

SUMMARY
It is common to think about and depict biological processes as being governed by fixed pathways with specific components interconnected by concrete positive and negative interactions. However, these models may fail to effectively capture the regulation of cell biological processes that are driven by chemical mechanisms that do not rely absolutely on specific metabolites or proteins. Here, we illustrate how ferroptosis, a non-apoptotic cell death mechanism with emerging links to disease, may be best understood as a highly flexible mechanism that can be executed and regulated by many functionally related metabolites and proteins. The inherent plasticity of ferroptosis has implications for how to define and study this mechanism in healthy and diseased cells and organisms.
Keywords: ferroptosis, iron, pathway, lipid, PUFA, peroxidation, cell death, ROS
eTOC:
Ferroptosis is a non-apoptotic cell death modality that is difficult to represent as a single biochemical pathway. Dixon and Pratt suggest that ferroptosis is best conceived and represented as a highly flexible mechanism where diverse metabolites, proteins and other molecules can contribute to ferroptosis regulation in a highly interchangeable manner.
Graphical Abstract
Introduction
How we conceive biological processes influences how we think about and investigate them. Advances in molecular genetics during the 20th century fostered an understanding of gene regulation in terms of defined networks of elements interconnected by positive and negative regulatory relationships1,2. Meanwhile, biochemical analyses identified numerous enzyme/substrate relationships and protein-protein interactions3,4. Blending these elements, it is typical to represent the molecular pathways that govern different cellular processes as networks of binary and non-quantitative on/off interactions, with the line between genetics, biochemistry and cell biology often blurred, and the restrictions of printed text appearing to carve a particular mechanism in stone (Figure 1).
Figure 1. A typical pathway map for a biological process.

The regulation of biological processes is often conceived and summarized using pathway maps. In these maps, individual elements like proteins and metabolites are represented as colored shapes and connections between elements are depicted by positive (arrow) or negative (T bar) interactions. Here, different shapes and colors are used to indicate different notional types of molecules. The strength and importance of individual connections are not easily captured or specified in such maps.
Cell death is an important process necessary for normal development and homeostasis in many organisms. Apoptosis, necroptosis, pyroptosis, and ferroptosis are different forms of cell death, each activated in unique circumstances5. Ferroptosis is of growing interest because it contributes to disease pathology and because it might be possible to activate this mechanism to selectively kill cancer cells6. The defining feature of ferroptosis is the accumulation of membrane lipid peroxides to toxic levels7,8. This accumulation eventually alters the physical integrity of the plasma membrane and triggers osmolytic processes that burst the cell9. Endogenous and synthetic lipophilic radical trapping antioxidants (RTAs) such as vitamin E and ferrostatin-1 can prevent lipid peroxide accumulation and inhibit ferroptosis but not other forms of cell death, offering a unique operational definition of this process7,10.
The study of ferroptosis may be reaching an inflection point. A decade of investigation has yielded numerous insights. However, it is proving difficult to define a single ferroptosis mechanism11. The translation of basic findings to clinical practice also appears to be slow. Here, we argue that one explanation for these surprising facts is that ferroptosis does not follow the classic pathway logic that we have come to expect from studying other mechanisms. This in turn likely contributes to confusion about what ferroptosis is and how to characterize and manipulate this process in vivo. We propose that there is no universal ferroptosis pathway that can be defined to the level of specific individual metabolites and proteins. Instead, we propose new ways to think about ferroptosis as a highly flexible mechanism involving a constellation of related biochemical mechanisms. We hope this conception will stimulate new lines of basic research and aid translation of these findings.
Understanding ferroptosis: fixed pathway or flexible mechanism?
Lipid peroxidation per se is not synonymous with ferroptosis. Lipid peroxidation inevitably occurs at a basal rate in the membranes of mammalian cells12. When lipid peroxidation exceeds some unknown threshold at the plasma membrane11,13,14, this physical barrier between the inside and outside of the cell becomes irreversibly permeabilized and ferroptosis is said to have occurred. Dozens of metabolites and proteins contribute to the toxic accumulation of membrane lipid peroxides, including those involved in redox metabolism, iron homeostasis, lipid metabolism, signal transduction, and transcription6. Multiple organelles, including lysosomes, endoplasmic reticulum, the Golgi apparatus, and mitochondria, may contribute to the lipid peroxidation process6. An attempt to present key elements of the ferroptosis mechanism in a traditional manner is shown in Figure 2A. Beyond the extensive literature on the subject6, it is challenging to conceive of and depict ferroptosis as a single pathway for at least two fundamental reasons. First, many different metabolites and proteins seem sufficient to initiate or regulate ferroptosis, without any being necessary. Second, some metabolites and proteins influence ferroptosis sensitivity very differently (or not at all) depending on the type of cell and how ferroptosis is induced.
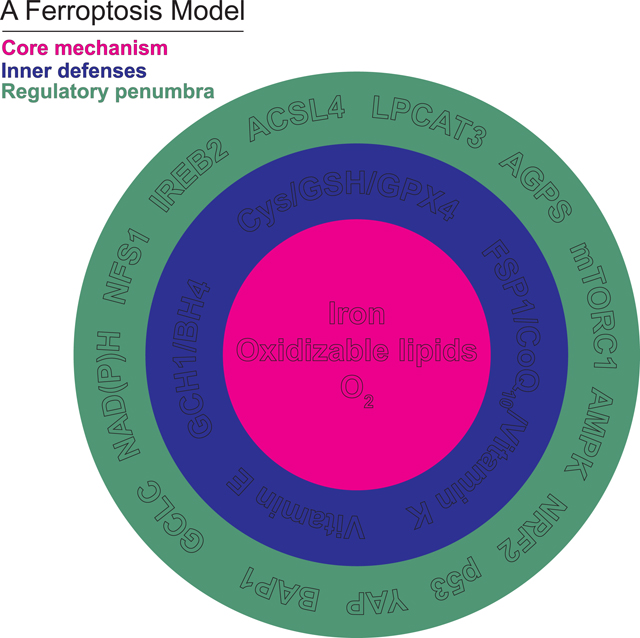
Figure 2. Old and new representations of ferroptosis.

(A) Several key elements of the ferroptosis mechanism, depicted using a traditional pathway map. This pathway map encompasses far less than is known about ferroptosis. Some key inputs are also not easily captured in this map due to conflicting evidence in the literature. This map also contains substantial redundancy, which makes it difficult to intuit how inhibition or activation of any one element modulates ferroptosis. Key small molecule inducers of ferroptosis are indicated in red. NOX: NAD(P)H oxidase, LOX: lipoxygenase, CI/CIII: complex I and complex III of the mitochondrial electron transport chain. POR: NAD(P)H-cytochrome P450 reductase. Notional peroxidized phospholipid acyl chains are denoted in red; purple indicates termination of the phospholipid peroxidation process.
(B) A new depiction of ferroptosis as a flexible mechanism involving a constellation of related biochemical mechanisms. The core mechanism of lipid peroxidation (magenta) involves iron, and different configurations of oxidizable lipids and lipid peroxidation initiating reactions and enzymes. The core lipid peroxidation mechanism is surrounded by several biochemically distinct inner defense mechanisms (blue). The core mechanism and the inner defenses are in turn modulated by a regulatory penumbra that may encompass dozens or hundreds of regulatory inputs, only some of which are depicted here (green).
(C) Mechanistic details of the core lipid peroxidation mechanism, illustrating the processes of (auto)initiation, propagation, and termination of the chain reaction. Key points of intervention by radical trapping antioxidants (RTAs), peroxidase mimics, and iron chelators, are indicated.
To address these challenges, we propose a view of ferroptosis that dispenses with fixed pathways and instead emphasizes flexibility, interchangeability, and mechanistic coherence at a level of abstraction beyond specific individual metabolites or proteins. We define a core mechanism that executes ferroptosis, a set of inner defenses that protect cells against ferroptosis, and a broader regulatory penumbra that influences ferroptosis sensitivity by modulating the core mechanism or the inner defenses (Figure 2B). The ferroptosis core mechanism is the process of membrane lipid peroxidation, which involves a series of chemical reactions between oxidizable lipids, O2 and iron. The inner defenses comprise enzymes, cofactors, and metabolites that guard against this toxic accumulation of membrane lipid peroxides. The regulatory penumbra includes signaling and metabolic pathways that impinge upon the core mechanism and/or the inner defenses to alter ferroptosis sensitivity positively or negatively, often in a context-specific manner. Below, we will elaborate on key elements of this model and provide evidence for flexibility at each level.
The ferroptosis core mechanism
Ferroptosis induction.
Ferroptosis requires – and is defined by – the overwhelming accumulation of lipid peroxides. Lipid peroxidation is a radical-mediated chain reaction that leads to the incorporation of molecular oxygen (O2) into lipids15 (Figure 2C). This process first involves the formation of a carbon-centered lipid radical on a polyunsaturated fatty acid (PUFA) acyl chain, such as those found in membrane phospholipids. The lipid radical then combines with O2 to yield a lipidperoxyl radical. The lipidperoxyl radical carries on the chain reaction by abstracting an H-atom from another lipid. Once the first H-atom is abstracted, the chain reaction can “auto-initiate” by reaction of a product lipid peroxide with labile (reduced) iron or iron-containing enzymes, to form a lipid-derived alkoxyl radical, which can continue the process by abstracting an H-atom from a neighboring PUFA chain (Figure 2B). In principle, once started by an initiating species, no further intervention is required to sustain the chain reaction. Direct interference with this lipid peroxidation mechanism by feeding cells deuterated (i.e., less-oxidizable) PUFAs is sufficient to prevent ferroptosis16.
