

Abstract
Vibrio cholerae colonizes the small intestine and releases cholera toxin into the extracellular space. The toxin binds to the apical surface of the epithelium, is internalized into the host endomembrane system, and escapes into the cytosol where it activates the stimulatory alpha subunit of the heterotrimeric G protein by ADP-ribosylation. This initiates a cAMP-dependent signalling pathway that stimulates chloride efflux into the gut, with diarrhea resulting from the accompanying osmotic movement of water into the intestinal lumen. G protein signalling is not the only host system manipulated by cholera toxin, however. Other cellular mechanisms and signalling pathways active in the intoxication process include endocytosis through lipid rafts, retrograde transport to the endoplasmic reticulum, the endoplasmic reticulum-associated degradation system for protein delivery to the cytosol, the unfolded protein response, and G protein de-activation through degradation or the function of ADP-ribosyl hydrolases. Although toxin-induced chloride efflux is thought to be an irreversible event, alterations to these processes could facilitate cellular recovery from intoxication. This review will highlight how cholera toxin exploits signalling pathways and other cell biology events to elicit a diarrheal response from the host.
Keywords: ADP-ribosyl hydrolase, cAMP, cystic fibrosis transmembrane regulator, lipid rafts, molecular ratchet, unfolded protein response
Pathogens are master cell biologists; practically every cellular process is exploited by one pathogen or another. Vibrio cholerae is a prime example of this phenomenon. It produces a virulence factor, cholera toxin (CT)1, which induces a diarrheal response that flushes the pathogen back into the environment for further spread. In order to elicit this effect, CT manipulates several cellular functions. This review will focus on the manipulation of host cell biology by CT, including how the toxin alters host signaling pathways.
1. Etiology and Epidemiology of Cholera
The profuse and potentially life-threatening diarrhea of cholera results from persistent activation of a cAMP-dependent signaling pathway. As summarized in a series of Lancet reviews [6–8], the primary signs of cholera are profuse diarrhea and vomiting of clear fluid. An individual who is untreated can produce up to 20 liters of diarrhea a day, with the stools containing flakes of mucus that generate a distinctive rice-water appearance. Without treatment, the patient can become extremely dehydrated and experience hypovolemic shock. Oral or intravenous rehydration to replace the lost fluids and electrolytes is used for treatment. Vomiting ceases after the patient has been properly hydrated, which allows the subsequent use of oral antibiotics to reduce persisting symptoms and bacterial shedding. Without antibiotics, a typical case of cholera will be cleared by the adaptive immune system in 7-10 days. The individual will then be immunoprotected against the disease for around three years. Prophylactic protection conferred by the three commonly used cholera vaccines will last about the same length of time.
V. cholerae, the causative agent of cholera, has an aquatic reservoir and is transmitted by a fecal-oral route through contaminated food or water. Asymptomatic and mildly symptomatic infections assist the spread of disease, along with the hyperinfectious state of V. cholerae shed from a host. The pathogen has a global distribution, but most infections occur in developing countries where infrastructure for water and sewage treatment is lacking. Cholera can be endemic in these regions, while epidemic spikes are frequently seen after natural disasters and in refugee camps [9, 10]. The global burden of cholera is estimated to be between 2-9 million annual cases with 95,000 yearly deaths [10, 11].
There are over 200 serotypes of V. cholerae that are based on variations in the lipopolysaccharide O antigen. However, only the O1 and O139 serotypes are associated with cholera epidemics [9]. These strains harbor a chromosomal copy of CTXΦ, a filamentous bacteriophage that encodes an operon with the ctxab genes of CT [12, 13]. This instance of lysogenic conversion allows O1 and O139 V. cholerae to elicit the profuse, fulminant diarrhea of cholera. The enhanced virulence of O1/O139 V. cholerae resulting from the production of phage-encoded CT also benefits CTXΦ, as its genome is replicated every time the bacterium divides.
CT does not kill the intoxicated cell. Instead, it alters a cAMP-dependent signaling pathway to promote chloride efflux through the cystic fibrosis transmembrane regulator (CFTR). The release of chloride and accompanying movement of water into the intestinal lumen is what generates the diarrheal disease of cholera. Other CT-triggered physiological responses may contribute to the disease state, but CFTR activity appears to be the main mechanism of action [14–17]. Intestinal biopsies from CFTR (−/−) individuals are thus non-responsive to CT [18]. Transgenic CFTR(−/−) mice are also completely resistant to CT-induced secretory diarrhea, while heterozygous CFTR(+/−) mice are partially resistant to the toxin [19]. Many investigators have accordingly focused on CFTR inhibitors as a therapeutic strategy to treat cholera [20–22]. The ability of a CFTR(+/−) genotype to withstand severe cases of cholera and other secretory diarrheal diseases may provide an evolutionary explanation for the unusually high frequency of CFTR heterozygosity and cystic fibrosis disease (Box 1).
Box 1. Cholera and cystic fibrosis: the confluence of infectious and genetic disease.
The first of seven modern cholera pandemics began in 1817, but Hippocrates described a cholera-like illness in the 4th century BCE [12, 23]. Even before this, early Indian texts dating back to the 5th century BCE described isolated instances of cholera-like illness [24]. Thus, over the course of human history, there has been a strong selective pressure to minimize the CFTR-driven effects of secretory diarrhea (i.e., diarrhea resulting from electrolyte imbalance in the gut) due to infection with V. cholerae or other enteric pathogens. For perspective, there are still 4 billion annual cases of diarrhea resulting in 2-4 million global deaths, mainly in young children from developing countries [17].
A CFTR (+/−) genotype reduces the CT-induced accumulation of intestinal fluid [19], which may explain why the frequency of CFTR heterozygosity is an unusually high 4% in the Caucasian population [25]: heterozygosity provides a survival advantage against cholera and other secretory diarrheas by lowering the chances of severe dehydration and death [18, 19, 26, 27]. However, the CFTR (−/−) genotype results in cystic fibrosis disease.
CFTR is a protein kinase A-responsive, ATP-dependent ion channel responsible for chloride efflux across the apical plasma membrane [28]. This process influences the movement of sodium and water into the extracellular space of respiratory, digestive, reproductive, and sweat gland epithelia. The hyperstimulation of CFTR generates an excess of extracellular water which causes secretory diarrhea, while the lack of CFTR produces a highly concentrated mucus that is difficult to clear and negatively impacts tissue function. In the digestive system, the hyperviscous mucus leads to bowel obstructions and nutrient malabsorption from the blockage of pancreatic ducts. In the respiratory system, the thick mucus layer promotes recurrent respiratory infections with chronic inflammation, breathing difficulties, and progressive lung damage [25, 29]. These complications limit the median lifespan of a cystic fibrosis patient to 48 years, which is a vast improvement over the median lifespan of 4-5 years in the 1950s [30].
The lethal outcome of secretory diarrhea, especially in young children, likely explains why mutant cftr alleles persist in the population despite the historical deaths of CFTR (−/−) individuals before child-bearing age. This would not be the first example of a link between infectious and genetic disease: malaria has provided a selective pressure in African populations to maintain the sickle cell trait, which provides a survival advantage against malaria in the heterozygous state but causes sickle cell anemia in the homozygous state [31]. Blood group antigens can also affect disease outcomes, with blood group O individuals having higher survival rates for malaria but more severe outcomes for cholera [31–34]. The latter observation explains the exceptionally low percentage of blood type O individuals in the Ganges Delta region where cholera has been endemic since at least the beginning of the modern pandemics [35]. The former observation suggests V. cholerae infections in Africa, where the majority of current pandemic cases occur [10, 11], may affect a large population of blood type O individuals who are more prone to severe cases of cholera.
2. Structure/Function of Cholera Toxin
CT belongs to the broad family of AB protein toxins that contain two functional components: an enzymatic A moiety and a cell-binding B moiety [36–38]. These toxins are released into the extracellular space but attack targets within the host cell. The toxins must therefore cross a membrane barrier in order to reach their intracellular targets. Translocation into the cytosol only occurs after endocytosis of the surface-bound toxin. Some AB toxins then access the cytosol from acidified endosomes. Other AB toxins travel by vesicle carriers from the endosomes, through the trans-Golgi network (TGN), and to the endoplasmic reticulum (ER) before passage into the cytoplasm. For both endosome and ER translocation sites, toxin disassembly occurs before or during A chain entry into the cytosol. In vivo toxin activity requires A chain dissociation from the B subunit. Furthermore, the A chain must attain an unfolded state in order to access the cytosol. The translocated toxin then returns to a folded conformation for the modification of its cytosolic target. An overview of the trafficking and translocation itinerary for CT is provided in Figure 1.
Figure 1. CT trafficking and translocation.
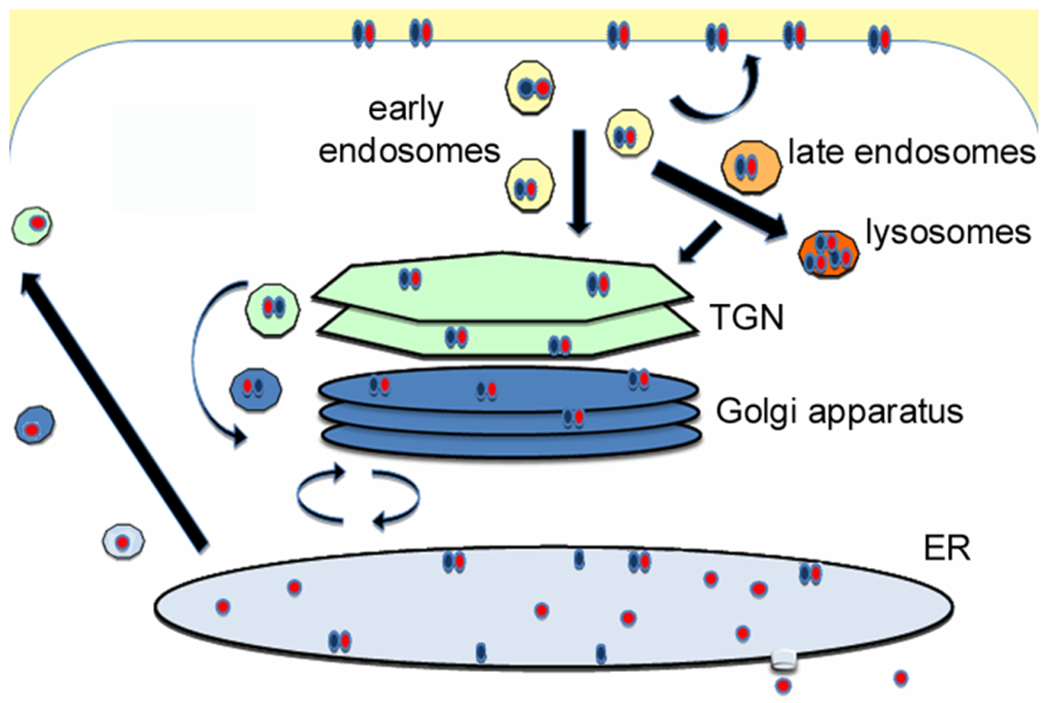
CT is internalized after its B moiety (blue) binds to GM1 gangliosides on the cell surface. The endocytosed toxin is then (i) recycled to the plasma membrane; (ii) directed to the lysosomes for degradation; or (iii) delivered to the TGN en route to the ER translocation site [39, 40]. The duration of toxin cycling between the cell surface and endosomes is unknown. At least 20-30% of cell-associated CT is degraded in the lysosomes after 6 h of intoxication [41–43], and only 1-10% of CT reaches the ER after 1-2 h [41, 44–47]. CT cycles between the Golgi and the ER for an indeterminate time until the catalytic A1 subunit (red) dissociates from the rest of the toxin in the ER and shifts to an unfolded conformation which triggers its export to the cytosol through the system of ER-associated degradation (ERAD) [48–50]. A substantial fraction of free, ER-localized CTA1 escapes ERAD and is secreted back into the medium [51]. Thus, less CTA1 reaches the cytosol than reaches the ER. The translocated A1 subunit regains an active conformation in the cytosol and evades proteasomal degradation long enough to modify its Gsα target [52]. Image from [53].
CT is an ER-translocating AB5 toxin [16]. A serine protease site spanning amino acids 192 to 195 in the CTA polypeptide2 is cleaved to generate separate A1 and A2 subunits that remain linked by a disulfide bond between C188 and C199. Catalytic activity resides in the CTA1 subunit, while CTA2 acts as a bridge that anchors CTA1 to the B moiety. Monomers of CTB assemble into a ring-like pentamer, with the C-terminal region of CTA2 occupying central pore of the structure (Fig. 2). The intact CT holotoxin is assembled in the bacterial periplasm and then secreted across the outer membrane to enter the intestinal lumen.
Figure 2. CT structure.
