

Abstract
SARS-CoV-2 entry into cells requires specific host proteases; however, no successful in vivo applications of host protease inhibitors have yet been reported for treatment of SARS-CoV-2 pathogenesis. Here we describe a chemically engineered nanosystem encapsulating CRISPR–Cas13d, developed to specifically target lung protease cathepsin L (Ctsl) messenger RNA to block SARS-CoV-2 infection in mice. We show that this nanosystem decreases lung Ctsl expression in normal mice efficiently, specifically and safely. We further show that this approach extends survival of mice lethally infected with SARS-CoV-2, correlating with decreased lung virus burden, reduced expression of proinflammatory cytokines/chemokines and diminished severity of pulmonary interstitial inflammation. Postinfection treatment by this nanosystem dramatically lowers the lung virus burden and alleviates virus-induced pathological changes. Our results indicate that targeting lung protease mRNA by Cas13d nanosystem represents a unique strategy for controlling SARS-CoV-2 infection and demonstrate that CRISPR can be used as a potential treatment for SARS-CoV-2 infection.
The prolonged coronavirus disease 2019 (COVID-19) outbreak has created enormous demand for the development of effective therapeutics against the causative agent, severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2). The Food and Drug Administration (FDA) has approved the nucleoside analog remdesivir as an antiviral drug that acts directly against SARS-CoV-2. While remdesivir shortens time to recovery, it does not provide a survival benefit for patients with COVID-19 (ref. 1). The FDA has also issued emergency use authorization for several monoclonal antibodies, including bamlanivimab plus etesevimab, casirivimab plus imdevimab and sotrovimab. Clinical studies have shown that these monoclonal antibodies can reduce hospitalization and death; however, the effectiveness of monoclonal antibodies can be attenuated or lost by the emergence of resistant mutations in the form of SARS-CoV-2 variants of concern such as the B.1.617.2 Delta and B.1.1.529 Omicron variants and may be undermined by their unknown bioavailability in affected tissues (for example, lungs)2–4. Emergency use authorizations were granted in December 2021 for two oral anti-SARS-CoV-2 drugs, molnupiravir and paxlovid. While these agents show great efficacy in reducing the risk of hospitalization and death from COVID-19 (refs. 5–7), patient numbers in trials have not yet been sufficiently high to reach a firm conclusion on whether they can prevent SARS-CoV-2 transmission5. In addition, safety concerns have been debated over the mutagenic potential of molnupiravir in human cells8 and the potential tolerance of paxlovid in some populations5,9. Thus, the development of new strategies to control SARS-CoV-2 infection remains an urgent requirement.
Recently, virologic and proteomic studies have revealed how SARS-CoV-2 hijacks the host cell machinery during infection10,11, offering new guidance on the development of host-directed therapies. Genome-wide CRISPR–Cas9 screening for host factors has identified the host protease cathepsin L (CTSL) as a top-ranked gene not only for infection by SARS-CoV-2, but also by related SARS-CoV-1 and Middle East Respiratory Syndrome coronavirus (MERS-CoV) infection in some cell models12,13. Indeed, CTSL has long been recognized as an important endosomal cysteine protease that mediates the spike S protein (S) priming and viral entry through virus–host cell endosome membrane fusion14. Several CTSL inhibitors have been reported to block entry of coronaviruses (for example, SARS-CoV-2 and SARS-CoV-1) in vitro10,15–18 and pseudotyped SARS-CoV-2 infection in vivo19. However, the antiviral activities of these CTSL inhibitors are lost or significantly reduced on cell surface expression of the serine proteases for cell entry such as TMPRSS2 (refs. 10,20). Moreover, a potent CTSL inhibitor against SARS-CoV-1 infection in vitro has failed to provide survival benefit in a lethal SARS-CoV-1 mouse model17. It remains unclear whether the lack of antiviral potency of CTSL inhibitors in vivo and in some in vitro models is due to the use of alternative coronavirus entry pathways and/or issues of drug efficacy/delivery. It is currently unknown whether targeting CTSL would block authentic SARS-CoV-2 infection in vivo.
Here we have overcome this problem by specifically targeting CTSL in the lung using a gene therapy strategy. Since cathepsins play important physiological roles in many cellular processes such as immune response, autophagy and development14,21, we have used the recently developed CRISPR–Cas13d RNA-targeting system22 to transiently knockdown Ctsl mRNA rather than using CRISPR–Cas9 to permanently delete the Ctsl gene. In contrast with the off-target effects associated with RNA interference (RNAi) strategies because of their essential role in some endogenous processes23,24, Cas13d mediates highly efficient and specific RNA knockdown22. In addition, no interference effects have been reported for Cas13 guide RNAs in mammalian cells25. To specifically target Ctsl mRNA in the lung, the primary organ of SARS-CoV-2 infection, we have constructed lung-selective lipid nanoparticles (LNPs) as a delivery vehicle for the Cas13d RNA-targeting system. We demonstrate that both prophylactic and therapeutic administration of this lung-specific Ctsl mRNA-targeting, CRISPR–Cas13d-based LNP therapy effectively inhibits the lethal SARS-CoV-2 infection in mice. This nanosystem also inhibits infection by SARS-CoV-1, SARS-CoV-2 and Delta variant of concern in vitro. Together, our data indicate a potential application of this nanosystem for the control of COVID-19 and related coronavirus-associated diseases.
Results
CasRx targeting CTSL inhibits coronavirus infection in vitro.
Previous reports have shown that pharmacological inhibition of CTSL blocks entry of coronaviruses (for example, SARS-CoV-2 and SARS-CoV-1) in some cultured cell models10,15–18. We asked whether knockdown of CTSL mRNA by CRISPR–CasRx (a Cas13d RNA-targeting enzyme from the Ruminococcus flavefaciens strain XPD3002)22 can inhibit coronavirus infection in vitro. We screened a series of unprocessed guide RNAs (pre-gRNAs) to identify one (pre-gCTSL) that mediates the most potent knockdown of CTSL (roughly 75%) mRNA by CasRx in 293FT cells with minimal off-target effects (Extended Data Fig. 1a–c). We then switched to more physiologically relevant cell lines, finding that CTSL mRNA knockdown by CasRx-pre-gCTSL inhibited not only pseudotyped SARS-CoV-2 infection, but also pseudotyped SARS-CoV-1 infection in Vero-ACE2-TMPRSS2 and/or Caco-2 cells (Fig. 1a–c and Extended Data Fig. 1d). CasRx-mediated knockdown of CTSL mRNA had an efficiency comparable to pharmacological inhibition of CTSL activity by E-64D (ref. 10) in blocking pseudotyped SARS-CoV-2 and SARS-CoV-1 infection (Fig. 1b,c). As a control, infection by vesicular stomatitis virus (VSV) was unaffected by CTSL knockdown or E-64d treatment (Fig. 1c). Notably, CasRx-mediated CTSL knockdown also significantly inhibited infection by the authentic SARS-CoV-2 B.1.617.2 Delta variant (Fig. 1a,d,e). Collectively, these data demonstrate that CasRx-mediated specific Ctsl knockdown effectively inhibits infection by multiple coronavirus strains in vitro.
Fig. 1 |. CasRx-mediated CTSL knockdown inhibits pseudotyped coronaviruses and authentic SARS-CoV-2 Delta variant (B.1.617.2) infection in cells.

a, Schematic depiction of the SARS-CoV-1 (SARS-1), SARS-CoV-2 (SARS-2) or B.1.617.2 Delta variant entry pathway, with CTSL cleavage sites on their respective spike proteins and mutation sites on the B.1.617.2 Delta variant spike protein shown. b,c, Relative infection of pseudotyped virus in Vero-ACE2-TMPRSS2 (b) and Caco-2 (c) cells. Cells were pretreated with CasRx-pre-gControl or CasRx-pre-gCTSL for 24 h or 25 μM E-64d for 1 h, followed by incubation with infectivity-normalized pseudovirus SARS-2 (b and c), SARS-1 (c) or VSV (c). n = 12 biologically independent replicates each group except n = 6 in the background (Bg) group for b; n = 12 (SARS-2, SARS-1) or 9 (VSV) biologically independent replicates each group except n = 5 in all E-64d groups for c. d,e, CasRx-pre-gCTSL treatment reduces live B.1.617.2 Delta variant infection in Vero-ACE2-TMPRSS2 cells. d, Viral N gene transcript level as normalized to 18S ribosomal RNA. e, Virus infectivity as determined by TCID50. n = 3 biologically independent replicates for d and e. Background cells were not challenged with virus and used as a negative control. Data are presented as mean ± s.e.m. P values were calculated by two-tailed Student’s t-tests. NS, not significant; **P < 0.01; ****P < 0.001.
Development of a CasRx-based nanosystem targeting lung Ctsl.
To investigate whether CasRx-mediated Ctsl knockdown can inhibit authentic SARS-CoV-2 infection in vivo, we first constructed a LNP system that can deliver CasRx and pre-gCtsl to the mouse lungs. Previous studies have reported that manipulating the internal and/or external charge of LNP formulations is one of the key factors in determining organ tropism26,27. We thus incorporated selective organ targeting nanotechnology27 into the traditional MC3-based LNP formulation used by the FDA-approved RNAi therapy named Patisiran/Onpattro28 by adding the supplementary cationic lipids DOTAP (1,2-dioleoyl-3-trimethylammonium-propane) at specific molar ratios (0–50%). This generated highly lung-selective LNPs with a final molar ratio of Dlin-MC3-DMA:DSPC (1,2-distearoyl-sn-glycero-3-phosphocholine):cholesterol: DMG-PEG:DOTAP at 25:5:19.3:0.8:50 (Extended Data Fig. 2). Empty LNPs (eLNPs) and LNPs encapsulating luciferase mRNA (LNP-Luc) had high reproducibility and were around 100 nm in size, spherical and evenly distributed, as determined by Zetasizer and transmission electron microscopy (Extended Data Fig. 3a,b). The particle surface was positively charged, that is, 12.2 ± 1.3 mV for eLNPs and 9.9 ± 0.7 mV for LNP-Luc, respectively (Extended Data Fig. 3c). The luciferase mRNA encapsulation rate was nearly 100% on days 2, 4, 6, 8 and 10 without change, indicating that these particles are stable for at least 10 days in Dulbecco’s PBS (DPBS) at 4 °C (Extended Data Fig. 3d).
Having optimized lung-selective LNP formulation, we next constructed Ctsl mRNA-targeting LNPs by using lung-selective LNPs to encapsulate CRISPR–CasRx and a pre-gRNA recognizing mouse Ctsl mRNA (Extended Data Fig. 4a–c). Since previous studies found that codelivery of Cas9 mRNA with gRNAs into cells produces faster gene editing kinetics and fewer off-target effects compared with a plasmid encoding them29,30, we engineered lung-targeting LNPs (referred to as LNP-CasRx-pre-gCtsl) to deliver CasRx mRNA and pre-gRNA oligos targeting Ctsl to the lungs. CasRx mRNA expression in the lungs peaked at 4 h after LNP-CasRx injection and then markedly decreased by 24 h (Extended Data Fig. 4d), similar to the fast and transient kinetics associated with Cas9 mRNA31. Compared with three control groups, including DPBS, eLNPs and LNP-CasRx-pre-gControl, intravenous administration of LNP-CasRx-pre-gCtsl significantly inhibited mRNA and protein levels of Ctsl in mouse lungs (Fig. 2a,b). These findings were further validated by immunohistochemistry (IHC) in lung sections. The strong Ctsl staining of bronchiolar epithelial cells, type II alveolar epithelial cells and macrophages was greatly decreased by LNP-CasRx-pre-gCtsl treatment (Fig. 2c). Notably, Ctsl expression was not changed in liver or spleen, and expression of the other cathepsin family members Ctsd, Ctsb and Ctss was not altered in the lungs after LNP-CasRx-pre-gCtsl treatment (Extended Data Fig. 4e,f), demonstrating the tissue and gene specificity of Ctsl targeting by LNP-CasRx-pre-gCtsl. Furthermore, no significant changes in cytokine/chemokine expression were observed in the lungs following LNP-CasRx-pre-gCtsl treatment (Extended Data Fig. 5a). Finally, mouse liver or kidney function and hematology (including red blood cell count, hemoglobin level, white blood cell count and platelet count) were normal (Fig. 2d and Extended Data Fig. 5b), with no histological toxicity in the main organ tissues of LNP-CasRx-pre-gCtsl treated mice (Extended Data Fig. 5c). Taken together, these data demonstrate that the lung-selective LNP-CasRx-pre-gCtsl system can decrease lung Ctsl expression efficiently, specifically and safely.
Fig. 2 |. Ctsl is specifically knocked down by a lung-selective LNP encapsulating CasRx-pre-gRNA system with minimal systemic toxicity in normal C57BL/6 mice.

