

Abstract
Mitochondria are principal metabolic organelles that are increasingly unveiled as immune regulators. However, it is currently not known whether mitochondrial-encoded peptides modulate T cells to induce changes in phenotype and function. Here, we found that MOTS-c prevented autoimmune β-cell destruction via phenotypical and functional changes of T cells in NOD mice, a type 1 diabetes (T1D) animal model. MOTS-c ameliorated the development of hyperglycemia and reduced islet-infiltrating immune cells. Furthermore, adoptive transfer of T cells from MOTS-c-treated NOD mice significantly decreased the incidence of diabetes in NOD-SCID mice. Metabolic and genomic analysis revealed that MOTS-c modulated T cell phenotype and function by regulating TCR/mTORC1 pathway. We observed that T1D patients had a lower serum MOTS-c level than healthy controls. Furthermore, MOTS-c reduced T cell activation by alleviating T cells from the glycolytic stress in T1D patients suggesting a potential therapeutic implication. Our findings indicate that the MOTS-c acts as a regulator of T cell phenotype and function in autoimmune diabetes.
Introduction
Type 1 diabetes (T1D) is a chronic autoimmune disease characterized by the destruction of insulin-secreting β-cells(Katsarou et al., 2017). Although the etiology of T1D is not fully understood, autoreactive T cells predominantly occupy the insulitis lesions and are considered key culprits of β-cell destruction(Magnuson et al., 2015; Pugliese, 2017). It is also becoming clearer that elevated glycolysis of autoreactive T cells is an important target in preventing autoimmune disorders. Several evidences suggest that alleviating glycolytic stress in autoimmune disorders have shown therapeutic effects in autoimmune diseases like multiple sclerosis (Kornberg et al., 2018). CD4+IFNγ+ T helper type 1 (TH1) cells are major effector cells exhibiting high glycolytic property and associated with T1D pathogenesis (Walker and von Herrath, 2016). Increased levels of IFNγ correlate with progression to diabetes in non-obese diabetic (NOD) mice and T1D patients (Walker and von Herrath, 2016). On the contrary, CD4+CD25+FOXP3+ regulatory T cells (Treg) exhibit low glycolytic character and are critical for regulating immune tolerance with demonstrated therapeutic potential in various experimental models of T1D and other autoimmune disorders (Bluestone et al., 2015; Dirice et al., 2019; Marek-Trzonkowska et al., 2014). In humans, adoptive immunotherapy with autologous Treg cells in T1D patients has shown to be safe and indicated some therapeutic potential(Bluestone et al., 2015; Marek-Trzonkowska et al., 2014). The differentiation of naïve CD4+ T cells into TH1 or Treg cells requires substantial metabolic rewiring in response to a shift in biosynthetic and bioenergetic needs(Pearce et al., 2013). Mitochondria, being chief metabolic organelles, have been strongly implicated in the regulation of T cell activation and differentiation(Bailis et al., 2019; Weinberg et al., 2015). Notably, mitochondrial signaling, in part, mediated by reactive oxygen species (ROS), has recently emerged as a key element of T cell activation and differentiation(Akkaya et al., 2018; Devadas et al., 2002; Jackson et al., 2004; Jones et al., 2007; Marcin et al., 2012; Michael and Richard, 2013; Sena et al., 2013).
Mitochondria communicate to other cellular compartments to coordinate a multitude of cellular functions (Antico Arciuch et al., 2012; Chandel, 2014; Chandel, 2015; da Cunha et al., 2015; Galluzzi et al., 2012; Lee et al., 2013; Quiros et al., 2016; Topf et al., 2016; Woo and Shadel, 2011; Xu et al., 2013; Yin and Cadenas, 2015; Zarse and Ristow, 2015). Traditionally, all gene-encoded mitochondrial signals have been thought to be encoded in the nuclear genome, but recently identified peptides that are encoded in the mitochondrial genome have been shown to mediate mitochondrial communication. MOTS-c (mitochondrial open reading frame of the twelve S rRNA type-c) is a mitochondrial-derived peptide (MDP) that promotes metabolic homeostasis in an AMPK-dependent manner(Lee et al., 2015). Upon metabolic stress, MOTS-c can dynamically translocate to the nucleus where it directly regulates adaptive nuclear gene expression(Kim et al., 2018b). Notably, the discovery of MOTS-c was, in part, influenced by previous work that reported strong interferon-related expression of transcripts from the mitochondrial rRNA loci(Tsuzuki et al., 1983), providing potential immunological implications of MOTS-c function. In fact, MOTS-c has been shown to have an anti-inflammatory effect in multiple systems and conditions(Hu and Chen, 2018; Lee et al., 2015; Li et al., 2018; Lu et al., 2019c; Ming et al., 2016; Qin et al., 2018; Raijmakers et al., 2019; Ramanjaneya et al., 2019; Xinqiang et al., 2020; Yin et al., 2020; Zhai et al., 2017).
Here, using the well-established NOD (non-obese diabetic) mouse model of T1D(Pearson et al., 2016), we show that MOTS-c treatment remarkably delays disease onset and prevents leukocyte infiltration of pancreatic islets. MOTS-c changes CD4+ T cell subset frequency of TH1 and/or Treg cells in the spleen, pancreas, and lymph nodes of NOD mice. Further, by layering microarray data, protein-protein docking simulation, and co-immunoprecipitation approaches, we find that MOTS-c can regulate IFNγ and FOXP3 expression, in part, by directly inhibiting mTORC1 signaling in T cells. The hydrophobic domain of MOTS-c allows binding to Raptor, a major component of mTORC1, and likely acts as an allosteric inhibitor of its substrates, including 4EBP1 and S6K. Using CD4+ T cells from mice (NOD) and humans (T1D subjects), we show that MOTS-c regulates glycolysis, cellular differentiation, and prevents autoimmune diabetes. Our study makes an unprecedented connection between inherent mitochondrial regulation of T cells and its therapeutic potential in T1D.
Results
MOTS-c prevents autoimmune diabetes in NOD mice
NOD mice exhibit T cell-mediated pancreatic β-cell destruction and consequent loss of insulin production(Delovitch and Singh, 1997). By 18.5 weeks of age, all NOD mice became diabetic (Figure 1A) with blood glucose levels > 500 mg/dL; the onset was as early as 8 weeks (Figure 1A). However, systemic MOTS-c treatment (0.5 mg/kg/day; IP), initiated at 7 weeks of age, significantly (i) delayed the onset of diabetes, whereby the first incidence occurred at 23 weeks of age (Figure 1A), (ii) delayed disease progression with 100% vs. 0% total incidence by 19 weeks of age, which was kept to 33.3% even at 30 weeks of age (Figure 1A), and (iii) considerably improved blood glucose levels compared to controls (251 ± 140 mg/dL vs 547 ± 90 mg/dL, respectively) (Figure 1B). These effects of MOTS-c were not attributable to altered body weight (Figure 1C). An intraperitoneal glucose tolerance test (IPGTT) in 18-week-old NOD mice revealed that MOTS-c treatment significantly improved glucose clearance (Figure 1D), concomitant with higher glucose-stimulated insulin levels in circulation (Figure 1E).
Figure 1. MOTS-c prevents autoimmune diabetes in NOD mice.
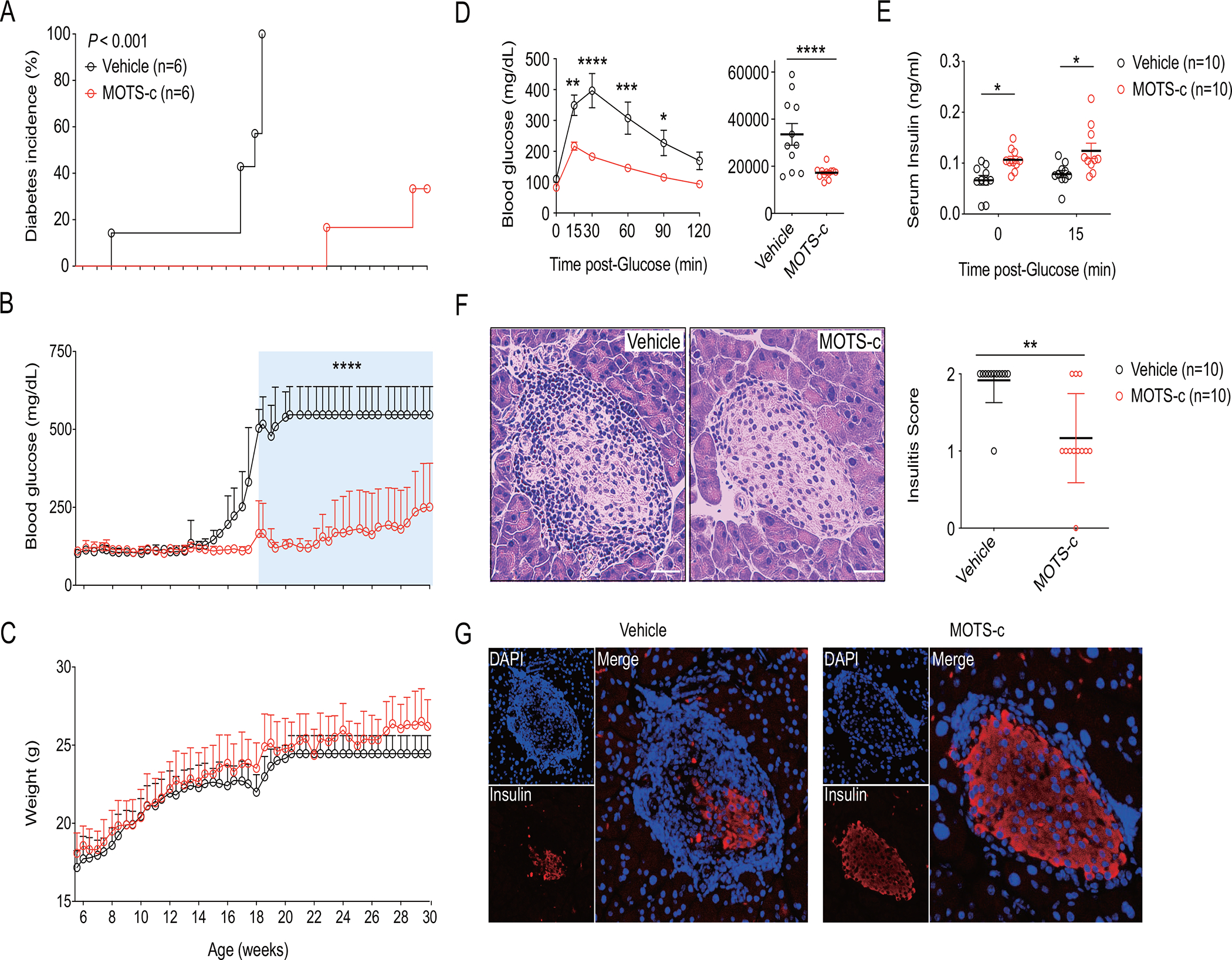
NOD mice were treated with MOTS-c daily (0.5 mg/kg/day; IP) for (A-C) 30 weeks (n=6) and (D-G) 18 weeks (n=10) of age. (A) Diabetes incidence was observed until 30 weeks of age. (B) Random blood glucose (RBG) level and (C) body weight were measured twice a week; two-way repeated measures ANOVA. A week before euthanasia, (D) IPGTT was performed by injecting ᴅ-glucose (2 g/kg; IP) in time-course and AUCs for blood glucose were calculated; two-tailed t-test (for AUC) and two-way (for time-dependent IPGTT) ANOVA. (E) Serum obtained before and after glucose injection in IPGTT were assessed for insulin concentration; two-tailed t-test. Paraffinized NOD pancreas were stained with (F) H&E for assessment of insulitis score and (G) IF staining of insulin. (F, G) Pancreatic sections were prepared as described and the representative data from ten pancreases with ten sections for each mouse (n=10); two-tailed t-test (for insulitis score), error bars are S.E.M. *p<0.05, **p<0.01, ***p<0.001, ****p<0.0001. Scale bars: 25 μm
T1D is driven by destructive autoimmune insulitis, an inflammatory islet pathology that is described by pancreatic infiltration of leukocytes with predominant presence of T and B lymphocytes(Magnuson et al., 2015), that leads to β-cell loss and insulin depletion(Katsarou et al., 2017). Histological analyses revealed that MOTS-c treatment significantly reduced (i) the presence of islet-infiltrating cells (Figure 1F) and (ii) the degree of insulitis (Figure 1F) in 18-week-old NOD mice. Insulin production in β-cells was preserved in MOTS-c-treated NOD mice (Figure 1G), whereas glucagon production in α-cells were unaffected (Figure S1A). Immunofluorescence (IF) staining showed that MOTS-c treatment significantly reduced the levels of pancreatic infiltration by CD4+ and CD8+ T cells in NOD mice (Figure S1B, S1C).