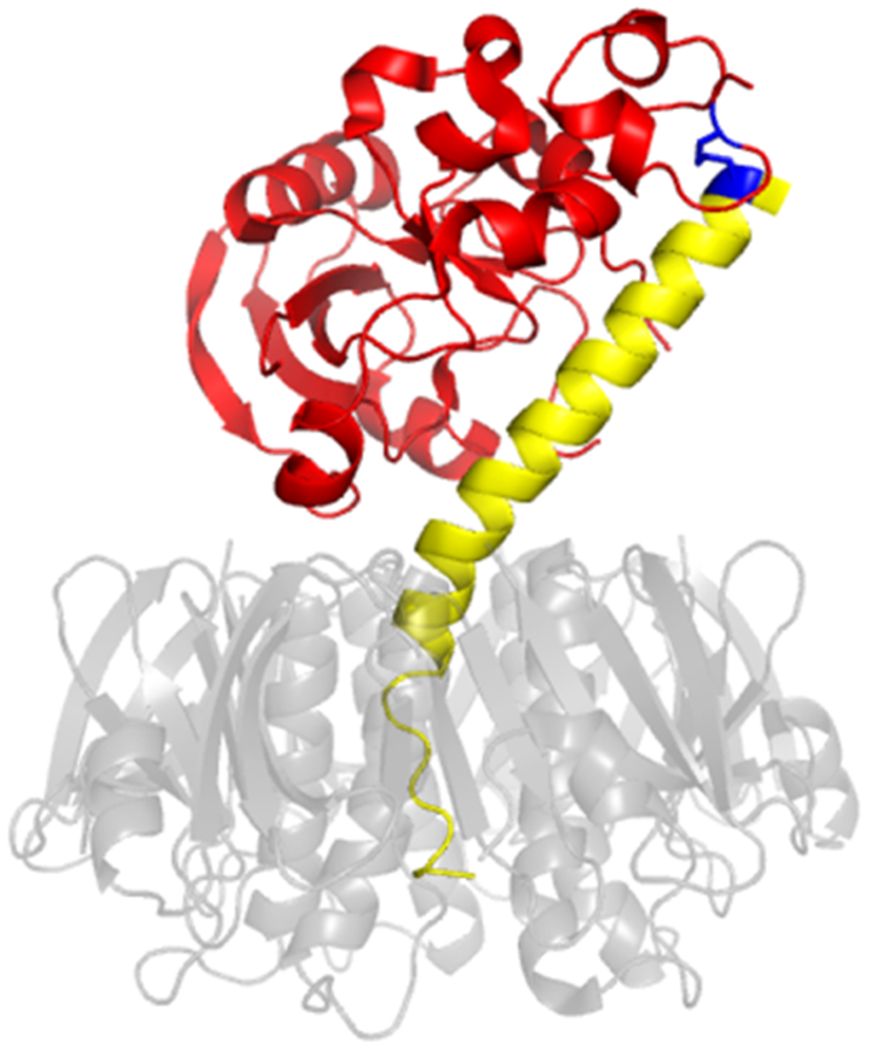
CT is composed of an A1 subunit with ADP-ribosyltransferase activity (red), an A2 linker (yellow), and a B homopentamer (translucent grey, in order to show extension of the A2 subunit into the central cavity of the ring-like B moiety). The paired cysteine residues of the CTA1/CTA2 disulfide bond are highlighted in blue. PDB entry 1S5F [54].
When CTA1 enters the host cytosol, it catalyzes the transfer of ADP-ribose from NAD to R2013 in the stimulatory alpha subunit of the heterotrimeric G protein (Gsα) [1, 55, 56]. This modification inhibits the intrinsic GTPase activity of Gsα, which locks the protein in an active, GTP-bound state and results in the prolonged production of cAMP through persistent stimulation of adenylate cyclase. Protein kinase A (PKA) is activated by the chronically high levels of cAMP, and its subsequent phosphorylation of CFTR promotes increased chloride secretion into the intestinal lumen [28]. Other proteins activated by PKA reduce the surface expression of sodium-hydrogen exchanger 3 (NHE3) through endocytosis and an inhibition of further NHE3 delivery to the plasma membrane [57, 58]. This effect, as well as direct inhibition of NHE3 by PKA [59], prevents sodium absorption into the intestinal epithelium. The resulting extracellular accumulation of sodium and chloride leads to the osmotic movement of large volumes of water into the intestinal lumen, thus generating the profuse watery diarrhea of cholera.
3. Intracellular Transport of CT
CTA1 must enter the cytosol in order to reach its Gsα target. CT does not have pore-forming capacity, so it utilizes a protein-conducting channel in the ER membrane to access the cytosol. Movement from the extracellular space to the ER is a multi-step process involving toxin binding to the plasma membrane, endocytosis, transfer from the endosomes to the TGN, retrograde transport to the ER, and toxin retention in the ER before CTA1 export to the cytosol. A substantial pool of internalized toxin is also routed to the lysosomes for degradation [41–43]. CT can be detected in the ER and cytosol as early as 45-60 minutes after binding to the cell surface, with a accompanying cAMP response and chloride efflux [42–44, 60, 61]. However, only a minor pool (1-10%) of cell-associated CT reaches the ER within 1-2 hours of its initial contact with the plasma membrane [41, 44–47]. Despite this inefficiency, CT still elicits a potent response from the intoxicated cell.
3.1. Surface binding and endocytosis of CT.
Lipid rafts play a critical role in the surface binding and intracellular transport of CT. These structures are enriched in cholesterol and sphingolipids which, based on their physical properties, promote the tight packing of saturated phospholipids into a liquid ordered phase that forms a floating platform within the larger, liquid disordered environment of the membrane [62–64]. The raft structure can serve as a site of endocytosis or recruit specific proteins to act as a signaling platform [64–66]. The former function is involved with the internalization of surface-bound CT. Alterations to membrane cholesterol or sphingomyelin consequently inhibit the endocytosis and cellular activity of CT [43, 60, 67].
CT initially interacts with the target cell by binding to GM1 gangliosides that can associate with lipid rafts [68–70]. Each monomer in the CTB pentamer has a GM1 binding site positioned at the bottom face of the pentamer, facing away from the A moiety [71]. Attachment to a single GM1 ganglioside is sufficient for the cellular activity of CT [72]. However, intoxication is enhanced when the CTB pentamer binds to three or more GM1 gangliosides [73, 74]. Multivalent binding stabilizes the association with lipid rafts through GM1 clustering [75–77] and, due to the circular nature of the pentamer, induces membrane curvature in preparation for endocytosis [63, 78]. The route of endocytosis varies depending on the cell type, yet all endocytic mechanisms converge on the sorting endosomes. From this early endosomal compartment, CT can return to the plasma membrane, move to the late endosomes and lysosomes, or traffic to the TGN.
3.2. Toxin transfer from the early endosomes to the TGN.
The sorting endosomes are tubulo-vesicular structures in which the majority of the membrane is found in narrow, elongated tubules [79]. The separation of transmembrane proteins and membrane-bound cargo from soluble cargo is based on the surface-to-volume ratio of the organelle: most membrane-associated cargo partitions into the tubular compartment, while soluble cargo remains in the lumenal volume of the vesicular compartment [80]. Lipid sorting to the tubular or vesicular components of the organelle is based on the physical properties of the lipid [81]. Vesicles derived from the tubules are directed back to the plasma membrane or to the TGN, in both cases either directly or through a recycling endosome intermediate. The lumenal content of the sorting endosome enters a pathway for delivery to the late endosomes and lysosomes [80]. CT, through its association with GM1, uses this lipid-based sorting mechanism to reach the TGN.
GM1 is an acidic glycosphingolipid with an extracellular oligosaccharide that is recognized by CTB and a membrane-associated ceramide that determines its route of intracellular transport. Different species of GM1 vary in the length and saturation of the ceramide moiety [63, 82, 83]. An unsaturated ceramide directs GM1 from the sorting endosomes to the TGN and ER, while GM1 species with saturated ceramides are distributed to the recycling or late endosomes. Binding to CT alters these outcomes, with the majority of all toxin-clustered GM1 species being routed to the late endosomes and lysosomes [83, 84]. This explains the extensive lysosomal degradation of cell-associated CT and its inefficient transport to the ER. Still, a minor pool of CT that is bound to unsaturated GM1 can move to the TGN and ER in a process that requires syntaxin 6, cholesterol, actin, and flotillin-1 [67, 83, 85–87]. The latter three factors are associated with lipid rafts [64, 65, 88], indicating these structures can be involved with both the endocytosis and intracellular transport of CT.
3.3. Retrograde toxin transport to the ER.
CT cannot be easily visualized in the ER by confocal or immunoelectron microscopy4, but its movement to the ER was eventually documented with the use of a recombinant CTB pentamer containing sites for tyrosine sulfation and N-linked glycosylation. Tyrosine sulfation occurs in the TGN and can be detected through the addition of radiolabeled sulfate to the target protein. N-linked glycosylation begins in the ER and continues in the Golgi apparatus; it can be detected by SDS-PAGE through the resulting mobility shift in the modified protein. With this strategy, it was reported that recombinant CTB is first sulfated in the TGN and then glycosylated in the ER. The core glycans are then modified by enzymes in the cis and medial Golgi stacks, which indicates CTB will cycle between the ER and Golgi. Fujinaga et. al. [48] used a CTB pentamer rather than the CT holotoxin for some of these studies. CTB, through its association with GM1 and lipid rafts, can thus reach the ER without a contribution from its A moiety.
A functional role for toxin transport to the ER was documented with toxicity assays that used brefeldin A (BfA) to block the retrograde trafficking pathway. BfA perturbs the formation of transport vesicles, causing the Golgi apparatus to collapse into the ER and the TGN to fuse with the early endosomes [89, 90]. The mixed ER/Golgi and TGN/endosomal systems do not communicate, so cargo in the TGN or endosomes cannot reach the ER. The CT-resistant phenotype of BfA-treated cells [41, 45, 91] thus established retrograde transport as an essential component of the intoxication process.
From the BfA work, it was initially thought that CT moves through the Golgi apparatus en route to the ER. The absence of a Golgi apparatus would consequently block CT movement into the ER. Yet an inhibition of toxin export from the fused TGN/endosomal system [4], rather than the absence of the Golgi apparatus, could also account for the inhibitory effect of BfA on CT. In support of this interpretation, it was later found that Exo2 – a drug that induces assimilation of the Golgi apparatus into the ER without affecting CT transport to or from the TGN – does not confer resistance to CT [61]. It thus appears that CT can bypass the Golgi apparatus as it moves from the TGN to the ER.
3.4. Toxin retention in the ER.
After CT is delivered to the ER, its A1 subunit is separated from the rest of the toxin and processed for export to the cytosol. Toxin disassembly is inefficient [92], so CT must be retained in the ER to ensure the release of CTA1 from CTA2/CTB5. A KDEL tetrapeptide at the C-terminus of the A2 subunit is used for this purpose.
A C-terminal KDEL tag serves as a retention/retrieval motif for many resident ER proteins. When these proteins escape the ER, they are recognized by a KDEL receptor in the Golgi apparatus and returned to the ER by a coatomer-dependent retrograde transport mechanism [93]. CT utilizes this pathway to move back from the Golgi to the ER [3]. This minimizes the time CT spends in the Golgi, as evidenced by the different patterns of N-linked glycosylation from various forms of recombinant toxins: KDEL-containing holotoxins do not receive Golgi-specific glycan modifications [44, 48], but glycans in the CTB pentamer are remodeled by cis and medial Golgi enzymes when a functional KDEL tag is missing [48]. Other studies with a recombinant CT lacking the KDEL sequence found that the tag is not required for intoxication but instead improves the potency of the toxin [94]. CTB is thus responsible for toxin transport to the ER, while CTA2 is responsible for the KDEL-driven retention/retrieval of ER-localized CT. Optimal intoxication requires efficient retention in the ER to complete the downstream disassembly and translocation events.
4. CTA1 Translocation from the ER to the cytosol
CT reaches the ER as a holotoxin with an intact CTA1/CTA2 disulfide bond. However, only the A1 subunit enters the cytosol. The ER-to-cytosol export of CTA1 thus requires toxin reduction, toxin disassembly, and chaperone-assisted delivery of the dissociated A1 subunit to a translocon pore in the ER membrane. The final event involves an interaction between CTA1 and ER-associated degradation (ERAD), an endogenous quality control system that recognizes misfolded or misassembled proteins in the ER and transfers them to the cytosol for degradation [95, 96].
4.1. Toxin reduction and disassembly.
The disulfide bridge between CTA1 and CTA2 is reduced in the ER. This occurs at the resident redox state of the ER [97] and can be further stimulated by protein disulfide isomerase (PDI) [98, 99] or, possibly, other ER-localized oxidoreductases. However, reduction alone does not trigger toxin disassembly: extensive non-covalent contacts between CTA1 and CTA2/CTB5 maintain a stable, intact holotoxin after cleavage of the A1/A2 disulfide bond [100, 101]. Disassembly requires an additional interaction with PDI [50, 102], which uses conditional disorder as a unique mechanism to displace reduced CTA1 from the rest of the toxin [103] (Fig. 3).
Figure 3. CT disassembly in the ER.
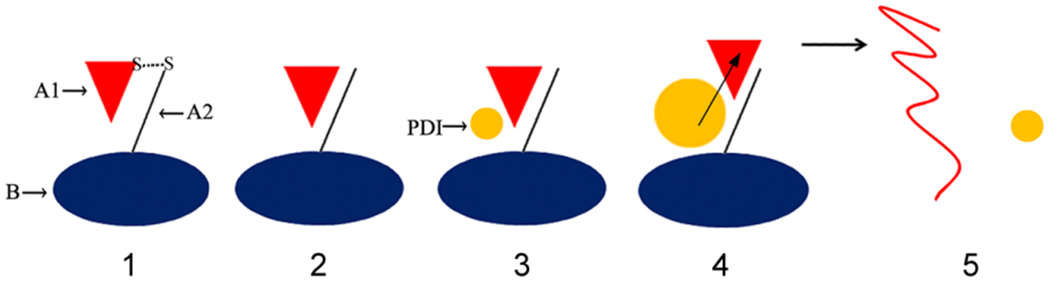
(1) CT travels from the cell surface to the ER as an intact, disulfide-linked holotoxin. (2) The CTA1/CTA2 disulfide bond is reduced in the ER [97, 98], but CTA1 remains associated with CTA2/CTB5 [50, 100–102] through extensive non-covalent contacts. (3) Reduced PDI binds to holotoxin-associated CTA1 [50, 102]. (4) PDI partially unfolds upon contact with CTA1, and the expanded hydrodynamic size of unfolded PDI acts as a wedge to dislodge CTA1 from CTA2/CTB5 [103]. (5) The dissociated CTA1 subunit unfolds spontaneously at 37°C [52], which consequently displaces its PDI binding partner [50]. PDI regains its native conformation after release from CTA1 [103], while disordered CTA1 is treated as a substrate for ERAD-mediated translocation to the cytosol [49, 51].
