

Abstract
While DNA and RNA helices often adopt the canonical B- or A-conformation, the fluid conformational landscape of nucleic acids allows for many higher energy states to be sampled. One such state is the Z-conformation of nucleic acids, which is unique in that it is left-handed and has a “zig-zagged” backbone. The Z-conformation is recognized and stabilized by Z-DNA/RNA binding domains called Zα domains. We recently demonstrated that a wide range of RNAs can adopt partial Z-conformations termed “A-Z junctions” upon binding to Zα and that the formation of such conformations may be dependent upon both sequence and context. In this Chapter, we present general protocols for characterizing the binding of Zα domains to A-Z junction-forming RNAs for the purpose of determining the affinity and stoichiometry of interactions as well as the extent and location of Z-RNA formation.
Keywords: A-Z junctions, Zα domains, Circular Dichroism, Ez score, Isothermal Titration Calorimetry, Analytical Ultracentrifugation, Nuclear Magnetic Resonance, Z-RNA
1. Introduction
The Z-conformation is a unique higher-energy helical structure adopted by DNA and RNA that is left-handed and comprised of dinucleotide repeats termed “Z-steps”1–3. Although the Z-form of nucleic acids was originally discovered in 19793, understanding the role of Z-DNA/RNA in biology has been a slow process with many ongoing areas of research. Z-DNA and Z-RNA have been shown to play important roles in many biological processes including DNA replication, RNA transcription, splicing, RNA editing, and the innate immune response4–8. Most of these processes involve proteins containing one or more winged helix-turn-helix Zα domains which recognize Z-RNA/DNA by binding to and stabilizing Z-form structures9–13 that form within the context of larger nucleic acid sequences4–6,14.
Despite the diversity of DNA and RNA sequences that interact with Zα-containing proteins9,15, most biochemical and biophysical characterization of RNA formation has been limited to studies involving (CpG)n sequences, the reason being that the CpG dinucleotide repeat is the sequence most prone to adopt the Z-conformation1,16. Significantly more progress has been made in this area for Z-DNA than for Z-RNA, with several studies demonstrating Z-DNA formation and Zα binding in TpA, CpA, GpC, and TpG steps9,17. In addition, work on understanding the role of Z-DNA formation under torsional stress has demonstrated that Z-forming sequences within the context of larger DNA sequences form B-Z junctions, where the B-DNA regions flanking the Z-DNA site become destabilized and the nucleotides in-between the B- and Z-DNA flip out of the helix in order to preserve the continuous base stacking18–22. The formation of such junctions can be conveniently monitored using 1D-1H NMR upon addition of Zα, as demonstrated for a variety of B-Z junction-forming DNAs18,23–25.
We recently applied what was known about B-Z junction formation to investigate a similar phenomenon in ribosomal hairpins15 and a predicted Z-forming regions within an Alu foldback5 created from the pairing of an AluSx1 and an AluJo within the 3’ UTR of the CTSS gene26. Using biophysical techniques including Circular Dichroism (CD), Analytical Ultracentrifugation (AUC), Isothermal Titration Calorimetry (ITC), and Nuclear Magnetic Resonance (NMR), we characterized the binding of Zα and the formation of A-Z junctions in these RNAs. We showed that the sequence dependence of Z-RNA formation may be more complex than originally thought26. In this Methods Chapter, we describe in detail our pipeline for how to carry out and analyze these biophysical experiments so they can be applied to characterize other potential A-Z junction-forming RNAs (Fig. 1).
Figure 1. Pipeline for the biophysical characterization of Zα-dependent A-Z junction adoption in RNAs.
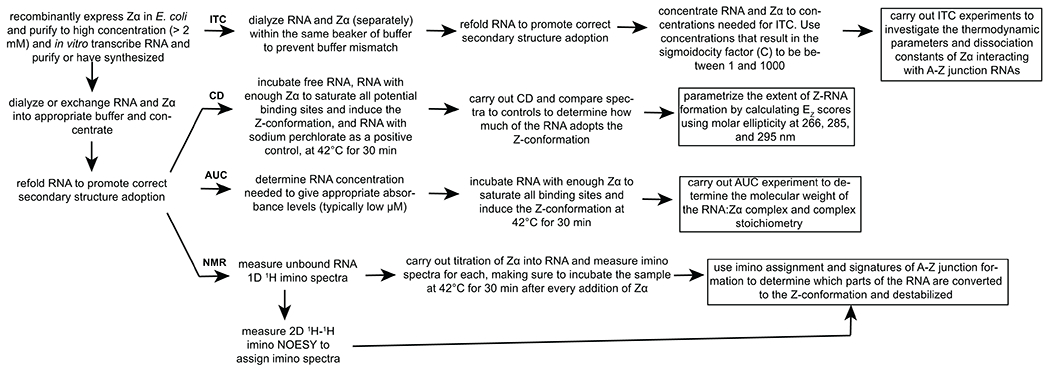
An overview of the steps required for characterization of A-Z junction adoption upon binding to Zα by Circular Dichroism (CD), Isothermal Titration Calorimetry (ITC), Analytical Ultracentrifugation (AUC), and Nuclear Magnetic Resonance (NMR) is shown.
2. Materials
2.1. General supplies needed.
Glass beakers (2 L are preferable).
500 mL and 1 L buffer bottles.
Magnetic stir bars.
1.5 mL microcentrifuge tubes.
0.2 mL PCR tubes.
15 and 50 mL conical tubes.
Micropipettes and micropipette tips.
Gel loading tips (not required but useful for loading samples into quartz cuvettes for CD and sample cells for AUC).
Potassium phosphate monobasic (KH2PO4).
Potassium phosphate dibasic (KHPO4).
Sodium chloride (NaCl).
Ethylenediaminetetraacetic acid (EDTA, [CH2N(CH2CO2H)2]2)
1,4-Dithiothreitol (DTT, C4H10O2S2).
Sodium perchlorate (NaClO4).
Heat block or water bath able to achieve 95°C and 42°C.
Tabletop microcentrifuge and swing-bucket rotor centrifuge (or any centrifuge that can spin 50 mL conicals).
3 kDa cutoff dialysis membrane or 2 kDa cutoff Slide-A-Lyzer cassettes (ThermoFisher, MA).
3 kDa cutoff Amicon Ultra-15 and Ultra-4 centrifugal filter units (MilliporeSigma, MO).
Nano UV-Vis spectrometer.
2.2. Circular Dichroism.
Access to a CD spectrometer (ours was a Jasco J-815 CD Spectropolarimeter, Jasco, RI).
Sample temperature controller (ours was a Jasco PTC-423L Peltier controller, Artisan Technology Group, IL).
Software for controlling CD spectrometer (we used Spectra Manager version 2, Jasco, RI).
Quartz cuvettes (Jasco, RI). We used 1 mm path length for our studies.
Single cuvette washer (Fireflysci Type P65S, ThermoFisher, MA).
Microsoft Excel or other data analysis software
2.3. Isothermal Titration Calorimetry.
Access to an Isothermal Titration Calorimeter (ours was a Malvern ITC200, Malvern, PA).
Software for controlling ITC machine (ours was ITC200 version 1.26.1, Malvern, PA).
Sample cell syringe (Malvern Panalytical INC ITC200 Syringe Filling, Malvern, PA).
Analysis software (we used Microcal Analysis version 7 SR4, Origin, MA)
2.4. Analytical Ultracentrifugation.
Access to an Analytical Ultracentrifugation instrument (ours was a XL-I Beckman Coulter, Beckman, CA).
An-60 Ti analytical 4-place titanium rotor (Beckman Coutler, CA).
Standard 12 mm EPON centerpieces with quartz windows (Beckman Coulter, CA).
Software for controlling AUC machine (we used ProteomeLab version 6.0, Beckman Coulter, CA).
Method for calculating buffer density. Can be readily done using SEDNTERP version 3 (by John S. Philo at Alliance Protein Laboratories, CA).
Software for analyzing AUC data (we used SEDFIT version 14.7g, NIH)
2.5. Nuclear Magnetic Resonance.
Access to a high-field NMR magnet with 1H/13C/15N cryoprobe and associated equipment (Bruker or Varian).
5 mm spinner for sample insertion (Bruker or Varian).
Regular 5 mm NMR tube or Shigemi tubes (Wilmad-LabGlass , IL).
Software for controlling NMR console and data analysis. We used VNMRJ version 4.2 Revision A (Agilent, CA) for collecting data from Varian spectrometers and TopSpin version 4.0.7 (Bruker, MA) for Bruker.
3. Methods
3.1. Acquiring and preparing protein Zα samples.
The recombinant expression and purification of the Zα protein have been covered thoroughly elsewhere12,26,27 and will not be discussed here. Once the Zα protein has been purified, it can be checked for its Z-DNA/RNA binding/stabilizing activity by incubating it with r/d(CpG)n repeats using CD (as discussed below). Note 1.
