of these proteases, active recombinant SARS-CoV-2 Mpro and PLpro were added to A549 and Jurkat human cell lysates, and subtiligase-mediated N-terminomics was used to capture and enrich protease substrate fragments. The precise location of each cleavage site was identified using mass spectrometry. Here, we report the identification of over 200 human host proteins that are potential substrates for SARS-CoV-2 Mpro and PLpro and provide a global mapping of proteolysis for these two viral proteases in vitro. Modulating proteolysis of these substrates will increase our understanding of SARS-CoV-2 pathobiology and COVID-19.
Keywords: Mpro, PLpro, SARS-CoV-2, COVID-19, N-terminomics, BRD2
Introduction
SARS-CoV-2 is an enveloped, positive-sense, single-stranded RNA virus in the family Coronaviridae, genus β-coronavirus. The genome of SARS-CoV-2 encodes at least 29 viral proteins including 4 structural proteins, 16 nonstructural proteins (NSPs), and 9 accessory proteins. Two of the viral proteins, NSP3 and NSP5, possess protease activity. They cleave two overlapping viral polyproteins (pp1a and pp1ab) translated in the major open reading frames ORF1a and ORF1b into 16 NSPs (NSP1-16) in their active form. The NSPs possess essential enzymatic activities in viral replication, including helicase and RNA-dependent RNA polymerase (see ref (1) for a review). Due to the critical role of the SARS-CoV-2 proteases, they are targets for antiviral drugs. GC376, a drug originally developed to treat feline coronavirus, also inhibits the main protease of SARS-CoV-2 and effectively blocks viral replication in cells.2 Currently, Paxlovid (oral antiviral drug nirmatrelvir/ritonavir, Pfizer) is the only approved COVID-19 treatment targeting the SARS-CoV-2 viral protease.3−6
The two SARS-CoV-2 proteases are named according to their catalytic and structural similarities to other known enzymes. NSP3pro is also known as papain-like protease (PLpro) and cleaves at only three sites in the polyproteins pp1a and pp1ab. NSP5pro or picornaviral 3C-like protease (3CLpro) cleaves at eleven sites and is thus also referred to as the main protease (Mpro). Both SARS-CoV-2 proteases are cysteine proteases. The active site of SARS-CoV-2 Mpro contains a Cys145-His41 catalytic dyad. Based on its native cleavage sequence consensus in the polyproteins and its crystal structure,7 Mpro preferentially cleaves after glutamine (P1 = Gln, Schechter and Berger nomenclature),8 which allows stabilization in its S1 pocket by three hydrogen bonds.9 Studies on SARS-CoV-1 Mpro show that cleavage can also occur after histidine but with a lower frequency.10 PLpro has a canonical cysteine protease catalytic triad Cys111-His272-Asp286 and is a multifunctional protein with both proteolytic and mainly deubiquitinating activities.11,12 It cleaves almost exclusively after residues GlyGly at P1 and P2 positions, with high preference for hydrophobic residues in P4 (Leu in particular) and broader specificity in P3.13
In addition to proteolytic processing of viral polyproteins, viral proteases can cleave host substrates to modulate immune evasion and host gene expression shutoff.14,15 Although the interactomes of SARS-CoV-2 viral proteins have been well studied,16−18 it is more challenging to characterize the entire range of substrates of viral proteases using conventional immunoprecipitation methods since proteolysis can lead to substrate release and the subsequent degradation of protein fragments. Even with a catalytically dead protease mutant, the protease–substrate interactions can be transient and difficult to detect.
A number of targeted studies have identified specific SARS-CoV-2 protease substrates in the human proteome. For example, Shin and co-workers hypothesized that the high-sequence homology between the SARS-CoV-1 and -2 proteases might contribute to common substrates and reported that the ubiquitin-like interferon-stimulated gene 15 protein is cleaved by SARS-CoV-2 PLpro.19 A systematic screening of interferon stimulatory genes and human innate immune pathway proteins showed that SARS-CoV-2 Mpro cleaves the E3 ligase BRE1A (RNF20 gene),20 NLRP12, TAB1,21 and CARD8,22 and PLpro also cleaves IRF3 to dysregulate the host innate immune response.21 Other researchers examined disrupted cellular apoptosis and autophagy pathways. Wenzel et al. found that Mpro cleaves NEMO, an essential modulator of NF-kappa-B signaling in brain endothelial cells,23 while Mohamud et al. reported that PLpro cleaves the protein kinase ULK1.24 Another method to identify potential viral protease targets is to search for short stretches of homologous host–pathogen sequences in the human proteome. Using this technique, Reynolds et al. showed that PLpro cleaves cardiac myosin proteins (MYH7 and MYH6), FOXP3, HER4, and PROS1 in vitro,25 and Miczi et al. showed that Mpro cleaves C-terminal-binding protein 1.26
N-Terminomics profiling of Mpro and PLpro can facilitate identification of human proteins potentially cleaved during SARS-CoV-2 infection on a greater scale. Meyer et al. characterized proteome-wide viral cleavage events occurring in both SARS-CoV-2-infected African green monkey kidney cells (Vero E6) and human lung carcinoma cells overexpressing the virus entry receptor (A549-ACE2).27 Refining the cleavage sites to match viral protease specificities, they identified 14 putative Mpro and PLpro substrates. Further biochemical analysis confirmed the Mpro cleavage of pinin, phosphoribosylaminoimidazole carboxylase (PAICS gene), and golgin A3 (GOLGA3 gene), whereas PLpro cleaves the protein kinase Src.
When a purified recombinant viral protease is incubated with human cell lysates, N-terminomics methods, such as terminal amine isotopic labeling of substrates (TAILS),28 can be used to identify protease cleavage sites. Using this method, Koudelka et al. identified 318 unique protein substrates of SARS-CoV-2 Mpro in lung epithelial carcinoma cells and pulmonary microvascular endothelial cells but did not validate these substrates in infected cells.29 Also, using TAILS, Pablos et al. profiled 101 Mpro substrates in human embryonic kidney cells and lung epithelial cells treated with antiviral type I interferons.30 They further characterized and performed functional studies on several of these Mpro substrates such as PTBP1 and the RNA polymerase, RPAP1, which are proteins involved in host transcription and translation. They confirmed the Mpro cleavage of proteins in the Hippo signaling pathway: the transcriptional coactivator YAP1, protein kinase MAP4K5, transcription factors CREB1 and ATF-1, as well as proteins involved in the antiviral response, such as galectin-8 and FYCO1.
Here, we employed an enzyme-mediated N-terminomics approach for the comprehensive identification of potential substrates of SARS-CoV-2 Mpro and PLpro in the human proteome. Using subtiligase-mediated N-terminomics in cell lysates, we identified 191 and 16 putative substrates of SARS-CoV-2 Mpro and PLpro, respectively. The enzymatic-labeling approach presented here is unique and complementary to the known SARS-CoV-2 degradome reported by other groups. By comparing our results to previous studies, we have generated a list of all current SARS-CoV-2 protease substrates reported thus far, thereby filling the gap of uncharacterized Mpro and PLpro interactomes. There is still a need for additional antivirals for COVID-19 patients, and the characterization of SARS-CoV-2 protease cellular targets will help us better understand the fundamental virology of SARS-CoV-2.
Results
To identify the host substrates of SARS-CoV-2 Mpro and PLpro and their corresponding cleavage sites, we used a subtiligase-mediated N-terminomics approach to positively enrich the newly generated N-termini from cleaved proteins in human cell lysates31,32 (Figure 1). Nascent N-termini were enzymatically labeled with a biotinylated peptide ester using subtiligase, allowing for the subsequent positive enrichment of biotinylated proteins on immobilized neutravidin beads. The proteins were further digested by trypsin, and the bound N-terminal peptides were released from the beads by cleavage at a tobacco etch viral (TEV) site engineered into the biotin ester tag. This leaves a unique N-terminal α-aminobutyric acid (Abu) modification on the peptides allowing for unambiguous and precise identification of SARS-CoV-2 protease cleavage sites using tandem mass spectrometry (LC–MS/MS).
Figure 1.
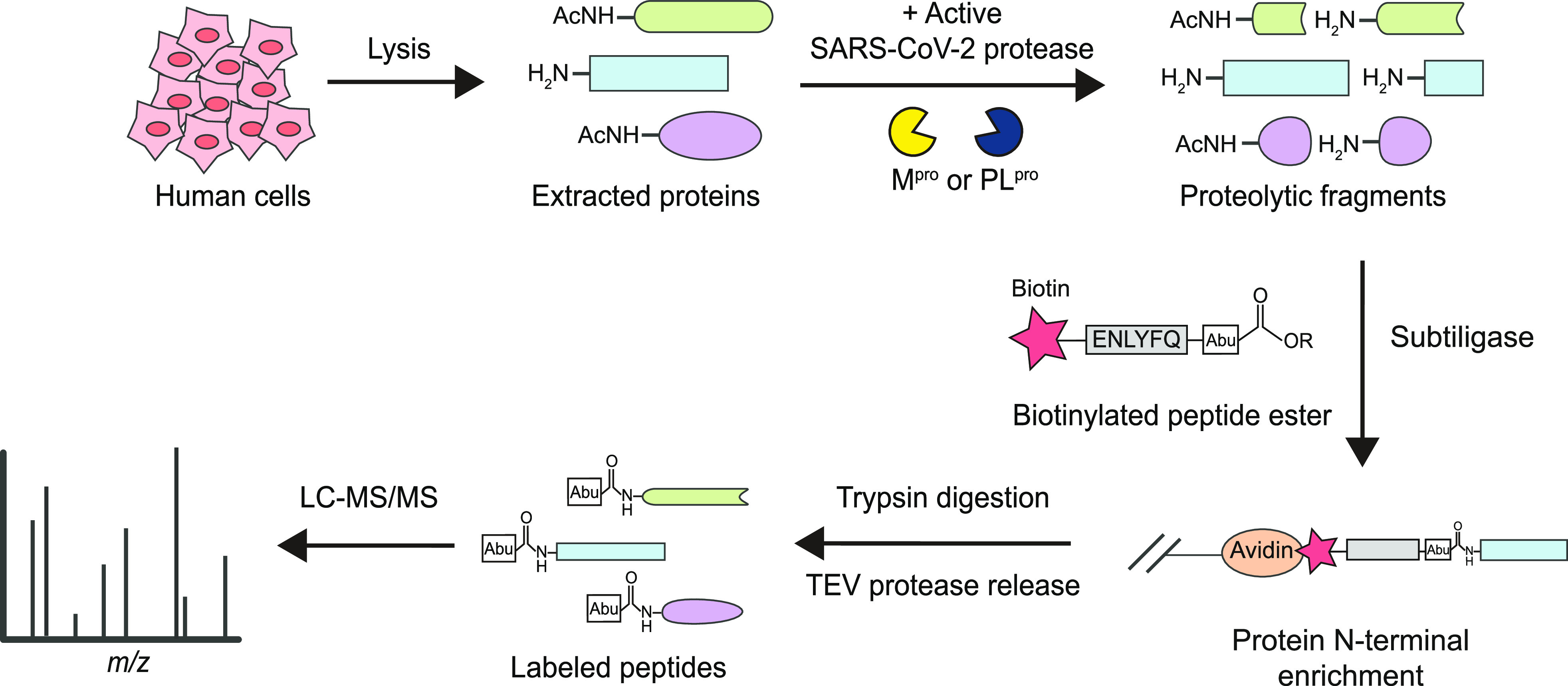
Identification of SARS-CoV-2 Mpro and PLpro host substrates in in vitro subtiligase N-terminomics. Active recombinant Mpro or PLpro was added to human cell lysate, generating protein cleavages that were labeled with a designed biotinylated peptide ester by subtiligase. After enrichment by neutravidin, trypsin and TEV protease were added for the release of labeled peptides with a unique N-terminal mass tag Abu (α-aminobutyric acid), allowing for identification of viral protease cleavage sites in LC–MS/MS.
