

Abstract
Background:
Dysregulation of airway smooth muscle cells (ASM) is central to the severity of asthma. Which molecules dominantly control ASM in asthmatics is unclear. High levels of the cytokine LIGHT (TNFSF14) have been linked to asthma severity and lower baseline FEV1 %predicted, implying signals through its receptors might directly control ASM dysfunction.
Objective:
To determine whether signaling via LTβR or HVEM from LIGHT dominantly drives ASM hyperreactivity induced by allergen.
Methods:
Conditional knockout mice deficient for LTβR or HVEM in smooth muscle cells were used to determine their role in ASM deregulation and airway hyperresponsiveness (AHR) in vivo. Human ASM were used to study signals induced by LTβR.
Results:
LTβR was strongly expressed in ASM from normal and asthmatic subjects compared to several other receptors implicated in smooth muscle deregulation. Correspondingly, conditional deletion of LTβR only in smooth muscle cells in smMHCCreLTβRfl/fl mice minimized changes in their numbers and mass, and AHR, induced by house dust mite allergen in a model of severe asthma. Intratracheal LIGHT administration independently induced ASM hypertrophy and AHR in vivo dependent on direct LTβR signals to ASM. LIGHT promoted contractility, hypertrophy, and hyperplasia of human ASM in vitro. Distinguishing LTβR from the receptors for IL-13, TNF, and IL-17 that have also been implicated in smooth muscle dysregulation, LIGHT promoted NIK-dependent non-canonical NF-κB in ASM in vitro, leading to sustained accumulation of F-actin, phosphorylation of myosin light chain kinase, and contractile activity.
Conclusion:
LTβR signals directly and dominantly drive airway smooth muscle hyperresponsiveness relevant for pathogenesis of airway remodeling in severe asthma.
Keywords: LTβR, Asthma, Airway Smooth Muscle, AHR, contractility, non-canonical NF-κB, TNF superfamily, LIGHT, TNFSF14
Capsule Summary:
Deletion of LTβR in smooth muscle cells limits allergen-driven airway smooth muscle remodeling and airway hyperresponsiveness, implying that LTβR may control excessive bronchial smooth muscle activity related to remodeling and lung function impairment in asthma.
INTRODUCTION
The pathological features of asthma are chronic airway inflammation, associated with an aberrant airway constriction response to allergen that is termed airway hypersensitivity or hyperresponsiveness (AHR)1. Deregulation of airway smooth muscle cells (ASM), which play a pivotal role in constriction and dilation of the airways, may be key to AHR2. ASM mass is greater in patients with severe asthma compared to those with moderate asthma3. Increased ASM mass is also observed in children with severe asthma, despite having a relatively short duration of asthma, suggesting that this change is directly linked with asthma severity4. Moreover, ASM isolated from asthmatics also exhibit enhanced contractile activity and enhanced proliferation2, 5. However, what drives these changes is still not fully appreciated.
Specifically, it has not been clear whether immune-mediated inflammatory signals into ASM are directly responsible for lung dysfunction and if so which signals may be essential. Several cytokines linked to asthma, notably IL-13, TNF, and IL-17, have been shown to promote contractility or other responses in ASM in vitro, and can contribute to AHR in vivo in mouse models6–11. Nevertheless, no data has yet shown that their direct actions on smooth muscle cells in vivo are essential for ASM changes or for AHR in response to allergen. In particular, studies of the receptor for IL-13, which has gained strong prominence in asthma pathogenesis from clinical trials of the IL-4Rα targeting antibody dupilumab12, 13, failed to find a defect in AHR and smooth muscle deregulation to allergen when IL-4Rα was conditionally deleted in mice only in smooth muscle cells14, 15. Thus, the key molecules directly and dominantly controlling asthma-related changes in ASM activity that are relevant for AHR are still not clear.
We previously linked the cytokine LIGHT (homologous to lymphotoxin, exhibits inducible expression and competes with HSV glycoprotein D for binding to HVEM, a receptor expressed on T lymphocytes), also known as TNF superfamily member 14 (TNFSF14), to the severity of lung inflammation and to changes in ASM mass with studies of LIGHT-deficient mice16. LIGHT can be expressed by several immune cell types including activated T cells, dendritic cells, and neutrophils, that are found in asthmatic lungs, and higher levels of soluble LIGHT or cell-associated LIGHT in the sputum of asthmatic patients are associated with severe disease17–19. Structural cells of the lung such as epithelial cells and fibroblasts express the receptors for LIGHT, namely HVEM (TNFRSF14) and LTβR (LTBR/TNFRSF3), and LIGHT can promote inflammatory activity in these cells20–24. However, whether LIGHT and its receptors act directly on airway smooth muscle cells in vivo to promote exaggerated responsiveness and play a direct role in AHR has not been demonstrated. In this study, we now show that LTβR is constitutively expressed on mouse and human airway smooth muscle cells. With conditional deletion of LTβR in smooth muscle cells in vivo, we demonstrate that LTβR activity in these cells is essential for smooth muscle remodeling, lung dysfunction, and airway hyperresponsiveness driven by inhaled allergen. This is explained by LTβR activating sustained signaling pathways involving NFκB-inducing kinase (NIK) and activation of non-canonical NF-κB, pathways not triggered by the other ASM-associated cytokines IL-13, TNF, and IL-17.
METHODS
Mice
C57BL/6J and smMHC (Myh11)/Cre/eGFP transgenic mice (B6.Cg-Tg(Myh11-cre,-EGFP)2Mik/J)25 were purchased from Jackson Laboratory. HVEM-floxed mice were generated in-house as previously described23, 26. LTβR-floxed mice were generated by Alexei Tumanov as previously described27. HVEM flox/flox or LTβR flox/flox mice crossed to smMHCCre transgenic mice were bred in-house on the C57BL/6 background. Animal experiments were performed with 6–8-wk-old female mice. All animals used in this study were maintained in specific pathogen-free conditions. All experiments were performed in compliance with the regulations of the La Jolla Institute for Immunology Animal Care Committee in accordance with guidelines of the Association for Assessment and Accreditation of Laboratory Animal Care.
Mouse models of airway remodeling and acute airway inflammation
For allergen airway remodeling experiments, mice were given intranasal administrations of HDM, Dermatophagoides pteronyssinus, extract (Greer Laboratories, Lenoir, NC, USA): 200 μg on day 0 and 100 μg on days 7 and 14, followed by 50 μg of HDM given twice per week for 4 weeks. For LIGHT experiments, mice were injected intratracheally with 10 μg of recombinant LIGHT (R&D, Mineapolis, MN, USA) or PBS on day 1 and 2 and analyzed on day 3. For acute airway inflammation, mice were sensitized by administration of 20 μg HDM protein in 2mg alum given intraperitoneally on day 0, and challenged with 10 μg HDM protein intranasally on days 10–13.
Confocal microscopy of lung tissue
Lungs were intubated, filled and embedded in the Cryomold with OCT compound, and frozen. The fresh frozen lung tissues were post-fixed in 4% PFA and lung sections were cut (30 μm) and stained with rabbit polyclonal antibody to αSMA (alpha smooth muscle actin: Abcam, Cambridge, UK), Phalloidin-AlexaFluor 568 (Thermo Fisher Scientific, Hampton, NJ, USA), and Hoechst33342 (BD Bioscience, San Jose, CA, USA). All 3D high resolution tertiary bronchi image stacks were acquired with an inverted Zeiss 780 or 880 Airyscan laser scanning confocal microscope (LSCM) using a 40x (1.4na) objective and the 32-channel GaAsP-PMT area detector (Zeiss Microscopy LLC, White Plains, NY, USA). Image stacks through 15–20 μm of lung tissue, on average 35 slices, were acquired with Nyquist resolution parameters using a 0.421 μm step size and optimal frame size of 2048×2048.
Lung tissue were further processed in imaris software using the isosurface module (Bitplane) to outline and quantify the volume and area of α-smooth muscle actin or Phalloidin around the mouse bronchioles. Phalloidin (F-actin) was more consistent in labeling the smooth muscle, based on the density of the signal and circular localization pattern in addition to the colocalization with αSMA. The actin labeling in all other non-muscle cells was omitted from analysis by masking only the bronchioles using imaris software and then thresholding the F-actin signal to omit very low to sparsely labeled F-actin in epithelial and other cells beyond the smooth muscle layer. 10–15 tertiary bronchi per 4–5 mouse lungs per condition and mouse model were analyzed.
Airway hyperresponsiveness
Airway resistance in response to different doses of Methacholine were measured using the FlexiVent system (SCIREQ Inc, Montreal, Canada) as previously described16. Peak airway resistance was analyzed by Scireq flexiWare software V8.
Flow cytometry
Lungs were dissociated using gentleMACS Dissociator (Miltenyi Biotec, Bergisch Gladbach, Germany) with Lung Dissociation Kit (Miltenyi Biotec). Single cells were stained with monoclonal antibodies to mouse CD45 (clone:30-F11), CD11b (clone:M1/70), CD11c (clone:HL3), SiglecF (clone: E50–2440) from BD Biosciences; EpCAM (clone:G8.8), CD31 (clone:390), PDGFRα (clone:1A4/asm-1), Mcam (clone:ME-9F1) from BioLegend; vimentin (clone:280618) from R&D; and αSMA (clone:1A4/asm-1) from Novus. Live/Dead cells were stained with Fixable Aqua Dead Cell Staining Kit (Thermo Fisher). For intracellular staining, Foxp3 Transcription Factor Staining Buffer Set (eBioscience, San Diego, CA, USA) were used for fixation and permeabilization. Flow analysis was performed on a Fortessa (BD Biosciences) and data were analyzed using FlowJo Software (version 10, FlowJo, LLC, Ashland, OR). Live+CD45+ lung immune cells were gated for neutrophils, eosinophils, alveolar macrophages, CD4+ T cells and CD8+ T cells following the gating strategy shown in the supplemental figure. Live+CD45− lung structural cells were gated as described in the figures into alternative populations by surface staining for Epcam, CD31, PDGFRα, Mcam, and intracellular staining for vimentin and αSMA.
Histology
Whole lung lobes were fixed with 10% formalin and embedded in paraffin. Sections were stained with hematoxylin and eosin or Periodic Acid-Schiff (PAS). Mucus production was assessed by measuring the percentage of PAS-positive cells airway epithelial cells in the bronchioles. More than 5 bronchi per section were randomly selected and used for quantification.
BAL cytokines
Cytokines in BAL fluid were assayed by sandwich ELISA with paired antibody sets, according to the manufacturers’ instructions. Mouse IL-4, IL-5 and IL-13 kits were from R&D.