Mice were administrated DPBS, empty LNPs, LNP-CasRx-pre-gControl or LNP-CasRx-pre-gCtsl, respectively, by tail vein. After 72 h, samples were collected for analysis. a, Ctsl mRNA level in lung. Ctsl transcript levels are normalized to Gapdh and expressed as a percentage of the DPBS control group. Each group consists of six females and six males, except for five females in the LNP-CasRx-pre-gControl group. The line shows the grand mean of each group, and each label represents an individual mouse. P values were calculated by one-tailed Mann–Whitney U-test, grand mean. ***P < 0.001, ****P < 0.0001. b, Representative western blot of Ctsl protein in lung. Samples were same as in a, but only one male and one female mouse were selected from each group for presenting. Calnexin was used as a loading control. c, Representative images for Ctsl immunostaining in lung. One lung section from one mouse of each control group and three mice from the LNP-CasRx-pre-gCtsl group were subjected to IHC analysis. Scale bars, 100 μm. d, Hepatic and renal functions were not impaired by LNP-CasRx-pre-Ctsl treatment. ALP, alkaline phosphatase; ALT, alanine transaminase; AST, aspartate aminotransferase; BUN, blood urea nitrogen; CREAT, creatinine. Data are presented as box and whisker plots of each group with n = 8 mice (four females and four males in each group except for three females and five males in LNP-CasRx-pre-gControl group). Box plots show the median (center line) with bounds of 25–75th percentiles. The whiskers are extended to 1.5× the interquartile range. P values were calculated by a two-tailed Mann–Whitney U-test.
CasRx nanosystem extends SARS-CoV-2-infected mouse survival.
The K18-hACE2 mouse model establishes a robust SARS-CoV-2 infection and recapitulates the main features of severe COVID-19 in humans, such as severe interstitial pneumonia and exuberant inflammatory response32–36. We thus considered this lethal challenge model for evaluation of the antiviral efficacy of our nanosystem in vivo (Fig. 3a). When challenged with 105 plaque-forming unit (PFU) of SARS-CoV-2 (USA-WA1/2020), the mice in the three control groups displayed a progressive weight loss until they all succumbed within 8 days after inoculation (Fig. 3b,c). In contrast, mice administered with LNP-CasRx-pre-gCtsl 2 days before and 1 day postinfection (DPI) with SARS-CoV-2 exhibited a delayed onset and reduced extent of the weight loss (Fig. 3b), contributing to an increased survival by 50% (Fig. 3c).
Fig. 3 |. LNP-CasRx-pre-gCtsl treatment protects K18-hACE2 mice against lethal SARS-CoV-2 infection.
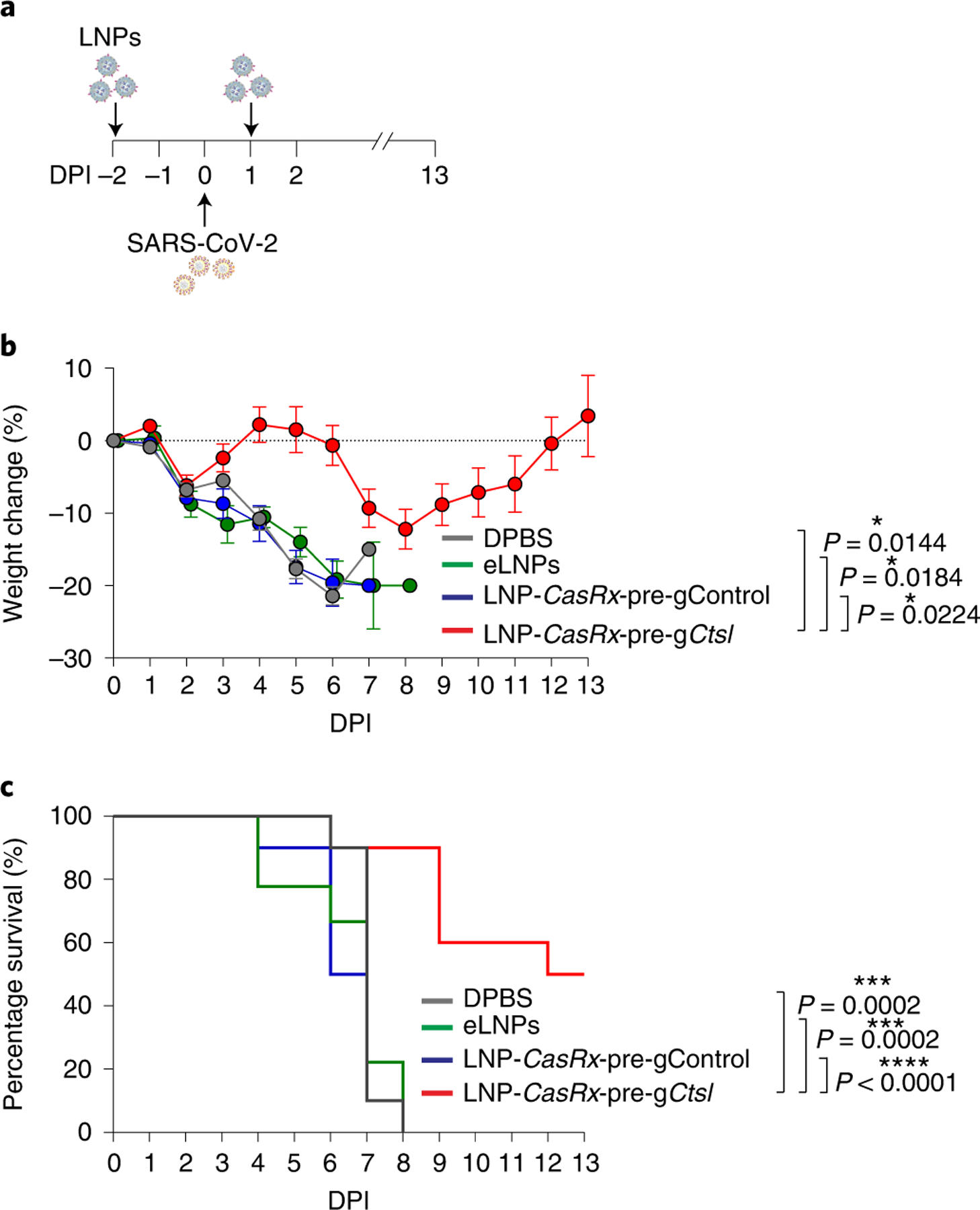
a, Schematic illustration of the experimental design. Seven-to eight-week-old K18-hACE2 female and male mice were intranasally infected with 105 PFU of SARS-CoV-2 (USA-WA1/2020). They were treated with DPBS, eLNPs, LNP-CasRx-pre-gControl or LNP-CasRx-pre-gCtsl through the retro-orbital injections 2 days before and 1 DPI. Each group consists of ten mice (five females and five males) except for nine mice in the eLNPs group (five females and four males). Mouse weights were monitored daily to DPI 13. b, Percentage of weight change. Data are presented as mean ± s.e.m. The dotted line represents the initial body weight. Mice that had lost >20% of their initial body weight were humanely euthanized. P values were calculated by two-tailed Student’s t-tests, *P < 0.05. c, Kaplan–Meyer survival curves. P values were determined by log-rank (Mantel–Cox) test. ***P < 0.001, ****P < 0.0001.
CasRx nanosystem reduces lung viral burden.
With an inoculation of 105 PFU of SARS-CoV-2, viral burden in K18-hACE2 mouse lungs has been shown to peak at 2 days postinfection (DPI)32,33. We thus collected the lung tissues at this time point to evaluate the efficacy of LNP-CasRx-pre-gCtsl treatment in reducing the lung viral load (Fig. 4a). Since there were no differences observed among the three control groups (DPBS, eLNPs and LNP-CasRx-pre-gControl) in mouse survival and weight loss (Fig. 3b,c), we pooled the three controls to create one ‘control’ group. We found that LNP-CasRx-pre-gCtsl treatment decreased the lung virus infectivity by two orders of magnitude compared with the control group (Fig. 4b). Consistently, we observed significantly lower mRNA and/or protein expression of viral nucleocapsid (N) and envelope (E) in the LNP-CasRx-pre-gCtsl treatment group than in the control group (Fig. 4c and Extended Data Fig. 6a left panel, b, c). Consistent with previous reports33,35, the lung sections of the control group showed strong diffuse N protein staining at 2 DPI, which was significantly decreased by LNP-CasRx-pre-gCtsl treatment (Fig. 4d and Extended Data Fig. 6d). As expected, LNP-CasRx-pre-gCtsl treatment significantly decreased mRNA and protein expression of Ctsl in lungs (Fig. 4c,e and Extended Data Fig. 6a right panel). Thus, LNP-CasRx-pre-gCtsl treatment ameliorated SARS-CoV-2 infection efficiently and rapidly.
Fig. 4 |. CasRx-mediated Ctsl knockdown markedly decreases SARS-CoV-2 burden in lungs of infected K18-hACE2 mice.

a, Schematic illustration of the experimental design, using the same treatment as in Fig. 3. Each group consists of two females and two males. Lungs were collected for analysis at 2 DPI. DPBS, eLNPs and LNP-CasRx-pre-gControl groups were pooled and referred to as control. b, Infectious viral titer in lung. Tissue homogenates were analyzed by plaque assay. The bar represents the average of the group, while each circle represents an individual animal. P values were calculated by one-tailed Mann–Whitney U-test, grand mean. ***P < 0.001. c, Western blot (WB) of viral N protein and Ctsl protein in lung. All animals were subjected to this analysis. Nonvirus, two mice (one female and one male) were not challenged with virus. Each lane represents an individual mouse. Calnexin was used as a loading control. The ratio of N or Ctsl protein over calnexin is listed under the blot. d, Representative images for viral N protein immunostaining in lung. One lung section per mouse for all animals were subjected to IHC analysis and only one image from each group was presented. Scale bar, 100 μm. e, Ctsl transcript levels as normalized by Gapdh. The bar represents the average of the group, while each circle represents an individual animal. P values were calculated by one-tailed Mann–Whitney U-test, grand mean. **P < 0.01.
CasRx nanosystem reduces lung cytokine levels and lesions.
During the development of severe COVID-19 disease, uncontrolled inflammation known as a cytokine storm is believed to be responsible for multi-organ damage and fatal outcome37. For example, elevated CXCL10 and TNF levels are correlated with increased disease severity and decreased survival of patients with COVID-19 (refs. 37,38). Here we have shown that a lethal dose of SARS-CoV-2 infection significantly increased the transcript levels of Cxcl10 and Tnf in mouse lung at 4 DPI in the control group relative to unchallenged mice (Fig. 5a,b). Cxcl10 and Tnf expression were almost the same in the LNP-CasRx-pre-gCtsl treated group as in unchallenged mice (Fig. 5b). Similarly, Ccl5, Ccl2 and Isg15 levels were similarly elevated by virus infection but decreased by LNP-CasRx-pre-gCtsl treatment (Extended Data Fig. 6e). Histologically, mice from the virus-infected control group had parenchymal lung lesions that were patchy in distribution at 4 DPI. Perivascular lymphoid infiltrates and interstitial thickening of alveolar septal regions with mixed mononuclear and polymorphonuclear leukocyte infiltrations were the most prominent features of the lung lesions. Notably, this pathogenesis was markedly decreased in the LNP-CasRx-pre-gCtsl group (Fig. 5c and Extended Data Fig. 6f). Similar to its effects at 2 DPI, LNP-CasRx-pre-gCtsl also reduced the mRNA and/or protein expression level of viral N and E in infected lungs at 4 DPI (Extended Data Fig. 7a–f). The efficient knockdown of Ctsl in mouse lung by LNP-CasRx-gCtsl treatment was also confirmed (Extended Data Fig. 7c,d lower panel, g). These data indicate that LNP-CasRx-pre-gCtsl treatment inhibited the expression of proinflammatory cytokines/chemokines and reduced lung disease.
Fig. 5 |. LNP-CasRx-pre-gCtsl reduces chemokine/cytokine levels and lung pathogenesis in SARS-CoV-2-infected K18-hACE2 mice.

a, Schematic illustration of the experimental design, using the same treatment as in Fig. 3. Each group consists of two females and two males except for the eLNPs group (one female and two males). Lungs were collected for analysis at 4 DPI. The DPBS, eLNPs and LNP-CasRx-pre-gControl groups were pooled and referred to as control. Nonvirus, two females and two males. b, Transcript levels of Cxcl10 and Tnf in lung. All transcript levels are normalized to Gapdh. The bar represents the average of the group, while each circle represents an individual animal. P values were calculated by one-tailed Mann–Whitney U-test, grand mean. **P < 0.01. c, Representative photomicrographs of H&E-stained lung sections demonstrate that the perivascular, interstitial and alveolar inflammatory lesions in virus-infected mice were reduced by LNP-CasRx-pre-gCtsl treatment. One section per mouse was subjected to histological evaluation and only one image from each group was shown. Annotations: BV, blood vessel; thin arrow, perivascular or interstitial inflammatory infiltrates; asterisk, intra-alveolar edema. Scale bars, 200 μm (left) and 50 μm (right).