Lipid peroxidation may be initiated in several ways and here we encounter the first evidence of mechanistic flexibility. Hydroxyl radical (HO●), formed from the reaction between hydrogen peroxide (H2O2) and labile (reduced) iron (the Fenton reaction), has been widely invoked as a lipid peroxidation-initiating species17. The more persistent hydroperoxyl radical (HOO●), the conjugate acid of superoxide (pKa ~ 5), may also serve as an initiating species for lipid peroxidation18. Indeed, O2●−/HOO● is formed in the vicinity of membrane phospholipids by multiple sources, including NAD(P)H oxidase (NOX) enzymes, oxidoreductases (e.g., POR, CYB5R1), and the mitochondrial electron transport chain (ETC). However, disrupting these individual enzymes, or disabling the entire mitochondrial ETC, has effects on ferroptosis that vary considerably between cells and contexts, from almost complete inhibition of cell death to minimal or no effect7,11,19–23. It is conceivable that each enzyme is sufficient to initiate ferroptosis, with none being necessary, and different configurations working in concert to initiate lipid peroxidation in different settings.
In addition to spontaneous chemical reactions, lipid peroxidation can be catalyzed in a regio- and stereospecific manner by lipoxygenase (LOX) enzymes16. A complex of LOX15 with PE binding protein 1 (PEBP1) promotes ferroptosis in some disease contexts16,24. However, genetic disruption of Alox15 in mice does not suppress ferroptosis in tissues where the key anti-ferroptosis enzyme Gpx4 (see below) has been deleted25. In human cancer cells, expression of the p53 tumor suppressor protein may sensitize cancer cells to ferroptosis through a mechanism that requires LOX12, but this is not universal26 (see below). Moreover, human cell lines that do not express any detectable LOX protein remain susceptible to ferroptosis, and transient overexpression of individual LOX enzymes only modestly sensitizes to this lethal process27. Thus, while LOX-catalyzed lipid peroxidation can initiate or perhaps accelerate lipid peroxidation, no single LOX enzyme appears necessary for ferroptosis in all cases.
The role of lipids in ferroptosis.
Peroxidation of PUFA-containing phospholipids (PUFA-PLs), as a class, may be a universal feature of ferroptosis and necessary for lethal membrane permeabilization. Mammalian cells contain dozens of distinct PUFA-PL species that vary by the nature of the head group and by the length and degree of unsaturation of the acyl chains28. PUFA-phosphatidylethanolamines (PUFA-PEs) represent an important target of oxidation during ferroptosis in fibroblasts, especially those PE species acylated with 20:4 (i.e., an acyl chain with 20 carbons and 4 double bonds) or 22:4 PUFAs29. However, oxidation of PUFA-containing phosphatidylinositols is predominant in other cells undergoing ferroptosis8. The loss of many PUFA-PEs, PUFA-phosphatidylcholines (PCs), and other diacyl phospholipids is also observed during ferroptosis and may be linked to cell death11,14,30,31. The oxidation of PUFA-containing ether-linked phospholipids (i.e., plasmalogens), which are distinct from diacyl phospholipids, may also drive ferroptosis in some – but not all – systems11,32–35. There is also evidence that ferroptosis-insensitive cells can sometimes be rendered ferroptosis-sensitive by incubation with any of several different PUFA free fatty acids21,36, and that different individual PUFA species alone may be sufficient to induce ferroptosis33,36,37. In sum, there may be no single PUFA-PL species whose oxidation is necessary for ferroptosis.
The role of iron in ferroptosis.
Iron can cycle between various oxidation states, donating or accepting electrons, thereby contributing to diverse redox reactions in the cell. Iron can participate in the core mechanism in at least two different ways. Mammalian cells contain a pool of intracellular iron (~10 μM) that is loosely coordinated to physiological ligands – the so-called labile iron pool (LIP)38. The LIP can contribute to spontaneous autoxidation and ferroptosis in some contexts39. The NOX and LOX enzymes that can help initiate lipid peroxidation (see above) also require iron or iron-containing cofactors to function. A role for iron in the core mechanism appears universal, although in principle other redox active metals like copper could conceivably generate lipid peroxides that drive ferroptosis under some circumstances. Also notable is that transcriptional and post-translational mechanisms respond dynamically to environmental factors or genetic alterations to change intracellular iron abundance and thereby influence ferroptosis sensitivity40,41. While iron is essential for ferroptosis in most or all cases, how iron specifically contributes to lethal membrane lipid peroxidation may easily differ between conditions.
The ferroptosis inner defenses
Several enzymes and metabolites function continuously to keep basal lipid peroxidation from spiraling destructively out of control and leading to ferroptosis. The glutathione-dependent phospholipid hydroperoxidase glutathione peroxidase 4 (GPX4) is a key selenoenzyme that protects against ferroptosis by converting potentially toxic lipid peroxides into non-toxic lipid alcohols42–45. In mice, genetic inactivation of Gpx4 is sufficient to cause rapid onset of ferroptosis in diverse tissues25,46. Likewise, humans born with severe GPX4 hypomorphic alleles suffer from debilitating developmental abnormalities that can be attributed to loss of control over membrane lipid peroxidation47. For unclear reasons, ferroptosis sensitivity varies by cell type and tissue. Thus, in mice a sub-set of GABAergic interneurons in the brain are most sensitive to partial Gpx4 inactivation43, while kidney cells are destroyed first by post-natal Gpx4 deletion25. In cultured cells, genetic GPX4 inactivation or covalent small molecule GPX4 inhibitors (e.g., RSL3, ML210) are potent inducers of ferroptosis in most – but not all – cells48–52. GPX4 is a glutathione (GSH)-dependent enzyme, and disruption of the system xc− antiporter responsible for the uptake of the GSH precursor cystine/cysteine (e.g., using the small molecule erastin) can also induce ferroptosis53.
Several non-peroxidase mechanisms operate in parallel to GPX4 to limit membrane lipid peroxidation and prevent ferroptosis. These mechanisms are generally based on the interception of propagating lipidperoxyl radicals by endogenous RTAs54 (Figure 2A,C). Here, beyond the fact that these systems operate in parallel to the GPX4-based mechanism, we encounter further mechanistic flexibility whereby several different RTAs appear capable of inhibiting membrane lipid peroxidation. The archetype cellular RTA is α-tocopherol, the most active form of vitamin E. Following its reaction with a lipidperoxyl radical, vitamin E can either combine with another lipidperoxyl radical or be regenerated by a water-soluble reductant, such as ascorbate. The generation and regeneration of reduced RTAs can also be enzyme-catalyzed. Ferroptosis suppressor protein 1 (FSP1) generates reduced forms of the endogenous electron carriers coenzyme Q10 (CoQ10) and vitamin K, which both possess significant RTA activity52,55,56. GTP cyclohydrolase I (GCH1) synthesizes a different RTA, tetrahydrobiopterin (BH4), which can be regenerated following its RTA reactions by dihydrofolate reductase31,57. FSP1 or GCH1 expression can render some cells highly resistant to GPX4 inhibitors31,52,55, demonstrating the parallel nature of these mechanisms. The expression of GPX4, FSP1, and other negative regulators of ferroptosis does not appear to be correlated between cells52. These redundant systems presumably act in concert to inhibit ferroptosis, with individual contributions that vary between cells and conditions.
We note that inactivation of GPX4 and other inner defense enzymes cannot increase lipid peroxidation and trigger ferroptosis if the core mechanism is incapable of generating sufficient lipid peroxides to begin with. For example, cells lacking GPX4 activity can survive and proliferate normally if an enzyme necessary for PUFA free fatty acid activation and PUFA-PL synthesis, acyl-CoA synthetase long chain family member 4 (Acsl4), is disrupted at the same time48,58. Incubation of cells with any of several different monounsaturated fatty acids (MUFA), which can displace more oxidizable PUFAs from membrane PLs, can also prevent lipid peroxidation and ferroptosis in cells lacking GPX4 function14,16,59,60. Expression of the phospholipase PLPL9 (PLA2G6) that cleaves oxidized PUFA acyl chains from membrane phospholipids is also sufficient to inhibit ferroptosis, even when GPX4 activity is lost61–63. In other words, the inner defenses are only needed to prevent ferroptosis in situations where runaway activation of the core mechanism is a threat in the first place: the inner defenses may not be needed to prevent ferroptosis when lipid peroxidation does not occur at high rates.
The ferroptosis regulatory penumbra
Ferroptosis sensitivity is governed by a regulatory penumbra that influences the likelihood that the core mechanism lethally breaches the inner defenses (Figure 2B). Any metabolite, protein, pathway, or organelle that regulates the core mechanism or the inner defenses has the potential to alter ferroptosis sensitivity and thereby contribute to the regulatory penumbra in one setting or another. Hundreds of entities that can modulate ferroptosis sensitivity by regulating what we are calling here the core mechanism and inner defenses have been catalogued to date64, including proteins that alter lipid or iron metabolism, signaling cascades that modulate enzyme function, and transcriptional regulators that govern the expression of these and other proteins. Upon close examination, many entities seem to play flexible roles in ferroptosis regulation that are hard to square with a universal ferroptosis mechanism11,34,35,57,65,66. Some specific examples are described, below.