ASM culture and analysis
Healthy donor human ASM or asthmatic diseased donor ASM were purchased from ScienCell (cat:3400; Carlsbad, CA, USA) or Lonza (cat: 00194850; Walkersville, MD, USA). Cells from several donors were used for reproducibility. Other asthmatic donor ASM were isolated from postmortem lungs provided by Richard Kurten from the Arkansas Regional Organ Recovery Agency as previously described28. Hl129: Asthma, 7 years old, Male, Caucasian, Non-smoker, COD CVA/Stroke; Hl227: Asthma, 21 years old, Male, Caucasian, Non-smoker, COD Head trauma. For flow analyses, cells were stained with monoclonal antibodies to human LTβR (clone:31G4D8), or HVEM (clone:122) from BioLegend. Cells were maintained in Smooth Muscle Cell Media (ScienCell) with supplied supplements and FBS added. Cells were used between passages 2–4 and cultured in smooth muscle basal media for 16 hours or 7 days before stimulation with recombinant LIGHT (100 ng/ml). In some experiments, inhibitors of NIK/non-canonical NF-κB (NIK-SMI, 25nM, MedChemExpress, Monmouth, NJ, USA), canonical NF-κB (BAY11–7082, 1nM, MedChemExpress), or Rac1 (NSC 23766, 10nM, Tocris, Birstol, UK) were added for 1 hour before stimulation with rLIGHT. For siRNA knockdown, ON-TARGETplus siRNA to human LTβR and NIK, and an NTC control, were purchased from Dharmacon (Pittsburg, PA, USA). siRNA Oligo Duplex siRNA to human HVEM was purchased from Origene (Rockville, MD, USA). 50 nM of siRNA was transfected into ASM using HiPerFect transfection reagent (Qiagen, Venlo, Netherlands) as described previously21 and according to the manufacturer’s instructions.
ASM gel contraction assay
3D Gel contraction assays were performed using collagen contraction assay kits purchased from Cell Biolabs (San Diego, CA, USA) according to the manufacturer’s instructions. In brief, suspensions of ASM were mixed with collagen solution at a density of 0.5–1×106 cells/well. After confirming the polymerization of collagen gels, smooth muscle basal media was added and incubated for 16 hours. Collagen gels were then stimulated with rLIGHT (100 ng/ml), rLTαβ (100 ng/ml) or PBS and analyzed for gel contraction at different timepoints (24, 48, 72 hours). Images were obtained with ImageLab (Bio-Rad Laboratories, Hercules, CA) and the area of collagen gel matrix measured using ImageJ software.
ASM intracellular protein analysis
For analyzing actin polymerization and focal adhesions, ASM were incubated in smooth muscle basal media without serum for 16 hours and seeded as monolayers at 70–80% confluency on coverslips coated with Collagen Type I solution (Sigma-Aldrich, St. Louis, MO, USA). Cells were stimulated with 100 ng/ml of rLIGHT or PBS with or without NIK-SMI or Rac inhibitors. After 6–12 hours, cells were washed and fixed with 4% Formalin and permeabilized with 0.1% TritonX-100 (Thermo Fisher Scientific). Cells were stained with Phalloidin-Alexa Fluor 568 (Thermo Fisher Scientific), anti-Vinculin antibody (Abcam) and Hoechst 33342 (BD Bioscience) and analyzed by confocal microscopy. Single ASM images were processed in imaris software using the isosurface module (Bitplane) to outline and quantify the volume of intracellular vinculin clusters and F-actin expression (Phalloidin). All 3D high resolution single ASM cell images were acquired with an inverted 880 Airyscan laser scanning confocal microscope (LSCM) using a 63x (1.4na) objective and the 32-channel GaAsP-PMT area detector (Zeiss Microscopy LLC). Image stacks through 3–5 μm of spreading ASM, on average 10–15 slices, were acquired with Nyquist resolution parameters using a 0.3 μm step size and optimal frame size of 4164×4164. 300–400 individual ASM, in two replicate experiments for the various conditions, were analyzed.
ASM migration/wound assay
ASM were seeded in monolayers on coverslips coated with Collagen Type I solution (Sigma-Aldrich) and cultured in smooth muscle media until reaching 90% confluency. Cells were incubated in smooth muscle basal media for 16 hours before making a scratch wound in a straight line followed by washing out of any debris. Smooth muscle basal media containing 100 ng/ml of rLIGHT or PBS was then added and incubated for 12 hours. Cells were washed and fixed with 4% Formalin and permeabilized with 0.1% TritonX-100 (Thermo Fisher Scientific). Cells were stained with Phalloidin-Alexa Fluor 568 (Thermo Fisher Scientific) and Hoechst 33342 (BD Bioscience) and analyzed by confocal microscopy. All 3D multi-stitched image panels were acquired on an inverted Zeiss LSM 780 LCSM using a 20x 0.8 NA objective, a z-step size of 0.85 microns and by using the automated tiling function of the Zen software with a 10% overlap between tiles. Images were stitched and 3D stacks were maximum intensity projected using the Zen software then imported into Image Pro Premier 10 (IPP10) Media Cybernetics) for further processing. Briefly, by using the phalloidin-AF555 signal to define the cells along the wound edge and the thresholding tool in IPP10 to define areas devoid of cells (set at a dynamic range of 0–50 for 8 bit images), the auto count tool automatically outlined and quantified the area all along the wound front, allowing quantification of the wound area and the extent of ASM migration into the wound.
ASM proliferation
ASM were cultured in the presence of 100 ng/ml rLIGHT or PBS. After 48 hours, cells were harvested and the proliferating cells were detected by BrdU incorporation using a FITC BrdU Flow Kit (BD Bioscience) according to the manufacturer’s protocol.
Western Blotting
Cells were lysed in RIPA Buffer containing protease inhibitor (Roche, Mannheim, Germany) Whole cell lysates were run in WedgeWell 8–16% Trys-Glysin gradient gels (Novex) and transferred to Nitrocellurose membrane (Bio-Rad). Membranes were incubated with 5% BSA (Sigma-Aldrich) or non-fat dry milk (R&D) in TBS-T for blocking, then incubated with primary antibodies to p-NFκB p65 (cat:3033, 1:1000), NFκB p65 (cat:8242, 1:1000), NFκB p100/52 (cat:3017, 1:1000), PAK1 (cat:2602,1:1000), p-MYPT(Thr696) (cat:5163, 1:1000), p-MLC2 (cat:3672, 1:1000), MLC2 (cat:8505, 1:500) from Cell Signaling Technology; to p-PAK1 (cat:135755, 1:200) and MYPT (cat:514261, 1:100) from Santa Cruz; followed by HRP-conjugated secondary antibodies to mouse IgG (cat:516102, Santa Cruz) or rabbit IgG (cat:2357, Santa Cruz). Chemiluminescence was enhanced with Amersham ECL Prime Western Blotting Detection Reagent (GE Health Care, Madison, WI, USA) and visualized after exposure to Gel Doc (Bio-Rad). Stripping and reblotting were performed with Restore Western Blot Stripping Buffer (Thermo Fisher Scientific). Immunoblotting for GAPDH (Santa Cruz, Dallas, TX, USA) was used as loading control. For Rac1 detection, whole cell lysates were used for immunoprecipitation using Active Rac1 detection kit (Cell Signaling Technology, Danvers, MA, USA) according to the manufacturer’s instruction.
RT-PCR
Total RNA from ASM was isolated using RNeasy kit (Qiagen). cDNA was prepared using a SuperScript IV reverse transcriptase kit (Thermo Fisher Scientific). Real-time PCR was performed with LightCycler (Roche, Indianapolis, Ind) using PowerUp SYBR Green master mixes (Applied Biosystems, Foster City, CA). Data were normalized with housekeeping gene GAPDH and presented as relative expression or relative quantification to control samples which were derived from the difference in cycle threshold (Ct) between the gene of interest and the housekeeping genes using the equation RQ = 2−ΔΔct. Designed primers for human MAP3K14 were purchased from Bio-Rad (Hercules, CA, USA). The oligonucleotide primer sequences were: GAPDH, sense AGCCAAAAGGGTCATCATCTCT, anti-sense AGGGGCCATCCACAGTCTT; HVEM, sense AGCAGCTCCCACTGGGTATG, anti-sense GATTAGGCCAACTGTGGAGCA; LTβR, sense CCGACACAACCTGCAAAAAT, anti-sense GAGCAGAAAGAAGGCCAGTG.
Analysis of published RNA-seq data
Bulk RNA-seq of healthy and asthma ASM were analyzed for transcripts of cytokine receptors. Fong et al29–31 used ASM from 11 asthmatic and 12 non-asthmatic donors over two GEO submissions: GSE119578 (GSE119578_GeneCount_rpkm_by_gene_RDB.txt.gz) and GSE119579 (GSE119579_GeneCount_rpkm_by_gene_SW.txt.gz). Himes et al29–31 used ASM from 6 donors with fatal asthma and 12 control donors. Only baseline (no treatment) measurements from GSE58434 were analyzed (GSE58434_All_Sample_FPKM_Matrix.txt.gz). Kan et al (Kan et al., 2019) used ASM from 9 fatal asthma and 8 non-asthma donors. Only vehicle control samples from GSE94335 were analyzed (GSE94335_PostQC_all_sample_TPM.txt.gz).
Statistical analysis
All results are presented as mean ±SEM. Statistical significance was analyzed by Mann-Whitney test for comparison of means between 2 independent groups or by 1-way ANOVA followed by Tukey’s post hoc multiple comparison test for differences of means between multiple groups using Prism 8 software (GraphPad Prism, San Diego, CA). P value <0.05 was considered statistically significant. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
RESULTS
LTβR and HVEM are expressed on ASM from healthy and asthmatic individuals
We first studied expression of the receptors for LIGHT, LTβR and HVEM, in published RNA-seq data from airway smooth muscle cells (ASM) isolated from healthy and asthmatic individuals29–31. Three separate data sets revealed strong expression of transcripts for LTβR and lower levels of transcripts for HVEM (Fig. 1A). Interestingly, compared to the receptors for the three primary asthma-related cytokines linked to activities in airway smooth muscle cells, namely TNFR1, IL4R, and IL17RA and IL17RC, only TNFR1 transcripts were as, or more, abundant compared to LTβR transcripts. No differences in transcript levels were observed for any of the receptors between healthy and asthmatic ASM (Fig. 1A). Flow cytometric analyses of two preparations of ASM from healthy individuals obtained from commercial sources confirmed that LTβR was more strongly expressed on the cell surface than HVEM. Analysis of ASM from asthmatic subjects, from a commercial source and from post-mortem lungs of asthmatics isolated in-house, furthermore showed equivalent expression of both molecules compared to healthy donor ASM, with LTβR being most highly expressed (Fig. 1B).
Figure 1. LTβR and HVEM are expressed by human healthy and asthmatic airway smooth muscle cells.
