

Abstract
Background and Aim:
Viruses are important components of the microbiome of ticks. Ticks are capable of transmitting several serious viral diseases to humans and animals. Hitherto, the composition of viral communities in Hyalomma dromedarii ticks associated with camels in the United Arab Emirates (UAE) remains unexplored. This study aimed to characterize the RNA virome diversity in male and female H. dromedarii ticks collected from camels in Al Ain, UAE.
Materials and Methods:
We collected ticks, extracted, and sequenced RNA, using Illumina (NovaSeq 6000) and Oxford Nanopore (MinION).
Results:
From the total generated sequencing reads, 180,559 (~0.35%) and 197,801 (~0.34%) reads were identified as virus-related reads in male and female tick samples, respectively. Taxonomic assignment of the viral sequencing reads was accomplished based on bioinformatic analyses. Further, viral reads were classified into 39 viral families. Poxiviridae, Phycodnaviridae, Phenuiviridae, Mimiviridae, and Polydnaviridae were the most abundant families in the tick viromes. Notably, we assembled the genomes of three RNA viruses, which were placed by phylogenetic analyses in clades that included the Bole tick virus.
Conclusion:
Overall, this study attempts to elucidate the RNA virome of ticks associated with camels in the UAE and the results obtained from this study improve the knowledge of the diversity of viruses in H. dromedarii ticks.
Keywords: camels, Hyalomma dromedarii, nanopore technology, UAE, viral diversity, virome analysis, whole genome sequencing
Introduction
Ticks are hematophagous ectoparasites that can transmit a wide range of microbial pathogens and substantially threaten animal and human health [1, 2]. Genomic analyses through next-generation sequencing platforms have proven valuable for investigating the diversity and composition of microbes in ticks [3]. The discovery of novel pathogens may be facilitated by the characterization of the tick microbiome, which is important to understand the relationship between microbes and their tick hosts [4]. Tick viromes have not been well-studied and high-throughput sequencing facilitates the investigation of diversity and circulation of existing and novel viruses in tick vectors [5]. In recent decades, several viral epidemics have been reported in the Arabian Peninsula [6–8], although the knowledge about the viruses in the region remains limited. Comprehensive surveillance and screening of vector populations, especially ticks, are essential to prepare for new viral outbreaks. Recently, the Middle East and North Africa (MENA) region has observed the emergence and re-emergence of several viruses [6, 9–15]. Notable examples include the Middle East Respiratory Syndrome virus [9, 10], Alkhurma hemorrhagic fever virus [11], Crimean-Congo hemorrhagic fever virus (CCHFV) [12, 13], and Kadam virus [6]. Recent studies screening for anti-CCHFV antibodies indicated that CCHFV likely persisted in the UAE, suggesting widespread surveillance of this tick-borne virus is needed [14, 15]. Several studies have reported novel viruses outside the MENA region in ticks. For example, new viruses were discovered from Ixodes scapularis, Dermacentor variabilis, and Amblyomma americanum collected in New York and Pennsylvania [16–18], which include monoegavirus, nairoviruses, and phleboviruses [16]. The pathogenicity and the impact of most of these viruses remain unstudied.
The UAE has a camel population of approximately 459,000 camel heads [19]. Hyalomma dromedarii ticks feed mainly on blood from camels and can transmit viruses such as the CCHFV, Dera–Ghazi–Khan virus, Dhori virus, and Kadam virus [6, 20, 21]. Recently, a new lineage of CCHFV was isolated from dromedary camels in UAE. S segment analysis of the CCHFV genome showed the highest percentage nucleotide (NT) identity at 89.8% with a CCHFV isolate (from humans) from South Africa (KJ682821) and was an outlier to the Africa 3 lineage. Furthermore, M segment analysis placed the UAE CCHFV into a distinct lineage, Asia 4, due to possible reassortment of the current sequence from camel and its nearest neighbor (76.7% NT identity) from a tick in Pakistan (KY484038). Furthermore, L segment analysis showed the highest percentage identity (NT identity at 87.7%) with a human CCHFV isolate from Russia (KX013486) [15]. Camels are frequently brought from different parts of Asia and Africa into the Middle East. This could result in widespread mixing of viral lineages leading to the emergence of novel variants [15]. The patterns of tick–host–virus interrelationships are changing due to changes in population densities of ticks, hosts, and other environmental factors [22]. An understanding of the epidemiology of tick-borne viral diseases, tick vector competence, and transmission dynamics in tick vectors is important for designing tick management strategies and curtailing the spread of viral infections.
The composition of viral communities in H. dromedarii ticks in the UAE is not adequately explored and therefore, there is a need for studies to quantify tick-borne viral diversity and abundance. This study aimed to characterize the virome diversity of male and female H. dromedarii ticks collected from Al Ain, UAE.
Materials and Methods
Ethical approval
Tick collection was carried out in accordance with the recommendations of the Animal Research Ethics Committee of the UAE University (ethical approval# ERA_2019_5953). In addition, the experimental protocol was approved by the UAE University Research Office.
Study period and location
Ticks were collected during the month of June 2021 from camel farms in Al Ain (24.1302° N, 55.8023° E), UAE. Ticks were processed in Animal Ecology and Entomology Laboratory at United Arab Emirates University, UAE.
Tick sampling and identification
Live ticks were collected from 15 camels in Al Ain, UAE and were frozen immediately in liquid nitrogen. From each camel, ticks were collected manually from the whole body and 10 ticks were collected per animal. Therefore, a total of 150 ticks were collected. All the collected ticks were taken to the Animal Ecology and Entomology Laboratory at UAE University and identified morphologically using tick taxonomic keys [23, 24]. For molecular identification, our previously published protocol was followed [25]. Briefly, legs from ticks were removed with a sterile scalpel blade and homogenized using liquid nitrogen in a 1.5 mL tube. The DNA extraction was accomplished by using DNA Mini Kit (Qiagen, Hilden, Germany) following the manufacturer’s protocol. The isolated DNA quality was confirmed using 1% agarose gel electrophoresis and DNA concentration was measured using a NanoDrop 2000 UV spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA). We used the 16S rRNA gene-specific oligonucleotide primer pair (16S + 1: CTGCTCAATGATTTTTTAAATTGCTGTGG, 16S − 1: CCGGTCTGAACTCAGATCAAGT) to amplify 460 bp based on a published protocol [26]. After polymerase chain reaction (PCR), sequencing of the amplicon was performed on Genetic Analyzer 3500 (using primers 16S + 1 and 16S − 1). Further, a sequenced amplicon similarity search was carried out using the online BLAST tool (https://blast.ncbi.nlm.nih.gov) in GenBank NCBI.