Activity Assay in Cell Lysates
We used two different cell lines, A549 (adenocarcinomic human alveolar basal epithelial cells) and Jurkat (human T lymphocyte cells) to compare the results across different cell origins and maximize substrate identification. To ensure that the purified recombinant protease was active in cell lysates, we monitored its proteolytic activity using a fluorescence activity assay. The optimal P4-P1 substrates of SARS-CoV-2 Mpro and PLpro were previously identified via substrate specificity screening.9,13 Coumarin probes based on these sequences were used to test recombinant protease activities: Ac-Abu-Tle-Leu-Gln-ACC was incubated with purified Mpro and Ac-Leu-Arg-Gly-Gly-ACC with PLpro in both cell-free environment and in cell lysates (Figures S1 and S2). Following optimization, the viral proteases were able to cleave the coumarin probes in the complex cellular environment, demonstrating comparable fluorescence signals to cell-free assays.
Identification of Mpro Substrates
Since primary T lymphocytes have been previously reported to be infected by SARS-CoV-2,33 we first performed two N-terminomics replicates in Jurkat cell lysates to identify human host substrates of Mpro. We discarded labeled peptides with N-termini located within the first four residues from the start of a protein sequence in order to focus our analysis on endoproteolytic sites (e.g., to avoid protein start sites and methionine removal). In the Jurkat proteome, we were able to identify 746 labeled unique cleavages in 600 host substrates. We then searched for cleavage sites with Gln and His residues at P1 position only (P1 = Q/H), which correspond to Mpro specificity.9 This yielded 154 unique cleavages in 146 substrates, exhibiting a 20.6% enrichment at P1 = Q/H from background protease activity (Figure S3a). To expand the host proteome targeted by Mpro, we performed two experimental replicates with lung epithelial cells (A549) and identified 2283 unique labeled cleavage sites. Of these, 210 cleavage sites in 196 substrates contained P1 = Q/H, corresponding to an enrichment rate of 9.0% (Figure S3a). Interestingly, the enrichment rate of Mpro-specific cleavage sites in A549 was lower than in Jurkat but still higher than untreated lysates with endogenous proteases that typically showed a P1 = Q/H at 3.9% (2.5 and 1.4%, respectively).34 This suggested that the added Mpro was active in cell lysates and cleaved human substrates. Combining labeled cleavages with P1 = Q/H in A549 and Jurkat, we found 334 unique sites (Figure 2a). However, we hypothesized that a new emerging virus, such as SARS-CoV-2, would cleave host substrates at new sites, i.e., sites not identified in previous N-terminomics experiments. Therefore, we used the DegraBase35 to eliminate sites previously observed in healthy and apoptotic cells in subtiligase-based N-terminomics experiments. These included background proteolysis in human cells due to the incomplete inhibition of endogenous proteases. In addition, we could narrow down the list of Mpro substrates by identifying cleavage sites found in the PLpro data set matching Mpro specificity, and vice versa. In total, we estimated that 39% of cleavage sites featuring P1 = Q/H may not be directly attributed to the Mpro activity (Figure 2a, 131/334 cleavage sites). While these cleavages may result from background proteolysis of the host cells, we also cannot rule out that the viral proteases may be targeting the same sites as the host proteins. Ultimately, we focused our analysis on the 157 cleavage sites where P1 = Q and 46 where P1 = H, found in 148 and 46 host substrates, respectively (for a total of 191 substrates as three substrates contain both P1 = Q and P1 = H cleavage sites). Overall, we observed 203 unique cleavage sites (P1 = Q/H) in 191 human substrates cleaved by the Mpro of SARS-CoV-2 in vitro (see Supporting Information, S1).
Figure 2.
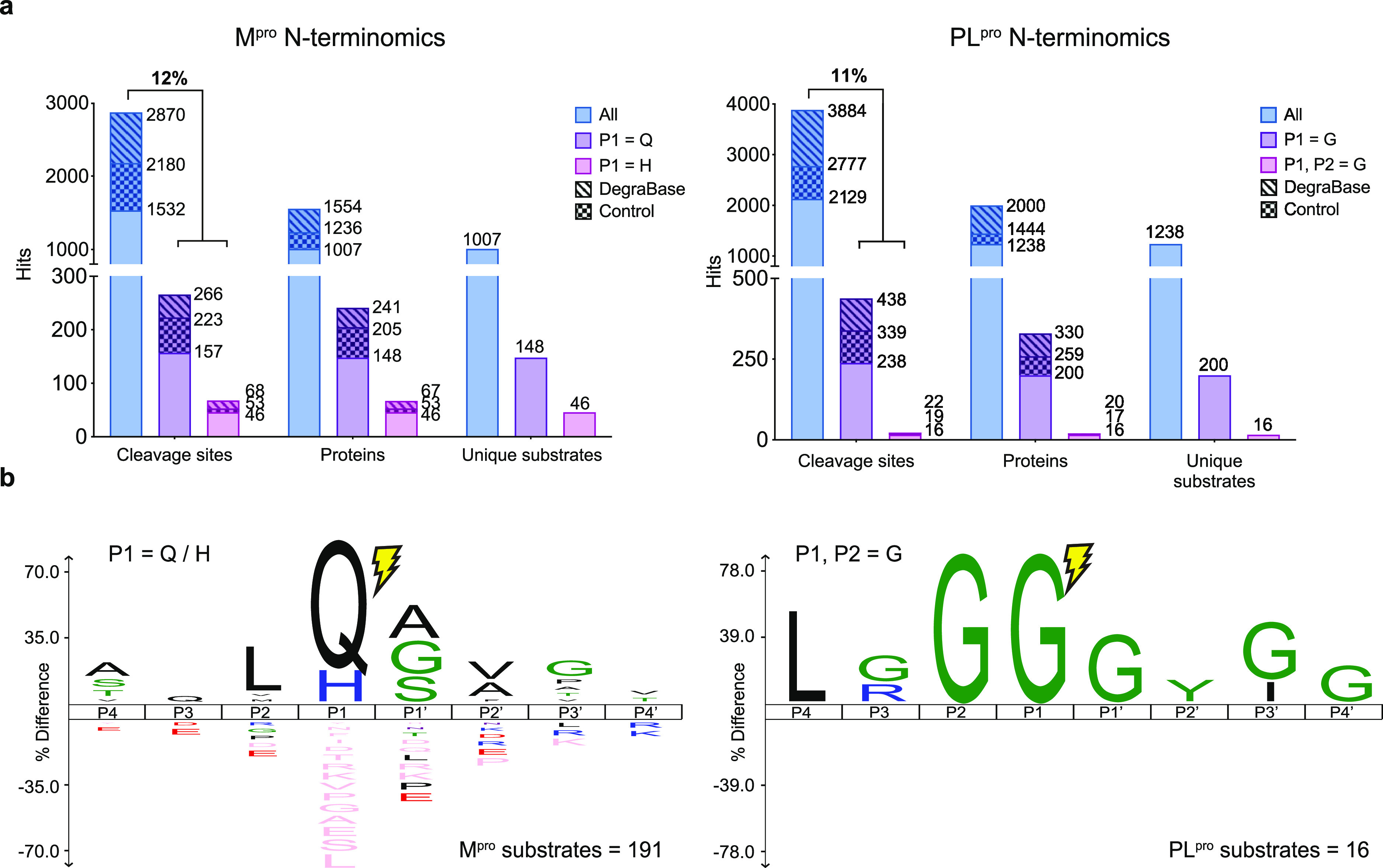
Identification of SARS-CoV-2 Mpro and PLpro substrates using subtiligase N-terminal labeling. (a) N-terminomics statistics of two Jurkat and two A549 replicates for each of Mpro (left) and PLpro (right). For Mpro, 2870 unique labeled cleavages were identified with 334 sites at P1 = Q/H in 308 proteins, showing an enhanced enrichment at 12%. For PLpro, 3884 unique labeled cleavages were identified with 438 sites at P1 = G and 22 sites at P1, P2 = G in 330 and 20 proteins, respectively, showing enrichment for P1 = G at 11%. For each viral protease, the number of Mpro and PLpro substrates unique to each protease is also reported. (b) IceLogo showing P4-P4′ residue enrichment for Mpro with P1 = Q/H (left) and for PLpro with P1, P2 = G (right).
Identification of PLpro Substrates
We conducted similar experiments with PLpro. We performed two replicates in A549 and Jurkat cell lysates and identified 3884 labeled unique cleavage sites in 2000 human proteins (Figure S4). We then looked for cleavage sites with Gly in the P1 position only or with Gly in both P1 and P2 positions, corresponding to the known PLpro specificity.13 We identified 438 unique cleavages in 330 host proteins corresponding to an 11.2% enrichment of P1 = G and 22 unique cleavages in 20 proteins corresponding to a 0.65% enrichment of P1, P2 = G (Figure 2a). Additionally, by comparing these results to the DegraBase and removing any cleavage sites with P1, P2 = G detected in the Mpro N-terminomics experiments, we identified 16 unique cleavage sites that have not been previously observed in healthy and apoptotic cells by subtiligase-based N-terminomics (Table 1). Of particular interest, 11 of these featured a Leu at P4 (LxGG motif). By comparison, only one LxGG cleavage site was observed in the Mpro data set. Overall, we identified 16 new cleavage sites at P1, P2 = G in 16 putative substrates in SARS-CoV-2 PLpro in vitro N-terminomics, with 11 featuring a LxGG motif (see Supporting Information, S2).
Table 1. Selected Putative Substrates of SARS-CoV-2 PLpro.