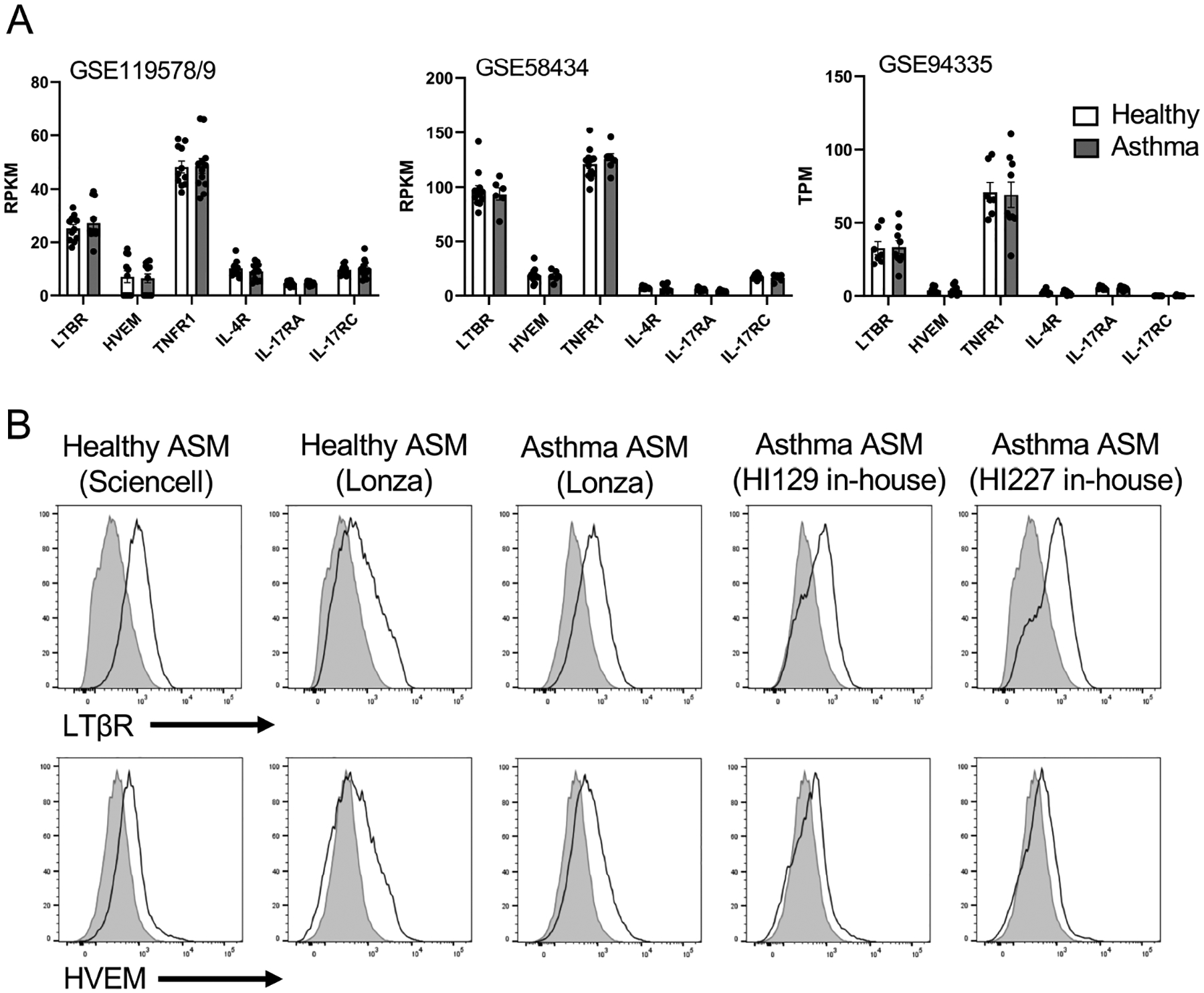
(A) Expression of transcripts of the receptors for LIGHT, TNF, IL-13/IL-4, and IL-17 from published RNA-seq data of human healthy and asthmatic ASM (GSE119578/9; GSE58434; GSE94335). (B) Flow analysis of LTβR and HVEM expression on healthy and asthma donor-derived ASM.
ASM-specific LTβR-deficient mice display reduced ASM mass and AHR in allergen-induced experimental asthma
To then assess the importance of LTβR or HVEM on ASM in vivo, we used smooth muscle Myosin Heavy Chain (smMHC)Cre (Myh11)-eGFP transgenic mice25. Similar to human cells, each receptor was expressed constitutively on ASM in the mouse lungs (Fig. 2A). We next generated smooth muscle cell-specific receptor knockout mice by crossing smMHCCre mice to HVEMfl/fl or LTβRfl/fl mice, and confirmed deletion of each receptor in ASM after gating on lung GFP-smMHC positive cells (Fig. 2A). Previous studies using scRNAseq have revealed several markers to identify structural cell subsets in the lung32, 33, however there is still a lack of an established gating strategy to identify ASM by conventional flow cytometric methods without a marker tag. We found that among the CD45− EpCAM− PDGFRα− and Vimentinlow population, that excludes epithelial and endothelial cells and fibroblasts, there are two distinct αSMA (alpha smooth muscle actin) positive populations subdivided based on MCAM (CD146) expression (Fig. E1A). MCAM is known to be highly expressed on pericytes and lower in smMHC+ mature smooth muscle cells34. By analyzing the smMHCCre conditional knockout mice, we found that both HVEM and LTβR were deficient in the αSMA+ MCAM− population, but not the αSMA+ MCAM+ population or in fibroblasts or epithelial cells. This further showed the specificity of the conditional deletion of the two receptors and revealed that ASM can be distinguished effectively as CD45− EpCAM− PDGFRα− Vimentinlow αSMA+ MCAM− cells in the lungs (Fig. E1B).
Figure 2. ASM-specific LTβR-deficient mice exhibit reduced ASM mass and AHR in allergen-induced experimental severe asthma.
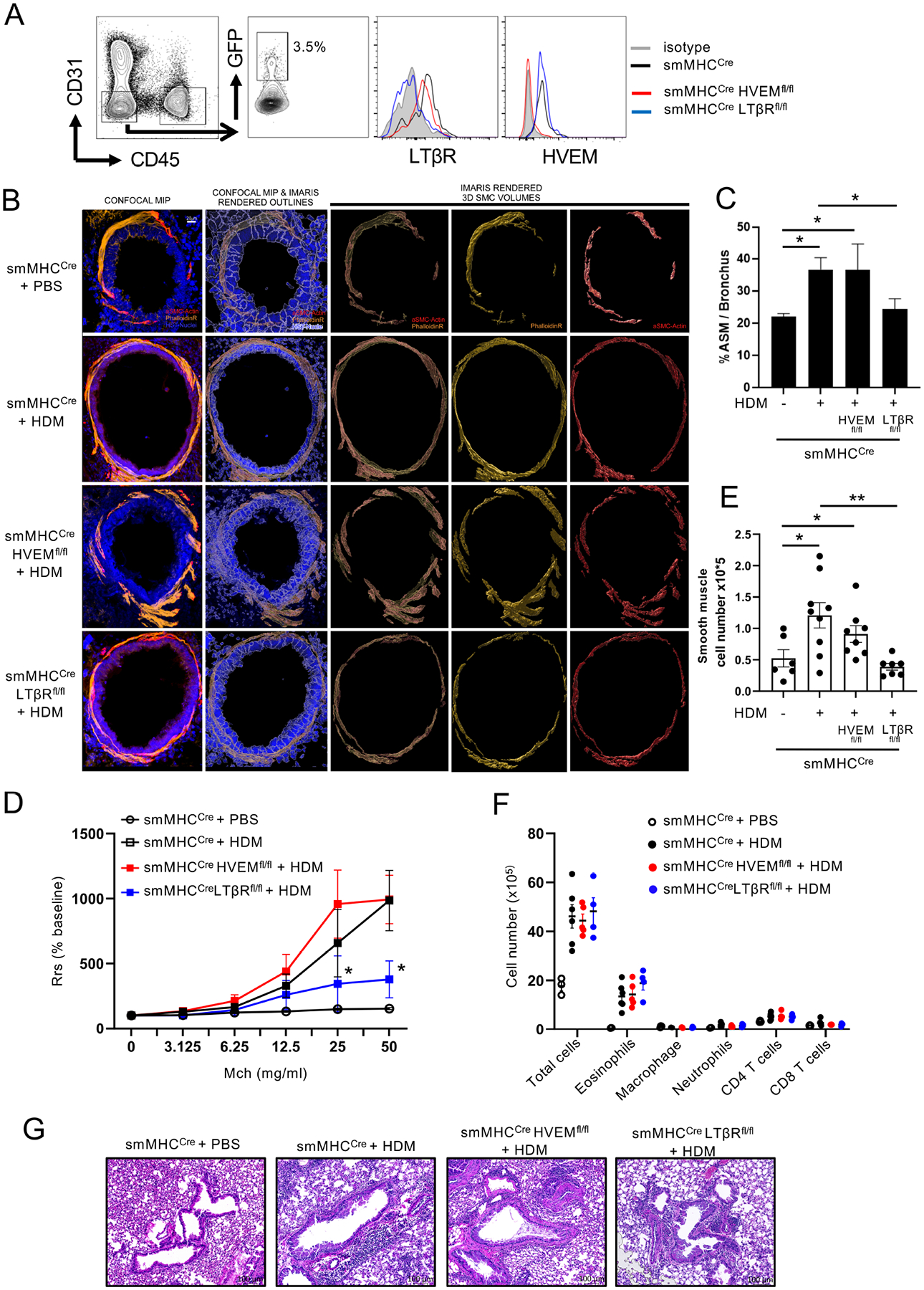
(A) HVEM and LTβR expression on ASM (GFP+) from smMHCCre(eGFP), smMHCCreHVEMfl/fl or smMHCCreLTβRfl/fl mice. Data from 3 experiments. (B-G) smMHCCre, smMHCCreHVEMfl/fl, or smMHCCreLTβRfl/fl mice were given intranasal challenges of HDM extract over 6 weeks (n = 4–6/group). (B) Confocal analysis of lung bronchi (scale 20 μm). F-actin (Phalloidin, yellow), alpha smooth muscle actin (red), DAPI (blue). Data representative of 3 experiments. (C) Quantitation of ASM volume based on phalloidin staining around the bronchi from (B) (n = 6–9/group, 10–15 tertiary bronchi per mouse). Data representative of 3 experiments. (D) Airway resistance (AHR) after methacholine challenge, measured by Flexivent (n = 5–8/group). Data representative of two experiments. *P < 0.05, smMHCCre vs. LTβRfl/fl. (E) Flow quantitation of total lung tissue smooth muscle cells (ASM). Data points, individual mice. (F) Flow quantitation of total lung inflammatory/immune cells. Data points, individual mice. (G) Representative H&E stained lung sections (scale 100 μm). n =4–6 mice/group. Data representative of 3 experiments. Means ± SEM. *P < 0.05, **P < 0.01.
The conditional knockout mice and control animals (smMHCCre) were then exposed to chronic repetitive intranasal challenges with house dust mite allergen extract over 6 weeks in a model of asthma that results in airway remodeling. Similar to what we previously reported16, the allergen drove an eosinophilic inflammatory response in the lungs of control animals, accompanied by a strong increase in bronchial ASM mass, as visualized and quantified by high-resolution 3-dimensional confocal microscopy of the immunofluorescent staining of αSMA and phalloidin (Filamentous actin). These changes were associated with profound airway hyperresponsiveness, reflected by increased lung resistance to methacholine when administered intranasally (Fig. 2B–G). Strikingly, we found that no significant increase in bronchial ASM mass was observed in ASM-specific LTβR-deficient mice compared to the approximate doubling in mass seen in control animals when quantified by histological analysis (Fig. 2B–C). In contrast, a normal smooth muscle response to allergen was found in mice specifically lacking HVEM in ASM. Importantly, AHR was strongly reduced in LTβR conditional knockout mice challenged with allergen whereas AHR was induced in HVEM conditional knockout mice comparable to control wild-type mice (Fig. 2D).