Whole RNA isolation, quality control, pooling, and sequencing
A total of 150 ticks were collected from 15 camels at a farm in Al Ain in the eastern region of Abu Dhabi, UAE (24°01’38.7”N 55°38’57.8”E). After the identification of ticks, male and female ticks were washed using Dulbecco’s phosphate-buffered saline solution by following the protocol detailed in Harvey et al. [27]. For each RNA extraction, two male and female ticks were used from each camel sample (10 ticks were collected from each camel). Accordingly, 30 male and 30 female ticks (out of 150 ticks from 15 animals) were subjected to RNA extraction. After RNA extraction from each tick, we created two pools, one for male ticks and one for female ticks (all RNAs for male ticks were pooled together in one tube and all RNAs from female ticks were pooled together in another tube). Ticks were homogenized and high-quality whole RNA was isolated using the Promega Maxwell RSC simplyRNA Tissue kit (Promega, Madison, WI, USA), based on the manufacturer’s instructions. Isolated RNA quality and quantity were confirmed using agarose gel electrophoresis, NanoDrop 2000 spectrophotometer (ThermoFisher Scientific™, Waltham, MA, USA), and Qubit 2.0 fluorometer (ThermoFisher Scientific™). Samples were pooled for sequencing into male and female pools. Furthermore, RNAs (from the same subset of ticks) was used for both Illumina and Oxford Nanopore Technology based library method. Before library preparation, the host rRNAs were removed using RiboMinus Eukaryote Kit (Cat no. A10837-08, Thermo Fisher Scientific), then the RNAs were converted into cDNA and Illumina compatible RNAseq library was prepared using NEBNext Ultra Directional RNA Library Prep Kit (New England Biolabs, Ipswich, MA, USA) and sequenced using Illumina Novaseq 6000 machine (150 bp PE chemistry). We prepared the Oxford Nanopore – MinION compatible libraries using cDNA-PCR sequencing Kit (Cat no. SQK-PCS 109, Oxford Nanopore, Abingdon, UK) and sequenced them in MinION sequencer (flow cell id: FLO-MIN106D R9.4 revision D chip).
Sequencing data quality control and trimming
The initial raw Illumina data quality was confirmed using FastQC tool (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/) and, adapter and low-quality regions present in the reads were trimmed using Trimmomatic v.0.39 software [28]. Raw data generated from the MinION were error corrected and trimmed using CANU v.2.1.1 [29].
Virus profiling based on illumina reads
Illumina reads from male and female tick samples were searched against the virus reference genome database using Kaiju v.1.8.0 (−z 50 −e 1 −m 20 −E 0.001) [30] and possible virus-related reads were taxonomically classified. Based on the virus taxonomy classification, virus family distribution Krono diagram was created with KronoTools v.2.7.1 [31]. Similarly, reads were searched against nr database and fungi database and used to identify the distribution of eukaryotic, fungi, and bacteria-related reads.
Virus genome assembly and annotation
Trimmed Illumina and Nanopore reads were used for hybrid de novo assembly using rnaSpades v.3.15.3 program (parameter: −t 80 −m 700) [32]. Assembled contigs were further error corrected using Pilon v.1.23 tool [33]. Corrected contigs were searched against the virus reference genome database using the kaiju program and contigs showing similarity with virus genomes were extracted for the downstream annotation.
Virus genome annotation
The complete genome found in our assembly was annotated using the GeneMarkS program [34]. The phylogenetic tree of the related virus was created using the neighbor-joining method using MEGA–X software (https://www.megasoftware.net/) with the bootstrap value of 1000 replicates.
Accession number(s)
A representative sequence of H. dromedarii was submitted to GenBank and acquired accession number ON937757. All sequencing data generated during this study were submitted to the NCBI-SRA database under the BioProject ids PRJNA827495 (female), and PRJNA844213 (male) (Table-1). All assembled virus sequences were deposited in the Zenodo data repository (https://zenodo.org/).
Table-1.
Information on the library constructed and data generated in this study.
| Library ID | Sample site | Host | Collection date | No. of ticks | No. of total reads | No. of bases | RNAseq technique | NCBI SRA accession no. |
|---|---|---|---|---|---|---|---|---|
| HDM1* | Al Ain | Camel ♀ | June-2021 | 30 ♂ | 51,994,138 | 15.6 Gb | Illumina NovaSeq 6000 (150bp×2) | SRR19500736 |
| HDM2** | Al Ain | Camel ♀ | June-2021 | 30 ♂ | 1,000,182 | 553.3 Mb | Oxford Nanopore (MinION) | SRR19500735 |
| HDF1*** | Al Ain | Camel ♀ | June-2021 | 30 ♀ | 58,119,014 | 17.4 Gb | Illumina NovaSeq 6000 (150bp×2) | SRR18797880 |
| HDF2**** | Al Ain | Camel ♀ | June-2021 | 30 ♀ | 707,067 | 267.3 Mb | Oxford Nanopore (MinION) | SRR18797879 |
HDM1: Hyalomma dromedarii Male 1 (for Illumina NovaSeq 6000),
HDM2: Hyalomma dromedarii Male 2 (for Oxford Nanopore [MinION]),
HDF1: Hyalomma dromedarii Female 1 (for Illumina NovaSeq 6000),
HDF2: Hyalomma dromedarii Female 2 (for Oxford Nanopore [MinION]). ♀: Female, ♂: Male
Results
Tick species identification
We analyzed 150 ticks collected at Al Ain, Abu Dhabi, UAE. All the ticks were identified as H. dromedarii morphologically. This identification was confirmed using the 16S rRNA gene which showed 99.75% similarity between the UAE sample and H. dromedarii GenBank samples from Pakistan (OL307008, coverage: 95% and E-valve: 0.0), India with 99.50% similarity (LC661680, coverage: 95% and E-valve 0.0), and UAE with 99.50% similarity (MZ976772, coverage: 95% and E-valve 0.0).