While COVID-19 is primarily a respiratory disease, patients may also suffer neurological complications ranging from loss of smell and headache to acute encephalopathy39. Here we have observed high expression levels of viral N and E genes in five out of 11 mice in the control group (Extended Data Fig. 8a–d), consistent with previous observations that some mice with high dose virus challenge may develop brain infections33–35. Extensive staining of the viral N protein was mainly located in the cerebral cortex and in the thalamic and hypothalamic regions (Extended Data Fig. 8d). Of note, the viral transcript expression and N protein staining levels were markedly reduced in the LNP-CasRx-pre-gCtsl group, although statistical significance was not reached (Extended Data Fig. 8b–d), possibly due to the heterogenous response to SARS-CoV-2 infection in the brain. Clinically, some patients with COVID-19 also displayed a significant increase in the levels of CCL and CXCL family chemokines in brain, possibly caused by inflammation indirectly relayed into the brain by SARS-CoV-2 infection40. We also observed an elevation of the Cxcl10, Ccl2 and Ccl5 transcripts, particularly in some control mice, whereas their expression in the LNP-CasRx-pre-gCtsl group remained at levels similar to those in unchallenged mice (Extended Data Fig. 8e). Therefore, LNP-CasRx-pre-gCtsl treatment likely protects the brain from direct and/or indirect damage by SARS-CoV-2 infection. Histological changes in the brain were limited to focal meningeal and perivascular accumulation of inflammatory cells in Virchow–Robin spaces. The limited tissue sampling scheme did not allow comparison of these lesions across the control and treatment groups (Extended Data Fig. 8f). Of note, no significant histological changes were present in other tissues examined (liver, spleen, kidney, jejunum and colon), regardless of whether mice were associated with control or treatment groups at 4 DPI (Supplementary Fig. 1).
Postinfection treatment by CasRx nanosystem.
Finally, we tested the therapeutic anti-SARS-CoV-2 potential of our nanosystem. K18-hACE2 mice were treated with LNP-CasRx-pre-gControl or LNP-CasRx-pre-gCtsl 2 h and 1 day after challenge with 105 PFU of SARS-CoV-2 (Fig. 6a). The lungs were harvested for analysis on 3 DPI. The LNP-CasRx-pre-gCtsl group showed significantly decreased levels of viral N and E gene transcripts, compared with the LNP-CasRx-pre-gControl group (Extended Data Fig. 9a,b). Viral N protein expression was also markedly reduced in the LNP-CasRx-pre-gCtsl group, as demonstrated by both immunostaining of lung sections and western blot analysis (Fig. 6b,c and Extended Data Fig. 9c,d left panel). Ctsl downregulation was confirmed in the LNP-CasRx-pre-gCtsl group (Fig. 6c,d and Extended Data Fig. 9d right panel). Viral lesions consisted of a patchy angiocentric pleocellular inflammatory infiltrate consisting of lymphoid cells and macrophages with occasional admixed polymorphonuclear leukocytes that extended into interstitial parenchyma, which was dramatically alleviated by the LNP-CasRx-pre-gCtsl treatment (Fig. 6e). We found that a single dose of LNP-CasRx-pre-gCtsl, administered 8 hours following infection of 104 PFU of SARS-CoV-2, also significantly lowered the virus burden and alleviated virus-induced pathological changes in the lungs (Extended Data Fig. 10). Together, these data demonstrate that postexposure treatment with LNP-CasRx-pre-gCtsl can effectively inhibit SARS-CoV-2 infection.
Fig. 6 |. Postexposure treatment with LNP-CasRx-pre-gCtsl reduces virus burden in lungs of SARS-Cov-2 infected K18-hACE2 mice.
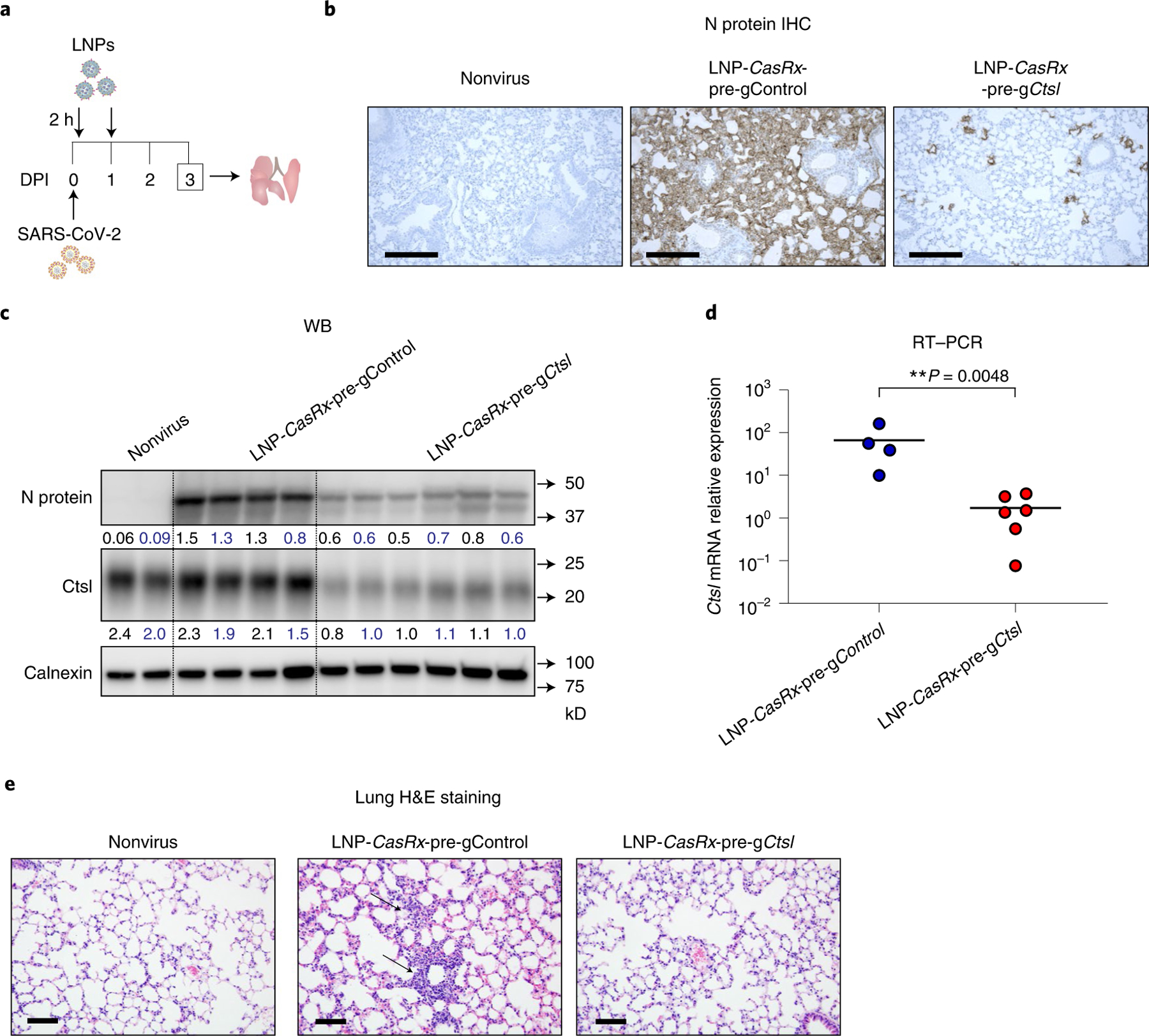
a, Schematic illustration of the experimental design. Female and male mice were intranasally infected with 105 PFU of SARS-CoV-2. They were treated with LNP-CasRx-pre-gControl or LNP-CasRx-pre-gCtsl through the retro-orbital injections 2 h and 1 DPI. The LNP-CasRx-pre-gControl group consisted of four mice (two females and two males), whereas the LNP-CasRx-pre-gCtsl group had six mice (three females and three males). The lungs were collected at 3 DPI for analysis. b, Representative images for viral N protein immunostaining in lung. One lung section per mouse was subjected to IHC analysis and only one image from each group was shown. Nonvirus, one female and one male mouse. Scale bar, 100 μm. c, Western blot (WB) of viral N protein and Ctsl protein in lung. All mice were subjected to this analysis. Each lane represents an individual mouse. Calnexin was used as a loading control. The ratio of N or Ctsl protein over calnexin is listed under the blot. d, Ctsl transcript levels as normalized by Gapdh. The bar represents the average of the group, while each circle represents an individual animal. P values were calculated by one-tailed Mann–Whitney U-test, grand mean. **P < 0.01. e, Representative photomicrographs of H&E-stained lung sections demonstrating that alveolar septal areas have essentially normal cellularity following LNP-CasRx-pre-gCtsl treatment. One lung section per animal was used for histological analysis and one image from each group was shown. Annotations: thin arrow, inflammatory infiltrate in interstitial regions causing hypercellularity. Scale bars, 100 μm.
Discussion
In this study, we report the development of an efficient, specific and safe lung Ctsl-targeted nanosystem in preventing and treating SARS-CoV-2 infection. We show that LNP-CasRx-pre-gCtsl treatment potently depletes Ctsl expression in the lungs (Fig. 2a–c) yet does not affect the expression of other Ctsl family members (Extended Data Fig. 4f) that are critical for physiological and pathological processes (for example, the function of Ctss in antigen processing/presentation and antibody production41,42). Furthermore, the LNP-CasRx-pre-gCtsl treatment does not alter the Ctsl expression in liver or spleen (Extended Data Fig. 4e) where it plays essential physiological roles (for example, Ctsl inhibition may impair CD4+ T cell selection in splenocytes and thymocytes43,44). Finally, the nonpathogenic Ruminococcus flavefaciens-derived CasRx delivered by lung-selective LNPs appears to be noninflammatory and nontoxic in vivo (Fig. 2d and Extended Data Fig. 5).
LNP-CasRx-pre-gCtsl treatment significantly protects K18-hACE2 transgenic mice from lethal infection by SARS-CoV-2 (Fig. 3). The survival benefit of this approach is believed to be mediated by reductions of viral load, cytokines/chemokines levels and lung pathology (Figs. 4 and 5). Notably, while recent studies have used Cas13 for blocking SARS-CoV-2 infection by targeting highly conserved regions in the SARS-CoV-2 genome (for example, the RNA-dependent RNA polymerase (RdRP) and N gene regions), those previous reports apply Cas13 either in vitro or prophylactically in vivo45,46. The present studies are a significant development in that we have found that postinfection delivery of LNP-CasRx-pre-gCtsl decreases the virus burden and reduces virus-induced pathological changes in the lungs (Fig. 6 and Extended Data Fig. 10). This study demonstrates that Cas13, and more broadly, CRISPR, can be used as a treatment for SARS-CoV-2 infection. Of note, our application of LNP-CasRx-pre-gCtsl to knockdown the Ctsl mRNA expression impairs the functions of both the catalytic and noncatalytic domains of Ctsl, where Ctsl small molecule inhibitors only inhibit the active sites of the Ctsl enzyme. This is an important distinction since previous studies have found that the noncatalytic domains of proteases may enhance substrate specificity, steer cellular localization and alter kinetic properties as well as the sensitivity to endogenous inhibitors47. In addition, while Ctsl small molecule inhibitors were systemically distributed, LNP-CasRx-pre-gCtsl was selectively delivered to the lungs. We reason that these factors could contribute to the markedly enhanced efficacy of our LNP-CasRx-pre-gCtsl treatment compared to the small molecule inhibitors.
SARS-CoV-2 is one of three novel coronaviruses (along with SARS-CoV-1 and MERS-CoV) that have caused large-scale pandemics in the 21st century48. Notably, our LNP-CasRx-pre-gCtsl treatment also inhibits infection of SARS-CoV-1 pseudovirus and the authentic B.1.617.2 SARS-CoV-2 Delta variant in vitro (Fig. 1c–e), suggesting that our lung Ctsl-targeted nanosystem may broadly inhibit infection by other related highly pathogenic coronaviruses in vivo. In addition, the findings that many coronaviruses circulating in bats are capable of replicating in human lung and airway cells demonstrate the need for the development of pan antiviral therapeutic strategies for future zoonotic coronavirus outbreaks48. Key considerations include the involvement of CTSL in mediating cellular entry by several coronaviruses as well as their mutant derivatives (for example, SARS-CoV-1, MERS-CoV, SARS-CoV-2 and its Delta and Omicron variants)12,13,49 (Fig. 1), and the fact that most coronavirus spike proteins do not frequently undergo mutations within the CTSL cleavage sites (S1/S2 border, S2′ position and between18,19). In this context, our lung Ctsl-targeted nanosystem may represent an attractive prophylactic and therapeutic approach against both existing and emerging coronavirus infection.