Flow cytometry analysis of dissociated lung tissue cells furthermore revealed another specific requirement for LTβR in controlling ASM. While the total number of smooth muscle cells was enhanced in the lungs of allergen-exposed smMHCCre and ASM-specific HVEM-deficient mice, no increase was found in LTβR-deficient mice (Fig. 2E). Moreover, demonstrating that this was not a bystander effect of a poor inflammatory response, the number of inflammatory cells, including eosinophils, that accumulated in the lung tissue, and the overall infiltrates around the bronchioles, as well as Th2 cytokine production in BAL fluid, were not different between the control and LTβR conditional knockout mice challenged with allergen (Fig. 2F–G, Fig. E2A–B). Mucus production determined by PAS staining of bronchial epithelium was also not different between the control and LTβR conditional knockout mice challenged with allergen (Fig. E2C).
Thus, LTβR controlled hyperplasia and hypertrophy of ASM in the context of chronic repetitive airway allergen exposure, and this strongly contributed to the development of AHR and lung dysfunction. Importantly, the latter was dissociated from generalized inflammation demonstrating the requirement for direct signals to smooth muscle cells.
We also assessed if LTβR played a role in AHR in an acute asthma model without chronic repetitive allergen exposure, where airway remodeling does not occur to any extent. In this case, only a weak AHR response resulted in control mice challenged with HDM. However, this was reduced in LTβR conditional knockout mice to the level seen in naïve unchallenged mice (Fig. E2D). Together, this suggests that the primary effect of LTβR on AHR is explained by its action on driving ASM remodeling, but that LTβR might also control ASM contractility in the absence of significant remodeling.
LIGHT-LTβR signals increase ASM mass and AHR in vivo
The above findings in allergen-induced airway remodeling correlated with the reduction in airway smooth muscle mass we found in LIGHT-deficient mice16. To directly relate the results to the activity of LIGHT as a cytokine, we then injected rLIGHT at a high dose intratracheally into unimmunized control and conditional knockout mice over 3 days. The smooth muscle layer was again quantified by high-resolution 3-dimensional confocal microscopy (Fig. E3A). Replicating our observations from two previous studies16, 24, rLIGHT administration to the lungs induced a significant increase in smooth muscle mass surrounding the bronchi in control mice (Fig. 3A–B). Importantly, this increased ASM mass was not observed in mice with conditional deletion of LTβR in ASM, whereas ASM specific-deletion of HVEM did not diminish the LIGHT-driven response compared to that in smMHCCre control mice (Fig. 3A–B).
Figure 3. LIGHT-LTβR signals increase airway smooth muscle mass and induce AHR in mice.
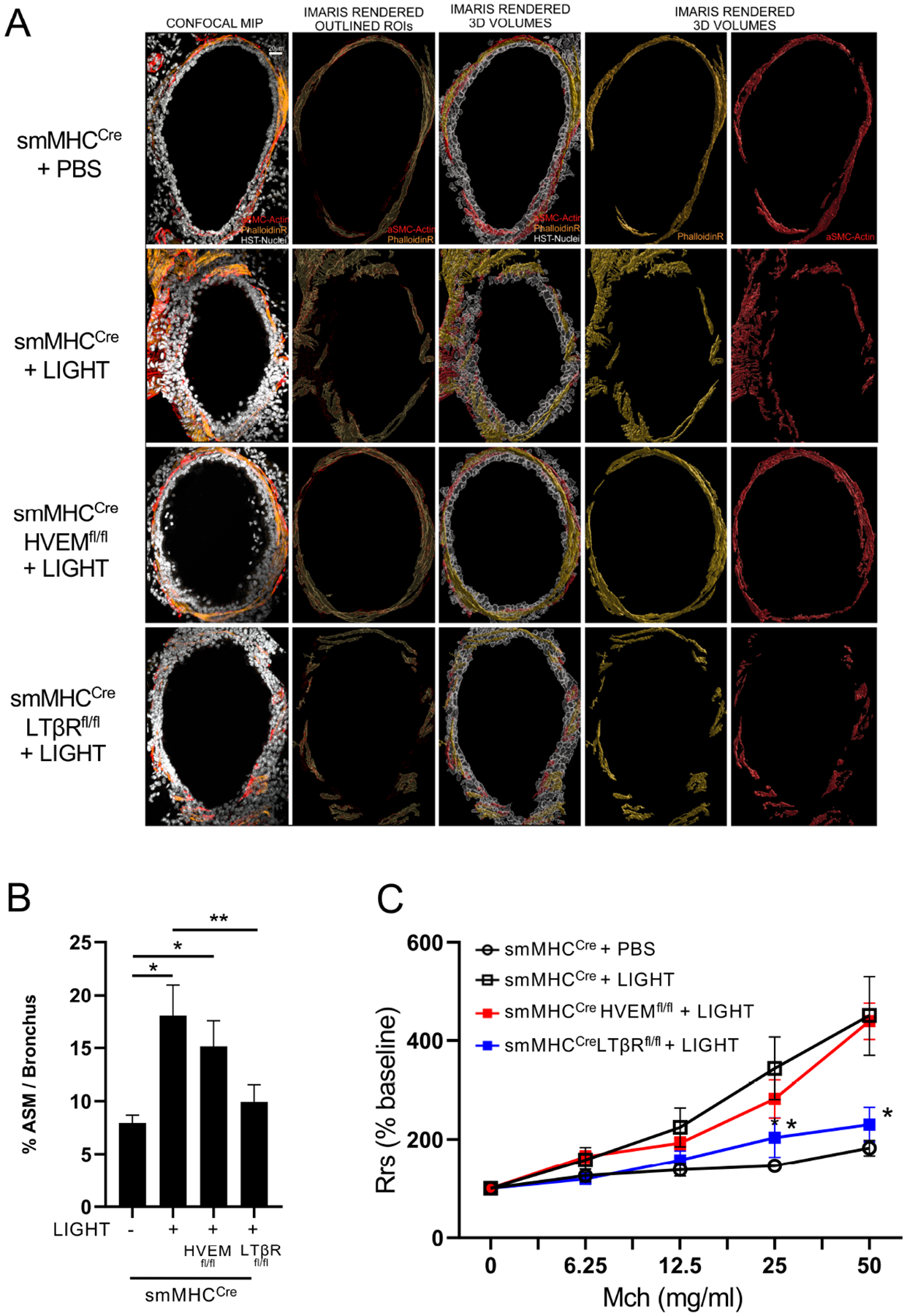
Mice received intratracheal recombinant LIGHT or PBS over 3 days. (A-B) Confocal microscopy of lung bronchial sections for expression of F-actin (Phalloidin, yellow) and α-smooth muscle actin (red). (A) Representative images of bronchi (scale 20 μm). (B) Imaris 3D imaging analysis quantification of ASM volume (phalloidin) around individual bronchi (n = 4–5 mice/group, 10–15 tertiary bronchi per mouse). Data means ± SEM and representative of 3 experiments. *P < 0.05, **P < 0.01. (C) Airway resistance (AHR) after methacholine challenge, measured by Flexivent (n = 4 mice/group). *P < 0.05, smMHCCre vs. LTβRfl/fl. Data means ± SEM and representative of 2 experiments.
To extend this data, we quantified the number of total lung smooth muscle cells by flow cytometry as before. In this case, intratracheal rLIGHT administration did not alter the number of ASM, as well as having no effect on promoting increased numbers of total cells and eosinophils in the lung tissue (Fig. E3B). Thus, the primary action of recombinant LIGHT in this setting in vivo was to promote ASM hypertrophy, suggesting that the LTβR-driven hyperplasia seen with repetitive allergen exposure (Fig. 2E) may have been dependent on other inflammatory factors elicited by the allergen acting together with LIGHT. However, regardless of the latter, rLIGHT injection still significantly induced AHR, albeit less than that see with repetitive allergen (Fig. 3C, compare to Fig. 2D). Most importantly, ASM-specific LTβR-deficient animals displayed strongly reduced AHR, unlike HVEM-deficient animals that showed similar AHR compared to the control mice treated with rLIGHT (Fig. 3C). These results collectively demonstrate that LIGHT can signal through LTβR on ASM in the lungs and directly drive smooth muscle hypertrophy and lung dysfunction (Fig. 3), and in the context of a response to allergen the LTβR signal is also critical and dominant for smooth muscle hyperplasia leading to even greater lung dysfunction associated with airway remodeling (Fig. 2).
LIGHT-LTβR interactions promotes contractile activity, hypertrophy, and hyperplasia in human ASM
To further investigate the direct actions of LIGHT-LTβR signals on airway smooth muscle cells, we analyzed normal human primary bronchial smooth muscle cells. Indicating that LTβR was active, it was downregulated after culture with LIGHT, a phenomenon previously associated with initiation of signaling through this receptor (Fig. 4A). The human ASM were then cultured in 3D collagen gels to make a net structure (Fig. 4B) and stimulated with LIGHT to assess contractile activity. Significantly, LIGHT induced strong activity in this assay shown by shrinking of the collagen gel, largely seen from 24–48 hours (Fig. 4C). In line with the notion that this was due to LTβR signaling, knockdown of this receptor with siRNA, but not knockdown of HVEM, prevented LIGHT-induced contractile activity (Fig. E4A–B). The membrane version of lymphotoxin, LTαβ, is also a ligand of LTβR. Recombinant LTαβ induced identical gel contraction compared to LIGHT (Fig. E4C), further supporting the dominant role of LTβR in driving smooth muscle cell activity.
Figure 4. LIGHT induces human ASM proliferation and contractile activity.