Virome analysis
Based on the morphological identification, the collected ticks were separated into two types of samples; male (30 ticks) and female (30 ticks). In total 51,994,138 (~15.6 Gb data) and 1,000,182 (553.3 Mb data) reads were generated from the male sample using Illumina and MinION, respectively. Similarly, 58,119,014 (17.4 Gb data) and 707,067 (267.3 Mb data) sequencing reads, respectively, were generated from the female sample (Table-1).
Taxonomic classification of the sequencing data resulted in 180,559 (~0.35%) and 197,801 (~0.34%) virus-related reads in male and female tick samples, respectively. Overall, ~5% bacterial, ~1.5% fungal, and ~93% of animal (ticks and camel) related reads were identified in the male sample. In the female sample, ~12% bacterial, ~1% fungal, and ~86% of animal (ticks and camel) related reads were profiled.
Analysis of the viral reads obtained from female tick samples revealed that 77.7% were similar to unclassified ssRNA viruses, 14.5% to classified dsDNA viruses, 4.12% were similar to classified ssRNA viruses, and <1% unclassified dsDNA viruses, dsRNA viruses or unclassified dsRNA viruses (Figure-1). However, the viral reads obtained from male ticks revealed that 52.9% were similar to classified dsDNA viruses, 24% to unclassified ssRNA viruses, 14.6% to classified ssRNA viruses, and <1% unclassified dsDNA viruses and classified dsRNA viruses (Figure-1).
Figure-1.
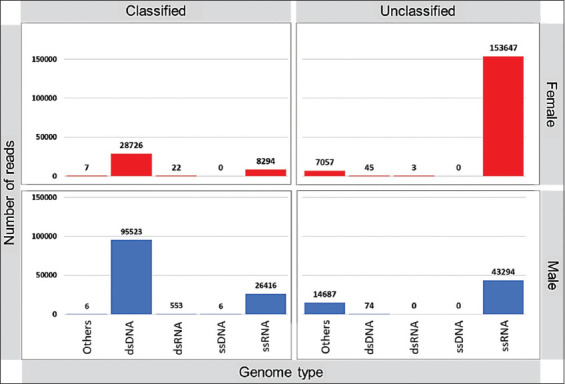
Overview of the number of viral reads grouped by viral type.
Taxonomic profiling of viruses revealed that at the kingdom, phylum, and class levels, the maximum viral reads belonged to Bamfordvirae, Nucleocytoviricota, and Megaviricetes, in both male and female ticks. However, Chitovirales was the most abundant order in male ticks and Algavirales in female ticks.
Viral sequences were classified into 39 viral families. Other viral sequences shared by all unclassified Riboviria, unclassified Varidnaviria, and unclassified Duplodnaviria. Poxviridae (21%), Phycodnaviridae (16.8%), Phenuiviridae (14.5%), Mimiviridae (8.1%), and Polydnaviridae (4.9%) were the most abundant families in the male tick viromes (Figure-2). Similarly, Phycodnaviridae (4%), Phenuiviridae (3.9%), and Poxviridae (3.7%) were the most abundant families in the female tick viromes (Figure-2). Siphoviridae, Autographiviridae, and Myoviridae were found abundant in female ticks as compared to male ticks, whereas Marseilleviridae and Reoviridae were abundant in male ticks (Figure-2). Most of these abundant families are dsDNA virus families.
Figure-2.
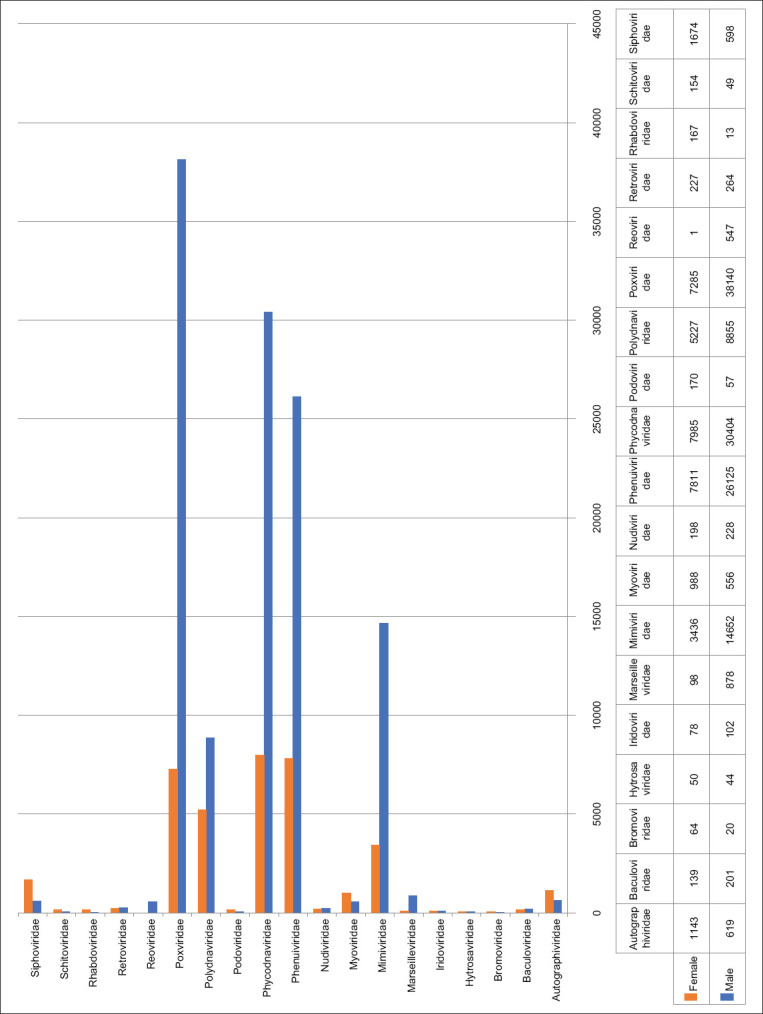
The number of viral reads at the family level obtained from classified viruses.