PDI-deficient cells are resistant to CT, which highlights the essential role of PDI in the intoxication process [50]. The efficiency of toxin disassembly by PDI also appears to influence toxin potency. This could explain differences in the cellular and physiological responses to CT vs. Escherichia coli heat-labile enterotoxin (LT), another AB5 toxin that is highly related to CT (Box 2). Both toxins bind the same GM1 receptor, have identical catalytic mechanisms, and follow the same intracellular trafficking pathway to their shared Gsα target [14–16]. Despite these similarities, CT is more potent than LT in cell culture [94, 104, 105]. We recently reported that PDI disassembles CT more efficiently than LT [105], which provides a possible explanation for the greater cellular potency of CT: the limited release of LTA1 from LTA2/LTB5 would restrict its entry into the cytosol and thus minimize the effects of LT intoxication in comparison to CT. The correlation between toxin disassembly and toxin potency was also observed with a CT/LT hybrid toxin that exhibited lower potency and less efficient PDI-driven disassembly than native LT [105].
Box 2. E. coli heat-labile enterotoxin.
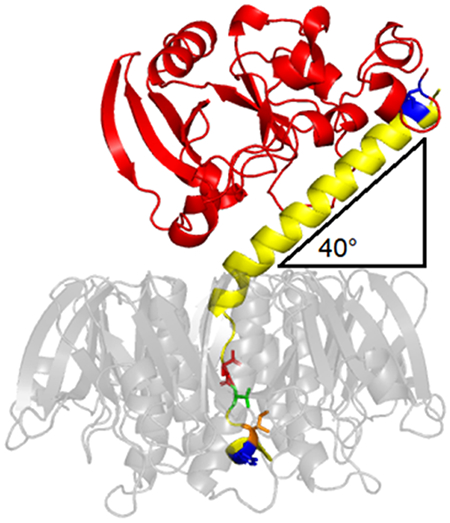
Enterotoxigenic E. coli can induce a diarrheal response through the production of LT, an AB5 protein toxin with 82% amino acid sequence identity to CT [15–17]. Despite similar mechanisms of toxicity, CT is more potent than LT. The difference in potency was previously mapped to four amino acid differences between the CTA2 and LTA2 linkers that anchor their A1 subunits to the B pentamer [104]. Those differences, which are highlighted in the LT ribbon diagram above, contribute to the altered interdomain architecture of CT and LT [106]: with respect to the plane of their respective B pentamers, there is a 9° difference between the tilt angles of CTA1 (49°) and LTA1 (40°) [105]. Docking simulations have suggested the tilt angle influences the mode of interaction with PDI [105], with corresponding effects on toxin disassembly, toxin potency, and disease outcomes. The A1 subunit is red; the A2 subunit is yellow; cysteines for the A1/A2 disulfide bond are blue; and the B pentamer is translucent grey to reveal the four amino acid differences between LTA2 and CTA2 that are responsible for the lower potency of LT (residues 229, 230, 232, and 233, shown in stick format). PDB entry 1LTS [107].
4.2. Toxin unfolding and activation of the ERAD system.
CTA1 is held in a stable conformation when associated with the CT holotoxin [108, 109]. However, the isolated CTA1 subunit is an unstable protein with a highly disordered conformation at the physiological temperature of 37°C [52, 110]. The PDI-driven release of CTA1 from CTA2/CTB5 thus results in the spontaneous unfolding of the dissociated A1 subunit. This prevents the potential unproductive reassembly of a CT holotoxin in the ER [111]. It also identifies CTA1 as an unfolded protein for ERAD-mediated export to the cytosol.
CHO cell lines that cannot efficiently export ERAD substrates to the cytosol are resistant to several ER-translocating toxins, including CT, LT, pertussis toxin, exotoxin A, and ricin [112–114]. This provided functional evidence for the role of ERAD in the intoxication process. All the aforementioned toxins contain labile A chains that can be stabilized and trapped in the ER through the application of chemical chaperones, resulting in a toxin-resistant phenotype [49, 51, 114]. Instability in the isolated A chain thus represents a common strategy for triggering ERAD-mediated toxin export to the cytosol [53].
After delivery to the cytosol, most ERAD substrates are rapidly degraded by the ubiquitin-proteasome system [95]. The A chains of ER-translocating toxins avoid this fate because they lack the lysine residues that serve as the attachment site for ubiquitin. Arginine residues are instead found throughout the protein sequence. For example, CTA1 contains fifteen arginine residues but only two lysine residues. This amino acid bias is not found in CTB or the B subunits of other ER-translocating toxins [115, 116], which indicates there was an evolutionary pressure on the A chain to replace lysine with a conserved amino acid substitution. In support of this possibility, recombinant toxins with one or more lysine-for-arginine replacements in the toxin A chain were found to be less potent than their cognate wild-type toxins. Proteasome inhibitors could restore wild-type levels of toxin activity to the lysine-enriched recombinant toxin, which indicated the loss of activity was due to effective proteasome-mediated clearance of the mutant A chain from the cytosol [117–119]. CTA1 and other toxin A chains thus exploit ERAD for passage into the cytosol but then evade the ubiquitin-dependent degradation that usually accompanies the processing of an ERAD substrate [36, 116].
4.3. CTA1 interactions with ER chaperones.
After its release from the holotoxin, CTA1 interacts with a series of ER-localized chaperones and/or ERAD components. This involves processing by Hsp40 and Hsp70 family members that act sequentially on a client protein. An Hsp40 chaperone initially binds to the misfolded/unfolded substrate and then transfers it to an Hsp70 chaperone which, in the open ATP-bound conformation, has low affinity for the client. ATP hydrolysis is stimulated by a J domain in the Hsp40 chaperone during substrate transfer, thus converting Hsp70 to an ADP-bound conformation that closes on the substrate for tight binding [120]. Following this general pattern, CTA1 has been reported to interact with two ER-localized Hsp40 family members: ERdj3 and ERdj5. ERdj3 specifically recognizes the disordered conformation of CTA1 and masks the exposed hydrophobic amino acids in the unfolded toxin. Its function is required for the ER-to-cytosol export of CTA1 and, thus, the cellular activity of CT [121]. ERdj5 also binds to CTA1 and is active in toxin translocation to the cytosol [122]. It has been shown to enhance CTA1 binding to BiP, an ER-localized Hsp70 family member that prevents the aggregation of CTA1 at 37°C and facilitates toxin passage into the cytosol [123]. CTA1 is released from BiP through the redundant actions of Grp170 and Sil1, two ER-localized nucleotide exchange factors that convert BiP back to its open, low-affinity conformation by facilitating the replacement of ADP with ATP in the nucleotide binding pocket of BiP [124]. As described below, these collective events are often coupled with substrate docking to a protein-conducting channel in the ER membrane. The intrinsic instability of free CTA1 thus co-opts the normal process of ERAD chaperone interactions for delivery to a translocon pore and subsequent passage into the cytosol.
4.4. CTA1 interactions with ER translocon pores.
The ER contains two translocon pores for protein egress to the cytosol: Sec61 and Hrd1. Both appear capable of transferring CTA1 to the cytosol. The Sec61 translocon is responsible for co-translational import into the ER, but it can also facilitate the removal of ERAD substrates from the ER [125]. CTA1 co-immunoprecipitated with Sec61 when synthesized directly in ER-derived microsomes, and occlusion of the Sec61 pore trapped CTA1 within the microsomes [126]. Its recruitment to the Sec61 channel could possibly be facilitated by BiP, which has established interactions with both Sec61 and CTA1 [123, 127].
BiP also interacts with Sel1, a core component of the multimeric Hrd1 translocon [128]. Knockdown of Sel1 by siRNA reduced the level of cytosolic CTA1 by 50%, thus establishing its functional role in CTA1 translocation [122]. Other siRNA studies recorded a similar 50% reduction in cytosolic CTA1 after the depletion of Derlin-1, another component of the Hrd1 complex, or Hrd1 itself [129, 130]. For these two studies, the lower levels of cytosolic CTA1 corresponded to a 2-fold drop in the toxin-induced cAMP response. Although CTA1 is not a target for ubiquitination, a dominant negative variant of Hrd1 that lacked the E3 ubiquitin ligase activity of its RING-finger domain inhibited CTA1 translocation to the cytosol and CT activity [130]. Dominant negative variants of Ube2g2, a Hrd1-associated E2 ubiquitin conjugating enzyme, also inhibited the cytosolic accumulation of CTA1 [130]. These collective observations were consistent with the proposed function of the Hrd1 translocon, which involves the input of multiple proteins and auto-ubiquitination as a trigger for cargo passage through the protein-conducting channel [131].
From the available data, it appears CTA1 can use both the Sec61 and Hrd1 translocons for entry into the cytosol. The Hrd1 pathway has been characterized in greater detail and fits the expected pattern of Hrd1-substrate interactions, but the loss-of-function studies have only documented a partial inhibition of CT activity. This stands in contrast to the complete inhibition of CT activity resulting from the loss of other ERAD factors [5, 50, 132] and suggests there is an alternative mechanism (i.e., Sec61) for CTA1 translocation to the cytosol. Cellular loss-of-function studies with Sec61 are problematic because of its essential nature, but other ER-translocating toxins also interact with Sec61 (exotoxin A, ricin) or both Sec61 and Hrd1 (Shiga toxin) [133–136].
5. Cytosolic Activity of CTA1
CTA1 will exit the ER in an unfolded state, which generates two problems: (i) a linear protein will not spontaneously move in a unidirectional manner through the translocon pore; and (ii) CTA1 will not spontaneously return to a folded, functional conformation in the cytosol. Extraction of CTA1 through the translocon pore and refolding of the exported toxin must therefore involve the contribution of host factors. An initial “push” into the translocon pore may be mediated by torsin A, an AAA+ ATPase located in the ER lumen [137]. This process is not well-characterized, but, as described below, some detail is available on the extraction and refolding of CTA1 by cytosolic factors.
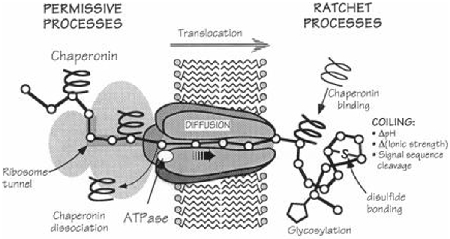
5.1. A ratchet mechanism for CTA1 extraction to the cytosol.
Most ERAD substrates are delivered to the cytosol through the action of the AAA+ ATPase p97, but this protein is not required for CTA1 export to the cytosol [138, 139]. CTA1 is extracted from the ER through a distinct mechanism involving the coordinated action of two cytosolic chaperones, Hsp90 and Hsc705. Loss-of-function studies have shown both chaperones are needed for CT activity and CTA1 translocation to the cytosol [5, 132]. An in vitro reconstitution system further demonstrated that the addition of Hsp90 to the buffer is sufficient for CTA1 export from ER-derived microsomes [140]. The ubiquitin-independent turnover of cytosolic CTA1 [52, 113, 141] may explain why Hsc70 is required for the cellular but not in vitro translocation of CTA1: we propose the efficiency of Hsp90-driven toxin translocation is enhanced by Hsc70, thus allowing the balance between toxin delivery to the cytosol and toxin degradation in the cytosol to favor the accumulation of cytosolic toxin. The slow rate of toxin export in the absence of Hsc70 would thus result in toxin clearance from the cytosol but not from an in vitro buffer where degradation does not occur.
Chaperones usually interact with the surface-exposed hydrophobic amino acids of a disordered protein, but Hsp90 and Hsc70 recognize defined amino acid sequences in CTA1: Hsp90 binds to an N-terminal RPPDEI motif (residues 11-16), while Hsc70 binds to an internal YYIYVI motif (residues 83-88 of the 192 amino acid protein) [5, 142]. The RPPDEI motif is also found at the N- or C-termini of other toxin A chains that move from the ER to the cytosol and function as ADP-ribosyltransferases [142]. The Hsc70 binding motif is unique to CTA1, as is a second Hsp90 binding motif (LDIAPA) spanning amino acid residues 153-158. The functional significance of the second Hsp90 binding motif has not yet been determined, but mutations to any of the RPP residues in the N-terminal Hsp90 binding motif will sequester CTA1 in the ER [142].
As expected for a typical chaperone-substrate interaction, binding to either Hsp90 or Hsc70 will induce a gain-of-structure in disordered CTA1 [5, 140]. The interaction with Hsp90 also follows a typical pattern where (i) toxin binding requires the chaperone to be complexed with ATP and (ii) toxin refolding requires ATP hydrolysis by the chaperone. We found that the Hsp90-driven extraction of CTA1 from ER-derived micrososomes also required ATP binding and hydrolysis [140]. This observation, combined with the location of the N-terminal Hsp90 binding motif in CTA1, led us to propose a ratchet mechanism (Box 3) for toxin translocation in which CTA1 refolding is coupled with its extraction from the translocon pore: the gain-of-structure induced by Hsp90 prevents folded CTA1 from sliding back into the translocon pore, thus ensuring its unidirectional movement out of the ER. The process begins when the N-terminal RPPDEI motif of CTA1 appears at the cytosolic face of the ER membrane. Extraction continues as the Hsc70 and second Hsp90 binding sites emerge from the translocon pore, thereby allowing further toxin refolding and a continued ratchet mechanism for full removal of CTA1 from the ER. This process may represent a variation on the Hsp90-dependent mechanism in dendritic cells that delivers extracellular proteins to the cytosol for antigen cross-presentation [143, 144].
Box 3. The Brownian Ratchet.