3.2. Preparation of RNA and RNA:Zα complexes.
We will not cover these methods here, but RNAs can either be produced in-house through in vitro T7 transcription28–32 or by solid-phase synthesis33 and purified through preparative denaturing polyacrylamide gel electrophoresis32,34 (we usually used 20% polyacrylamide as most of the A-Z junction RNAs we tested were ~30 nts or less) or chromatography35,36. Because RNAs comprising a few dozen nucleotides at most may be difficult to in vitro transcribe, they may more conveniently be obtained by chemical synthesis, which can be carried out for example by Integrated DNA Technologies (IDT) or Horizon Discovery (formerly known as Dharmacon). Note 2.
Prepare anion exchange binding and equilibration buffers. Binding/wash buffer: 20 mM potassium phosphate (pH 6.4), 150 mM NaCl, 0.2 mM EDTA. Elution buffer: 20 mM potassium phosphate (pH 6.4), 2000 mM NaCl, 0.2 mM EDTA.
Equilibrate the DEAE-sepharose into anion exchange binding buffer by pumping at least two column volumes (CV) of buffer through the column. A ~20 mL column should be more than sufficient. We purchase the pack of five 5 mL DEAE columns from MilliporeSigma and attach them together.
Inject the RNA sample onto the column and wash with at least 3 CVs of wash buffer.
Elute the RNA using elution buffer and monitor the ultraviolet signal at 260 nm (preferred) or 280 nm to determine when all of the RNA has been collected.
Prepare 20 mM potassium phosphate (pH 6.4), 25 mM NaCl, 0.5 mM EDTA, and 1mM DTT for NMR or 20 mM potassium phosphate (pH 7.0), 25 mM NaCl, 0.5 mM EDTA, and 1 mM DTT for all other measurements (Note 3 and 4).
Dialyze RNA and Zα protein into the correct buffer. This can be done either by standard dialysis through dialysis membrane (SpectraPOR) in a 2 L beaker with buffer, or by buffer exchange using Amicon centricons (MilliporeSigma). For centricon dialysis, running about 50 mL of buffer is usually sufficient to effectively dialyze the sample. Keep everything at 4°C prior to measurement. Note 5.
Concentrate RNA and Zα using 3kDa Amicon centrifugal filter units (MilliporeSigma) to concentrations required for subsequent experiments (concentrations can be determined using a nanodrop or other methods). For NMR, aim for concentrations in excess of 1 mM in order to minimize dilution of the sample upon addition of titrant. RNA and Zα can be frozen at this point at −20°C (−80°C preferred) for long term storage.
Heat-anneal RNAs by incubating them at 95°C for 5 min followed by cooling on ice for 20 min (or the RNA can be cooled slowly at room temperature for 45–60 min). Note 6 and 7.
Combine Zα with RNA at proper concentrations and molar ratios for planned experiment (see below for details depending on which experiment is being performed).
Important: For any equilibrium measurement, such as NMR, CD, or AUC, incubate Zα with the RNA at 42°C for at least 30 min before measuring to ensure complete conversion of the RNA to the Z-conformation. See Note 8.
3.3. Circular Dichroism for quantification of Z-form.
The concentration of RNA needed to acquire good signal-to-noise for CD will depend on both the size of the RNA (longer RNAs have a higher absorbance), the type of cuvette being used (larger cuvettes will have a longer path length and thus require more volume but less concentration), and the sensitivity of the spectrometer. See Note 9.
There are two ways to carry out the CD titration. To conserve the RNA sample, the Zα stock can be concentrated to high levels so that the dilution of the RNA is minimal and can be titrated into the RNA sample for each titration point. Note that this method requires that the sample be incubated at 42°C for 30 min following each titration. The other method is to make a separate sample for each titration point (which is what we usually do).
Calculate out the volume needed from the concentrated RNA stock to reach 50 μM RNA in 175 μL of 20 mM potassium phosphate (pH 7.0), 25 mM NaCl, 0.5 mM EDTA, 1 mM DTT. Then, calculate how much volume from the Zα stock is needed to reach the desired molar ratios for each of the titration points. For example, if we were going to measure five different points (free RNA, 6M sodium perchlorate, 6:1, 1:1, and 1:6 RNA:Zα), and the RNA was 1 mM and Zα stock was 2 mM, then (Table 1) would describe the different volumes needed from each. Also prepare the proper controls needed for proper interpretation of the data (Note 10).
Incubate the samples at 42°C for 30 min. While waiting, turn on the nitrogen gas to the instrument followed by starting the Jasco J-815 CD spectropolarimeter (Jasco), as well as the Jasco PTC-423L Peltier controller (Artisan Technology Group) and start equilibrating the sample cell holder to 25°C. The temperature will also need to be set within Spectra Manager version 2 (Jasco).
Set up the spectral measurement parameters within Spectral Manager. The measurement range should be set to be 320 nm to 220 nm (the range which reports on the secondary structure of nucleic acids), the step size to 1 nm, and the number of scans to at least two.
Once the samples have incubated, pipette (using gel loading pipettes here is helpful) the entire 175 μL of the first sample to be measured into a 1 mm quartz cuvette, making sure that the sample settles all the way to the bottom of the cuvette and that there are no bubbles.
Insert the cuvette containing the sample into the sample holder and begin the measurement. Once the collection is complete, transfer the absorbance versus wavelength data into an excel sheet or other data analysis software.
Wash the cuvette with buffer (or water, but buffer is preferred) two times, followed by ethanol once, and then with water four times before the next measurement. Remove as much of the residual water as possible before preceding to the next measurement. It is useful to have a cuvette washer (ThermoFisher) for this step.
Repeat for the remaining samples. Export all the data into an excel sheet.
- Convert the CD absorbance molar ellipticity using the following equation:
where [Θ] is the molar ellipticity, Θobs is the measured raw ellipticity in mdeg, M is the molecular weight of the molecule in gmol−1, l is the path length of the cell in cm, and c is the concentration in g.L−1. Plot the molar ellipticity of the different measurements on the y-axis versus the wavelength in nm on the x-axis to obtain the final result (Fig. 2a).
Table 1.
Example setup for circular dichroism titration of RNA with Zα.
| CD experiment / titration point | RNA stock (1 mM) | Zα stock (2 mM) | Buffer (20 mM potassium phosphate (pH 6.4), 25 mM NaCl, 0.5 mM EDTA, 1mM DTT) | Sodium perchlorate (8 M) |
|---|---|---|---|---|
| Buffer control | 175 μL | |||
| free RNA | 8.75 μL | 166.25 μL | ||
| free Zα control | 26.25 μL | 148.75 μL | ||
| Z-RNA control | 8.75 μL | 35 μL | 131.25 μL | |
| 6:1 RNA:Zα | 8.75 μL | 0.73 μL | 165.52 μL | |
| 1:1 RNA:Zα | 8.75 μL | 4.38 μL | 153.12 μL | |
| 1:6 RNA:Zα | 8.75 μL | 26.25 μL | 140 μL |
Figure 2. Circular Dichroism to investigate A-Z junction adoption and determine the extent of Z-RNA formation.
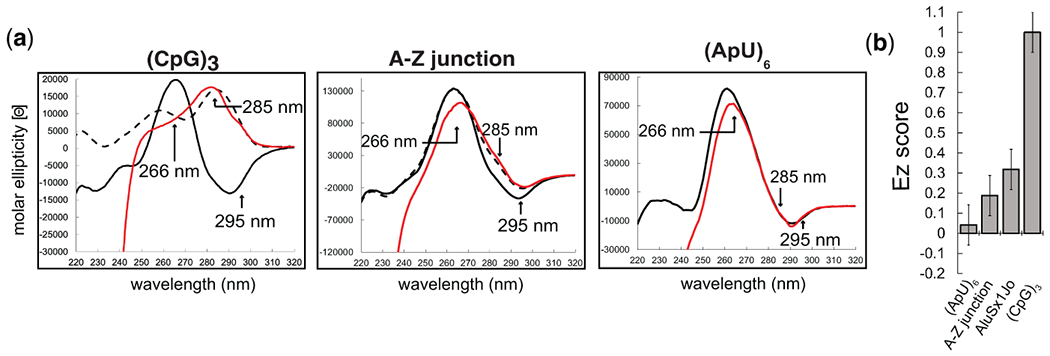
(a) CD spectra of the (CpG)3, an A-Z junction positive control, and an (ApU)6 negative control RNA in the absence of protein (black), with 6 M sodium perchlorate (dotted black line), and with a molar ratio of 1:6 RNA:Zα (red) at which binding is saturated. (b) Extent of Z-RNA (EZ) scores quantifying the extent of Z-conformation for the (ApU)6, A-Z junction control, AluSx1Jo, and (CpG)3 RNAs.