The richness of each genus differed considerably in the male and female tick pools containing multiple viral sequences. Prasinovirus (9.4%) was found with high relative abundance in the male tick pool, followed by Bracovirus (4.9%). However, in the female tick pool, Bracovirus (2.8%) had high relative abundance followed by Prasinovirus (2.5%) (Figure-3).
Figure-3.
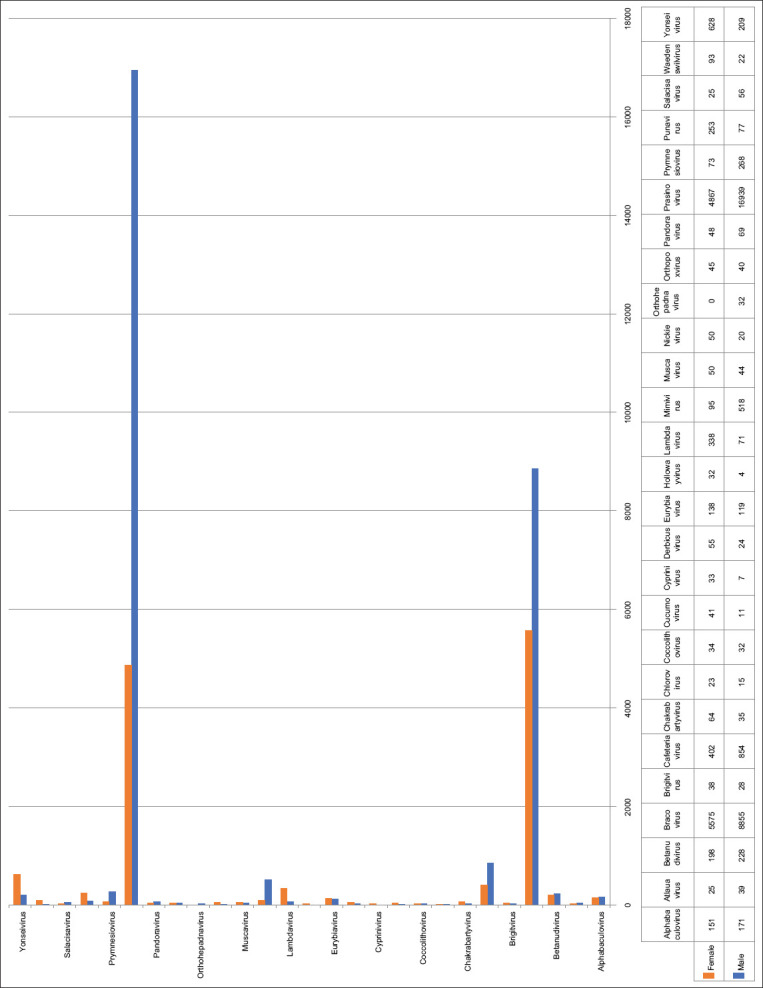
The number of viral reads at the genus level obtained from classified viruses.
De novo assembly of male and female samples resulted in three virus whole genomes. Among the three virus genomes in this study, two genomes have 90% similarity with unclassified Bole tick virus 4 (KR902736.1), and the third genome has 98.45% similarity with Iftin tick virus (MZ567078.1) that is Phlebovirus belonging to the family Phenuiviridae (Table-2). Therefore, our data contain two viruses. The viral genome sequences were deposited in the Zenodo data repository for both female and male tick viruses. The polyprotein tree (Figure-4) [35] was constructed with two virus species of the present study and the polyprotein from the GenBank showed the highest similarity. In the present study, polyproteins of the viruses showed the highest similarity with a Bole tick virus 4 strain detected in China (QYW06822) (Figure-4).
Table-2.
Identified viral species with similarity to the known virus sequences from BLASTn and BLASTp databases.
| Ticks (Hyalomma dromedarii) | Taxonomy | Species (Closest Match) | Query Cover (%) | Amino Acid/polyprotein P-Identity (%) | Contig (n) | Genome length |
|---|---|---|---|---|---|---|
| Female | Ribovirea/Unclassified | Bole tick virus 4 | 99 | 90.35/93.58 | 1 | 16305 |
| Male | Ribovirea/Unclassified | Bole tick virus 4 | 99 | 90.36/93.76 | 1 | 15454 |
| Male | Phenuiviridae/Phlebovirus | Iftin tick virus | 99/100 | 98.45/99.75 | 1 | 4905 |
Figure-4.
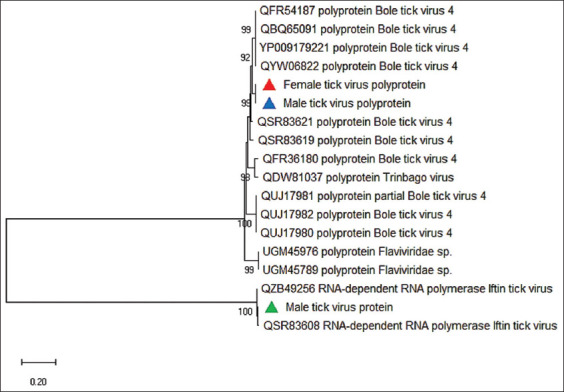
Phylogenetic tree based on viruses’ polyprotein detected in female (red triangle), male (blue triangle) ticks and protein (green triangle) in male ticks. For each sequence, we provided the accession number and virus name/strain. The tree was constructed using the Neighbor-Joining method, bootstrap test (1000 replicates), and the JTT matrix-based method [35].
