

Visual Abstract
Abstract
The era of genomic medicine has allowed acute myeloid leukemia (AML) researchers to improve disease characterization, optimize risk-stratification systems, and develop new treatments. Although there has been significant progress, AML remains a lethal cancer because of its remarkably complex and plastic cellular architecture. This degree of heterogeneity continues to pose a major challenge, because it limits the ability to identify and therefore eradicate the cells responsible for leukemogenesis and treatment failure. In recent years, the field of single-cell genomics has led to unprecedented strides in the ability to characterize cellular heterogeneity, and it holds promise for the study of AML. In this review, we highlight advancements in single-cell technologies, outline important shortcomings in our understanding of AML biology and clinical management, and discuss how single-cell genomics can address these shortcomings as well as provide unique opportunities in basic and translational AML research.
Edited by Associate Editor Berthold Göttgens, this Review Series focuses on how the use of single-cell genomic and multiomic analyses are broadening our understanding of the complexity of leukemias and myeloid neoplasms. For acute myeloid leukemia, acute lymphoblastic leukemia, chronic lymphocytic leukemia, and myeloproliferative neoplasm, leading experts bring us up to date with recent data and speculate how these rapidly developing technologies may inform the directions of clinical care.
Introduction
Acute myeloid leukemia (AML), an aggressive blood and bone marrow cancer, was first described as an acute leukemia in the 1850s. The disease was initially categorized as myeloid or lymphoid in the early 1900s1 and for many decades was predominantly classified morphologically.2,3 This paradigm has failed to accurately capture the underlying complexity of AML,4, 5 which has in turn limited the ability to design effective therapies. It was not until the 1970s that viable treatment options were identified, which included cytotoxic chemotherapies6 and allogeneic hematopoietic cell transplantation (alloHCT).7 For >40 years, the standard treatment has subsequently been induction chemotherapy followed by either consolidation chemotherapy or alloHCT. Using this approach, patients often achieve complete remission, defined as normalization of peripheral blood counts and <5% blasts identified in the bone marrow. However, most patients either are too frail to receive aggressive upfront therapy or experience relapse with highly refractory disease, and therefore, only 30.5% of patients survive past 5 years,8 and only 5% to 15% of patients age >60 years are cured9.
These poor outcomes may be partly explained by the AML stem cell model,10 which suggests that a rare population of leukemic stem cells (LSCs), residing at the apex of the AML cellular hierarchy, are the primary drivers of leukemia initiation and maintenance (Figure 1).10 Since Lapidot et al11 established the foundations for identifying and evaluating AML LSCs in the 1990s, there have been several studies characterizing the immunophenotype,12, 13, 14, 15, 16, 17, 18, 19 biology,20, 21, 22, 23, 24 and clinical relevance of AML LSCs.25, 26, 27 By definition, LSCs can transplant and maintain leukemia in immunodeficient mice and are capable of self-renewing, avoiding apoptosis, and increasing drug efflux.10 This model holds that LSCs can resist standard antileukemia therapies, persist during complete remission, and promote disease relapse (Figure 2A). It has been historically difficult to isolate, study, and target LSCs, because they constitute a minor fraction of cells within the bulk leukemic population. Moving forward, AML researchers will need to reliably differentiate LSCs from bulk leukemia, which can potentially be addressed with new single-cell technologies.
Figure 1.

Stem cell model of normal and malignant hematopoiesis. Hematopoietic stem cells (HSCs) are capable of self-renewal (semicircular arrow) and reside at the apex of healthy polyclonal hematopoiesis (black). The acquisition of mutations in HSCs (m1) can create preleukemic HSCs (pHSCs; purple) that can drive clonal hematopoiesis (shaded purple). Clonal evolution through additional mutation acquisition (m2, 3, …x) or other molecular processes can further transform pHSCs into LSCs (red). These LSCs reside at the apex of the AML cellular hierarchy and are capable of (re)generating frank leukemia (brown).
Figure 2.

Clinical course for a patient with AML who is refractory or eventually relapses. (A) Patients with AML present with high leukemia blast burden (brown), receive induction chemotherapy, and often enter complete remission. Some patients have refractory disease, and most patients who achieve complete remission ultimately relapse. Patients who are refractory to induction therapies never clear their disease, as assessed by morphology. Those who enter complete remission often have persistent measurable residual disease (MRD), below levels detectable by morphologic analysis. These patients relapse, with variable clonal dynamics, but often have an expanded LSC (red) compartment. Those who relapse late may have had persistent pHSCs (purple) that reevolved into AML. (B) MRD can be assessed by morphology using microscopy (detection limit, 10−2 cells), cytogenetics (10−2 cells), multiparameter flow cytometry (10−4 cells), or mutation-specific polymerase chain reaction (PCR) or next-generation sequencing (NGS; 10−6 cells). FISH, fluorescence in situ hybridization; FITC, fluorescein isothiocyanate; PE, phycoerythrin.
AML LSCs and their diverse progeny contribute to the underlying molecular complexity and cellular heterogeneity of AML22 in manners not apparent from standard morphologic assessment.4,5 For example, genomic studies have demonstrated that human AML is associated with mutations in multiple genes occurring at different allelic frequencies and undergoing variable patterns of clonal evolution on treatment.28 These observations suggest that AML contains a complex clonal architecture with several subclones of variable frequency. Moreover, each subclone is potentially organized as a cellular hierarchy initiated and maintained by LSCs with varying properties of self-renewal, quiescence, and therapeutic vulnerability.10,22,23,29 Although recent advancements in both characterizing and treating AML have occurred in parallel with advancements in genomic and biomolecular profiling,30 identifying effective monitoring and therapeutic strategies has been challenging. Since 2012, multiple studies have evaluated the genomic landscape of AML,31, 32, 33 which has led to new AML subgroups,34 risk-stratification systems,35,36 and targeted therapies.37, 38, 39, 40 Despite these advancements, patients continue to have variable and limited treatment responses to both standard and novel therapies.30 Furthermore, it is not always clear which patients will benefit from cytotoxic chemotherapies, targeted agents, or alloHCT.
International society guidelines provide clinicians assistance with AML risk stratification, disease monitoring, and treatment strategies. For example, the European LeukemiaNet (ELN), an international expert panel, published updated recommendations on the diagnosis and management of AML in 2017 (ELN-2017). The ELN-2017 stratifies patients with AML into adverse-, intermediate-, and favorable-risk groups based on common genetic abnormalities, which guides clinicians on prognosis and therapeutic strategies. There are several limitations, including, for instance, the impact of cooccurring signaling mutations on core binding factor AML41,42 and the FLT3-ITD allelic ratio threshold43 on prediction of survival and relapse. Additionally, the ELN-2017 was less effective in predicting outcomes in older patients, because survival was similar between intermediate- and adverse-risk patients. This deficiency was improved by incorporating mutation cooccurrence patterns from NGS.44 An extensive genomic evaluation of 1540 patients emphasized this point by noting the importance of broadening the genomic features considered prognostic per the ELN-2017 and incorporating gene-gene interactions.33 Currently, the ELN-2017 provides hematologists with a helpful tool for risk stratification; however, there are several shortcomings that will need to be addressed with future research and clinical trials.
Emerging evidence suggests that detecting and characterizing MRD may improve our ability to determine which patients can benefit from further treatment and, if so, which therapy will be most effective (Figure 2B). MRD is defined as persistent leukemia below the 5% of total bone marrow cells detectable by morphologic evaluation and has been shown to independently predict treatment outcome.45 MRD can be detected by using multiparameter flow cytometry or other molecular assays.46 Flow cytometry–based MRD detection greater than the 0.1% threshold is associated with significantly shorter relapse-free survival and overall survival.47,48 Most cases of AML contain mutations that can serve as clonal markers for MRD, and therefore, genomic profiling using real-time quantitative PCR or NGS can provide specific assays for MRD detection. In fact, detection of molecular MRD during complete remission is independently prognostic in regard to relapse and survival rates49 and has subsequently been incorporated in the ELN-2017 risk-stratification system.35,45 However, bulk genomic profiling methods fail to accurately resolve the clonal architecture of AML MRD and are limited in their ability to identify AML clones that drive disease relapse.28 Of interest, several groups have observed improvements in predicting prognosis by incorporating basic LSC flow cytometry markers into MRD assessment.45 It will therefore be critical to design LSC-specific MRD assays, because LSCs may need to be eradicated in order to achieve cure.