Remarkably, different metabolites and proteins can have more or less of an effect on ferroptosis sensitivity depending on how ferroptosis is triggered57. For example, in cancer cells, inhibition of mRNA translation prevents ferroptosis triggered by system xc- inhibition (i.e., cystine deprivation) but not by direct GPX4 inhibition7,67,68. One model is that inhibition of mRNA translation specifically limits ferroptosis in response cystine deprivation because it allows for intracellular cysteine to be diverted away from protein synthesis and towards the synthesis of GSH that sustains downstream GPX4 activity69,70, although other explanations are possible. Adding further complexity, mutation-induced loss of hematopoetic stem cells in vivo appears to be caused by reduced synthesis of anti-ferroptotic proteins GPX4, SLC7A11, and proteins involved in iron storage71.
Context-specific effects are also observed with respect to the function of lipid metabolic enzymes. Here, disruption of ACSL4 potently suppresses ferroptosis in response to direct GPX4 inhibition but not in response to cystine deprivation11,57. The mechanistic explanation for this conditional effect is unclear. Direct GPX4 inhibition should inhibit only this enzyme. By contrast, cystine deprivation will limit the synthesis of the GPX4 cofactor GSH, and also prevent the synthesis of coenzyme A and iron/sulfur clusters, and elevate intracellular glutamate levels, all of which may contribute to ferroptosis40,72–75. Of note, ACSL4 is also not required when ferroptosis is induced by photodynamic therapy or by the combination of p53 expression and tert-butyl hydroperoxide26,76. Finally, LOX12 is essential for ferroptosis triggered by p53 expression and exposure to tert-butyl hydroperoxide but dispensable for ferroptosis induced by system xc− inhibition or direct GPX4 inhibition26. Thus, different lethal stimuli seem to require different configurations of the regulatory penumbra to effectively engage the core mechanism and induce ferroptosis.
Some further illustrations of complexity in the ferroptosis regulatory penumbra concern metabolites and pathways that appear to have highly distinct effects on ferroptosis regulation depending on cell types or other circumstances.
NAD(P)H.
NAD(P)H is required for the generation of reduced endogenous RTAs, like CoQ10, and for the regeneration of GSH (from GSSG) used by GPX4 (Refs.31,52,55,57). Indeed, NAD(P)H abundance generally correlates with ferroptosis resistance77. However, NAD(P)H also donates electrons to NOX and POR enzymes that can contribute to the initiation of lipid peroxidation. Furthermore, NAD(P)H may be needed for the synthesis of more oxidizable longer chain PUFAs from shorter chain precursors. Thus, NAD(P)H may both limit and promote ferroptosis (Figure 3A). Which effect dominates in one cell or another may vary. Indeed, inhibition of NAD(P)H synthesis suppresses ferroptosis in cells where NOX activity is important for ferroptosis initiation7 despite presumably also decreasing the abundance of various reduced RTAs. From the point of view of ferroptosis pathway construction, NAD(P)H synthesis presents additional complexity. NAD(P)H can be synthesized by the oxidative pentose phosphate pathway (oxPPP), malic enzyme 1 (ME1), and isocitrate dehydrogenase 1 (IDH1)78,79. These mechanisms are partially redundant, and the relative contribution of each pathway to total NAD(P)H synthesis varies by cell78,79. Consequently, all three mechanisms are likely to contribute to the ferroptosis regulatory penumbra in a cell-type specific way.
Figure 3. Examples of complexity and contradiction in ferroptosis regulation.

(A) Several different enzymes and pathways can contribute redundantly to synthesis of the reducing agent NAD(P)H, including the oxidative pentose phosphate pathway (oxPPP), malic enzyme 1 (ME1), and isocitrate dehydrogenase 1 (IDH1). NAD(P)H is used by enzymes that both promote and inhibit ferroptosis.
(B) Mechanistic target of rapamycin complex 1 (mTORC1) can regulate ferroptosis both positively and negatively through effects on the expression of glutathione peroxidase 4 (GPX4) or sterol response element binding protein 1 (SREBP1), or by diverting cysteine flux into protein synthesis that competes with the synthesis of glutathione.
(C) The p53 transcription factor can inhibit ferroptosis through transcriptional induction of CDKN1A or interaction with the dipeptidyl peptidase 4 (DPP4). P53 can also induce ferroptosis through transcriptional repression of SLC7A11; SLC7A11 protein can inhibit LOX12 activity through direct interaction.
The mTORC1 pathway.
The mechanistic target of rapamycin complex I (mTORC1) pathway can influence ferroptosis sensitivity in different ways (Figure 3B). In cystine-deprived fibrosarcoma cells, mTORC1 activity promotes ferroptosis, possibly by diverting cysteine away from GSH synthesis and towards cap-dependent mRNA translation70. Thus, genetic and pharmacological mTORC1 inhibition suppresses ferroptosis. By contrast, in renal carcinoma cells mTORC1 inhibits ferroptosis by promoting GPX4 protein expression80. In yet other cell types, mTORC1 inhibits ferroptosis by boosting the synthesis of anti-ferroptotic MUFA lipids81. These three mechanisms of mTORC1- dependent ferroptosis modulation are distinct, plausible, and contradictory. No universal role for mTORC1 is obvious, and one effect may dominate over another depending on the cell or situation.
The p53 pathway.
The tumor suppressor p53 regulates diverse processes in the cell, including redox metabolism82. In some cancer cells, expression of wild-type p53 sensitizes to ferroptosis induced by tert-butyl hydroperoxide (but not erastin or RSL3) by repressing the expression of SLC7A11 (encoding the antiporter subunit of system xc-), which in turn physically interacts with and inhibits LOX12 (Ref.26,83). Here, ferroptosis induction does not require ACSL4 but is suppressed by treatment with the synthetic RTA ferrostatin-1, implying that this combination (i.e., p53 expression + tert-butyl hydroperoxide treatment) can access the core lethal mechanism. In other cancer cells, stabilization of wild-type p53 suppresses ferroptosis through physical interaction with the protease DPP4 or by transcriptional induction of CDKN1A (p21), which can in turn inhibit GSH-dependent nucleotide synthesis84–86. Thus, p53 can seemingly either enhance or suppress ferroptosis through distinct mechanisms (Figure 3C), possibly depending on how ferroptosis is initiated. To add even greater complexity, in yet other cells p53 expression has little or no effect on ferroptosis sensitivity87.
Autophagy.
Genetic screening and targeted studies suggest that autophagy or the more specialized version, ferritinophagy, promotes ferroptosis by enhancing the supply of labile iron within the cell88,89. This is also a plausible mechanism to promote activation of the core mechanism and likely important in some cells. However, core autophagy genes are rarely identified in large-scale ferroptosis genetic suppressor screens, and targeted deletion of key autophagy genes like ATG7 have no effect on ferroptosis in some cells11,90. Whether autophagy is strictly necessary for ferroptosis may be dependent on the cell type.
Summarizing ferroptosis mechanistic flexibility
The above observations illustrate the challenge of understanding and depicting ferroptosis as a single mechanism. We have proposed above that this challenge can be met by allowing for the possibility that ferroptosis is governed by a flexible constellation of functionally interchangeable molecules (Figure 2B). We further envision that all cells fall somewhere across two general axes that correspond with the abstract quantities of “total lipid peroxidation susceptibility” and “total inner defense capacity”, which sum up the direct and indirect contribution of these different molecules to enhancing or suppressing the rate of lipid peroxidation in one setting (Figure 4A). In general, total lipid peroxidation susceptibility may be dictated by factors such as the size of the labile iron pool, the expression, localization and activity of NOX, LOX, POR, and mitochondrial enzymes, O2 availability, and the abundance of different PUFA-PL species. By contrast, total inner defense capacity will reflect GSH abundance, GPX4 and FSP1 expression, CoQ10 levels, and so on. The influence of any one metabolite, protein, or mechanism to either of these quantities and the ultimate sensitivity of the cell to ferroptosis will depend on the cell and the lethal trigger, as described above (Figure 4B). Only cells with the appropriate configuration of high total lipid peroxidation susceptibility and low total inner defense capacity will succumb to ferroptosis. Expressing total lipid peroxidation susceptibility and total inner defense capacity quantitatively using chemical rate equations may help to conceive of this mechanism more formally (see Box item 1).
Figure 4. Visualizing the contribution of different molecules to ferroptosis.

(A) The total inner defense capacity and total lipid peroxidation susceptibility of the cell can be influenced by many metabolites and proteins, some of which are listed here. The relative contribution of each molecule to ferroptosis sensitivity will vary between contexts.
(B) Quadrant diagram summarizing four different possible states in which a cell may exist. Cells with low total inner defense capacity and high total lipid peroxidation susceptibility with be most sensitive to ferroptosis. The boundaries between these states are likely to be fluid.
(C) Different treatments, such as iron chelation, or overexpression of GPX4, will reduce ferroptosis sensitivity by lowering total lipid peroxidation susceptibility or increasing total inner defense capacity.
(D) Cells may exist in different states of ferroptosis sensitivity and resistance over time, for example due to induction of NRF2, epithelial-to-mesenchymal transition (EMT), or transit in the body through a lymph fluid rich in monounsaturated fatty acids.
Box 1. Towards a quantitative model of ferroptosis susceptibility.