Our study demonstrates that a simple modification of the FDA-approved MC3 LNP28 by adding a cationic lipid (DOTAP) can effectively deliver Cas13 mRNA/gRNA to nonliver tissues where it can specifically knockdown a target gene. It is noteworthy that this RNA-targeting system allows for the flexible design of alternative gRNAs for encapsulation in Cas13-based LNPs, providing an opportunity to target other essential host factors and viral genes relevant to infection by coronavirus and other RNA viruses12,13,45,46,50. This nanosystem can be easily adapted in the future to target infection by a DNA virus (for example, hepatitis B virus50) by modification of the LNP components and cargo (for example, using liver-selective MC3 LNPs (Extended Data Fig. 2a,b) to deliver Cas9 for targeting the hepatitis B virus genome).
Methods
Cell culture.
Human embryonic kidney 293T (HEK293T) (ATCC no. CRL-11268, RRID: CVCL_1926), 293FT (Invitrogen, no. R70007) and Vero-ACE2 (Vero-E6 expressing high endogenous ACE2, BEI, NR-53726) cells were grown in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 1% penicillin/streptomycin and 10% fetal bovine serum (FBS, Thermo). Caco-2 (ATCC no. HTB-37, RRID: CVCL_0025) cells were grown in DMEM supplemented with 1% penicillin/streptomycin and 20% FBS. Vero-ACE2 cells stably expressing TMPRSS2 were generated by transduction of Vero-ACE2 cells with a lentiviral vector expressing human TMPRSS2, followed by blasticidin S HCl selection (7.5 μg ml−1) for 7 d. KLN 205 cells were grown in minimum essential media supplemented with 1× sodium pyruvate (Gibco, no. 11360070), 1× minimum essential media nonessential amino acids solution (Gibco, no. 11140050) and 10% FBS. All cell lines were maintained at 37 °C with 5% CO2. The cell lines were routinely screened using VenorTM GeM Mycoplasma Detection Kit (Sigma-Aldrich, no. MP0025) following the manufacturer’s protocol.
RNA isolation and PCR with reverse transcription (RT–PCR).
Total RNA of cells was isolated by RNeasy mini kit (Qiagen, no. 74104). Tissues were first homogenized by a BeadBug microtube homogenizer (for virus-infected tissues) or a power homogenizer (for normal tissues), total RNA was then extracted using Ambion PARIS system (Invitrogen, no. AM192) and reverse transcribed by transcriptase (Applied Biosystems, no. 4368813). Transcript level was determined by SYBR Green gene expression assays (Applied Biosystems, no. A25742). Real-time PCR reactions were carried out in triplicate using a qTOWER3G system (Analytik Jena) and data was collected by qPCRsoft3.4. Primers were synthesized by IDT and their sequences are listed in Supplementary Table 1. The expression was calculated with comparative Ct method and the raw data was normalized by the internal control Gapdh or 18 s.
RNA-sequencing (RNA-seq) and data analysis.
RNA-seq analysis was carried out as previously described in ref. 51. Briefly, 293FT cells were transfected with pre-gControl or pre-CTSL vectors in combination with a CasRx expression plasmid and incubated for 24 h. RNA was extracted using the RNeasy Mini Kit (Qiagen, no. 74106). For library preparation, mRNA was first enriched with NEBNext Poly(A) mRNA Magnetic Isolation Module (NEB, no. E7490L). After two rounds of processing, the mRNA was recovered for library generation, which was performed with NEBNext Ultra II Directional RNA Library Prep Kit for Illumina (NEB, no. E7765S) following the manufacturer’s instructions. The complementary DNA molecules were amplified by eight cycles of polymerase chain reaction (PCR). Nonsize-selected libraries were sequenced using an Illumina NEXTSEQ 500 platform at the Duke Sequencing and Genomic Technologies Shared Resource. Read alignment to human hg19 was conducted using HISAT2 v.2.1.0 and the expression counts were computed by htseq-count v.0.11.2. Differentially expressed genes were identified with DESeq2 v.1.26.0.
In vitro virus infection and assessment assays.
Pseudotyped virus.
Lentiviral pseudotyped virus was produced as previously described52. Briefly, HEK293T cells were transfected with HIV-1 NL4.3-inGluc (a gift from M. Johnson at the University of Missouri) and pcDNA3.1-SARS-CoV-2-S-C9 (obtained from F. Li at the University of Minnesota) constructs in a 2:1 ratio using polyelthylenimine. Supernatants were harvested 24, 48 and 72 h postransfection and were pooled, aliquoted and stored at −80 °C.
Pseudotyped virus infection.
Here, 3 × 105 of Vero-ACE2-TMPRSS2 or Caco-2 were seeded into six-well plates and transfected with CasRx mRNA (0.75 μg ml−1) and pre-gControl oligo (pre-gControl, 0.75 μg ml−1) or pre-gCTSL oligo (pre-gCTSL, 0.75 μg ml−1) by Lipofectamine MessengerMAX (Invitrogen, no. LMRNA015). Cells were reseeded into 24-well plate 12 h posttransfection and infected with pseudotyped virus for 6 h. The media was then replaced with 500 μl of fresh media. Then 20 μl of media was collected at 72 h and incubated with 20 μl of Gaussia luciferase substrate (0.1 M Tris (MilliporeSigma, no. T6066) pH 7.4, 0.3 M sodium ascorbate (Spectrum, no. S1349) and 10 μM coelenterazine (GoldBio, no. CZ2.5)). For inhibitor treatment, cells were treated with 25 μM E-64d (Sigma, no. 330005) 1 h before virus infection, then read by BioTek Cytation5 plate-reader using BioTek Gen 5 v.3.03.
Authentic SARS-CoV-2 B.1.617.2 Delta variant and cell infection.
The variant strain (USA/PHC658/2021) was obtained from BEI Resources NR-55611. 3 × 105 of Vero-ACE2-TMPRSS2 cells were seeded into six-well plates and transfected with CasRx-pre-gControl (1.5 μg ml−1) or CasRx-pre-gCTSL (1.5 μg ml−1) by Lipofectamine MessengerMAX. Cells were reseeded into 12-well plate 12 h posttransfection and infected with the SARS-CoV-2, Isolate hCoV-19/USA/PHC658/2021 at a multiplicity of infection of 0.01.
Tissue culture infection dose 50 (TCID50) assay.
Cell culture supernatants were collected 20 h after infection. Samples were serially diluted tenfold and used to determine live virus titer by TCID50. Briefly, 4 × 104 Vero-E6 cells were placed in each well of a 96-well tissue culture plate and allowed to adhere overnight. Growth medium was removed, and serial dilutions of experimental supernatant were added. Samples in duplicate were incubated for 96 h, followed by being fixed with 10% neutral buffered formalin and stained with 0.1% crystal violet. The cytopathic effect was examined by eye, and TCID50 per ml was calculated by the Reed–Muench method.
Preparation of lung-targeting LNPs encapsulating CasRx mRNA and gRNA oligos.
LNPs were generated by simultaneously mixing one volume of lipid mixture (25:5:19.3:0.8:50 molar ratio) of DLin-MC3-DMA (MedKoo Biosciences, no. 555308), DSPC (Avanti Polar Lipids, no. 850365), cholesterol (Sigma, no. C3045), DMG-PEG2000 (NOF America Corporation) and DOTAP (Avanti Polar Lipids, no. 890890) in ethanol and two volumes of RNAs in citrate buffer (10 mM, pH 4.0) in a microfluidic device (TheNanoAssemblr Spark, Precision Nanosystems) to satisfy a final weight ratio of 20:1 for total lipids:RNAs. CasRx mRNA with pseudouridine modification (pseudouridine-5′-triphosphate, TriLink) was synthesized using in vitro transcription (AmpliScribe T7-Flash Transcription Kit, Lucigen) and was then installed with 5′ cap (Vaccinia Capping System, NEB; Cap 2′-O-methyltransferase, NEB) and 3′ poly(A) tail structures by Y. Dong’s laboratory at The Ohio State University53,54. Pre-gControl and pre-gCtsl oligos with 2′OMe and phosphorothioate modification were synthesized by Exonano RNA, LLC, and their sequences are listed in Supplementary Table 2. The formulation was then dialyzed using Slide-A-Lyzer Dialysis cassette (WMCO 3.5 kDa, Thermo Fisher, no. 66330) against 1× DPBS for 1 h and diluted to 2 μg μl−1 of lipids with DPBS for injections. The RNA (CasRx mRNA:pre-gRNA, 1:1.5, wt:wt) dose was 1 mg kg−1.
Physicochemical characterization of LNPs.
Empty LNPs (eLNPs) and luciferase mRNA (TriLink, no. L-7202)-loaded LNPs (LNP-Luc) were characterized by their size, morphology, zeta potential and stability. Size and zeta potential were measured using capillary cells (Malvern, no. DTS1070) by Zetasizer Nano ZS (Malvern Panalytical) using Zetasizer Software v.7.03. Transmission electron microscopy examination was performed by Chapel Hill Analytical and Nanofabrication Laboratory, University of North Carolina-Chapel Hill. Briefly, LNPs were stained with 2% uranyl acetate for 2 min and imaged by Talos F200X (Thermo Fisher) at an accelerating voltage of 200 kV. Data was collected by Velox (v.2.8.0.898-bd6c8535e0). We also stored the LNPs in DPBS at 4 °C for 10 d to study their stability. Aliquots were taken every other day for determination of their encapsulation efficiency by RiboGreen assay (Thermo Fisher, no. R11490) and Spectra Max M3 (Molecular Devices) with SoftMax pro6.4.
Mouse experiments.
Animal studies were approved by the Institutional Animal Care and Use Committee of Duke University. C57BL/6 mice (Stock no. B6-MPF, 5–8 weeks old, female or male) were purchased from Taconic. Transgenic mice expressing human ACE2 under control of cytokeratin 18 promoter K18-hACE2 (Stock no. 034860, 7–8 weeks old, female or male) were obtained from Jackson Laboratory. Mice were maintained on a 12 h light/dark cycle with food and water ad libitum at an average room temperature of 22.5 °C and humidity of 51%. Mice were acclimatized for 1 week before the beginning of an experiment. All animal experiments with SARS-CoV-2 were performed in the Duke Regional Biocontainment Laboratory (RBL), a Biosafety Level 3 (BSL3) facility.
In vivo imaging software imaging.
LNPs encapsulating luciferase mRNA were administered to C57BL/6 mice by intravenal bolus through either a retro-orbital route (0.5 mg kg−1) or tail vein (0.4 or 0.5 mg kg−1) injection. After 3 h, mice were intraperitoneally injected with the substrate d-luciferin (150 mg kg−1, Perkin Elmer, no. 770505). Whole body and major organs (heart, liver, spleen, lung, kidney) were then imaged for fluorescence using an IVIS Lumina XR system with Living Image Software v.4.3.1 (Caliper Life Science).
Toxicity profiling.
C57BL/6 mice were used for in vivo toxicity studies. Mice were injected through tail vein with DPBS, empty LNP, LNP-CasRx-pre-gControl or LNP-CasRx-pre-gCtsl, respectively. Each group had eight mice with four female and four males. Blood was collected 72 h after the injection. Complete blood cell count was assessed by IDEXX Procyte DX (IDEXX Laboratories) and serum level of alkaline phosphatase, alanine aminotransferase, aspartate aminotransferase, blood urea nitrogen and creatinine were analyzed by the Animal Histopathology and Laboratory Medicine Core, University of North Carolina-Chapel Hill using Alfawassermann Vet Axcel (Alfawassermann Diagnostic Technologies). Selected tissues, including lung, liver, spleen, heart and kidney, were collected for histopathological evaluation.
Survival study.
K18-hACE2 mice were lightly anesthetized with isoflurane and infected intranasally with 105 PFU of SARS-CoV-2 (USA-WA1/2020) in a total volume of 50 μl of DMEM on day 0. This input virus was expanded from a seed stock from BEI Resources (the National Institutes of Health (NIH) Repository). After passaging in the Duke RBL, it has been sequenced several times to confirm the absence of mutations in the Spike protein (that is, the S1/S2 site is intact). Mice were given with DPBS (n = 10), empty LNP (n = 9), LNP-CasRx-pre-gControl (n = 10) or LNP-CasRx-pre-gCtsl (n = 10), respectively, through a retro-orbital route 2 d before infection and 1 day postinfection (DPI). Animal weight, temperature and clinical signs were monitored daily. The criteria for symptom scoring were: (0) normal, (1) ruffled fur and hunched, (2) respiration-labored breathing and sneezing, (3) discharge from nose, (4) lethargic and (5) moribund or dead. Mice were humanely euthanized when weight loss exceeded 20% and/or symptom score reached 5.