Proteins that move through a translocon pore are thought to be unfolded or largely unstructured. However, a linear polypeptide will not spontaneously move in unidirectional fashion through the pore. Thermal fluctuations will move the protein back-and-forth within the pore, but there will be no net progress for movement in either direction. Oster and colleagues proposed directionality can be accomplished with a “Brownian ratchet” in which random diffusion becomes biased through asymmetric interactions at one face of the pore [145, 146]. For co-translational import into the ER, this asymmetry takes the form of folding events that produce a three-dimensional protein structure incapable of re-entering the pore. The linear portion of the protein can still fluctuate out of the pore by Brownian motion, but, once exposed to the ER lumen, its folding will prevent back-sliding into the pore. This repetitive process will eventually deliver the entire polypeptide through the translocon pore and into the ER. The ubiquitination of an ERAD substrate as it exits the ER has been proposed to act in a similar manner for retro-translocation to the cytosol [147, 148], but CTA1 dislocation does not involve its ubiquitination [117]. Toxin extraction from the ER may instead be mediated by the refolding that occurs when Hsp90 binds to the N-terminal RPPDEI sequence of CTA1.
With a Brownian ratchet, exposure of the RPPDEI motif at the cytosolic face of the ER membrane is based on the probability of random thermal motions resulting in the short-term directional movement of CTA1 through the translocon pore. N-terminal amino acid extensions to CTA1 would make it less likely that the back-and-forth thermal fluctuations within the translocon pore would push the toxin far enough into the cytosol for the appearance of its RPPDEI tag. Longer extensions would accordingly be more deleterious than shorter extensions. This result was reported for CTA1 variants with N-terminal extensions containing 6, 16, or 23 amino acids from the E. coli heat-stable enterotoxin: the extensions did not have a significant effect on in vitro toxin activity but greatly reduced cellular potency, with a 1,000-fold drop in the toxin-driven cAMP response resulting from the 6 amino acid extension and a 100,000-fold drop resulting from the 23 amino acid extension [149]. All ER-translocating toxins with the RPPDEI motif contain the sequence within the first 18 amino acids of the N- or C-terminus [142], which would be expected from the Brownian ratchet model. Image from [146], reprinted with permission.
5.2. Additional roles for Hsp90 and Hsc70 in the cellular activity of CTA1.
The refolding of a client protein usually results in its dissociation from the chaperone, but both Hsp90 and Hsc70 remain associated with the refolded CTA1 polypeptide [5, 140]. This is critical for the cellular activity of CTA1, as the chaperone-free toxin would revert to a disordered, inactive state. The lack of CTA1 activity at 37°C has been confirmed with in vitro ADP-ribosylation assays [150, 151]. However, Hsp90-ATP can induce a gain-of-function when added to the in vitro assay either before or after heating CTA1 to 37°C. The continued association with Hsp90 and/or Hsc70 likely prolongs the half-life of cytosolic CTA1 as well, given that its ubiquitin-independent proteasomal degradation by the core 20S proteasome requires an unfolded substrate [51, 52]. CTA1 thus exploits both typical and atypical interactions with cytosolic chaperones to reach the cytosol in a functional state.
5.3. CTA1 activation by other host factors.
The cytosolic activity of CTA1 is stimulated through an essential interaction with host ADP-ribosylation factors (ARFs) [151]. These proteins normally function in vesicle transport, but they are named ARFs because they were first identified as allosteric activators of CTA1 (an ADP-ribosyltransferase) [152, 153]. The interaction with ARF generates conformational changes in CTA1 that optimize its enzymatic activity [54, 154], but ARF does not induce a gain-of-structure in disordered CTA1 [151]. As such, CTA1 exhibits no in vitro activity when exposed to an ARF at 37°C [140, 151]. ARF-stimulated enzymatic activity can be attained at physiological temperature if CTA1 is first placed in a folded conformation through its association with Hsp90-ATP [140]. The lipid raft environment where Gsα is located also exhibited a “lipochaperone” effect on CTA1, inducing both a gain-of-structure and ARF-stimulated gain-of-function when added to the unfolded toxin [151, 155]. The greatest level of activity was obtained when Hsp90-ATP was incubated with CTA1 at 37°C before exposure to both ARF and large unilamellar vesicles with the composition of a lipid raft [140]. This mirrored the sequence of events that would occur in the host cell and indicated the order of host factor interactions is important for CTA1 to achieve its optimal cytosolic activity.
6. Atypical Activation of the Unfolded Protein Response (UPR) by CT
We recently documented an additional CT-linked signalling pathway that leads to induction of the UPR [156]. The UPR acts in concert with ERAD to manage the load of misfolded proteins in the ER [96, 157, 158]. When stress conditions overwhelm the ERAD system, a compensatory UPR (i) reduces overall protein synthesis to limit the accumulation of misfolded proteins, (ii) enhances the production of ERAD factors to clear the misfolded proteins, and (iii) stimulates the production of ER membrane to enlarge the compartment and dilute the pool of misfolded proteins. Components of the vesicle trafficking machinery are also up-regulated during the UPR [159]. This final effect likely accounts for the increased efficiency of CTA1 delivery to the cytosol and additional sensitization to CT when the UPR is operational.
6.1. Characteristics of UPR activation.
CT induction of the UPR occurred in a dose-dependent manner and was not replicated with enzymatically inert toxins [156]. Elevated levels of cAMP could be detected within 90 min of toxin exposure, but a transcriptional reporter for the UPR was not active until 4 h of intoxication. A disconnect between cAMP production and UPR activation was further emphasized by the lack of UPR in cells treated with forskolin, an agonist of adenylate cyclase. In contrast, chemical activation of the heterotrimeric G protein led to an UPR. Initiation of the UPR thus required a functional toxin and appeared to involve the G protein target of CT, yet the cAMP response that results from CT activity against Gsα was not responsible for the effect.
The Draper lab also described a toxin-induced signalling pathway that up-regulates the expression of proteins in the ERAD system, but this effect was caused by relatively high concentrations of the CTB pentamer (~10 nM) rather than the 1 nM concentration of CT required for UPR activation [160]. It also appeared to alter the translation, rather than transcription, of ERAD factors and occurred within 15 min of exposure to CTB. CT thus appears to exploit multiple, distinct signalling mechanisms for sensitizing the target cell to intoxication.
6.2. UPR-enhanced delivery of CTA1 to the cytosol.
A remarkable increase in the cytosolic pool of CTA1 occurs between 4 and 5 h of intoxication [51, 156], which coincides with the toxin-induced activation of the UPR [156] and the maximal cAMP response that occurs between 3.5 and 6.5 hours of intoxication [149]. The jump in cytosolic CTA1 is not seen with enzymatically inert toxins that do not trigger the UPR [156]. Rapid toxin delivery to the cytosol, within 15 minutes of toxin exposure, can also be accomplished through chemical activation of either the heterotrimeric G protein or the UPR [156]. Furthermore, toxin sensitization results from chemical activation of the UPR [156, 161]. These collective observations suggest CT intoxication is a two-stage event in which the initially slow and inefficient delivery of CTA1 to the cytosol is enhanced by the UPR activation that occurs after the first pool of CTA1 reaches the cytosol and its G protein target.
The elevated levels of cytosolic CTA1 resulting from UPR activation could be attributed to enhanced ERAD processing of the toxin, more efficient toxin transport to the ER, or both. The cellular mechanisms at play remain to be established, but it has been reported that CT transport to the ER is more efficient when the ER calcium store is depleted [161]. This resulted in a toxin-sensitive phenotype. Transport was monitored by electron microscopy and ER-localized reduction of the CTA1/CTA2 disulfide bond in cells treated with thapsigargin, an inhibitor of the ER Ca2+-ATPase. Thapsigargin also induces an UPR through the inactivation of calcium-dependent ER chaperones and resulting accumulation of misfolded proteins. It was one of the methods used to document the rapid accumulation of cytosolic CTA1 [156]. The UPR thus appears to accelerate retrograde toxin transport to the ER, leading to a larger pool of ER-localized toxin for passage into the cytosol.
6.3. Mechanism of UPR activation.
The toxin-triggered UPR is a cAMP-independent event, yet it still appears to involve an active heterotrimeric G protein. It is therefore possible that the signaling pathway leading to UPR activation involves the dissociated βγ subunits rather than ADP-ribosylated Gsα. Other cAMP-independent outcomes resulting from exposure to an active CT could involve a Gβγ signaling mechanism as well [162–165]. Gβγ subunits can regulate the UPR in Arabidopsis thaliana [166, 167] and can interact with IP3 receptors to induce calcium release from the mammalian ER [168]. The Gβγ-stimulated depletion of ER calcium could thus, like thapsigargin, trigger an UPR through the inactivation of ER chaperones. With this model, an atypical G protein signaling pathway leads to UPR activation through the typical route of protein misfolding in the ER.
7. Potential Down-Regulation of the Toxin-Induced cAMP Response
It is thought that recovery from CT-induced diarrhea can only occur after the intoxicated enterocytes, which have a 3-5 day life span, are sloughed from the intestinal epithelium [12, 15, 19, 169–172]. As considered below, a number of observations indicate this is not the case. The collective evidence suggests it may instead be possible to reverse the effects of intoxication.
7.1. Removal of toxin-modified Gsα.
The ADP-ribosylation of Gsα is commonly referenced as an irreversible modification [12, 15, 19, 82]. Gsα would thus be locked in an active conformation for the lifetime of the cell, leading to an unending stimulation of adenylate cyclase and persistently high levels of cAMP. However, two mechanisms have been established for the clearance of ADP-ribosylated Gsα.
The Bourne and Milligan laboratories both demonstrated that Gsα is rapidly degraded after its modification by CT. Elevated levels of cAMP did not mimic this effect in unintoxicated cells [173, 174], but a short half-life was reported for a constitutively active mutant of Gsα [175]. The active, GTP-bound conformation of Gsα thus appears to be specifically targeted for degradation. Still, there is a residual pool of ADP-ribosylated G protein that evades degradation and is sufficient to propagate the cAMP response.
In addition to the turnover of ADP-ribosylated (active) Gsα, there is an ADP-ribosylarginine hydrolase that can remove ADP-ribose from the modified G protein. ADP-ribosylation is a normal cellular process mediated by a large superfamily of mammalian enzymes. Several cellular events are affected by the mono- or poly-ADP-ribosylation of proteins and nucleic acids [176, 177]. This regulatory mechanism also involves removal of the ADP-ribose moiety by ADP-ribosyl hydrolases, with ARH1 representing the family member that releases ADP-ribose from the arginine residue of a modified protein [177, 178]. The addition and removal of ADP-ribose as a regulatory mechanism was not fully appreciated when the mono-ADP-ribosylation activities of bacterial toxins were first elucidated around the 1970s [179–181], which likely explains why toxin-catalyzed ADP-ribosylation has traditionally been viewed as an irreversible event. However, Moss and colleagues have documented enhanced CT activity in the cells and intestinal loops of transgenic mice lacking ARH1 [182, 183]. This indicates that ARH1 limits the effects of intoxication by removing ADP-ribose from the modified G protein. Gsα could then hydrolyze its bound GTP and shift to an inactive state, thus shutting down the cAMP signaling cascade. Interestingly, female ARH1(−/−) mice displayed greater sensitivity to CT than male ARH1(−/−) mice. This correlated with clinical reports of more severe cholera outcomes in women than men [183] and suggested there is an additional, gender-specific mechanism to limit the effects of CT.
7.2. Removal of cytosolic toxin.
The rapid turnover of ADP-ribosylated Gsα and the action of ARH1 suggest the toxin-activated form of Gsα can be effectively cleared from the intoxicated cell, provided that CTA1 is also removed from the cytosol. The R-over-K amino acid bias in CTA1 protects it from rapid ubiquitin-dependent proteasomal degradation, but the cytosolic toxin still exhibits a two hour half-life that reflects its turnover by a ubiquitin-independent proteasomal mechanism [52]. Conditions that enhance the turnover of CTA1 reduce its potency, which indicates the amount of cytosolic toxin determines the extent of intoxication [117, 141, 184]. Likewise, differences in the efficiency of toxin disassembly and resulting toxin delivery to the cytosol may explain why CT is more potent than LT in cell culture [105]. These observations suggest endogenous cellular mechanisms are more effective at counteracting the effects of intoxication when there is less toxin in the cytosol. With this model, CT would ensure effective intoxication of the host cell by activating the UPR for enhanced CTA1 delivery to the cytosol [156].
7.3. Intoxication may require a threshold quantity of cytosolic toxin.
Early models of CT activity proposed a single molecule of cytosolic toxin was sufficient for irreversible intoxication of the target cell. This model was apparently influenced by early discussions on whether CT even had an enzymatic function, the supposedly irreversible nature of ADP-ribosylation, the presumed stability of the intracellular toxin, and the low levels of exogenously applied toxin that are required for a cellular response. In contrast, it now appears that a threshold concentration of cytosolic toxin is required to initiate the cAMP signaling cascade. Several studies have provided experimental support for this possibility. For example, we reported the depletion of Hsp90 by RNAi completely blocks CT intoxication but does not completely block CTA1 passage into the cytosol [132]. Likewise, mutant cell lines with attenuated ERAD translocation mechanisms are highly resistant to CT and LT despite releasing low levels of toxin into the cytosol [112, 113]. More recently, we have shown it is possible to detect low levels of cytosolic CTA1 without a corresponding increase in cAMP [185]. It thus appears that CT must exceed a threshold quantity of cytosolic toxin in order to elicit a cAMP response from the intoxicated cell.