These observations highlight the underlying biomolecular complexity of AML that has limited our ability to effectively study, monitor, and treat patients. It is important to accurately characterize this underlying heterogeneity in order to improve AML research and disease management. Recent advancements in single-cell genomics have provided a remarkable toolset to begin addressing these limitations.50, 51, 52 Here, we review how single-cell genomics has enhanced the investigation of AML by discussing the current state and future directions of single-cell genomics in AML research, important gaps in our understanding of AML pathophysiology and disease management, and potential clinical translations of these technologies.
Clonal heterogeneity, clonal evolution, MRD, and the utility of scDNA-seq
NGS can provide an important method for MRD detection, which has been shown to be prognostically relevant.49,53,54 It is also an important tool for characterizing the clonal architecture of AML that may assist in risk stratification and disease management. Indeed, clonality inferred by bulk NGS is heterogenous in AML and highly dynamic at relapse.32,55 An evaluation of the clonal architecture across 2829 patients using bulk NGS identified specific clonal abundances and gene-gene interactions that were associated with clinical outcomes and drug sensitivities.56 However, we know that individual clones exist in a complex ecosystem, and analogous to individual and population genomics, single-cell genomes can be quite different from bulk populations of cells.57 An important example is that several mutations, including DNMT3A, TET2, and ASXL1, detected at remission may represent preleukemic clonal hematopoiesis58 (Figure 1) and not relapse-causing leukemic cells.49 Studies incorporating single-cell DNA sequencing (scDNA-seq) observed distinct clones at remission that contain variants associated with clonal hematopoiesis and further delineated which clone harboring these mutations contributed to relapsed disease.28 Because bulk NGS is unable to accurately resolve clonal architecture, especially with rare variants, it is inherently limited in its ability to identify relapse-fated MRD.
Accurately characterizing the clonal context of MRD is critical in determining its clinical relevance. Modern paradigms of preleukemic clonal evolution, developed from rigorous xenotransplantation models of leukemia and high-throughput sequencing studies, suggest that early preleukemic mutations arise in self-renewing cells or promote self-renewal in differentiated cells.59 Therefore, given the low mutation rate in hematopoietic cells, latency of leukemogenesis, and low mutation burden in AML, it is believed that leukemogenesis arises from the accumulation of mutations in functionally normal HSCs. These HSCs, containing early leukemia-specific mutations, are considered pHSCs, which can drive clonal hematopoiesis, and will, through further evolution, transform into frank AML17,21,60,61 (Figure 1). Numerous studies have evaluated the patterns of clonal evolution in AML using bulk NGS on highly purified cell types17,21,59 and showed that in certain cases, AML arises from sequential mutation acquisition in pHSCs. Subsequent analyses revealed that these pHSCs can survive posttreatment,21 and clones expanding at relapse either retain or develop stem cell properties.23
Ordering of mutation acquisition and cooccurrence analyses using bulk NGS have provided insight into not only the prevalence of pHSCs17,59 but also the treatment vulnerabilities and outcomes.33,56 However, these patterns are often missed by bulk NGS. Several groups have addressed these limitations using scDNA-seq.28,62,63 Studies by Miles et al62 and Morita et al63 demonstrated that cooccurring mutations in epigenetic regulators commonly occur in ancestral clones, whereas mutations in signaling pathways occur later, in distinct subclones, possibly through convergent evolution. Additionally, our group has demonstrated that scDNA-seq could identify clinically relevant MRD by differentiating preleukemic clonal hematopoiesis from relapse-causing clones. We also observed that in certain cases, relapse-causing clones were not present at diagnosis or during remission.28 Dillon et al64 subsequently showed that scDNA-seq could be used to distinguish clonal hematopoiesis from leukemic cells by designing targeted gene panels that also captured breakpoints in AML patients with cytogenetic abnormalities.
Furthermore, the incorporation of cell-surface protein expression with scDNA-seq analyses has provided interesting insight into the significance of underlying clonal architectures. Both Miles et al62 and Morita et al63 were able to determine the specific contributions of clonal hematopoietic mutations to mature leukocyte lineages, providing additional evidence that these preleukemic mutations may exist in pHSCs and uniquely contribute to hematopoiesis. Additionally, they were able to determine specific alterations in immunophenotype depending on clonality in AML.
The ability to accurately resolve clonal architectures has also enhanced the ability to identify clonal origins of drug resistance. Bulk NGS analyses have implicated mutations in RAS/RTK/MAPK signaling pathways in the acquired resistance to targeted therapies.65,66 These observations have been supported by multiple scDNA-seq analyses, because subclones with RAS/RTK/MAPK mutations expanded during treatment with enasidenib,63 ivosidenib,66 FLT3 inhibitor,67,68 and venetoclax.69,70 The phylogenies of these resistant clones were highly variable, illuminating a complicating factor that was not apparent from bulk NGS.
Limitations of scDNA-seq
Although scDNA-seq has provided greater resolution for evaluating clonal architectures in AML, there are several limitations, including cost, limited single-cell throughput, small mutation panels, and allelic dropout.28 The limitation in throughput is a critical barrier for developing meaningful MRD detection assays, because >10 000 cells are required for accurate MRD assessment below the 0.01% threshold.71 Advancements in microfluidic technologies may allow the analysis of >50 000 cells, which decreases the detection limit to levels similar to error-corrected NGS and droplet digital PCR. Improvements are still needed to increase throughput to >100 000 cells, which would be comparable to MRD detection using multiparameter flow cytometry72 (Figure 2B), without impairing performance through, for example, doublet formation or incurring significant costs.
Many of the technical challenges associated with scDNA-seq are a consequence of the limited DNA material within a cell and subsequent difficulties associated with genomic amplification within confined droplets.57,73 scDNA-seq workflows need to carefully address errors associated with genome amplification, including allelic dropout, copying mistakes, and unbalanced amplifications. Allelic dropout occurs when 1 of the 2 alleles of diploid genomic DNA fails to amplify in PCR-based assays. This phenomenon can either be random or result from target specific features that impair polymerase function. Therefore, amplification bias and allelic dropout, in both the wild-type and variant alleles, can occur either biochemically or computationally and will significantly obscure the downstream reconstruction of clonal architectures. Additionally, the formation of doublets, which is often inferred by the Poisson statistic and is therefore limited by cell loading, can further confound these analyses. Although several downstream74, 75, 76 and standalone77 analysis packages incorporate allelic dropout and doublet detection, best practices for addressing these limitations are not well established.
Popular high-throughput scDNA-seq methods in AML research often incorporate targeted gene panels.28,62,63,67 Although there are several benefits to using targeted genomic amplification rather than whole-genome amplification,57 a targeted approach inherently limits the ability to completely resolve the underlying clonal architecture and identify novel variants that are biologically relevant. Furthermore, targeted panels are restricted to regions containing commonly mutated variants, termed hotspot regions, and not all regions are amplified with similar efficiency. Difficulty in detecting variants in commonly mutated genes, including NPM1, ASXL1, TP53, and SRSF2, highlights these limitations.28,63 It is therefore important to address these shortcomings when deriving conclusions from scDNA-seq analyses. New analysis methods have been developed to integrate bulk NGS with scDNA-seq78 in order to accurately infer clonal structures and evolution. Until the technical limitations of scDNA-seq are addressed, future work will need to acknowledge the complementary roles of bulk NGS and scDNA-seq in characterizing clonal compositions and evolution.