MOTS-c exhibits immunoregulatory effects in splenic CD4+ T cells
To test whether MOTS-c acted on T cells in a tissue-specific manner (Figure 2A), we measured T cell frequency in T1D-relevant tissues, including the spleen, pancreatic lymph nodes, and thymus(D’Alise et al., 2008; Yeh et al., 2013). Only the spleen exhibited an altered T cell profile upon MOTS-c treatment with increased CD4+, but decreased CD8+, populations (Figure 2B; Figure S2A); no significant changes were observed in pancreatic lymph nodes (LN) nor the thymus (Figure 2C, 2D; Figure S2B, S2C). We then tested if MOTS-c affected specific subsets of CD4+ T cells. In the spleen, the frequency of TH1 (CD4+IFNγ+) declined whereas Treg (CD4+CD25+FOXP3+) population increased in MOTS-c-treated NOD mice (Figure 2E, Figure S2D, S2K). Notably, in the LN, although the total CD4+ T cell frequency was largely unchanged, Treg subset was enriched (Figure 2F, Figure S2M). The thymus did not show any changes (Figure 2G). To test if the organ-specific actions of systemically injected MOTS-c is related to targeted tissue distribution, we intravenously injected FLAG-tagged MOTS-c (MOTS-c-FLAG) in C57BL/6J mice. MOTS-c-FLAG was detected in the spleen and the pancreas within an hour of injection, but not in the thymus or pancreatic LN, consistent with splenic and pancreatic effects of MOTS-c (Figure S3A).
Figure 2. MOTS-c exhibits immunoregulatory effects in splenic CD4+ T cells.
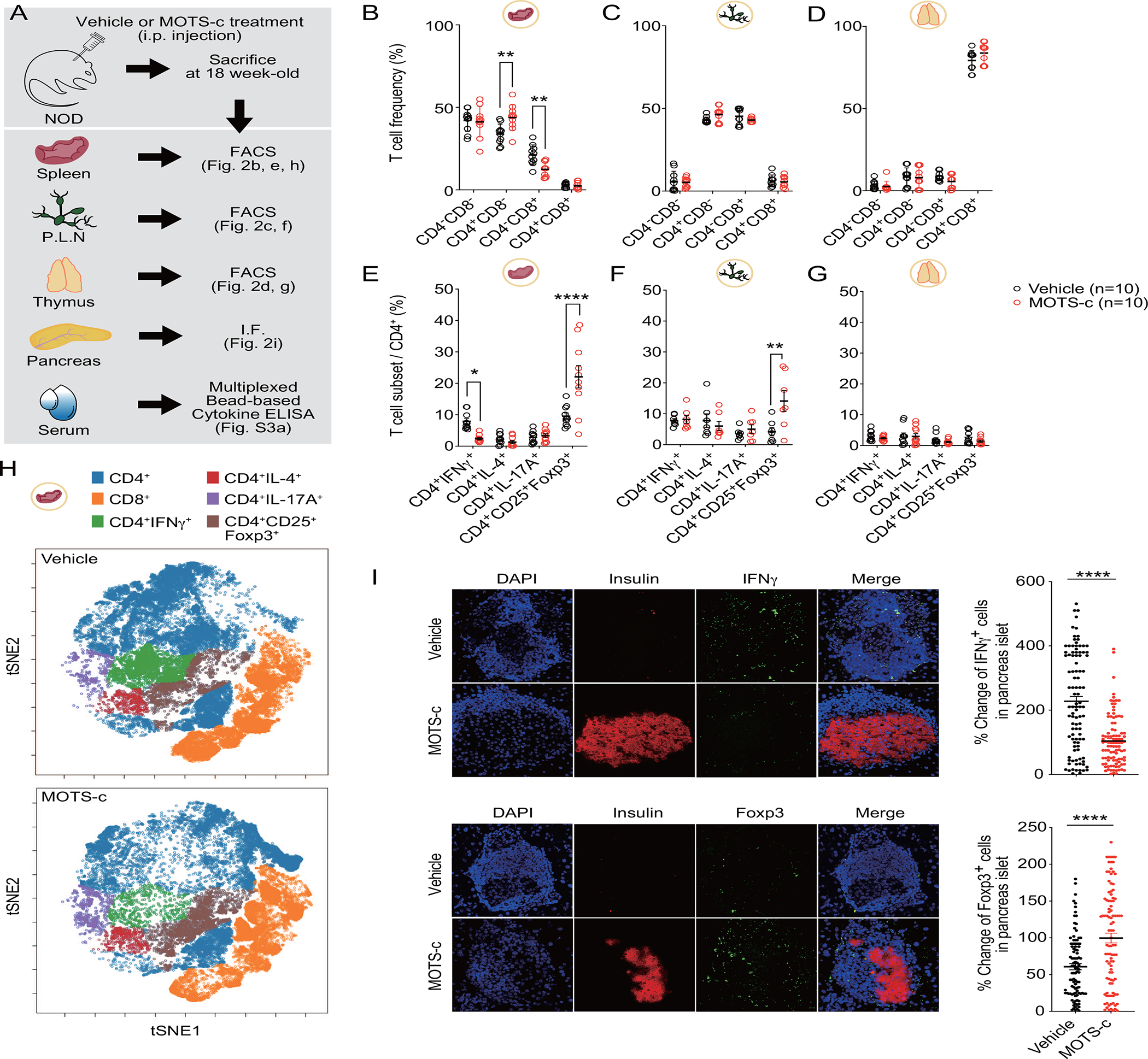
(A) NOD mice were treated with MOTS-c daily (0.5 mg/kg/day; IP) for 18 weeks (n=10) of age to isolate (B, E, H) spleen, (C, F) pancreatic lymph nodes (PLN), (D, G) thymus, (I) pancreas, and (Figure S3A) serum. Cells obtained from each organ were stained for (B, C, D) CD4− CD8−, CD4+CD8−, CD4−CD8+, CD4+CD8+; and (E, F, G) IFNγ+, CD25+FOXP3+, IL-4+, and IL-17A+ in CD4+ cells; two-way ANOVA. (H) t-SNE was performed for CD4+, CD8+, IFNγ+, CD25+FOXP3+, IL-4+, and IL-17A+ after down-sampling of splenocytes. The indicated positive cells were overlaid in color onto the overall t-SNE map. Immunohistological analysis for T cell subsets were performed in the pancreas of NOD mice. IF staining (I) shows for insulin (red) and IFNγ, and FOXP3 (green). IF staining on pancreatic sections were prepared as described and the representative data from ten pancreases with ten sections for each group (n=100); two-tailed t-test, error bars are S.E.M. *p<0.05, **p<0.01, ****p<0.0001.
To better visualize the effects of MOTS-c on splenic T cells, we performed t-SNE analysis and found that MOTS-c treatment targeted TH1 (CD4+IFNγ+) and Treg (CD4+CD25+FOXP3+) cells (Figure 2H). This is consistent with the fact that TH1 cells accelerate T1D, whereas Treg cells maintain immunological tolerance to islet antigens in NOD mice(Chaudhry et al., 2009; Dirice et al., 2019; Feuerer et al., 2009; Koch et al., 2009; Zheng et al., 2009). We then examined if the MOTS-c-dependent T cell subset modulation in the spleen was reflected in the pancreas of 18-week old NOD mice. Indeed, using fluorescence imaging, we observed a decrease in IFNγ+ and IL-17A+ cells, but an increase in FOXP3+ cells in the pancreas (Figure 2I, Figure S1E). GATA3+ cells did not show any changes (Figure S1D). Notably, the serum levels of IFNγ was 8-fold lower and IL-10 was 5-fold higher in MOTS-c-treated NOD mice compared to the vehicle group (Figure S3B). To find whether the change of cytokine production is associated with T cell proliferation, we stained 6-week-old non-diabetic NOD T cells with CFSE. MOTS-c did not alter the proliferation of both CD4+ and CD8+ T cells (Figure S3C). Overall, MOTS-c treatment significantly reduced TH1 cells and enriched Treg cells in the spleen and pancreas of NOD mice.
MOTS-c binds to Raptor and regulates mTORC1 signaling in T cells
To obtain an overall molecular perspective on the cellular context of MOTS-c-treated T cells, we performed gene microarray analyses on splenocytes from 18-week-old NOD mice treated with or without MOTS-c. Principal component analysis (PCA) revealed a clear shift in gene expression between vehicle and MOTS-c-treated groups derived from multiple components (Figure 3A). Hierarchical clustering and enrichment analyses further accentuated the shift in gene expression pattern by MOTS-c treatment (Figure 3B). Using the KEGG database, we found that many MOTS-c-dependent genes that were differentially down-regulated were strongly associated with suspected cellular functions such as diabetes, T cell, and metabolism related pathways (Figure 3C, Figure S4A). Furthermore, mTOR, EIF4EBP1, and genes associated with T cell differentiation (Dnmt1, Cdk2, Smad4, Bcl2, Foxo1)(Chen and Konkel, 2010; Corn et al., 2003; Gabriel et al., 2016; Jones and Pearce, 2017; Long et al., 2009; Ouyang et al., 2010; Yu et al., 2006) were significantly differentially up- and down-regulated with MOTS-c treatment (Figure S4B). mTOR and EIF4EBP1 are major components of mTOR complex 1 (mTORC1) signaling pathway, which is a prominent sensor and regulator of nutrient and differentiation in T cells, especially regarding FOXP3+ Treg differentiation(Kim and Guan, 2019). High mTOR signaling or mTORC1 activation promotes TH1 differentiation, whereas low mTOR signaling or mTORC1 inhibition is favored by Treg(Angelin et al., 2017; Buck et al., 2016; Chornoguz et al., 2017; Shi et al., 2011). Raptor is a key component of mTORC1 that binds to 4EBP-1 and S6K via their TOR signaling (TOS) motifs and supports their phosphorylation(Kim et al., 2002; Yang et al., 2017). Notably mTORC1 can be directly inhibited by another master metabolic regulator AMPK (Gwinn et al., 2008; Inoki et al., 2006), which is a key mediator of MOTS-c function (Kim et al., 2018a, b; Lee et al., 2015; Lu et al., 2019a; Lu et al., 2019b; Ming et al., 2016; Wei et al., 2020; Xinqiang et al., 2020; Yan et al., 2019; Yin et al., 2020). Thus, we asked if MOTS-c may regulate mTORC1 signaling, either directly or indirectly. Prior to the protein homology modeling of MOTS-c using the full Raptor structure (PDB:5WBI), we assessed the reliability of the predicted protein structure of MOTS-c and Raptor using Ramachandran plot. It revealed a distribution of 89.8% of Raptor residues and 92.9% of MOTS-c residues in the favored region (Figure S4C, 4D), which suggests that the predicted structures of MOTS-c and Raptor are reliable for subsequent docking studies. The alpha helical MOTS-c likely docks to a groove between the caspase homology domain of Raptor and the α-solenoid through side chain-side chain interaction (Figure 3D), specifically between the hydrophobic 8YIFY11 residues of MOTS-c and Q333-D518 residues of Raptor (Figure 3E), also known to bind to the TOS motifs of 4EBP-1, S6K, and PRAS(Yang et al., 2017). Indeed, superimposing MOTS-c on the Raptor-TOS motif of 4EBP-1 (PDB:5WBJ) and S6K (PDB:5WBK) revealed remarkable overlap and interaction with identical Raptor residues (i.e., Y475, L341, L441) (Figure 3F). We confirmed direct binding of MOTS-c and Raptor by co-immunoprecipitation studies using MOTS-c-EGFP and Myc-Raptor (Figure 3G). The overexpression of Raptor (mTORC1), but not Rictor (mTORC2), increased the phosphorylation of its downstream proteins 4EBP-1 and p70S6K, concomitantly decreasing MOTS-c detection (Figure 3H). Conversely, MOTS-c treatment inhibited the expression of mTORC1-related genes (Figure S4B). Conversely, knocking down Raptor, but not Rictor, using shRNA increased MOTS-c expression (Figure 3I). Together, these data support direct interaction between MOTS-c and Raptor (mTORC1) and an inverse functional relation between MOTS-c and mTORC1 signaling in T cells.
Figure 3. MOTS-c binds to Raptor and regulates mTORC1 signaling in T cells.