Treatment regimens and sample collection.
(1) For mixed therapy, K18-hACE2 mice were intranasally inoculated with 105 PFU of SARS-CoV-2. Treatments were administered 2 d before and 1 DPI. Organ tissues were collected on 2 or 4 DPI as indicated. Left lung lobe, left brain, liver, spleen, kidney, jejunum and colon were fixed by immersion in 10% neutral buffered formalin for histological analysis. Right lung lobe and right brain were homogenized and used for extraction of total RNA and proteins and plaque assay. (2) For postexposure therapy, K18-hACE2 mice were treated with LNP-CasRx-pre-gControl or LNP-CasRx-pre-gCtsl either 2 h and 1 d postinfection with 105 PFU of SARS-CoV-2 or 8 h postinfection with 104 PFU of SARS-CoV-2. Lung tissues were collected on 3 DPI for biochemical and histological analysis.
Western blot.
Cells were collected and lysed in RIPA buffer (Boston BioProducts, no. BP-115) with proteinase inhibitor cocktail (Roche). Homogenized tissues were lysed using cell disruption buffer in Ambion PARIS system. After 30 min incubation on ice and centrifugation, the supernatant was separated and used. Its protein concentration was determined by BCA protein assay (Thermo, no. 23225) using Spectra Max M3 with SoftMax pro6.4. Samples were then resolved on a 4–15% polyacrylamide gradient gel (BIO-RAD, no. 4568084 or 5671085), transferred to 0.2-μm polyvinyl difluoride membrane (Thermo, no. 88520 or BIO-RAD, no. 1704157) and probed with corresponding antibodies. Antibody against CTSL was purchased from R&D SYSTEMS (no. AF1515), against N protein from Sino Biological (no. 40143-R019, Monoclonal rabbit IgG Clone no. 19, 1:1,000) and against calnexin from Enzo (no. ADI-SPA-860-F, 1:1,000). The specificity and sensitivity of the antibodies were validated by the manufacturers. CTSL antibody was further validated by our laboratory using small-interfering RNA transfection. Immunoblots were developed using SuperSignal West Pico PLUS chemiluminescent substrate (Thermo, no. 34580) and visualized using the LI-COR Odyssey Fc Imaging System with Image Studio v.5.2. The protein expression was quantified by densitometry (ImageJ v.1.52a/Java v.1.8.0_112) and normalized to calnexin.
Histology and IHC.
Tissues were processed routinely in paraffin, sectioned at 4 μm, stained with hematoxylin and eosin (H&E) and evaluated by a board-certified veterinary pathologist in a masked manner without knowledge of study allocation group. IHC was performed by the Research Immunohistology Shared Resource at Duke University. Antibodies targeting the CTSL (R&D SYSTEMS, no. AF1515, 1:800) and the SARS-CoV-2 nucleoprotein (N) (Sino Scientific, no. 40143-R019, 1:20,000) were used. The IHC score of N protein expression in lung sections ranged from 0 to 2, which was performed by the product of intensity (graded as 0, absent; 1, weak positive and 2, strong positive) and proportion of staining in pneumocytes showing maximum staining intensity.
Tissue virus titer by plaque assay.
SARS-CoV-2 plaque assays were performed in the Duke Regional Biocontainment BSL3 Laboratory as previously described55. Serial dilutions of lung homogenate were incubated with Vero-E6 cells in a standard plaque assay. Homogenate and cells were incubated at 37 °C, 5% CO2 for 1 h. At the end of the incubation, 1 ml of a viscous overlay (1:1 2× DMEM and 1.2% methylcellulose) was added to each well. Plates were incubated for 4 d. After fixation, staining and washing, plates were dried and plaques from each dilution of sample were counted. Data are reported as PFU per g of lung tissue.
Statistical analysis.
All data were analyzed by GraphPad Prism v.9.0 software. Differences in mean between groups were determined by Student’s t-test or by Mann–Whitney U-test as applicable. Differences in survival were analyzed by log-rank test. P values <0.05 were considered statistically significant.
Reporting summary.
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Extended Data
Extended Data Fig. 1 |. Screening for a pre-gRNA targeting human CTSL and evaluation of its knockdown efficiency and specificity in cultured cells.
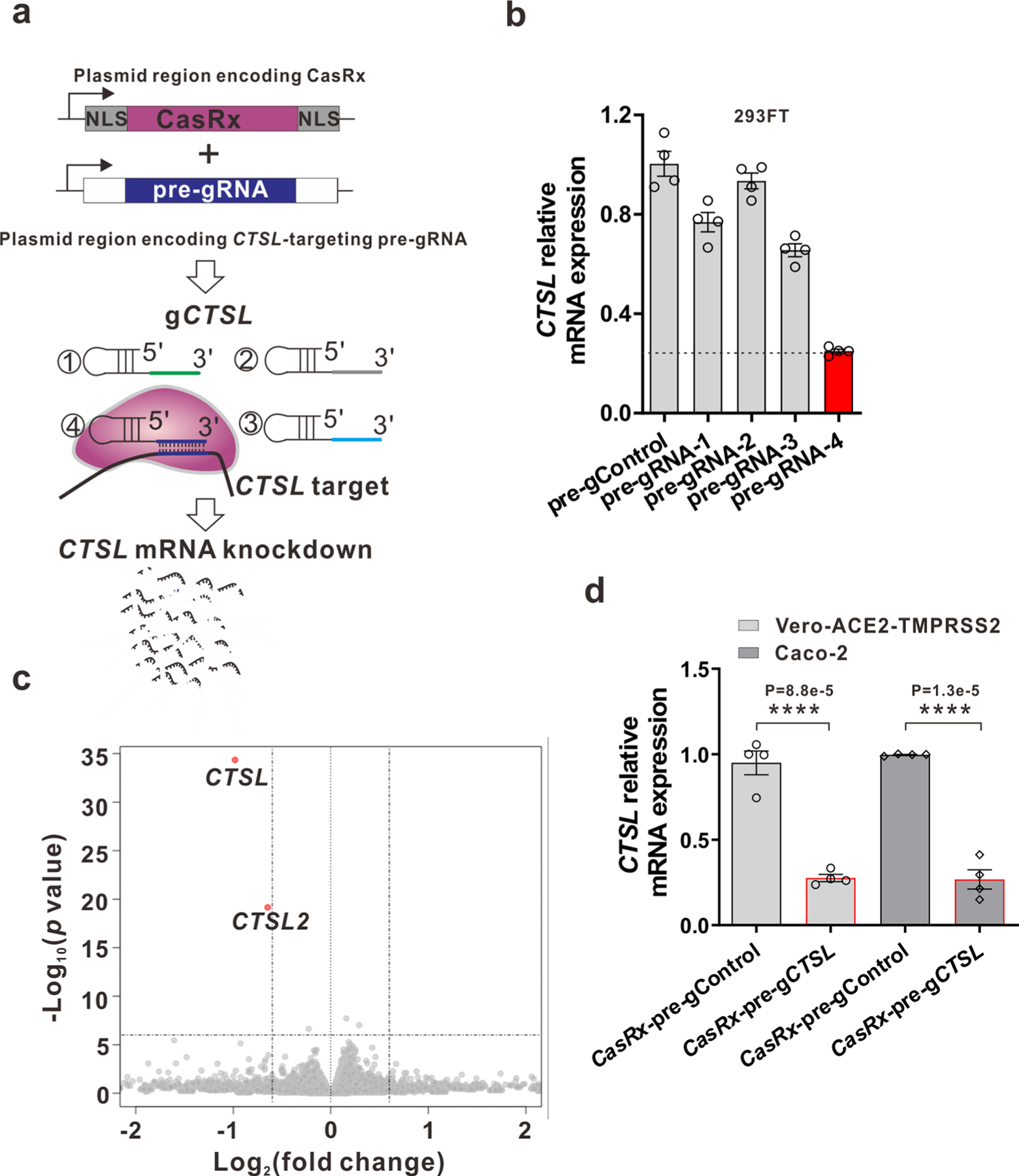
a, Schematic of CasRx and pre-gRNA plasmid system for screening candidate pre-gRNA targeting human CTSL. b, Relative CTSL mRNA expression was measured by RT-PCR after 48 h after transfection with CasRx plasmid and different pre-gRNAs designed to target CTSL in 293 FT cells. c, Volcano plot of gene expression changes between CasRx-pre-gCTSL and CasRx-pre-gControl transfected 293FT cells for 24 h. RNA sequencing was performed in biological triplicates. Significantly differentially expressed genes (Fold change >1.5, P-value<1E-6) are shown in red color. d, CTSL knockdown efficiency in Vero-ACE2-TMPRSS2 and Caco-2 cells. Of note, the pre-gCTSL targeting sequence in CTSL mRNA is same for human and monkey. Data in plots b and d were shown with n = 4 biologically independent replicates and presented as mean ± SEM. Transcript levels are normalized to 18 s rRNA. P values were calculated by two-tailed Student’s t-test. **** P < 0.0001.
Extended Data Fig. 2 |. IVIS imaging for the LNPs biodistribution.
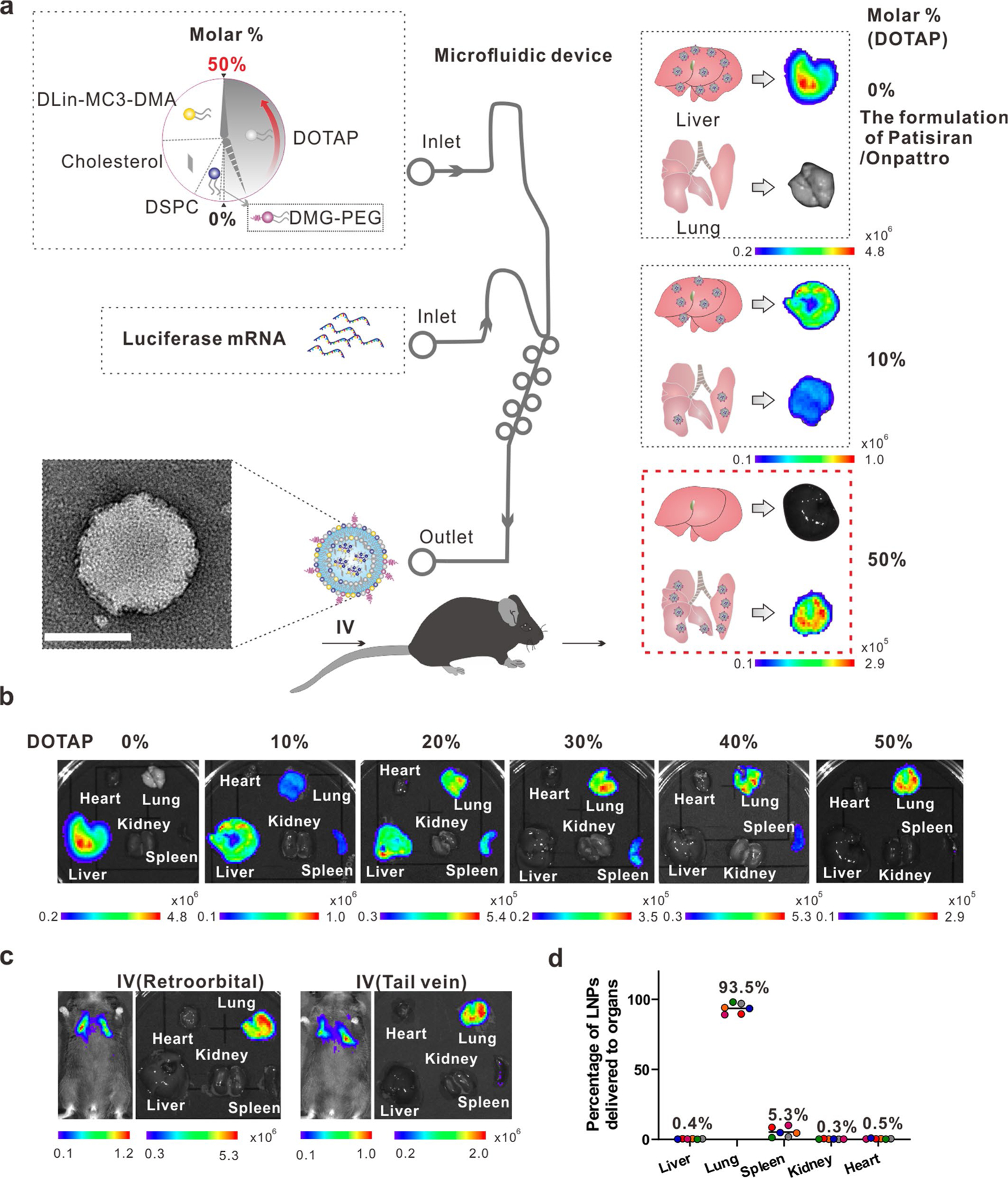
a, Schematic illustration for the construction of lung-targeting LNPs. The lipids are injected into the upper inlet of a microfluidic device (SPARKTM CARTRIDGE), whereas the cargo luciferase mRNA is loaded into the lower inlet to generate the LNPs from the outlet. Lung-selectivity of LNPs is achieved by adding 50% (molar%) supplementary cationic lipid DOTAP (a SORT molecule) to the traditional LNPs formulation employed by the FDA-approved RNAi therapy Patisiran/Onpattro. Scale bar, 100 nm. b, Mouse major organs were imaged 3 h following tail vein injection of luciferase mRNA-loaded LNPs (LNP-Luc, 0.4 mg/kg) formulated with a series of molar % of DOTAP (0–50 %). c, The animal’s whole body and organs were imaged 3 h after IV injection (retro-orbital or tail vein) of 50% DOTAP-containing LNPs loaded with 0.5 mg/kg luciferase mRNA. d, The average percentages of LNPs with 50% DOTAP delivered to each mouse organ. n = 6 mice.