Nongenetic factors, transcriptional evolution, and single-cell multiomics
Cancer is classically defined by genomic alterations that transform cells into a variety of malignant states through complex multistep processes. The ongoing study of these malignant states has led to the description of classic hallmarks of cancer.79 Hanahan80 recently proposed 2 new emerging hallmarks, phenotypic plasticity and nonmutational epigenetic reprogramming. Cellular plasticity is the ability to bypass or disrupt normal differentiation potencies. Epigenetic reprogramming and alterations in gene and histone composition, chromatin structure, and in turn gene expression can lead to a multitude of cell states within genomically identical clones. Stochastic gene expression within isogenic cells may not only lead to variable cellular priming and subsequent cell fate decisions but also influence drug sensitivities.81 This realm of heterogeneity is not captured by current AML risk-stratification systems35 and may contribute to their shortcomings. New single-cell technologies are enabling more sophisticated analyses of cell states (Figure 3). Fennell et al82 leveraged such advancements by developing SPLINTR (single-cell profiling and lineage tracing) to track isogenic clones in murine AML and identified nongenetic factors that influence clonal fitness. Indeed, advancements in single-cell multiomics, including scRNA-seq,52,83 assay for transposase-accessible chromatin using sequencing,84, 85, 86 single-cell mass cytometry,87 and surface proteomics with antibody-derived tags,88, 89, 90 are describing cellular landscapes and continuums that have been incompletely described with bulk high-throughput biomolecular assays.91, 92, 93, 94
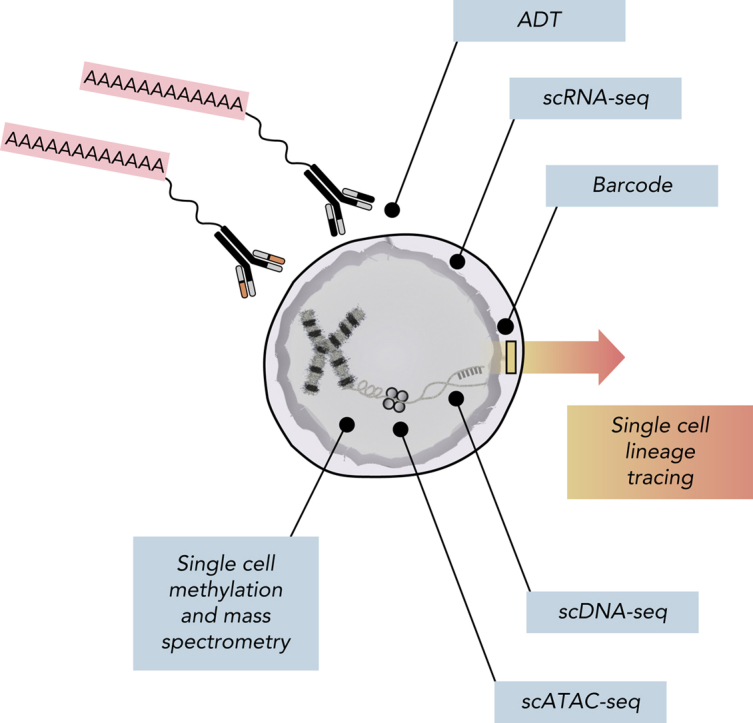
Figure 3.

Multiomic analysis of single cells. Current single-cell methods can quantify numerous analytes, including scDNA-seq, messenger RNA (scRNA-seq), chromosome accessibility (assay for transposase-accessible chromatin using sequencing [scATAC-seq]), DNA methylation, intracellular proteins using mass spectrometry, and surface proteins using antibody-derived tags (ADTs). Additionally, barcoding methods coupled with both scRNA-seq and scATAC-seq can facilitate lineage-tracing studies.
AML stem cell biology, heterogeneity, and evolution
The stem cell model of leukemogenesis (Figure 1) can partly explain the epigenomic and transcriptional hierarchy of AML. There remains controversy as to the clinical relevance of LSCs because of the significant variability in LSC content in primary patient samples and the nature of xenograft assays. Klco et al22 evaluated the functional heterogeneity of AML clones and observed variable engraftment potencies within AML subclones. Although they observed clonal restriction within the engrafted populations, there was no clear relationship between clonal mutations and engraftment. Furthermore, the engrafting clone was not consistently the clone that expanded during clinical relapse, nor was it the founding clone from the primary leukemia as defined by bulk genetic analysis. To add to the complexity, Sarry et al95 observed a degree of phenotypic plasticity in LSC subpopulations that has been corroborated by other groups.19,27
There is clearly a significant degree of heterogeneity within LSC subpopulations, both at the intra- and interpatient levels. Regardless, AML cells may converge on a similar transcriptional state, enriched for stem cell programs, in the face of underlying genomic and immunophenotypic differences.19,27,29 These observations are supported by the identification of gene expression programs that are upregulated in functionally defined LSCs and are associated with poor outcome25, 26, 27 and disease relapse.23 Therefore, characterizing LSCs may be enhanced by using transcriptional signatures in addition to mutation profiling and immunophenotyping. A primary limitation of prior studies is that LSC gene signatures and immunophenotyping have been derived from populations of cells enriched for LSCs, but still at low frequency. It is therefore difficult to determine which specific LSC subpopulations are responsible for treatment resistance or disease relapse.
Single-cell technologies can enhance our understanding of AML biology, including the stem cell compartment (Figure 1). Recent scRNA-seq studies in AML revealed that the presence of primitive and differentiated cell states in AML is highly variable and may be associated with certain genomic features. van Galen et al83 observed that primitive stem cell signatures derived from their analysis were associated with worse survival, and differentiated AML cells may modulate the immune microenvironment. These studies provide preliminary evidence of cell-specific gene expression programs and cell-cell interactions that may be associated with AML prognosis and pathogenesis.
It is becoming apparent that novel AML therapies, including hypomethylating agents such as azacitidine and the oral BCL2 inhibitor venetoclax, have differential effects on AML subpopulations. The recent landmark VIALE-A trial showed that the addition of venetoclax to azacitidine improved survival in older and unfit patients with AML.96 These observations are practice changing, because they provide an effective treatment for a patient population that has traditionally been high risk with limited therapeutic options. Despite these improvements, certain patients are resistant to these novel agents, and many relapse. Pei et al97 hypothesized that therapy resistance may be cell type specific and performed concurrent scRNA-seq and surface marker profiling to evaluate resistance mechanisms for patients who do relapse after azacitidine and venetoclax treatment. They observed that monocytic AML subpopulations preferentially expand compared with phenotypically primitive AML cells at relapse. The expanded population had not only lower BCL2 expression but also increased expression of MCL1, which encodes for an alternative antiapoptotic protein and is the target of novel selective inhibitors currently being evaluated in clinical trials.98 Pei et al also noticed that the relapsed monocytic subpopulation upregulated stem cell–associated gene expression programs, despite having a different immunophenotype, consistent with prior observations.23 This study provides important insights into cell-specific vulnerabilities to novel agents and subsequently, patient-specific therapeutic strategies.
Understanding the importance of transcriptional heterogeneity, Petti et al99 evaluated its role in AML relapse by performing paired whole-genome sequencing and ultradeep scRNA-seq on samples from patients with AML obtained pretreatment and at relapse. They observed significant transcriptional evolution at relapse that was inconsistently associated with genetic changes. In their cohort, relapsed AML was enriched for a quiescent cell state that upregulated unique expression programs, including an LSC signature.27 They performed a case-by-case analysis, revealing the importance of both the genome and transcriptome in leukemia evolution, as well as potential contributions from the immune microenvironment. Although their analysis was limited by cohort size, their observations highlight important cell-intrinsic and -extrinsic properties that modulate AML heterogeneity and are effectively captured with single-cell assays.