The CD results can be used to judge how much of an RNA adopts the Z-conformation when bound by Zα. For RNAs that fully adopt the Z-form, such as (CpG)n repeat sequences, the CD spectra upon being saturated with Zα are characterized by a complete shift of the peak from ~266 nm to ~285 nm (Fig. 2a, left). In contrast, RNAs that only adopt a partial Z-conformation, such as the A-Z junction control (which is a sequence that contains a (CpG)6 followed by an A-forming region), are characterized by a population-weighted shift in the CD absorbance towards the Z-form values (Fig. 2a, middle), compared to an (ApU)6 RNA which has a minor dip at 266 nm but otherwise no growth at 285 and 295 nm (Fig. 2a, right). To obtain a quantitative description of this shift, we parametrize the different wavelengths that inform on Z-RNA formation as described in section 3.4.
3.4. Calculation of Ez-scores from CD data to determine extent of Z-RNA formation.
Certain wavelengths within CD spectra inform on whether the RNA is in the A- or Z-form and therefore can be tracked upon addition of Zα to determine how much of an RNA adopts the Z-conformation. In particular, a decrease in the positive molar ellipticity centered around ~266 nm (which is indicative of the A-form) and a growth at 285 and 295 nm report on formation of Z-RNA11,37 (Fig. 2a). By parametrizing the change in the molar ellipticity at these wavelengths, we calculate what we have termed as an “EZ score” which gives a single number that is useful for comparing different Z-adopting RNAs to each other (Fig. 2b)26.
Take the molar ellipticity values at 266, 285, and 295 nm for both the free form and the fully saturated complex (usually 1:6 RNA:Zα) and paste them into a new column as shown (Table 2, example taken from (CpG)3).
- Next, the decay of the molar ellipticity at 266 nm is calculated using the following equation:
where decay266 is the decay of the molar ellipticity at 266 nm, Intfree266 is the CD signal of the free RNA at 266 nm, and Intbound266 is the CD signal of the bound RNA at 266 nm. -
The growth at 285 and 295 nm is then calculated as:
where growth285/growth295 is the growth of the molar ellipticity at 285 and 295 nm, respectively, Intfree266 is the CD signal of the free RNA at 266 nm, Intbound266 is the CD signal of the bound RNA at 266 nm, Intfree285/Intfree295 are the CD signals of the free RNA at 295 and 295 nm respectively, and Intbound285/Intbound295 are the CD signals of the bound RNA at 295 and 295 nm, respectively.Since the CD signal will vary for different RNA sequence contexts, it is important to always normalize the growth/decay to the signal of the free form, which is why each growth/decays are divided by Intfree266.
Next, the growth/decays are calibrated to the control (CpG)3 RNA since this RNA fully adopts the Z-conformation when bound by Zα. This means that we take the growths/decays at the three wavelengths and multiply them by a constant that makes them equal to the number 1 for the (CpG)3 case. We then use the same constants for the other tested RNAs.
Multiply the decay266 by 1.11, the growth285 by 0.718 and the growth295 by 1.80 (which are the values determined empirically to calibrate the EZ score to the (CpG)3 RNA). See Table 3 for an example.
The final Ez score of the RNA is the average of the three calibrate growth/decay values. Example A-Z scores from an (ApU)6 negative control, A-Z junction positive control, AluSx1Jo foldback, and (CpG)3 positive control RNA are shown (Fig. 2b).
Table 2.
Example of excel sheet containing final data from CD titration.
| Wavelength (nm) | [Θ] (free RNA) | [Θ] (saturated RNA) | growth/decay of [Θ] | calibrated growth/decay |
|---|---|---|---|---|
| 295 | −10799.72 | 7059.54 | ||
| 285 | −10834.9 | 16748.64 | ||
| 266 | 19807.86 | 8806.2 |
Table 3.
Example of excel sheet with calculated growth/decays of titration data.
| Wavelength (nm) | [Θ] (free RNA) | [Θ] (saturated RNA) | Growth/decay of [Θ] | calibrated growth/decay |
|---|---|---|---|---|
| 295 | −10799.72 | 7059.54 | 0.90162491 | 1 |
| 285 | −10834.9 | 16748.64 | 1.39255528 | 1 |
| 266 | 19807.86 | 8806.2 | 0.55541891 | 1 |
3.5. Isothermal Titration Calorimetry to investigate affinity and thermodynamics of binding.
The optimal amount of macromolecule in the cell usually needs to be determined to acquire quality results and a sigmoidal binding curve. Ideally, we want the sigmoidicity factor (C) to be between 10 and 1000, which can be determined through the following equation:
where N is the stoichiometry, [M]T is the biomolecule concentration in the ITC cell, and KD is the dissociation constant of the interaction.
For example, with an injection of Zα into (CpG)3 RNA at a concentration 50 μM:
While the KD of the interaction between Zα and an RNA may be unknown, an initial pilot experiment can be carried out assuming a KD similar to the literature values and then adjusting the concentration from there. The stoichiometry is also usually an unknown but can be determined readily by AUC methods as described below.
Aim for a titrant concentration of ~10x the concentration of the biomolecule in the cell, so if the cell concentration is 50 μM, then the concentration of titrant in the syringe should be 500 μM. Due to the often many Zα binding sites present on some A-Z junction forming RNAs, it is not uncommon for the binding curve to be unsaturated by the end of the experiment. If this happens, it is possible to reload the syringe with additional titrant and carry out another injection series and then concatenate the two data sets together (Microcal Concat ITC software, Microcal). Another option is to use a higher concentration of titrant and do smaller volumes per injection. Keep in mind that the volumes and best practices will depend on the type of ITC instrument used. In our case, we used a Malvern ITC200 (Malvern).
Dialyze Zα and the RNA(s) together (within separate 3 kDa cutoff dialysis bags (SpectrPor) or Slide-a-Lyzers (ThermoFisher) so that the components do not mix) overnight at 4°C in 2 L of 20 mM potassium phosphate, 25 mM NaCl, 0.5 mM EDTA. For ITC, avoid DTT as it can cause erratic baselines (2-Mercaptoethanol (BME) can be used as a replacement). Make sure to dialyze enough Zα and RNA to reach the concentrations in the volumes required for ITC (Note 11). For example, 50 μM of RNA in > 280 μL (for the sample cell) and > 40 μL of 500 μM Zα (for the injection syringe).
Concentrate Zα and the RNA(s) using 3 kDa cutoff Amicon centrifugal filters to concentrations required for ITC. Make sure to save some buffer from the 2 L dialysis for dilutions and for the buffer control titration (ligand into buffer).
Make sure the sample cell is thoroughly washed with buffer before measurement. Use the sample cell syringe to completely wash the cell with at least 3x with buffer before loading the ~280 μL of RNA into the cell. Avoid introducing bubbles.
Wash the injection syringe with 3x ~40 μL of buffer and then load the 40 μL of 500 μM Zα titrant.
Prepare the experimental settings in ITC200 version 1.26.1 by setting the sample cell reference temperature at 25°C, the stirring speed at 750 rpm, the reference power to 10 μcals−1, and inputting the sample cell and syringe concentrations. Edit the injection parameters so that there are 19 total injections of 2 μL and one injection (the first one in the list) with a volume of 0.4 μL. Set the initial delay to 6 s and the spacing between each injection to be 180 s.
Run the experiment to collect the ITC thermogram.
The ITC data can be fit and analyzed using Microcal Analysis version 7 SR4 (Origin). Load the data into the software and remove the first point from the data (the 0.4 μL injection). Next, perform a baseline correction so that the ΔH for the points at the end of the titration (when the RNA is saturated with Zα) are close to zero.
Choose a binding model (whether it is one-site, two-site, etc) and fit the data to extract the ΔH, ΔS, ΔG, stoichiometry, and KD. The appropriate model to choose depends on the complexity of the interaction. The (CpG)3 RNA for example has two binding sites for Zα but they are equivalent, and therefore the data fits best to a one-site model. When we measured ITC for the AluSx1Jo RNA26, there were two non-equivalent binding sites and therefore a two-site model was more appropriate for that case. More details about fitting and analysis of ITC data can be found here38.
Example ITC thermograms are shown for the (CpG)3 RNA as well as two A-Z junction-forming RNAs, AluSx1Jo, and h41 E. coli (Fig. 3a) together with fitted binding parameters (Fig. 3b). Binding of Zα to the (CpG)3 RNA is characterized by single-site exothermic binding in the nanomolar range suggesting that the two binding sites for Zα are equivalent and that the overall reaction is favorable (Fig. 3a, left). In contrast, Zα binding to the AluSx1Jo and h41 E. coli RNAs are characterized by two-site binding in the nanomolar-micromolar range, with both events being endothermic for the h41 E. coli RNA and the first binding event being endothermic for the AluSx1Jo RNA and the second being exothermic (Fig. 3a, middle, right). These data suggest that the adoption of A-Z junctions by binding to Zα is a complex process relative to the adoption of Z-RNA in the (CpG)3 repeat, involving multiple binding events and opposing thermodynamic processes. The nature of these processes can be investigated further through higher-resolution techniques such as NMR (discussed below), see Note 12.