| acc # | P4-P1|P1′-P4′ | gene | protein name | subcellular localization |
|---|---|---|---|---|
| O00487 | LGGG10|MPGL | PSMD14 | 26S proteasome non-ATPase regulatory subunit 14 | cytosol, extracellular region, nucleoplasm, and nucleus |
| Q9NVZ3 | AVGG211|SLVQ | NECAP2 | adaptin ear-binding coat-associated protein 2 | cytoplasmic vesicle (clathrin-coated vesicle membrane) and cell membrane |
| P04632 | LKGG11|GGGG | CAPNS1 | calpain small subunit 1 | cytoplasm and cell membrane |
| P22626 | NQGG281|GYGG | HNRNPA2B1 | heterogeneous nuclear ribonucleoproteins A2/B1 | nucleus, nucleoplasm, cytoplasm, cytoplasmic granule, and secreted (extracellular exosome) |
| P05787 | YAGG422|LSSA | KRT8 | keratin and type II cytoskeletal 8 | cytoplasm, nucleus (nucleoplasm), and nucleus matrix |
| Q6PKG0 | LPGG12|ATLL | LARP1 | La-related protein 1 | cytoplasm and cytoplasmic granule |
| Q9UJU2 | LSGG7|GGGG | LEF1 | lymphoid enhancer-binding factor 1 | nucleus |
| P22059 | LGGG23|GAGP | OSBP | oxysterol-binding protein 1 | cytoplasm (cytosol, perinuclear region), Golgi apparatus membrane, endoplasmic reticulum membrane, and Golgi apparatus (trans-Golgi network) |
| O14908 | LGGG38|GSGG | GIPC1 | PDZ domain-containing protein GIPC1 | cytoplasm and membrane |
| Q7L014 | LRGG884|TILA | DDX46 | probable ATP-dependent RNA helicase DDX46 | nucleus speckle, nucleus (Cajal body), and membrane |
| O15234 | LRGG33|GSCS | CASC3 | protein CASC3 | cytoplasm (perinuclear region, stress granule, cytoplasmic ribonucleoprotein granule), nucleus, nucleus speckle, and cell projection (dendrite) |
| O60610 | LPGG624|VCIS | DIAPH1 | protein diaphanous homologue 1 | cell membrane, cell projection (ruffle membrane), cytoplasm (cytoskeleton, microtubule organizing center, centrosome, and spindle), and nucleus |
| A0A0B4J2F0 | IAGG21|VYIF | PIGBOS1 | protein PIGBOS1 | mitochondrion outer membrane |
| P35637 | GSGG192|GYGN | FUS | RNA-binding protein FUS | nucleus |
| P23246 | LGGG637|GGIG | SFPQ | splicing factor, and proline- and glutamine-rich | nucleus speckle, nucleus matrix, and cytoplasm |
| P62987 | LRGG76|IIEP | UBA52 | ubiquitin-60S ribosomal protein L40 | cytoplasm and nucleus |
SARS-CoV-2 Mpro Cleaves BRD2
The bromodomain and extra-terminal (BET) domain family of proteins is known to (1) regulate gene expression by interacting with acetylated histones and (2) facilitate RNA polymerase II transcription (see ref (36) for a review). In SARS-CoV-2, multiple studies reported that BET proteins can have both pro- and anti-viral effects.16,37,38 In our N-terminomics experiment, we observed bromodomain-containing protein 2 (BRD2) cleavage by Mpro after Q206 (AALQ ↓ GSVT) in Jurkat cell lysates. We investigated BRD2 cleavage by immunoblot in both Jurkat cell lysates and lysates of HEK293T-ACE2 cells overexpressing GFP-tagged BRD2. We observed the appearance of a cleavage product matching the molecular weight of the N-terminal fragment of BRD2 after cleavage at Q206 following a 2 h incubation with Mpro (23 kDa, 50 kDa with GFP) (Figure 3b,c). To confirm this cleavage site, we overexpressed the mutant GFP-BRD2 Q206A and did not observe the cleavage product (Figure 3c). In addition, no cleavage product was observed for GFP-BRD2 when incubated with Mpro in the presence of the Mpro inhibitor GC376 (Figure S5).2 Collectively, these results confirmed that BRD2 is a Mpro substrate. To examine the effect of viral infection on the level of host BRD2, we infected three cell lines (A549-ACE2, HEK293T-ACE2, and H23-ACE2) with SARS-CoV-2 and saw a decrease in full-length BRD2 levels compared to uninfected control (Figures 3d,e and S6–S9) and the presence of a cleavage product in infected HEK293T-ACE2 and H23-ACE2 (Figures S6b and S9b).
Figure 3.
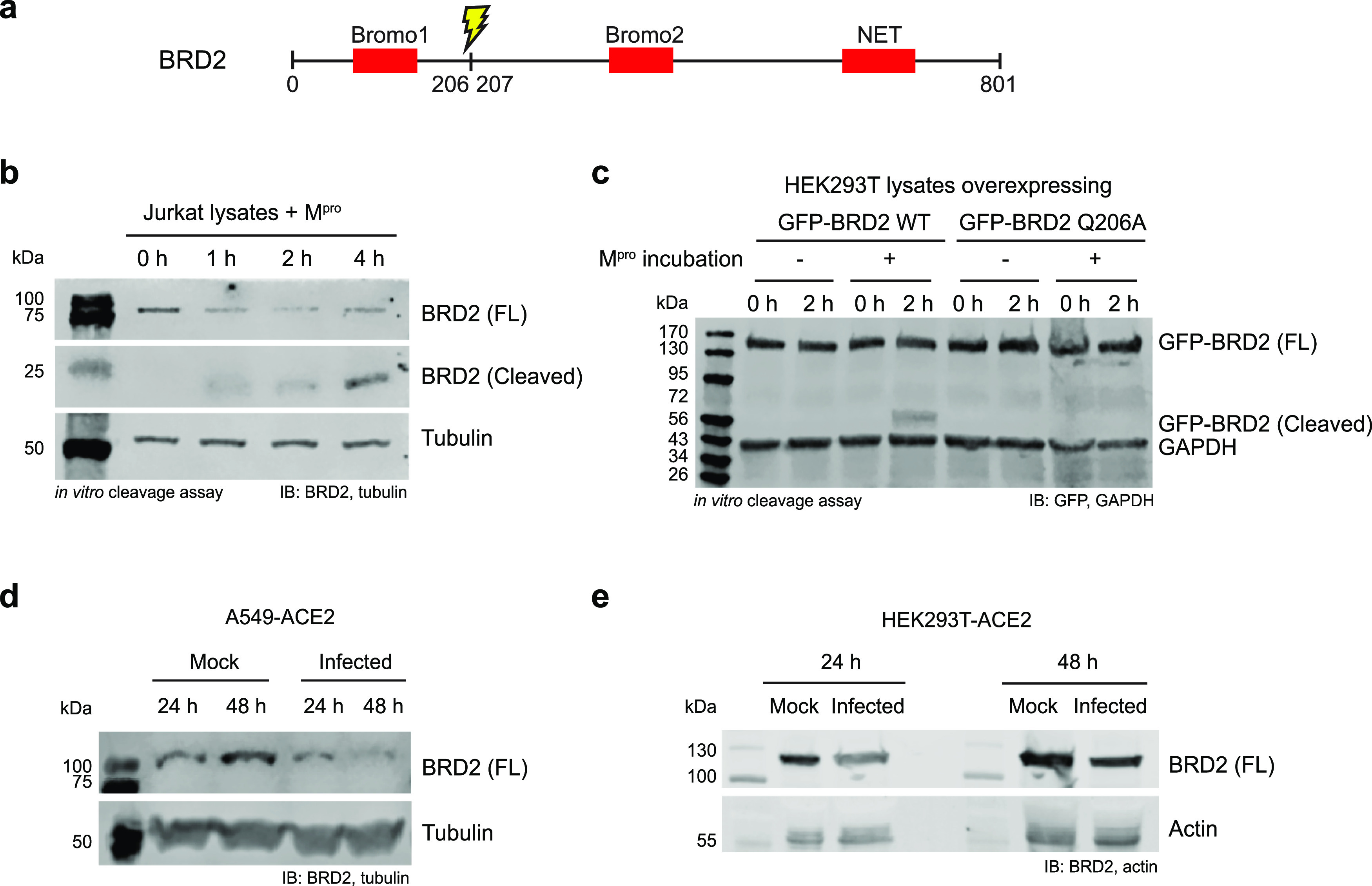
Proteolysis of BRD2 by Mpro in vitro and in SARS-CoV-2 infected cells. (a) BRD2 contains Bromo 1, Bromo 2, and N-terminal extra terminal (NET) domains. Our N-terminomics study identified Mpro cleavage site in BRD2 after Q206, cleaving off the Bromo 1 domain. (b) BRD2 was cleaved by recombinant Mpro in Jurkat cell lysates. Jurkat cell lysates were incubated with recombinant Mpro for 0–4 h and immunoblotted against BRD2. A cleavage product appeared with incubation time as the full length BRD2 level decreased. (c) GFP-BRD2 WT and Q206A mutant overexpression in HEK293T-ACE2 and in vitro cleavage by recombinant SARS-CoV-2 Mpro. HEK293T-ACE2 cells overexpressing GFP-BRD2 were lysed, and the cell lysates were incubated with Mpro for 2 h and immunoblotted against GFP. Cleavage was only observed with GFP-BRD2 WT. Depletion of full-length BRD2 was also observed in SARS-CoV-2 infected (d) A549-ACE2 and (e) HEK293T-ACE2 at 24 and 48 h.p.i.
SFPQ Is Cleaved in SARS-CoV-2 Infected Cells
Among the 16 putative substrates identified for SARS-CoV-2 PLpro, we further investigated the splicing factor, proline- and glutamine-rich (SFPQ). SFPQ is a DNA- and RNA-binding protein found in paraspeckles. SFPQ was shown to play a proviral role in Influenza A virus transcription, with its downregulation resulting in reduced viral replication.39 SFPQ is also exploited by several RNA viruses during infection, including the encephalomyocarditis virus40 and hepatitis delta virus41 and even targeted for proteolysis in human rhinovirus A16 to promote viral replication.42 Immunoblotting of SARS-CoV-2 infected A549-ACE2 lysates showed the full-length SFPQ at 76 kDa in both the mock and infected cells at 24 and 48 h.p.i. and the expected 69 kDa SFPQ cleavage product only in infected lysates (Figure 4b). This was consistent with our in vitro studies and suggests that PLpro cleaves SFPQ during infection. However, when uninfected Jurkat and A549-ACE2 cell lysates were incubated with SARS-CoV-2 PLpro, no corresponding cleavage product was observed on immunoblot (Figures 4c and S10a). Additionally, no clear in vitro cleavage product was observed in PLpro incubation with overexpressed FLAG-SFPQ in HEK293T-ACE2 cells (Figure S10b), even after immunoprecipitation (Figure S10c). Expression of full-length NSP3 in HEK293T-ACE2 cells (24 h post-transfection) also did not induce detectable endogenous SFPQ proteolysis by immunoblot (Figure S10d). While SFPQ is clearly cleaved during viral infection, it is also likely involving host proteases.
Figure 4.
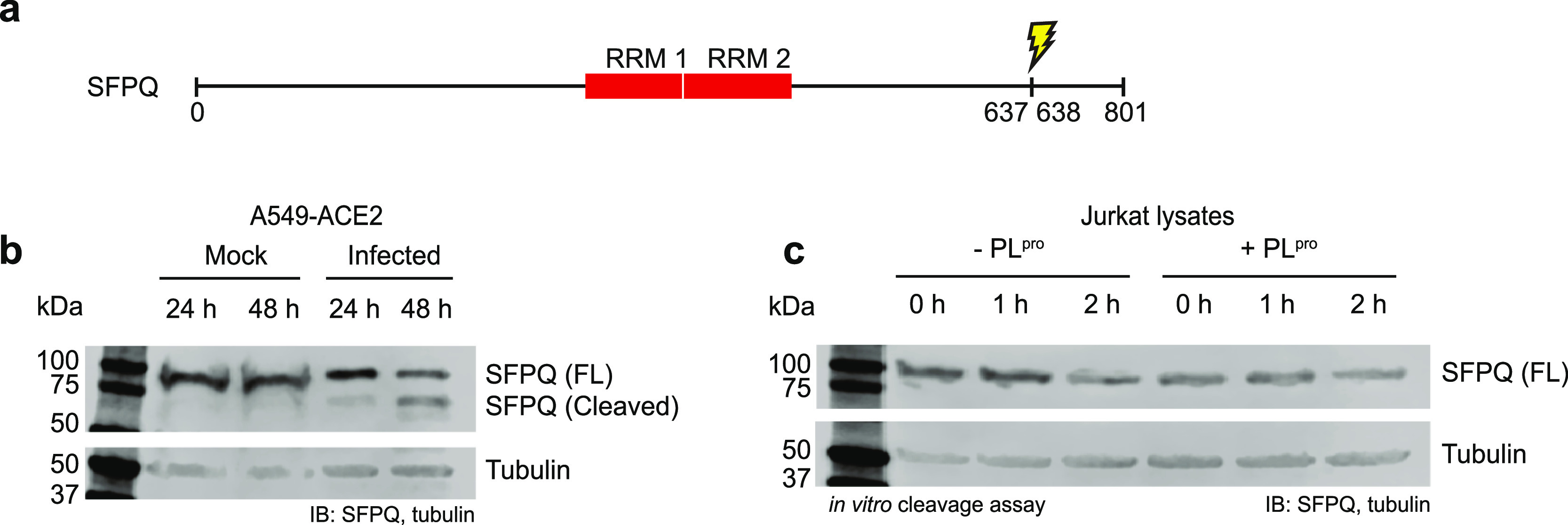
Proteolysis of SFPQ in SARS-CoV-2 infected cells. (a) PLpro cleaves SFPQ after G637, C-terminal to the RNA recognition motifs. (b) SFPQ was cleaved in A549-ACE2 cells infected with SARS-CoV-2. (c) SFPQ cleavage by PLpro could not be detected using immunoblotting in Jurkat cell lysates.