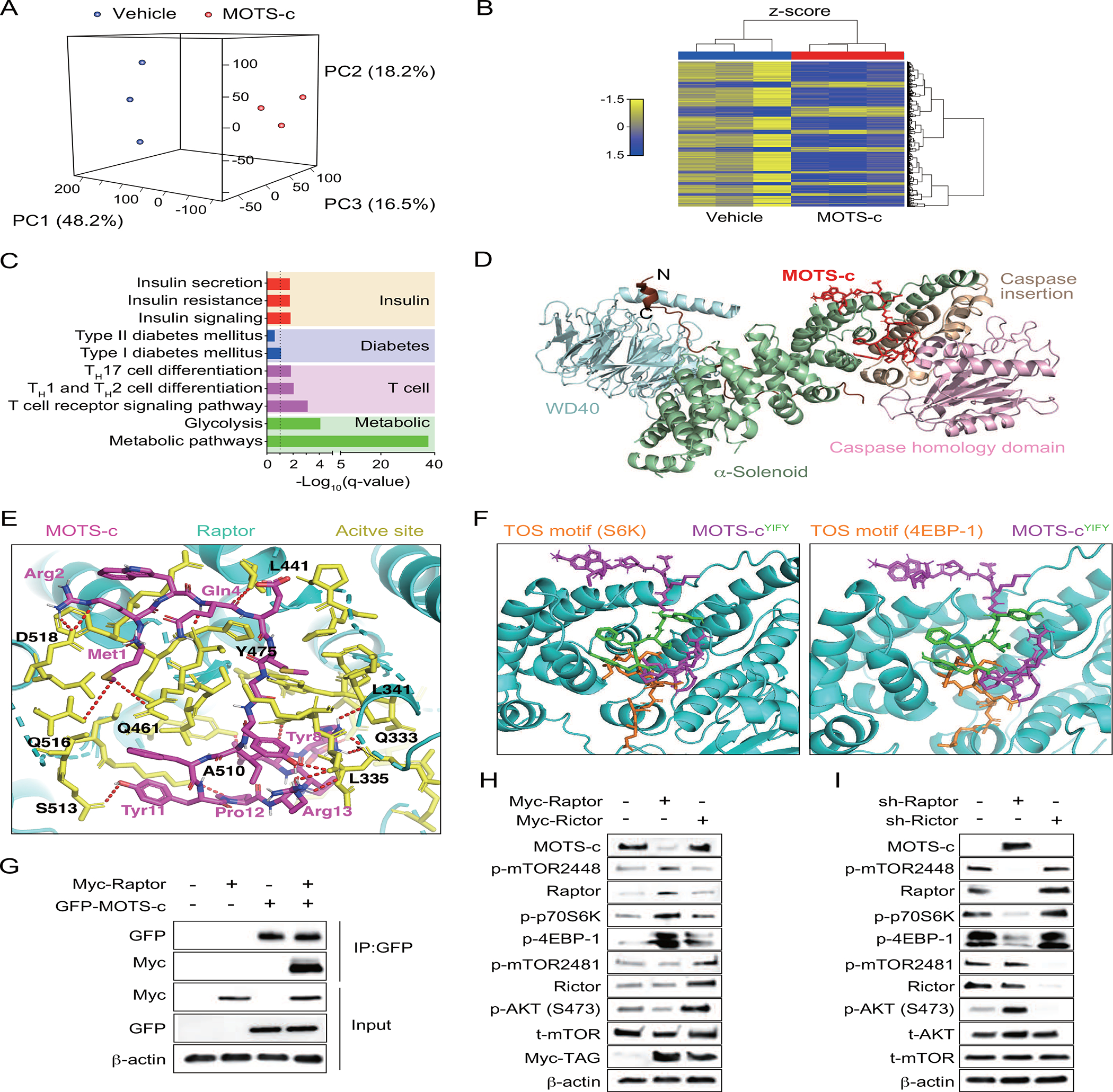
(A-D) Microarray analyses on NOD splenocytes treated with either vehicle or MOTS-c for 18 weeks of age (n=3). (A) Principal component analysis (PCA), (B) Hierarchical clustering, and (C) KEGG analysis on insulin-related, diabetes-related, T-cell related, metabolic-related pathways. (D) Raptor and MOTS-c (magenta) structures are superimposed. MOTS-c interacts with a groove in between the α-solenoid and caspase homology domain of Raptor. (E) Close-up views of the interface between Raptor (cartoon, cyan), Raptor active site (sticks, yellow), and MOTS-c (sticks, magenta) with residue numbers and interaction (red-dotted lines). (F) MOTS-c (sticks, magenta) and its YIFY hydrophobic core (sticks, green) were superimposed on Raptor (cyan)-TOS motifs of 4EBP-1 and S6K (sticks, orange). Immunoblotting of MOTS-c and mTORC1 were assessed by (G) Co-immunoprecipitation of EGFP-MOTS-c and Myc-tagged Raptor (Myc-Raptor) in Jurkat cells, (H) overexpression of Myc-Raptor and Myc-Rictor in Jurkat cells, and (I) shRNA of Raptor and Rictor in Jurkat cells. All western data are representative of at least 3 independent experiments.
mTOR (mammalian target of rapamycin) signaling in T cells is central to the pathogenesis of autoimmune diseases and rapamycin was thought to cause immunosuppression by inhibiting T cell activation(Perl, 2015). We stimulated mTOR signaling by TCR ligation, IGF-1, or glutamine to phenocopy activated T cells in autoimmune disorders(Chi, 2012; Haxhinasto et al., 2008; Powell and Delgoffe, 2010; Sauer et al., 2008) using primary T cells from C57BL/6J mice. Consistent with our previous results, MOTS-c expression and mTORC1 signaling showed a strong negative correlation in CD3+ T cells (Figure S5A). Similar results were observed in CD4+ and CD8+ T cells and Jurkat cells activated with TCR ligation (Figure S5B, S5C). Conversely, mTORC1 inhibition using rapamycin(Battaglia et al., 2006a; Choi et al., 1996; Monti et al., 2008), increased MOTS-c levels in Jurkat cells (Figure S5D). To visualize the expression pattern of MOTS-c in T cells, we overexpressed MOTS-c-EGFP in Jurkat cells. In time lapse experiment, we used a marker-free holotomographic and confocal microscopy to observe MOTS-c-EGFP expression during Jurkat cell activation. We found that MOTS-c-EGFP levels diminished upon T cell activation (Figure S5E, S5F; Movie S1). These results confirm the opposing functional relationship between MOTS-c and mTORC1 signaling.
MOTS-c regulates TCR/mTORC1 signaling to differentiate CD4+ T cells
T cell receptor signaling plays a crucial role in immune responses (Sprent and Surh, 2011). The interaction between a peptide-MHC complex on antigen presenting cells and T cell receptors is a critical process during the pathogenesis of autoimmune disorders including T1D (Bhattacharyya and Feng, 2020; Katsarou et al., 2017). TCR signaling is known to activate mTORC1 signaling in T cells for survival, proliferation, and differentiation (Chapman and Chi, 2014). MOTS-c has shown a high association with both TCR and mTORC1 signaling pathway (Figure 3A–I). To assess whether MOTS-c could regulate TCR/mTORC1 signaling-mediated differentiation, we treated cells with αCD3 and αCD28 in MOTS-cWT or MOTS-cMut overexpressed Jurkat cells. Because the tyrosine residues in the hydrophobic domain of MOTS-c are critical for its interaction with Raptor, we generated a mutant in which the hydrophobic core of MOTS-c (8YIFY11 to 8AAAA11, MOTS-cMut) substituted with alanine residues (Figure 4A). Using 3D protein docking simulations, MOTS-cMut was predicted to have fewer interaction (red-dotted lines) with Raptor (Figure 4B, 4C), which diminished its ability to inhibit mTORC1 signaling activated by αCD3 and αCD28 (Figure 4D). Furthermore, the loss-of-expression of MOTS-c using actinonin, which specifically degrades mitochondrial RNA (Kim et al., 2018b; Lee et al., 2015; Richter et al., 2013), increased mTORC1 signaling in MOTS-cWT-overexpressed Jurkat cells under resting condition. Notably, the TCR activation further strengthened this negative correlation of mTORC1 signaling and MOTS-c expression (Figure 4E). mTORC1 regulates T cell fate, in part, by differentially regulating the expression of FOXP3 and IFNγ (Chi, 2012). Thus, we assessed whether the regulation of TCR/mTORC1 signaling by MOTS-c could change the fate of T cell and differentiation. TCR activation increased both IFNγ and FOXP3 expression in empty-vector transfected Jurkat cells. However, MOTS-cMut showed diminished effects on regulating T cell differentiation compared to MOTS-cWT. Indeed, MOTS-cWT, but not MOTS-cMut, exhibited lowered IFNγ expression level and increased FOXP3 expression level compared to empty vector transfected Jurkat cells (Figure 4F). Next, we assessed whether the synthetic MOTS-c peptide treatment to T cells exhibit similar results as the genetic modification of MOTS-c. Before testing this, we firstly assessed whether MOTS-c peptide could be directly delivered into T cells using flagged-MOTS-c peptide. Data showed that MOTS-c peptide enters both CD4+ and CD8+ T cells only under activated condition. Also, MOTS-c peptide favors CD4+ T cells than CD8+ T cells. (Figure 4G). Finally, we assessed whether MOTS-c treatment could regulate T cell differentiation. MOTS-c alone did not increase the frequency of CD4+IFNγ+ (TH1) and CD4+CD25+FOXP3+ (Treg) cells (Figure 4H). However, when MOTS-c was given under T cell polarizing conditions, it lowers the differentiation into CD4+IFNγ+ cells and strengthens CD4+CD25+FOXP3+ cell frequency in comparison to TCR alone condition (Figure 4G; Figure S6A, S6B). Overall, these data correlate with the NOD spleen data (Figure 2–4) and confirm that MOTS-c can inhibit TCR/mTORC1 signaling and regulate T cell differentiation to favor Treg cells.
Figure 4. MOTS-c regulates TCR/mTORC1 signaling to differentiate CD4+ T cell.
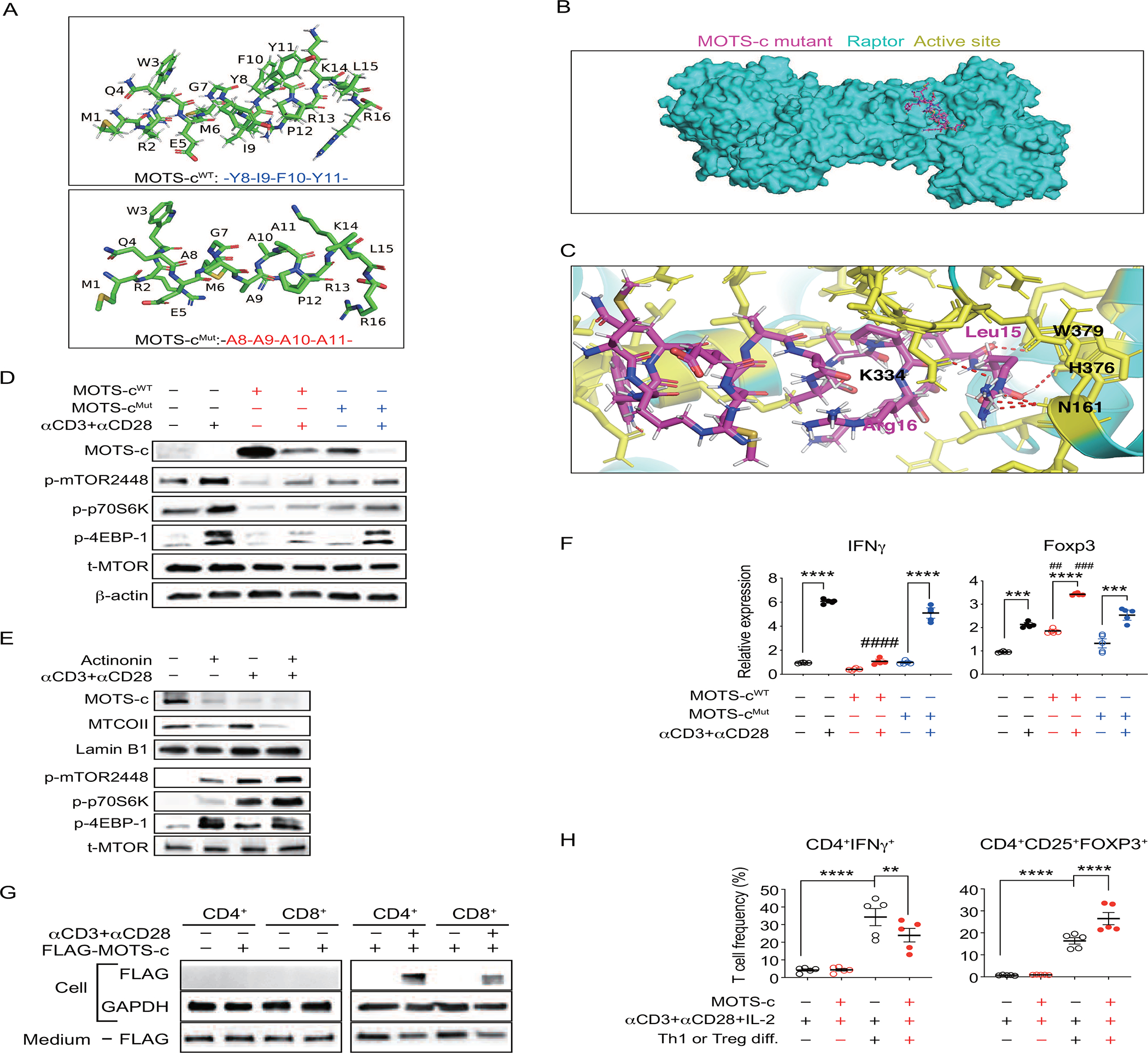
(A) 3D Structure of MOTS-cWT and MOTS-cMut. (B) Raptor and MOTS-cMut are superimposed. (C) Close-up views of the interface between Raptor (cyan), active site (sticks, yellow), and MOTS-cMut (sticks, magenta) with residue numbers. Interaction between MOTS-cMut and Raptor active site is shown in red dotted line.
Empty vector plasmid, MOTS-c wild type (MOTS-cWT), and MOTS-c mutant (MOTS-cMut) were overexpressed in Jurkat cells and then activated with soluble human αCD3 (4 μg/ml) and αCD28 (10 μg/ml) for 3 hours to analyze (D) MOTS-c and mTORC1 signaling proteins; (E) For loss-of-expression of MOTS-c, mitochondrial RNA depletion was induced by actinonin (50 μM, 24h) in MOTS-cWT overexpressed Jurkat cells. (F) CD4+ and CD8+ T cells were obtained from 7-week-old C56BL/6 mice splenocytes. FLAG-MOTS-c (10 uM) was treated from 3 hours with or without αCD3 (4 μg/ml) and αCD28 (10 μg/ml). Cells and medium were collected from immunoblotting on FLAG. All western data are representative of at least 3 independent experiments.