Figure 3. Isothermal Titration Calorimetry to characterize the thermodynamics of A-Z junction formation by Zα binding.
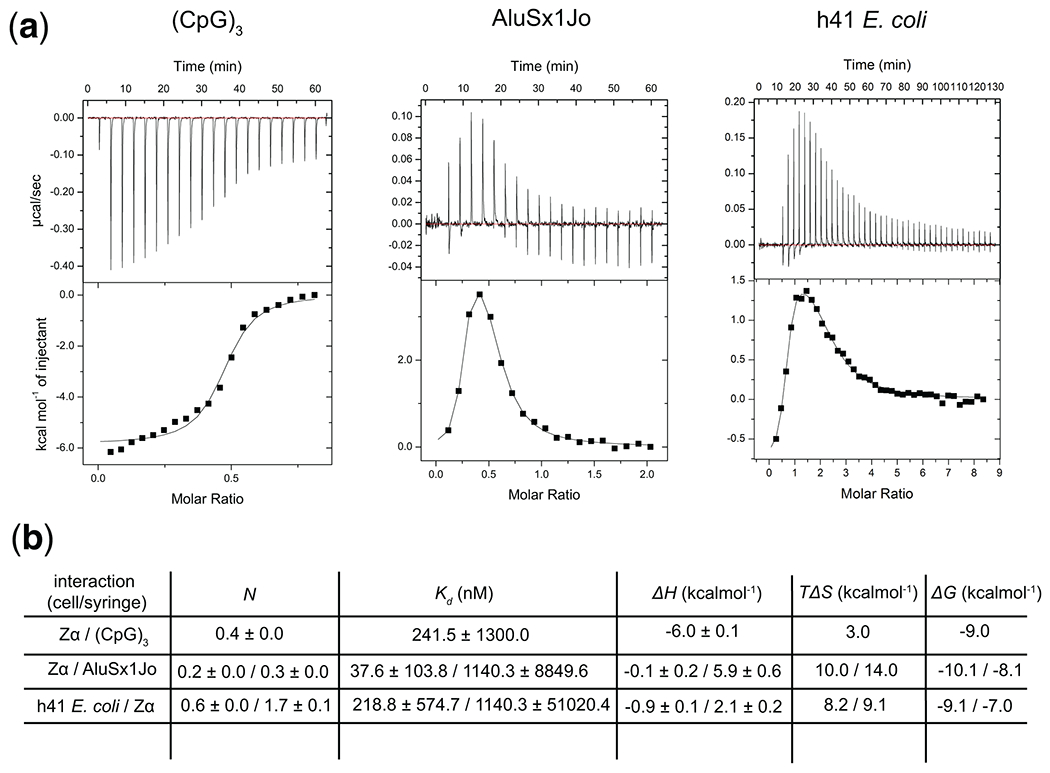
(a) ITC thermograms and fits from titrating Zα into the (CpG)3 and AluSx1Jo, and h41 E. coli RNAs. (b) Fitted thermodynamic parameters for the three titrations.
3.6. Sedimentation velocity Analytical Ultracentrifugation of Z-conformation-containing RNA/DNAs bound to Zα.
AUC is a powerful and versatile method for the quantitative analysis of macromolecules in solution39. While the setup is tedious and low throughput, it can provide unrivaled information about the stoichiometry of Zα:RNA complexes. AUC measures the absorbance of the sample as it is spun at high speeds, and so it is important not to use buffers which absorb in the UV range. Since the absorbance of RNA is significantly higher than Zα in the majority of cases, the sedimentation of the RNA can be tracked without interference from free protein.
Estimate the concentration of RNA needed to perform the AUC experiment. On our AUC instrument (XL-I Beckman Coulter, Beckman), the ideal absorbance at 260 nm is 0.8-1.2 so that the detector is not saturated before and during the run after the sample starts to sediment. An RNA concentration of 2 μM was usually sufficient on our instrument, but this is highly dependent upon the length of the RNA. Prepare enough RNA in 20 mM potassium phosphate (pH 7.0), 25 mM NaCl, 0.5 mM EDTA, 1 mm DDT at the concentration needed to fill the total volume of the AUC cell which is ~420 μL. Make sure to also bring enough buffer (20 mM potassium phosphate (pH 7.0), 25 mM NaCl, 0.5 mM EDTA, 1 mM DTT) to fill the reference cell.
Refold the RNA as mentioned in section 3.2.
Add to the RNA sample enough Zα from the concentrated stock to reach a molar ratio of 1:6 RNA:Zα. If more than 6 binding sites are predicted, adjust the molar ratio to be above the total number of sites. For example, if the RNA concentration is 2 μM, then add 12 μM Zα (which would be μL of 2 M Zα into a 500 μL volume).
Incubate the sample at 42°C for 30 min. During the incubation, proceed to step 5 (assembly of the AUC cells).
Assemble the Standard 12 mm EPON centerpieces with quartz windows (Beckman Coulter) and load the sample (this can be done with a gel loading pipette or syringe). A good reference for how to do this can be found here40.
Insert the sample cells (including the buffer only cell) into the An-60 Ti Analytical 4-Place titanium rotor (Beckman Coutler) and measure one scan at 3000 rpm to check for leaks and determine whether the absorbance is at an acceptable level. A leak is easily spotted by an absorbance versus radius plot that tails off too quickly (the absorbance should stay level from a radius of ~7 to ~6 cm. If you notice it dip to zero at values significantly before 6 cm, there is a leak and the cell will need to be rebuilt). See Note 13. If no leaking is detected and the absorbance falls within the expected range, move onto step 7.
Begin pulling the vacuum and wait for it to drop below 100 microns (13.3 Pa). After this point, set the temperature to 25°C and make sure that the temperature is fully equilibrated before starting the run. Note that this process can take several hours, so the equilibration step can be performed overnight with a scheduled sample run the following morning.
Setup the experimental parameters. The rotor speed should be set to 50,000 rpm (this is the recommended rpm for complexes between 30 and 300 kDa, which is the range most Zα:RNA complexes will be within). The measurement time for a sedimentation velocity experiment should be 2–12 hrs depending on the protein size. Zα:RNA complexes are fairly small (ranging from ~18 kDa to 100 kDa, but the complexes we studied were usually below 50 kDa) so we generally plan for longer run times to ensure full sedimentation of the sample. See Note 14.
Once all parameters have been setup, the experiment can be run.
-
Calculate the buffer density using SEDNTERP version 3 (by John S. Philo at Alliance Protein Laboratories). SEDNTERP will determine the density and/or viscosity of a buffer to be calculated after entering the buffer composition.
Since the partial-specific volume of RNA and protein is different, it may be necessary to calculate an average value corresponding to the composition of the Zα:RNA complex (See Note 15).
Load the scans from the sedimentation velocity runs and fit the data using SEDFIT version 14.7g, NIH. Resources for how to do this can be found on the SEDFIT website (https://sedfitsedphat.nibib.nih.gov/software/default.aspx) or here (http://www.analyticalultracentrifugation.com/sedfit.htm). Remember to input the buffer density calculated from SEDNTERB and the partial-specific volume calculated from percentage of Zα and RNA in the complex.
Once the data is fit, compare the measured molecular weight to the theoretical molecular weight from different Zα:RNA complexes to determine the stoichiometry. For example, the major peak measured from AUC for the (CpG)3 RNA bound to Zα was 19.6 kDa confirming the 2:1 complex with a theoretical molecular weight of 18.8 kDa (Fig. 4). If the fitted molecular weight suggests a different complex stoichiometry than was assumed for the calculation of the partial-specific volume, recalculate it using the new data and redo the analysis in SEDFIT.
Figure 4. AUC of heat annealed and unannealed (CpG)3 RNA bound to Zα.