A recurring objective in AML research is to distinguish LSCs not only from mature blasts but also from residual healthy cells. A general strategy is to use immunophenotypic or transcriptional signatures to identify primitive cells and genomic mutations to differentiate leukemic and healthy cells. Several groups are developing strategies to make these distinctions with single-cell genomics by inferring mutation status through detection of variant transcripts,83,99, 100, 101, 102 and a few groups have been successful in applying these methods to AML research.50,83,99,100,102 Significant limitations include low expression of variant transcripts and limited throughput in current scRNA-seq methods. Velten et al102 attempted to address these shortcomings by incorporating mitochondrial variant detection; however, limitations persist because of low coverage of both genomic and mitochondrial mutations and mitochondrial somatic mutation variability. Future improvements in these methods will be critical for accurately integrating cellular states with the clonal architecture of AML.
Limitations and challenges of current single-cell technologies
Although the clinical care of AML has entered a renaissance, with 9 new therapeutic options approved by the US Food and Drug Administration since 2017,96,103 AML remains a highly lethal cancer. Previously, the ability to accurately delineate and monitor the cellular heterogeneity of AML has limited its study and management. With recent advancements in single-cell technologies, AML research has reached a new frontier, because we can begin to accurately reconstruct both the clonal architecture and cellular states of AML during treatment. The rapid development of these assays is unprecedented, and researchers will need to critically evaluate the data gathered, not only for accuracy but also for functional and clinical relevance.
As we move forward, integrating the measurement and computational analyses of multiple single-cell analytes will be increasingly important (Figure 3). Mutation status differentiates leukemic cells from their residual healthy counterparts, transcriptomic and epigenomic programs delineate cell states and potencies, and surface immunophenotype enables prospective isolation and functional interrogations. Although the multiomic evaluation of single cells is informative, the data provide only a snapshot of the cellular composition. Determining cellular potencies and their ultimate fate is vital for understanding the functional relevance of the captured cells. Advanced computational tools are available to infer cellular trajectories104,105; however, they provide only a prediction of the true fate of each cell. New tools are being developed to assess cell fates using single-cell barcoding strategies.82,106,107 Weinreb et al106 developed a clonal labeling system, LARRY (Lineage And RNA RecoverY),106 similar to SPLINTR,82 and a bioinformatic pipeline108 to facilitate lineage-tracing experiments using scRNA-seq. An important caveat of barcoding systems using viral delivery is that the process of transduction and in vitro culture can dramatically alter relevant cell biology. Therefore, natural barcoding using mitochondrial variants is a promising alternative method,109, 110, 111 although it is limited by mitochondrial somatic mutation variability and RNA editing. Of interest, Bowling et al112 developed an in vivo system for inducible lineage tracing capabilities allowing for simultaneous characterization of lineage history and gene expression. Future work may consider crossing their engineered mouse line, CARLIN (CRISPR array repair lineage tracing), with established genetically engineered mouse models of AML.10 These new lineage-tracing tools are early in development but can further enhance our understanding of the functional relevance of important AML subpopulations.
Limitations of single-cell assays include both cost and throughput. Although numerous research laboratories are generating single-cell data and analyses, the cost associated with these approaches for large AML cohorts remains prohibitive. A concerted effort is needed to make these data and relevant clinical annotations publicly available for all researchers. For now, it will be important to externally validate key observations from single-cell analyses with bulk biomolecular profiling data sets that are available for numerous AML patient samples.113 Several pipelines are in place to rigorously perform these analyses, which can include cell label transfer114 and bulk expression deconvolution.115, 116, 117 Zeng et al29 performed such an analysis by reannotating leukemia stem and progenitor cell populations from published scRNA-seq data83 in order to characterize changes in the cellular composition of AML during treatment. By deconvolving published gene expression data sets across >1000 patients, they provide an interesting framework for evaluating responses to a variety of therapeutic agents, which can be adapted to future studies in AML.
Future perspectives of single-cell genomics in AML
Advanced single-cell methods can allow us to answer critical questions in AML research. For instance, can we reliably distinguish pHSCs from LSCs? What is the true degree of clonal heterogeneity within these important AML subsets, and what are the molecular processes that transform pHSCs to LSCs? Furthermore, how do AML cells interact with one another and the microenvironment, and how do they change with time and treatment? Granja et al86,118 have developed a framework for integrating assay for transposase-accessible chromatin using sequencing and scRNA-seq that can facilitate the identification of differentially regulated gene expression networks relevant in cellular transitions. It will also be important to incorporate surface marker expression using antibody-derived tags into these analyses to enable the necessary functional correlates and lineage-tracing experiments in vivo. Investigating the hypothesis that leukemia stem cells and/or pHSCs do in fact persist and generate relapsed disease will be critical for designing curative therapies in AML.
The potential for translating single-cell technologies into viable clinical assays is exciting, and evaluating MRD for its cellular composition and clinical relevance may be a promising application (Figure 4).28 Although single-cell assays cannot reliably detect MRD because of throughput, it is clear that MRD, when detected, is not homogenous.119 Therefore, it will be important to characterize the cellular composition of that MRD to determine which cells are truly responsible for treatment failure and relapse. For instance, if the cellular composition at a late relapse is the same as that at diagnosis, should clinicians rechallenge with the same initial induction regimen? If there has been evolution, which new treatment should clinicians recommend? It will therefore be critical to incorporate markers that could differentiate relapse-fated clones from clonal hematopoiesis and, similarly, bona fide leukemia cells (ie, LSCs and their progeny) from pHSCs (Figure 1). Furthermore, incorporating transcriptomic or epigenomic features in addition to mutation status may be important for identifying nongenetic factors that can contribute to resistant and relapsed AML.119 Using targeted multiomic profiling will likely be more feasible than whole-genomic or transcriptomic assays; however, the features used may need to be relatively broad or dynamic, because markers present at diagnosis and relapse may be different. Ultimately, prospective clinical trials will need to compare single-cell assays with bulk biomolecular profiling methods in order to reliably understand their utility.
Figure 4.

Single-cell assessment of MRD. Multiomic evaluation of MRD at remission may provide not only the underlying clonal architecture but also the cellular state of each clone, potential cellular fates, and cell-cell ecosystems within the bone marrow environment. Comparing these observations with those from diagnosis and relapse and the patient’s eventual clinical outcome may provide valuable insight into the clinical significance of the detected MRD.
Summary
Many fundamental questions in AML biology and disease management remain unanswered, driven in part by the diversity of cells within the leukemia. Prior research has relied on bulk analysis methods, which have been limited by their inability to accurately deconvolve the cellular heterogeneity of the disease. Characterizing the cellular composition of AML and underlying cell-specific processes is proving to be vital in addressing these important questions. Recent advances in the development and implementation of single-cell methods for the analysis of AML are starting to advance our understanding of this heterogeneity. Although there are limitations to these single-cell methods, these techniques may provide the resolution needed to drive AML research into a new era. It will be critical to not only establish rigorous benchmarks as we continue to analyze these complex datasets but also perform the necessary functional assessments of compelling single-cell observations. In the upcoming years, the careful implementation of these methods can provide the insights necessary to advance personalized and effective medicine in AML.
Conflict-of-interest disclosure: R.M. is on the board of directors of CircBio, Inc., and the advisory boards of Kodikaz Therapeutic Solutions, Inc., and Syros Pharmaceuticals and is an inventor on a number of patents related to CD47 cancer immunotherapy licensed to Gilead Sciences, Inc.; receives research support from Gilead Sciences, Inc.; and is a cofounder of and equity holder in CircBio, Inc., Pheast Therapeutics, MyeloGene, Inc., and RNAC Therapeutics, Inc. The remaining authors declare no competing financial interests.