(F) IFNγ and FOXP3 mRNA level (n=5); one-way ANOVA, error bars are S.E.M. ##p<0.01, ###p<0.001, ####p<0.0001, ***p<0.001, ****p<0.0001. #indicates difference between MOTS-cWT vs empty vector transfected group. *indicates difference within the group. (G) CD4+ T cells were negatively isolated from C57BL/6J mice spleen (n=5). TH1 or Treg polarization were performed, as described in the method, in the presence or absence of MOTS-c (10 μM). Then, cells were stained with adequate antibodies (CD4, IFNγ, CD25, FOXP3) for FACS analysis; one-way ANOVA, error bars are S.E.M. p**0.01, ****p<0.0001.
MOTS-c lowers CD4+ T cells glycolysis
Treg responses require FOXP3-mediated metabolic reprogramming that engages mitochondria and favors oxidative phosphorylation (OXPHOS) over glycolysis, which can be turned off by mTORC1 activation(Chapman et al., 2020). Because MOTS-c promoted the expansion of Treg cells in 18-week-old NOD spleen (Figure 2B–G; Figure S2A–F), we first measured metabolic flux in splenocytes collected from MOTS-c-treated NOD mice (Figure 5A) and observed relatively higher oxidative and reduced glycolytic capacity (Figure 5B, 5C). To confirm this effect was driven by T cells, we isolated CD4+ T cells by negative selection. Indeed, purified splenic CD4+ T cells from MOTS-c-treated NOD mice showed significantly higher respiratory capacity (Figure 5E) and decreased glycolysis (Figure 5D), which was proportionally reflected in their contribution to total ATP production (Figure S7A). Similar results were seen in CD8+ T cells (Figure S7B). Alternatively, we also directly tested the effect of MOTS-c on T cells. We measured metabolic flux in Jurkat cells that overexpressed Empty Vector, MOTS-cWT, or MOTS-cMut (Figure 5F). We found that MOTS-cWT, but not MOTS-cMut, effectively reduced glycolytic capacity and increased respiratory capacity in Jurkat cells (Figure 5G, 5H). These data add metabolic support to the MOTS-c-dependent enrichment of Treg cells in the NOD spleen.
Figure 5. MOTS-c lowers CD4+ T cells glycolysis.
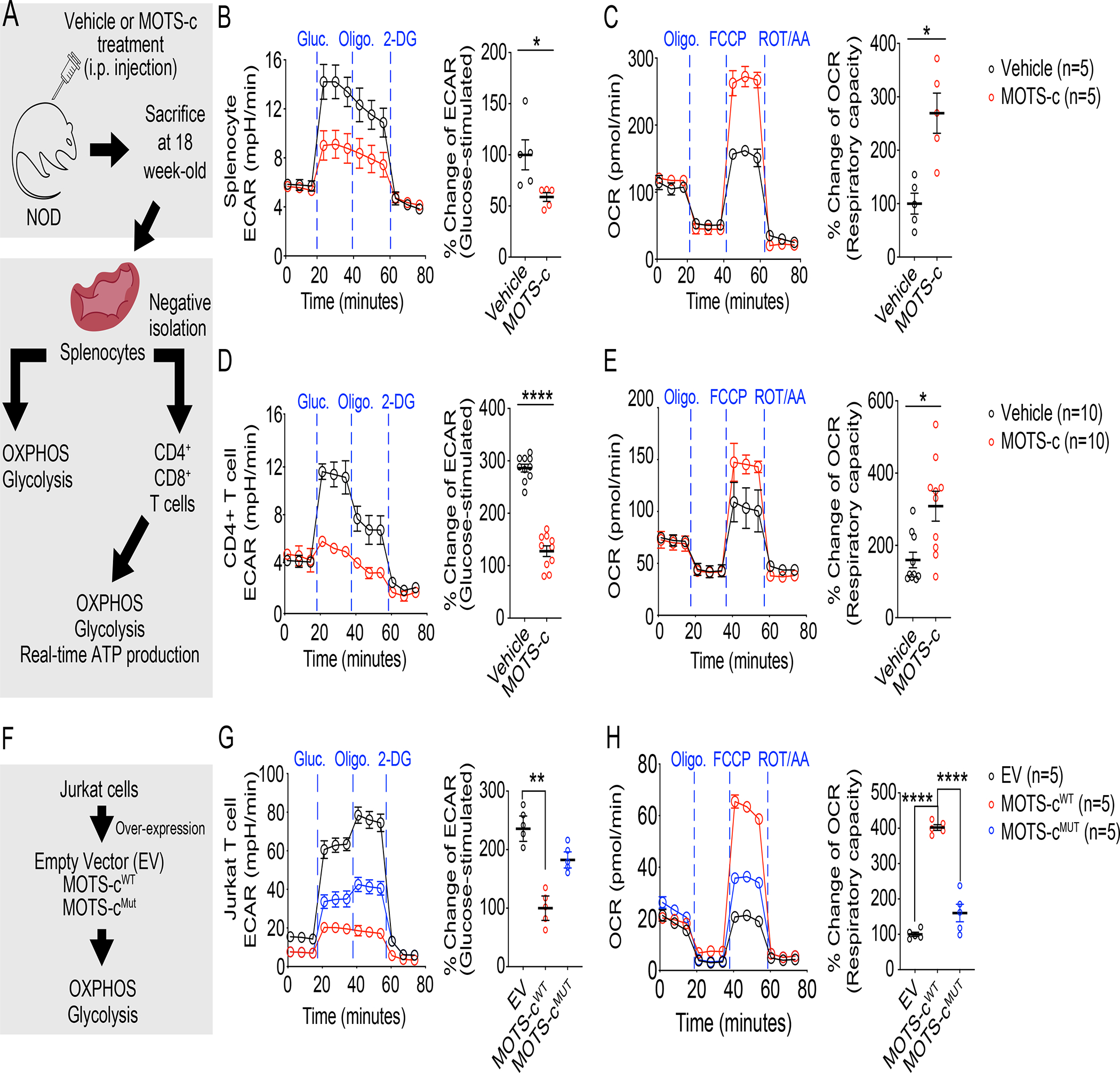
(A) NOD mice were treated with either vehicle or MOTS-c daily (0.5 mg/kg/day; IP) for 18 weeks of age. Then, NOD splenocytes (n=5) and CD4+ T cells were assessed for (B, D) glycolysis after glucose stimulation (n=10) and (C, E) respiratory capacity (n=10); two-tailed t-test (B, C, D, E), error bars are S.E.M. *p<0.05, **p<0.01, ****p<0.0001.
(F) Jurkat cells were overexpressed with empty vector, MOTS-cWT, and MOTS-cMut. These cells were assessed for (G) glycolysis after glucose stimulation (n=5) and (H) respiratory capacity (n=5); one-way ANOVA (G, H), error bars are S.E.M. **p<0.01, ****p<0.0001.
MOTS-c prevents diabetes in NOD-SCID adoptive transfer model
Adoptive transfer of splenocytes from NOD to the immunocompromised NOD-SCID (T and B cell deficient) mice can transmit T1D pathology, including β-cell destruction, in the otherwise non-diabetogenic strain owing to CD4+ T cells, but not CD8+ T cells or B lymphocytes(Bendelac et al., 1988; Christianson et al., 1993; Prochazka et al., 1992a, b; Thivolet et al., 1991; Wang et al., 1991b). MOTS-c increased splenic Treg population, which confers immunotolerance against islet antigens. Thus, we hypothesized that adoptive transfer of splenocytes from MOTS-c-treated NOD mice would not elicit T1D in the NOD-SCID background. To test this, we transferred splenocytes from NOD mice that were treated/untreated with MOTS-c (18-week-old) to NOD-SCID (6-week-old) mice. We transferred total splenocytes, which are predominantly T and B lymphocytes(Magnuson et al., 2015), as it transfers the disease much more efficiently than using isolated diabetic CD4+ or CD8+ cells alone(Christianson et al., 1993), likely because islet antigen exposed CD4+ T cells are required for self-recognizing CD8+ T cells to target the pancreas(Christianson et al., 1993; Fuchtenbusch et al., 2005; Thivolet et al., 1991; Wang et al., 1991a). B cells are not required for disease transfer to NOD-SCID mice(Bendelac et al., 1988). Other leukocytes, such as macrophages and NK cells, are associated with autoimmune diabetes, but they are already present in NOD-SCID mice(Prochazka et al., 1992a). Splenocyte transfer from control NOD mice induced diabetes in NOD-SCID mice within 10~14 weeks (Figure 6A–C), whereas those from MOTS-c-treated NOD mice showed significantly (i) delayed disease onset and (ii) reduced hyperglycemia compared to controls (Figure 6A–C). Notably, splenic T cell frequency in recipient NOD-SCID mice at termination showed that those that received splenocytes from MOTS-c-treated NOD mice had much lower CD4+IFNγ+ and higher CD4+FOXP3+ cell frequency (Figure 6D; Figure S8A). Furthermore, CD4+ T cells isolated from pancreas islets showed reduced IFNγ and increased FOXP3 gene expression (Figure 6E). Overall, these results confirmed that MOTS-c modulates CD4+ T cell and prevents diabetes in NOD mice.
Figure 6. MOTS-c prevents diabetes in NOD-SCID adoptive transfer model.
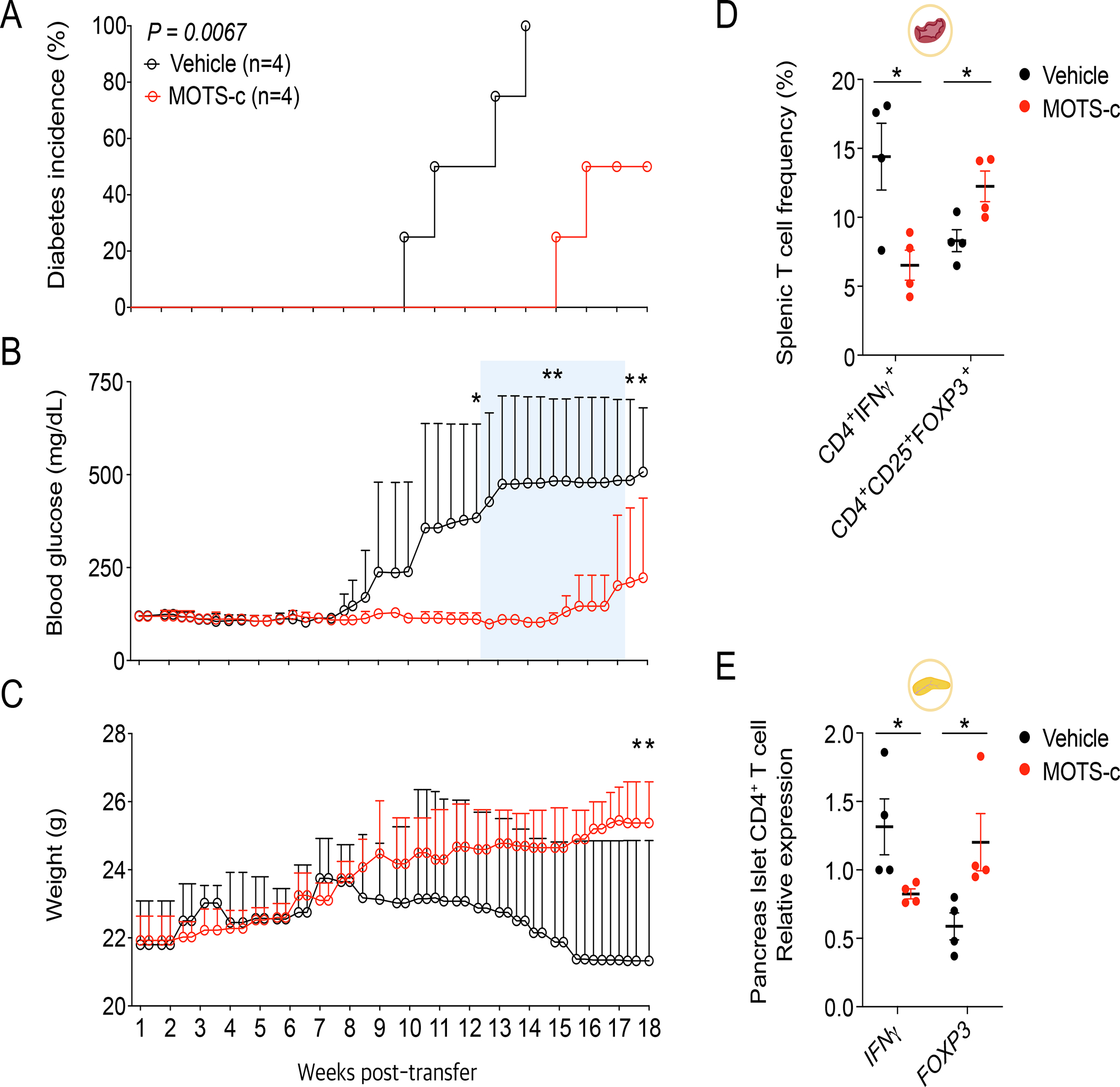
18-week-old NOD splenocytes (1 × 107 cells) from each group were i.v. injected to 6-week-old NOD/SCID mice (n=4) for measurement of (A) diabetes incidence, (B) RBG level, and (C) body weight; two-way repeated measures ANOVA, error bars are S.E.M. *p<0.05, **p<0.01.
(D, E) 18-weeks after adoptive transfer, NOD-SCID spleen and pancreas were isolated from each group at termination (n=4). Then, splenocytes were stained with appropriate antibodies to analyze (D) CD4+IFNγ+ and CD4+CD25+FOXP3+ cells (n=4) using FACS. Also, CD4+ cells were negatively isolated from pancreas islets to analyze (E) mRNA expression level of IFNγ (n=4) and FOXP3 (n=4); two-tailed t-test, error bars are S.E.M. *p<0.05.