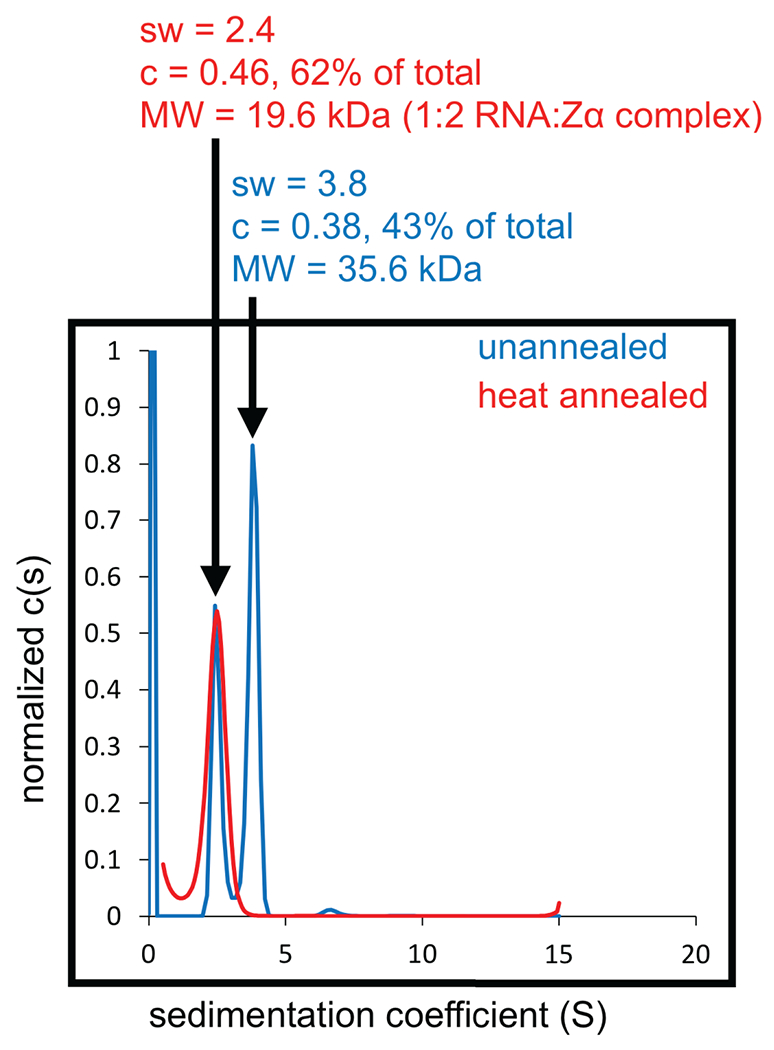
Sedimentation coefficient distributions obtained by AUC for the (CpG)3 RNA with a molar ratio of 1:6 RNA:Zα. The blue and red plots show the distributions acquired when the (CpG)3 RNA was not heat annealed and heat annealed prior to measurement, respectively.
The same process can be used to investigate the stoichiometry of any Zα:RNA complex amenable to AUC, and can be particularity useful when attempting to determine the stoichiometry of Zα:RNA complexes involving complex sequence contexts where the binding sites may not be obvious. Example AUC data from Zα binding to the (CpG)3, an A-Z junction control, and AluSx1Jo RNA are shown (Fig. 5a). The expected molecular weight of the Zα:RNA complexes versus the measure molecular weight is indicated (Fig. 5b).
Figure 5. Analytical Ultracentrifugation to determine the stoichiometry of A-Z junction:Zα complexes.
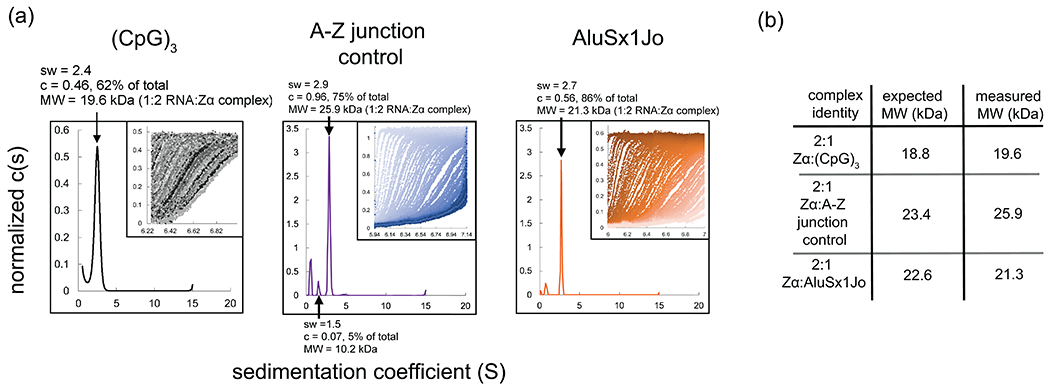
(a) Sedimentation coefficient distributions obtained by AUC for (CpG)3, A-Z junction control, and AluSx1Jo RNA. (b) The insets show the raw data from the AUC run with the window position on the x-axis and the absorbance on the y-axis, and individual scans over time going from left to right. (b) Predicted molecular weights for the different Zα:RNA complexes and measured molecular weights from AUC.
3.7. Nuclear Magnetic Resonance to monitor Zα-dependent switch from A- to Z-form.
While CD, ITC, and AUC can be used to determine whether a RNA adopts a Z-conformation, the stoichiometry, and thermodynamics of binding, relatively simple NMR experiments can be used to determine exactly what part of an RNA is adopting the Z-conformation in addition to the adjacent regions which are destabilized to accommodate A-Z junction formation26. For these experiments, we rely heavily on the foundational NMR characterization of B-Z junctions carried out previously which demonstrated specific NMR signatures for Z-DNA formation and junction destabilization18,25. Specifically, it was shown that at early titration points, specific imino peaks within a 1D-1H spectra would begin to disappear into the noise indicating destabilization of the base pairs adjacent to the Z-DNA forming region18,25. This was then followed by chemical shift perturbations (CSPs) and line broadening at later titration points indicating Z-DNA formation18,25. We observed similar signatures when titrating Zα into our test ribosomal hairpins and AluSx1Jo RNA foldback fragment26, suggesting a conformational process was occurring from A- to Z-form. We were then able to determine which regions within the secondary structure of the RNAs were being bound by Zα along with the neighboring destabilized regions26.
Access to a high field solution-state NMR spectrometer is needed, preferably with a 1H/13C/15N cryoprobe and associated equipment (Bruker or Varian).
Prepare the RNA sample at a reasonable concentration in 300 μL (for Shigemi tubes, Wilmad-LabGlass) or 500 μL (for 5 mm NMR tubes, Wilmad-LabGlass). For assignment of the imino peaks, a sample concentration of at least 500 μM is recommended to acquire enough signal-to-noise to observe all the cross peaks in the 2D [1H,1H]-NOESY spectra.
Concentrate Zα to at least 2 mM (the higher the better to limit dilution of the RNA sample upon titration) if it is not already so.
Measure a 2D [1H,1H]-NOESY making sure to not use a water suppression method that saturates the water signal as the imino proton signal will also be killed. W5, WATERGATE, or flip-back water suppression schemes are what we usually use. Make sure that the spectral widths are set so that they that will cover the entire imino spectrum range, which is about 9–16 ppm. The NOESY mixing time is dependent upon the size of the RNA, but generally the range of 160 to 320 ms is appropriate. A detailed description for measuring this experiment can be found in reference #41.
The 2D [1H,1H]-NOESY data can be processed using NMRpipe42 (a detailed tutorial for how to do this can be found here: https://spin.niddk.nih.gov/NMRPipe/doc1/) and the cross-peaks between imino peaks can be used to assign the base-pairing pattern of the RNA through what is known as a “NOESY walk”41 (Fig. 6a). Note that less stable parts of the RNA helix such as terminal and non-canonical base pairs exchange with water on a faster timescale than stable ones and therefore will have less signal. The signal of such base pairs may be improved by decreasing the temperature and increasing the concentration.
Once the imino peaks have been assigned, they can be used to track A-Z junction formation upon binding to Zα through an 1D imino titration (Fig. 6b). Since only 1D 1H spectra are needed for this, the concentration of the RNA can be decreased to ~100 μM, but we recommend using a 5 mm tube to limit volume loss during the titration (some volume is lost each time the plunger is removed from the Shigemi tube).
Measure a 1D 1H imino spectrum of the free RNA at the new concentration. Again, make sure to use a W5, WATERGATE, or flip-back water suppression scheme and that the spectral width is large enough to cover the entire imino proton region.
The choice of titration points is somewhat subjective (make sure to choose a good range of RNA:Zα ratios), but we do recommend including these ratios: free, 4:1, 2:1, 1:1. 1:2, and 1:4 RNA:Zα. Titrate Zα into the RNA to reach the selected molar ratio, making sure to incubate the sample at 42°C for 30 min afterwards. Start with the largest ratio of RNA:Zα first (for example, 8:1 RNA:Zα) and move downwards to the smallest ratio.
Continue the process until 1D 1H imino spectra are measured for each titration point.
Load the 1D 1H imino spectra data into a data analysis software such as TopSpin version 4.0.7 (Bruker), VNMRJ version 4.2 Revision A (Agilent), NMRpipe42, or others, to analyze the data.