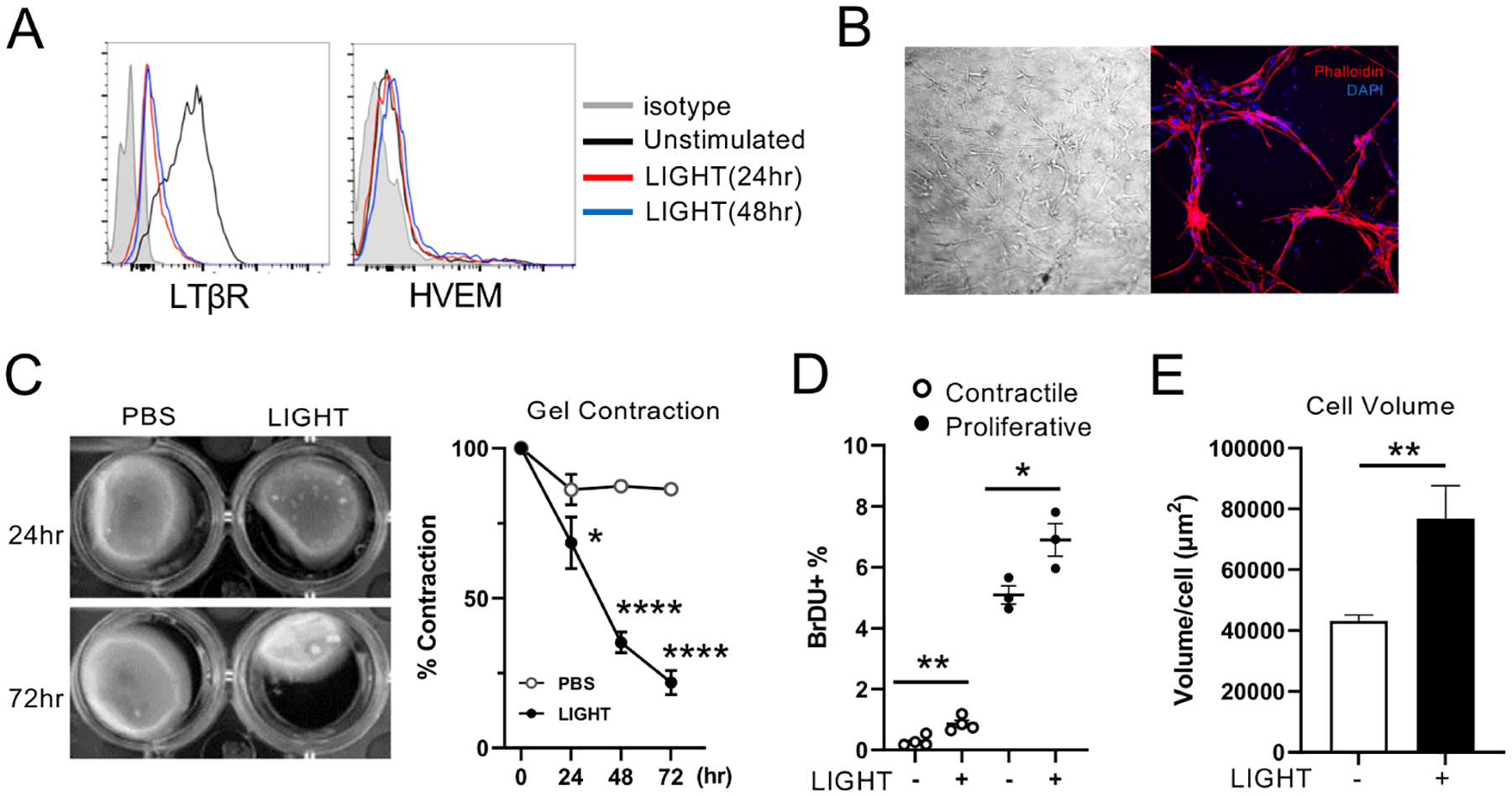
(A) HVEM and LTβR expression on human ASM assessed by flow after stimulation with LIGHT for 24 or 48hr. Similar data in 3 experiments. (B) Images of ASM in collagen 3D gels. Left: Bright field, Right: Phalloidin (red), DAPI (blue). (C) ASM collagen gel contraction at 24–72hr after stimulation with LIGHT. Data combined means from triplicates from 3 experiments. (D) Percentages of BrdU+ ASM stimulated with LIGHT for 48hr. Cells either first incubated in serum free media for 16 hours for proliferative ASM, or for 7 days for contractile ASM. Data means of triplicates and representative of 3 experiments. (E) ASM volume after stimulation with LIGHT, measured by confocal microscopy. 300–400 ASM, in two replicate experiments were analyzed. Data means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
We next assessed whether LIGHT-LTβR interactions can promote proliferation of ASM. Functional changes in cultured ASM have been studied previously with both contractile and synthetic/proliferative phenotypes observed dependent on culture conditions. Synthetic/proliferative cells can mature into contractile cells by prolonged serum deprivation2. We thus compared early passage human ASM with a proliferative phenotype to those with a contractile phenotype cultured without serum for 7 days. By assessing BrdU incorporation, proliferative ASM exhibited a high background rate of BrdU uptake (Fig. 4D), and this was enhanced significantly by LIGHT stimulation. Contractile ASM weakly incorporated BrdU, which was less strongly, albeit significantly, affected by LIGHT (Fig. 4D). Lastly, human ASM hypertrophy was assessed in vitro, and LIGHT significantly increased the volume of individual cells (Fig. 4E), again corresponding to the in vivo phenotype observed in the mouse (Figs. 2 and 3).
LIGHT increases actin polymerization in ASM
Contractile force in smooth muscle cells is generated by ATP-dependent interaction between F-actin and myosin, regulated in part by phosphorylation of myosin light chain by myosin light chain kinase (MLCK). In asthmatic patients, in addition to increased numbers of the contractile actin/myosin-expressing ASM subset, upregulation of MLCK and other proteins associated with MLCK activity has been reported5. Furthermore, cell hypertrophy is also directly related to several factors associated with contractility, including actin polymerization, as is migration of ASM, another process thought relevant to ASM mass changes in asthma35.
We therefore first assessed by RNA-seq whether LIGHT might promote responsiveness in ASM by regulating the expression of contractile molecules and signaling molecules associated with contraction. We did not detect any upregulation of transcripts for such molecules including alpha smooth muscle actin, contractile myosin heavy chain 11 isoform, and RhoA (Fig. 5A). In contrast, by GSEA, we found significant enrichment by LIGHT of transcriptional machinery associated with the cell membrane and actin rearrangement (Fig. 5B). This led us to focus on the quantitative analysis of intracellular F-actin and vinculin expression which reflect cytoskeletal macromolecular changes associated with mechanical force, migration, and hypertrophy. By confocal imaging analysis, human ASM stimulated with LIGHT were found to significantly increase the volume of vinculin rich focal adhesions and polymerized F-actin rich stress fibers compared to unstimulated cells within 6–12 hr (Fig. 5C). Further in line with this, migration of ASM was enhanced by LIGHT stimulation in migration/wound assays of monolayers assayed at 12hr, correlating with increased expression of actin filaments (Fig. 5D). Thus, overall, these results suggested that actin polymerization might have been central to many or all of the activities of LTβR on driving ASM dysregulation.
Figure 5. LIGHT induces actin polymerization and migratory ability in human ASM.
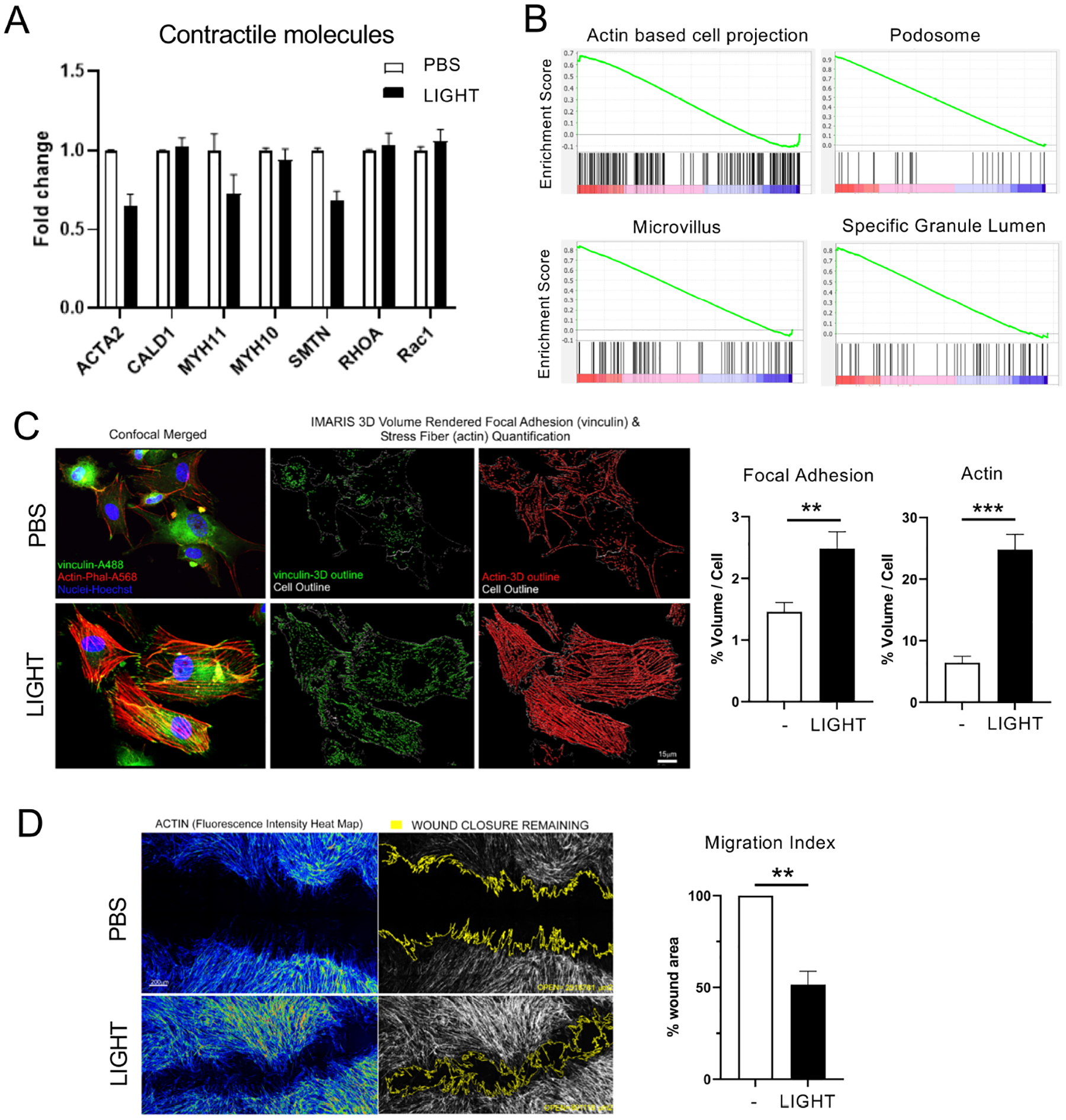
(A) RNA-seq analysis of contractile molecules in human ASM treated with LIGHT. Log2 fold change after 4hr compared to PBS treated cells. (B) GSEA of cell structure-related genes from RNA-seq of ASM stimulated with LIGHT compared to PBS for 4hr. (C) Confocal images of ASM treated with LIGHT for 6hr and stained with Phalloidin (red), Vinculin (green), DAPI (blue). Graphs show volume of F-actin (phalloidin) and focal adhesions (vinculin) normalized to individual cell sizes (scale 15 μm). 300–400 ASM, in two replicate experiments were analyzed. (D) Images of wounded monolayers of ASM stimulated with LIGHT for 12hr; left, fluorescence intensity of F-actin shown (yellow to red); right, remaining wound highlighted in yellow outline. Percentage of wound area remaining after LIGHT stimulation compared to PBS (scale 200 μm). Data combined from 3 independent experiments. Data means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001.
LIGHT-induced non-canonical NF-κB signaling induces actin polymerization in ASM
As LTβR signals are known to induce both early onset canonical NF-κB activation, as well as late onset non-canonical NF-κB activation, in other cell types, we asked if one or both of these pathways were activated by LIGHT in human ASM. Indeed, LIGHT stimulation induced a small increase in phosphorylation of p65 (canonical NF-κB) after 15 mins. However, most interestingly, LIGHT resulted in processing of p100 to p52 (the signature of non-canonical NF-κB pathway activation) starting ~4 hrs after stimulation and extending to 24 hrs (Fig. 6A). We then assessed whether LIGHT-induced non-canonical NF-κB signaling was required for actin rearrangement and contractile activity using NIK-SMI, an inhibitor of NF-κB-inducing kinase (NIK), the kinase that is central to p100 processing into p5236. Treatment with NIK-SMI significantly reduced LIGHT-induced actin polymerization although it did not block the increase in focal adhesions (Fig. 6B). Moreover, NIK-SMI also effectively prevented LIGHT-induced contractile activity visualized in 3D collagen gels, whereas in contrast, BAY11–7082, a canonical NF-κB inhibitor, had no effect on LIGHT-induced gel contraction (Fig. 6C). Knockdown of NIK (Map3k14) with siRNA further supported the inhibitor studies and prevented LIGHT-driven gel contraction (Fig. E4A–B). Notably, non-canonical signaling, initiated from 4hr and sustained at least up to 24hr (Fig. 6A), preceded the initiation of contraction at 24hr (Fig. 4C and E4), and preceded or coincided with changes in actin polymerization and migration related to actin polymerization (Fig. 5C and D), reinforcing the conclusion regarding the relevance of this pathway from the NIK inhibition assays. Thus, LIGHT-LTβR dependent non-canonical NF-κB signaling is central to dysregulation of ASM. Moreover, distinguishing LIGHT from other cytokines reported to be capable of acting on ASM, namely IL-13, TNF, and IL-17, none of these cytokines induced non-canonical NFκB activation in ASM (p52 processing), even though canonical NF-κB (pp65) was induced, particularly by TNF, as previously reported (Fig. E5).