Discussion
Whole genome sequencing (WGS) enabled us to characterize the RNA virome diversity of male and female H. dromedarii ticks collected from camels in Al Ain region in the UAE. Even though the WGS is a good tool for compiling and assembling genomes of different organisms, there are cases where the taxonomic assignment of sequences to the species level is not straightforward. We provided the genomes of three viruses, two of which were close to the Bole tick virus and the third showed similarity with Iftin tick virus. In the UAE, camels are important animals and have been associated with tick-borne viruses such as CCHFV [14]. Whether camels are the main reservoir of various viral disease-causing agents of humans and veterinary importance is yet to be studied. Hyalomma dromedarii has been found with a high prevalence in camels in various studies [25, 36]; therefore, the knowledge about the diversity of viruses in these ticks is crucial for surveillance strategies and for the management of any future tick-borne viral disease outbreak in the country. It should be mentioned that despite the fact that H. dromedarii is the most prevalent tick species on camels in the UAE, its microbiome is still poorly explored, despite the publication of few studies [37, 38] that characterized the bacterial communities associated with it. Therefore, the current work sheds light on unexplored viral communities, which are the other important component in its microbiome.
In this study, for molecular identification of tick species, we used the 16S rRNA sequences of H. dromedarii from UAE that showed 99.75% similarity with H. dromedarii identified from Pakistan (OL307008), 99.50% similarity with sequences of H. dromedarii from India (LC661680), UAE (MZ976772) [25]. The H. dromedarii virome revealed numerous RNA viral families. However, the data were dominated by unclassified sequences as compared to classified ones. We found a large proportion of unclassified ssRNA viruses in females compared to male ticks. This could be due to differences in the biology and feeding patterns of sexes. The amount of blood meal consumed by males is considerably less than that consumed by females and the body mass of males increases only about 1.5–2 times [39]. These findings are partially consistent with the camel tick virome characterized in a study from Saudi Arabia [40], where Phenuiviridae and Nairoviridae were detected as common families. However, in our study Poxviridae, Phycodnaviridae, and Phenuiviridae were the most abundant families. Furthermore, Zakham et al. [40] reported Phlebovirus as the most common genus whereas in our study the genus Prasinovirus was found with high relative abundance. No coronavirus was detected in either study. The differences in tick viromes between our study and Zakham et al. [40] could simply be due to a temporal mismatch since their samples were collected later in the year (between July and October) while ours was collected during June. Evidence from camel tick microbiomes indicates that microbial communities in ticks differ substantially during the course of a year, with notable increases in certain microbes at specific times in the year [38]. Since ecological conditions influence viruses as well, it is expected that certain groups or genera of viruses will dominate at certain times of the year. Tick-borne encephalitis virus (TBEV) in Ixodes ricinus ticks in Europe indicates that different tick stages, such as larvae, nymphs or adults, host a greater abundance of TBEV at different times in the year [41]. These periodic outbreaks in TBEV are associated with small mammals, such as rodents, that serve as local reservoirs of the virus during periods when no transmission occurs in humans. Some of the differences between our study and Zakham et al. [40] could also be because of inherent differences in regional climate and microclimate differences between the farms in UAE and Mekkah Province, Saudi Arabia [40]. It is worth noting that the camel tick virome analysis of this study showed diverse sequences related to animals and viruses, fungi, and bacteria that may be due to host-parasite relationship and their interaction with biotic and abiotic factors. The temporal and spatial ecology of tick-borne viruses are likely to be linked with the abundance of different microbial groups, as viruses are often associated with specific bacterial communities [41]. Since temporal and spatial aspects of H. dromedarii have not been studied, there is an urgent need to quantify patterns of high or low abundance of viruses and their diversity during the course of a year and across different sites [38]. The Poxviridae family was detected in our study with high relative abundance, whereas, the Orthopoxvirus genus was identified with some reads in both male and female ticks. Orthopoxvirus genus comprises a number of species that can infect animals and humans, including variola virus (the causative agent of smallpox), and monkeypox virus [42]. Recently, Orf, ecthyma contagiosum, caused by a parapox virus, has been reported in a 42-year-old man from Saudi Arabia who came into contact with an infected camel and developed a typical orf lesion [43]. Our results differed from a previous study on the characterization of tick viromes collected from dogs in China, where Circoviridae was detected with high abundance [44]. However, overall this study reported the highest percentage of ssRNA viruses, and similarly, we found a high abundance of unclassified ssRNA viruses in female ticks.
Whole genome sequencing provided the genomes of three viruses in the present study; two viruses showed 90.18% similarity with Bole tick virus 4 (KR902736.1, NCBI GenBank) detected in Hyalomma asiaticum [45] and 81.50% (MW561135.1), and 81.47% (MW561133.1) similarity with Bole tick virus 4 detected in the virome analysis of Rhipicephalus, Dermacentor, and Haemaphysalis ticks collected from the vegetation or engorged small ruminants from Eastern Romania [46]. For both viral genomes in our study, we found that the phylogenetic analyses placed them in clades with Bole tick virus isolates, confirming similarity and association with ticks. This relatively low level (90.18%) of sequence similarity with other GenBank genomes is likely due to the lack of viral genomes collected from ticks in the Arabian Peninsula. This demonstrates the urgent need for further studies on tick-borne viral communities in this part of the world.
The second virus showed 98.45% similarity with the Iftin tick virus (MZ567078.1, NCBI GenBank) detected in viral RNA metagenomics of Hyalomma ticks collected from dromedary camels in Makkah Province, Saudi Arabia [40], and 77.08% similarity with the Iftin tick virus (KM817664.1, NCBI GenBank) detected in genomic diversity of RNA viruses in arthropods reveals the ancestry of negative-sense RNA viruses [47]. A Bracovirus (Polydnaviridae) was detected as another abundant genus after Prasinovirus. We also identified a few sequences reads of Reoviridae that have been previously reported from different tick species in Asia, Europe, and Africa [21], and recently from Ixodes uriae [48]. Our study was limited in geographic scope to Al Ain and we suggest that future studies should consider a larger geographical distribution, which must parallel the expanding distribution of camels and camel ticks in the UAE. This would increase the chance of identifying novel tick-borne viruses that might have a potential zoonotic risk for humans or domestic animals.