Discussion
Comparative Analysis of All Known Mpro Substrates
We compared our data with SARS-CoV-2 Mpro substrates reported using TAILS or tandem mass tag labeling (Figure 5b).27,29,30 TAILS and subtiligase-based labeling are complementary N-terminomics methods used to identify the N-termini of proteins; the protease-induced neo-N-termini are identified by negative and positive enrichment, respectively (see review ref (32)). The two methods each have their own advantages, such as the ability to identify cleavages in low-abundance proteins and different biases in P1′ sites. Thus, a compilation of all N-terminomics data on the SARS-CoV-2 Mpro can expand our understanding of how viral proteases function to regulate the host cell environment. We provide here a global analysis of all reported cleavage sites of SARS-CoV-2 Mpro identified: our subtiligase-mediated N-terminomics study, the TAILS experiments from Pablos et al. and Koudelka et al., and the N-terminomics study in SARS-CoV-2 infection-induced proteolysis from Meyer et al. (Figure 5a) (see Supporting Information S3 for the complete list). For consistency across the data sets, cleavage sites with P1 = Q/H in any cell lines, that passed the authors’ statistical evaluation in the case of negative enrichment techniques and were not within the first four residues of the start of a protein sequence, were included in this comparative analysis. In total, there were 742 unique cleavage sites in 604 human proteins attributed to SARS-CoV-2 Mpro activity. Of these, 59 new substrate cleavages were identified by two or more studies (Figure 5b). Interestingly, one protein called NUP107 was identified in all four data sets, with the cleavage site between residues Q35 and A36 (VLLQ35 ↓ ASQD). NUP107 is a nucleoporin and a member of the nuclear pore complex that mediates the transport between the cytoplasm and nucleus. Many nuclear pore complex proteins are cleaved during RNA viral infections, a strategy that is employed by picornaviruses such as polioviruses and rhinoviruses.43 However, the NUP107 cleavage site was also found in the PLpro data set, suggesting that perhaps this substrate is targeted by both Mpro and an unidentified human host protease. Similarly, stathmin and XRCC1 were also identified as putative Mpro substrates by three groups but were found in our PLpro data set. Stathmin regulates the cell cycle by re-organization of the microtubule cytoskeleton. Downregulation of phosphorylation on stathmin and other cytoskeleton assembly proteins was also observed in SARS-CoV-2 infected cells.44 The DNA repair protein XRCC1 is required for the repair of DNA single-strand breaks and interacts with DNA viruses such as human papillomavirus,45 as well as hepatitis B and C viruses.46 There are also two new protein cleavages that were commonly identified in three groups but not observed in the DegraBase or in the PLpro N-terminomics experiments: the 26S proteasome non-ATPase regulatory subunit 8 (PSMD8) cleaved after Q89 and a bifunctional enzyme involved in de novo purine biosynthesis called PAICS that is cleaved after Q34. PSMD8 is a part of the 26S proteasome complex and is a host restriction factor in HIV-1.47 PAICS is identified as an oncogene in several tumor types, but it has also been shown to bind to influenza A virus nucleoprotein.48 Although the link between virology and the two proteins have not been well studied, their cleavages could imply a significant role in the SARS-CoV-2 pathogenesis.
Figure 5.

Comparative analysis of SARS-CoV-2 Mpro putative substrates. (a) Overlap in identified cleavage sites among SARS-CoV-2 Mpro N-terminomics in vitro studies.29,30 (b) 59 common cleavage sites were identified by different Mpro studies and have not been observed in the PLpro N-terminomics data set nor in the DegraBase.35
Possible Roles of BRD2 Cleavage in SARS-CoV-2
BET proteins interact with many viral proteins and modulate viral infections. In particular, bromodomain-containing protein 4 (BRD4) forms a complex with E2 for transcriptional silencing in human papillomaviruses,49 and BRD2 interacts with latency-associated nuclear antigen 1 (LANA1) in Kaposi’s sarcoma-associated herpesvirus.50 BRD2 binds the SARS-Cov-2 envelope E protein16 and is required for ACE2 transcription which likely benefits SARS-CoV-2 replication in human lung epithelial cells. It also acts as a host antiviral factor by promoting the transcription of genes involved in type I interferon response.37 In another recent study, BRD2, 3, and 4 inactivation was shown to aggravate viral infection in cells and mice, overexpressing ACE2.38 Our N-terminomics and immunoblot studies showed BRD2 cleavage by Mpro after Q206 (AALQ ↓ GSVT). This viral protease cleavage removes the bromodomain I (BDI) (Figure 3a), potentially disrupting BRD2 binding to the acetylated histones and thereby affecting host gene transcription.51 Our N-terminomics study also detected proteolysis of BRD4, which is another member of the BET family that binds to SARS-CoV-2 E protein.16,38 The cleavage of BRD4 after Q1077 (SQFQ ↓ SLTH) in the C-terminal region could interfere with the formation of the P-TEFb transcriptional complex, preventing the activation of interferon-stimulated genes.52 Thus, the BET proteins have a sophisticated role during SARS-CoV-2 viral pathogenesis that may interact with multiple viral proteins and fine-tune the gene expression of key proteins involved in biological pathways.
Noncanonical Specificity of Mpro
Previous biochemical analyses and N-terminomics studies on SARS-CoV-2 Mpro placed a rather stringent selection filter for Mpro substrates, where only cleavage sites with a P1 = Q are considered as potential Mpro targets. Indeed, based on the crystal structure of both SARS-CoV-1 and -2, Gln can occupy the S1 pocket by stable interactions with His163, Phe140, and Glu166.7,9,53 However, the ability for SARS-CoV-1 Mpro to recognize other residues at the P1 site, specifically His and Met, and incorporate them into its active site has been reported by peptide library screening.10 In Koudelka et al.’s in vitro SARS-CoV-2 Mpro N-terminomics experiment, a strong enrichment of His in P1 position was also observed in the identified cleavage sites.29 Pablos et al. explored the noncanonical cleavage sequences of Mpro in detail using peptide libraries derived from N-terminomics substrates combined with molecular docking simulations and showed that Mpro can cleave after P1 = G/H/M.30 Similarly, while Leu is the preferred residue at the P2 position, Mpro can also recognize other hydrophobic residues at P2, such as Val, Phe, Met, Ala, and Ile, and has even broader specificities at positions P3 and P4. Hence, we selected potential Mpro substrates featuring a Gln or His at the P1 position in our in vitro N-terminomics (P1 = Q/H only) to allow some selectivity at the P1 site but relaxed restrictions on other sites. We did not include cleavage sites P1 = M for further investigation (3.8% of total labeled cleavages) as these were not enriched over typical background proteolysis.34 We also looked at the secondary structure of the Mpro cleavage site locations, where we found that 10 cuts occurred in α-helices, 6 in β-strands, 1 in turn, and the rest in uncharacterized or disordered regions.
Potential Activation of Other Cellular Proteases in Cell Lysates
The putative SARS-CoV-2 Mpro and PLpro cellular targets were subjected to pathway analyses using Metascape54 to reveal how viral proteases potentially disrupt cellular processes during infection. Mpro cleavage of host substrates is predicted to affect the cell cycle and cellular gene expression (Figure S11a), and the enriched processes of PLpro substrates highlight metabolism of RNA (Figure S11b). In our in vitro N-terminomics experiments, there are also many labeled cleavages in the human proteome that do not fall under the specificity profiles of the viral proteases Mpro and PLpro. While we cannot rule out exogenous co-purified protease activity from Escherichia coli, we believe that by adding a cocktail of protease inhibitors (targeting metallo- and serine proteases) and focusing on substrates matching Mpro and PLpro specificities, we have minimized the identification of nonrelated protease substrates. In addition, we can exclude substrates found in the PLpro data set matching Mpro specificity and vice versa, using each data set to identify unique cleavage sites to the viral proteases. However, the nonselective inhibition of other proteases is not 100% efficient. The observed cleavage sites that do not fall under the specificity profiles of Mpro and PLpro may be due to cellular protease activation and may still be of interest. The activity of the host proteases can be attributed to a few possibilities, such as the direct activation by the viral proteases to initiate proteolysis of other proteins or indirectly resulted from the viral protease incubation in the cellular proteome. Therefore, we searched all labeled cleavage sites from our N-terminomics data sets on TopFind 4.155 to investigate which endogenous proteases account for those cleavages (Figure S12). A majority of the cleavage sites correspond to granzyme M specificity (P1 = L/M).56 As the viral-infection-induced activation of granzyme M is characteristic in cytotoxic T lymphocytes, it is interesting to find its activation by viral proteases in an in vitro environment.
Up- and Downregulation of Viral Protease Substrates during Viral Infection
We initially hypothesized that viral proteases would cleave host restriction factors to improve replication efficiency and that the cleaved host protein fragments, due to their low stability, could be subsequently targeted for degradation by the host cell machinery. However, when we used the list of all putative substrates identified by N-terminomics and compared it to the reported proteome changes during SARS-CoV-2 infection17,57 or CRISPR screens,58,59 we found that the protease substrates we identified did not correlate with lower protein levels in infected cells. It is possible that these were not targeted for degradation by the cell. Alternatively, some of these proteolytic fragments could potentially lead to a gain-of-function, such as is the case of SFPQ, where a proviral factor can be cleaved by a viral protease.42
Limitations of the Study
We acknowledge that a large number of substrates identified in our in vitro N-terminomics may not actually be cleaved during infection and could be bystanders. We performed studies on the protein substrates known to play a role in host antiviral defense, such as transcription intermediary factor 1-beta (TRIM28) and the zinc finger antiviral protein (ZC3HAV1, also known as ZAP). However, we did not see depletion of TRIM28 and ZAP in the in vitro cleavage assays or in infected cells using immunoblot (Figure S13). Similarly, proteolysis in protein mono-ADP-ribosyltransferase PARP10 and nuclear pore complex protein Nup98-Nup96 could not be detected via immunoblot. This could be in part due to inability of the proteases to access substrates during infection and/or the fact that high concentrations of viral proteases were used in the in vitro studies (0.5 and 5 μM for Mpro and PLpro, respectively). It can also be challenging to precisely detect substrate proteolysis or degradation via immunoblot of infected cell lysates, where depending on the cell line, only a fraction of the cell population is infected, and a subfraction of those infected cells has only low levels of proteolysis in the corresponding host proteins. Many commercial antibodies also failed to detect protease-cleaved substrates in immunoblots. There are many reasons that could explain this discrepancy: substrate degradation, proteolysis by host proteases or E. coli protease contaminants, suboptimal time points, subcellular localization, and interferon-induced protein expression. It is also possible that the epitope could also be damaged by proteases in the lysate or that post-translational modifications (ubiquitination, phosphorylation, etc.) of the substrates could prevent antibody recognition. Furthermore, the overexpression of ACE2 receptor improves cellular susceptibility to viral infection in human cell lines, such as HEK293T and A549, but since many host proteins are involved in ACE2-mediated pathways, the overexpressed ACE2 might affect the degradation of these substrates in vivo, such as TRIM2860 and BRD2.37 When we compared the results from this study to other subtiligase-based N-terminomics studies on human proteases such as caspase-3 and -9, most identified substrates showed robust cleavage by immunoblot in in vitro cleavage assays and in apoptotic cells.61 The drastic difference in detection between the studies demonstrates that proteolysis in host proteins by viral proteases may occur only at very low levels. As a host cell is infected, even though many cellular pathways are disrupted, the virus prevents cell death in order to sustain viral replication. Hence, the low level of cellular protein proteolysis by viral proteases can be interpreted as a mechanism for the virus to maximize replication efficiency while maintaining cell viability.