7.4. Reversal of intoxication.
The cellular response to intoxication is influenced by (i) the extent of CTA1 translocation to the cytosol; (ii) the rate of CTA1 degradation in the cytosol; (iii) CTA1 activity against Gsα; (iv) the de-activation of ADP-ribosylated Gsα by ARH1 or proteolysis; and (v) the clearance of cAMP by cAMP phosphodiesterases. The balance between these factors usually favors CTA1 activity against Gsα and productive intoxication. However, as shown experimentally in cultured cells, this balance can be shifted to protect cells from intoxication without completely eliminating the cytosolic pool of toxin. An inhibition of toxin delivery to the cytosol, enacted after toxin exposure, could thus reduce the cytosolic pool of CTA1 to the point where the ongoing de-activation of Gsα would effectively reverse the cellular effects of intoxication.
Toxin delivery from the cell surface to the cytosol is an extremely inefficient process, and the small pool of cytosolic toxin is cleared from the host cell [185]. However, we hypothesize that a portion of internalized CT that is bound to unsaturated isoforms of GM1 can reside for prolonged intervals in the host endomembrane system. This pool of CT would be sequestered from lysosomal transport and would consequently act as a reservoir for continual delivery of CTA1 into the cytosol. Therapeutic strategies based on the inhibition of toxin binding to the cell surface [20, 22, 186] could thus prevent, but not treat, cholera. In contrast, a drug-induced inhibition of toxin movement from the ER to the cytosol could mitigate the effects of intoxication even after exposure to the toxin: when toxin translocation is blocked, the cytosolic pool of CTA1 would be degraded and would not be replenished from the endomembrane-localized reservoir of toxin. The host cell could then de-activate ADP-ribosylated Gsα and consequently recover from intoxication. Proof-of-principle for this therapeutic approach has been generated using glycerol-treated cells. Glycerol acts as a chemical chaperone to block the thermal unfolding of CTA1, thus trapping the toxin in the ER [51]. When applied to cells after an exposure to CT, glycerol lowered the initial quantity of cytosolic CTA1 and prevented the toxin-induced rise in cAMP. Cells treated with both glycerol and a proteasome inhibitor had higher levels of cytosolic CTA1 than cells treated with glycerol alone, which indicated the loss of cytosolic toxin in the presence of glycerol alone was due to (i) proteasome-mediated clearance of the cytosolic toxin and (ii) an inhibition of further toxin delivery to the cytosol [185]. Preliminary experiments indicate glycerol can also reverse the effects of intoxication after a cAMP response has already begun (Fig. 4). Long-term exposure to glycerol could not be used as an actual therapeutic, but there are other feasible options for translocation inhibitors that could be examined in the future (e.g., geldanamycin derivatives to block Hsp90-driven extraction from the ER [132]; rutin hydrate to block PDI-driven toxin disassembly [99]).
Figure 4. A post-exposure block of toxin translocation can reverse the effects of intoxication.
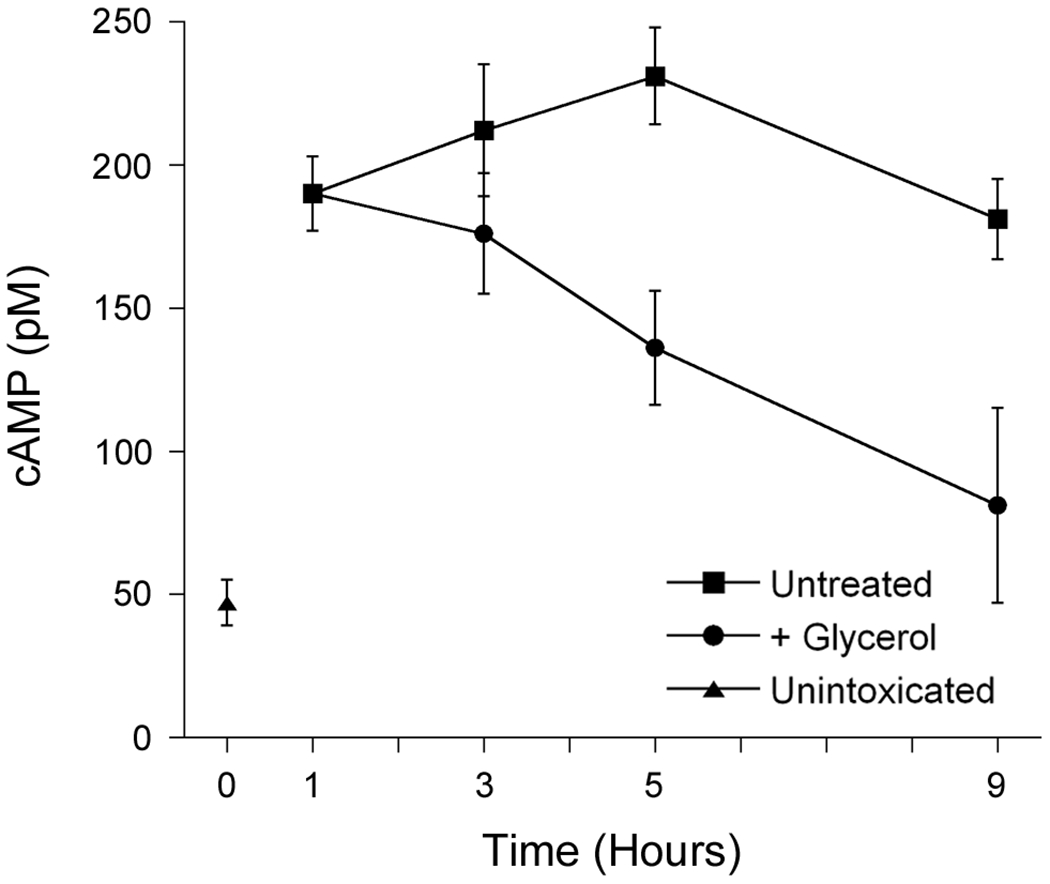
Following our general protocols for toxin pulse-chase experiments and intracellular cAMP calculations [114, 187], HeLa cells were surface-labeled with 1 µg/mL of CT at 4°C and chased at 37°C in toxin-free medium for the indicated intervals before cAMP levels were quantified (squares). After 1 h of chase, 10% glycerol was added to the medium of one subset of cells (circles). The basal level of cAMP from unintoxicated cells was also determined (triangle). Values represent the averages ± standard deviations from triplicate samples. One of two representative experiments is shown.
8. Summary
Cholera arises from a deceptively simple process. The few organisms of V. cholerae that survive the gastric barrier colonize the apical face of the small intestine and undergo explosive growth. The production of CT then induces a diarrheal response that flushes the pathogen back into the environment for another round of fecal-oral transmission to a new host. Yet, each step of the intoxication process manipulates an aspect of host cell biology:
CTB-induced clustering of GM1 in lipid rafts generates the membrane curvature to facilitate endocytosis
CTB association with unsaturated GM1 isoforms uses the natural process of lipid partitioning for toxin routing from the endosomes to the retrograde transport pathway
A KDEL tag at the C-terminus of CTA2 ensures efficient retention of the toxin in the ER
An interaction with ER resident protein PDI releases CTA1 from the rest of the toxin
The intrinsic instability of the dissociated A1 subunit leads to its processing by the ERAD system
Cytosolic chaperones recognize specific amino acid motifs in CTA1 for its ratchet-driven extraction from the ER
The R-over-K amino acid bias in CTA1 protects it from ubiquitin-dependent proteasomal degradation
ARF proteins, normally involved in vesicle transport, act as allosteric activators of CTA1
cAMP-independent activation of the UPR by the cytosolic toxin enhances further toxin delivery to the cytosol
These collective events result in the ADP-ribosylation of Gsα by CTA1, activation of a cAMP/PKA signalling pathway, and chloride efflux through apical CFTR channels. A standard signal transduction pathway is reversible. Under the right conditions, it may also be possible to eliminate the toxin-induced elevation of cAMP and thereby reverse the diarrheal effects of intoxication.
ACKNOWLEDGEMENTS
We thank Albert Serrano for preparing the toxin ribbon diagrams and Dr. Patrick Cherubin for preparing the schematic diagram in Figure 3. Work in the Teter lab on the cell biology of CT is currently funded by the National Institutes of Health under Award Number R01AI137056 to KT. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Footnotes
Abbreviations: cholera toxin, CT; cystic fibrosis transmembrane regulator, CFTR; trans-Golgi network, TGN; endoplasmic reticulum, ER; protein kinase A, PKA; sodium-hydrogen exchanger 3, NHE3; brefeldin A, BfA; ER-associated degradation, ERAD; protein disulfide isomerase; PDI; Escherichia coli heat-labile enterotoxin, LT; ADP-ribosylation factors, ARFs; unfolded protein response, UPR.
CTA1 amino acid residue numbers throughout the manuscript refer to the mature toxin sequence, after removal of the N-terminal signal peptide that delivers it to the bacterial periplasm.
The Gsα residue targeted for ADP-ribosylation by CTA1 is also referenced as R187; this reflects a difference in the particular splice variant of Gsα that is under consideration rather than a difference in the target residue [1].
In contrast to most cell types, CT is efficiently delivered to the ER of Vero cells and can be readily visualized in the compartment by immunofluorescence microscopy [2–4]. The basis for this phenomenon has not been established, but it could be related to the particular GM1 isoforms synthesized by Vero cells.
Hsc70 and Hsp70, the stress-inducible variant of Hsc70, play redundant roles in CT intoxication [5].
REFERENCES
- 1.Freissmuth M, Gilman AG (1989) Mutations of GS alpha designed to alter the reactivity of the protein with bacterial toxins. Substitutions at ARG187 result in loss of GTPase activity. J Biol Chem 264(36): 21907–21914. [PubMed] [Google Scholar]
- 2.Chen A, Hu T, Mikoryak C, Draper RK (2002) Retrograde transport of protein toxins under conditions of COPI dysfunction. Biochim Biophys Acta 1589(2): 124–139. [DOI] [PubMed] [Google Scholar]
- 3.Majoul I, Sohn K, Wieland FT, Pepperkok R, Pizza M, Hillemann J, Soling HD (1998) KDEL receptor (Erd2p)-mediated retrograde transport of the cholera toxin A subunit from the Golgi involves COPI, p23, and the COOH terminus of Erd2p. J Cell Biol 143(3): 601–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Richards AA, Stang E, Pepperkok R, Parton RG (2002) Inhibitors of COP-mediated transport and cholera toxin action inhibit simian virus 40 infection. Mol Biol Cell 13(5):1750–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Burress H, Kellner A, Guyette J, Tatulian SA, Teter K (2019) HSC70 and HSP90 chaperones perform complementary roles in translocation of the cholera toxin A1 subunit from the endoplasmic reticulum to the cytosol. J Biol Chem 294(32): 12122–12131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Harris JB, LaRocque RC, Qadri F, Ryan ET, Calderwood SB (2012) Cholera. Lancet 379(9835): 2466–2476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Clemens JD, Nair GB, Ahmed T, Qadri F, Holmgren J (2017) Cholera. Lancet 390(10101): 1539–1549. [DOI] [PubMed] [Google Scholar]
- 8.Kanungo A, Azman AS, Ramamurthy T, Deen J, Dutta S (2022) Cholera. Lancet 399(1429–1440. [DOI] [PubMed] [Google Scholar]
- 9.Davies HG, Bowman C, Luby SP (2017) Cholera - management and prevention. J Infect 74(S66–S73. [DOI] [PubMed] [Google Scholar]
- 10.Zuckerman JN, Rombo L, Fisch A (2007) The true burden and risk of cholera: implications for prevention and control. Lancet Infect Dis 7(521–530. [DOI] [PubMed] [Google Scholar]
- 11.Ali M, Nelson AR, Lopez AL, Sack DA (2015) Updated global burden of cholera in endemic countries. PLoS Negl Trop Dis 9(6): e0003832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Reidl J, Klose KE (2002) Vibrio cholerae and cholera: out of the water and into the host. FEMS Microbiol Rev 26(2): 125–139. [DOI] [PubMed] [Google Scholar]
- 13.Davis BM, Waldor MK (2003) Filamentous phages linked to virulence of Vibrio cholerae. Curr Opin Microbiol 6(35–42. [DOI] [PubMed] [Google Scholar]
- 14.Sanchez J, Holmgren J (2008) Cholera toxin structure, gene regulation and pathophysiological and immunological aspects. Cell Mol Life Sci 65(9): 1347–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.De Haan L, Hirst TR (2004) Cholera toxin: a paradigm for multi-functional engagement of cellular mechanisms (Review). Mol Membr Biol 21(2): 77–92. [DOI] [PubMed] [Google Scholar]
- 16.Heggelund JE, Bjørnestad VA, Krengel U, Vibrio cholerae and Escherichia coli heat-labile enterotoxins and beyond, in The Comprehensive Sourcebook of Bacterial Protein Toxins, 4th edition, Alouf JE, Ladant D, and Popoff MR, Editors. 2015, Elsevier: Waltham, MA. p. 195–229. [Google Scholar]
- 17.Kopic S, Geibel JP (2010) Toxin mediated diarrhea in the 21st century: the pathophysiology of intestinal ion transport in the course of ETEC, V. cholerae and rotavirus infection. Toxins 2(2132–2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Baxter PS, Goldhill J, Hardcastle J, Hardcastle PT, Taylor CJ (1988) Accounting for cystic fibrosis. Nature 335(211. [DOI] [PubMed] [Google Scholar]
- 19.Gabriel SE, Brigman KN, Koller BH, Boucher RC, Stutts MJ (1994) Cystic fibrosis heterozygote resistance to cholera toxin in the cystic fibrosis mouse model. Science 266(5182): 107–109. [DOI] [PubMed] [Google Scholar]
- 20.Muanprasat C, Chatsudthipong V (2013) Cholera: pathophysiology and emerging therapeutic targets. Future Med Chem 5(7): 781–798. [DOI] [PubMed] [Google Scholar]
- 21.Thiagarajah JR, Verkman AS (2012) CFTR inhibitors for treating diarrheal disease. Clin Pharmacol Ther 92(3): 287–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sousa FBM,Noleto IRSG, Chaves LS, Pacheco G, Oliveira AP, Fonseca MMV, Medeiros JVR (2020) A comprehensive review of therapeutic approaches available for the treatment of cholera. J Pharm Pharmacol 72(12): 1715–1731. [DOI] [PubMed] [Google Scholar]
- 23.Blake PA, Historical perspectives on pandemic cholera, in Vibrio cholerae and cholera: molecular to global perspectives, Wachsmuth IK, Blake PA, and Olsvik O, Editors. 1994, ASM Press: Washington, D.C. p. 293–295. [Google Scholar]
- 24.Barua D, History of cholera, in Cholera, Barua D and Greenough WB, Editors. 1992, Springer Science + Business Media: New York. p. 1–36. [Google Scholar]
- 25.Radlovic N (2012) Cystic fibrosis. Srp Arh Celok Lek 140(3-4): 244–249. [PubMed] [Google Scholar]
- 26.Rodman DM, Zamudio S (1991) The cystic fibrosis heterozygote - advantage in surviving cholera? Med Hypotheses 36(3): 253–258. [DOI] [PubMed] [Google Scholar]
- 27.Romeo G, Devoto M, Galietta LJV (1989) Why is the cystic fibrosis gene so frequent? Hum Genet 84(1–5. [DOI] [PubMed] [Google Scholar]
- 28.Sheppard DN, Welsh MJ (1999) Structure and function of the CFTR cholride channel. Physiol Rev 79(1 Suppl): S23–45. [DOI] [PubMed] [Google Scholar]