In the desert ecosystems of the UAE, the camel tick life cycle can span over 2–3 years [49], depending on host availability and temperature. The expansion of camel farming due to the abundance of resources in the oil-rich Middle Eastern countries means that novel opportunities are available for the associated tick vectors [12]. Thus, the traditional camel farming industry has facilitated the geographic spread of ticks, which in turn could have promoted the maintenance and spread of tick-borne viruses in these extreme environments [12]. We found that H. dromedarii harbors a diversity of viruses belonging to several families of RNA and DNA viruses that can be transferred to and from neighboring countries. This diversity of tick-borne viruses needs to be properly studied to understand better the risks associated with certain groups of viruses.
Conclusion
Characterizing a tick virome is a key factor for understanding its microbiome and knowing its pathogenic potential. In this study, we showed that the H. dromedarii virome contained several families and genera with some potentially pathogenic virus groups. This emphasizes the need for continued camel tick surveillance in the UAE to circumvent any future outbreak of tick-borne viruses. The current study is a pioneering work in the UAE that characterized RNA virome diversity of H. dromedarii and provided preliminary data that could be used as a reference for future work to detect any change in viral community composition. This work also serves as a basis for monitoring and surveillance of the prominent tick-borne virus families that are of human and animal importance.
Data Availability
The supplementary data can be available from the corresponding author upon a reasonable request.
Authors’ Contributions
MAA, NP, and SBM: Conceptualization. MAA, NP, BK, NS, and RSA: Methodology. NP, BK, NS, and RSA: Investigation. NS: Data curation. NS, BK, and NP: Formal analysis. NS, NP, and MAA: Visualization. MAA and SBM: Supervision and project administration. NP, NS, BK, and MAA: Writing – original draft preparation. NP, MAA, NS, SBM, and BK: Writing – review and editing. All authors have read and approved the final manuscript.
Acknowledgments
We thank Amjad Saeed for his help in tick collection and the UAE University Transportation Department for providing vehicles for fieldwork. The funding for this study was provided by the UAE University, UAE, through UPAR Grant # G00002604.
Footnotes
The funding for this study was provided by the UAE University, UAE, through UPAR Grant # G00002604.
Competing Interests
The authors declare that they have no competing interests.
Publisher’s Note
Veterinary World remains neutral with regard to jurisdictional claims in published institutional affiliation.
References
- 1.De la Fuente J, Estrada-Pena A, Venzal J.M, Kocan K.M, Sonenshine D.E. Overview:Ticks as vectors of pathogens that cause disease in humans and animals. Front. Biosci. 2008;13(18):6938–6946. doi: 10.2741/3200. [DOI] [PubMed] [Google Scholar]
- 2.Pfäffle M, Littwin N, Muders S.V, Petney T.N. The ecology of tick-borne diseases. Int. J. Parasitol. 2013;43(12–13):1059–1077. doi: 10.1016/j.ijpara.2013.06.009. [DOI] [PubMed] [Google Scholar]
- 3.Greay T.L, Gofton A.W, Paparini A, Ryan U.M, Oskam C.L, Irwin P.J. Recent insights into the tick microbiome gained through next-generation sequencing. Parasit. Vectors. 2018;11(1):12. doi: 10.1186/s13071-017-2550-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cabezas-Cruz A, Pollet T, Estrada-Peña A, Allain E, Bonnet S.I, Moutailler S. Handling the microbial complexity associated to ticks. In:Ticks and Tick-borne Pathogens. IntechOpe, London. 2018 [Google Scholar]
- 5.Tokarz R, Sameroff S, Tagliafierro T, Jain K, Williams S.H, Cucura D.M, Rochlin I, Monzon J, Carpi G, Tufts D, Diuk-Wasser M, Brinkerhoff J, Lipkin W.I. Identification of novel viruses in Amblyomma americanum, Dermacentor variabilis, and Ixodes scapularis ticks. mSphere. 2018;3(2):e00614–e00617. doi: 10.1128/mSphere.00614-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Wood O.L, Moussa M.I, Hoogstraal H, Büttiker W. Kadam virus (Togaviridae, Flavivirus) infecting camel-parasitizing Hyalomma dromedarii ticks (Acari:Ixodidae) in Saudi Arabia. J. Med. Entomol. 1982;19(2):207–208. doi: 10.1093/jmedent/19.2.207. [DOI] [PubMed] [Google Scholar]
- 7.Zakham F, Al-habal M, Taher R, Alaoui A, El Mzibri M. Viral hemorrhagic fevers in the Tihamah region of the western Arabian Peninsula. PLoS Negl. Trop. Dis. 2017;11(4):e0005322. doi: 10.1371/journal.pntd.0005322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zakham F, Alaloui A, Levanov L, Vapalahti O. Viral haemorrhagic fevers in the Middle East. Rev. Sci. Tech. 2019;38(1):185–198. doi: 10.20506/rst.38.1.2952. [DOI] [PubMed] [Google Scholar]
- 9.Sabir J.S.M, Ahmed M.M.M, Li L, Shen Y, Abo-Aba S.E.M, Qureshi M.I, Abu-Zeid M, Zhang Y, Khiyami M.A, Alharbi N.S, Hajrah N.H, Sabir M.J, Mutwakil M.H.Z, Kabli S.A, Alsulaimany F.A.S, Obaid A.Y, Zhou B, Smith D.K, Holmes E.C, Zhu H, Guan Y. Co-circulation of three camel coronavirus species and recombination of MERS-CoVs in Saudi Arabia. Science. 2016;351(6268):81–84. doi: 10.1126/science.aac8608. [DOI] [PubMed] [Google Scholar]
