

Abstract
Background
Although genome-wide association studies (GWAS) have identified many genomic regions associated with idiopathic pulmonary fibrosis (IPF), the causal genes and functions remain largely unknown. Many single-cell expression data have become available for IPF, and there is increasing evidence suggesting a shared genetic basis between IPF and other diseases.
Methods
We conducted integrative analyses to improve the power of GWAS. First, we calculated global and local genetic correlations to identify IPF genetically associated traits and local regions. Then, we prioritised candidate genes contributing to local genetic correlation. Second, we performed transcriptome-wide association analysis (TWAS) of 44 tissues to identify candidate genes whose genetically predicted expression level is associated with IPF. To replicate our findings and investigate the regulatory role of the transcription factors (TF) in identified candidate genes, we first conducted the heritability enrichment analysis in TF binding sites. Then, we examined the enrichment of the TF target genes in cell-type-specific differentially expressed genes (DEGs) identified from single-cell expression data of IPF and healthy lung samples.
Findings
We identified 12 candidate genes across 13 genomic regions using local genetic correlation, including the POT1 locus (p value=0.00041), which contained variants with protective effects on lung cancer but increasing IPF risk. We identified another 13 novel genes using TWAS. Two TFs, MAFK and SMAD2, showed significant enrichment in both partitioned heritability and cell-type-specific DEGs.
Interpretation
Our integrative analysis identified new genes for IPF susceptibility and expanded the understanding of the complex genetic architecture and disease mechanism of IPF.
INTRODUCTION
Idiopathic pulmonary fibrosis (IPF) is a rare and fatal disease. In recent years, several common and rare genetic variants, implicating genes involved in alveolar stability, telomere biology, host defence and cellular barrier function,1 2 have been associated with IPF. The identification and interpretation of genetic risk factors will facilitate the understanding of molecular mechanisms involved in the pathogenesis of IPF, which could potentially lead to new treatments. However, due to limited sample size, genome-wide association studies (GWAS) have only identified tens of risk loci for IPF,2–4 and the biological interpretations behind GWAS signals remain largely unknown.
Increasing evidence suggests that pleiotropy exists in complex traits, and most trait-associated loci can influence multiple traits.5 Genes such as TERT, DSP and FAM13A have been consistently identified for IPF, chronic obstructive pulmonary disease (COPD) and lung cancer.6 Some transcriptomic pathways and metabolite regulations are also shared between COPD and IPF.7 8 These findings suggest that the novel IPF genetic risk factors could be identified by leveraging shared genetics between traits. Recent developments in multitrait analysis have led to the emergence of new methods that study the shared genetic basis across multiple phenotypes.9–14 In particular, global genetic correlation10 and local genetic correlation13 14 measure genetic similarity from different angles to understand the shared genetic architecture between traits. Multitrait association analysis,11 15–17 such as multitrait association mapping (MTAG),11 can substantially improve GWAS power by leveraging genetic correlation.
Transcriptomic studies provide abundant resources to identify novel biomarkers and biological interpretations for IPF risk loci. Data generated from large consortium efforts such as the genotype-tissue expression (GTEx) project have provided comprehensive functional annotations for single nucleotide polymorphisms (SNPs). Integrating these data help biological interpretation of IPF GWAS results and prioritise potential effector genes. For example, transcriptome-wide association studies (TWAS)18 map genetic effects to gene expression and test for the association between predicted gene expression and trait. Methods like UTMOST (Unified Test for MOlecular Signa-Tures)19 jointly impute gene expression from multiple tissues to improve power and accuracy.
This manuscript aims to identify novel genetic risk factors for IPF through multitrait modelling and TWAS to improve statistical power (figure 1). First, we investigated the genetic correlation between IPF and other traits.20 Then, we estimated local genetic correlation14 of IPF top-correlated traits to further identify correlated local regions and prioritised 12 candidate genes from the identified regions. Second, we applied UTMOST19 using expression quantitative trait loci (eQTL) data from GTEx to identify 13 additional candidate risk genes. To replicate our findings, we investigated the expression patterns of candidate genes in single-cell RNA sequencing data (scRNA-Seq). 21 22 We demonstrated the regulatory role of two transcription factors (TFs), MAFK and SMAD2, in IPF through heritability enrichment analysis and cell-type-specific differential expression analysis on their target genes.