- 29.Shteinberg M, Haq IJ, Polineni D, Davies JC (2021) Cystic fibrosis. Lancet 397(2195–2211. [DOI] [PubMed] [Google Scholar]
- 30.McBennett KA, Davis PB, Konstan MW (2022) Increasing life expectancy in cystic fibrosis: advances and challenges. Pediatr Pulmonol 57(Suppl 1): S5–S12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Anstee DJ (2010) The relationship between blood groups and disease. Blood 115(23):4635–4643. [DOI] [PubMed] [Google Scholar]
- 32.Rowe JA, Handel IG, Thera MA, Deans A-M, Lyke KE, Kone A, Diallo DA, Raza A, Kai O, Marsh K, Plowe CV, Doumbo OK, Moulds JM (2007) Blood group O protects against severe Plasmodium falciparum malaria through the mechanism of reduced rosetting. Proc Natl Acad Sci U S A 104(44): 17471–17476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Heggelund JE, Burschowsky D, Bjornestad VA, Hodnik V, Anderluh G, Krengel U (2016) High-resolution crystal structures elucidate the molecular basis of cholera blood group dependence. PLoS Pathog 12(4): e1005567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kuhlmann FW, Santhanam S, Kumar P, Luo Q, Ciorba MA, Fleckenstein JM (2016) Blood group O-dependent cellular responses to cholera toxin: parallel clinical and epidemiological links to severe cholera. Am J Trop Med Hyg 95(2): 440–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Glass RI, Holmgren J, Haley CE, Khan MR, Svennerholm A, Stoll BJ, Belayet Hossain KM, Black RE, Yunus M, Barua D (1985) Predisposition for cholera of individuals with O blood group. Possible evolutionary significance. Am J Epidemiol 121(6): 791–796. [DOI] [PubMed] [Google Scholar]
- 36.Nowakowska-Gołacka J, Sominka H, Sowa-Rogozinska N, Slominska-Wojewodzka M (2019) Toxins utilize the endoplasmic reticulum-associated protein degradation pathway in their intoxication process. Int J Mol Sci 20(6): pii: E1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zuverink M, Barbieri JT (2018) Protein toxins that utilize gangliosides as host receptors. Prog Mol Biol Transl Sci. 156(325–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Ernst K, Schnell L, Barth H (2017) Host cell chaperones Hsp70/Hsp90 and peptidyl-prolyl cis/trans isomerases are required for the membrane translocation of bacterial ADP-ribosylating toxins. Curr Top Microbiol Immunol 406(163–198. doi: 110.1007/1082_2016_1014. [DOI] [PubMed] [Google Scholar]
- 39.Cho JA, Chinnapen DJ, Aamar E, Welscher YM, Lencer WI, Massol R (2012) Insights on the trafficking and retro-translocation of glycosphingolipid-binding bacterial toxins. Frontiers in Cellular and Infection Microbiology 2(51): doi: 10.3389/fcimb.2012.00051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smith DC, Lord JM, Roberts LM, Johannes L (2004) Glycosphingolipids as toxin receptors. Semin Cell Dev Biol 15(4): 397–408. [DOI] [PubMed] [Google Scholar]
- 41.Orlandi PA, Curran PK, Fishman PH (1993) Brefeldin A blocks the response of cultured cells to cholera toxin. Implications for intracellular trafficking in toxin action. J Biol Chem 268(16): 12010–12016. [PubMed] [Google Scholar]
- 42.Fishman PH (1982) Internalization and degradation of cholera toxin by cultured cells: relationship to toxin action. J Cell Biol 93(3): 860–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Orlandi PA, Fishman PH (1998) Filipin-dependent inhibition of cholera toxin: evidence for toxin internalization and activation through caveolae-like domains. J Cell Biol 141(4): 905–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Guimaraes CP, Carette JE, Varadarajan M, Antos J, Popp MW, Spooner E, Brummelkamp TR, Ploegh HL (2011) Identification of host cell factors required for intoxication through use of modified cholera toxin. J Cell Biol 195(5): 751–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lencer WI, de Almeida JB, Moe S, Stow JL, Ausiello DA, Madara JL (1993) Entry of cholera toxin into polarized human intestinal epithelial cells. Identification of an early brefeldin A sensitive event required for A1-peptide generation. J Clin Invest 92(6): 2941–2951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lencer WI, Delp C, Neutra MR, Madara JL (1992) Mechanism of cholera toxin action on a polarized human intestinal epithelial cell line: role of vesicular traffic. J Cell Biol 117(6): 1197–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kassis S, Hagmann J, Fishman PH, Chang PP, Moss J (1982) Mechanism of action of cholera toxin on intact cells. Generation of A1 peptide and activation of adenylate cyclase. J Biol Chem 257(20): 12148–12152. [PubMed] [Google Scholar]
- 48.Fujinaga Y, Wolf AA, Rodighiero C, Wheeler H, Tsai B, Allen L, Jobling MG, Rapoport T, Holmes RK, Lencer WI (2003) Gangliosides that associate with lipid rafts mediate transport of cholera and related toxins from the plasma membrane to endoplasmic reticulm. Mol Biol Cell 14(12): 4783–4793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Taylor M, Banerjee T, Navarro-Garcia F, Huerta J, Massey S, Burlingame M, Pande AH, Tatulian SA, Teter K (2011) A therapeutic chemical chaperone inhibits cholera intoxication and unfolding/translocation of the cholera toxin A1 subunit. PLoS ONE 6(4): e18825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Taylor M, Banerjee T, Ray S, Tatulian SA, Teter K (2011) Protein disulfide isomerase displaces the cholera toxin A1 subunit from the holotoxin without unfolding the A1 subunit. J Biol Chem 286(25): 22090–22100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Massey S, Banerjee T, Pande AH, Taylor M, Tatulian SA, Teter K (2009) Stabilization of the tertiary structure of the cholera toxin A1 subunit inhibits toxin dislocation and cellular intoxication. J Mol Biol 393(5): 1083–1096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pande AH, Scaglione P, Taylor M, Nemec KN, Tuthill S, Moe D, Holmes RK, Tatulian SA, Teter K (2007) Conformational instability of the cholera toxin A1 polypeptide. J Mol Biol 374(4): 1114–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Teter K (2013) Toxin instability and its role in toxin translocation from the endoplasmic reticulum to the cytosol. Biomolecules 3(4): 997–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.O’Neal CJ, Amaya EI, Jobling MG, Holmes RK, Hol WGJ (2004) Crystal structures of an intrinsically active cholera toxin mutant yield insight into the toxin activation mechanism. Biochemistry 43(13): 3772–3782. [DOI] [PubMed] [Google Scholar]
- 55.Van Dop C, Tsubokawa M, Bourne HR, Ramachandran J (1984) Amino acid sequence of retinal transducin at the site ADP-ribosylated by cholera toxin. J Biol Chem 259(2): 696–698. [PubMed] [Google Scholar]
- 56.Landis CA, Masters SB, Spada A, Pace AM, Bourne HR, Vallar L (1989) GTPase inhibiting mutations activate the α chain of Gs and stimulate adenylyl cyclase in human pituitary tumours. Nature 340(692–696. [DOI] [PubMed] [Google Scholar]
- 57.Musch MW, Arvans DL, Walsh-Reitz MM, Uchiyama K, Fukuda M, Chang EB (2007) Synaptotagmin I binds intestinal epithelial NHE3 and mediates cAMP- and Ca2+-induced endocytosis by recruitment of AP2 and clathrin. Am J Physiol Gastrointest Liver Physiol 292(G1549–G1558. [DOI] [PubMed] [Google Scholar]
- 58.Singh V, Yang J, Yin J, Cole R, Tse M, Berman DE, Small SA, Petsko G, Donowitz M (2018) Cholera toxin inhibits SNX27-retromer-mediated delivery of cargo proteins to the plasma membrane. J Cell Sci 131(jcs218610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Moe OW, Amemiya M, Yamaji Y (1995) Activation of protein kinase A acutely inhibits and phosphorylates Na/H exchanger NHE-3. J Clin Invest 96(5): 2187–2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Saslowsky DE, Lencer WI (2008) Conversion of apical plasma membrane sphingomyelin to ceramide attenuates the intoxication of host cells by cholera toxin. Cell Microbiol 10(1): 67–80. [DOI] [PubMed] [Google Scholar]
- 61.Feng Y, Jadhav AP, Rodighiero C, Fujinaga Y, Kirchhausen T, Lencer WI (2004) Retrograde transport of cholera toxin from the plasma membrane to the endoplasmic reticulum requires the trans-Golgi network but not the Golgi apparatus in Exo2-treated cells. EMBO Rep 5(6): 596–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Lingwood D, Simons K (2010) Lipid rafts as a membrane-organizing principle. Science 327(5961): 46–50. [DOI] [PubMed] [Google Scholar]
- 63.Kenworthy AK, Schmieder SS, Raghunathan K, Tiwari A, Wang T, Kelly CV, Lencer WI (2021) Cholera toxin as a probe for membrane biology. Toxins 13(543): 10.3390/toxins13080543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Brown DA, London E (2000) Structure and function of sphingolipid- and cholesterol-rich membrane rafts. J Biol Chem 275(23): 17221–17224. [DOI] [PubMed] [Google Scholar]
- 65.Lajoie P, Nabi IR (2010) Lipid rafts, caveolae, and their endocytosis. Int Rev Cell Mol Biol 282(135–163. [DOI] [PubMed] [Google Scholar]
- 66.Simons K, Toomre D (2000) Lipid rafts and signal transduction. Nat Rev Mol Cell Biol 1(1): 31–39. [DOI] [PubMed] [Google Scholar]
- 67.Wolf AA, Fujinaga Y, Lencer WI (2002) Uncoupling of the cholera toxin-G(M1) ganglioside receptor complex from endocytosis, retrograde Golgi trafficking, and downstream signal transduction by depletion of membrane cholesterol. J Biol Chem 277(18): 16249–16256 Epub 12002 Feb 16221. [DOI] [PubMed] [Google Scholar]
- 68.Holmgren J, Lonnroth I, Mansson J-E, Svennerholm L (1975) Interaction of cholera toxin and membrane GM1 ganglioside of small intestine. Proc Natl Acad Sci U S A 72(7): 2520–2524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Holmgren J, Lonnroth I, Svennerholm L (1973) Tissue receptor for cholera exotoxin: postulated structure from studies with GM1 ganglioside and related glycolipids. Infect Immun 8(2): 208–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Fujita A, Cheng J-S, Hirakawa M, Furukawa K, Kusunoki S, Fujimoto T (2007) Gangliosides GM1 and GM3 in the living cell membrane form clusters susceptible to cholesterol depletion and chilling. Mol Biol Cell 18(6): 2112–2122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Merritt EA, Sarfaty S, van den Akker F, L’Hoir C, Martial JA, Hol WG (1994) Crystal structure of cholera toxin B-pentamer bound to receptor GM1 pentasaccharide. Protein Sci 3(2): 166–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jobling MG, Yang Z, Kam WR, Lencer WI, Holmes RK (2012) A single native ganglioside GM1-binding site is sufficient for cholera toxin to bind to cells and complete the intoxication pathway. MBio 3(6): e00401–00412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.De Wolf MJ, Dams E, Dierick WS (1994) Interaction of a cholera toxin derivative containing a reduced number of receptor binding sites with intact cells in culture. Biochim Biophys Acta 1223(2): 296–305. [DOI] [PubMed] [Google Scholar]
- 74.Wolf AA, Jobling MG, Saslowsky DE, Kern E, Drake KR, Kenworthy AK, Holmes RK, Lencer WI (2008) Attenuated endocytosis and toxicity of a mutant cholera toxin with decreased ability to cluster ganglioside GM1 molecules. Infect Immun 76(4): 1476–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hagmann J, Fishman PH (1982) Detergent extraction of cholera toxin and gangliosides from cultured cells and isolated membranes. Biochim Biophys Acta 720(2): 181–187. [DOI] [PubMed] [Google Scholar]
- 76.Bacia K, Schwille P, Kurzchalia T (2005) Sterol structure determines the separation of phases and the curvature of the liquid-ordered phase in model membranes. Proc Natl Acad Sci U S A 102(9): 3272–3277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Raghunathan K, Wong TH, Chinnapen DJ, Lencer WI, Jobling MG, Kenworthy AK (2016) Glycolipid crosslinking is required for cholera toxin to partition into and stabilize ordered domains. Biophys J 111(12): 2547–2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kabbani AM, Raghunathan K, Lencer WI, Kenworthy AK, Kelly CV (2020) Structured clustering of the glycosphingolipid GM1 is required for membrane curvature induced by cholera toxin. Proc Natl Acad Sci U S A 117(26): 14978–14986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mukherjee S, Ghosh RN, Maxfield FR (1997) Endocytosis. Physiol Rev 77(3): 759–803. [DOI] [PubMed] [Google Scholar]
- 80.Maxfield FR, McGraw TE (2004) Endocytic recycling. Nat Rev Mol Cell Biol 5(2): 121–132. [DOI] [PubMed] [Google Scholar]
- 81.Mukherjee S, Maxfield FR (2000) Role of membrane organization and membrane domains in endocytic lipid trafficking. Traffic 1(203–211. [DOI] [PubMed] [Google Scholar]