Total lipid peroxidation susceptibility and total inner defense capacity can in principle be quantified using standard rate equations for lipid peroxidation115. Viewing ferroptosis sensitivity in this way is conceptually useful because it provides a formal basis to understand how changes in lipid composition may modulate sensitivity to, as well as the rate of, ferroptosis. The kinetic expression associated with the radical chain reaction associated with lipid peroxidation is shown in Eq 1.
| (1) |
The rate at which the process occurs is a function of the concentration of the lipid ([LH]) and the rate constant for its reaction with propagating lipidperoxyl radicals. This rate law is generally associated with a single oxidizable lipid. However, replacing with a series, where x represents each relevant lipid species, would yield a corresponding expression for a lipid mixture. In practice, solving this equation for a cell – or even a sub-cellular compartment is a Herculean task: information regarding the concentrations of all possible oxidizable lipids, exact values, and termination rate constants () for lipids found in a biological membrane are sparse – not to mention that there are multiple independent (i.e. physically-separated) membrane systems throughout the cell! Nonetheless, this formalism helps us grasp key mechanistic points. Any biochemical mechanism that can contribute to a pool of oxidizable lipid (LH) will impact the [LH] concentration term, and this could vary substantially between cells or contexts without altering the fundamental rate equation. The rate of chain initiation also helps interpret the role of the inner defenses and regulatory penumbra in dictating ferroptosis sensitivity. is characteristic of a single reaction in simple chemical systems116. However, in the cell, there are multiple potential radical initiators. Virtually any reaction x that may result in the formation of a lipid-based radical can contribute to, that is: , including those catalyzed by NOX, P450, or other enzymes. Also, since the lipid peroxidation products can decompose to yield initiating radicals, GPX4 is key to reducing by minimizing hydroperoxide concentration available for auto-initiation.
The position of individual cells on the two-dimensional map of total lipid peroxidation susceptibility and total inner defense capacity can be shifted by treatments or conditions that alter either the lipid peroxidation susceptibility or total inner defense capacity, such as inhibitor treatment or alterations in gene expression (Figure 4C). In vivo, cells are likely to traverse this two-dimensional landscape in different directions in a dynamic manner, resulting in the gain or loss of ferroptosis sensitivity in space and/or over time. For example, this may occur in response to gene induction (e.g., NRF2 overexpression)91, cell state changes (e.g., epithelial-to-mesenchymal transition)92, or exposure to an altered environment (e.g., metastatic cancer cell transit transit through MUFA-rich lymph fluid)59 (Figure 4D). The position of a cell on this map will also be relative to the inducing stimulus, as different stimuli appear to trigger ferroptosis in ways that only converge at the level of the core membrane lipid peroxidation mechanism11,57.
Implications of the model for our understanding of ferroptosis
Thinking about ferroptosis as a flexible mechanism responsive to many inputs may have important implications for how we study this process and apply basic findings to medicine.
Depicting ferroptosis.
The core lipid peroxidation mechanism requires iron, O2, and PUFA-PLs. The inner defenses comprise a series of enzymes and metabolites, including GPX4, FSP1, and CoQ10. ACSL4, p53, NRF2, mitochondria and other pathways and organelles lying in the regulatory penumbra alter ferroptosis sensitivity, either by impinging on the total lipid peroxidation susceptibility and/or the total inner defense capacity of the cell (Figure 2B). In short, anything in the cell that contributes to the core mechanism or inner defenses can alter ferroptosis sensitivity in a quantitative manner, and this will vary for different cells, conditions, and lethal triggers (Figure 4B). By contrast, depictions of a ferroptosis pathway that simply recapitulates known metabolic, signaling and transcriptional regulatory machinery of the cell in a fixed manner (e.g., Figure 2A) cannot easily account for contradictory and context-dependent effects of different molecules, and may have less value in helping to understand this mechanism.
The ferroptosis phenotype as a discovery tool.
Ferroptosis sensitivity is dictated by many elements of the intracellular metabolic and signaling network. Ferroptosis can therefore be used to help discover and study a broad swath of fundamental biology that may be important for cell function beyond ferroptosis. For example, ferroptosis can be used as a phenotypic readout to help elucidate mechanisms relating to NADPH sensing93, lipid metabolic94, cell-cell communication66, CoQ10 transport95, selenoprotein translation42, and protein sequence editing96, among other things. Using ferroptosis to probe fundamental mechanisms around iron, lipid, and redox metabolism is likely to remain fruitful in the years ahead.
Modulating ferroptosis to treat disease.
Ferroptosis contributes to acute and chronic pathologies in the brain, kidney, heart, and other tissues, and may also be a good target for new cancer therapies6. However, the flexible nature of ferroptosis means that findings from one system may not automatically apply elsewhere. This may in part underlie the seemingly slow translation of basic findings. Thinking about ferroptosis as described above leads to a refined view of how to target this pathway therapeutically. To prevent ferroptosis, the most plausible approach is to directly reduce the total lipid peroxidation susceptibility and/or increase the total inner defense capacity of the cell, thereby inhibiting the core membrane lipid peroxidation mechanism. One approach would be to remove key components necessary for engagement of the core mechanism (e.g., iron, using iron chelators). Another and perhaps less toxic approach is to enhance the total inner defense capacity of the cell through the addition of synthetic RTAs, peroxidase mimics, or other agents, some of which have already shown promise in pre-clinical models for a range of diseases7,25,67,97–100 (Figure 2C, 4C). The flexible nature of the inner defenses and regulatory penumbra suggest that specific targets in these regions might need to be approached with caution as they could act redundantly or in a manner that is very tissue- and/or lethal trigger-specific.
Inducing ferroptosis for cancer treatment may be more difficult. It is possible to imagine increasing the total lipid peroxidation susceptibility of the cell and/or decreasing the total inner defense capacity of the cell. For example, dietary treatment with PUFAs may directly engage the core lipid peroxidation mechanism36,37. Small molecule inhibitors of key inner defense enzymes like GPX4 or FSP1 could also be optimized into anti-cancer agents55,101. Here, targets in the regulatory penumbra involved in iron or lipid metabolism, or that control the expression of key inner defense enzymes, could also be envisioned as potential targets. However, even within the same broad tumor type there are highly heterogeneous responses to the induction of ferroptosis102,103, and the ability of isogenic tumor cells to adopt different epigenetic states with unique ferroptosis sensitivity profiles is concerning92,104–106. Indeed, diverse metabolites and proteins can impinge upon the core mechanism and inner defenses to reduce the rate of lipid peroxide initiation and thereby cause resistance to ferroptosis14,16,59,65,66,90. Identifying individual tumors with a stable and favorable combination of high total lipid peroxidation susceptibility and/or low the total inner defense capacity will be challenging yet essential (for a quantitative representation of this challenge, see Box 2).
Box 2. Modelling overall ferroptosis sensitivity.
In addition to suppressing, components of the inner defenses can interfere with propagation of lipid peroxidation by acting as RTAs. When peroxidation of a single lipid takes place in the presence of an RTA, the kinetics reflect the competition for a propagating lipidperoxyl radical between the oxidizable lipids and the RTA, as in Eq 2.
| (2) |
While lipids contribute to the numerator via the sum of the series (Box 1), RTAs contribute to the denominator via the sum of the series. Here, n and are the radical-trapping stoichiometries and reactivities for each RTA. Using this kinetic competition as a fundamental consideration that determines whether a cell lives or dies by ferroptosis, one could quantify the relative contribution of different biochemical pathways on ferroptosis sensitivity by changing the numerator or denominator of Eq 2. Very simply, this ratio reflects the total lipid peroxidation susceptibility and total inner defense capacity of the cell. Thus, enzymes that can increase oxidizable PUFA-PLs (e.g., ACSL4) impact the numerator via individual LH concentration terms; pathways that increase the availability of RTAs (e.g., biosynthetic pathways, such as the GCH1-controlled BH4 pathway) impact the denominator via individual RTA concentration terms. Pathways that modulate radical generation impact the numerator; pathways that modulate the regeneration of RTAs (e.g., FSP1, DHFR) impact the denominator via individual stoichiometry terms n. Different cells or contexts may involve different contributions from individual elements to dictate the overall ferroptosis sensitivity.
Molecular markers of ferroptosis.
Having molecules or other readouts that uniquely indicate when a cell may be sensitive to or is undergoing ferroptosis would be highly valued to study this process and monitor responses in vivo6,107. High levels of lipid peroxide accumulation are the defining characteristic of ferroptosis itself and in relation to other forms of cell death8. Oxidized PUFA-PLs or their derivatives (e.g., 4-hydroxynonenal) may therefore be the most characteristic marker of this process. However, lipid peroxides are generated at some level under physiological conditions12 and it remains unclear at what threshold lipid peroxidation is associated with ferroptotic cell death in vivo. Moreover, dozens of different oxidized lipid species are generated during ferroptosis29, and these may vary between cells depending on the starting lipid content, the predominant initiating mechanism, and other factors. Although progress is being made towards defining other cell biological and molecular markers that are modulated in cells undergoing ferroptosis108,109, it is possible that no marker will be entirely specific to ferroptosis and generalizable between conditions. For example, expression of the marker gene CHAC1, sometimes used in ferroptosis studies30, is strongly induced by cystine deprivation17, but also by other amino acid deprivation conditions that do not induce ferroptosis110. Moreover, in the same cells CHAC1 is induced by cystine deprivation but not by another canonical ferroptosis trigger: direct GPX4 inhibition17. It will probably be important to validate candidate markers for each setting where ferroptosis is thought to take place, with functional inhibition of cell death by synthetic RTAs or other agents remaining the ultimate proof that ferroptosis occurred.