- 10.Meyer B, Müller M.A, Corman V.M, Reusken C.B.E, Ritz D, Godeke G.J, Lattwein E, Kallies S, Siemens A, van Beek J, Drexler J.F, Muth D, Bosch B.J, Wernery U, Koopmans M.P.G, Wernery R, Drosten C. Antibodies against MERS Coronavirus in dromedary camels, United Arab Emirates, 2003 and 2013. Emerg. Infect. Dis. 2014;20(4):552–559. doi: 10.3201/eid2004.131746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Memish Z.A, Charrel R.N, Zaki A.M, Fagbo S.F. Alkhurma haemorrhagic fever--a viral haemorrhagic disease unique to the Arabian Peninsula. Int. J. Antimicrob. Agents. 2010;36(Suppl 1):S53–S57. doi: 10.1016/j.ijantimicag.2010.06.022. [DOI] [PubMed] [Google Scholar]
- 12.Perveen N, Muzaffar S.B, Al-Deeb M.A. Ticks and tick-borne diseases of livestock in the middle east and North Africa:A review. Insects. 2021;12(1):83. doi: 10.3390/insects12010083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Al-Abri S.S, Al Abaidani I, Fazlalipour M, Mostafavi E, Leblebicioglu H, Pshenichnaya N, Memish Z.A, Hewson R, Petersen E, Mala P, Nguyen T.M.N, Malik M.R, Formenty P, Jeffries R. Current status of Crimean-Congo haemorrhagic fever in the World Health Organization Eastern Mediterranean Region:Issues, challenges, and future directions. Int. J. Infect. Dis. 2017;58:82–89. doi: 10.1016/j.ijid.2017.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Camp J.V, Kannan D.O, Osman B.M, Shah M.S, Howarth B, Khafaga T, Weidinger P, Karuvantevida N, Kolodziejek J, Mazrooei H, Wolf N, Loney T., Nowotny N. Crimean-Congo hemorrhagic fever virus endemicity in United Arab Emirates, 2019. Emerg. Infect. Dis. 2020;26(5):1019–1021. doi: 10.3201/eid2605.191414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Khalafalla A.I, Li Y, Uehara A, Hussein N.A, Zhang J, Tao Y, Bergeron E, Ibrahim I.H, Al Hosani M.A, Yusof M.F, Alhammadi Z.M, Alyammahi S.M, Gasim E.F, Ishag H.Z.A, Hosani F.A.L, Gerber S.I, Almuhairi S.A, Tong S. Identification of a novel lineage of Crimean-Congo haemorrhagic fever virus in dromedary camels, United Arab Emirates. J. Gen. Virol. 2021;102(2):001473. doi: 10.1099/jgv.0.001473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tokarz R, Williams S.H, Sameroff S, Leon M.S, Jain K, Lipkin W.I. Virome analysis of Amblyomma americanum, Dermacentor variabilis, and Ixodes scapularis ticks reveals novel highly divergent vertebrate and invertebrate Viruses. J. Virol. 2014;88(19):11480–11492. doi: 10.1128/JVI.01858-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shi J, Hu Z, Deng F, Shen S. Tick-borne viruses. Virol. Sin. 2018;33(1):21–43. doi: 10.1007/s12250-018-0019-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sakamoto J.M, Ng T.F.F, Suzuki Y, Tsujimoto H, Deng X, Delwart E, Rasgon J.L. Bunyaviruses are common in male and female Ixodes scapularis ticks in central Pennsylvania. PeerJ. 2016;4:e2324. doi: 10.7717/peerj.2324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.FCSA. Livestock Statistics. 2017. [Last accessed on 2019 Jul 23]. Available from: https://www.fcsa.gov.ae/en-us/pages/statistics/statistics.aspx .
- 20.Hoogstraal H, Wassef H.Y, Buttiker W. Ticks (Acarina) of Saudi Arabia fam argasidae ixodidae. Fauna Saudi Arab. 1981;3:25–110. [Google Scholar]
- 21.Hoogstraal H. The epidemiology of tick-borne Crimean-Congo hemorrhagic fever in Asia, Europe, and Africa. J. Med. Entomol. 1979;15(4):307–417. doi: 10.1093/jmedent/15.4.307. [DOI] [PubMed] [Google Scholar]
- 22.Estrada-Peña A, de la Fuente J. The ecology of ticks and epidemiology of tick-borne viral diseases. Antiviral Res. 2014;108:104–128. doi: 10.1016/j.antiviral.2014.05.016. [DOI] [PubMed] [Google Scholar]
- 23.Walker A.R, Bouattour A, Camicas J.L, Estrada-Peña A, Horak I.G, Latif A.A, Pegram R.G, Preston P.M. Ticks of Domestic Animals in Africa:A Guide to Identification of Species;Bioscience Reports. Edinburg., United Kingdom. 2003 [Google Scholar]
- 24.Apanaskevich D.A, Schuster A.L, Horak I.G. The genus Hyalomma:VII. redescription of all parasitic stages of H. (Euhyalomma) dromedarii and H. (E.) schulzei (Acari:Ixodidae) J. Med. Entomol. 2008;45(5):817–831. doi: 10.1603/0022-2585(2008)45[817:tghvro]2.0.co;2. [DOI] [PubMed] [Google Scholar]
- 25.Perveen N, Muzaffar S.B, Al-deeb M.A. Prevalence, distribution, and molecular record of four hard ticks from livestock in the United Arab Emirates. Insects. 2021;12(11):1016. doi: 10.3390/insects12111016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Black W.C, 4th, Piesman J. Phylogeny of hard-and soft-tick taxa (Acari:Ixodida) based on mitochondrial 16S rDNA sequences. Proc. Natl. Acad. Sci. U. S. A. 1994;91(21):10034–10038. doi: 10.1073/pnas.91.21.10034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harvey E, Rose K, Eden J.S, Lo N, Abeyasuriya T, Shi M, Doggett S.L, Holmes E.C. Extensive diversity of RNA viruses in Australian ticks. J. Virol. 2019;93(3):e01358–e01318. doi: 10.1128/JVI.01358-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bolger A.M, Lohse M, Usadel B. Trimmomatic:A flexible trimmer for Illumina sequence data. Bioinformatics. 2014;30(15):2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Koren S, Walenz B.P, Berlin K, Miller J.R, Bergman N.H, Phillippy A.M. Canu:Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017;27(5):722–736. doi: 10.1101/gr.215087.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Menzel P, Ng K.L, Krogh A. Fast and sensitive taxonomic classification for metagenomics with Kaiju. Nat. Commun. 