Acknowledgments
R.M. was supported by the National Heart, Lung, and Blood Institute (NHLBI) and the National Cancer Institute (NCI) of the National Institutes of Health (NIH) under awards 1R01HL142637 and 1R01CA251331 respectively, the Stanford Ludwig Center for Cancer Stem Cell Research and Medicine, the Blood Cancer Discoveries Grant Program through the Leukemia and Lymphoma Society, the Mark Foundation for Cancer Research, the Paul G. Allen Frontiers Group, the RUNX1 Research Program, and a Discovery Research Grant through the Edward P. Evans Foundation. A.J.G. was supported by the NCI under awards R21CA238971, U24CA224309, and U54CA209971. A.E. was supported by the NCI under award F32CA250304, the Advanced Residency Training Program at Stanford, and the American Society of Hematology Scholar Award.
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Authorship
Contribution: A.E., A.J.G., and R.M. wrote the paper.
References
- 1.Forkner CE. Clinical and pathological differentiation of acute leukemias: with special reference to acute monocytic leukemia. Arch Intern Med (Chic) 1934;53(1):1–34. [Google Scholar]
- 2.Vardiman JW, Thiele J, Arber DA, et al. The 2008 revision of the World Health Organization (WHO) classification of myeloid neoplasms and acute leukemia: rationale and important changes. Blood. 2009;114(5):937–951. doi: 10.1182/blood-2009-03-209262. [DOI] [PubMed] [Google Scholar]
- 3.Bennett JM, Catovsky D, Daniel MT, et al. Proposals for the classification of the acute leukaemias. French-American-British (FAB) co-operative group. Br J Haematol. 1976;33(4):451–458. doi: 10.1111/j.1365-2141.1976.tb03563.x. [DOI] [PubMed] [Google Scholar]
- 4.Griffin JD, Löwenberg B. Clonogenic cells in acute myeloblastic leukemia. Blood. 1986;68(6):1185–1195. [PubMed] [Google Scholar]
- 5.Fialkow PJ, Singer JW, Adamson JW, et al. Acute nonlymphocytic leukemia: heterogeneity of stem cell origin. Blood. 1981;57(6):1068–1073. [PubMed] [Google Scholar]
- 6.Yates JW, Wallace HJ, Jr., Ellison RR, Holland JF. Cytosine arabinoside (NSC-63878) and daunorubicin (NSC-83142) therapy in acute nonlymphocytic leukemia. Cancer Chemother Rep. 1973;57(4):485–488. [PubMed] [Google Scholar]
- 7.Thomas ED, Buckner CD, Clift RA, et al. Marrow transplantation for acute nonlymphoblastic leukemia in first remission. N Engl J Med. 1979;301(11):597–599. doi: 10.1056/NEJM197909133011109. [DOI] [PubMed] [Google Scholar]
- 8.National Cancer Institute Surveillance, Epidemiology, and End Results: Cancer Stat Facts: Leukemia—Acute Myeloid Leukemia. https://seer.cancer.gov/statfacts/html/amyl.html Available at:
- 9.Döhner H, Weisdorf DJ, Bloomfield CD. Acute myeloid leukemia. N Engl J Med. 2015;373(12):1136–1152. doi: 10.1056/NEJMra1406184. [DOI] [PubMed] [Google Scholar]
- 10.Thomas D, Majeti R. Biology and relevance of human acute myeloid leukemia stem cells. Blood. 2017;129(12):1577–1585. doi: 10.1182/blood-2016-10-696054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lapidot T, Sirard C, Vormoor J, et al. A cell initiating human acute myeloid leukaemia after transplantation into SCID mice. Nature. 1994;367(6464):645–648. doi: 10.1038/367645a0. [DOI] [PubMed] [Google Scholar]
- 12.Bonnet D, Dick JE. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat Med. 1997;3(7):730–737. doi: 10.1038/nm0797-730. [DOI] [PubMed] [Google Scholar]
- 13.Blair A, Hogge DE, Ailles LE, Lansdorp PM, Sutherland HJ. Lack of expression of Thy-1 (CD90) on acute myeloid leukemia cells with long-term proliferative ability in vitro and in vivo. Blood. 1997;89(9):3104–3112. [PubMed] [Google Scholar]
- 14.Blair A, Hogge DE, Sutherland HJ. Most acute myeloid leukemia progenitor cells with long-term proliferative ability in vitro and in vivo have the phenotype CD34(+)/CD71 (-)/HLA-DR- Blood. 1998;92(11):4325–4335. [PubMed] [Google Scholar]
- 15.Majeti R, Chao MP, Alizadeh AA, et al. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell. 2009;138(2):286–299. doi: 10.1016/j.cell.2009.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jan M, Chao MP, Cha AC, et al. Prospective separation of normal and leukemic stem cells based on differential expression of TIM3, a human acute myeloid leukemia stem cell marker. Proc Natl Acad Sci USA. 2011;108(12):5009–5014. doi: 10.1073/pnas.1100551108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Jan M, Snyder TM, Corces-Zimmerman MR, et al. Clonal evolution of preleukemic hematopoietic stem cells precedes human acute myeloid leukemia. Sci Transl Med. 2012;4(149) doi: 10.1126/scitranslmed.3004315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pabst C, Bergeron A, Lavallée VP, et al. GPR56 identifies primary human acute myeloid leukemia cells with high repopulating potential in vivo. Blood. 2016;127(16):2018–2027. doi: 10.1182/blood-2015-11-683649. [DOI] [PubMed] [Google Scholar]
- 19.Quek L, Otto GW, Garnett C, et al. Genetically distinct leukemic stem cells in human CD34- acute myeloid leukemia are arrested at a hemopoietic precursor-like stage. J Exp Med. 2016;213(8):1513–1535. doi: 10.1084/jem.20151775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sutherland HJ, Blair A, Zapf RW. Characterization of a hierarchy in human acute myeloid leukemia progenitor cells. Blood. 1996;87(11):4754–4761. [PubMed] [Google Scholar]
- 21.Shlush LI, Zandi S, Mitchell A, et al. HALT Pan-Leukemia Gene Panel Consortium. Identification of pre-leukaemic haematopoietic stem cells in acute leukaemia. Nature. 2014;506(7488):328–333. doi: 10.1038/nature13038. [published correction appears in Nature. 2014;508(7496):420] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Klco JM, Spencer DH, Miller CA, et al. Functional heterogeneity of genetically defined subclones in acute myeloid leukemia. Cancer Cell. 2014;25(3):379–392. doi: 10.1016/j.ccr.2014.01.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shlush LI, Mitchell A, Heisler L, et al. Tracing the origins of relapse in acute myeloid leukaemia to stem cells. Nature. 2017;547(7661):104–108. doi: 10.1038/nature22993. [DOI] [PubMed] [Google Scholar]
- 24.Paczulla AM, Rothfelder K, Raffel S, et al. Absence of NKG2D ligands defines leukaemia stem cells and mediates their immune evasion. Nature. 2019;572(7768):254–259. doi: 10.1038/s41586-019-1410-1. [published correction appears in Nature. 2019;572(7770):E19]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gentles AJ, Plevritis SK, Majeti R, Alizadeh AA. Association of a leukemic stem cell gene expression signature with clinical outcomes in acute myeloid leukemia. JAMA. 2010;304(24):2706–2715. doi: 10.1001/jama.2010.1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Eppert K, Takenaka K, Lechman ER, et al. Stem cell gene expression programs influence clinical outcome in human leukemia. Nat Med. 2011;17(9):1086–1093. doi: 10.1038/nm.2415. [DOI] [PubMed] [Google Scholar]
- 27.Ng SW, Mitchell A, Kennedy JA, et al. A 17-gene stemness score for rapid determination of risk in acute leukaemia. Nature. 2016;540(7633):433–437. doi: 10.1038/nature20598. [DOI] [PubMed] [Google Scholar]
- 28.Ediriwickrema A, Aleshin A, Reiter JG, et al. Single-cell mutational profiling enhances the clinical evaluation of AML MRD. Blood Adv. 2020;4(5):943–952. doi: 10.1182/bloodadvances.2019001181. [published correction appears in Blood Adv. 2020;4(7):1218]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zeng AGX, Bansal S, Jin L, et al. A cellular hierarchy framework for understanding heterogeneity and predicting drug response in acute myeloid leukemia. Nat Med. 2022;28(6):1212–1223. doi: 10.1038/s41591-022-01819-x. [DOI] [PubMed] [Google Scholar]