Extended Data Fig. 3 |. Characterization of empty LNPs (eLNPs) and LNPs encapsulating luciferase mRNA.

a, Size distribution. The size of two batches of eLNPs and LNP-Luc were analyzed by Zetasizer. b, Representative transmission electron microscopic images. The experiment was repeated twice with similar results. c, Zeta potential. eLNPs and LNP-Luc were diluted to 0.8 μg/μl with 1X DPBS for measurement. Data are presented as mean ± SD of two batches of samples. d, Encapsulation rate and stability of LNP-Luc. The encapsulation efficiency of LNP-Luc was determined by RiboGreen assay on days 0, 2, 4, 6, 8 and 10 following its storage at 4°C.
Extended Data Fig. 4 |. Screening of candidate pre-gRNAs targeting mouse Ctsl in vitro and examination of the editing specificity of LNP-CasRx-pre-gCtsl in vivo.
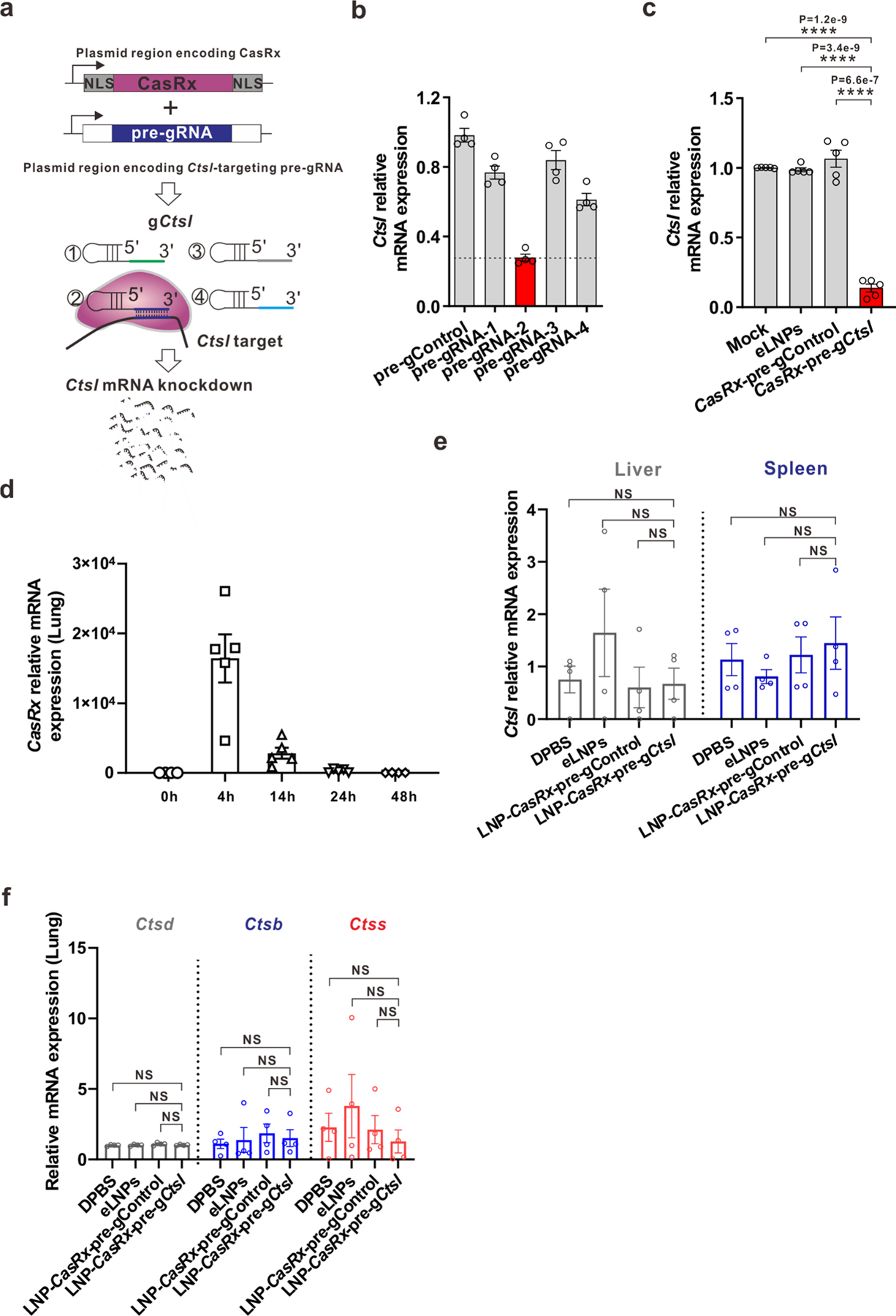
a, Schematic of CasRx and pre-gRNA plasmid system for screening candidate pre-gRNA targeting mouse Ctsl. Relative Ctsl mRNA expression was measured by RT-PCR after 48 h transfection with b, Plasmids encoding CasRx with different pre-gRNAs designed for targeting Ctsl; or c, CasRx mRNA and selected pre-gCtsl from (b) in mouse KLN205 cells. n = 4 biologically independent samples for b; n = 5 biologically independent samples for c. Data are presented as mean ± SEM. P values were calculated by two-tailed Student’s t-tests. ****P < 0.0001. d, Kinetics of CasRx mRNA in lung. C57BL/6 mice were administered LNPs encapsulating CasRx mRNA at 0.5 mg/kg by tail vein. Lungs were collected at 0, 4, 14, 24 or 48 h, respectively, after the injection for qRT-PCR analysis. Data are presented as mean ± SEM for each time point (n = 4 for 0, 48 h; n = 5 for 4 h, 14 h, 24 h). e-f, C57BL/6 mice were administered DPBS, eLNPs, LNP-CasRx-pre-gControl or LNP-CasRx-pre-gCtsl by tail vein. After 72 h, the lung, liver and spleen were collected for analysis. e, Ctsl mRNA levels in liver and spleen remained unchanged by LNP-CasRx-pre-gCtsl treatment. f, mRNA levels of isoform Ctsd, Ctsb and Ctss in lung are unchanged by LNP-CasRx-pre-gCtsl treatment. Transcript levels were normalized to Gapdh and presented as mean ± SEM. n = 4 per group, 2 females and 2 males. P values were calculated by one-tailed Mann-Whitney U test. NS, not significant.
Extended Data Fig. 5 |. Immunogenicity, hematology and histological evaluation of LNP-CasRx-pre-gCtsl treatment in C57BL/6 mice.

a, Cytokine and chemokine transcript levels in mouse lung were unchanged by LNP-CasRx-pre-gCtsl treatment. C57BL/6 mice lung issues were collected at 72 h following the treatments. All transcript levels are normalized to Gapdh. Data are presented as mean ± SEM for each group (n = 4, 2 females and 2 males). P values were calculated by two-tailed Mann-Whitney U test, NS, not significant. b, Hematology was not impaired by LNP-CasRx-pre-Ctsl treatment. WBC, white blood cells; PLT, platelets; RBC, red blood cells; HGB, hemoglobin. Data are presented as box and whisker plots of each group with n = 8 (4 females and 4 males in each group except for 3 females and 5 males in LNP-CasRx-pre-gControl group). Whiskers are min to max. P values were calculated by two-tailed Mann-Whitney U test, NS, not significant. c, No substantial histopathological changes were produced by LNP-CasRx-pre-gCtsl treatment in indicated organ tissues. Two sections of each organ from 3 mice per group were evaluated after H&E staining and the representative images are presented.
Extended Data Fig. 6 |. CasRx-mediated Ctsl knockdown reduces viral burden and chemokines/cytokines in lungs of K18-hACE2 mice on Day 2 or 4 after SARS-CoV-2 infection.
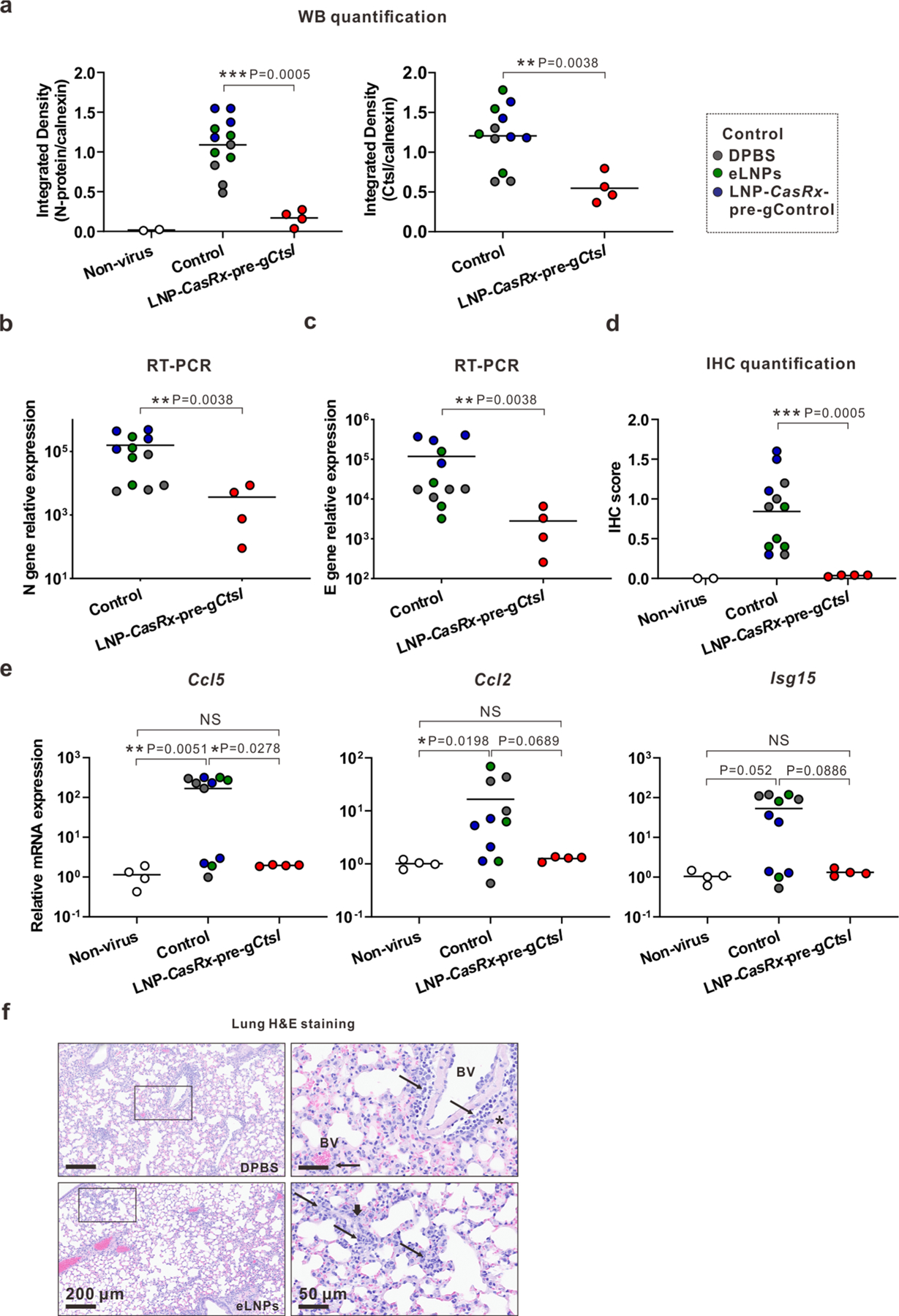
The experimental designs are same as in Fig. 4a. or 5a, respectively. a, Integrated density of N protein (left panel) and Ctsl (right panel) over the loading control on 2 DPI. b, Viral N gene transcript level on 2 DPI. c, Viral E gene transcript level on 2 DPI. d, IHC score for N protein on 2 DPI. e, Ccl5, Ccl2 and Isg15 transcript levels on 4 DPI. b, c, e, All transcript levels were determined by RT-PCR and normalized to Gapdh. P values were calculated by one-tailed Mann-Whitney U test, grand mean. NS, not significant, * P < 0.05, ** P < 0.01. f, Representative photomicrographs of H&E-stained lung sections for the controls of DPBS and eLNPs on 4 DPI. Annotations: BV, blood vessel; thin arrow, perivascular or interstitial inflammatory infiltrates; thick arrow, karyorrhexic nuclei and region of diffuse alveolar damage; asterisk, intra-alveolar edema.