Concluding thoughts
Based on the evidence presented here, we propose three principles of ferroptosis regulation to foster debate and guide future experimentation: (i) there is no single universal ferroptosis pathway; (ii) many different metabolites and proteins are individually sufficient to initiate, promote and regulate ferroptosis, with none being necessary; and (iii) ferroptosis sensitivity can be altered by many signaling and transcriptional networks in the cell. Thus, we have argued that a general understanding and depiction of the ferroptosis mechanism can only be made at a level of abstraction beyond individual metabolites and proteins. Approaches that emphasize mechanistic flexibility over rigidity, and eventually quantitation of diverse metabolites and reaction rates over static nodes in a pathway diagram, may be most useful (Figure 2B, 4D and Box 1,2).
Moving from a fixed model of ferroptosis to one that encompasses a more flexible constellation of possible effectors could help think about the large number of related yet not identical ferroptosis mechanisms that seem to exist in different cells and in response to different triggers. Carefully defining these different mechanisms will be important, but their potential lack of generalizability may need to be more widely acknowledged. Viewing ferroptosis in a highly flexible way may make it easier, conceptually, to integrate findings from diverse organisms where a variety of “ferroptosis-like” processes have been identified33,111–113. Similarities and differences between ferroptosis and other forms of oxidative cell death (e.g., oxeiptosis)114 in mammalian systems may also be easier to specify. The new concepts elaborated here may also encourage efforts to develop new quantitative models of ferroptosis. Pursuing this line of thinking may also stimulate efforts to re-think how best to successfully translate our burgeoning basic knowledge into effective therapies targeting this process in disease.
ACKNOWLEDGEMENTS
We thank L. Magtanong and members of the Dixon lab for comments. This work was supported by awards from the National Institutes of Health (R01GM122923) and the American Cancer Society (RSG-21-017-01-CCG) to S.J.D., and the Natural Sciences and Engineering Research Council of Canada (RGPIN-2022-05058) to D.A.P.
Footnotes
DECLARATION OF INTERESTS
S.J.D. and D.A.P. are co-founders of Prothegen Inc. S.J.D. is a member of the scientific advisory board of Hillstream BioPharma. S.J.D. and D.A.P. hold patents related to ferroptosis.
REFERENCES
- 1.Botstein D. (2004). Ira Herskowitz: 1946–2003. Genetics 166, 653–660. 10.1534/genetics.166.2.653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Herskowitz I. (1973). Control of gene expression in bacteriophage lambda. Annu Rev Genet 7, 289–324. 10.1146/annurev.ge.07.120173.001445. [DOI] [PubMed] [Google Scholar]
- 3.Kanehis M., Got S., Sat Y., Kawashim M., Furumich M., and Tanab M. (2014). Data, information, knowledge and principle: back to metabolism in KEGG. Nucleic Acids Res 42, D199–205. 10.1093/nar/gkt1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pawson T. (2004). Specificity in signal transduction: from phosphotyrosine-SH2 domain interactions to complex cellular systems. Cell 116, 191–203. 10.1016/s0092-8674(03)01077-8. [DOI] [PubMed] [Google Scholar]
- 5.Green DR (2019). The Coming Decade of Cell Death Research: Five Riddles. Cell 177, 1094–1107. 10.1016/j.cell.2019.04.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stockwell BR (2022). Ferroptosis turns 10: Emerging mechanisms, physiological functions, and therapeutic applications. Cell 185, 2401–2421. 10.1016/j.cell.2022.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, et al. (2012). Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072. 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wiernicki B, Dubois H, Tyurina YY, Hassannia B, Bayir H, Kagan VE, Vandenabeele P, Wullaert A, and Vanden Berghe T. (2020). Excessive phospholipid peroxidation distinguishes ferroptosis from other cell death modes including pyroptosis. Cell Death Dis 11, 922. 10.1038/s41419-020-03118-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Riegman M., Sagie L., Galed C., Levin T., Steinberg N., Dixon SJ., Wiesner U., Bradbury MS., Niethammer P., Zaritsky A., and Overholtzer M. (2020). Ferroptosis occurs through an osmotic mechanism and propagates independently of cell rupture. Nat Cell Biol 22, 1042–1048. 10.1038/s41556-020-0565-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shah R, Margison K, and Pratt DA (2017). The Potency of Diarylamine Radical-Trapping Antioxidants as Inhibitors of Ferroptosis Underscores the Role of Autoxidation in the Mechanism of Cell Death. ACS Chem Biol 12, 2538–2545. 10.1021/acschembio.7b00730. [DOI] [PubMed] [Google Scholar]
- 11.Magtanong L, Mueller GD, Williams KJ, Billmann M, Chan K, Armenta DA, Pope LE, Moffat J, Boone C, Myers CL, et al. (2022). Context-dependent regulation of ferroptosis sensitivity. Cell Chem Biol 29, 1409–1418 e1406. 10.1016/j.chembiol.2022.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Greene LE, Lincoln R, and Cosa G. (2017). Rate of Lipid Peroxyl Radical Production during Cellular Homeostasis Unraveled via Fluorescence Imaging. J Am Chem Soc 139, 15801–15811. 10.1021/jacs.7b08036. [DOI] [PubMed] [Google Scholar]
- 13.von Krusenstiern AN, Robson RN, Qian N, Qiu B, Hu F, Reznik E, Smith N, Zandkarimi F, Estes VM, Dupont M, et al. (2023). Identification of essential sites of lipid peroxidation in ferroptosis. Nat Chem Biol. 10.1038/s41589-022-01249-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Magtanong L., Ko PJ., To M., Cao JY., Forcina GC., Tarangelo A., Ward CC., Cho K., Patti GJ., Nomura DK., et al. (2019). Exogenous Monounsaturated Fatty Acids Promote a Ferroptosis-Resistant Cell State. Cell Chem Biol 26, 420–432 e429. 10.1016/j.chembiol.2018.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Conrad M, and Pratt DA (2019). The chemical basis of ferroptosis. Nat Chem Biol 15, 1137–1147. 10.1038/s41589-019-0408-1. [DOI] [PubMed] [Google Scholar]
- 16.Yang WS, Kim KJ, Gaschler MM, Patel M, Shchepinov MS, and Stockwell BR (2016). Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proc Natl Acad Sci U S A 113, E4966–4975. 10.1073/pnas.1603244113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Dixon SJ, and Stockwell BR (2014). The role of iron and reactive oxygen species in cell death. Nat Chem Biol 10, 9–17. 10.1038/nchembio.1416. [DOI] [PubMed] [Google Scholar]
- 18.Aikens J, and Dix TA (1991). Perhydroxyl radical (HOO.) initiated lipid peroxidation. The role of fatty acid hydroperoxides. J Biol Chem 266, 15091–15098. [PubMed] [Google Scholar]
- 19.Gaschler MM, Hu F, Feng H, Linkermann A, Min W, and Stockwell BR (2018). Determination of the Subcellular Localization and Mechanism of Action of Ferrostatins in Suppressing Ferroptosis. ACS Chem Biol 13, 1013–1020. 10.1021/acschembio.8b00199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yan B, Ai Y, Sun Q, Ma Y, Cao Y, Wang J, Zhang Z, and Wang X. (2021). Membrane Damage during Ferroptosis Is Caused by Oxidation of Phospholipids Catalyzed by the Oxidoreductases POR and CYB5R1. Mol Cell 81, 355–369 e310. 10.1016/j.molcel.2020.11.024. [DOI] [PubMed] [Google Scholar]
- 21.Zou Y, Li H, Graham ET, Deik AA, Eaton JK, Wang W, Sandoval-Gomez G, Clish CB, Doench JG, and Schreiber SL (2020). Cytochrome P450 oxidoreductase contributes to phospholipid peroxidation in ferroptosis. Nat Chem Biol 16, 302–309. 10.1038/s41589-020-0472-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gao M, Yi J, Zhu J, Minikes AM, Monian P, Thompson CB, and Jiang X. (2019). Role of Mitochondria in Ferroptosis. Mol Cell 73, 354–363 e353. 10.1016/j.molcel.2018.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yang WH, Ding CC, Sun T, Rupprecht G, Lin CC, Hsu D, and Chi JT (2019). The Hippo Pathway Effector TAZ Regulates Ferroptosis in Renal Cell Carcinoma. Cell Rep 28, 2501–2508 e2504. 10.1016/j.celrep.2019.07.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wenzel SE, Tyurina YY, Zhao J, St Croix CM, Dar HH, Mao G, Tyurin VA, Anthonymuthu TS, Kapralov AA, Amoscato AA, et al. (2017). PEBP1 Wardens Ferroptosis by Enabling Lipoxygenase Generation of Lipid Death Signals. Cell 171, 628–641 e626. 10.1016/j.cell.2017.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Friedmann Angeli JP., Schneider M., Proneth B., Tyurina YY., Tyurin VA., Hammond VJ., Herbach N., Aichler M., Walch A., Eggenhofer E., et al. (2014). Inactivation of the ferroptosis regulator Gpx4 triggers acute renal failure in mice. Nat Cell Biol 16, 1180–1191. 