- 82.Masserini M, Palestini P, Pitto M, Chigorno V, Tomasi M, Tettamanti G (1990) Cyclic AMP accumulation in HeLa cells induced by cholera toxin. Involvement of the ceramide moiety of GM1 ganglioside. Biochem J 271(1): 107–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chinnapen DJ-F, Hsieh W-T, te Welscher YM, Saslowsky DE, Kaoutzani L, Brandsma E, D’Auria L, Park H, Wagner JS, Drake KR, Kang M, Benjamin T, Ullman MD, Costello CE, Kenworthy AK, Baumgart T, Massol RH, Lencer WI (2012) Lipid sorting by ceramide structure from plasma membrane to ER for the cholera toxin receptor ganglioside GM1. Dev Cell 23(3): 573–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Iglesias-Bartolome R, Trenchi A, Comin R, Moyano AL, Nores GA, Daniotti JL (2009) Differential endocytic trafficking of neuropathy-associated antibodies to GM1 ganglioside and cholera toxin in epithelial and neural cells. Biochim Biophys Acta 1788(2526–2540. [DOI] [PubMed] [Google Scholar]
- 85.Shogomori H, Futerman AH (2001) Cholesterol depletion by methyl-beta-cyclodextrin blocks cholera toxin transport from endosomes to the Golgi apparatus in hippocampal neurons. J Neurochem 78(5): 991–999. [DOI] [PubMed] [Google Scholar]
- 86.Badizadegan K, Wheeler HE, Fujinaga Y, Lencer WI (2004) Trafficking of cholera toxin-ganglioside GM1 complex into Golgi and induction of toxicity depend on actin cytoskeleton. Am J Physiol Cell Physiol 287(C1453–C1462. [DOI] [PubMed] [Google Scholar]
- 87.Saslowsky DE, Cho JA, Chinnapen H, Massol RH, Chinnapen DJ, Wagner JS, De Luca HE, Kam W, Paw BH, Lencer WI (2010) Intoxication of zebrafish and mammalian cells by cholera toxin depends on the flotillin/reggie proteins but not Derlin-1 or -2. J Clin Invest 120(12): 4399–4409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Otto GP, Nichols BJ (2011) The roles of flotillin microdomains -- endocytosis and beyond. J Cell Sci 124(Pt 23): 3933–3940. [DOI] [PubMed] [Google Scholar]
- 89.Lippincott-Schwartz J, Yuan LC, Bonifacino JS, Klausner RD (1989) Rapid redistribution of Golgi proteins into the ER in cells treated with brefeldin A: evidence for membrane cycling from Golgi to ER. Cell 56(5): 801–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lippincott-Schwartz J, Yuan L, Tipper C, Amherdt M, Orci L, Klausner RD (1991) Brefeldin A’s effects on endosomes, lysosomes, and the TGN suggest a general mechanism for regulating organelle structure and membrane traffic. Cell 67(3): 601–616. [DOI] [PubMed] [Google Scholar]
- 91.Nambiar MP, Oda T, Chen C, Kuwazuru Y, Wu HC (1993) Involvement of the Golgi region in the intracellular trafficking of cholera toxin. J Cell Physiol 154(2): 222–228. [DOI] [PubMed] [Google Scholar]
- 92.Cherubin P, Guyette J, Taylor M, O’Donnell M, Herndon L, Burress H, Riad A, Tatulian SA, Teter K (2018) Protein disulfide isomerase does not act as an unfoldase in the disassembly of cholera toxin. Biosci Rep 38(5): pii: BSR20181320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Newstead S, Barr F (2020) Molecular basis for KDEL-mediated retrieval of escaped ER-resident proteins - SWEET talking the COPs. J Cell Sci 133(jcs250100): doi: 10.1242/jcs.250100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Lencer WI, Constable C, Moe S, Jobling MG, Webb HM, Ruston S, Madara JL, Hirst TR, Holmes RK (1995) Targeting of cholera toxin and Escherichia coli heat labile toxin in polarized epithelia: role of COOH-terminal KDEL. J Cell Biol 131(4): 951–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Needham PG, Guerriero CJ, Brodsky JL (2019) Chaperoning endoplasmic reticulum-associated degradation (ERAD) and protein conformational diseases. Cold Spring Harb Perspect Med 11(a033928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Wiseman RL, Mesgarzadeh JS, Hendershot LM (2022) Reshaping endoplasmic reticulum quality control through the unfolded protein response. Mol Cell 82(1477–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Majoul I, Ferrari D, Soling HD (1997) Reduction of protein disulfide bonds in an oxidizing environment. The disulfide bridge of cholera toxin A-subunit is reduced in the endoplasmic reticulum. FEBS Lett 401(2-3): 104–108. [DOI] [PubMed] [Google Scholar]
- 98.Orlandi PA (1997) Protein-disulfide isomerase-mediated reduction of the A subunit of cholera toxin in a human intestinal cell line. J Biol Chem 272(7): 4591–4599. [PubMed] [Google Scholar]
- 99.Guyette J, Cherubin P, Serrano A, Taylor M, Abedin F, O’Donnell M, Burress H, Tatulian SA, Teter K (2019) Quercetin-3-rutinoside blocks the disassembly of cholera toxin by protein disulfide isomerase. Toxins 11(8): pii: E458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Mekalanos JJ, Collier RJ, Romig WR (1979) Enzymic activity of cholera toxin. II. Relationships to proteolytic processing, disulfide bond reduction, and subunit composition. J Biol Chem 254(13): 5855–5861. [PubMed] [Google Scholar]
- 101.Tomasi M, Battistini A, Araco A, Roda LG, D’Agnolo G (1979) The role of the reactive disulfide bond in the interaction of cholera-toxin functional regions. Eur J Biochem 93(3): 621–627. [DOI] [PubMed] [Google Scholar]
- 102.Tsai B, Rodighiero C, Lencer WI, Rapoport TA (2001) Protein disulfide isomerase acts as a redox-dependent chaperone to unfold cholera toxin. Cell 104(6): 937–948. [DOI] [PubMed] [Google Scholar]
- 103.Taylor M, Burress H, Banerjee T, Ray S, Curtis D, Tatulian SA, Teter K (2014) Substrate-induced unfolding of protein disulfide isomerase displaces the cholera toxin A1 subunit from its holotoxin. PLoS Pathogens 10(2): e1003925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Rodighiero C, Aman AT, Kenny MJ, Moss J, Lencer WI, Hirst TR (1999) Structural basis for the differential toxicity of cholera toxin and Escherichia coli heat-labile enterotoxin. Construction of hybrid toxins identifies the A2-domain as the determinant of differential toxicity. J Biol Chem 274(7): 3962–3969. [DOI] [PubMed] [Google Scholar]
- 105.Serrano A, Guyette JL, Heim JB, Taylor M, Cherubin P, Krengel U, Teter K, Tatulian SA (2022) Holotoxin disassembly by protein disulfide isomerase is less efficient for Escherichia coli heat-labile enterotoxin than cholera toxin. Sci Rep 12(34): 10.1038/s41598-41021-03939-41599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Craft JW Jr, Shen TW, Brier LM, Briggs JM (2015) Biophysical characteristics of cholera toxin and Escherichia coli heat-labile enterotoxin structure and chemistry lead to differential toxicity. J Phys Chem B 119(3): 1048–1061. [DOI] [PubMed] [Google Scholar]
- 107.Sixma TK, Kalk KH, van Zanten BA, Dauter Z, Kingma J, Witholt B, Hol WG (1993) Refined structure of Escherichia coli heat-labile enterotoxin, a close relative of cholera toxin. J Mol Biol 230(3): 890–918. [DOI] [PubMed] [Google Scholar]
- 108.Goins B, Freire E (1988) Thermal stability and intersubunit interactions of cholera toxin in solution and in association with its cell-surface receptor ganglioside GM1. Biochemistry 27(6): 2046–2052. [DOI] [PubMed] [Google Scholar]
- 109.Surewicz WK, Leddy JJ, Mantsch HH (1990) Structure, stability, and receptor interaction of cholera toxin as studied by Fourier-transform infrared spectroscopy. Biochemistry 29(35): 8106–8111. [DOI] [PubMed] [Google Scholar]
- 110.Ampapathi RS, Creath AL, Lou DI, Craft JW Jr., Blanke SR, Legge GB (2008) Order-disorder-order transitions mediate the activation of cholera toxin. J Mol Biol 377(3): 748–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Taylor M, Curtis D, Teter K (2015) A conformational shift in the dissociated cholera toxin A1 subunit prevents reassembly of the cholera holotoxin. Toxins 7(2674–2684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Geden SE, Gardner RA, Fabbrini MS, Ohashi M, Phanstiel Iv O, Teter K (2007) Lipopolyamine treatment increases the efficacy of intoxication with saporin and an anticancer saporin conjugate. FEBS J 274(18): 4825–4836. [DOI] [PubMed] [Google Scholar]
- 113.Teter K, Holmes RK (2002) Inhibition of endoplasmic reticulum-associated degradation in CHO cells resistant to cholera toxin, Pseudomonas aeruginosa exotoxin A, and ricin. Infect Immun 70(11): 6172–6179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Banerjee T, Cilenti L, Taylor M, Showman A, Tatulian SA, Teter K (2016) Thermal unfolding of the pertussis toxin S1 subunit facilitates toxin translocation to the cytosol by the mechanism of endoplasmic reticulum-associated degradation. Infect Immun 84(12): 3388–3398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.London E, Luongo CL (1989) Domain-specific bias in arginine/lysine usage by protein toxins. Biochem Biophys Res Commun 160(1): 333–339. [DOI] [PubMed] [Google Scholar]
- 116.Hazes B, Read RJ (1997) Accumulating evidence suggests that several AB-toxins subvert the endoplasmic reticulum-associated protein degradation pathway to enter target cells. Biochemistry 36(37): 11051–11054. [DOI] [PubMed] [Google Scholar]
- 117.Rodighiero C, Tsai B, Rapoport TA, Lencer WI (2002) Role of ubiquitination in retro-translocation of cholera toxin and escape of cytosolic degradation. EMBO Rep 3(12): 1222–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Deeks ED, Cook JP, Day PJ, Smith DC, Roberts LM, Lord JM (2002) The low lysine content of ricin A chain reduces the risk of proteolytic degradation after translocation from the endoplasmic reticulum to the cytosol. Biochemistry 41(10): 3405–3413. [DOI] [PubMed] [Google Scholar]
- 119.Worthington ZE, Carbonetti NH (2007) Evading the proteasome: absence of lysine residues contributes to pertussis toxin activity by evasion of proteasome degradation. Infect Immun 75(6): 2946–2953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Brodsky JL (2007) The protective and destructive roles played by molecular chaperones during ERAD (endoplasmic-reticulum-associated degradation). Biochem J 404(3): 353–363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Massey S, Burress H, Taylor M, Nemec KN, Ray S, Haslam DB, Teter K (2011) Structural and functional interactions between the cholera toxin A1 subunit and ERdj3/HEDJ, a chaperone of the endoplasmic reticulum. Infect Immun 79(11): 4739–4747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Williams JM, Inoue T, Banks L, Tsai B (2013) The ERdj5-Sel1L complex facilitates cholera toxin retrotranslocation. Mol Biol Cell 24(6): 785–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Winkeler A, Godderz D, Herzog V, Schmitz A (2003) BiP-dependent export of cholera toxin from endoplasmic reticulum-derived microsomes. FEBS Lett 554(3): 439–442. [DOI] [PubMed] [Google Scholar]
- 124.Williams JM, Inoue T, Chen G, Tsai B (2015) The nucleotide exchange factors Grp170 and Sil1 induce cholera toxin release from BiP to enable retrotranslocation. Mol Biol Cell 26(2181–2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Romisch K (2017) A case for Sec61 channel involvement in ERAD. Trends Biochem Sci 42(3): 171–179. [DOI] [PubMed] [Google Scholar]
- 126.Schmitz A, Herrgen H, Winkeler A, Herzog V (2000) Cholera toxin is exported from microsomes by the Sec61p complex. J Cell Biol 148(6): 1203–1212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Alder NN, Shen Y, Brodsky JL, Hendershot LM, Johnson AE (2005) The molecular mechanisms underlying BiP-mediated gating of the Sec61 translocon of the endoplasmic reticulum. J Cell Biol 168(389–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Preston GM, Brodsky JL (2017) The evolving role of ubiquitin modification in endoplasmic reticulum-associated degradation. Biochem J 474(445–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Bernardi KM, Forster ML, Lencer WI, Tsai B (2008) Derlin-1 facilitates the retro-translocation of cholera toxin. Mol Biol Cell 19(3): 877–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Bernardi KM, Williams JM, Kikkert M, van Voorden S, Wiertz EJ, Ye Y, Tsai B (2010) The E3 ubiquitin ligases Hrd1 and gp78 bind to and promote cholera toxin retro-translocation. Mol Biol Cell 21(1): 140–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Wu X, Rapoport TA (2018) Mechanistic insights into ER-associated protein degradation. Curr Opin Cell Biol 53(22–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Taylor M, Navarro-Garcia F, Huerta J, Burress H, Massey S, Ireton K, Teter K (2010) Hsp90 is required for transfer of the cholera toxin A1 subunit from the endoplasmic reticulum to the cytosol. J Biol Chem 285(41): 31261–31267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Koopmann JO, Albring J, Huter E, Bulbuc N, Spee P, Neefjes J, Hammerling GJ, Momburg F (2000) Export of antigenic peptides from the endoplasmic reticulum intersects with retrograde protein translocation through the Sec61p channel. Immunity 13(1): 117–127. [DOI] [PubMed] [Google Scholar]