Extended Data Fig. 7 |. CasRx-mediated Ctsl knockdown reduces viral burden in the lungs of K18-hACE2 mice on Day 4 after SARS-CoV-2 infection.

The experiment design was same as in Fig. 5a. a, Viral N gene transcript level. b, Viral E gene transcript level. c, Western blot of viral N protein and Ctsl in mouse lung. All mice were subjected to this analysis. Each lane represents an individual mouse. Calnexin was used as a loading control. The ratio of N protein or Ctsl to calnexin signal is listed under the blot. d, Integrated density of N protein (upper panel) and Ctsl (lower panel) over the loading control. e, Representative images for N protein immunostaining in the lungs. One section per mouse was subjected to IHC analysis and only one representative image from each group was presented. f, IHC score for N protein. g, Ctsl transcript levels. a, b, g, All transcript levels are normalized to Gapdh. P values were calculated by one-tailed Mann-Whitney U test, grand mean. * P < 0.05, ** P < 0.01, *** P < 0.001.
Extended Data Fig. 8 |. Brain responses in SARS-CoV-2-infected K18-hACE2 mice at 4 DPI following LNP-CasRx-pre-gCtsl treatment.
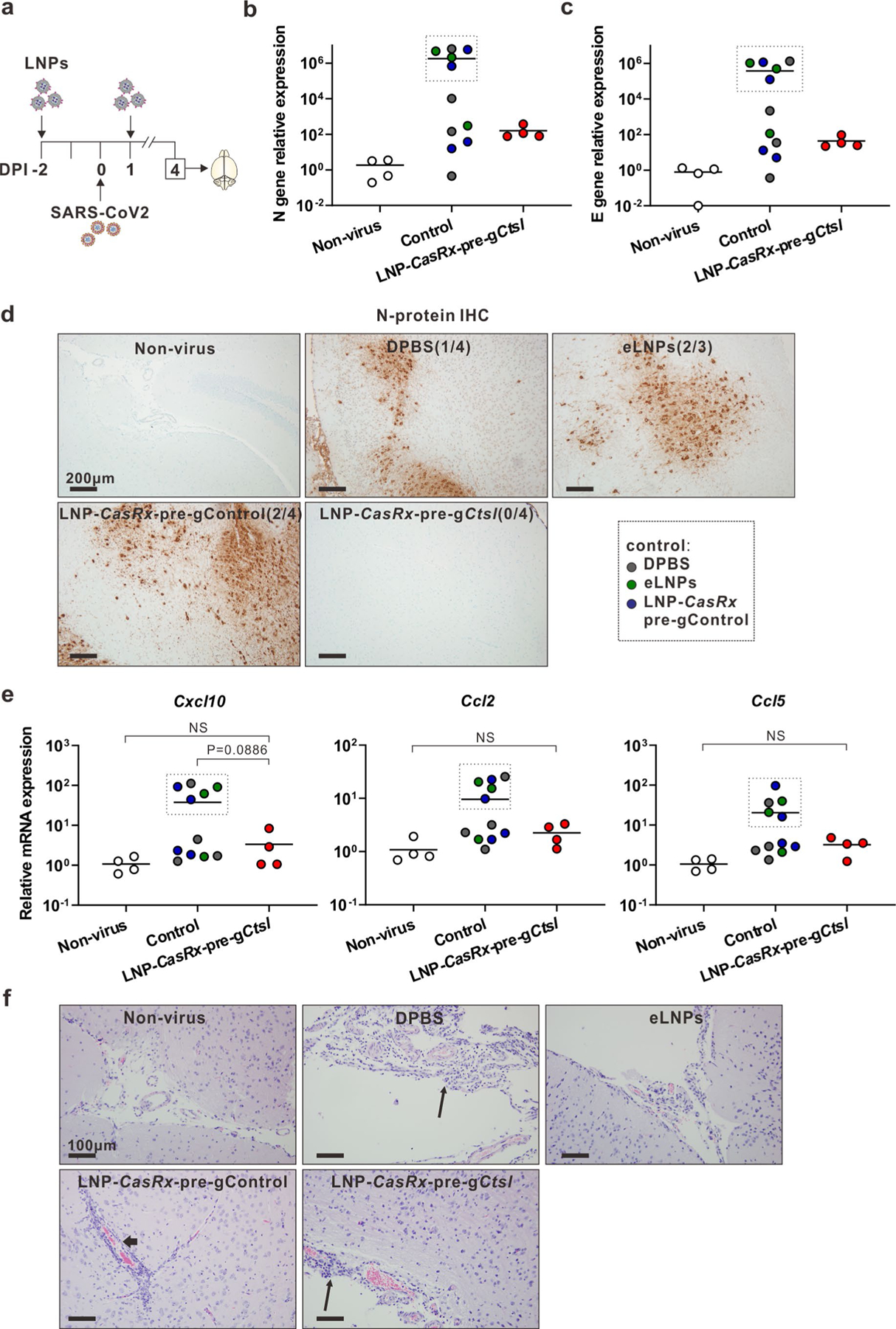
a, Schematic illustration of the experimental design. The treatments and virus challenge are same as in Fig. 5a. Brains were collected at 4 DPI for analysis. The right lobes were subjected to RT-PCR, whereas the left lobes were sectioned for staining. b, Viral N gene transcript level. c, Viral E gene transcript level. d, Representative photomicrographs of N-protein immunostaining show strong intracytoplasmic immunoreactivity of neurons in the cerebral cortex and the thalamus and hypothalamus regions of SARS-CoV-2 infected mice. Two sections of brain tissues per mouse from 4 mice of each group (3 for eLNPs group) were subjected to IHC analysis. The occurrence of strong positive staining was indicated in brackets for each group. e, Cxcl10, Ccl2 and Ccl5 transcript levels. b, c, e, all transcript levels were normalized to Gapdh. P values were calculated by one-tailed Mann-Whitney U test, grand mean. NS, not significant. As boxed in the graphs, 5 out of 11 mice in the control group express high transcript levels of viral N gene, E gene and chemokines in the brain. f, Representative photomicrographs of perivascular lymphoid infiltrate (thin arrows) in meninges and Virchow-Robin spaces (thick arrow) show vascular inflammatory changes in all virus-infected groups. Two brain sections per mouse for all mice were subjected to histological analysis and one representative image from each group was shown.
Extended Data Fig. 9 |. Postexposure treatment with LNP-CasRx-pre-gCtsl reduces virus burden in lungs of SARS-Cov-2 infected K18-hACE2 mice.

The experiment design was same as in Fig. 6a. LNP-CasRx-pre-gControl group consisted of 4 mice (2 females and 2 males), whereas LNP-CasRx-pre-gCtsl group had 6 mice (3 females and 3 males). Non-virus, 1 female and 1 male mouse. The lungs were collected on 3 DPI for analysis. a, Viral nucleocapsid N gene transcript levels. b, Viral envelope E gene transcript levels. All transcript levels are normalized to Gapdh. c, IHC score for the viral N protein. d, Integrated density of N protein (left panel) and Ctsl (right panel) over the loading control calnexin. P values were calculated by one-tailed Mann-Whitney U test, grand mean. ** P <0.01.
Extended Data Fig. 10 |. A single 8-hour post-exposure treatment with LNP-CasRx-pre-gCtsl decreases virus burden in the lungs of SARS-Cov-2 infected K18-hACE2 mice.
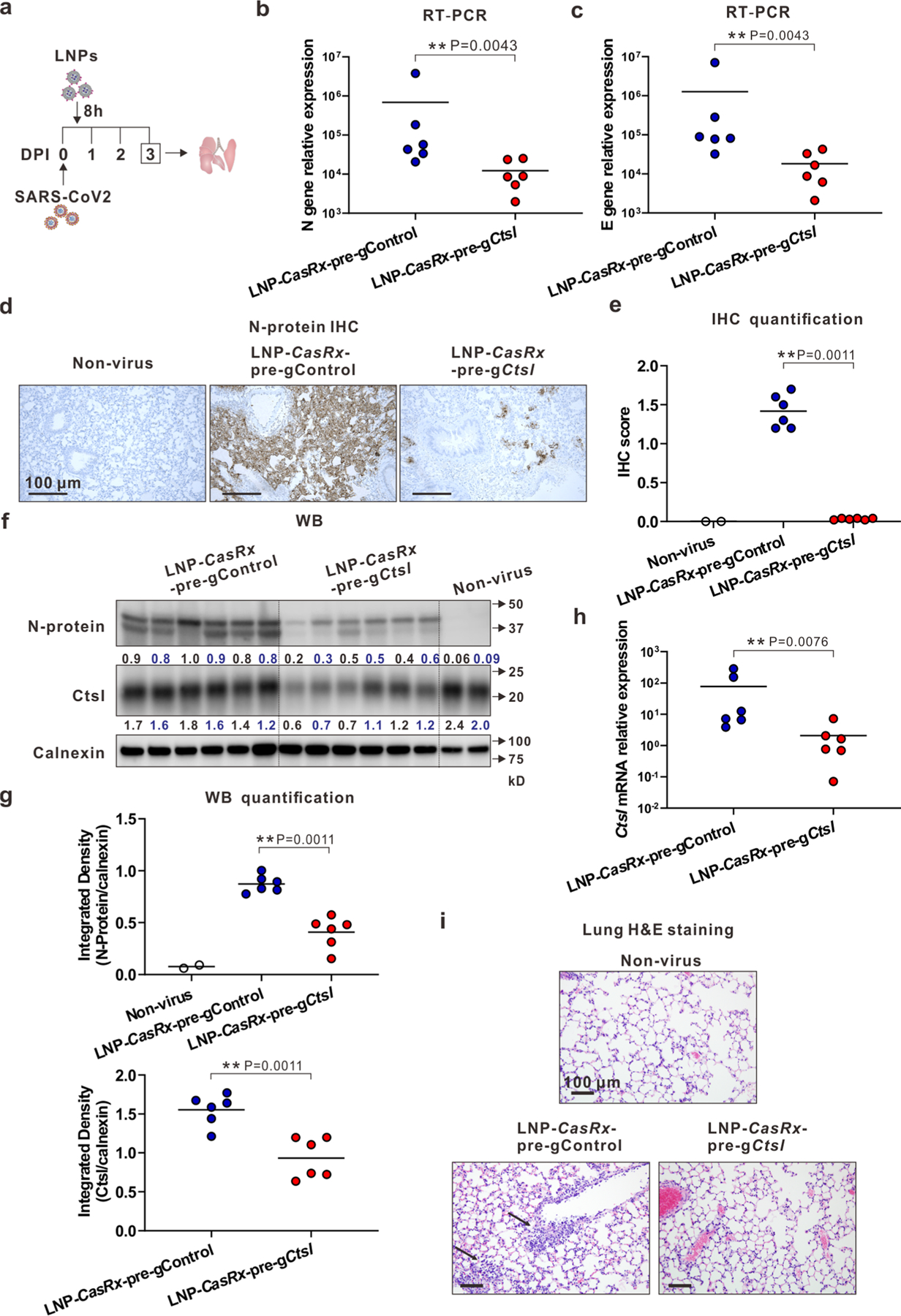
a, Schematic illustration of the experimental design. Mice were intranasally infected with 104 PFU of SARS-CoV-2. They were treated with LNP-CasRx-pre-gControl or LNP-CasRx-pre-gCtsl through retro-orbital injections 8 hours post infection. Each group consisted of 6 mice (3 females and 3 males). Non-virus, 1 female and 1 male mouse. Lungs were collected on 3 DPI for analysis. b, Viral nucleocapsid N gene transcript levels. c, Viral envelope E gene transcript levels. d, Representative images for N protein immunostaining in the lungs. One section per mouse for all mice were subjected for IHC analysis and only one representative image from each group is shown. e, IHC score for N protein. f, Western blot of viral N protein and Ctsl protein in mouse lung. All mice were subjected to this analysis. Each lane represents an individual mouse. Calnexin was used as a loading control. The density ratio of N protein or Ctsl to calnexin is listed under the blot. g, Integrated density of N protein (upper panel) and Ctsl (lower panel) over the loading control. h, Ctsl transcript levels. b, c, h, All transcript levels are normalized to Gapdh. P values were calculated by one-tailed Mann-Whitney U test, grand mean. **P < 0.01. i, Representative photomicrographs of H&E-stained lung sections demonstrating normal cellularity of parenchymal lung by LNP-CasRx-pre-gCtsl treatment. One lung section per mouse for all animals was used for histological analysis and only one representative image from each group is presented. The thin arrow indicates especially severe interstitial inflammatory infiltrate within perivascular regions.