10.1038/ncb3064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chu B, Kon N, Chen D, Li T, Liu T, Jiang L, Song S, Tavana O, and Gu W. (2019). ALOX12 is required for p53-mediated tumour suppression through a distinct ferroptosis pathway. Nat Cell Biol 21, 579–591. 10.1038/s41556-019-0305-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shah R, Shchepinov MS, and Pratt DA (2018). Resolving the Role of Lipoxygenases in the Initiation and Execution of Ferroptosis. ACS Cent Sci 4, 387–396. 10.1021/acscentsci.7b00589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Harayama T, and Shimizu T. (2020). Roles of polyunsaturated fatty acids, from mediators to membranes. J Lipid Res 61, 1150–1160. 10.1194/jlr.R120000800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kagan VE, Mao G, Qu F, Angeli JP, Doll S, Croix CS, Dar HH, Liu B, Tyurin VA, Ritov VB, et al. (2017). Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nat Chem Biol 13, 81–90. 10.1038/nchembio.2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang Y, Tan H, Daniels JD, Zandkarimi F, Liu H, Brown LM, Uchida K, O’Connor OA, and Stockwell BR (2019). Imidazole Ketone Erastin Induces Ferroptosis and Slows Tumor Growth in a Mouse Lymphoma Model. Cell Chem Biol 26, 623–633 e629. 10.1016/j.chembiol.2019.01.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kraft VAN., Bezjian CT., Pfeiffer S., Ringelstetter L., Muller C., Zandkarimi F., Merl-Pham J., Bao X., Anastasov N., Kossl J., et al. (2020). GTP Cyclohydrolase 1/Tetrahydrobiopterin Counteract Ferroptosis through Lipid Remodeling. ACS Cent Sci 6, 41–53. 10.1021/acscentsci.9b01063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Perez MA, Clostio AJ, Houston IR, Ruiz J, Magtanong L, Dixon SJ, and Watts JL (2022). Ether lipid deficiency disrupts lipid homeostasis leading to ferroptosis sensitivity. PLoS Genet 18, e1010436. 10.1371/journal.pgen.1010436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Perez MA, Magtanong L, Dixon SJ, and Watts JL (2020). Dietary Lipids Induce Ferroptosis in Caenorhabditiselegans and Human Cancer Cells. Dev Cell 54, 447–454 e444. 10.1016/j.devcel.2020.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zou Y, Henry WS, Ricq EL, Graham ET, Phadnis VV, Maretich P, Paradkar S, Boehnke N, Deik AA, Reinhardt F, et al. (2020). Plasticity of ether lipids promotes ferroptosis susceptibility and evasion. Nature 585, 603–608. 10.1038/s41586-020-2732-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Reed A, Ware T, Li H, Fernando Bazan J, and Cravatt BF (2023). TMEM164 is an acyltransferase that forms ferroptotic C20:4 ether phospholipids. Nat Chem Biol. 10.1038/s41589-022-01253-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dierge E., Debock E., Guilbaud C., Corbet C., Mignolet E., Mignard L., Bastien E., Dessy C., Larondelle Y., and Feron O. (2021). Peroxidation of n-3 and n-6 polyunsaturated fatty acids in the acidic tumor environment leads to ferroptosis-mediated anticancer effects. Cell Metab 33, 1701–1715 e1705. 10.1016/j.cmet.2021.05.016. [DOI] [PubMed] [Google Scholar]
- 37.Beatty A, Singh T, Tyurina YY, Tyurin VA, Samovich S, Nicolas E, Maslar K, Zhou Y, Cai KQ, Tan Y, et al. (2021). Ferroptotic cell death triggered by conjugated linolenic acids is mediated by ACSL1. Nat Commun 12, 2244. 10.1038/s41467-021-22471-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kakhlon O, and Cabantchik ZI (2002). The labile iron pool: characterization, measurement, and participation in cellular processes(1). Free Radic Biol Med 33, 1037–1046. 10.1016/s0891-5849(02)01006-7. [DOI] [PubMed] [Google Scholar]
- 39.Han S, Lin F, Qi Y, Liu C, Zhou L, Xia Y, Chen K, Xing J, Liu Z, Yu W, et al. (2022). HO-1 Contributes to Luteolin-Triggered Ferroptosis in Clear Cell Renal Cell Carcinoma via Increasing the Labile Iron Pool and Promoting Lipid Peroxidation. Oxid Med Cell Longev 2022, 3846217. 10.1155/2022/3846217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Alvarez SW, Sviderskiy VO, Terzi EM, Papagiannakopoulos T, Moreira AL, Adams S, Sabatini DM, Birsoy K, and Possemato R. (2017). NFS1 undergoes positive selection in lung tumours and protects cells from ferroptosis. Nature 551, 639–643. 10.1038/nature24637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Anandhan A., Dodson M., Shakya A., Chen J., Liu P., Wei Y., Tan H., Wang Q., Jiang Z., Yang K., et al. (2023). NRF2 controls iron homeostasis and ferroptosis through HERC2 and VAMP8. Sci Adv 9, eade9585. 10.1126/sciadv.ade9585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Li Z, Ferguson L, Deol KK, Roberts MA, Magtanong L, Hendricks JM, Mousa GA, Kilinc S, Schaefer K, Wells JA, et al. (2022). Ribosome stalling during selenoprotein translation exposes a ferroptosis vulnerability. Nat Chem Biol. 10.1038/s41589-022-01033-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ingold I, Berndt C, Schmitt S, Doll S, Poschmann G, Buday K, Roveri A, Peng X, Porto Freitas F, Seibt T, et al. (2018). Selenium Utilization by GPX4 Is Required to Prevent Hydroperoxide-Induced Ferroptosis. Cell 172, 409–422 e421. 10.1016/j.cell.2017.11.048. [DOI] [PubMed] [Google Scholar]
- 44.Seiler A, Schneider M, Forster H, Roth S, Wirth EK, Culmsee C, Plesnila N, Kremmer E, Radmark O, Wurst W, et al. (2008). Glutathione peroxidase 4 senses and translates oxidative stress into 12/15-lipoxygenase dependent- and AIF-mediated cell death. Cell Metab 8, 237–248. 10.1016/j.cmet.2008.07.005. [DOI] [PubMed] [Google Scholar]
- 45.Ursini F, and Maiorino M. (2020). Lipid peroxidation and ferroptosis: The role of GSH and GPx4. Free Radic Biol Med 152, 175–185. 10.1016/j.freeradbiomed.2020.02.027. [DOI] [PubMed] [Google Scholar]
- 46.Yant LJ., Ran Q., Rao L., Van Remmen H., Shibatani T., Belter JG., Motta L., Richardson A., and Prolla TA. (2003). The selenoprotein GPX4 is essential for mouse development and protects from radiation and oxidative damage insults. Free Radic Biol Med 34, 496–502. 10.1016/s0891-5849(02)01360-6. [DOI] [PubMed] [Google Scholar]
- 47.Liu H, Forouhar F, Seibt T, Saneto R, Wigby K, Friedman J, Xia X, Shchepinov MS, Ramesh SK, Conrad M, and Stockwell BR (2022). Characterization of a patient-derived variant of GPX4 for precision therapy. Nat Chem Biol 18, 91–100. 10.1038/s41589-021-00915-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Doll S, Proneth B, Tyurina YY, Panzilius E, Kobayashi S, Ingold I, Irmler M, Beckers J, Aichler M, Walch A, et al. (2017). ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nat Chem Biol 13, 91–98. 10.1038/nchembio.2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Shimada K, Skouta R, Kaplan A, Yang WS, Hayano M, Dixon SJ, Brown LM, Valenzuela CA, Wolpaw AJ, and Stockwell BR (2016). Global survey of cell death mechanisms reveals metabolic regulation of ferroptosis. Nat Chem Biol 12, 497–503. 10.1038/nchembio.2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Yang WS, SriRamaratnam R, Welsch ME, Shimada K, Skouta R, Viswanathan VS, Cheah JH, Clemons PA, Shamji AF, Clish CB, et al. (2014). Regulation of ferroptotic cancer cell death by GPX4. Cell 156, 317–331. 10.1016/j.cell.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zou Y., Palte MJ., Deik AA., Li H., Eaton JK., Wang W., Tseng YY., Deasy R., Kost-Alimova M., Dancik V., et al. (2019). A GPX4-dependent cancer cell state underlies the clear-cell morphology and confers sensitivity to ferroptosis. Nat Commun 10, 1617. 10.1038/s41467-019-09277-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, Roberts MA, Tong B, Maimone TJ, Zoncu R, et al. (2019). The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 575, 688–692. 10.1038/s41586-019-1705-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Dixon SJ, Patel DN, Welsch M, Skouta R, Lee ED, Hayano M, Thomas AG, Gleason CE, Tatonetti NP, Slusher BS, and Stockwell BR (2014). Pharmacological inhibition of cystine-glutamate exchange induces endoplasmic reticulum stress and ferroptosis. Elife 3, e02523. 10.7554/eLife.02523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ingold KU, and Pratt DA (2014). Advances in radical-trapping antioxidant chemistry in the 21st century: a kinetics and mechanisms perspective. Chem Rev 114, 9022–9046. 10.1021/cr500226n. [DOI] [PubMed] [Google Scholar]
- 55.Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I, Goya Grocin A, Xavier da Silva TN, Panzilius E, Scheel CH, et al. (2019). FSP1 is a glutathione-independent ferroptosis suppressor. Nature 575, 693–698. 10.1038/s41586-019-1707-0. [DOI] [PubMed] [Google Scholar]