- 134.Simpson JC, Roberts LM, Romisch K, Davey J, Wolf DH, Lord JM (1999) Ricin A chain utilises the endoplasmic reticulum-associated protein degradation pathway to enter the cytosol of yeast. FEBS Lett 459(1): 80–84. [DOI] [PubMed] [Google Scholar]
- 135.Yu M, Haslam DB (2005) Shiga toxin is transported from the endoplasmic reticulum following interaction with the luminal chaperone HEDJ/ERdj3. Infect Immun 73(4): 2524–2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Li S, Spooner RA, Hampton RY, Lord JM, Roberts LM (2012) Cytosolic entry of Shiga-like toxin a chain from the yeast endoplasmic reticulum requires catalytically active Hrd1p. PLoS One 7(7): e41119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Nery FC, Armata IA, Farley JE, Cho JA, Yaqub U, Chen P, Carla da Hora C, Wang Q, Tagaya M, Klein C, Tannous B, Caldwell KA, Caldwell GA, Lencer WI, Ye Y, Breakefield XO (2011) TorsinA participates in endoplasmic reticulum-associated degradation. Nat Commun 2(393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Kothe M, Ye Y, Wagner JS, De Luca HE, Kern E, Rapoport TA, Lencer WI (2005) Role of p97 AAA-ATPase in the retrotranslocation of the cholera toxin A1 chain, a non-ubiquitinated substrate. J Biol Chem 280(30): 28127–28132. [DOI] [PubMed] [Google Scholar]
- 139.McConnell E, Lass A, Wojcik C (2007) Ufd1-Npl4 is a negative regulator of cholera toxin retrotranslocation. Biochem Biophys Res Commun 355(4): 1087–1090. [DOI] [PubMed] [Google Scholar]
- 140.Burress H, Taylor M, Banerjee T, Tatulian SA, Teter K (2014) Co- and post-translocation roles for Hsp90 in cholera intoxication. J Biol Chem 289(48): 33644–33654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Teter K, Jobling MG, Holmes RK (2003) A class of mutant CHO cells resistant to cholera toxin rapidly degrades the catalytic polypeptide of cholera toxin and exhibits increased endoplasmic reticulum-associated degradation. Traffic 4(4): 232–242. [DOI] [PubMed] [Google Scholar]
- 142.Kellner A, Taylor M, Banerjee T, Britt CBT, Teter K (2019) A binding motif for Hsp90 in the A chains of ADP-ribosylating toxins that move from the endoplasmic reticulum to the cytosol. Cell Microbiol 21(10): e13074. doi: 13010.11111/cmi.13074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Imai T, Kato Y, Kajiwara C, Mizukami S, Ishige I, Ichiyanagi T, Hikida M, Wang J-Y, Udono H (2011) Heat shock protein 90 (HSP90) contributes to cytosolic translocation of extracellular antigen for cross-presentation by dendritic cells. Proc Natl Acad Sci U S A 108(39): 16363–16368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Grotzke JE, Cresswell P (2015) Are ERAD components involved in cross-presentation? Mol Immunol 68(112–115. [DOI] [PubMed] [Google Scholar]
- 145.Simon SM, Peskin CS, Oster GF (1992) What drives the translocation of proteins? Proc Natl Acad Sci U S A 89(3770–3774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Peskin CS, Odell GM, Oster GF (1993) Cellular motions and thermal fluctuations: the Brownian ratchet. Biophys J 65(1): 316–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Shamu CE, Flierman D, Ploegh HL, Rapoport TA, Chau V (2001) Polyubiquitination is required for US11-dependent movement of MHC class I heavy chain from endoplasmic reticulum into cytosol. Mol Biol Cell 12(8): 2546–2555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Jarosch E, Geiss-Friedlander R, Meusser B, Walter J, Sommer T (2002) Protein dislocation from the endoplasmic reticulum -- pulling out the suspect. Traffic 3(8): 530–536. [DOI] [PubMed] [Google Scholar]
- 149.Sanchez J, Wallerstrom G, Fredriksson M, Angstrom J, Holmgren J (2002) Detoxification of cholera toxin without removal of its immunoadjuvanticity by the addition of (STa-related) peptides to the catalytic subunit. A potential new strategy to generate immunostimulants for vaccination. J Biol Chem 277(36): 33369–33377. [DOI] [PubMed] [Google Scholar]
- 150.Murayama T, Tsai SC, Adamik R, Moss J, Vaughan M (1993) Effects of temperature on ADP-ribosylation factor stimulation of cholera toxin activity. Biochemistry 32(2): 561–566. [DOI] [PubMed] [Google Scholar]
- 151.Banerjee T, Taylor M, Jobling MG, Burress H, Yang Z, Serrano A, Holmes RK, Tatulian SA, Teter K (2014) ADP-ribosylation factor 6 acts as an allosteric activator for the folded but not disordered cholera toxin A1 polypeptide. Mol Microbiol 94(4): 898–912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Kahn RA, Gilman AG (1984) Purification of a protein cofactor required for ADP-ribosylation of the stimulatory regulatory component of adenylate cyclase by cholera toxin. J Biol Chem 259(10): 6228–6234. [PubMed] [Google Scholar]
- 153.Moss J, Vaughan M (1995) Structure and function of ARF proteins: activators of cholera toxin and critical components of intracellular vesicular transport processes. J Biol Chem 270(21): 12327–12330. [DOI] [PubMed] [Google Scholar]
- 154.O’Neal CJ, Jobling MG, Holmes RK, Hol WG (2005) Structural basis for the activation of cholera toxin by human ARF6-GTP. Science 309(5737): 1093–1096. [DOI] [PubMed] [Google Scholar]
- 155.Ray S, Taylor M, Banerjee T, Tatulian SA, Teter K (2012) Lipid rafts alter the stability and activity of the cholera toxin A1 subunit. J Biol Chem 287(36): 30395–30405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Banerjee T, Grabon A, Taylor M, Teter K (2021) cAMP-independent activation of the unfolded protein response by cholera toxin. Infect Immun 89(2): e00447–00420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Bernales S, Papa FR, Walter P (2006) Intracellular signaling by the unfolded protein response. Annu Rev Cell Dev Biol 22(487–508. [DOI] [PubMed] [Google Scholar]
- 158.Mori K (2009) Signalling pathways in the unfolded protein response: development from yeast to mammals. J Biochem 146(6): 743–750. [DOI] [PubMed] [Google Scholar]
- 159.Travers KJ, Patil CK, Wodicka L, Lockhart DJ, Weissman JS, Walter P (2000) Functional and genomic analyses reveal an essential coordination between the unfolded protein response and ER-associated degradation. Cell 101(3): 249–258. [DOI] [PubMed] [Google Scholar]
- 160.Dixit G, Mikoryak C, Hayslett T, Bhat A, Draper RK (2008) Cholera toxin up-regulates endoplasmic reticulum proteins that correlate with sensitivity to the toxin. Exp Biol Med (Maywood) 233(2): 163–175. [DOI] [PubMed] [Google Scholar]
- 161.Sandvig K, Garred O, van Deurs B (1996) Thapsigargin-induced transport of cholera toxin to the endoplasmic reticulum. Proc Natl Acad Sci U S A 93(22): 12339–12343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.McCloskey MA (1988) Cholera toxin potentiates IgE-coupled inositol phospholipid hydrolysis and mediator secretion by RBL-2H3 cells. Proc Natl Acad Sci U S A 85(19): 7260–7264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Narasimhan V, Holowka D, Fewtrell C, Baird B (1988) Cholera toxin increases the rate of antigen-stimulated calcium influx in rat basophilic leukemia cells. J Biol Chem 263(36): 19626–19632. [PubMed] [Google Scholar]
- 164.Qureshi SA, Alexandropoulos K, Joseph CK, Spangler R, Foster DA (1991) Cholera toxin induces expression of the immediate-early response gene JE via a cyclic AMP-independent signaling pathway. Mol Cell Biol 11(1): 102–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Aksamit RR, Backlund PS Jr., Cantoni GL (1985) Cholera toxin inhibits chemotaxis by a cAMP-independent mechanism. Proc Natl Acad Sci U S A 82(22): 7475–7479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Chen Y, Brandizzi F (2012) AtIRE1A/AtIRE1B and AGB1 independently control two essential unfolded protein response pathways in Arabidopsis. Plant J 69(2): 266–277. [DOI] [PubMed] [Google Scholar]
- 167.Wang S, Narendra S, Fedoroff N (2007) Heterotrimeric G protein signaling in the Arabidopsis unfolded protein response. Proc Natl Acad Sci U S A 104(10): 3817–3822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Zeng W, Mak D-OD, Li Q, Shin DM, Foskett JK, Muallem S (2003) A new mode of Ca2+ signaling by G protein-coupled receptors: gating of IP3 receptor Ca2+ release channels by Gβγ. Curr Biol 13(872–876. [DOI] [PubMed] [Google Scholar]
- 169.Ding QM, Ko TC, Evers BM (1998) Caco-2 intestinal cell differentiation is associated with G1 arrest and suppression of CDK2 and CDK4. Am J Physiol 275(5 Pt 1): C1193–1200. [DOI] [PubMed] [Google Scholar]
- 170.Kaper JB, Fasano A, Trucksis M, Toxins of Vibrio cholerae, in Vibrio cholerae and Cholera: Molecular to Global Perspectives, Wachsmuth IK, Blake PA, and Olsvik O, Editors. 1994, ASM Press: Washington, D.C. p. 145–176. [Google Scholar]
- 171.Kaper JB, Morris JG Jr., Levine MM (1995) Cholera. Clin Microbiol Rev 8(1): 48–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Rodighiero C, Lencer WI, Trafficking of cholera toxin and related bacterial enterotoxins: pathways and endpoints, in Microbial Pathogenesis and the Intestinal Epithelial Cell, Hecht G, Editor. 2003, ASM Press: Washington, D.C. p. 385–401. [Google Scholar]
- 173.Chang FH, Bourne HR (1989) Cholera toxin induces cAMP-independent degradation of Gs. J Biol Chem 264(10): 5352–5357. [PubMed] [Google Scholar]
- 174.Milligan G, Unson CG, Wakelam MJO (1989) Cholera toxin treatment produces down-regulation of the α-subunit of the stimulatory guanine-nucleotide-binding protein (Gs). BIochem J 262(643–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Levis MJ, Bourne HR (1992) Activation of the α subunit of Gs in intact cells alters its abundance, rate of degradation, and membrane avidity. J Cell Biol 119(5): 1297–1307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Palazzo L, Mikoc A, Ahel I (2017) ADP-ribosylation: new facets of an ancient modification. FEBS J 284(18): 2932–2946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Rack JGM, Palazzo L, Ahel I (2020) (ADP-ribosyl)hydrolases: structure, function, and biology. Genes Dev 34(5–6): 263–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Mashimo M, Kato J, Moss J (2014) Structure and function of the ARH family of ADP-ribosyl-acceptor hydrolases. DNA Repair 23(88–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Gill DM, Meren R (1978) ADP-ribosylation of membrane proteins catalyzed by cholera toxin: basis of the activation of adenylate cyclase. Proc Natl Acad Sci U S A 75(7): 3050–3054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Honjo T, NIshizuka Y, Hayaishi O (1968) Diphtheria toxin-dependent adenosine diphosphate ribosylation of aminoacyl transferase II and inhibition of protein synthesis. J Biol Chem 243(12): 3553–3555. [PubMed] [Google Scholar]
- 181.Moss J, Stanley SJ, Burns DL, Hsia JA, Yost DA, Myers GA, Hewlett EL (1983) Activation by thiol of the latent NAD glycohydrolase and ADP-ribosyltransferase activities of Bordetella pertussis toxin (islet-activating protein). J Biol Chem 258(19): 11879–11882. [PubMed] [Google Scholar]
- 182.Kato J, Zhu J, Liu C, Moss J (2007) Enhanced sensitivity to cholera toxin in ADP-ribosylarginine hydrolase-deficient mice. Mol Cell Biol 27(15): 5534–5543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Watanabe K, Kato J, Zhu J, Oda H, Ishiwata-Endo H, Moss J (2018) Enhanced sensitivity to cholera toxin in female ADP-ribosylarginine hydrolase (ARH1)-deficient mice. PLOS ONE 13(11): e0207693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Wernick NL, De Luca H, Kam WR, Lencer WI (2010) N-terminal extension of the cholera toxin A1-chain causes rapid degradation after retrotranslocation from endoplasmic reticulum to cytosol. J Biol Chem 285(9): 6145–6152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Bader C, Taylor M, Banerjee T, Teter K (2022) The cytopathic activity of cholera toxin requires a threshold quantity of cytosolic toxin. submitted. [DOI] [PMC free article] [PubMed]
- 186.Kumar V, Turnbull WB (2018) Carbohydrate inhibitors of cholera toxin. Beilstein J Org Chem 14(484–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Reddy S, Taylor M, Zhao M, Cherubin P, Geden S, Ray S, Francis D, Teter K (2013) Grape extracts inhibit multiple events in the cell biology of cholera intoxication. PLoS One 8(9): e73390. [DOI] [PMC free article] [PubMed] [Google Scholar]