MOTS-c restricts glycolysis in CD4+ T cells from T1D patients
T1D patients showed significantly lower levels of circulating MOTS-c than healthy controls (HC) (Figure 7B), consistently reflecting the function of MOTS-c in promoting Treg cells and suppressing TH1 cells. Indeed, CD4+ T cells from T1D patients showed significantly reduced OXPHOS and relied more on glycolysis to produce cellular ATP than those from healthy individuals (Figure 7C, 7D; Figure S9A). Further analyses between serum MOTS-c level and CD4+ glycoATP showed negative correlation, whereas serum MOTS-c level and mitoATP showed positive correlation. These data suggest that MOTS-c is highly associated with CD4+ T cell metabolism (Figure 7E, 7F). Interestingly, aerobic glycolysis is a key mTOR-dependent metabolic pathway that enables CD4+ T cells to exhibit effector functions(Buck et al., 2016; Chang et al., 2013; Cheng et al., 2014). We tested whether MOTS-c could suppress glycolysis in CD4+ T cell activation. We firstly treated cells with either vehicle or MOTS-c and then, activated CD4+ T cells in real-time by stimulating them with human antibodies against CD3 and CD28. MOTS-c significantly blunted activation-induced glycolytic increase in CD4+ T cells from both HC and T1D, yet the effect was less pronounced in cells from T1D patients (Figure 7G); basal glycolytic rate was higher in T cells from T1D patients. Similar results were observed in CD8+ T cells in T1D patients (Figure S9B, S9C). These results suggest that MOTS-c can inhibit T cell activation in T1D patients and may provide a novel therapeutic target.
Figure 7. MOTS-c restricts glycolysis in CD4+ T cells from T1D patients.
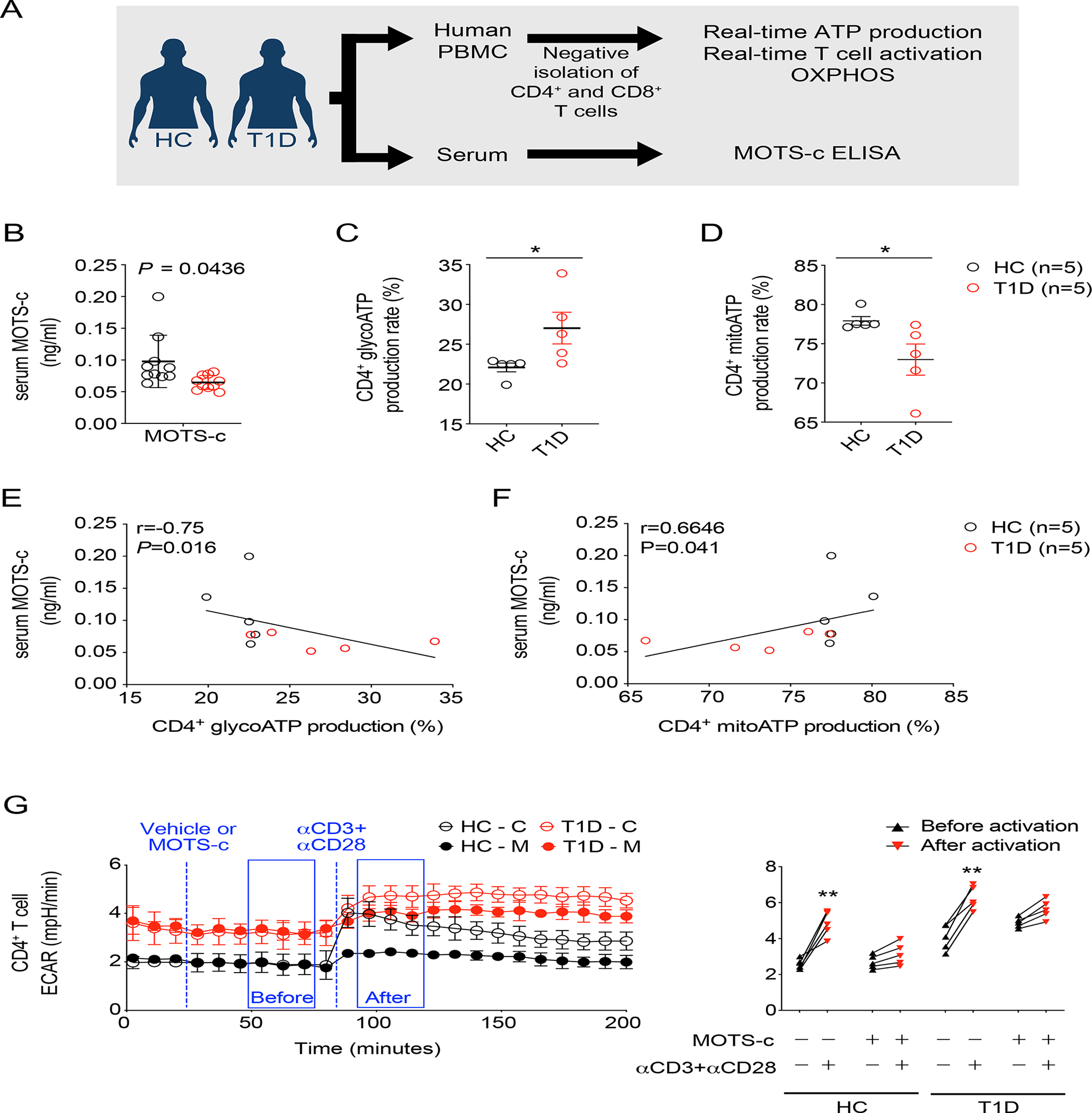
(A) Human T cells were isolated from PBMCs and serum were obtained from T1D and healthy controls (HC). (B) Human serum MOTS-c level in T1D patients and HC (n=10) were assessed. Negatively isolated human CD4+ T cells were assessed for (C) glycoATP production and (D) mitoATP production in percentage (n=5); two-tailed t-test, *p<0.05.
The relationship between serum MOTS-c level and CD4+ glycoATP or mitoATP were evaluated (n=5 from each group). Serum MOTS-c showed (E) negative correlation with CD4+ glycoATP (r=−0.75, P=0.016), whereas (F) positive correlation with CD4+ mitoATP (r=0.67, P=0.041). Relationships were evaluated by Spearman correlation coefficient (r).
For the real-time human T cell activation, T1D patients and healthy controls (HC) (G) CD4+ T cells were analyzed by XFe96 Seahorse analyzer (n=5). T cells were activated with human αCD3 (4 μg/ml) and αCD28 (20 μg/ml) in the presence or absence of MOTS-c (10 μM); two-way ANOVA, **p<0.01.
Discussion
In this study, we have evaluated the role of MOTS-c in regulating splenic T cells to prevent autoimmune diabetes. MOTS-c altered CD4+ T cell subsets by enriching FOXP3+ Treg while suppressing IFNγ+ TH1 cells. Notably, the discovery of MOTS-c was strongly influenced by prior work that demonstrated considerably expression of transcripts from the mitochondrial rRNA loci under interferon-inducing conditions(Tsuzuki et al., 1983). MOTS-c broadly inhibited the expression of IFN-related genes, including interferon regulatory factors (IRFs) and interferon-stimulated response element (ISRE)(Kim et al., 2018a), indicating a feedback regulation between IFN and MOTS-c. The effect of MOTS-c on autoimmune tolerance may not be limited to T1D as Treg cells are highly involved in multiple autoimmune diseases, such as multiple sclerosis, rheumatoid arthritis, and Graves’ disease, which needs further investigation.
To date, interventions that inhibit CD8+ T cells, such as anti-CD3 monoclonal antibodies, have shown varying degrees of efficacy in delaying the onset of T1D or decline in β-cell function with no long-term success(Herold et al., 2019; Kolb and von Herrath, 2017). T cell metabolism is an emerging therapeutic target for autoimmune diseases(Pearce et al., 2013). Several drugs including 2-deoxy-D-glucose, metformin, and rapamycin, which regulate metabolic pathways in immune cells are actively investigated as potential therapeutic agents for the treatment of autoimmune diseases(Bjornstad et al., 2018; Sukumar et al., 2013). Notably, rapamycin, mTOR inhibitor, may have therapeutic potential to prevent T1D by promoting the expansion of FOXP3+ Treg cells in NOD mice and in T1D patients(Battaglia et al., 2006b; Monti et al., 2008). Rapamycin showed therapeutic efficacy in other autoimmune disease models such as experimental autoimmune encephalomyelitis (EAE)(Donia et al., 2009), autoimmune hepatitis(Montano Loza and Czaja, 2007), and Crohn’s disease(Massey et al., 2008). These studies are compatible with our data and supports MOTS-c as an attractive therapeutic target to modulate T cell metabolism and suppress its activation in T1D and likely other autoimmune conditions. Taken together, a mitochondrial-encoded peptide MOTS-c may be leveraged to unveil an unprecedented relationship between T cell function and mitochondria in T1D and shed new light on the development of novel mitochondrial-based therapeutic strategies.
Methods
Cell culture.
Jurkat (source: male) and HEK293FT (source: female) were grown in RPMI supplemented with 10% fetal bovine serum (FBS), respectively. All cells were incubated in a humidity-controlled environment at 37°C, 5% CO2 (Thermo Fisher Scientific Forma 371), were utilized up to 10 passages, and were purchased from ATCC.
Transfection
Jurkat cell was transfected in 1.5 × 105 cells in 12 well-plate using Neon transfection system (Thermo Fisher Scientific). MOTS-c wild type (MOTS-cWt), MOTS-c Mutant (MOTS-cMut), MOTS-c-EGFP, EGFP-vectors, and empty-vectors were acquired as previous reports (Kim et al., 2018b; Lee et al., 2015). After transfection, cells were sorted, selected, and maintained in G418 (Gibco) in RPMI with 10% FBS. Raptor and Rictor shRNA were purchased from Addgene (#1857, 1853)(Sarbassov et al., 2005), which were transfected into HEK293FT using Lipofectamine 3000 (Thermo). Virus-containing supernatants were collected 24 and 48 hours after transfection. Jurkat cells were infected in the presence of 10 ug/ml of polybrene, selected for puromycin resistance and analyzed on the 7th day after transfection.
Mice.
6-week-old NOD (female, n=56) and 5-week-old NOD-SCID mice (female, n=8) were purchased from the Jackson laboratory (Bar Harbor, USA), Animal Resource Centre (Canning Vale, Australia), respectively. 6-week-old C57BL/6 (female, n=30) were purchased from Koatech (Seoul, Korea). C57BL/6J, NOD, and NOD-SCID mice had a week of stabilization before any experimental procedure. Mice were bred and housed in a specific pathogen-free environment in the animal facility at the Seoul National University Hospital (IACUC no. 17-0095-C1A1, 18-0144-S1A1).
MOTS-c treatment in mice.
MOTS-c peptides were synthesized as previously(Lee et al., 2015) and flagged-MOTS-c peptides were synthesized from Cosmogentech (Seoul, Korea). NOD mice were treated with either vehicle (non-specific scrambled MOTS-c peptide) or MOTS-c (0.5 mg/kg/day; IP) daily in the morning up to 18 or 30 weeks of age. Random blood glucose (RBG) level and body weights were measured in the morning twice every week for all types of mice (Accu-Chek). Blood glucose levels>300 mg/dL for two consecutive times in RBG level were consider as diabetes. Intraperitoneal glucose tolerance test was performed a week before euthanasia. For euthanasia, NOD mice aged 18 weeks were fasted overnight (16–18 hrs), weighed and random blood glucose were measured. Blood was collected with heparinized capillary tubes from the retro-orbital sinus before the glucose injection and 15 minutes after the glucose injection, as previously reported (Chong et al., 2006). The collected blood was centrifuged for plasma separation, stored at −8°C, and analyzed insulin and cytokine content within 2 weeks (Alpco). For adoptive transfer, 6-week-old NOD/SCID mice were used as recipients and received i.v. injection of 1.0 × 107 cells/ml splenocytes from 18-week-old NOD mice donors. For flagged MOTS-c injection, 7-week-old C57BL/6J were i.v. injected with 10 mg/kg of flagged-MOTS-c in time dependent manner. After sacrifice, tissues were collected from C57BL/6J, NOD, and NOD-SCID mice to be stored in RNAlater (Thermo), fixed in paraformaldehyde, or homogenized for further analysis. All animal experiments were repeated by 3 times.
T1D patients and healthy controls
10 patients with Type 1 diabetes (T1D) fulfilling diagnostic criteria for T1D were enrolled from the Division of Endocrinology and Metabolism, Department of Internal Medicine, Seoul National University Hospital (IRB no. 1808-151-967). Ten age- and sex-matched healthy controls were also recruited for blood donation. Demographic and clinical characteristics of two groups and basic information of each patient are summarized in Supplemental Table 2. Peripheral blood was obtained by venipuncture and processed immediately for T cell purification as described below.
T cell proliferation
To assess proliferation, CD4+ and CD8+ T cells were isolated from NOD mice (6-week-old) fluorescein-labed with 1 μM CFSE for 5 min at 37°C. Then, they were treated with soluble αCD3 and αCD28 (2 μg/ml each) for 3 days with or without IL-2 (10 ng/ml) in RPMI media supplemented with 10% fetal bovine serum (FBS).