- 30.Pollyea DA. New drugs for acute myeloid leukemia inspired by genomics and when to use them. Hematology Am Soc Hematol Educ Program. 2018;2018:45–50. doi: 10.1182/asheducation-2018.1.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Patel JP, Gönen M, Figueroa ME, et al. Prognostic relevance of integrated genetic profiling in acute myeloid leukemia. N Engl J Med. 2012;366(12):1079–1089. doi: 10.1056/NEJMoa1112304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ley TJ, Miller C, Ding L, et al. Cancer Genome Atlas Research Network Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N Engl J Med. 2013;368(22):2059–2074. doi: 10.1056/NEJMoa1301689. [published correction appears in N Engl J Med. 2013;369(1):98] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Papaemmanuil E, Gerstung M, Bullinger L, et al. Genomic classification and prognosis in acute myeloid leukemia. N Engl J Med. 2016;374(23):2209–2221. doi: 10.1056/NEJMoa1516192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Arber DA, Orazi A, Hasserjian R, et al. The 2016 revision to the World Health Organization classification of myeloid neoplasms and acute leukemia. Blood. 2016;127(20):2391–2405. doi: 10.1182/blood-2016-03-643544. [DOI] [PubMed] [Google Scholar]
- 35.Döhner H, Estey E, Grimwade D, et al. Diagnosis and management of AML in adults: 2017 ELN recommendations from an international expert panel. Blood. 2017;129(4):424–447. doi: 10.1182/blood-2016-08-733196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gerstung M, Papaemmanuil E, Martincorena I, et al. Precision oncology for acute myeloid leukemia using a knowledge bank approach. Nat Genet. 2017;49(3):332–340. doi: 10.1038/ng.3756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stone RM, Mandrekar SJ, Sanford BL, et al. Midostaurin plus chemotherapy for acute myeloid leukemia with a FLT3 mutation. N Engl J Med. 2017;377(5):454–464. doi: 10.1056/NEJMoa1614359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Perl AE, Altman JK, Cortes J, et al. Selective inhibition of FLT3 by gilteritinib in relapsed or refractory acute myeloid leukaemia: a multicentre, first-in-human, open-label, phase 1-2 study. Lancet Oncol. 2017;18(8):1061–1075. doi: 10.1016/S1470-2045(17)30416-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stein EM, DiNardo CD, Pollyea DA, et al. Enasidenib in mutant IDH2 relapsed or refractory acute myeloid leukemia. Blood. 2017;130(6):722–731. doi: 10.1182/blood-2017-04-779405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.DiNardo CD, Stein EM, de Botton S, et al. Durable remissions with ivosidenib in IDH1-mutated relapsed or refractory AML. N Engl J Med. 2018;378(25):2386–2398. doi: 10.1056/NEJMoa1716984. [DOI] [PubMed] [Google Scholar]
- 41.Chen W, Xie H, Wang H, et al. Prognostic significance of KIT mutations in core-binding factor acute myeloid leukemia: a systematic review and meta-analysis. PLoS One. 2016;11(1) doi: 10.1371/journal.pone.0146614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jahn N, Terzer T, Sträng E, et al. Genomic heterogeneity in core-binding factor acute myeloid leukemia and its clinical implication. Blood Adv. 2020;4(24):6342–6352. doi: 10.1182/bloodadvances.2020002673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Harada Y, Nagata Y, Kihara R, et al. Japan Adult Leukemia Study Group JALSG Prognostic analysis according to the 2017 ELN risk stratification by genetics in adult acute myeloid leukemia patients treated in the Japan Adult Leukemia Study Group (JALSG) AML201 study. Leuk Res. 2018;66:20–27. doi: 10.1016/j.leukres.2018.01.008. [DOI] [PubMed] [Google Scholar]
- 44.Eisfeld A-K, Kohlschmidt J, Mrózek K, et al. Mutation patterns identify adult patients with de novo acute myeloid leukemia aged 60 years or older who respond favorably to standard chemotherapy: an analysis of Alliance studies. Leukemia. 2018;32(6):1338–1348. doi: 10.1038/s41375-018-0068-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Heuser M, Freeman SD, Ossenkoppele GJ, et al. 2021 update on MRD in acute myeloid leukemia: a consensus document from the European LeukemiaNet MRD Working Party. Blood. 2021;138(26):2753–2767. doi: 10.1182/blood.2021013626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hourigan CS, Gale RP, Gormley NJ, Ossenkoppele GJ, Walter RB. Measurable residual disease testing in acute myeloid leukaemia. Leukemia. 2017;31(7):1482–1490. doi: 10.1038/leu.2017.113. [DOI] [PubMed] [Google Scholar]
- 47.Freeman SD, Virgo P, Couzens S, et al. Prognostic relevance of treatment response measured by flow cytometric residual disease detection in older patients with acute myeloid leukemia. J Clin Oncol. 2013;31(32):4123–4131. doi: 10.1200/JCO.2013.49.1753. [DOI] [PubMed] [Google Scholar]
- 48.Terwijn M, van Putten WL, Kelder A, et al. High prognostic impact of flow cytometric minimal residual disease detection in acute myeloid leukemia: data from the HOVON/SAKK AML 42A study. J Clin Oncol. 2013;31(31):3889–3897. doi: 10.1200/JCO.2012.45.9628. [DOI] [PubMed] [Google Scholar]
- 49.Jongen-Lavrencic M, Grob T, Hanekamp D, et al. Molecular minimal residual disease in acute myeloid leukemia. N Engl J Med. 2018;378(13):1189–1199. doi: 10.1056/NEJMoa1716863. [DOI] [PubMed] [Google Scholar]
- 50.Nam AS, Chaligne R, Landau DA. Integrating genetic and non-genetic determinants of cancer evolution by single-cell multi-omics. Nat Rev Genet. 2021;22(1):3–18. doi: 10.1038/s41576-020-0265-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Povinelli BJ, Rodriguez-Meira A, Mead AJ. Single cell analysis of normal and leukemic hematopoiesis. Mol Aspects Med. 2018;59:85–94. doi: 10.1016/j.mam.2017.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zheng GXY, Terry JM, Belgrader P, et al. Massively parallel digital transcriptional profiling of single cells. Nat Commun. 2017;8 doi: 10.1038/ncomms14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Klco JM, Miller CA, Griffith M, et al. Association between mutation clearance after induction therapy and outcomes in acute myeloid leukemia. JAMA. 2015;314(8):811–822. doi: 10.1001/jama.2015.9643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rothenberg-Thurley M, Amler S, Goerlich D, et al. Persistence of pre-leukemic clones during first remission and risk of relapse in acute myeloid leukemia. Leukemia. 2018;32(7):1598–1608. doi: 10.1038/s41375-018-0034-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ding L, Ley TJ, Larson DE, et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature. 2012;481(7382):506–510. doi: 10.1038/nature10738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Benard BA, Leak LB, Azizi A, Thomas D, Gentles AJ, Majeti R. Clonal architecture predicts clinical outcomes and drug sensitivity in acute myeloid leukemia. Nat Commun. 2021;12(1):7244. doi: 10.1038/s41467-021-27472-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gawad C, Koh W, Quake SR. Single-cell genome sequencing: current state of the science. Nat Rev Genet. 2016;17(3):175–188. doi: 10.1038/nrg.2015.16. [DOI] [PubMed] [Google Scholar]