Figure 6. Nuclear Magnetic Resonance to determine regions adopting A-Z junctions, Z-conformation, or being destabilized by Zα binding.
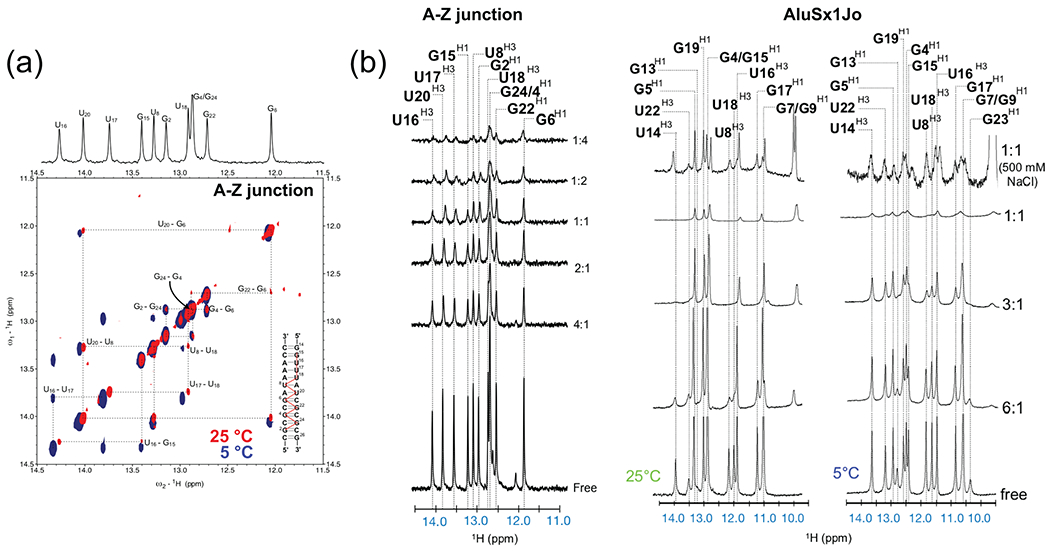
(a) The imino regions of the 2D [1H,1H]-NOESY spectrum with a mixing time of 300 ms for the A-Z junction control RNA is shown. Imino proton connectivities (the “NOESY walk”) and assignments are shown with dashed lines and illustrated as red lines on the 2D secondary structure of the A-Z control. (b) The imino regions of the 1D 1H titration are shown for the A-Z RNA control and AluSx1Jo RNAs. Imino proton assignments are indicated with dashed lines. The ratio of RNA:Zα is indicated on the right-hand side of each trace.
At early titration points, the base pairs adjacent to the Z-forming region become destabilized to allow the adoption of the A-Z junction conformation. Due to this, they disappear into the noise with increasing ratios of Zα (Fig. 6b). At later titration points (1:1, 1:2, 1:4, RNA:Zα, etc) chemical shift perturbations and line broadening of the imino peaks are observed indicating Z-RNA formation. By using the assignment of the imino peaks, these changes can be directly correlated to the RNA 2D structure and the regions which adopt the Z-conformation and regions which are destabilized can be determined26.
Figure 7.
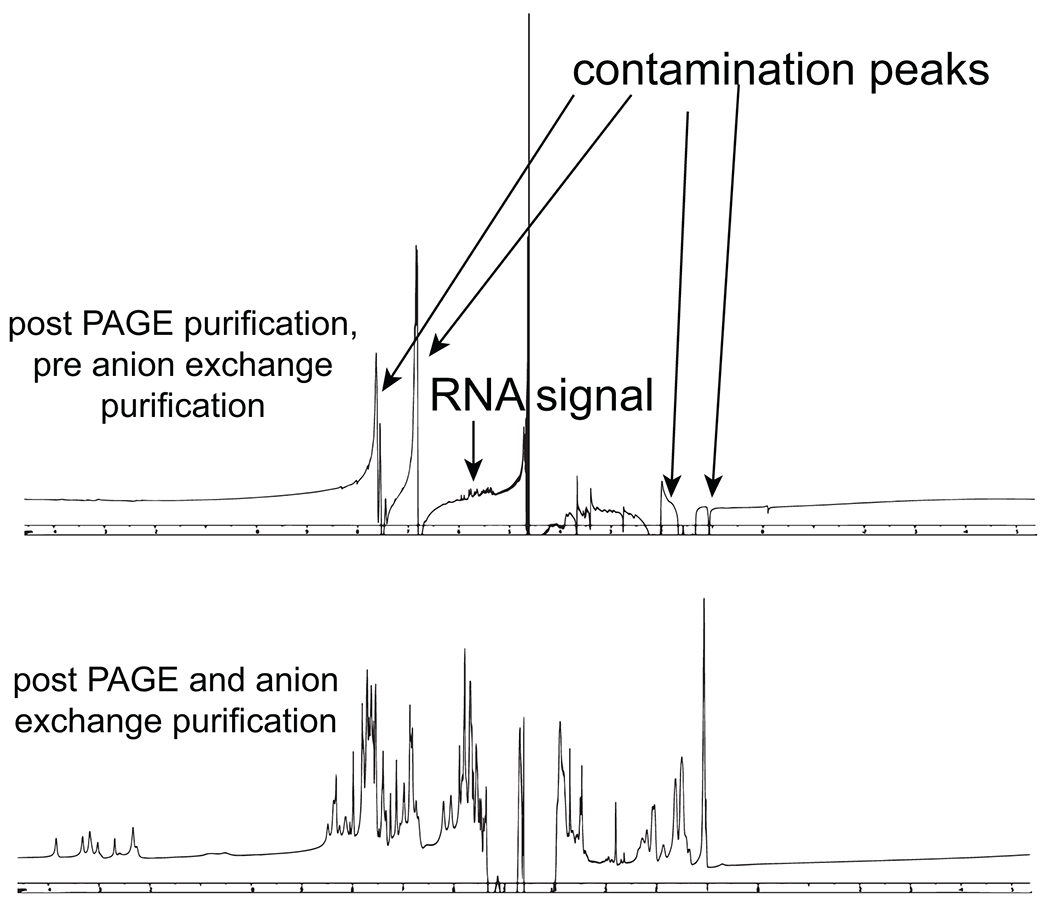
1D NMR spectra from RNA post denaturing PAGE purification and post denaturing PAGE and anion exchange chromatography purification.
4. Notes
For the most part, Zα domains are stable and therefore relatively forgiving proteins to work with. In our hands, the Zα domain from human ADAR1 (recombinantly expressed in BL21(DE3) E. coli) can be stored in the freezer in low salt buffers (we usually use 20 mM potassium phosphate at pH 6.4, 25 mM NaCl) at high concentration (ZαADAR1 can be concentrated past 10 mM if desired) for long periods of time (~ a year) without significant degradation. In addition, freeze-thaw cycles do not appear to degrade ZαADAR1 or alter its structure or function (as judged by CD and NMR). ZαADAR1 alone and bound to RNA is stable at room temperature for several weeks (as monitored by NMR) but it will begin to degrade after 3–6 months if kept unfrozen. Owing to the high sequence and structural homology of Zα domains, it is likely that the observed properties of ZαADAR1 apply to other Zα domains from other species and proteins, but this should not be assumed.
Purifying RNA by denaturing PAGE often involves an ultraviolet (UV) shadowing step to mark the band of interest and excise it from the gel. This step can cause significant chemical damage to the RNA43 and therefore should be avoided if possible (through chromatography purification methods) or the exposure of the RNA band to the UV light should be kept as short as possible. RNAs that are purified by denaturing PAGE32,34 should be subjected to an additional round of chromatography purification before high-sensitivity experiments such as NMR in order to remove any residual contamination from the gel itself (Fig. 7). This can be done in 2-3 hours via weak anion exchange chromatography (using DEAE-sepharose, MilliporeSigma) followed by buffer exchange to remove the high salt from the elution buffer.
CD, AUC, and NMR are or involve forms of absorbance spectroscopy and therefore some buffer compositions are not appropriate. For CD and AUC, buffering and reducing agents which absorb in the UV range should be avoided, such as TRIS, MOPS, citrates, imidazole, and DTT. For NMR, any buffer component at high concertation that contains protons will yield extremely strong peaks in the spectrum that can interfere with the analysis of the NMR data. In addition, high salt concentrations (> 200 mM) will severely attenuate the NMR signals.