Methods
SARS-CoV-2 Mpro Expression and Purification
The recombinant His6-GST-dual-tagged SARS-CoV-2 Mpro expression plasmid in the pGEX-6P-1 vector was cloned and kindly gifted by Dr. Rolf Hilgenfeld’s lab.7 The plasmid was transformed into E. coli strain BL21-Gold (DE3) cells (Novagen). Cells were grown in LB media supplemented with 100 μg/mL ampicillin at 37 °C to an OD600 at 0.8. Protease expression was induced with 0.5 mM isopropyl ß-D-1-thiogalactopyranoside (IPTG) at 37 °C for 5 h. The cells were harvested by centrifugation and lysed by Emulsiflex (Avestin). The cell lysates were clarified by centrifugation, and the soluble fraction was purified by HisTrap FF column (5 mL; Cytiva). The eluants were pooled and dialyzed with 10 units of PreScission protease (Cytiva) per mg of target protein. The cleaved proteins were applied to connected GSTrap FF (1 mL; Cytiva) and Talon (1 mL; Cytiva) columns. The flow-through was collected and concentrated using Amicon Ultra 15 centrifugal filters (10 kDa). The purified untagged proteins were diluted with glycerol, flash frozen in liquid nitrogen, and stored at −80 °C.
SARS-CoV-2 PLpro Expression and Purification
The GST-tagged SARS-CoV-2 PLpro expression plasmid in the pGEX-6P-1 vector was cloned and graciously gifted by Dr. Shaun K. Olsen’s lab.13 The plasmid was transformed in the E. coli strain BL21(DE3)pLysS. Cells were grown in LB media supplemented with 100 μg/mL ampicillin and 25 μg/mL chloramphenicol at 37 °C with 250 rpm shaking to an OD600 at 0.8. The media was supplemented with 0.1 mM zinc sulfate, and protein expression was induced with 0.5 mM IPTG 18 °C with 200 rpm shaking for 16 h. The cells were harvested by centrifugation at 7000 rpm for 15 min at 4 °C and subsequently lysed in a binding buffer (50 mM Tris pH 7.5, 300 mM NaCl, 2 mM 2-mercaptoethanol) by Emulsiflex (Avestin). The lysates were clarified by centrifugation, and the soluble fraction was purified GSTrap HP column (5 mL; Cytiva). The eluants in the elution buffer (50 mM Tris pH 7.5, 300 mM NaCl, 2 mM 2-mercaptoethanol, 100 mM reduced glutathione) were pooled and dialyzed for 12 h with 10 units GST-PreScission protease (Cytiva) per mg of target protein, or 1 mg protease per 50 mg target protein, in a dialysis buffer (50 mM Tris pH 7.5, 300 mM NaCl, 1 mM EDTA, 1 mM DTT). The cleaved proteins were purified by a GSTrap HP column (5 mL; Cytiva). The flow-through and wash fractions were pooled and concentrated by Amicon Ultra 15 centrifugal filters (10 kDa). The purified untagged protease was diluted to 10% glycerol, flash frozen in liquid nitrogen, and stored at −80 °C.
Synthesis of Coumarin Fluorescent Probe
A total of 200 mg of Rink Amide AM resin (0.89 mmol/g) was transferred to the reaction cartridge (Poly-Prep Chromatography Column, Bio-Rad), and 6 mL of DCM was added to the resin for swelling (30 min with constant mixing). DCM was removed by vacuum filtration, and the resin was washed three times with DMF, one time with methanol, one time with DCM, and a final wash with DMF (6 mL per wash).
The Fmoc-group was removed with 6 mL of 20% (v/v) piperidine in DMF. The resin and deprotection solution were gently agitated for 30 min. After that, the solution was removed by vacuum filtration, and the resin was washed five times with DMF (6 mL per wash). After the final wash, a Kaiser test (ninhydrin test) was performed to confirm the removal of the Fmoc-group. The Kaiser test reagents were prepared according to AAPPTec recommendations. For reagent A, 16.5 mg of KCN was dissolved in 25 mL of distilled water. A 1:50 dilution was made with 1 mL of the KCN solution and 49 mL of pyridine. For reagent B, 1 g of ninhydrin was dissolved in 20 mL of butanol. Reagent C contained 20 g of phenol in 10 mL of n-butanol. A few beads were transferred to a 1.5 mL Eppendorf tube. Three drops of each reagent were added. The mixture was heated for 3 min at 95 °C in a heating block. The presence of a blue color indicates deprotection of the resin. Addition of the Fmoc-ACC group was carried out according to Poreba et al. with a few modifications.62 A mixture of Fmoc-ACC-OH (0.35 mmol, 2 equiv), HATU (0.35 mmol, 2 equiv), and Collidine (0.53 mmol, 3 equiv) in 3 mL of DMF was added to the resin. The cartridge was protected from light with aluminum foil and incubated with gentle agitation for 24 h. Next day, the mixture was removed by vacuum filtration, and the resin was washed five times with DMF (6 mL per wash). Two extra ACC additions were carried out under the same conditions. Kaiser test was performed at the end to confirm coupling completion.
The sequence used for the probe corresponds to the most preferred substrate for Mpro, Ac-Abu-Tle-Leu-Gln-ACC.9 Each step addition was done for 2 h with constant mixing using the Fmoc-protected version of each residue (1.75 mmol, 10 equiv), HATU (1.75 mmol, 10 equiv), and Collidine (1.75 mmol, 10 equiv) in 5 mL of DMF. This was followed by Fmoc-group removal for 15 min with 20% (vol/vol) piperidine in DMF (1.2 mL of piperidine in 4.8 mL of DMF). Five DMF washes at the end of addition and deprotection steps were done (6 mL each). The completion of the reaction was monitored with the Kaiser test. After the final deprotection, capping of the N-termini was done with 6 mL of the acetylation mixture (acetic anhydride, pyridine, and DMF in 20:20:60% v/v/v) for 30 min with constant mixing. Once the Kaiser test was negative (yellow color in solution and beads), the resin was washed five times with 6 mL of DMF and three times with 6 mL of DCM. The resin was dried by vacuum filtration for 1 h. Cleavage of the final product was carried out for 2 h with constant mixing with 5 mL of the cleavage solution, TFA/H2O/tripropylsilane (95:2.5:2.5% v/v/v). The solution was recovered and precipitated in 40 mL of cold diethyl ether for 1 h. Tube was spun down at 8000g for 20 min. The pellet was resuspended in ACN/H2O (50/50% v/v) and lyophilized until fully dry. The purity of the substrate was confirmed by MALDI-TOF (Autoflex speed MALDI-TOF, Bruker). The final ACC probe was dissolved in DMSO at a final 10 mM concentration and stored at −80 °C. Similarly, the probe of sequence Ac-Leu-Arg-Gly-Gly-ACC was synthesized for PLpro13 and stored at −80 °C in DMSO at a final concentration of 1 mM.
SARS-CoV-2 Mpro Activity Assay
Activity assays were performed in 96-well standard opaque plates using a microplate reader (SpectraMax M3; Molecular Devices) in assay volumes of 100 μL. In cell-free assays, 20 mM Tris–HCl, pH 7.5, 1 mM ethylenediaminetetraacetic acid (EDTA), and 10 mM dithiothreitol (DTT) were used as the assay buffer. To assay in cell lysates, cells were lysed by probe sonication in a lysis buffer [20 mM Tris–HCl, pH 7.5, 0.1% Triton x-100, and 10 mM DTT with protease inhibitors (5 mM EDTA, 1 mM 4-(2-aminoethyl) benzenesulfonyl fluoride hydrochloride (AEBSF), 1 mM phenylmethanesulfonyl fluoride (PMSF), and 20 μM z-VAD-fmk (N-benzyloxycarbonyl-Val-Ala-Asp(O-Me) fluoromethyl ketone))]. Cell lysates were clarified by centrifugation, and the soluble fraction was taken as the assay buffer. Final concentrations of 0.5 μM purified SARS-CoV-2 Mpro and 2 μM of coumarin probe Ac-Abu-Tle-Leu-Gln-ACC dissolved in DMSO were added to the buffer with a final [DMSO] of 0.2%. The PMT gain was set to low with reads in 30 s intervals for 1 h at λexcitation of 355 nm and λemission of 460 nm.
SARS-CoV-2 PLpro Activity Assay
Activity assays were performed in 96-well standard opaque plates using a microplate reader (SpectraMax M3; Molecular Devices) in assay volumes of 100 μL. In cell-free assays, 20 mM Tris–HCl, pH 8.0, 5 mM NaCl, and 5 mM DTT was used as the assay buffer. To assay in cell lysates, cells were lysed by probe sonication in a lysis buffer [20 mM Tris–HCl, pH 8.0, and 0.1% Triton x-100 with protease inhibitors (5 mM EDTA, 1 mM AEBSF, 1 mM PMSF, and 4 mM iodoacetamide (IAM) with 30 min incubation in the dark)]. IAM was quenched with 20 mM DTT; then the cell lysates were clarified by centrifugation, and the soluble fraction was taken as the assay buffer. Final concentrations of 5 μM purified SARS-CoV-2 PLpro and 10 μM of coumarin probe Ac-Leu-Arg-Gly-Gly-ACC dissolved in DMSO were added to the buffer, with a final [DMSO] of 1%. The PMT gain was set to low with reads in 30 s intervals for 3 h at λexcitation of 355 nm and λemission of 460 nm.
Cell Culture
A549 and Jurkat (ATCC) were cultured, respectively, in Dulbecco’s Modified Eagle Medium (DMEM) (Gibco #11995-065) and Roswell Park Memorial Institute (RPMI) 1640 medium (Gibco #11875-093), supplemented with 10% fetal bovine serum, 100 U/mL penicillin and 100 μg/mL streptomycin, and 2 mM l-glutamine.
N-Terminal Labeling and Enrichment
The expression constructs for subtiligase expression (WT and M222A mutants) were a gift from Jim Wells and Amy Weeks.31 Jurkat (5 × 109) and A549 (2.5 × 109) cells were, respectively, used in each corresponding replicate. Cells were harvested by centrifugation and lysed by gentle probe sonication in lysis buffer to maintain native protein fold [20 mM Tris–HCl, pH 7.5, 0.1% Triton x-100, and 10 mM DTT with protease inhibitors (5 mM EDTA, 1 mM AEBSF, and 1 mM PMSF) for Mpro and 20 mM Tris–HCl, pH 8.0, and 0.1% Triton x-100 with protease inhibitors (5 mM EDTA, 1 mM AEBSF, 1 mM PMSF, and 4 mM IAM subsequently quenched with 20 mM DTT prior to addition of PLpro) for PLpro]. In Jurkat cell lysates, we also added 20 μM z-VAD-fmk to irreversibly inhibit endogenous caspases prior to adding SARS-CoV-2 Mpro. Cell lysates were clarified by centrifugation. For PLpro, 10× assay buffer (200 mM Tris–HCl, pH 8.0, 50 mM NaCl, 50 mM DTT) was added 1:10 to clarify the lysate. 0.5 μM of purified SARS-CoV-2 Mpro or 5 μM of purified SARS-CoV-2 PLpro was added to the soluble cell lysates for 2 h incubation, with aliquots taken out to monitor protease activity in the activity assay as a function of time. N-terminal labeling was then performed with 1 μM stabiligase WT, 1 μM subtiligase M222A, and 1 mM TEVest631 for 1 h. Tagged protein fragments were precipitated using acetonitrile, then denatured (8 M Gdn-HCl) and reduced (5 mM TCEP), and thiols were alkylated (10 mM IAM), before ethanol precipitation. Biotinylated N-terminal peptides were then captured with NeutrAvidin agarose beads (Thermo Fisher) for 24 h. The beads were washed using 4 M Gdn-HCl, trypsinized, and peptides were released from the beads using TEV protease. The TEV protease was precipitated using 2.5% TFA, and the peptides were desalted with using C18 Ziptips (Rainin).