- 58.Jaiswal S, Fontanillas P, Flannick J, et al. Age-related clonal hematopoiesis associated with adverse outcomes. N Engl J Med. 2014;371(26):2488–2498. doi: 10.1056/NEJMoa1408617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Corces-Zimmerman MR, Hong W-J, Weissman IL, Medeiros BC, Majeti R. Preleukemic mutations in human acute myeloid leukemia affect epigenetic regulators and persist in remission. Proc Natl Acad Sci USA. 2014;111(7):2548–2553. doi: 10.1073/pnas.1324297111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Corces-Zimmerman MR, Majeti R. Pre-leukemic evolution of hematopoietic stem cells: the importance of early mutations in leukemogenesis. Leukemia. 2014;28(12):2276–2282. doi: 10.1038/leu.2014.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Köhnke T, Majeti R. Clonal hematopoiesis: from mechanisms to clinical intervention. Cancer Discov. 2021;11(12):2987–2997. doi: 10.1158/2159-8290.CD-21-0901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Miles LA, Bowman RL, Merlinsky TR, et al. Single-cell mutation analysis of clonal evolution in myeloid malignancies. Nature. 2020;587(7834):477–482. doi: 10.1038/s41586-020-2864-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Morita K, Wang F, Jahn K, et al. Clonal evolution of acute myeloid leukemia revealed by high-throughput single-cell genomics. Nat Commun. 2020;11(1):5327. doi: 10.1038/s41467-020-19119-8. [published corrections appear in Nat Commun. 2020;11(1):5996 and Nat Commun. 2021;12(1):2823] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Dillon LW, Ghannam J, Nosiri C, et al. Personalized single-cell proteogenomics to distinguish acute myeloid leukemia from non-malignant clonal hematopoiesis. Blood Cancer Discov. 2021;2(4):319–325. doi: 10.1158/2643-3230.BCD-21-0046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Schmalbrock LK, Dolnik A, Cocciardi S, et al. Clonal evolution of acute myeloid leukemia with FLT3-ITD mutation under treatment with midostaurin. Blood. 2021;137(22):3093–3104. doi: 10.1182/blood.2020007626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Choe S, Wang H, DiNardo CD, et al. Molecular mechanisms mediating relapse following ivosidenib monotherapy in IDH1-mutant relapsed or refractory AML. Blood Adv. 2020;4(9):1894–1905. doi: 10.1182/bloodadvances.2020001503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.McMahon CM, Ferng T, Canaani J, et al. Clonal selection with Ras pathway activation mediates secondary clinical resistance to selective FLT3 inhibition in acute myeloid leukemia. Cancer Discov. 2019;9(8):1050–1063. doi: 10.1158/2159-8290.CD-18-1453. [DOI] [PubMed] [Google Scholar]
- 68.Peretz CAC, McGary LHF, Kumar T, et al. Single-cell DNA sequencing reveals complex mechanisms of resistance to quizartinib. Blood Adv. 2021;5(5):1437–1441. doi: 10.1182/bloodadvances.2020003398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.DiNardo CD, Tiong IS, Quaglieri A, et al. Molecular patterns of response and treatment failure after frontline venetoclax combinations in older patients with AML. Blood. 2020;135(11):791–803. doi: 10.1182/blood.2019003988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang Q, Riley-Gillis B, Han L, et al. Activation of RAS/MAPK pathway confers MCL-1 mediated acquired resistance to BCL-2 inhibitor venetoclax in acute myeloid leukemia. Signal Transduct Target Ther. 2022;7(1):51. doi: 10.1038/s41392-021-00870-3. [published correction appears in Signal Transduct Target Ther. 2022;7(1):110] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schuurhuis GJ, Heuser M, Freeman S, et al. Minimal/measurable residual disease in AML: a consensus document from the European LeukemiaNet MRD Working Party. Blood. 2018;131(12):1275–1291. doi: 10.1182/blood-2017-09-801498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Schmitt MW, Kennedy SR, Salk JJ, Fox EJ, Hiatt JB, Loeb LA. Detection of ultra-rare mutations by next-generation sequencing. Proc Natl Acad Sci USA. 2012;109(36):14508–14513. doi: 10.1073/pnas.1208715109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Pellegrino M, Sciambi A, Treusch S, et al. High-throughput single-cell DNA sequencing of acute myeloid leukemia tumors with droplet microfluidics. Genome Res. 2018;28(9):1345–1352. doi: 10.1101/gr.232272.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kuipers J, Jahn K, Raphael BJ, Beerenwinkel N. Single-cell sequencing data reveal widespread recurrence and loss of mutational hits in the life histories of tumors. Genome Res. 2017;27(11):1885–1894. doi: 10.1101/gr.220707.117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Roth A, McPherson A, Laks E, et al. Clonal genotype and population structure inference from single-cell tumor sequencing. Nat Methods. 2016;13(7):573–576. doi: 10.1038/nmeth.3867. [DOI] [PubMed] [Google Scholar]
- 76.Zafar H, Navin N, Chen K, Nakhleh L. SiCloneFit: Bayesian inference of population structure, genotype, and phylogeny of tumor clones from single-cell genome sequencing data. Genome Res. 2019;29(11):1847–1859. doi: 10.1101/gr.243121.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Weber LL, Sashittal P, El-Kebir M. doubletD: detecting doublets in single-cell DNA sequencing data. Bioinformatics. 2021;37(suppl 1):i214–i221. doi: 10.1093/bioinformatics/btab266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Malikic S, Jahn K, Kuipers J, Sahinalp SC, Beerenwinkel N. Integrative inference of subclonal tumour evolution from single-cell and bulk sequencing data. Nat Commun. 2019;10(1):2750. doi: 10.1038/s41467-019-10737-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1):57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 80.Hanahan D. Hallmarks of cancer: new dimensions. Cancer Discov. 2022;12(1):31–46. doi: 10.1158/2159-8290.CD-21-1059. [DOI] [PubMed] [Google Scholar]
- 81.Emert BL, Cote CJ, Torre EA, et al. Variability within rare cell states enables multiple paths toward drug resistance. Nat Biotechnol. 2021;39(7):865–876. doi: 10.1038/s41587-021-00837-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Fennell KA, Vassiliadis D, Lam EYN, et al. Non-genetic determinants of malignant clonal fitness at single-cell resolution. Nature. 2022;601(7891):125–131. doi: 10.1038/s41586-021-04206-7. [DOI] [PubMed] [Google Scholar]
- 83.van Galen P, Hovestadt V, Wadsworth Ii MH, et al. Single-cell RNA-seq reveals AML hierarchies relevant to disease progression and immunity. Cell. 2019;176(6):1265–1281. doi: 10.1016/j.cell.2019.01.031. e24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Buenrostro JD, Wu B, Litzenburger UM, et al. Single-cell chromatin accessibility reveals principles of regulatory variation. Nature. 2015;523(7561):486–490. doi: 10.1038/nature14590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Buenrostro JD, Corces MR, Lareau CA, et al. Integrated single-cell analysis maps the continuous regulatory landscape of human hematopoietic differentiation. Cell. 2018;173(6):1535–1548. doi: 10.1016/j.cell.2018.03.074. e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Granja JM, Klemm S, McGinnis LM, et al. Single-cell multiomic analysis identifies regulatory programs in mixed-phenotype acute leukemia. Nat Biotechnol. 2019;37(12):1458–1465. doi: 10.1038/s41587-019-0332-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Levine JH, Simonds EF, Bendall SC, et al. Data-driven phenotypic dissection of AML reveals progenitor-like cells that correlate with prognosis. Cell. 2015;162(1):184–197. doi: 10.1016/j.cell.2015.05.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Mair F, Erickson JR, Voillet V, et al. A targeted multi-omic analysis approach measures protein expression and low-abundance transcripts on the single-cell level. Cell Rep. 2020;31(1) doi: 10.1016/j.celrep.2020.03.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Stoeckius M, Hafemeister C, Stephenson W, et al. Simultaneous epitope and transcriptome measurement in single cells. Nat Methods. 