Real-time PCR
Total RNA was purified using the RNeasy Mini Kit (Qiagen) as per manufacturer’s protocol. cDNA was synthesized by reverse transcription of 1 μg of total RNA using AMV Reverse Transcriptase (Promega) following manufacturer’s instructions. Quantitative real-time PCR was performed in 20 μl of reaction mixture containing 1 μl of cDNA, 10 pM of each primer, and 10 μl of SYBR Select Master Mix (Thermo Fisher Scientific) using Applied Biosystems 7500 Real-Time PCR System (Thermo Fisher Scientific). Products were amplified with primers listed in Supplemental Table 1 and 18S ribosomal RNA was used as a reference control for each reaction.
Western blotting
Splenocytes and Jurkat cell whole lysates were extracted with lysis buffer containing a protease/phosphatase inhibitor cocktail as previously reported to assess mTORC1(Kim et al., 2002) and MOTS-c(Kim et al., 2018b; Lee et al., 2015). The lysates were subjected to electrophoresis using 4–15% precast gels (Bio-rad). The resolved gels from precast SDS-PAGE were transferred to PVDF membranes, blocked with 5% skimmed milk in tris-buffered saline containing 0.05% Tween-20 (TBS-T) and incubated with primary antibody at 4°C overnight. The membranes were incubated with HRP-conjugated secondary antibodies at room temperature for 2 hrs and developed using Pico chemiluminescent substrate (Thermo) and imaged using Amersham imager 600 (GE Healthcare).
Co-immunoprecipitation
Cells were lysed in lysis buffer as previously reported(Kim et al., 2002). Dynabeads (per mg) were coated with 5 μg GFP antibody at room temperature overnight. Cell lysates were incubated with antibody-conjugated Dynabeads and rotated at 4°C for 3 hr. The immunoprecipitation complex was washed and eluted. The eluted samples were suspended with SDS sample buffer and subjected to western blotting.
Pancreas histology, scoring of insulitis and immunofluorescence analyses on tissue samples
NOD mice were sacrificed and pancreas were obtained and preserved either in 4% paraformaldehyde or in OCT compound for frozen cryotome sectioning. Pancreas embedded in paraffin were used for hematoxylin and eosin staining to score insulitis, as previously described(Zhang et al., 2007). Insulin-secreting islets were scored as follows: 0, no insulitis (free of infiltration); 1, periinsulitis (a few to many inflammatory cells outside or in the immediate vicinity of the islets); 2, insulitis a clear and often extensive islet infiltrate that shows direct lymphocyte-B cell contact). The percentage of no insulitis, periinsulitis, and insulitis among total islets in each mouse was calculated. Pancreas embedded in frozen block were prepared in 5 μM cross sections for immunofluorescence staining. Sections were blocked with 5% goat serum (Jackson ImmunoResearch) in antibody diluent (Dako) for 30 minutes and incubated overnight at 4°C with following antibodies: Insulin, Glucagon, and lymphocyte markers (1:100 of antibody concentration). After washing with TBST, slides were incubated for 1 hr with appropriate fluorochrome-attached secondary antibody (Jackson ImmunoResearch). Slides were rinsed and analyzed after mounting cover-glass. Then, images were acquired by using DMi8 (Leica).
Mouse pancreas islet CD4+ T cell isolation
NOD-SCID mice islets were isolated were isolated, as previously reported(Li et al., 2009). Then, islets were further processed to negatively isolate CD4 T cells using MACS microbeads (Miltenyi) for real-time PCR.
Flow cytometry and intracellular cytokine staining
All antibodies were purchased BD Biosciences or Biolegend. To evaluate T cell subsets, monocytes, and macrophages frequency, spleen, lymph nodes, and thymus were isolated and mashed with nylon mesh (Spectrum). Red blood cells were removed using RBC lysis buffer (eBioscience), and cells were washed, and stained with adequate antibodies and isotype controls for FACS analysis. For intracellular cytokines staining, cells were stimulated at 37°C for 6 hrs in the presence of Golgistop (BD Biosciences), PMA (Sigma), and ionomycin (Sigma). CD4−CD8−, CD4+CD8−, CD4−CD8+, CD4+CD8+, TH1 (CD4+IFNγ+), TH2 (CD4+IL-4+), TH17(CD4+IL-17A+), Treg (CD4+CD25+FOXP3+) cells were gated for FACS analysis. All FACS data was acquired using FACS Canto, and were analyzed with Flowjo software (Treestar, Ashland).
NOD serum analysis using multiplex cytokine assay
NOD mice serum was assessed using mouse TH1/TH2/TH17 cytokine CBA kit (BD Biosciences) and were analyzed using FCAP array software (BD Biosciences). All FACS data was acquired using FACS Canto, and were analyzed with Flowjo software (Treestar, Ashland).
t-SNE analysis
For t-SNE analysis, data from exported FCS files were analyzed using Cytobank (www.cytobank.org).
TH1 and Treg differentiation.
CD4+ T cells were negatively isolated from C57BL/6J mice (7-week-old) using MACS microbeads (Miltenyi). TH1 polarization was performed by the addition of recombinant IL-2 (10 ng/ml; Peprotech), anti-mouse IL-4 (10 μg/ml, R&D systems) and IL-12 (10 ng/ml; Peprotech). Treg polarization was performed by the addition of recombinant human TGF-β (10ng/ml, Peprotech) and IL-2 (10ng/ml) in the presence of mouse αCD3 and αCD28 (2 μg/ml each).
Metabolic flux analyses
Murine splenocytes were obtained from NOD spleen after the treatment of either vehicle or MOTS-c for 18 weeks. Human PBMCs were isolated using a Ficoll density gradient (GE Healthcare) of buffy coats from blood of healthy donors or patients. Then, CD4+ or CD8+ T cells were negatively isolated using T cell isolation kit (MACS, Miltenyi Biotec). Isolated CD4+, CD8+ T cells, or splenocytes from each group were plated onto Seahorse cell plates coated with Cell-Tak (Corning) to enhance T cell attachment. Cells were analyzed with the Seahorse XFe24/96 Extracellular Flux analyzer (Agilent) to determine real-time oxygen consumption rate (OCR), extracellular acidification rate (ECAR), and Real-time ATP production assay. Real-time ATP production assay measures and quantifies mitoATP and glycoATP, simultaneously, by using oligomycin and Rotenone/Antimycin A. To monitor CD4+ and CD8+ T cell activation in real-time, αCD3 (4 μg/ml; BD Biosciences) and αCD28 (20 μg/ml; BD Biosciences) were mixed and directly applied onto plated cells via the instrument’s multi-injection ports. Both antibodies were injected 30 minutes after the experiment was initiated for murine T cells, as previously reported (Gubser et al., 2013). For human T cells, antibodies were introduced 60 minutes after the injection of MOTS-c (10μM) or vehicle.
In-house MOTS-c ELISA test
MOTS-c-BSA conjugation was synthesized in Youngin Frontier (Seoul, Korea). ELISA method was used and modified from the previous study to test MOTS-c level in human serum (Lee et al., 2015).
Jurkat T cell imaging using Nanolive.
MOTS-c EGFP transfected Jurkat cells treated with or without human αCD3 (4 μg/ml, 3 hours) and αCD28 (10μg/ml, 3 hours). For live cell imaging of T cells, MOTS-c EGFP transfected Jurkat cells were seeded (1 × 106 cells) in a confocal dish and placed in a culture chamber of Nanolive, which is a marker-free phase holotomographic microscope. Then, cells were either untreated or treated with αCD3 (4 μg/ml; BD Biosciences) and αCD28 (20 μg/ml; BD Biosciences) for 1 hr. Images were analyzed using STEVE software (Nanolive).
MOTS-c-Raptor molecular docking and refinement.
3D structure of MOTS-cWT and MOTS-cMut peptide was acquired by using Pepfold 3.0 (https://mobyle.rpbs.univ-paris-diderot.fr/cgi-bin/portal.py#forms::PEP-FOLD3) and Raptor (PDB ID:5WBI), Raptor-TOS motifs of 4EBP-1 (PDB ID:5WBJ), and Raptor-TOS motifs of S6K (PDB:5WBK) from previous report (Yang et al., 2017). These structures were analyzed with Ramachandran plot to verify the protein structure is reliable for subsequent docking studies(Lovell et al., 2003). The Ramachandran plot also helps to determine whether the protein will form α helices (ϕ, −60° and Ψ, −45°), β sheets (ϕ, −120° and ψ, 115°), or left-handed α helices (ϕ, 60° and ψ, 45°). Majority of MOTS-c residues are α helical, but Raptor shows more complicated structure. For the sampling method, hierarchical optimization (number of clusters:250) was set to optimize side chains within 5.0 Å of the ligand-binding site. The sampling algorithm was set to Monte Carlo minimization (number of steps: 500) to consider backbone flexibility and find the global minima within 10.0 Å of the ligand-binding site. Other parameters were set to default. Computational studies were carried out using PyRX 2.0 and molecular visualization of MOTS-c-Raptor complex was acquired by using PyMOL 2.3 (http://www.pymol.org).
Microarray.
RNA purity and integrity were evaluated by ND-1000 Spectrophotometer (NanoDrop), Agilent 2100 Bioanalyzer (Agilent Technologies). RNA labeling and hybridization were performed by using the Agilent One-Color Microarray-Based Gene Expression Analysis protocol (Agilent Technology, V 6.5, 2010). Briefly, 100ng of total RNA from each sample was linearly amplified and labeled with Cy3-dCTP. The labeled cRNAs were purified by RNAeasy Mini Kit (Qiagen). The concentration and specific activity of the labeled cRNAs (pmol Cy3/μg cRNA) were measured by NanoDrop ND-1000(NanoDrop, Wilmington, USA). 600ng of each labeled cRNA was fragmented by adding 5 μl 10 × blocking agent and 1 μl of 25 × fragmentation buffer, and then heated at 60°C for 30 min. Finally, 25 μl 2 × GE hybridization buffer was added to dilute the labeled cRNA. 40μl of hybridization solution was dispensed into the gasket slide and assembled to the Agilent SurePrint G3 Mouse GE 8X60K, V2 Microarrays (Agilent®). The slides were incubated for 17 h at 65°C in an Agilent hybridization oven. then washed at room temperature by using the Agilent One-Color Microarray-Based Gene Expression Analysis protocol (Agilent Technology, V 6.5, 2010). The hybridized array was immediately scanned with an Agilent Microarray Scanner D (Agilent Technologies, Inc.). Microarray results were extracted using Agilent Feature Extraction software v11.0 (Agilent Technologies). Array probes that have Flag A in samples were filtered out. Selected gProcessedSignal value was transformed by logarithm and normalized by quantile method. Statistical significance of the expression data was determined using independent t-test and fold change in which the null hypothesis was that no difference exists among groups. False discovery rate (FDR) was controlled by adjusting p value using Benjamini-Hochberg algorithm. For a DEG set, Hierarchical cluster analysis was performed using complete linkage and Euclidean distance as a measure of similarity. Gene-Enrichment and Functional Annotation analysis for significant probe list was performed using KEGG (www.genome.jp/kegg/)(Kanehisa and Goto, 2000) (Supplemental Table 3). All data analysis and visualization of differentially expressed genes was conducted using R 3.1.2 (www.r-project.org).
Statistical analysis.
Data are presented as mean ± standard error of mean (SEM). Graphpad Prism was used for two-tailed t-test, one-way or two-way ANOVA to assess significance between groups and Bonferroni’s multiple comparison was performed.
Supplementary Material
Acknowledgements
The authors would like to thank the patients and donors who participated in this study and Kyunghee Kim, RN, who helped in coordination. Also, we thank Dr. Chang Ho Ahn, Mi Na Kim, and Eun Hye Choi for the assistance in this study. This research was supported by Basic Science Research program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2017R1A2B4007166) and Seoul National University Hospital (0320160040).
Footnotes
Declarations of Interests The authors declare no competing interests.
Ethics approval This study was approved by the Ethics Committee of Seoul National University Hospital, Korea and all methods were performed in accordance with the relevant guidelines and regulations. Written informed consent was obtained from all participants.
Availability of data and material
Data are available on request due to privacy or other restrictions.