Figure 6. LIGHT-dependent non-canonical NF-κB signaling induces actin polymerization and ASM contractility.
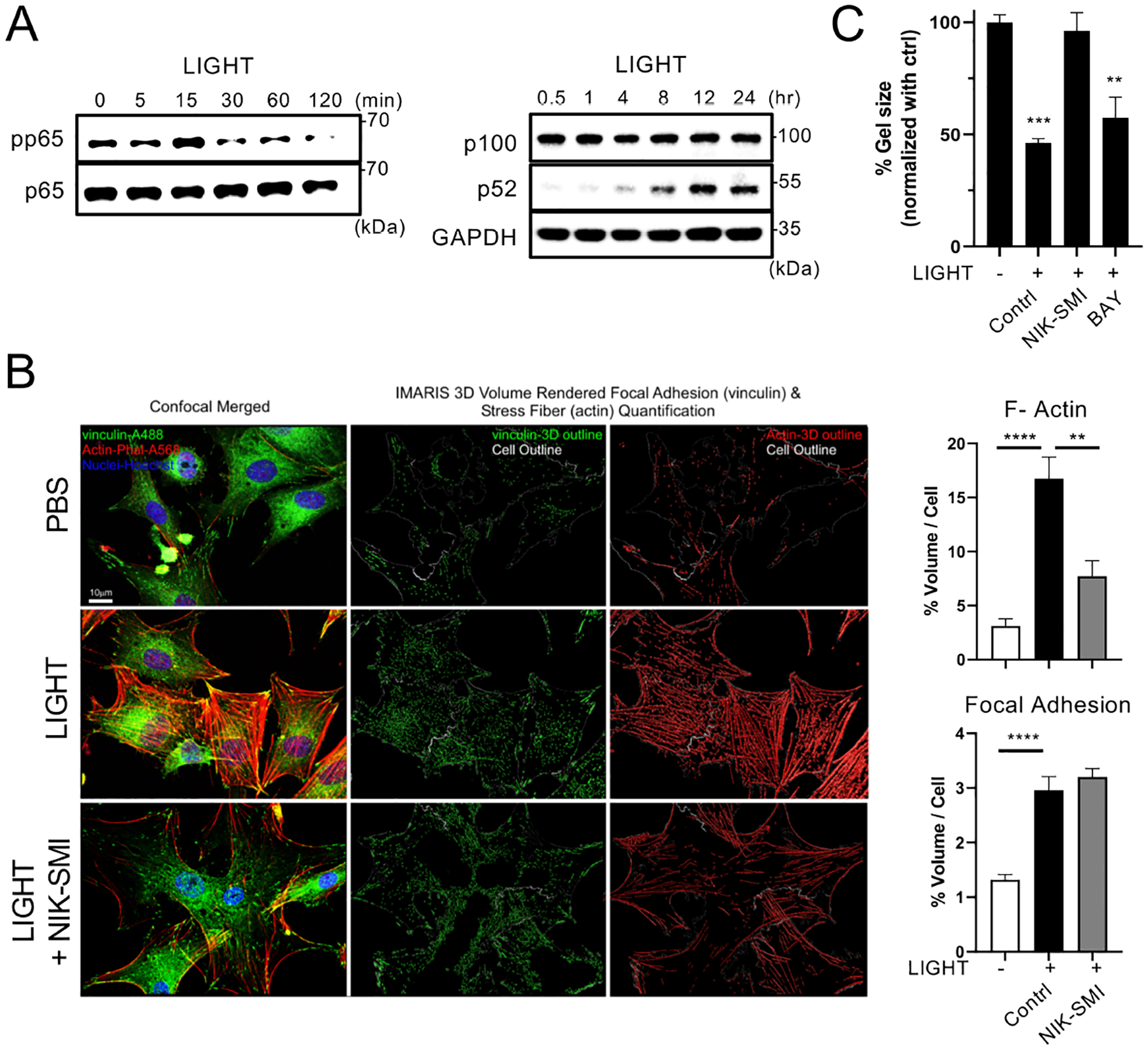
(A) Activation of canonical NF-κB (pp65) and non-canonical NF-κB (processing of p100 to p52) assessed in human ASM stimulated with LIGHT over 120 mins or 24hr. Data representative of 3 experiments. (B) Confocal analysis of ASM treated with LIGHT with or without NIK-SMI, an inhibitor of non-canonical NF-κB. Volume of phalloidin and vinculin normalized to cell size from triplicates (scale 10 μm). 300–400 ASM, in two replicate experiments were analyzed. (C) Gel contraction assay of LIGHT-stimulated ASM with or without inhibitors of canonical (BAY11–7082) and non-canonical (NIK-SMI) NF-κB. Percentage gel contraction compared to PBS in triplicates. Data representative of 2 experiments. Data means ± SEM. **P < 0.01, ***P < 0.001, ****P < 0.0001.
LIGHT induces actin polymerization and sustained phosphorylation of MLC via non-canonical NF-κB activation of Rac1/PAK1
To further understand the role of non-canonical NF-κB signaling, we examined other changes associated with activity in ASM. Phosphorylation of myosin light chain (MLC) is regulated directly by MLC kinase and MLC phosphatase, and a Rho-dependent pathway inhibits MLC phosphatase whereas calcium dependent calmodulin signaling activates MLC kinase5, 37. LIGHT promoted phosphorylation of MLC in human ASM within 5 minutes but importantly it sustained MLC phosphorylation over 24 hrs (Fig. 7A). We also detected late and sustained phosphorylation of the serine threonine kinase PAK1 (p21-activated kinase 1) over 24 hrs, that is a target of the GTP binding protein Rac1, and has been implicated in cytoskeleton/actin reorganization as well as being upstream of MLC kinase (Fig. 7B). Consistent with this, the active (GTP) form of Rac1, bound to PAK1, was detected between 1–4 hours after LIGHT stimulation (Fig. 7C). We also assessed phosphorylation of MYPT (a subunit of MLC phosphatase) downstream of the Rho-dependent pathway38, but in this case LIGHT induced only early transient and weak phosphorylation of MYPT at 15 mins (Fig. 7B).
Figure 7. LIGHT induces non-canonical NF-κB-dependent activation of Rac1/PAK1 and phosphorylation of MLC in ASM.
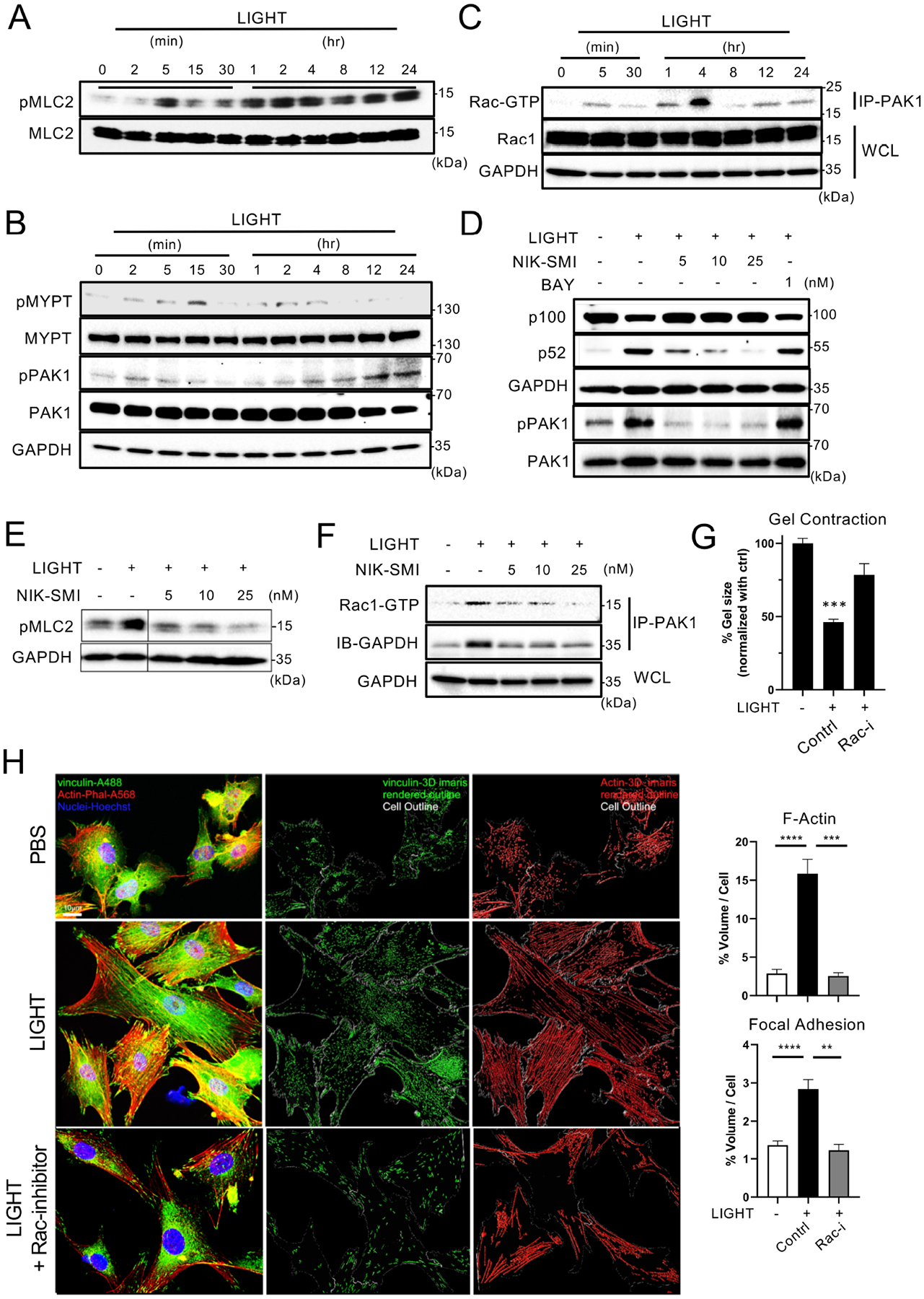
(A-C) pMLC2 (A), pMYPT and pPAK1 (B), and active Rac1-GTP immunoprecipitated with PAK1 (C) in human ASM stimulated with LIGHT for the indicated times. (D-E) Processing of p52, and pPAK1 (D) and pMLC2 (E) in ASM stimulated with LIGHT for 12hr, with or without inhibitors of canonical (BAY) and non-canonical NF-κB (NIK-SMI). (F) Active Rac1-GTP immunoprecipitated with PAK1 in ASM stimulated with LIGHT for 4hr with or without NIK-SMI. All data in A-F representative of 3 experiments. (G) Gel contraction of ASM stimulated with LIGHT, with or without an inhibitor of Rac1. Percentage gel contraction compared to PBS in triplicates. (H) Confocal analysis of ASM treated with LIGHT with or without a Rac1 inhibitor. Volume of phalloidin and vinculin normalized to cell size from triplicates (scale 10 μm). 300–400 ASM, in two replicate experiments were analyzed. Data representative of 2 experiments. Data means ± SEM. **P < 0.01, ***P < 0.001, ****P < 0.0001.