- 56.Mishima E., Ito J., Wu Z., Nakamura T., Wahida A., Doll S., Tonnus W., Nepachalovich P., Eggenhofer E., Aldrovandi M., et al. (2022). A non-canonical vitamin K cycle is a potent ferroptosis suppressor. Nature 608, 778–783. 10.1038/s41586-022-05022-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Soula M, Weber RA, Zilka O, Alwaseem H, La K, Yen F, Molina H, Garcia-Bermudez J, Pratt DA, and Birsoy K. (2020). Metabolic determinants of cancer cell sensitivity to canonical ferroptosis inducers. Nat Chem Biol 16, 1351–1360. 10.1038/s41589-020-0613-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dixon SJ, Winter GE, Musavi LS, Lee ED, Snijder B, Rebsamen M, Superti-Furga G, and Stockwell BR (2015). Human Haploid Cell Genetics Reveals Roles for Lipid Metabolism Genes in Nonapoptotic Cell Death. ACS Chem Biol 10, 1604–1609. 10.1021/acschembio.5b00245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Ubellacker JM, Tasdogan A, Ramesh V, Shen B, Mitchell EC, Martin- Sandoval MS, Gu Z, McCormick ML, Durham AB, Spitz DR, et al. (2020). Lymph protects metastasizing melanoma cells from ferroptosis. Nature 585, 113–118. 10.1038/s41586-020-2623-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bartolacci C, Andreani C, Vale G, Berto S, Melegari M, Crouch AC, Baluya DL, Kemble G, Hodges K, Starrett J, et al. (2022). Targeting de novo lipogenesis and the Lands cycle induces ferroptosis in KRAS-mutant lung cancer. Nat Commun 13, 4327. 10.1038/s41467-022-31963-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen D., Chu B., Yang X., Liu Z., Jin Y., Kon N., Rabadan R., Jiang X., Stockwell BR., and Gu W. (2021). iPLA2beta-mediated lipid detoxification controls p53-driven ferroptosis independent of GPX4. Nat Commun 12, 3644. 10.1038/s41467-021-23902-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Beharier O, Tyurin VA, Goff JP, Guerrero-Santoro J, Kajiwara K, Chu T, Tyurina YY, St Croix CM, Wallace CT, Parry S, et al. (2020). PLA2G6 guards placental trophoblasts against ferroptotic injury. Proc Natl Acad Sci U S A 117, 27319–27328. 10.1073/pnas.2009201117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sun WY, Tyurin VA, Mikulska-Ruminska K, Shrivastava IH, Anthonymuthu TS, Zhai YJ, Pan MH, Gong HB, Lu DH, Sun J, et al. (2021). Phospholipase iPLA2beta averts ferroptosis by eliminating a redox lipid death signal. Nat Chem Biol. 10.1038/s41589-020-00734-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Zhou N, Yuan X, Du Q, Zhang Z, Shi X, Bao J, Ning Y, and Peng L. (2023). FerrDb V2: update of the manually curated database of ferroptosis regulators and ferroptosis-disease associations. Nucleic Acids Res 51, D571–D582. 10.1093/nar/gkac935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Garcia-Bermudez J, Baudrier L, Bayraktar EC, Shen Y, La K, Guarecuco R, Yucel B, Fiore D, Tavora B, Freinkman E, et al. (2019). Squalene accumulation in cholesterol auxotrophic lymphomas prevents oxidative cell death. Nature 567, 118–122. 10.1038/s41586-019-0945-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wu J., Minikes AM., Gao M., Bian H., Li Y., Stockwell BR., Chen ZN., and Jiang X. (2019). Intercellular interaction dictates cancer cell ferroptosis via NF2- YAP signalling. Nature 572, 402–406. 10.1038/s41586-019-1426-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yang WS, and Stockwell BR (2008). Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chem Biol 15, 234–245. 10.1016/j.chembiol.2008.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yagoda N, von Rechenberg M, Zaganjor E, Bauer AJ, Yang WS, Fridman DJ, Wolpaw AJ, Smukste I, Peltier JM, Boniface JJ, et al. (2007). RAS-RAF-MEK-dependent oxidative cell death involving voltage-dependent anion channels. Nature 447, 864–868. 10.1038/nature05859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ratan RR, Murphy TH, and Baraban JM (1994). Macromolecular synthesis inhibitors prevent oxidative stress-induced apoptosis in embryonic cortical neurons by shunting cysteine from protein synthesis to glutathione. J Neurosci 14, 4385–4392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Conlon M, Poltorack CD, Forcina GC, Armenta DA, Mallais M, Perez MA, Wells A, Kahanu A, Magtanong L, Watts JL, et al. (2021). A compendium of kinetic modulatory profiles identifies ferroptosis regulators. Nat Chem Biol. 10.1038/s41589-021-00751-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhao J, Jia Y, Mahmut D, Deik AA, Jeanfavre S, Clish CB, and Sankaran VG. (2023). Human hematopoietic stem cell vulnerability to ferroptosis. Cell 186, 732–747 e716. 10.1016/j.cell.2023.01.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Badgley MA, Kremer DM, Maurer HC, DelGiorno KE, Lee HJ, Purohit V, Sagalovskiy IR, Ma A, Kapilian J, Firl CEM, et al. (2020). Cysteine depletion induces pancreatic tumor ferroptosis in mice. Science 368, 85–89. 10.1126/science.aaw9872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Leu JI, Murphy ME, and George DL (2020). Functional interplay among thiol- based redox signaling, metabolism, and ferroptosis unveiled by a genetic variant of TP53. Proc Natl Acad Sci U S A 117, 26804–26811. 10.1073/pnas.2009943117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Leu JI, Murphy ME, and George DL (2019). Mechanistic basis for impaired ferroptosis in cells expressing the African-centric S47 variant of p53. Proc Natl Acad Sci U S A 116, 8390–8396. 10.1073/pnas.1821277116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Kang YP, Mockabee-Macias A, Jiang C, Falzone A, Prieto-Farigua N, Stone E, Harris IS, and DeNicola GM (2021). Non-canonical Glutamate-Cysteine Ligase Activity Protects against Ferroptosis. Cell Metab 33, 174–189 e177. 10.1016/j.cmet.2020.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shui S, Zhao Z, Wang H, Conrad M, and Liu G. (2021). Non-enzymatic lipid peroxidation initiated by photodynamic therapy drives a distinct ferroptosis-like cell death pathway. Redox Biol 45, 102056. 10.1016/j.redox.2021.102056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Shimada K., Hayano M., Pagano NC., and Stockwell BR. (2016). Cell-Line Selectivity Improves the Predictive Power of Pharmacogenomic Analyses and Helps Identify NADPH as Biomarker for Ferroptosis Sensitivity. Cell Chem Biol 23, 225–235. 10.1016/j.chembiol.2015.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ghergurovich JM, Garcia-Canaveras JC, Wang J, Schmidt E, Zhang Z, TeSlaa T, Patel H, Chen L, Britt EC, Piqueras-Nebot M, et al. (2020). A small molecule G6PD inhibitor reveals immune dependence on pentose phosphate pathway. Nat Chem Biol 16, 731–739. 10.1038/s41589-020-0533-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Chen L, Zhang Z, Hoshino A, Zheng HD, Morley M, Arany Z, and Rabinowitz JD (2019). NADPH production by the oxidative pentose-phosphate pathway supports folate metabolism. Nat Metab 1, 404–415. [PMC free article] [PubMed] [Google Scholar]
- 80.Zhang Y, Swanda RV, Nie L, Liu X, Wang C, Lee H, Lei G, Mao C, Koppula P, Cheng W, et al. (2021). mTORC1 couples cyst(e)ine availability with GPX4 protein synthesis and ferroptosis regulation. Nat Commun 12, 1589. 10.1038/s41467-021-21841-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yi J, Zhu J, Wu J, Thompson CB, and Jiang X. (2020). Oncogenic activation of PI3K-AKT-mTOR signaling suppresses ferroptosis via SREBP-mediated lipogenesis. Proc Natl Acad Sci U S A 117, 31189–31197. 10.1073/pnas.2017152117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Maddocks OD., Berkers CR., Mason SM., Zheng L., Blyth K., Gottlieb E., and Vousden KH. (2013). Serine starvation induces stress and p53-dependent metabolic remodelling in cancer cells. Nature 493, 542–546. 10.1038/nature11743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Jiang L, Kon N, Li T, Wang SJ, Su T, Hibshoosh H, Baer R, and Gu W. (2015). Ferroptosis as a p53-mediated activity during tumour suppression. Nature 520, 57–62. 10.1038/nature14344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tarangelo A, Rodencal J, Kim JT, Magtanong L, Long JZ, and Dixon SJ (2022). Nucleotide biosynthesis links glutathione metabolism to ferroptosis sensitivity. Life Sci Alliance 5. 10.26508/lsa.202101157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Tarangelo A, Magtanong L, Bieging-Rolett KT, Li Y, Ye J, Attardi LD, and Dixon SJ (2018). p53 Suppresses Metabolic Stress-Induced Ferroptosis in Cancer Cells. Cell Rep 22, 569–575. 10.1016/j.celrep.2017.12.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Xie Y, Zhu S, Song X, Sun X, Fan Y, Liu J, Zhong M, Yuan H, Zhang L, Billiar TR, et al. (2017). The Tumor Suppressor p53 Limits Ferroptosis by Blocking DPP4 Activity. Cell Rep 20, 1692–1704. 10.1016/j.celrep.2017.07.055. [DOI] [PubMed] [Google Scholar]