Mass Spectrometry Analyses
Peptides were separated using a nanoflow-HPLC (Thermo Scientific EASY-nLC 1200 System) coupled to an Orbitrap Fusion Lumos Tribrid mass spectrometer (Thermo Fisher Scientific). A trap column (5 μm, 100 Å, 100 μm × 2 cm, Acclaim PepMap 100 nanoViper C18; Thermo Fisher Scientific) and an analytical column (2 μm, 100 Å, 50 μm × 15 cm, PepMap RSLC C18; Thermo Fisher Scientific) were used for the reverse-phase separation of the peptide mixture. Peptides were eluted over a linear gradient over the course of 120 min (or 90 min for the PLpro Jurkat data set) from 3.85 to 36.8% acetonitrile in 0.1% formic acid. 2 replicates of Mpro N-terminomics in Jurkat lysates were injected on the MS with and without the installation of FAIMS Pro interface (field asymmetric ion mobility spectrometry) to broaden protein coverage. Data were analyzed using ProteinProspector (v5.22.1) against the concatenated database of the human proteome (SwissProt.2017.11.01.random.concat), with maximum false discovery rate 1% for peptides. The peptides were searched at a maximum of 3 missed trypsin cleavages with TrypsinPro digest specificity relaxed at peptide N-termini. Search parameters included a precursor mass tolerance of 15 ppm, a fragment mass tolerance of 0.8 Da, precursor charge range of 2–5, with the constant modification carbamidomethylation (C) and variable modifications of Abu (N-term), deamidated (N/Q), and oxidation (M). The maximum number of variable modifications was set to 3. MS data are available through MASSIVE: MSV000088583 and MSV000088584 (Mpro) and MSV000090124 and MSV000090125 (PLpro).
In Vitro Cleavage Assays of Putative Substrates
HEK293T and HEK293T-ACE2 cells were transiently transfected with plasmid GFP-BRD2 (Addgene #65376) or FLAG-SFPQ (Addgene #166960) using Polyplus jetOPTIMUS DNA transfection reagent and harvested using 0.5 mM EDTA. Jurkat and A549 cells were cultured, harvested using 0.5 mM EDTA, and lysed in the same lysis buffer used in the N-terminomics protocol discussed above. The cell lysates were incubated with or without the active recombinant SARS-CoV-2 Mpro and PLpro, with activity assays to monitor protease activity in parallel. Aliquots of the cell lysates were taken at time points 0, 1, and 2 h, and reactions were quenched by boiling with the 5× Laemmli buffer for 5 min. The GFP-BRD2 Q206A plasmid was generated using site-directed mutagenesis with the forward oligo: GCCAAGTTGGCAGCGCTCGCGGGCAGTGTTACCAGTG and reverse oligo: CACTGGTAACACTGCCCGCGAGCGCTGCCAACTTGGC to mutate codon CAG to GCG (oligos purchased from IDT). The thermocycle was performed on 50 ng of GFP-BRD2 (Addgene, #65376) and pfu (Truin Science Ltd., #ETS4020) with 5 min initial denaturation at 95 °C, 17 cycles of 50 s denaturation at 95 °C, 50 s of annealing at 50 °C, and 16 min of extension of 68 °C, and final extension at 68 °C for 10 min. The PCR product was incubated with DpnI and transformed in DH5α cells. The final extracted plasmid was Sanger sequenced.
Stable Cell Line Generation and Viral Infection
HEK293T-ACE2, A549-ACE2, and H23-ACE2 stable cell lines and SARS-CoV-2 infection were performed as described previously.63 SARS-CoV-2 (hCoV-19/Canada/ON-VIDO-01/2020; GISAID accession no. EPI_ISL_425177) was kindly provided by Darryl Falzarano (Vaccine and Infectious Disease Organization, Saskatoon, Canada). HEK 293T-ACE2 and A549-ACE2 cells were developed by electroporating a human ACE2 encoding plasmid (Addgene #1786; a gift from Hyeryun Choe). The cells were passaged six times in culture, surface-stained for ACE2 (goat anti-ACE2; AF933-SP; R&D Systems), and the highest 2% of cells expressing ACE2 were sorted from the bulk population. Virus culture and experiments were performed according to level-3 containment procedures. Virus stocks were generated and titrated (by plaque assay) in Vero E6 cells, and HEK293T-ACE2, A549-ACE2, and H23-ACE2 cells were infected using MOI = 1.
Immunoblot
SARS-CoV-2 infected cell lysates and cell lysates incubated with SARS-CoV-2 Mpro and PLpro were loaded on 7.5% or 10% SDS-PAGE gels. After separation, proteins were transferred onto 0.45 μm nitrocellulose membranes (BioRad), blocked in 2.5% fish skin gelatin in TBS at RT for 1 h, then incubated with primary antibodies diluted in 2.5% fish skin gelatin in TBST at 4 °C overnight. The membrane was washed 3× with TBS for 5 min and incubated with secondary antibodies diluted in 2.5% fish skin gelatin in TBST at RT for 1 h. The membrane was washed again 2× with TBST and 1× with TBS for 5 min before viewing on the LI-COR Odyssey imaging system. Antibodies and mammalian plasmids used in this study are presented in Table S1. Uncropped images can be found in Supporting Information (Figures S6 and S14).
Acknowledgments
We would like to thank Rolf Hilgenfeld’s group for the Mpro plasmid used in this study and Shaun Olsen’s group for the PLpro plasmid. We also thank Joanne Lemieux and Elena Aruyunova for their help and support, as well as Anthony O’Donoghue, Danielle Skinner, and Marcin Drag for providing reagents. This work was supported by funding from the Li Ka Shing Institute of Virology at the University of Alberta, the Canadian Institutes of Health Research to O.J., A.K., and T.C.H. (OV3-172302; GA1-177707) and Tonix Pharmaceuticals. No competing financial interests have been declared.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsinfecdis.2c00458.
Compiled list of Mpro substrates (XLSX)
Compiled list of PLpro substrates (XLSX)
Comparative analysis of Mpro substrates (XLSX)
SARS-CoV-2 Mpro and PLpro expression and purification; Mpro and PLpro N-terminomics statistics; cleavage assay of GFP-BRD2 by Mpro with and without GC376; immunoblot images of BRD2 in infected cells; cleavage of SFPQ by PLpro additional validation; Metascape analysis of Mpro and PLpro putative substrates; TopFind analysis of Mpro and PLpro putative substrates; cleavage of Mpro substrates not detectable by immunoblots; and antibodies and plasmids used in the study (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Ullrich S.; Nitsche C. The SARS-CoV-2 main protease as drug target. Bioorg. Med. Chem. Lett. 2020, 30, 127377. 10.1016/j.bmcl.2020.127377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuong W.; Khan M. B.; Fischer C.; Arutyunova E.; Lamer T.; Shields J.; Saffran H. A.; McKay R. T.; van Belkum M. J.; Joyce M. A.; Young H. S.; Tyrrell D. L.; Vederas J. C.; Lemieux M. J.; Lemieux M. J. Feline coronavirus drug inhibits the main protease of SARS-CoV-2 and blocks virus replication. Nat. Commun. 2020, 11, 4282. 10.1038/s41467-020-18096-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vries M.; Mohamed A. S.; Prescott R. A.; Valero-Jimenez A. M.; Desvignes L.; O’Connor R.; Steppan C.; Devlin J. C.; Ivanova E.; Herrera A.; Schinlever A.; Loose P.; Ruggles K.; Koralov S. B.; Anderson A. S.; Binder J.; Dittmann M. A comparative analysis of SARS-CoV-2 antivirals characterizes 3CLpro inhibitor PF-00835231 as a potential new treatment for COVID-19. J. Virol. 2021, 95, e01819-20 10.1128/jvi.01819-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boras B.; Jones R. M.; Anson B. J.; Arenson D.; Aschenbrenner L.; Bakowski M. A.; Beutler N.; Binder J.; Chen E.; Eng H.; Hammond H.; Hammond J.; Haupt R. E.; Hoffman R.; Kadar E. P.; Kania R.; Kimoto E.; Kirkpatrick M. G.; Lanyon L.; Lendy E. K.; Lillis J. R.; Logue J.; Luthra S. A.; Ma C.; Mason S. W.; McGrath M. E.; Noell S.; Obach R. S.; O’ Brien M. N.; O’Connor R.; Ogilvie K.; Owen D.; Pettersson M.; Reese M. R.; Rogers T. F.; Rosales R.; Rossulek M. I.; Sathish J. G.; Shirai N.; Steppan C.; Ticehurst M.; Updyke L. W.; Weston S.; Zhu Y.; White K. M.; García-Sastre A.; Wang J.; Chatterjee A. K.; Mesecar A. D.; Frieman M. B.; Anderson A. S.; Allerton C. Preclinical characterization of an intravenous coronavirus 3CL protease inhibitor for the potential treatment of COVID19. Nat. Commun. 2021, 12, 6055. 10.1038/s41467-021-26239-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen D. R.; Allerton C. M. N.; Anderson A. S.; Aschenbrenner L.; Avery M.; Berritt S.; Boras B.; Cardin R. D.; Carlo A.; Coffman K. J.; Dantonio A.; Di L.; Eng H.; Ferre R.; Gajiwala K. S.; Gibson S. A.; Greasley S. E.; Hurst B. L.; Kadar E. P.; Kalgutkar A. S.; Lee J. C.; Lee J.; Liu W.; Mason S. W.; Noell S.; Novak J. J.; Obach R. S.; Ogilvie K.; Patel N. C.; Pettersson M.; Rai D. K.; Reese M. R.; Sammons M. F.; Sathish J. G.; Singh R. S. P.; Steppan C. M.; Stewart A. E.; Tuttle J. B.; Updyke L.; Verhoest P. R.; Wei L.; Yang Q.; Zhu Y. An oral SARS-CoV-2 Mpro inhibitor clinical candidate for the treatment of COVID-19. Science 2021, 374, 1586–1593. 10.1126/science.abl4784. [DOI] [PubMed] [Google Scholar]
- Abdelnabi R.; Foo C. S.; Jochmans D.; Vangeel L.; De Jonghe S.; Augustijns P.; Mols R.; Weynand B.; Wattanakul T.; Hoglund R. M.; Tarning J.; Mowbray C. E.; Sjö P.; Escudié F.; Scandale I.; Chatelain E.; Neyts J. The oral protease inhibitor (PF-07321332) protects Syrian hamsters against infection with SARS-CoV-2 variants of concern. Nat. Commun. 2022, 13, 719. 10.1038/s41467-022-28354-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L.; Lin D.; Sun X.; Curth U.; Drosten C.; Sauerhering L.; Becker S.; Rox K.; Hilgenfeld R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science 2020, 368, 409–412. 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schechter I.; Berger A. On the size of the active site in proteases. I. Papain. Biochem. Biophys. Res. Commun. 1967, 27, 157–162. 10.1016/s0006-291x(67)80055-x. [DOI] [PubMed] [Google Scholar]