2017;14(9):865–868. doi: 10.1038/nmeth.4380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Triana S, Vonficht D, Jopp-Saile L, et al. Single-cell proteo-genomic reference maps of the hematopoietic system enable the purification and massive profiling of precisely defined cell states. Nat Immunol. 2021;22(12):1577–1589. doi: 10.1038/s41590-021-01059-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Karamitros D, Stoilova B, Aboukhalil Z, et al. Single-cell analysis reveals the continuum of human lympho-myeloid progenitor cells. Nat Immunol. 2018;19(1):85–97. doi: 10.1038/s41590-017-0001-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Velten L, Haas SF, Raffel S, et al. Human haematopoietic stem cell lineage commitment is a continuous process. Nat Cell Biol. 2017;19(4):271–281. doi: 10.1038/ncb3493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Weinreb C, Wolock S, Tusi BK, Socolovsky M, Klein AM. Fundamental limits on dynamic inference from single-cell snapshots. Proc Natl Acad Sci USA. 2018;115(10):E2467–E2476. doi: 10.1073/pnas.1714723115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Laurenti E, Göttgens B. From haematopoietic stem cells to complex differentiation landscapes. Nature. 2018;553(7689):418–426. doi: 10.1038/nature25022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Sarry J-E, Murphy K, Perry R, et al. Human acute myelogenous leukemia stem cells are rare and heterogeneous when assayed in NOD/SCID/IL2Rγc-deficient mice. J Clin Invest. 2011;121(1):384–395. doi: 10.1172/JCI41495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.DiNardo CD, Jonas BA, Pullarkat V, et al. Azacitidine and venetoclax in previously untreated acute myeloid leukemia. N Engl J Med. 2020;383(7):617–629. doi: 10.1056/NEJMoa2012971. [DOI] [PubMed] [Google Scholar]
- 97.Pei S, Pollyea DA, Gustafson A, et al. Monocytic subclones confer resistance to venetoclax-based therapy in patients with acute myeloid leukemia. Cancer Discov. 2020;10(4):536–551. doi: 10.1158/2159-8290.CD-19-0710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wang H, Guo M, Wei H, Chen Y. Targeting MCL-1 in cancer: current status and perspectives. J Hematol Oncol. 2021;14(1):67. doi: 10.1186/s13045-021-01079-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Petti AA, Williams SR, Miller CA, et al. A general approach for detecting expressed mutations in AML cells using single cell RNA-sequencing. Nat Commun. 2019;10(1):3660. doi: 10.1038/s41467-019-11591-1. [published correction appears in Nat Commun. 2022;13(1):4216] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Nam AS, Kim KT, Chaligne R, et al. Somatic mutations and cell identity linked by Genotyping of Transcriptomes. Nature. 2019;571(7765):355–360. doi: 10.1038/s41586-019-1367-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Rodriguez-Meira A, Buck G, Clark SA, et al. Unravelling intratumoral heterogeneity through high-sensitivity single-cell mutational analysis and parallel RNA sequencing. Mol Cell. 2019;73(6):1292–1305. doi: 10.1016/j.molcel.2019.01.009. e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Velten L, Story BA, Hernández-Malmierca P, et al. Identification of leukemic and pre-leukemic stem cells by clonal tracking from single-cell transcriptomics. Nat Commun. 2021;12(1):1366. doi: 10.1038/s41467-021-21650-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.DiNardo CD, Perl AE. Advances in patient care through increasingly individualized therapy. Nat Rev Clin Oncol. 2019;16(2):73–74. doi: 10.1038/s41571-018-0156-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gulati GS, Sikandar SS, Wesche DJ, et al. Single-cell transcriptional diversity is a hallmark of developmental potential. Science. 2020;367(6476):405–411. doi: 10.1126/science.aax0249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Saelens W, Cannoodt R, Todorov H, Saeys Y. A comparison of single-cell trajectory inference methods. Nat Biotechnol. 2019;37(5):547–554. doi: 10.1038/s41587-019-0071-9. [DOI] [PubMed] [Google Scholar]
- 106.Weinreb C, Rodriguez-Fraticelli A, Camargo FD, Klein AM. Lineage tracing on transcriptional landscapes links state to fate during differentiation. Science. 2020;367(6479) doi: 10.1126/science.aaw3381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Oren Y, Tsabar M, Cuoco MS, et al. Cycling cancer persister cells arise from lineages with distinct programs. Nature. 2021;596(7873):576–582. doi: 10.1038/s41586-021-03796-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Wang S-W, Herriges MJ, Hurley K, Kotton DN, Klein AM. CoSpar identifies early cell fate biases from single-cell transcriptomic and lineage information. Nat Biotechnol. 2022;40(7):1066–1074. doi: 10.1038/s41587-022-01209-1. [DOI] [PubMed] [Google Scholar]
- 109.Miller TE, Lareau CA, Verga JA, et al. Mitochondrial variant enrichment from high-throughput single-cell RNA sequencing resolves clonal populations. Nat Biotechnol. 2022;40(7):1030–1034. doi: 10.1038/s41587-022-01210-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ludwig LS, Lareau CA, Ulirsch JC, et al. Lineage tracing in humans enabled by mitochondrial mutations and single-cell genomics. Cell. 2019;176(6):1325–1339. doi: 10.1016/j.cell.2019.01.022. e22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lareau CA, Ludwig LS, Muus C, et al. Massively parallel single-cell mitochondrial DNA genotyping and chromatin profiling. Nat Biotechnol. 2021;39(4):451–461. doi: 10.1038/s41587-020-0645-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Bowling S, Sritharan D, Osorio FG, et al. An engineered CRISPR-Cas9 mouse line for simultaneous readout of lineage histories and gene expression profiles in single cells. Cell. 2020;181(6):1410–1422.e27. doi: 10.1016/j.cell.2020.04.048. [published correction appears in Cell. 2020;181(7):1693-1694] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Gentles AJ, Newman AM, Liu CL, et al. The prognostic landscape of genes and infiltrating immune cells across human cancers. Nat Med. 2015;21(8):938–945. doi: 10.1038/nm.3909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Abdelaal T, Michielsen L, Cats D, et al. A comparison of automatic cell identification methods for single-cell RNA sequencing data. Genome Biol. 2019;20(1):194. doi: 10.1186/s13059-019-1795-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Luca BA, Steen CB, Matusiak M, et al. Atlas of clinically distinct cell states and ecosystems across human solid tumors. Cell. 2021;184(21):5482–5496. doi: 10.1016/j.cell.2021.09.014. e28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Sutton GJ, Poppe D, Simmons RK, et al. Comprehensive evaluation of deconvolution methods for human brain gene expression. Nat Commun. 2022;13(1):1358. doi: 10.1038/s41467-022-28655-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Newman AM, Steen CB, Liu CL, et al. Determining cell type abundance and expression from bulk tissues with digital cytometry. Nat Biotechnol. 2019;37(7):773–782. doi: 10.1038/s41587-019-0114-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Granja JM, Corces MR, Pierce SE, et al. ArchR is a scalable software package for integrative single-cell chromatin accessibility analysis. Nat Genet. 2021;53(3):403–411. doi: 10.1038/s41588-021-00790-6. [published correction appears in Nat Genet. 2021;53(6):935] [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Marine J-C, Dawson S-J, Dawson MA. Non-genetic mechanisms of therapeutic resistance in cancer. Nat Rev Cancer. 2020;20(12):743–756. doi: 10.1038/s41568-020-00302-4. [DOI] [PubMed] [Google Scholar]