These buffers can be made by making 500 mM stocks of potassium phosphate dibasic (KHPO4) and potassium phosphate monobasic (KH2PO4), and then diluting each into the desired buffer volume in a ratio that gives the correct pH (as reported within potassium phosphate pH tables). For example, to get 20 mM potassium phosphate at pH 6.4 in 1 L, the ratio is 72.2% monobasic and 27.8% dibasic, so the final amount to add from the 500 mM stocks would be 28.88 mL of monobasic and 11.12 mL of dibasic. NaCl in powder form (or from a concentrated stock) can then be added in to reach the desired concentration. A concentrated stock of EDTA should be made separately and pH adjusted (using NaOH) to either 6.4 or 7.0 and diluted to reach the final concentration of 0.5 mM. Finally, add water until 1L is reached.
When preparing for ITC experiments, we do not recommend dialyzing using Amicon centricons due to the high sensitivity of the technique to buffer mismatches between the cell and injection samples. Also, it is best to dialyze both the Zα protein and RNA in the same 2 L beaker overnight (in separate dialysis bags) to ensure that the buffers are completely matched.
Before proceeding to any experiment, it is important to make sure that the RNAs to be tested are in the correct buffer and heat annealed to promote the correct formation of secondary structure and tertiary structure (especially if RNAs are thawed from frozen stocks). For example, we ran AUC experiments for Zα bound to the RNA (CpG)3 repeat, one where the RNA was not heat annealed before incubating with protein and the other where we denatured it at 95°C for 5 min followed by cooling on ice for 20 min. For the heat-annealed condition, we observed a peak centered at 19.6 kDa which corresponds to a 2:1 protein:RNA complex (theoretical molecular weight of 18.8 kDa), whereas for the unannealed condition, two peaks were present, with the peak corresponding to a molecular weight of 35.6 kDa making up the majority of the sample (Fig. 4). This higher molecular weight complex was likely formed from tiling of the (CpG)3 RNAs to form longer helical structures, which were prevented by the refolding step. Refolding of RNAs is case-dependent and may need to be optimized. For small RNA hairpins and duplexes, it is usually sufficient to heat at 95°C for 5 min and cool rapidly (~ 2 min) on ice.
For longer RNAs and those involving complex tertiary interactions, a slower annealing process is recommended, such as allowing the RNA to slowly cool to the correct temperature over a period of 45 min, for example, in a thermocycler. The buffer conditions may need to be optimized in addition to the annealing process to ensure good results, including optimizing divalent metal and salt concentrations44. In addition to AUC, RNA folding can be monitored through native polyacrylamide gels (PAGE)45.
It is critical for any equilibrium measurement that the Zα:RNA mixtures are incubated at 42°C for at least 30 min before measurement in order to promote formation and stabilization of the Z-conformation within the RNA. Z-RNA formation is highly temperature dependent due to the fact that the conformational flip from A- to Z-form has a high activation energy barrier (for RNA, not DNA)11. At 25°C, conversion of the r(CpG)6 sequence from the A- to the Z-form proceeds slowly at ~12.5% conversion per hour. In comparison, ~100% conversion is observed in about 10 min after incubation at 42°C11. Note that the temperature and time dependence of non-(CpG)n Z-forming sequences has not been investigated and therefore could deviate from the standard 42°C for 30 min. It may be important to investigate and optimize incubation times and temperatures when working with unique sequences. For example, incubating the AluSx1Jo and h43 E. coli RNAs at 42°C for 10 min was enough to promote complex formation.
Keep in mind that for CD experiments, a higher concentration will only help the signal-to-noise ratio to a certain point, beyond which the difference between the right- and left-polarized light (Δε, which is measured) will become so small compared to the absorbance of the sample that it will be dominated by the noise. We carried out all our measurements on a Jasco J-815 CD Spectropolarimeter (Jasco) using a 1 mm quartz cuvette (Jasco). We found that an RNA concentration of 50 µM in a ~175 μL volume was usually sufficient to yield good data, but this will likely have to be optimized on a case-by-case basis.
For circular dichroism experiments, three controls are needed in order to properly interpret the data. First, make sure that the buffer by itself contributes minorly to the CD spectrum by measuring it in the same range as the actual experiment. If there is some minor absorbance, baseline correction can be done by subtracting the buffer spectrum from the experimental one to correct for as much of the buffer absorbance as possible. Second, an 8 M stock of sodium perchlorate (NaClO4) is needed to create the positive control sample which has the RNA in 6 M sodium perchlorate in order to induce the Z-conformation46. Finally, a sample with Zα by itself at the highest concentration to be used in the titration is also important as Zα has a minor contribution to the signal in 220–320 nm range (although this is usually negligible up to concentrations of 300 μM.
ITC can either be carried out with RNA in the ITC cell and Zα at a high concentration in the syringe or with the opposite configuration. Depending on availability of sample, it is usually easier and less expensive to have RNA in the cell and Zα at high concentration in the syringe since Zα can be made through recombinant expression.
We would like to stress that the complex thermodynamics from the binding of Zα to A-Z junctions uncovered by ITC also incur a risk of erroneously fitting the data and should be interpreted cautiously if used in isolation. It is best to confirm the KD and stoichiometry using other techniques, such as AUC and NMR.
Leaky cells are most often a problem with the contact between the centerpiece and the windows. Any small amount of dust, scratches, oils, etc, can easily compromise this seal, leading to leaking of the sample from the cell. If the absorbance is too high, the sample can be removed from the cell using a syringe (with a high gauge needle), diluted with buffer to obtain an appropriate absorbance, and reloaded into the cell.
Thus, the number of scans to measure is dependent upon how many samples are being measured. For a single sample, we usually measure about 270 scans (as each scan takes about 0.8 min, the duplicate measurement takes 1.6 min multiplied by 270 gives a total measurement time of ~8 hrs). If two samples are being measured, the number of scans needs to be halved to achieve the same measurement time. For three samples, we would measure 1/3 of the scans.
Since the stoichiometry is likely unknown going into an AUC experiment, start with a best guess and then adjust the partial-specific volume depending on the results of the fit. The partial-specific volumes for proteins and RNA are 0.73 mL.g−1 and 0.61 mL.g−1, respectively. The partial-specific volume for a Zα:RNA complex can therefore be calculated by weighting the partial-specific volumes of RNA and protein alone by the percentage of RNA and Zα that makes up the complex. As an example for the (CpG)3. The fully saturated Zα:RNA complex for the (CpG)3 RNA is 2:1, meaning one complex has two Zα proteins and one RNA. The molecular weights of Zα and the (CpG)3 RNA are 7.3 kDa and 4.2 kDa, respectively for a total combined molecular weight of 18.8 kDa. Therefore, Zα makes up 78% of the complex while the (CpG)3 RNA makes up 22%. Multiply the partial specific volumes for protein and RNA by these percentages and take the average ( (0.78*0.73 mL.g−1 + 0.22*0.61 mL.g−1)/2 ) to acquire the final partial-specific volume, which for this case would be 0.703 mLg−1.