- Rut W.; Groborz K.; Zhang L.; Sun X.; Zmudzinski M.; Pawlik B.; Wang X.; Jochmans D.; Neyts J.; Młynarski W.; Hilgenfeld R.; Drag M. SARS-CoV-2 Mpro inhibitors and activity-based probes for patient-sample imaging. Nat. Chem. Biol. 2021, 17, 222–228. 10.1038/s41589-020-00689-z. [DOI] [PubMed] [Google Scholar]
- Chuck C. P.; Chong L. T.; Chen C.; Chow H. F.; Wan D. C. C.; Wong K. B. Profiling of Substrate Specificity of SARS-CoV 3CLpro. PLoS One 2010, 5, e13197 10.1371/journal.pone.0013197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freitas B. T.; Durie I. A.; Murray J.; Longo J. E.; Miller H. C.; Crich D.; Hogan R. J.; Tripp R. A.; Pegan S. D. Characterization and Noncovalent Inhibition of the Deubiquitinase and deISGylase Activity of SARS-CoV-2 Papain-Like Protease. ACS Infect. Dis. 2020, 6, 2099–2109. 10.1021/acsinfecdis.0c00168. [DOI] [PubMed] [Google Scholar]
- Gao X.; Qin B.; Chen P.; Zhu K.; Hou P.; Wojdyla J. A. Crystal structure of SARS-CoV-2 papain-like protease. Acta Pharm. Sin. B 2021, 11, 237. 10.1016/j.apsb.2020.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rut W.; Lv Z.; Zmudzinski M.; Patchett S.; Nayak D.; Snipas S. J.; El Oualid F.; Huang T. T.; Bekes M.; Drag M.; Olsen S. K. Activity profiling and crystal structures of inhibitor-bound SARS-CoV-2 papain-like protease: A framework for anti-COVID-19 drug design. Sci. Adv. 2020, 6, eabd4596 10.1126/sciadv.abd4596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill M. E.; Kumar A.; Wells J. A.; Hobman T. C.; Julien O.; Hardy J. A. The Unique Cofactor Region of Zika Virus NS2B–NS3 Protease Facilitates Cleavage of Key Host Proteins. ACS Chem. Biol. 2018, 13, 2398–2405. 10.1021/acschembio.8b00508. [DOI] [PubMed] [Google Scholar]
- Jagdeo J. M.; Dufour A.; Klein T.; Solis N.; Kleifeld O.; Kizhakkedathu J.; Luo H.; Overall C. M.; Jan E. N-Terminomics TAILS Identifies Host Cell Substrates of Poliovirus and Coxsackievirus B3 3C Proteinases That Modulate Virus Infection. J. Virol. 2018, 92, e02211–e02217. 10.1128/jvi.02211-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon D. E.; Jang G. M.; Bouhaddou M.; Xu J.; Obernier K.; White K. M.; O’Meara M. J.; Rezelj V. V.; Guo J. Z.; Swaney D. L.; Tummino T. A.; Hüttenhain R.; Kaake R. M.; Richards A. L.; Tutuncuoglu B.; Foussard H.; Batra J.; Haas K.; Modak M.; Kim M.; Haas P.; Polacco B. J.; Braberg H.; Fabius J. M.; Eckhardt M.; Soucheray M.; Bennett M. J.; Cakir M.; McGregor M. J.; Li Q.; Meyer B.; Roesch F.; Vallet T.; Mac Kain A.; Miorin L.; Moreno E.; Naing Z. Z. C.; Zhou Y.; Peng S.; Shi Y.; Zhang Z.; Shen W.; Kirby I. T.; Melnyk J. E.; Chorba J. S.; Lou K.; Dai S. A.; Barrio-Hernandez I.; Memon D.; Hernandez-Armenta C.; Lyu J.; Mathy C. J. P.; Perica T.; Pilla K. B.; Ganesan S. J.; Saltzberg D. J.; Rakesh R.; Liu X.; Rosenthal S. B.; Calviello L.; Venkataramanan S.; Liboy-Lugo J.; Lin Y.; Huang X. P.; Liu Y.; Wankowicz S. A.; Bohn M.; Safari M.; Ugur F. S.; Koh C.; Savar N. S.; Tran Q. D.; Shengjuler D.; Fletcher S. J.; O’Neal M. C.; Cai Y.; Chang J. C. J.; Broadhurst D. J.; Klippsten S.; Sharp P. P.; Wenzell N. A.; Kuzuoglu-Ozturk D.; Wang H. Y.; Trenker R.; Young J. M.; Cavero D. A.; Hiatt J.; Roth T. L.; Rathore U.; Subramanian A.; Noack J.; Hubert M.; Stroud R. M.; Frankel A. D.; Rosenberg O. S.; Verba K. A.; Agard D. A.; Ott M.; Emerman M.; Jura N.; von Zastrow M.; Verdin E.; Ashworth A.; Schwartz O.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468. 10.1038/s41586-020-2286-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stukalov A.; Girault V.; Grass V.; Karayel O.; Bergant V.; Urban C.; Haas D. A.; Huang Y.; Oubraham L.; Wang A.; Hamad M. S.; Piras A.; Hansen F. M.; Tanzer M. C.; Paron I.; Zinzula L.; Engleitner T.; Reinecke M.; Lavacca T. M.; Ehmann R.; Wölfel R.; Jores J.; Kuster B.; Protzer U.; Rad R.; Ziebuhr J.; Thiel V.; Scaturro P.; Mann M.; Pichlmair A. Multilevel proteomics reveals host perturbations by SARS-CoV-2 and SARS-CoV. Nature 2021, 594, 246–252. 10.1038/s41586-021-03493-4. [DOI] [PubMed] [Google Scholar]
- May D. G.; Martin-Sancho L.; Anschau V.; Liu S.; Chrisopulos R. J.; Scott K. L.; Halfmann C. T.; Diaz Pena R.; Pratt D.; Campos A. R.; et al. A BioID-Derived Proximity Interactome for SARS-CoV-2 Proteins. Viruses 2022, 14, 611. 10.3390/v14030611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin D.; Mukherjee R.; Grewe D.; Bojkova D.; Baek K.; Bhattacharya A.; Schulz L.; Widera M.; Mehdipour A. R.; Tascher G.; Geurink P. P.; Wilhelm A.; et al. Papain-like protease regulates SARS-CoV-2 viral spread and innate immunity. Nature 2020, 587, 657–662. 10.1038/s41586-020-2601-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang S.; Wang J.; Cheng G. Protease cleavage of RNF20 facilitates coronavirus replication via stabilization of SREBP1. Proc. Natl. Acad. Sci. U. S. A. 2021, 118, e2107108118 10.1073/pnas.2107108118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moustaqil M.; Ollivier E.; Chiu H. P.; Van Tol S.; Rudolffi-Soto P.; Stevens C.; Bhumkar A.; Hunter D. J. B.; Freiberg A. N.; Jacques D.; Lee B.; Sierecki E.; Gambin Y. SARS-CoV-2 proteases PLpro and 3CLpro cleave IRF3 and critical modulators of inflammatory pathways (NLRP12 and TAB1): implications for disease presentation across species. Emerg. Microb. Infect. 2021, 10, 178–195. 10.1080/22221751.2020.1870414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsu B. V.; Agarwal R.; Gokhale N. S.; Kulsuptrakul J.; Ryan A. P.; Castro L. K.; Beierschmitt C. M.; Turcotte E. A.; Fay E. J.; Vance R. E.; Hyde J. L.; Savan R.; Mitchell P. S.; Daugherty M. D.. Host specific sensing of coronaviruses and picornaviruses by the CARD8 inflammasome. 2022, 2022.09.21.508960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wenzel J.; Lampe J.; Müller-Fielitz H.; Schuster R.; Zille M.; Müller K.; Krohn M.; Körbelin J.; Zhang L.; Ozorhan U.; et al. The SARS-CoV-2 main protease Mpro causes microvascular brain pathology by cleaving NEMO in brain endothelial cells. Nat. Neurosci. 2021, 24, 1522–1533. 10.1038/s41593-021-00926-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohamud Y.; Xue Y. C.; Liu H.; Ng C. S.; Bahreyni A.; Jan E.; Luo H. The papain-like protease of coronaviruses cleaves ULK1 to disrupt host autophagy. Biochem. Biophys. Res. Commun. 2021, 540, 75–82. 10.1016/j.bbrc.2020.12.091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds N. D.; Aceves N. M.; Liu J. L.; Compton J. R.; Leary D. H.; Freitas B. T.; Pegan S. D.; Doctor K. Z.; Wu F. Y.; Hu X.; Legler P. M. The SARS-CoV-2 SSHHPS Recognized by the Papain-like Protease. ACS Infect. Dis. 2021, 7, 1483–1502. 10.1021/acsinfecdis.0c00866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miczi M.; Golda M.; Kunkli B.; Nagy T.; Tőzsér J.; Mótyán J. A. Identification of Host Cellular Protein Substrates of SARS-COV-2 Main Protease. Int. J. Mol. Sci. 2020, 21, E9523 10.3390/ijms21249523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer B.; Chiaravalli J.; Gellenoncourt S.; Brownridge P.; Bryne D. P.; Daly L. A.; Grauslys A.; Walter M.; Agou F.; Chakrabarti L. A.; Craik C. S.; Eyers C. E.; Eyers P. A.; Gambin Y.; Jones A. R.; Sierecki E.; Verdin E.; Vignuzzi M.; Emmott E. Characterising proteolysis during SARS-CoV-2 infection identifies viral cleavage sites and cellular targets with therapeutic potential. Nat. Commun. 2021, 12, 5553. 10.1038/s41467-021-25796-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kleifeld O.; Doucet A.; auf dem Keller U.; Prudova A.; Schilling O.; Kainthan R. K.; Starr A. E.; Foster L. J.; Kizhakkedathu J. N.; Overall C. M. Isotopic labeling of terminal amines in complex samples identifies protein N-termini and protease cleavage products. Nat. Biotechnol. 2010, 28, 281–288. 10.1038/nbt.1611. [DOI] [PubMed] [Google Scholar]
- Koudelka T.; Boger J.; Henkel A.; Schönherr R.; Krantz S.; Fuchs S.; Rodríguez E.; Redecke L.; Tholey A. N-Terminomics for the Identification of In Vitro Substrates and Cleavage Site Specificity of the SARS-CoV-2 Main Protease. Proteomics 2021, 21, e2000246 10.1002/pmic.202000246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pablos I.; Machado Y.; de Jesus H. C. R.; Mohamud Y.; Kappelhoff R.; Lindskog C.; Vlok M.; Bell P. A.; Butler G. S.; Grin P. M.; Cao Q. T.; Nguyen J. P.; Solis N.; Abbina S.; Rut W.; Vederas J. C.; Szekely L.; Szakos A.; Drag M.; Kizhakkedathu J. N.; Mossman K.; Hirota J. A.; Jan E.; Luo H.; Banerjee A.; Overall C. M. Mechanistic insights into COVID-19 by global analysis of the SARS-CoV-2 3CLpro substrate degradome. Cell Rep. 2021, 37, 109892. 10.1016/j.celrep.2021.109892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weeks A. M.; Wells J. A. Engineering peptide ligase specificity by proteomic identification of ligation sites. Nat. Chem. Biol. 2018, 14, 50–57. 10.1038/nchembio.2521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo S. Y.; Araya L. E.; Julien O. Protease Substrate Identification Using N-terminomics. ACS Chem. Biol. 2019, 14, 2361–2371. 10.1021/acschembio.9b00398. [DOI] [PubMed] [Google Scholar]