Since PAK1 has been reported to interact with NIK39, we hypothesized that the late phosphorylation of MLC and activation of Rac1/PAK1 was also related to non-canonical NF-κB signaling. Indeed, LIGHT-induced phosphorylation of PAK1 (Fig. 7D) and sustained phosphorylation of MLC (Fig. 7E) was reduced by NIK inhibition in a dose-dependent manner. Correspondingly, inhibiting NIK also prevented LIGHT from promoting the binding of active Rac1 to PAK1 (Fig. 7F). Lastly, to further consolidate the significance of this signaling pathway to ASM dysfunction, we found that a Rac1 inhibitor blocked LIGHT-induced contraction in 3D gels (Fig. 7G). Correspondingly treatment with the Rac1 inhibitor reduced actin polymerization, and in this case also blocked the increase in focal adhesions driven by LIGHT (Fig. 7H).
Together with our in vivo data, these results indicate that LTβR-induced non-canonical NF-κB signaling is a primary and dominant driver of airway smooth muscle cell hyperresponsiveness.
DISCUSSION
Airway smooth muscle cells have long been considered to be central for lung dysfunction in asthma due to their ability to narrow the airways. However, whether there are dominant receptors on ASM that explain ASM remodeling that is seen in severe asthma and explain airway hyperresponsiveness has not been clear. Our present study now reveals the novel finding that LTβR is essential for allergen-driven ASM remodeling and the resultant increased airway hyperresponsiveness that is associated with remodeling. Importantly, to our knowledge, out of the primary cytokine receptors that have been associated with asthma and ASM activity, namely those for IL-13, TNF, and IL-17, this is the first study to show that deletion of a single cytokine receptor only on smooth muscle cells can strongly impact ASM deregulation and AHR in response to allergen in vivo. This correlates with the unique ability of LTβR to drive non-canonical NF-κB signaling, an activity not shared by these other cytokine receptors. The results have major implications for our understanding of the control of airway remodeling and hyperresponsiveness in asthma.
We favor the notion that LIGHT, rather than LTαβ the other ligand for LTβR, is the primary factor relevant for smooth muscle remodeling. Higher expression of LIGHT has been reported in the sputum of severe asthmatic patients17–19, and in previous studies of LIGHT-deficient mice we showed that this cytokine participated in remodeling of the ASM mass that was induced by repetitive exposure to allergen16. LIGHT-deficient mice were protected from ASM remodeling at a similar level as WT mice treated with LTβR.Ig that blocks both LIGHT and LTαβ16, further implying that at least in our mouse model systems LIGHT is the primary ligand. However, it is possible that LTαβ could be important in human asthmatics as it is capable of inducing the same signals as LIGHT through the LTβR, and thus we cannot definitively rule out a contribution of LTαβ.
Increased ASM mass has previously been related to the severity and baseline lung function in patients with asthma3 with ASM hyperplasia and an increased number of bronchial ASM being well established as features of asthmatic airways35. Although ASM hypertrophy in asthma is less discussed35, asthmatic bronchial biopsies have been found to contain smooth muscle cells of larger diameter compared to control subjects, and severe asthmatics presented with the highest smooth muscle cell size40. Our data here show that LIGHT-LTβR promoted both ASM hyperplasia and hypertrophy in vivo and in vitro. LIGHT drove only moderate proliferation in cell culture but LTβR signals to ASM were essential for the increased number of lung smooth muscle cells seen in vivo in response to allergen. In addition, LIGHT directly promoted an increase in ASM size in vitro accompanied by increased actin polymerization, and in vivo, recombinant LIGHT administration without other inflammatory signals increased peribronchial ASM mass without affecting the total number of lung ASM. Thus, these observations strongly imply that LTβR signals could be central to both smooth muscle cell hyperplasia and hypertrophy seen in asthma.
The finding that LTβR activates the non-canonical NF-κB pathway in ASM and drives sustained changes in actin polymerization and rearrangement distinguishes it from the other primary cytokine receptors that have been described to also induce ASM responses in vitro, namely those for IL-13, TNF, and IL-1741. Non-canonical NF-κB signaling is a late-onset and more continual inflammatory signal reliant on recruitment of the NF-κB-inducing kinase, NIK. TNF via canonical NF-κB was found to induce activation of RhoA in ASM leading to inhibition of MLC phosphatase9, 42. IL-13 was shown to upregulate the expression of RhoA or enhance Ca2+ signaling dependent on STAT6, MAPK, and PI3K activity7, 43–46. Moreover, IL-17 was also found to drive migration, contraction, or other activities in ASM, dependent on several MAPK pathways, canonical NF-κB, and PKC47–49. Thus, while these cytokine receptors might then promote other signaling pathways compared to LTβR, or in the case of the receptors for TNF and IL-17 impact canonical NF-κB activation similar to LTβR, they have not been described to activate NIK, and we show they do not drive p100 processing to p52 in ASM. Therefore, they are more likely to trigger early and transient activities compared to LTβR.
Collectively, this suggests that LTβR signaling can differ from the actions of these other cytokine receptors in engendering sustained increases in ASM mass and a contractile phenotype. This could explain the dominant effect of deleting the LTβR in smooth muscle cells that we observed in vivo. Our data however do not rule out important contributions for these other cytokine receptors on ASM in asthma, simply that LTβR signaling in the context of allergen is essential. In fact, recent results with mice with conditional deletion of IL-4Rα in both epithelial cells and smooth muscle cells supports a role for IL-13 and IL-4R in control of ASM albeit dependent on epithelial cell activity50. We suggest that LIGHT might act on ASM in concert with these other cytokines, dependent on their production and availability within the lung, given that IL-13, IL-17 and TNF are also products of T cells and other inflammatory cells that can make LIGHT. We previously reported that LIGHT stimulated IL-13 production from eosinophils in experimental asthma models16, and that injection of LIGHT into the naive lungs also upregulated IL-13, potentially from ILC, coincident with a partial requirement for IL-4Rα in promoting ASM mass24, both observations that are in line with LIGHT working together with IL-13 to amplify smooth muscle responsiveness. Whether LIGHT co-operates with TNF or IL-17 to regulate ASM activities has not been shown yet, but it will be interesting in future studies to fully understand the extent of cross-talk and synergy between these cytokines on ASM.
In summary, our study reveals direct evidence that LTβR, most likely activated by LIGHT, can drive ASM hypertrophy, hyperplasia, and contractile activity, and is essential for airway hyperreactivity that results after chronic exposure to airway allergen. The data suggest that signaling through LTβR is critically required for smooth muscle changes and AHR regardless of the potential for other cytokines to be active, as shown by the almost complete lack of ASM hypertrophy, hyperplasia, and airway hyperresponsiveness in allergen-challenged mice where LTβR was specifically deleted in smooth muscle cells. Existing asthma treatments largely target inflammatory cells such as eosinophils or mast cells, and airway constriction is only transiently controlled by beta agonist inhalers. Blocking LIGHT-LTβR interactions, and possibly LTαβ-LTβR interactions, or disrupting non-canonical NF-κB signaling in smooth muscle cells, could represent important therapeutic approaches for persistent and chronic airway hyperresponsiveness in severe asthmatic patients or patients with similar lung dysfunction.
Supplementary Material
Key Messages:
LTβR interactions on smooth muscle cells in vivo control airway smooth muscle mass and airway hyperresponsiveness driven by inhaled allergen
LIGHT-LTβR non-canonical NF-κB signaling promotes contractile activity, hyperplasia and hypertrophy in airway smooth muscle cells
Acknowledgements
This study was supported by NIH grant AI070535 to MC.
HM was supported by an JSPS Overseas Research Fellowship, and JSPS KAKENHI Grant Number JP22K20924.
We would like to thank Jaqueline Miller for technical support, Richard Kurten for providing post-mortem lung slices for ASM isolation, Alexa Pham for assistance with ASM isolation, and Mitch Kronenberg for originally providing HVEM-floxed mice.
Abbreviations
- HDM
house dust mite
- ASM
airway smooth muscle cells
- AHR
airway hyperresponsiveness
- LIGHT
homologous to lymphotoxin, exhibits inducible expression and competes with HSV glycoprotein D for binding to HVEM, a receptor expressed on T lymphocytes
- HVEM
herpes virus entry mediator
- LTβR
lymphotoxin beta receptor
- LTαβ
lymphotoxin alpha beta
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest statement: M.C has patents related to LIGHT and lung inflammation.