- 87.Valente LJ, Tarangelo A, Li AM, Naciri M, Raj N, Boutelle AM, Li Y, Mello SS, Bieging-Rolett K, DeBerardinis RJ, et al. (2020). p53 deficiency triggers dysregulation of diverse cellular processes in physiological oxygen. J Cell Biol 219. 10.1083/jcb.201908212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gao M, Monian P, Pan Q, Zhang W, Xiang J, and Jiang X. (2016). Ferroptosis is an autophagic cell death process. Cell Res 26, 1021–1032. 10.1038/cr.2016.95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Hou W, Xie Y, Song X, Sun X, Lotze MT, Zeh HJ 3rd, Kang R, and Tang D. (2016). Autophagy promotes ferroptosis by degradation of ferritin. Autophagy 12, 1425–1428. 10.1080/15548627.2016.1187366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Armenta DA, Laqtom NN, Alchemy G, Dong W, Morrow D, Poltorack CD, Nathanson DA, Abu-Remalieh M, and Dixon SJ (2022). Ferroptosis inhibition by lysosome-dependent catabolism of extracellular protein. Cell Chem Biol 29, 1588–1600 e1587. 10.1016/j.chembiol.2022.10.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Sun X, Ou Z, Chen R, Niu X, Chen D, Kang R, and Tang D. (2016). Activation of the p62-Keap1-NRF2 pathway protects against ferroptosis in hepatocellular carcinoma cells. Hepatology 63, 173–184. 10.1002/hep.28251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Viswanathan VS, Ryan MJ, Dhruv HD, Gill S, Eichhoff OM, Seashore-Ludlow B, Kaffenberger SD, Eaton JK, Shimada K, Aguirre AJ, et al. (2017). Dependency of a therapy-resistant state of cancer cells on a lipid peroxidase pathway. Nature 547, 453–457. 10.1038/nature23007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Nguyen KT, Mun SH, Yang J, Lee J, Seok OH, Kim E, Kim D, An SY, Seo DY, Suh JY, et al. (2022). The MARCHF6 E3 ubiquitin ligase acts as an NADPH sensor for the regulation of ferroptosis. Nat Cell Biol 24, 1239–1251. 10.1038/s41556-022-00973-1. [DOI] [PubMed] [Google Scholar]
- 94.Zhang H-L., Hu B-X., Li Z-L., Du T., Shan J-L., Ye Z-P., Peng X-D., Li X., Huang Y., Zshu X-Y., Chen Y-H., Feng G-K.,Yang D., Deng R., Zhu X-F. (2022). PKCβII phosphorylates ACSL4 to amplify lipid peroxidation to induce ferroptosis. Nature Cell Biology In press. [DOI] [PubMed] [Google Scholar]
- 95.Deshwal S, Onishi M, Tatsuta T, Bartsch T, Cors E, Ried K, Lemke K, Nolte H, Giavalisco P, and Langer T. (2023). Mitochondria regulate intracellular coenzyme Q transport and ferroptotic resistance via STARD7. Nat Cell Biol 25, 246–257. 10.1038/s41556-022-01071-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Forcina GC, Pope L, Murray M, Dong W, Abu-Remaileh M, Bertozzi CR, and Dixon SJ (2022). Ferroptosis regulation by the NGLY1/NFE2L1 pathway. Proc Natl Acad Sci U S A 119, e2118646119. 10.1073/pnas.2118646119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Zilka O, Shah R, Li B, Friedmann Angeli JP, Griesser M, Conrad M, and Pratt DA (2017). On the Mechanism of Cytoprotection by Ferrostatin-1 and Liproxstatin-1 and the Role of Lipid Peroxidation in Ferroptotic Cell Death. ACS Cent Sci 3, 232–243. 10.1021/acscentsci.7b00028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Skouta R, Dixon SJ, Wang J, Dunn DE, Orman M, Shimada K, Rosenberg PA, Lo DC, Weinberg JM, Linkermann A, and Stockwell BR (2014). Ferrostatins inhibit oxidative lipid damage and cell death in diverse disease models. J Am Chem Soc 136, 4551–4556. 10.1021/ja411006a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Alim I., Caulfiel JT., Chen Y., Swarup V., Geschwind DH., Ivanova E., Seravalli J., Ai Y., Sansing LH., Ste Marie EJ., et al. (2019). Selenium Drives a Transcriptional Adaptive Program to Block Ferroptosis and Treat Stroke. Cell 177, 1262–1279 e1225. 10.1016/j.cell.2019.03.032. [DOI] [PubMed] [Google Scholar]
- 100.Farmer LA, Wu Z, Poon JF, Zilka O, Lorenz SM, Huehn S, Proneth B, Conrad M, and Pratt DA (2022). Intrinsic and Extrinsic Limitations to the Design and Optimization of Inhibitors of Lipid Peroxidation and Associated Cell Death. J Am Chem Soc 144, 14706–14721. 10.1021/jacs.2c05252. [DOI] [PubMed] [Google Scholar]
- 101.Eaton JK, Furst L, Ruberto RA, Moosmayer D, Hilpmann A, Ryan MJ, Zimmermann K, Cai LL, Niehues M, Badock V, et al. (2020). Selective covalent targeting of GPX4 using masked nitrile-oxide electrophiles. Nat Chem Biol 16, 497–506. 10.1038/s41589-020-0501-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Yang F, Xiao Y, Ding JH, Jin X, Ma D, Li DQ, Shi JX, Huang W, Wang YP, Jiang YZ, and Shao ZM (2023). Ferroptosis heterogeneity in triple- negative breast cancer reveals an innovative immunotherapy combination strategy. Cell Metab 35, 84–100 e108. 10.1016/j.cmet.2022.09.021. [DOI] [PubMed] [Google Scholar]
- 103.Wang F, Graham ET, Naowarojna N, Shi Z, Wang Y, Xie G, Zhou L, Salmon W, Jia JM, Wang X, et al. (2022). PALP: A rapid imaging technique for stratifying ferroptosis sensitivity in normal and tumor tissues in situ. Cell Chem Biol 29, 157–170 e156. 10.1016/j.chembiol.2021.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Tsoi J., Robert L., Paraiso K., Galvan C., Sheu KM., Lay J., Wong DJL., Atefi M., Shirazi R., Wang X., et al. (2018). Multi-stage Differentiation Defines Melanoma Subtypes with Differential Vulnerability to Drug-Induced Iron-Dependent Oxidative Stress. Cancer Cell 33, 890–904 e895. 10.1016/j.ccell.2018.03.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hangauer MJ, Viswanathan VS, Ryan MJ, Bole D, Eaton JK, Matov A, Galeas J, Dhruv HD, Berens ME, Schreiber SL, et al. (2017). Drug-tolerant persister cancer cells are vulnerable to GPX4 inhibition. Nature 551, 247–250. 10.1038/nature24297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kalkavan H, Chen MJ, Crawford JC, Quarato G, Fitzgerald P, Tait SWG, Goding CR, and Green DR (2022). Sublethal cytochrome c release generates drug-tolerant persister cells. Cell 185, 3356–3374 e3322. 10.1016/j.cell.2022.07.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hadian K, and Stockwell BR (2021). A roadmap to creating ferroptosis-based medicines. Nat Chem Biol 17, 1113–1116. 10.1038/s41589-021-00853-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Jin J, Schorpp K, Samaga D, Unger K, Hadian K, and Stockwell BR (2022). Machine Learning Classifies Ferroptosis and Apoptosis Cell Death Modalities with TfR1 Immunostaining. ACS Chem Biol 17, 654–660. 10.1021/acschembio.1c00953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Feng H., Schorpp K., Jin J., Yozwiak CE., Hoffstrom BG., Decker AM., Rajbhandari P., Stokes ME., Bender HG., Csuka JM., et al. (2020). Transferrin Receptor Is a Specific Ferroptosis Marker. Cell Rep 30, 3411–3423 e3417. 10.1016/j.celrep.2020.02.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wallis KF, Morehead LC, Bird JT, Byrum SD, and Miousse IR (2021). Differences in cell death in methionine versus cysteine depletion. Environ Mol Mutagen 62, 216–226. 10.1002/em.22428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Conrad M, Kagan VE, Bayir H, Pagnussat GC, Head B, Traber MG, and Stockwell BR (2018). Regulation of lipid peroxidation and ferroptosis in diverse species. Genes Dev 32, 602–619. 10.1101/gad.314674.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Bogacz M, and Krauth-Siegel RL (2018). Tryparedoxin peroxidase-deficiency commits trypanosomes to ferroptosis-type cell death. Elife 7. 10.7554/eLife.37503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Distefano AM, Martin MV, Cordoba JP, Bellido AM, D’Ippolito S, Colman SL, Soto D, Roldan JA, Bartoli CG, Zabaleta EJ, et al. (2017). Heat stress induces ferroptosis-like cell death in plants. J Cell Biol 216, 463–476. 10.1083/jcb.201605110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Holze C, Michaudel C, Mackowiak C, Haas DA, Benda C, Hubel P, Pennemann FL, Schnepf D, Wettmarshausen J, Braun M, et al. (2018). Oxeiptosis, a ROS-induced caspase-independent apoptosis-like cell-death pathway. Nat Immunol 19, 130–140. 10.1038/s41590-017-0013-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Helberg J, and Pratt DA (2021). Autoxidation vs. antioxidants - the fight for forever. Chem Soc Rev 50, 7343–7358. 10.1039/d1cs00265a. [DOI] [PubMed] [Google Scholar]
- 116.Shah R., Farmer LA., Zilka O., Van Kessel ATM., and Pratt DA. (2019). Beyond DPPH: Use of Fluorescence-Enabled Inhibited Autoxidation to Predict Oxidative Cell Death Rescue. Cell Chem Biol 26, 1594–1607 e1597. 10.1016/j.chembiol.2019.09.007. [DOI] [PubMed] [Google Scholar]