References
- 1.D’Ascenzo L, Leonarski F, Vicens Q & Auffinger P ‘Z-DNA like’ fragments in RNA: A recurring structural motif with implications for folding, RNA/protein recognition and immune response. Nucleic Acids Res. (2016). doi: 10.1093/nar/gkw388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Harvey SC DNA structural dynamics: longitudinal breathing as a possible mechanism for the B in equilibrium Z transition. Nucleic Acids Res. 11, 4867–4878 (1983). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang AHJ et al. Molecular structure of a left-Handed double helical DNA fragment at atomic resolution. Nature (1979). doi: 10.1038/282680a0 [DOI] [PubMed] [Google Scholar]
- 4.Rich A & Zhang S Z-DNA: The long road to biological function. Nature Reviews Genetics (2003). doi: 10.1038/nrg1115 [DOI] [PubMed] [Google Scholar]
- 5.Herbert A Z-DNA and Z-RNA in human disease. Communications Biology (2019). doi: 10.1038/s42003-018-0237-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Herbert A & Rich A The biology of left-handed Z-DNA. Journal of Biological Chemistry (1996). doi: 10.1074/jbc.271.20.11595 [DOI] [PubMed] [Google Scholar]
- 7.Chiang DC, Li Y & Ng SK The Role of the Z-DNA Binding Domain in Innate Immunity and Stress Granules. Frontiers in Immunology (2021). doi: 10.3389/fimmu.2020.625504 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lushnikov AY et al. Interaction of the Zα domain of human ADAR1 with a negatively supercoiled plasmid visualized by atomic force microscopy. Nucleic Acids Res. (2004). doi: 10.1093/nar/gkh810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Herbert A et al. The Zα domain from human ADAR1 binds to the Z-DNA conformer of many different sequences. Nucleic Acids Res. (1998). doi: 10.1093/nar/26.15.3486 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Schwartz T, Rould MA, Lowenhaupt K, Herbert A & Rich A Crystal structure of the Zalpha domain of the human editing enzyme ADAR1 bound to left-handed Z-DNA. Science (80-. ). 11, 1841–1845 (1999). [DOI] [PubMed] [Google Scholar]
- 11.Brown BA, Lowenhaupt K, Wilbert CM, Hanlon EB & Rich A The Zα domain of the editing enzyme dsRNA adenosine deaminase binds left-handed Z-RNA as well as Z-DNA. Proc. Natl. Acad. Sci. U. S. A. (2000). doi: 10.1073/pnas.240464097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Placido D, Brown BA, Lowenhaupt K, Rich A & Athanasiadis A A Left-Handed RNA Double Helix Bound by the Zα Domain of the RNA-Editing Enzyme ADAR1. Structure (2007). doi: 10.1016/j.str.2007.03.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kruse H, Mrazikova K, D’Ascenzo L, Sponer J & Auffinger P Short but Weak: The Z-DNA Lone-Pair⋯π Conundrum Challenges Standard Carbon Van der Waals Radii. Angew. Chemie - Int. Ed. (2020). doi: 10.1002/anie.202004201 [DOI] [PubMed] [Google Scholar]
- 14.Chung H et al. Human ADAR1 Prevents Endogenous RNA from Triggering Translational Shutdown. Cell (2018). doi: 10.1016/j.cell.2017.12.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Feng S et al. Alternate rRNA secondary structures as regulators of translation. Nat. Struct. Mol. Biol. (2011). doi: 10.1038/nsmb.1962 [DOI] [PubMed] [Google Scholar]
- 16.Dickerson RE et al. The anatomy of A-, B-, and Z-DNA. Science (80-. ). (1982). doi: 10.1126/science.7071593 [DOI] [PubMed] [Google Scholar]
- 17.Ha SC et al. The structures of non-CG-repeat Z-DNAs co-crystallized with the Z-DNA-binding domain, hZαADAR1. Nucleic Acids Res. (2009). doi: 10.1093/nar/gkn976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee YM et al. NMR investigation on the DNA binding and B-Z transition pathway of the Zα domain of human ADAR1. Biophys. Chem. (2013). doi: 10.1016/j.bpc.2012.12.002 [DOI] [PubMed] [Google Scholar]
- 19.Lee YM et al. NMR study on the B-Z junction formation of DNA duplexes induced by Z-DNA binding domain of human ADAR1. J. Am. Chem. Soc. (2012). doi: 10.1021/ja211581b [DOI] [PubMed] [Google Scholar]
- 20.Kim D et al. Sequence preference and structural heterogeneity of BZ junctions. Nucleic Acids Res. (2018). doi: 10.1093/nar/gky784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sung CH, Lowenhaupt K, Rich A, Kim YG & Kyeong KK Crystal structure of a junction between B-DNA and Z-DNA reveals two extruded bases. Nature (2005). doi: 10.1038/nature04088 [DOI] [PubMed] [Google Scholar]
- 22.Kim D et al. Base extrusion is found at helical junctions between right- and left-handed forms of DNA and RNA. Nucleic Acids Res. (2009). doi: 10.1093/nar/gkp364 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lee EH et al. NMR study of hydrogen exchange during the B-Z transition of a DNA duplex induced by the Zα domains of yatapoxvirus E3L. FEBS Lett. (2010). doi: 10.1016/j.febslet.2010.10.003 [DOI] [PubMed] [Google Scholar]
- 24.Lee AR et al. NMR Dynamics Study Reveals the Zα Domain of Human ADAR1 Associates with and Dissociates from Z-RNA More Slowly than Z-DNA. ACS Chem. Biol. (2019). doi: 10.1021/acschembio.8b00914 [DOI] [PubMed] [Google Scholar]
- 25.Jeong M et al. NMR study of the Z-DNA binding mode and B-Z transition activity of the Zα domain of human ADAR1 when perturbed by mutation on the α3 helix and β-hairpin. Arch. Biochem. Biophys. (2014). doi: 10.1016/j.abb.2014.06.026 [DOI] [PubMed] [Google Scholar]
- 26.Nichols PJ et al. Recognition of non-CpG repeats in Alu and ribosomal RNAs by the Z-RNA binding domain of ADAR1 induces A-Z junctions. Nat. Commun. (2021). doi: 10.1038/s41467-021-21039-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Schwartz T et al. Proteolytic dissection of Zab, the Z-DNA-binding domain of human ADAR1. J. Biol. Chem. (1999). doi: 10.1074/jbc.274.5.2899 [DOI] [PubMed] [Google Scholar]
- 28.Brunelle JL & Green R In vitro transcription from plasmid or PCR-amplified DNA. in Methods in Enzymology (2013). doi: 10.1016/B978-0-12-420037-1.00005-1 [DOI] [PubMed] [Google Scholar]
- 29.Scott LG & Hennig M RNA structure determination by NMR. Methods Mol. Biol. (2008). doi: 10.1007/978-1-60327-159-2_2 [DOI] [PubMed] [Google Scholar]
- 30.Jeng S, Gardnerq J & Gumport R Transcription Termination in Vitro by Bacteriophage T7 RNA Polymerase. J. Biol. Chem. (1992). [PubMed] [Google Scholar]
- 31.Beckert B & Masquida B Synthesis of RNA by in vitro transcription. Methods Mol. Biol. (2011). doi: 10.1007/978-1-59745-248-9_3 [DOI] [PubMed] [Google Scholar]
- 32.Edwards AL, Garst AD & Batey RT Determining structures of RNA aptamers and riboswitches by X-ray crystallography. Methods Mol. Biol. (2009). doi: 10.1007/978-1-59745-557-2_9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Francis AJ & Resendiz MJE Protocol for the solid-phase synthesis of oligomers of RNA containing a 2’-othiophenylmethyl modification and characterization via circular dichroism. J. Vis. Exp. (2017). doi: 10.3791/56189 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Petrov A, Wu T, Puglisi EV & Puglisi JD RNA purification by preparative polyacrylamide gel electrophoresis. in Methods in Enzymology (2013). doi: 10.1016/B978-0-12-420037-1.00017-8 [DOI] [PubMed] [Google Scholar]
- 35.Easton LE, Shibata Y & Lukavsky PJ Rapid, nondenaturing RNA purification using weak anion-exchange fast performance liquid chromatography. RNA (2010). doi: 10.1261/rna.1862210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kim I, Mckenna SA, Puglisi EV & Puglisi JD Rapid purification of RNAs using fast performance liquid chromatography (FPLC). RNA (2007). doi: 10.1261/rna.342607 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Miyahara T, Nakatsuji H & Sugiyama H Similarities and Differences between RNA and DNA Double-Helical Structures in Circular Dichroism Spectroscopy: A SAC-CI Study. J. Phys. Chem. A (2016). doi: 10.1021/acs.jpca.6b08023 [DOI] [PubMed] [Google Scholar]
- 38.Freyer MW & Lewis EA Isothermal Titration Calorimetry: Experimental Design, Data Analysis, and Probing Macromolecule/Ligand Binding and Kinetic Interactions. Methods in Cell Biology (2008). doi: 10.1016/S0091-679X(07)84004-0 [DOI] [PubMed] [Google Scholar]
- 39.Cole JL, Lary JW, Moody, T P. & Laue TM Analytical Ultracentrifugation: Sedimentation Velocity and Sedimentation Equilibrium. Methods in Cell Biology (2008). doi: 10.1016/S0091-679X(07)84006-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Balbo A, Zhao H, Brown PH & Schuck P Assembly, loading, and alignment of an analytical ultracentrifuge sample cell. J. Vis. Exp. (2010). doi: 10.3791/1530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Fürtig B, Richter C, Wöhnert J & Schwalbe H NMR spectroscopy of RNA. ChemBioChem 4, 936–962 (2003). [DOI] [PubMed] [Google Scholar]
- 42.Delaglio F et al. NMRPipe: A multidimensional spectral processing system based on UNIX pipes. J. Biomol. NMR 6, 277–293 (1995). [DOI] [PubMed] [Google Scholar]
- 43.Kladwang W, Hum J & Das R Ultraviolet shadowing of RNA can cause significant chemical damage in seconds. Sci. Rep. (2012). doi: 10.1038/srep00517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Edelmann FT, Niedner A & Niessing D Production of pure and functional RNA for in vitro reconstitution experiments. Methods (2014). doi: 10.1016/j.ymeth.2013.08.034 [DOI] [PubMed] [Google Scholar]
- 45.Woodson SA & Koculi E Analysis of RNA folding by native polyacrylamide gel electrophoresis. Methods Enzymol. (2009). doi: 10.1016/s0076-6879(09)69009-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Klump HH & Jovin TM Formation of a left-handed RNA double helix: energetics of the A-Z transition of poly[r(G-C)] in concentrated sodium perchlorate solutions. Biochemistry 26, 5186–5190 (1987). [DOI] [PubMed] [Google Scholar]