2016;7:11257. doi: 10.1038/ncomms11257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ondov B.D, Bergman N.H, Phillippy A.M. Interactive metagenomic visualization in a Web browser. BMC Bioinformatics. 2011;12(1):385. doi: 10.1186/1471-2105-12-385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bushmanova E, Antipov D, Lapidus A, Prjibelski A.D. rnaSPAdes:A de novo transcriptome assembler and its application to RNA-Seq data. Gigascience. 2019;8(9):giz100. doi: 10.1093/gigascience/giz100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Walker B.J, Abeel T, Shea T, Priest M, Abouelliel A, Sakthikumar S, Cuomo C.A, Zeng Q, Wortman J, Young S.K, Earl A.M. Pilon:An integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One. 2014;9(11):e112963. doi: 10.1371/journal.pone.0112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Besemer J, Lomsadze A, Borodovsky M. GeneMarkS:A self-training method for prediction of gene starts in microbial genomes. Implications for finding sequence motifs in regulatory regions. Nucleic Acids Res. 2001;29(12):2607–2618. doi: 10.1093/nar/29.12.2607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kumar S, Stecher G, Li M, Knyaz C, Tamura K. MEGA X:Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018;35(6):1547–1549. doi: 10.1093/molbev/msy096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Al-Deeb M.A, Muzaffar S.B. Prevalence, distribution on host's body, and chemical control of camel ticks Hyalomma dromedarii in the United Arab Emirates. Vet. World. 2020;13(1):114–120. doi: 10.14202/vetworld.2020.114-120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Perveen N, Muzaffar S.B, Vijayan R, Al-Deeb M.A. Microbial communities associated with the camel tick, Hyalomma dromedarii:16S rRNA gene-based analysis. Sci. Rep. 2020;10(17035):1–11. doi: 10.1038/s41598-020-74116-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Perveen N, Muzaffar S.B, Vijayan R, Al-Deeb M.A. Assessing temporal changes in microbial communities in Hyalomma dromedarii collected from camels in the UAE Using high-throughput sequencing. Front. Vet. Sci. 2022;9:861233. doi: 10.3389/fvets.2022.861233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Apanaskevich D.A, Oliver J.H. Life cycles and natural history of ticks. In: Sonenshin D.E, Roe R.M, editors. Biology of Ticks. Vol. 1. New York: Oxford University Press; 2013. pp. 59–73. [Google Scholar]
- 40.Zakham F, Albalawi A.E, Alanazi A.D, Nguyen P.T, Alouffi A.S, Alaoui A, Sironen T, Smura T, Vapalahti O. Viral RNA metagenomics of Hyalomma ticks collected from dromedary camels in Makkah province, Saudi Arabia. Viruses. 2021;13(7):1396. doi: 10.3390/v13071396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Bournez L, Umhang G, Moinet M, Richomme C, Demerson J.M, Caillot C, Devillers E, Boucher J.M, Hansmann Y, Boué F, Moutailler S. Tick-borne encephalitis virus:Seasonal and annual variation of epidemiological parameters related to nymph-to-larva transmission and exposure of small mammals. Pathogens. 2020;9(7):518. doi: 10.3390/pathogens9070518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shchelkunov S.N. An increasing danger of zoonotic orthopoxvirus infections. PLoS Pathog. 2013;9(12):e1003756. doi: 10.1371/journal.ppat.1003756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Alajlan A.M, Alsubeeh N.A. Orf (Ecthyma contagiosum) transmitted from a camel to a human:A case report. Am. J. Case Rep. 2020;21:e927579. doi: 10.12659/AJCR.927579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang S, Zhao T, Yu X, Lin Z, Hua X, Cui L. Characterization of tick viromes collected from dogs in China. Biosaf. Health. 2020;2(2):79–88. [Google Scholar]
- 45.Shi M, Lin X.D, Vasilakis N, Tian J.H, Li C.X, Chen L.J, Eastwood G, Diao X.N, Chen M.H, Chen X, Qin X.C, Widen S.G, Wood T.G, Tesh R.B, Xu J, Holmes E.C, Zhang Y.Z. Divergent viruses discovered in arthropods and vertebrates revise the evolutionary history of the Flaviviridae and related viruses. J. Virol. 2016;90(2):659–669. doi: 10.1128/JVI.02036-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bratuleanu B.E, Temmam S, Chrétien D, Regnault B, Pérot P, Bouchier C, Bigot T, Savuţa G, Eloit M. The virome of Rhipicephalus, Dermacentor and Haemaphysalis ticks from Eastern Romania includes novel viruses with potential relevance for public health. Transbound. Emerg. Dis. 2021;69(3):1387–1403. doi: 10.1111/tbed.14105. [DOI] [PubMed] [Google Scholar]
- 47.Li C.X, Shi M, Tian J.H, Lin X.D, Kang Y.J, Chen L.J, Qin X.C, Xu J, Holmes E.C, Zhang Y.Z. Unprecedented genomic diversity of RNA viruses in arthropods reveals the ancestry of negative-sense RNA viruses. Elife. 2015;4:e05378. doi: 10.7554/eLife.05378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Pettersson J.H.O, Ellström P, Ling J, Nilsson I, Bergström S, González-Acuña D, Olsen B, Holmes E.C. Circumpolar diversification of the Ixodes uriae tick virome. PLoS Pathog. 2020;16(8):e1008759. doi: 10.1371/journal.ppat.1008759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Perveen N, Muzaffar S.B, Al-Deeb M.A. Population dynamics of Hyalomma dromedarii on camels in the United Arab Emirates. Insects. 2020;11(5):320. doi: 10.3390/insects11050320. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The supplementary data can be available from the corresponding author upon a reasonable request.