Reference
- Akkaya B, Roesler AS, Miozzo P, Theall BP, Al Souz J, Smelkinson MG, Kabat J, Traba J, Sack MN, Brzostowski JA, et al. (2018). Increased Mitochondrial Biogenesis and Reactive Oxygen Species Production Accompany Prolonged CD4+ T Cell Activation. The Journal of Immunology 201, 3294–3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angelin A, Gil-de-Gomez L, Dahiya S, Jiao J, Guo L, Levine MH, Wang Z, Quinn WJ 3rd, Kopinski PK, Wang L, et al. (2017). Foxp3 Reprograms T Cell Metabolism to Function in Low-Glucose, High-Lactate Environments. Cell Metab 25, 1282–1293 e1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antico Arciuch VG, Elguero ME, Poderoso JJ, and Carreras MC (2012). Mitochondrial regulation of cell cycle and proliferation. Antioxidants & redox signaling 16, 1150–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bailis W, Shyer JA, Zhao J, Canaveras JCG, Al Khazal FJ, Qu R, Steach HR, Bielecki P, Khan O, Jackson R, et al. (2019). Distinct modes of mitochondrial metabolism uncouple T cell differentiation and function. Nature 571, 403–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battaglia M, Stabilini A, Draghici E, Migliavacca B, Gregori S, Bonifacio E, and Roncarolo MG (2006a). Induction of tolerance in type 1 diabetes via both CD4+CD25+ T regulatory cells and T regulatory type 1 cells. Diabetes 55, 1571–1580. [DOI] [PubMed] [Google Scholar]
- Battaglia M, Stabilini A, Migliavacca B, Horejs-Hoeck J, Kaupper T, and Roncarolo MG (2006b). Rapamycin promotes expansion of functional CD4+CD25+FOXP3+ regulatory T cells of both healthy subjects and type 1 diabetic patients. J Immunol 177, 8338–8347. [DOI] [PubMed] [Google Scholar]
- Bendelac A, Boitard C, Bedossa P, Bazin H, Bach JF, and Carnaud C (1988). Adoptive T cell transfer of autoimmune nonobese diabetic mouse diabetes does not require recruitment of host B lymphocytes. J Immunol 141, 2625–2628. [PubMed] [Google Scholar]
- Bhattacharyya ND, and Feng CG (2020). Regulation of T Helper Cell Fate by TCR Signal Strength. Front Immunol 11, 624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bjornstad P, Schafer M, Truong U, Cree-Green M, Pyle L, Baumgartner A, Garcia Reyes Y, Maniatis A, Nayak S, Wadwa RP, et al. (2018). Metformin Improves Insulin Sensitivity and Vascular Health in Youth With Type 1 Diabetes Mellitus. Circulation 138, 2895–2907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bluestone JA, Buckner JH, Fitch M, Gitelman SE, Gupta S, Hellerstein MK, Herold KC, Lares A, Lee MR, Li K, et al. (2015). Type 1 diabetes immunotherapy using polyclonal regulatory T cells. Sci Transl Med 7, 315ra189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buck MD, O’Sullivan D, Klein Geltink RI, Curtis JD, Chang CH, Sanin DE, Qiu J, Kretz O, Braas D, van der Windt GJ, et al. (2016). Mitochondrial Dynamics Controls T Cell Fate through Metabolic Programming. Cell 166, 63–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandel NS (2014). Mitochondria as signaling organelles. BMC biology 12, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandel NS (2015). Evolution of Mitochondria as Signaling Organelles. Cell Metab. [DOI] [PubMed] [Google Scholar]
- Chang CH, Curtis JD, Maggi LB Jr., Faubert B, Villarino AV, O’Sullivan D, Huang SC, van der Windt GJ, Blagih J, Qiu J, et al. (2013). Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 153, 1239–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman NM, Boothby MR, and Chi H (2020). Metabolic coordination of T cell quiescence and activation. Nature Reviews Immunology 20, 55–70. [DOI] [PubMed] [Google Scholar]
- Chapman NM, and Chi H (2014). mTOR Links Environmental Signals to T Cell Fate Decisions. Front Immunol 5, 686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhry A, Rudra D, Treuting P, Samstein RM, Liang Y, Kas A, and Rudensky AY (2009). CD4+ regulatory T cells control TH17 responses in a Stat3-dependent manner. Science 326, 986–991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen W, and Konkel JE (2010). TGF-beta and ‘adaptive’ Foxp3(+) regulatory T cells. J Mol Cell Biol 2, 30–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng SC, Quintin J, Cramer RA, Shepardson KM, Saeed S, Kumar V, Giamarellos-Bourboulis EJ, Martens JH, Rao NA, Aghajanirefah A, et al. (2014). mTOR- and HIF-1alpha-mediated aerobic glycolysis as metabolic basis for trained immunity. Science 345, 1250684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chi H (2012). Regulation and function of mTOR signalling in T cell fate decisions. Nat Rev Immunol 12, 325–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi J, Chen J, Schreiber SL, and Clardy J (1996). Structure of the FKBP12-rapamycin complex interacting with the binding domain of human FRAP. Science 273, 239–242. [DOI] [PubMed] [Google Scholar]
- Chong AS, Shen J, Tao J, Yin D, Kuznetsov A, Hara M, and Philipson LH (2006). Reversal of diabetes in non-obese diabetic mice without spleen cell-derived beta cell regeneration. Science 311, 1774–1775. [DOI] [PubMed] [Google Scholar]
- Chornoguz O, Hagan RS, Haile A, Arwood ML, Gamper CJ, Banerjee A, and Powell JD (2017). mTORC1 Promotes T-bet Phosphorylation To Regulate Th1 Differentiation. J Immunol 198, 3939–3948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christianson SW, Shultz LD, and Leiter EH (1993). Adoptive transfer of diabetes into immunodeficient NOD-scid/scid mice. Relative contributions of CD4+ and CD8+ T-cells from diabetic versus prediabetic NOD.NON-Thy-1a donors. Diabetes 42, 44–55. [DOI] [PubMed] [Google Scholar]
- Corn RA, Aronica MA, Zhang F, Tong Y, Stanley SA, Kim SR, Stephenson L, Enerson B, McCarthy S, Mora A, et al. (2003). T cell-intrinsic requirement for NF-kappa B induction in postdifferentiation IFN-gamma production and clonal expansion in a Th1 response. J Immunol 171, 1816–1824. [DOI] [PubMed] [Google Scholar]
- D’Alise AM, Auyeung V, Feuerer M, Nishio J, Fontenot J, Benoist C, and Mathis D (2008). The defect in T-cell regulation in NOD mice is an effect on the T-cell effectors. Proc Natl Acad Sci U S A 105, 19857–19862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Cunha FM, Torelli NQ, and Kowaltowski AJ (2015). Mitochondrial Retrograde Signaling: Triggers, Pathways, and Outcomes. Oxidative medicine and cellular longevity 2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delovitch TL, and Singh B (1997). The nonobese diabetic mouse as a model of autoimmune diabetes: immune dysregulation gets the NOD. Immunity 7, 727–738. [DOI] [PubMed] [Google Scholar]
- Devadas S, Zaritskaya L, Rhee SG, Oberley L, and Williams MS (2002). Discrete Generation of Superoxide and Hydrogen Peroxide by T Cell Receptor Stimulation. The Journal of experimental medicine 195, 59–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirice E, Kahraman S, De Jesus DF, El Ouaamari A, Basile G, Baker RL, Yigit B, Piehowski PD, Kim M-J, Dwyer AJ, et al. (2019). Increased β-cell proliferation before immune cell invasion prevents progression of type 1 diabetes. Nature Metabolism 1, 509–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Donia M, Mangano K, Amoroso A, Mazzarino MC, Imbesi R, Castrogiovanni P, Coco M, Meroni P, and Nicoletti F (2009). Treatment with rapamycin ameliorates clinical and histological signs of protracted relapsing experimental allergic encephalomyelitis in Dark Agouti rats and induces expansion of peripheral CD4+CD25+Foxp3+ regulatory T cells. J Autoimmun 33, 135–140. [DOI] [PubMed] [Google Scholar]
- Feuerer M, Shen Y, Littman DR, Benoist C, and Mathis D (2009). How punctual ablation of regulatory T cells unleashes an autoimmune lesion within the pancreatic islets. Immunity 31, 654–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchtenbusch M, Larger E, Thebault K, and Boitard C (2005). Transfer of diabetes from prediabetic NOD mice to NOD-SCID/SCID mice: association with pancreatic insulin content. Horm Metab Res 37, 63–67. [DOI] [PubMed] [Google Scholar]
- Gabriel SS, Bon N, Chen J, Wekerle T, Bushell A, Fehr T, and Cippa PE (2016). Distinctive Expression of Bcl-2 Factors in Regulatory T Cells Determines a Pharmacological Target to Induce Immunological Tolerance. Front Immunol 7, 73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galluzzi L, Kepp O, Trojel-Hansen C, and Kroemer G (2012). Mitochondrial control of cellular life, stress, and death. Circulation research 111, 1198–1207. [DOI] [PubMed] [Google Scholar]
- Gubser PM, Bantug GR, Razik L, Fischer M, Dimeloe S, Hoenger G, Durovic B, Jauch A, and Hess C (2013). Rapid effector function of memory CD8+ T cells requires an immediate-early glycolytic switch. Nat Immunol 14, 1064–1072. [DOI] [PubMed] [Google Scholar]
- Gwinn DM, Shackelford DB, Egan DF, Mihaylova MM, Mery A, Vasquez DS, Turk BE, and Shaw RJ (2008). AMPK phosphorylation of raptor mediates a metabolic checkpoint. Mol Cell 30, 214–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haxhinasto S, Mathis D, and Benoist C (2008). The AKT-mTOR axis regulates de novo differentiation of CD4+Foxp3+ cells. J Exp Med 205, 565–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herold KC, Bundy BN, Long SA, Bluestone JA, DiMeglio LA, Dufort MJ, Gitelman SE, Gottlieb PA, Krischer JP, Linsley PS, et al. (2019). An Anti-CD3 Antibody, Teplizumab, in Relatives at Risk for Type 1 Diabetes. N Engl J Med 381, 603–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu BT, and Chen WZ (2018). MOTS-c improves osteoporosis by promoting osteogenic differentiation of bone marrow mesenchymal stem cells via TGF-beta/Smad pathway. Eur Rev Med Pharmacol Sci 22, 7156–7163. [DOI] [PubMed] [Google Scholar]
- Inoki K, Ouyang H, Zhu T, Lindvall C, Wang Y, Zhang X, Yang Q, Bennett C, Harada Y, Stankunas K, et al. (2006). TSC2 Integrates Wnt and Energy Signals via a Coordinated Phosphorylation by AMPK and GSK3 to Regulate Cell Growth. 126, 955–968. [DOI] [PubMed] [Google Scholar]
- Jackson SH, Devadas S, Kwon J, Pinto LA, and Williams MS (2004). T cells express a phagocyte-type NADPH oxidase that is activated after T cell receptor stimulation. Nat Immunol 5, 818–827. [DOI] [PubMed] [Google Scholar]
- Jones RG, Bui T, White C, Madesh M, Krawczyk CM, Lindsten T, Hawkins BJ, Kubek S, Frauwirth KA, Wang YL, et al. (2007). The Proapoptotic Factors Bax and Bak Regulate T Cell Proliferation through Control of Endoplasmic Reticulum Ca2+ Homeostasis. Immunity 27, 268–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones RG, and Pearce EJ (2017). MenTORing Immunity: mTOR Signaling in the Development and Function of Tissue-Resident Immune Cells. Immunity 46, 730–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kanehisa M, and Goto S (2000). KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res 28, 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katsarou A, Gudbjornsdottir S, Rawshani A, Dabelea D, Bonifacio E, Anderson BJ, Jacobsen LM, Schatz DA, and Lernmark A (2017). Type 1 diabetes mellitus. Nat Rev Dis Primers 3, 17016. [DOI] [PubMed] [Google Scholar]
- Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, Tempst P, and Sabatini DM (2002). mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 110, 163–175. [DOI] [PubMed] [Google Scholar]
- Kim J, and Guan K-L (2019). mTOR as a central hub of nutrient signalling and cell growth. Nature Cell Biology 21, 63–71. [DOI] [PubMed] [Google Scholar]
- Kim KH, Son JM, Benayoun BA, and Lee C (2018a). The Mitochondrial-Encoded Peptide MOTS-c Translocates to the Nucleus to Regulate Nuclear Gene Expression in Response to Metabolic Stress. Cell metabolism 28, 516–524 e517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KH, Son JM, Benayoun BA, and Lee C (2018b). The Mitochondrial-Encoded Peptide MOTS-c Translocates to the Nucleus to Regulate Nuclear Gene Expression in Response to Metabolic Stress. Cell Metab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koch MA, Tucker-Heard G, Perdue NR, Killebrew JR, Urdahl KB, and Campbell DJ (2009). The transcription factor T-bet controls regulatory T cell homeostasis and function during type 1 inflammation. Nat Immunol 10, 595–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolb H, and von Herrath M (2017). Immunotherapy for Type 1 Diabetes: Why Do Current Protocols Not Halt the Underlying Disease Process? Cell Metab 25, 233–241. [DOI] [PubMed] [Google Scholar]
- Kornberg MD, Bhargava P, Kim PM, Putluri V, Snowman AM, Putluri N, Calabresi PA, and Snyder SH (2018). Dimethyl fumarate targets GAPDH and aerobic glycolysis to modulate immunity. Science 360, 449–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C, Yen K, and Cohen P (2013). Humanin: a harbinger of mitochondrial-derived peptides? Trends Endocrinol Metab 24, 222–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C, Zeng J, Drew BG, Sallam T, Martin-Montalvo A, Wan J, Kim SJ, Mehta H, Hevener AL, de Cabo R, et al. (2015). The mitochondrial-derived peptide MOTS-c promotes metabolic homeostasis and reduces obesity and insulin resistance. Cell metabolism 21, 443–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li DS, Yuan YH, Tu HJ, Liang QL, and Dai LJ (2009). A protocol for islet isolation from mouse pancreas. Nat Protoc 4, 1649–1652. [DOI] [PubMed] [Google Scholar]