Supplementary Material
Acknowledgements
We thank A. Karlsson, K. Riebe, R. Asrican and T. Oguin III from the Duke RBL for performing authentic SARS-CoV-2 infection experiments in vitro and in vivo. We thank Z. Su from the Duke BioRepository and Precision Pathology Center for performing the IHC assays. We also thank L. Wang from the Animal Histopathology and Laboratory Medicine Core at UNC-Chapel Hill for performing animal hematological and clinical chemistry tests. We thank M. Sokolsky and A. Shankar Kumbhar from Nanomedicines Characterization Core Facility and Nanopformulation Core Facility at UNC-Chapel Hill for TEM and limulus test (to determine endotoxin level in LNPs). We thank M. Wiesner (Duke) for allowing us to use his Zetasizer. The following reagents were deposited by the Centers for Disease Control and Prevention and obtained through BEI Resources, NIAID, NIH: SARS-Related Coronavirus 2, Isolate USA-WA1/2020, NR-52281; SARS-Related Coronavirus 2, Isolate hCoV-19/USA/PHC658/2021 (Lineage B.1.617.2; Delta Variant), NR-55611, contributed by R. Webby and A. Patel. This work was supported by the Department of Pathology, Duke University School of Medicine (to Q.W.); NIH/NIAID (grant no. UC6-AI058607) (to Duke RBL); NIH/NIAID (grant no. R01 AI150473), NIH/NCI (grant no. U54 CA260582) and an anonymous private donor to The Ohio State University (to S.-L.L.) and NIH/NIGMS (grant no. R35 GM144117) (to Y.D.).
Footnotes
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41589-022-01094-4.
Competing interests
Q.W., Z.C. and Y.D. are inventors on a patent (US Patent Application no. 17/626,482 entitled ‘Nanoparticle Systems for Targeted Delivery of CRISPR–Cas13 and Methods of Using Same’ filed 12 January, 2022, patent pending; European Patent Application no. 20840456.6 entitled ‘Nanoparticle Systems for Targeted Delivery of CRISPR–Cas13 and Methods of Using Same’ filed 12 January, 2022, patent pending) filed by Duke University that relates to the research reported in this paper. J.H. is a consultant for or owns shares in the following companies: Kingmed, MoreHealth, OptraScan, Genetron, Omnitura, Vetonco, York Biotechnology, Genecode, VIVA Biotech and Sisu Pharma and received grants from Zenith Epigenetics, BioXcel Therapeutics, Inc. and Fortis Therapeutics. The remaining authors declare no competing interests.
Additional information
Extended data are available for this paper at https://doi.org/10.1038/s41589-022-01094-4.
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41589-022-01094-4.
Peer review information Nature Chemical Biology thanks Nigel McMillan and the other, anonymous, reviewer(s) for their contribution to the peer review of this work.
Reprints and permissions information is available at www.nature.com/reprints.
Data availability
Source data for Figs. 1–6 and Extended Data Fig. 1–10 are provided with this paper. The RNA-seq data generated in this study have been deposited in the Gene Expression Omnibus database under accession number GSE203052. All the other data that support this study are provided in the Supplementary Information files. The LNP reagents are available from the corresponding authors upon reasonable request.
References
- 1.Beigel JH et al. Remdesivir for the treatment of COVID-19—final report. N. Engl. J. Med 383, 1813–1826 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Taylor PC et al. Neutralizing monoclonal antibodies for treatment of COVID-19. Nat. Rev. Immunol 21, 382–393 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mlcochova P et al. SARS-CoV-2 B.1.617.2 Delta variant replication and immune evasion. Nature 599, 114–119 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Planas D et al. Considerable escape of SARS-CoV-2 Omicron to antibody neutralization. Nature 602, 671–675 (2022). [DOI] [PubMed] [Google Scholar]
- 5.Ledford H COVID antiviral pills: what scientists still want to know. Nature 599, 358–359 (2021). [DOI] [PubMed] [Google Scholar]
- 6.Jayk Bernal A et al. Molnupiravir for oral treatment of COVID-19 in nonhospitalized patients. N. Engl. J. Med 386, 509–520 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hammond J et al. Oral nirmatrelvir for high-risk, nonhospitalized adults with COVID-19. N. Engl. J. Med 386, 1397–1408 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhou S et al. beta-d-N4-hydroxycytidine inhibits SARS-CoV-2 through lethal mutagenesis but is also mutagenic to mammalian cells. J. Infect. Dis 224, 415–419 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lamb YN Nirmatrelvir plus ritonavir: first approval. Drugs 82, 585–591 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hoffmann M et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell 181, 271–280.e8 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gordon DE et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 583, 459–468 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wei J et al. Genome-wide CRISPR screens reveal host factors critical for SARS-CoV-2 Infection. Cell 184, 76–91 e13 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Daniloski Z et al. Identification of required host factors for SARS-CoV-2 infection in human cells. Cell 184, 92–105.e16 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu T, Luo S, Libby P & Shi GP Cathepsin L-selective inhibitors: a potentially promising treatment for COVID-19 patients. Pharmacol. Ther 213, 107587 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ou X et al. Characterization of spike glycoprotein of SARS-CoV-2 on virus entry and its immune cross-reactivity with SARS-CoV. Nat. Commun 11, 1620 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Smieszek SP, Przychodzen BP & Polymeropoulos MH Amantadine disrupts lysosomal gene expression: a hypothesis for COVID19 treatment. Int. J. Antimicrob. Agents 55, 106004 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhou Y et al. Protease inhibitors targeting coronavirus and filovirus entry. Antivir. Res 116, 76–84 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Simmons G et al. Inhibitors of cathepsin L prevent severe acute respiratory syndrome coronavirus entry. Proc. Natl Acad. Sci. USA 102, 11876–11881 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhao MM et al. Cathepsin L plays a key role in SARS-CoV-2 infection in humans and humanized mice and is a promising target for new drug development. Signal Transduct. Target Ther 6, 134 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Steuten K et al. Challenges for targeting SARS-CoV-2 proteases as a therapeutic strategy for COVID-19. ACS Infect. Dis 7, 1457–1468 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yadati T, Houben T, Bitorina A & Shiri-Sverdlov R The ins and outs of cathepsins: physiological function and role in disease management. Cells 9, 1679 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Konermann S et al. Transcriptome engineering with RNA-targeting type VI-D CRISPR effectors. Cell 173, 665–676.e14 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Birmingham A et al. 3′ UTR seed matches, but not overall identity, are associated with RNAi off-targets. Nat. Methods 3, 199–204 (2006). [DOI] [PubMed] [Google Scholar]
- 24.Sigoillot FD et al. A bioinformatics method identifies prominent off-targeted transcripts in RNAi screens. Nat. Methods 9, 363–366 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cox DBT et al. RNA editing with CRISPR–Cas13. Science 358, 1019–1027 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kranz LM et al. Systemic RNA delivery to dendritic cells exploits antiviral defence for cancer immunotherapy. Nature 534, 396–401 (2016). [DOI] [PubMed] [Google Scholar]
- 27.Cheng Q et al. Selective organ targeting (SORT) nanoparticles for tissue-specific mRNA delivery and CRISPR-Cas gene editing. Nat. Nanotechnol 15, 313–320 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mullard A FDA approves landmark RNAi drug. Nat. Rev. Drug Disco 17, 613 (2018). [DOI] [PubMed] [Google Scholar]
- 29.Eoh J & Gu L Biomaterials as vectors for the delivery of CRISPR–Cas9. Biomater. Sci 7, 1240–1261 (2019). [DOI] [PubMed] [Google Scholar]
- 30.Tong S, Moyo B, Lee CM, Leong K & Bao G Engineered materials for in vivo delivery of genome-editing machinery. Nat. Rev. Mater 4, 726–737 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yin H et al. Therapeutic genome editing by combined viral and non-viral delivery of CRISPR system components in vivo. Nat. Biotechnol 34, 328–333 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Oladunni FS et al. Lethality of SARS-CoV-2 infection in K18 human angiotensin-converting enzyme 2 transgenic mice. Nat. Commun 11, 6122 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zheng J et al. COVID-19 treatments and pathogenesis including anosmia in K18-hACE2 mice. Nature 589, 603–607 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yinda CK et al. K18-hACE2 mice develop respiratory disease resembling severe COVID-19. PLoS Pathog 17, e1009195 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Winkler ES et al. SARS-CoV-2 infection of human ACE2-transgenic mice causes severe lung inflammation and impaired function. Nat. Immunol 21, 1327–1335 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Arce VM & Costoya JA SARS-CoV-2 infection in K18-ACE2 transgenic mice replicates human pulmonary disease in COVID-19. Cell Mol. Immunol 18, 513–514 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Coperchini F, Chiovato L, Croce L, Magri F & Rotondi M The cytokine storm in COVID-19: an overview of the involvement of the chemokine/chemokine-receptor system. Cytokine Growth Factor Rev 53, 25–32 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Del Valle DM et al. An inflammatory cytokine signature predicts COVID-19 severity and survival. Nat. Med 26, 1636–1643 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chou SH et al. Global incidence of neurological manifestations among patients hospitalized with COVID-19—a report for the GCS-NeuroCOVID Consortium and the ENERGY Consortium. JAMA Netw. Open 4, e2112131 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Yang AC et al. Dysregulation of brain and choroid plexus cell types in severe COVID-19. Nature 595, 565–571 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Beers C et al. Cathepsin S controls MHC class II-mediated antigen presentation by epithelial cells in vivo. J. Immunol 174, 1205–1212 (2005). [DOI] [PubMed] [Google Scholar]
- 42.Shi GP et al. Cathepsin S required for normal MHC class II peptide loading and germinal center development. Immunity 10, 197–206 (1999). [DOI] [PubMed] [Google Scholar]
- 43.Honey K, Nakagawa T, Peters C & Rudensky A Cathepsin L regulates CD4+ T cell selection independently of its effect on invariant chain: a role in the generation of positively selecting peptide ligands. J. Exp. Med 195, 1349–1358 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Nakagawa T et al. Cathepsin L: critical role in Ii degradation and CD4 T cell selection in the thymus. Science 280, 450–453 (1998). [DOI] [PubMed] [Google Scholar]
- 45.Abbott TR et al. Development of CRISPR as an antiviral strategy to combat SARS-CoV-2 and influenza. Cell 181, 865–876.e12 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Blanchard EL et al. Treatment of influenza and SARS-CoV-2 infections via mRNA-encoded Cas13a in rodents. Nat. Biotechnol 39, 717–726 (2021). [DOI] [PubMed] [Google Scholar]
- 47.Lopez-Otin C & Bond JS Proteases: multifunctional enzymes in life and disease. J. Biol. Chem 283, 30433–30437 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Meganck RM & Baric RS Developing therapeutic approaches for twenty-first-century emerging infectious viral diseases. Nat. Med 27, 401–410 (2021). [DOI] [PubMed] [Google Scholar]
- 49.Meng B et al. Altered TMPRSS2 usage by SARS-CoV-2 Omicron impacts infectivity and fusogenicity. Nature 603, 706–714 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Baddeley HJE & Isalan M The application of CRISPR/Cas systems for antiviral therapy. Front Genome Ed 3, 745559 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen Z et al. Diverse AR-V7 cistromes in castration-resistant prostate cancer are governed by HoxB13. Proc. Natl Acad. Sci. USA 115, 6810–6815 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zeng C et al. Neutralizing antibody against SARS-CoV-2 spike in COVID-19 patients, health care workers, and convalescent plasma donors. JCI Insight 5, e743213 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li B, Luo X & Dong Y Effects of chemically modified messenger RNA on protein expression. Bioconjug Chem 27, 849–853 (2016). [DOI] [PubMed] [Google Scholar]
- 54.Li B, Zeng C & Dong Y Design and assessment of engineered CRISPR-Cpf1 and its use for genome editing. Nat. Protoc 13, 899–914 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Saunders KO et al. Neutralizing antibody vaccine for pandemic and pre-emergent coronaviruses. Nature 594, 553–559 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Source data for Figs. 1–6 and Extended Data Fig. 1–10 are provided with this paper. The RNA-seq data generated in this study have been deposited in the Gene Expression Omnibus database under accession number GSE203052. All the other data that support this study are provided in the Supplementary Information files. The LNP reagents are available from the corresponding authors upon reasonable request.