References
- 1.Busse WW, Lemanske RF Jr. Asthma. N Engl J Med 2001; 344:350–62. [DOI] [PubMed] [Google Scholar]
- 2.Zuyderduyn S, Sukkar MB, Fust A, Dhaliwal S, Burgess JK. Treating asthma means treating airway smooth muscle cells. Eur Respir J 2008; 32:265–74. [DOI] [PubMed] [Google Scholar]
- 3.Pepe C, Foley S, Shannon J, Lemiere C, Olivenstein R, Ernst P, et al. Differences in airway remodeling between subjects with severe and moderate asthma. J Allergy Clin Immunol 2005; 116:544–9. [DOI] [PubMed] [Google Scholar]
- 4.Kaminska M, Foley S, Maghni K, Storness-Bliss C, Coxson H, Ghezzo H, et al. Airway remodeling in subjects with severe asthma with or without chronic persistent airflow obstruction. J Allergy Clin Immunol 2009; 124:45–51. e1–4. [DOI] [PubMed] [Google Scholar]
- 5.Mahn K, Ojo OO, Chadwick G, Aaronson PI, Ward JP, Lee TH. Ca(2+) homeostasis and structural and functional remodelling of airway smooth muscle in asthma. Thorax 2010; 65:547–52. [DOI] [PubMed] [Google Scholar]
- 6.Lee JH, Kaminski N, Dolganov G, Grunig G, Koth L, Solomon C, et al. Interleukin-13 induces dramatically different transcriptional programs in three human airway cell types. Am J Respir Cell Mol Biol 2001; 25:474–85. [DOI] [PubMed] [Google Scholar]
- 7.Tliba O, Deshpande D, Chen H, Van Besien C, Kannan M, Panettieri RA Jr., et al. IL-13 enhances agonist-evoked calcium signals and contractile responses in airway smooth muscle. Br J Pharmacol 2003; 140:1159–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Amrani Y, Panettieri RA, Jr., Frossard N, Bronner C. Activation of the TNF alpha-p55 receptor induces myocyte proliferation and modulates agonist-evoked calcium transients in cultured human tracheal smooth muscle cells. Am J Respir Cell Mol Biol 1996; 15:55–63. [DOI] [PubMed] [Google Scholar]
- 9.Hunter I Tumor Necrosis Factor-alpha -Induced Activation of RhoA in Airway Smooth Muscle Cells: Role in the Ca2+ Sensitization of Myosin Light Chain20 Phosphorylation. Molecular Pharmacology 2003; 63:714–21. [DOI] [PubMed] [Google Scholar]
- 10.Chang Y, Al-Alwan L, Risse PA, Roussel L, Rousseau S, Halayko AJ, et al. TH17 cytokines induce human airway smooth muscle cell migration. J Allergy Clin Immunol 2011; 127:1046–53. e1–2. [DOI] [PubMed] [Google Scholar]
- 11.Kudo M, Melton AC, Chen C, Engler MB, Huang KE, Ren X, et al. IL-17A produced by alphabeta T cells drives airway hyper-responsiveness in mice and enhances mouse and human airway smooth muscle contraction. Nat Med 2012; 18:547–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wechsler ME, Ford LB, Maspero JF, Pavord ID, Papi A, Bourdin A, et al. Long-term safety and efficacy of dupilumab in patients with moderate-to-severe asthma (TRAVERSE): an open-label extension study. Lancet Respir Med 2022; 10:11–25. [DOI] [PubMed] [Google Scholar]
- 13.Wenzel S, Ford L, Pearlman D, Spector S, Sher L, Skobieranda F, et al. Dupilumab in persistent asthma with elevated eosinophil levels. N Engl J Med 2013; 368:2455–66. [DOI] [PubMed] [Google Scholar]
- 14.Kirstein F, Horsnell WG, Kuperman DA, Huang X, Erle DJ, Lopata AL, et al. Expression of IL-4 receptor alpha on smooth muscle cells is not necessary for development of experimental allergic asthma. J Allergy Clin Immunol 2010; 126:347–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Perkins C, Yanase N, Smulian G, Gildea L, Orekov T, Potter C, et al. Selective stimulation of IL-4 receptor on smooth muscle induces airway hyperresponsiveness in mice. J Exp Med 2011; 208:853–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Doherty TA, Soroosh P, Khorram N, Fukuyama S, Rosenthal P, Cho JY, et al. The tumor necrosis factor family member LIGHT is a target for asthmatic airway remodeling. Nat Med 2011; 17:596–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hastie AT, Moore WC, Meyers DA, Vestal PL, Li H, Peters SP, et al. Analyses of asthma severity phenotypes and inflammatory proteins in subjects stratified by sputum granulocytes. J Allergy Clin Immunol 2010; 125:1028–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Frossing L, Silberbrandt A, Von Bulow A, Kjaersgaard Klein D, Ross Christensen M, Backer V, et al. Airway gene expression identifies subtypes of type 2 inflammation in severe asthma. Clin Exp Allergy 2021; 52:59–69. [DOI] [PubMed] [Google Scholar]
- 19.Hirano T, Matsunaga K, Oishi K, Doi K, Harada M, Suizu J, et al. Abundant TNF-LIGHT expression in the airways of patients with asthma with persistent airflow limitation: Association with nitrative and inflammatory profiles. Respir Investig 2021; 59:651–60. [DOI] [PubMed] [Google Scholar]
- 20.da Silva Antunes R, Madge L, Soroosh P, Tocker J, Croft M. The TNF Family Molecules LIGHT and Lymphotoxin alphabeta Induce a Distinct Steroid-Resistant Inflammatory Phenotype in Human Lung Epithelial Cells. J Immunol 2015; 195:2429–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.da Silva Antunes R, Mehta AK, Madge L, Tocker J, Croft M. TNFSF14 (LIGHT) Exhibits Inflammatory Activities in Lung Fibroblasts Complementary to IL-13 and TGF-beta. Front Immunol 2018; 9:576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Manresa MC, Chiang AWT, Kurten RC, Dohil R, Brickner H, Dohil L, et al. Increased Production of LIGHT by T Cells in Eosinophilic Esophagitis Promotes Differentiation of Esophageal Fibroblasts Toward an Inflammatory Phenotype. Gastroenterology 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Herro R, Shui JW, Zahner S, Sidler D, Kawakami Y, Kawakami T, et al. LIGHT-HVEM signaling in keratinocytes controls development of dermatitis. J Exp Med 2018; 215:415–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Herro R, Da Silva Antunes R, Aguilera AR, Tamada K, Croft M. Tumor necrosis factor superfamily 14 (LIGHT) controls thymic stromal lymphopoietin to drive pulmonary fibrosis. J Allergy Clin Immunol 2015; 136:757–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Xin HB, Deng KY, Rishniw M, Ji G, Kotlikoff MI. Smooth muscle expression of Cre recombinase and eGFP in transgenic mice. Physiol Genomics 2002; 10:211–5. [DOI] [PubMed] [Google Scholar]
- 26.Seo GY, Shui JW, Takahashi D, Song C, Wang Q, Kim K, et al. LIGHT-HVEM Signaling in Innate Lymphoid Cell Subsets Protects Against Enteric Bacterial Infection. Cell Host Microbe 2018; 24:249–60.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang Y, Koroleva EP, Kruglov AA, Kuprash DV, Nedospasov SA, Fu YX, et al. Lymphotoxin beta receptor signaling in intestinal epithelial cells orchestrates innate immune responses against mucosal bacterial infection. Immunity 2010; 32:403–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pham AK, Miller M, Rosenthal P, Das S, Weng N, Jang S, et al. ORMDL3 expression in ASM regulates hypertrophy, hyperplasia via TPM1 and TPM4, and contractility. JCI Insight 2021; 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Fong V, Hsu A, Wu E, Looney AP, Ganesan P, Ren X, et al. Arhgef12 drives IL17A-induced airway contractility and airway hyperresponsiveness in mice. JCI Insight 2018; 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Himes BE, Koziol-White C, Johnson M, Nikolos C, Jester W, Klanderman B, et al. Vitamin D Modulates Expression of the Airway Smooth Muscle Transcriptome in Fatal Asthma. PLoS One 2015; 10:e0134057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kan M, Koziol-White C, Shumyatcher M, Johnson M et al. Airway Smooth Muscle-Specific Transcriptomic Signatures of Glucocorticoid Exposure. Am J Respir Cell Mol Biol 2019; 61:110–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tsukui T, Sun KH, Wetter JB, Wilson-Kanamori JR, Hazelwood LA, Henderson NC, et al. Collagen-producing lung cell atlas identifies multiple subsets with distinct localization and relevance to fibrosis. Nat Commun 2020; 11:1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zepp JA, Zacharias WJ, Frank DB, Cavanaugh CA, Zhou S, Morley MP, et al. Distinct Mesenchymal Lineages and Niches Promote Epithelial Self-Renewal and Myofibrogenesis in the Lung. Cell 2017; 170:1134–48.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kumar A, D’Souza SS, Moskvin OV, Toh H, Wang B, Zhang J, et al. Specification and Diversification of Pericytes and Smooth Muscle Cells from Mesenchymoangioblasts. Cell Rep 2017; 19:1902–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Bara I, Ozier A, Tunon de Lara JM, Marthan R, Berger P. Pathophysiology of bronchial smooth muscle remodelling in asthma. Eur Respir J 2010; 36:1174–84. [DOI] [PubMed] [Google Scholar]
- 36.Brightbill HD, Suto E, Blaquiere N, Ramamoorthi N, Sujatha-Bhaskar S, Gogol EB, et al. NF-kappaB inducing kinase is a therapeutic target for systemic lupus erythematosus. Nat Commun 2018; 9:179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sakai H, Suto W, Kai Y, Chiba Y. Mechanisms underlying the pathogenesis of hypercontractility of bronchial smooth muscle in allergic asthma. J Smooth Muscle Res 2017; 53:37–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Andre-Gregoire G, Dilasser F, Chesne J, Braza F, Magnan A, Loirand G, et al. Targeting of Rac1 prevents bronchoconstriction and airway hyperresponsiveness. J Allergy Clin Immunol 2018; 142:824–33 e3. [DOI] [PubMed] [Google Scholar]
- 39.Tong L, Tergaonkar V. Rho protein GTPases and their interactions with NFkappaB: crossroads of inflammation and matrix biology. Biosci Rep 2014; 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Benayoun L, Druilhe A, Dombret MC, Aubier M, Pretolani M. Airway structural alterations selectively associated with severe asthma. Am J Respir Crit Care Med 2003; 167:1360–8. [DOI] [PubMed] [Google Scholar]
- 41.Lauzon AM, Martin JG. Airway hyperresponsiveness; smooth muscle as the principal actor. F1000Res 2016; 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chiba Y, Ueno A, Shinozaki K, Takeyama H, Nakazawa S, Sakai H, et al. Involvement of RhoA-mediated Ca2+ sensitization in antigen-induced bronchial smooth muscle hyperresponsiveness in mice. Respir Res 2005; 6:4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Eum SY, Maghni K, Tolloczko B, Eidelman DH, Martin JG. IL-13 may mediate allergen-induced hyperresponsiveness independently of IL-5 or eotaxin by effects on airway smooth muscle. Am J Physiol Lung Cell Mol Physiol 2005; 288:L576–84. [DOI] [PubMed] [Google Scholar]
- 44.Moynihan B, Tolloczko B, Michoud MC, Tamaoka M, Ferraro P, Martin JG. MAP kinases mediate interleukin-13 effects on calcium signaling in human airway smooth muscle cells. Am J Physiol Lung Cell Mol Physiol 2008; 295:L171–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Farghaly HS, Blagbrough IS, Medina-Tato DA, Watson ML. Interleukin 13 increases contractility of murine tracheal smooth muscle by a phosphoinositide 3-kinase p110delta-dependent mechanism. Mol Pharmacol 2008; 73:1530–7. [DOI] [PubMed] [Google Scholar]
- 46.Chiba Y, Nakazawa S, Todoroki M, Shinozaki K, Sakai H, Misawa M. Interleukin-13 augments bronchial smooth muscle contractility with an up-regulation of RhoA protein. Am J Respir Cell Mol Biol 2009; 40:159–67. [DOI] [PubMed] [Google Scholar]
- 47.Chang Y, Al-Alwan L, Risse PA, Halayko AJ, Martin JG, Baglole CJ, et al. Th17-associated cytokines promote human airway smooth muscle cell proliferation. FASEB J 2012; 26:5152–60. [DOI] [PubMed] [Google Scholar]
- 48.Nakajima M, Kawaguchi M, Ota K, Fujita J, Matsukura S, Huang SK, et al. IL-17F induces IL-6 via TAK1-NFκB pathway in airway smooth muscle cells. Immun Inflamm Dis 2017; 5:124–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Bulek K, Chen X, Parron V, Sundaram A, Herjan T, Ouyang S, et al. IL-17A Recruits Rab35 to IL-17R to Mediate PKCalpha-Dependent Stress Fiber Formation and Airway Smooth Muscle Contractility. J Immunol 2019; 202:1540–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McKnight CG, Potter C, Finkelman FD. IL-4Ralpha expression by airway epithelium and smooth muscle accounts for nearly all airway hyperresponsiveness in murine allergic airway disease. Mucosal Immunol 2020; 13:283–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.