- Li H, Ren K, Jiang T, and Zhao GJ (2018). MOTS-c attenuates endothelial dysfunction via suppressing the MAPK/NF-kappaB pathway. Int J Cardiol 268, 40. [DOI] [PubMed] [Google Scholar]
- Long M, Park SG, Strickland I, Hayden MS, and Ghosh S (2009). Nuclear factor-kappaB modulates regulatory T cell development by directly regulating expression of Foxp3 transcription factor. Immunity 31, 921–931. [DOI] [PubMed] [Google Scholar]
- Lovell SC, Davis IW, Arendall WB 3rd, de Bakker PI, Word JM, Prisant MG, Richardson JS, and Richardson DC (2003). Structure validation by Calpha geometry: phi,psi and Cbeta deviation. Proteins 50, 437–450. [DOI] [PubMed] [Google Scholar]
- Lu Tang, Xue Liu, Wang Zhang, Luo, and Chen (2019a). Mitochondrial-Derived Peptide MOTS-c Increases Adipose Thermogenic Activation to Promote Cold Adaptation. International journal of molecular sciences 20, 2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu H, Wei M, Zhai Y, Li Q, Ye Z, Wang L, Luo W, Chen J, and Lu Z (2019b). MOTS-c peptide regulates adipose homeostasis to prevent ovariectomy-induced metabolic dysfunction. J Mol Med (Berl). [DOI] [PubMed] [Google Scholar]
- Lu H, Wei M, Zhai Y, Li Q, Ye Z, Wang L, Luo W, Chen J, and Lu Z (2019c). MOTS-c peptide regulates adipose homeostasis to prevent ovariectomy-induced metabolic dysfunction. J Mol Med (Berl) 97, 473–485. [DOI] [PubMed] [Google Scholar]
- Magnuson AM, Thurber GM, Kohler RH, Weissleder R, Mathis D, and Benoist C (2015). Population dynamics of islet-infiltrating cells in autoimmune diabetes. Proc Natl Acad Sci U S A 112, 1511–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcin Sven, Kamiński M, Opp S, Ruppert T, Grigaravičius P, Grudnik P, Gröne H-J, Peter, and Gülow K. (2012). T cell Activation Is Driven by an ADP-Dependent Glucokinase Linking Enhanced Glycolysis with Mitochondrial Reactive Oxygen Species Generation. Cell reports 2, 1300–1315. [DOI] [PubMed] [Google Scholar]
- Marek-Trzonkowska N, Mysliwiec M, Dobyszuk A, Grabowska M, Derkowska I, Juscinska J, Owczuk R, Szadkowska A, Witkowski P, Mlynarski W, et al. (2014). Therapy of type 1 diabetes with CD4(+)CD25(high)CD127-regulatory T cells prolongs survival of pancreatic islets - results of one year follow-up. Clin Immunol 153, 23–30. [DOI] [PubMed] [Google Scholar]
- Massey DC, Bredin F, and Parkes M (2008). Use of sirolimus (rapamycin) to treat refractory Crohn’s disease. Gut 57, 1294–1296 [DOI] [PubMed] [Google Scholar]
- Michael, and Richard, (2013). Mitochondrial ROS Fire Up T Cell Activation. Immunity 38, 201–202. [DOI] [PubMed] [Google Scholar]
- Ming W, Lu G, Xin S, Huanyu L, Yinghao J, Xiaoying L, Chengming X, Banjun R, Li W, and Zifan L (2016). Mitochondria related peptide MOTS-c suppresses ovariectomy-induced bone loss via AMPK activation. Biochemical and biophysical research communications 476, 412–419. [DOI] [PubMed] [Google Scholar]
- Montano Loza AJ, and Czaja AJ (2007). Current therapy for autoimmune hepatitis. Nat Clin Pract Gastroenterol Hepatol 4, 202–214. [DOI] [PubMed] [Google Scholar]
- Monti P, Scirpoli M, Maffi P, Piemonti L, Secchi A, Bonifacio E, Roncarolo MG, and Battaglia M (2008). Rapamycin monotherapy in patients with type 1 diabetes modifies CD4+CD25+FOXP3+ regulatory T-cells. Diabetes 57, 2341–2347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang W, Beckett O, Ma Q, Paik JH, DePinho RA, and Li MO (2010). Foxo proteins cooperatively control the differentiation of Foxp3+ regulatory T cells. Nat Immunol 11, 618–627. [DOI] [PubMed] [Google Scholar]
- Pearce EL, Poffenberger MC, Chang CH, and Jones RG (2013). Fueling immunity: insights into metabolism and lymphocyte function. Science 342, 1242454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pearson JA, Wong FS, and Wen L (2016). The importance of the Non Obese Diabetic (NOD) mouse model in autoimmune diabetes. Journal of autoimmunity 66, 76–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perl A (2015). mTOR activation is a biomarker and a central pathway to autoimmune disorders, cancer, obesity, and aging. Ann N Y Acad Sci 1346, 33–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell JD, and Delgoffe GM (2010). The mammalian target of rapamycin: linking T cell differentiation, function, and metabolism. Immunity 33, 301–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prochazka M, Gaskins HR, Shultz LD, and Leiter EH (1992a). The nonobese diabetic scid mouse: model for spontaneous thymomagenesis associated with immunodeficiency. Proc Natl Acad Sci U S A 89, 3290–3294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prochazka M, Gaskins HR, Shultz LD, and Leiter EH (1992b). The nonobese diabetic scid mouse: model for spontaneous thymomagenesis associated with immunodeficiency. Proceedings of the National Academy of Sciences 89, 3290–3294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pugliese A (2017). Autoreactive T cells in type 1 diabetes. Journal of Clinical Investigation 127, 2881–2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin Q, Delrio S, Wan J, Jay Widmer R, Cohen P, Lerman LO, and Lerman A (2018). Downregulation of circulating MOTS-c levels in patients with coronary endothelial dysfunction. International journal of cardiology 254, 23–27. [DOI] [PubMed] [Google Scholar]
- Quiros PM, Mottis A, and Auwerx J (2016). Mitonuclear communication in homeostasis and stress. Nat Rev Mol Cell Biol 17, 213–226. [DOI] [PubMed] [Google Scholar]
- Raijmakers RPH, Jansen AFM, Keijmel SP, Ter Horst R, Roerink ME, Novakovic B, Joosten LAB, van der Meer JWM, Netea MG, and Bleeker-Rovers CP (2019). A possible role for mitochondrial-derived peptides humanin and MOTS-c in patients with Q fever fatigue syndrome and chronic fatigue syndrome. J Transl Med 17, 157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramanjaneya M, Jerobin J, Bettahi I, Bensila M, Aye M, Siveen KS, Sathyapalan T, Skarulis M, Abou-Samra AB, and Atkin SL. (2019). Lipids and insulin regulate mitochondrial-derived peptide (MOTS-c) in PCOS and healthy subjects. Clinical Endocrinology. [DOI] [PubMed] [Google Scholar]
- Richter U, Lahtinen T, Marttinen P, Myohanen M, Greco D, Cannino G, Jacobs HT, Lietzen N, Nyman TA, and Battersby BJ (2013). A mitochondrial ribosomal and RNA decay pathway blocks cell proliferation. Curr Biol 23, 535–541. [DOI] [PubMed] [Google Scholar]
- Sarbassov DD, Guertin DA, Ali SM, and Sabatini DM (2005). Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307, 1098–1101. [DOI] [PubMed] [Google Scholar]
- Sauer S, Bruno L, Hertweck A, Finlay D, Leleu M, Spivakov M, Knight ZA, Cobb BS, Cantrell D, O’Connor E, et al. (2008). T cell receptor signaling controls Foxp3 expression via PI3K, Akt, and mTOR. Proc Natl Acad Sci U S A 105, 7797–7802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sena LA, Li S, Jairaman A, Prakriya M, Ezponda T, Hildeman DA, Wang CR, Schumacker PT, Licht JD, Perlman H, et al. (2013). Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity 38, 225–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi LZ, Wang R, Huang G, Vogel P, Neale G, Green DR, and Chi H (2011). HIF1alpha-dependent glycolytic pathway orchestrates a metabolic checkpoint for the differentiation of TH17 and Treg cells. J Exp Med 208, 1367–1376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprent J, and Surh CD (2011). Normal T cell homeostasis: the conversion of naive cells into memory-phenotype cells. Nat Immunol 12, 478–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sukumar M, Liu J, Ji Y, Subramanian M, Crompton JG, Yu Z, Roychoudhuri R, Palmer DC, Muranski P, Karoly ED, et al. (2013). Inhibiting glycolytic metabolism enhances CD8+ T cell memory and antitumor function. J Clin Invest 123, 4479–4488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thivolet C, Bendelac A, Bedossa P, Bach JF, and Carnaud C (1991). CD8+ T cell homing to the pancreas in the nonobese diabetic mouse is CD4+ T cell-dependent. J Immunol 146, 85–88. [PubMed] [Google Scholar]
- Topf U, Wrobel L, and Chacinska A (2016). Chatty Mitochondria: Keeping Balance in Cellular Protein Homeostasis. Trends in cell biology 26, 577–586. [DOI] [PubMed] [Google Scholar]
- Tsuzuki T, Nomiyama H, Setoyama C, Maeda S, Shimada K, and Pestka S (1983). The majority of cDNA clones with strong positive signals for the interferon-induction-specific sequences resemble mitochondrial ribosomal RNA genes. Biochemical and biophysical research communications 114, 670–676. [DOI] [PubMed] [Google Scholar]
- Walker LS, and von Herrath M (2016). CD4 T cell differentiation in type 1 diabetes. Clin Exp Immunol 183, 16–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Pontesilli O, Gill RG, La Rosa FG, and Lafferty KJ (1991a). The role of CD4+ and CD8+ T cells in the destruction of islet grafts by spontaneously diabetic mice. Proc Natl Acad Sci U S A 88, 527–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Pontesilli O, Gill RG, La Rosa FG, and Lafferty KJ (1991b). The role of CD4+ and CD8+ T cells in the destruction of islet grafts by spontaneously diabetic mice. Proceedings of the National Academy of Sciences 88, 527–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei M, Gan L, Liu Z, Liu L, Chang JR, Yin DC, Cao HL, Su XL, and Smith WW (2020). Mitochondrial-Derived Peptide MOTS-c Attenuates Vascular Calcification and Secondary Myocardial Remodeling via Adenosine Monophosphate-Activated Protein Kinase Signaling Pathway. Cardiorenal Med 10, 42–50. [DOI] [PubMed] [Google Scholar]
- Weinberg SE, Sena LA, and Chandel NS (2015). Mitochondria in the regulation of innate and adaptive immunity. Immunity 42, 406–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo DK, and Shadel GS (2011). Mitochondrial stress signals revise an old aging theory. Cell 144, 11–12. [DOI] [PubMed] [Google Scholar]
- Xinqiang Y, Quan C, Yuanyuan J, and Hanmei X (2020). Protective effect of MOTS-c on acute lung injury induced by lipopolysaccharide in mice. International immunopharmacology 80, 106174. [DOI] [PubMed] [Google Scholar]
- Xu X, Duan S, Yi F, Ocampo A, Liu GH, and Izpisua Belmonte JC (2013). Mitochondrial regulation in pluripotent stem cells. Cell metabolism 18, 325–332. [DOI] [PubMed] [Google Scholar]
- Yan Z, Zhu S, Wang H, Wang L, Du T, Ye Z, Zhai D, Zhu Z, Tian X, Lu Z, et al. (2019). MOTS-c inhibits Osteolysis in the Mouse Calvaria by affecting osteocyte-osteoclast crosstalk and inhibiting inflammation. Pharmacological Research 147, 104381. [DOI] [PubMed] [Google Scholar]
- Yang H, Jiang X, Li B, Yang HJ, Miller M, Yang A, Dhar A, and Pavletich NP (2017). Mechanisms of mTORC1 activation by RHEB and inhibition by PRAS40. Nature 552, 368–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeh LT, Miaw SC, Lin MH, Chou FC, Shieh SJ, Chuang YP, Lin SH, Chang DM, and Sytwu HK (2013). Different modulation of Ptpn22 in effector and regulatory T cells leads to attenuation of autoimmune diabetes in transgenic nonobese diabetic mice. J Immunol 191, 594–607. [DOI] [PubMed] [Google Scholar]
- Yin F, and Cadenas E (2015). Mitochondria: the cellular hub of the dynamic coordinated network. Antioxidants & redox signaling 22, 961–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin X, Jing Y, Chen Q, Abbas AB, Hu J, and Xu H (2020). The intraperitoneal administration of MOTS-c produces antinociceptive and anti-inflammatory effects through the activation of AMPK pathway in the mouse formalin test. European journal of pharmacology 870, 172909. [DOI] [PubMed] [Google Scholar]
- Yu J, Wei M, Becknell B, Trotta R, Liu S, Boyd Z, Jaung MS, Blaser BW, Sun J, Benson DM Jr., et al. (2006). Pro- and antiinflammatory cytokine signaling: reciprocal antagonism regulates interferon-gamma production by human natural killer cells. Immunity 24, 575–590. [DOI] [PubMed] [Google Scholar]
- Zarse K, and Ristow M (2015). A mitochondrially encoded hormone ameliorates obesity and insulin resistance. Cell metabolism 21, 355–356. [DOI] [PubMed] [Google Scholar]
- Zhai D, Ye Z, Jiang Y, Xu C, Ruan B, Yang Y, Lei X, Xiang A, Lu H, Zhu Z, et al. (2017). MOTS-c peptide increases survival and decreases bacterial load in mice infected with MRSA. Mol Immunol 92, 151–160. [DOI] [PubMed] [Google Scholar]
- Zhang C, Todorov I, Lin CL, Atkinson M, Kandeel F, Forman S, and Zeng D (2007). Elimination of insulitis and augmentation of islet beta cell regeneration via induction of chimerism in overtly diabetic NOD mice. Proc Natl Acad Sci U S A 104, 2337–2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, Chaudhry A, Kas A, deRoos P, Kim JM, Chu TT, Corcoran L, Treuting P, Klein U, and Rudensky AY (2009). Regulatory T-cell suppressor program co-opts transcription factor IRF4 to control T(H)2 responses. Nature 458, 351–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are available on request due to privacy or other restrictions.