- Shen X. R.; Geng R.; Li Q.; Chen Y.; Li S. F.; Wang Q.; Min J.; Yang Y.; Li B.; Jiang R. D.; Wang X.; Zheng X. S.; Zhu Y.; Jia J. K.; Yang X. L.; Liu M. Q.; Gong Q. C.; Zhang Y. L.; Guan Z. Q.; Li H. L.; Zheng Z. H.; Shi Z. L.; Zhang H. L.; Peng K.; Zhou P. ACE2-independent infection of T lymphocytes by SARS-CoV-2. Signal Transduction Targeted Ther. 2022, 7, 83. 10.1038/s41392-022-00919-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seaman J. E.; Julien O.; Lee P. S.; Rettenmaier T. J.; Thomsen N. D.; Wells J. A. Cacidases: caspases can cleave after aspartate, glutamate and phosphoserine residues. Cell Death Differ. 2016, 23, 1717–1726. 10.1038/cdd.2016.62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford E. D.; Seaman J. E.; Agard N.; Hsu G. W.; Julien O.; Mahrus S.; Nguyen H.; Shimbo K.; Yoshihara H. A.; Zhuang M.; Chalkley R. J.; Wells J. A. The DegraBase: a database of proteolysis in healthy and apoptotic human cells. Mol. Cell. Proteomics 2013, 12, 813–824. 10.1074/mcp.o112.024372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taniguchi Y. The Bromodomain and Extra-Terminal Domain (BET) Family: Functional Anatomy of BET Paralogous Proteins. Int. J. Mol. Sci. 2016, 17, E1849 10.3390/ijms17111849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samelson A. J.; Tran Q. D.; Robinot R.; Carrau L.; Rezelj V. V.; Kain A. M.; Chen M.; Ramadoss G. N.; Guo X.; Lim S. A.; Lui I.; Nuñez J. K.; Rockwood S. J.; Wang J.; Liu N.; Carlson-Stevermer J.; Oki J.; Maures T.; Holden K.; Weissman J. S.; Wells J. A.; Conklin B. R.; TenOever B. R.; Chakrabarti L. A.; Vignuzzi M.; Tian R.; Kampmann M. BRD2 inhibition blocks SARS-CoV-2 infection by reducing transcription of the host cell receptor ACE2. Nat. Cell Biol. 2022, 24, 24–34. 10.1038/s41556-021-00821-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen I. P.; Longbotham J. E.; McMahon S.; Suryawanshi R. K.; Khalid M. M.; Taha T. Y.; Tabata T.; Hayashi J. M.; Soveg F. W.; Carlson-Stevermer J.; Gupta M.; Zhang M. Y.; Lam V. L.; Li Y.; Yu Z.; Titus E. W.; Diallo A.; Oki J.; Holden K.; Krogan N.; Fujimori D. G.; Ott M. Viral E protein neutralizes BET protein-mediated post-entry antagonism of SARS-CoV-2. Cell Rep. 2022, 40, 111088. 10.1016/j.celrep.2022.111088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landeras-Bueno S.; Jorba N.; Pérez-Cidoncha M.; Ortín J. The splicing factor proline-glutamine rich (SFPQ/PSF) is involved in influenza virus transcription. PLoS Pathog. 2011, 7, e1002397 10.1371/journal.ppat.1002397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou B.; Wu F.; Han J.; Qi F.; Ni T.; Qian F. Exploitation of nuclear protein SFPQ by the encephalomyocarditis virus to facilitate its replication. Biochem. Biophys. Res. Commun. 2019, 510, 65–71. 10.1016/j.bbrc.2019.01.032. [DOI] [PubMed] [Google Scholar]
- Greco-Stewart V. S.; Thibault C. S. L.; Pelchat M. Binding of the polypyrimidine tract-binding protein-associated splicing factor (PSF) to the hepatitis delta virus RNA. Virology 2006, 356, 35–44. 10.1016/j.virol.2006.06.040. [DOI] [PubMed] [Google Scholar]
- Flather D.; Nguyen J. H. C.; Semler B. L.; Gershon P. D. Exploitation of nuclear functions by human rhinovirus, a cytoplasmic RNA virus. PLoS Pathog. 2018, 14, e1007277 10.1371/journal.ppat.1007277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park N.; Katikaneni P.; Skern T.; Gustin K. E. Differential targeting of nuclear pore complex proteins in poliovirus-infected cells. J. Virol. 2008, 82, 1647–1655. 10.1128/jvi.01670-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouhaddou M.; Memon D.; Meyer B.; White K. M.; Rezelj V. V.; Correa Marrero M.; Polacco B. J.; Melnyk J. E.; Ulferts S.; Kaake R. M.; et al. The Global Phosphorylation Landscape of SARS-CoV-2 Infection. Cell 2020, 182, 685–712.e19. 10.1016/j.cell.2020.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iftner T.; Elbel M.; Schopp B.; Hiller T.; Loizou J. I.; Caldecott K. W.; Stubenrauch F. Interference of papillomavirus E6 protein with single-strand break repair by interaction with XRCC1. EMBO J. 2002, 21, 4741–4748. 10.1093/emboj/cdf443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leite S. T. A. P.; Marques-Guimarães N.; Silva-Oliveira J. C.; Dutra-Souto F. J.; Alves-dos-Santos R.; Bassi-Branco C. L. The X-ray repair cross complementing protein 1 (XRCC1) rs25487 polymorphism and susceptibility to cirrhosis in Brazilian patients with chronic viral hepatitis. Ann. Hepatol. 2013, 12, 733–739. 10.1016/s1665-2681(19)31314-6. [DOI] [PubMed] [Google Scholar]
- Li Z.; Wu J.; Chavez L.; Hoh R.; Deeks S. G.; Pillai S. K.; Zhou Q. Reiterative Enrichment and Authentication of CRISPRi Targets (REACT) identifies the proteasome as a key contributor to HIV-1 latency. PLoS Pathog. 2019, 15, e1007498 10.1371/journal.ppat.1007498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Generous A.; Thorson M.; Barcus J.; Jacher J.; Busch M.; Sleister H. Identification of putative interactions between swine and human influenza A virus nucleoprotein and human host proteins. Virol. J. 2014, 11, 228. 10.1186/s12985-014-0228-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu S. Y.; Lee A. Y.; Hou S. Y.; Kemper J. K.; Erdjument-Bromage H.; Tempst P.; Chiang C. M. Brd4 links chromatin targeting to HPV transcriptional silencing. Genes Dev. 2006, 20, 2383–2396. 10.1101/gad.1448206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viejo-Borbolla A.; Ottinger M.; Brüning E.; Bürger A.; König R.; Kati E.; Sheldon J. A.; Schulz T. F. Brd2/RING3 interacts with a chromatin-binding domain in the Kaposi’s Sarcoma-associated herpesvirus latency-associated nuclear antigen 1 (LANA-1) that is required for multiple functions of LANA-1. J. Virol. 2005, 79, 13618–13629. 10.1128/jvi.79.21.13618-13629.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Filippakopoulos P.; Picaud S.; Mangos M.; Keates T.; Lambert J. P.; Barsyte-Lovejoy D.; Felletar I.; Volkmer R.; Müller S.; Pawson T.; Gingras A. C.; Arrowsmith C. H.; Knapp S. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell 2012, 149, 214–231. 10.1016/j.cell.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisgrove D. A.; Mahmoudi T.; Henklein P.; Verdin E. Conserved P-TEFb-interacting domain of BRD4 inhibits HIV transcription. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 13690–13695. 10.1073/pnas.0705053104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anand K.; Ziebuhr J.; Wadhwani P.; Mesters J. R.; Hilgenfeld R. Coronavirus main proteinase (3CLpro) structure: basis for design of anti-SARS drugs. Science 2003, 300, 1763–1767. 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- Zhou Y.; Zhou B.; Pache L.; Chang M.; Khodabakhshi A. H.; Tanaseichuk O.; Benner C.; Chanda S. K. Metascape provides a biologist-oriented resource for the analysis of systems-level datasets. Nat. Commun. 2019, 10, 1523. 10.1038/s41467-019-09234-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortelny N.; Yang S.; Pavlidis P.; Lange P. F.; Overall C. M. Proteome TopFIND 3.0 with TopFINDer and PathFINDer: database and analysis tools for the association of protein termini to pre- and post-translational events. Nucleic Acids Res. 2015, 43, D290–D297. 10.1093/nar/gku1012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahrus S.; Kisiel W.; Craik C. S. Granzyme M is a regulatory protease that inactivates proteinase inhibitor 9, an endogenous inhibitor of granzyme B. J. Biol. Chem. 2004, 279, 54275–54282. 10.1074/jbc.m411482200. [DOI] [PubMed] [Google Scholar]
- Bojkova D.; Klann K.; Koch B.; Widera M.; Krause D.; Ciesek S.; Cinatl J.; Münch C. Proteomics of SARS-CoV-2-infected host cells reveals therapy targets. Nature 2020, 583, 469–472. 10.1038/s41586-020-2332-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebendenne A.; Roy P.; Bonaventure B.; Chaves Valadao A. L.; Desmarets L.; Arnaud-Arnould M.; Rouille Y.; Tauziet M.; Giovannini D.; Touhami J.; et al. Bidirectional genome-wide CRISPR screens reveal host factors regulating SARS-CoV-2, MERS-CoV and seasonal HCoVs. Nat. Genet. 2022, 54, 1090–1102. 10.1038/s41588-022-01110-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mac Kain A.; Maarifi G.; Aicher S. M.; Arhel N.; Baidaliuk A.; Munier S.; Donati F.; Vallet T.; Tran Q. D.; Hardy A.; Chazal M.; Porrot F.; OhAinle M.; Carlson-Stevermer J.; Oki J.; Holden K.; Zimmer G.; Simon-Lorière E.; Bruel T.; Schwartz O.; van der Werf S.; Jouvenet N.; Nisole S.; Vignuzzi M.; Roesch F. Identification of DAXX as a restriction factor of SARS-CoV-2 through a CRISPR/Cas9 screen. Nat. Commun. 2022, 13, 2442. 10.1038/s41467-022-30134-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y.; Fan Y.; Huang Y.; Du T.; Liu Z.; Huang D.; Wang Y.; Wang N.; Zhang P. TRIM28 regulates SARS-CoV-2 cell entry by targeting ACE2. Cell Signal. 2021, 85, 110064. 10.1016/j.cellsig.2021.110064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araya L. E.; Soni I. V.; Hardy J. A.; Julien O. Deorphanizing Caspase-3 and Caspase-9 Substrates In and Out of Apoptosis with Deep Substrate Profiling. ACS Chem. Biol. 2021, 16, 2280–2296. 10.1021/acschembio.1c00456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poreba M.; Salvesen G. S.; Drag M. Synthesis of a HyCoSuL peptide substrate library to dissect protease substrate specificity. Nat. Protoc. 2017, 12, 2189–2214. 10.1038/nprot.2017.091. [DOI] [PubMed] [Google Scholar]
- Kumar A.; Ishida R.; Strilets T.; Cole J.; Lopez-Orozco J.; Fayad N.; Felix-Lopez A.; Elaish M.; Evseev D.; Magor K. E.; Mahal L. K.; Nagata L. P.; Evans D. H.; Hobman T. C. SARS-CoV-2 Nonstructural Protein 1 Inhibits the Interferon Response by Causing Depletion of Key Host Signaling Factors. J. Virol. 2021, 95, e0026621 10.1128/jvi.00266-21. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.