Figure 1.
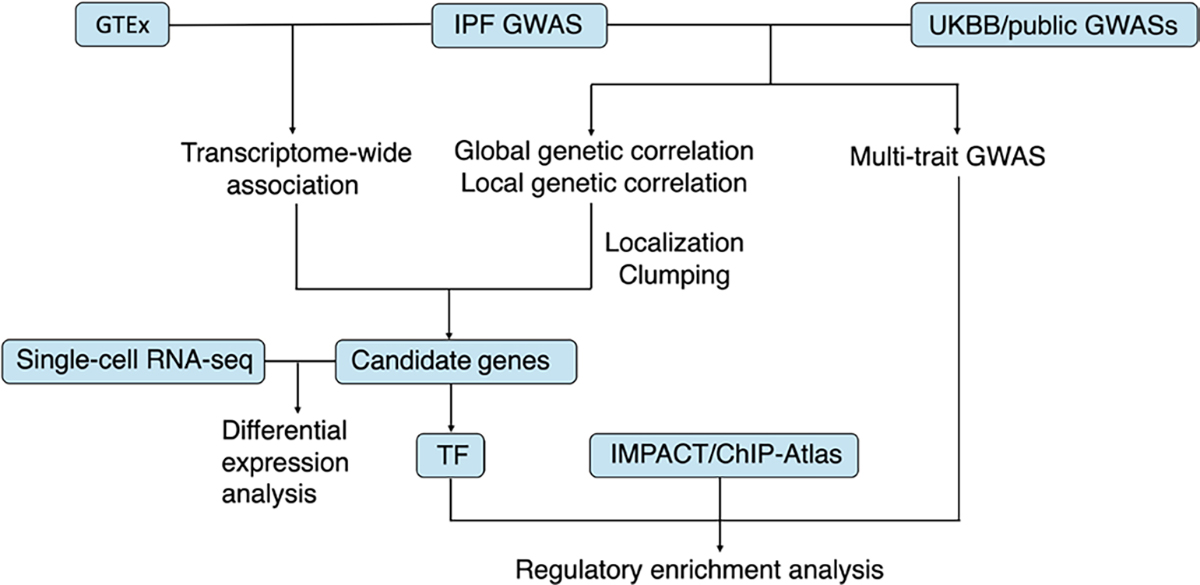
A schematic illustration of the integrated workflow. GTEx is a public database for genotype and tissue expression data. IMPACT is a genomic annotation tool of cell-state-specific regulatory elements inferred from the epigenome of bound transcription factors. ChIP-Atlas is a public database for ChIP-seq data. GTEx, The Genotype-Tissue Expression project; GWAS, genome-wide association study; TF, transcription factor; UKBB, UK Biobank.
METHODS
Analytic strategies
Figure 1 shows the overall workflow of our study. We employed two integrative frameworks to improve the power of the original IPF GWAS.2 For the first framework, we integrated IPF and another trait’s GWAS data to leverage the shared genetic effect. First, we investigated the global genetic correlation between IPF and traits from the UK Biobank (UKBB) (figure 2A). Then, we selected top correlated traits to identify genomic regions having significant local genetic correlations (figure 2B; online supplemental figures 1–9). Finally, we identified genes harbouring the leading SNPs in the correlated local regions as IPF candidate genes (online supplemental table 3). For the second framework, we integrated GWAS and eQTL data of 44 tissues from the GTEx database. We used UTMOST joint test to identify genes whose genetically regulated expressions showed associations with IPF (online supplemental table 5). To replicate our findings and understand the biological implications of the newly identified genes, we investigated the differential expression pattern of candidate genes using scRNA-Seq data from IPF and healthy distal lung parenchyma samples (figure 3). For two TFs, MAFK and SMAD2, among our identified genes, we validated their regulatory role in the pathogenesis of IPF through heritability enrichment analysis and target gene enrichment analysis. For heritability enrichment analysis, we applied multitrait analysis to improve the power of IPF GWAS and tested for disease heritability enrichment in the binding site of the TF. For target gene enrichment analysis, we hypothesise that if the TF is related to IPF, their target genes should likely exhibit differential expression patterns in IPF versus control samples. We showed that target genes were significant enriched in differentially expressed genes (DEGs) in most cell types of scRNA-Seq data (figure 4).
Figure 2.
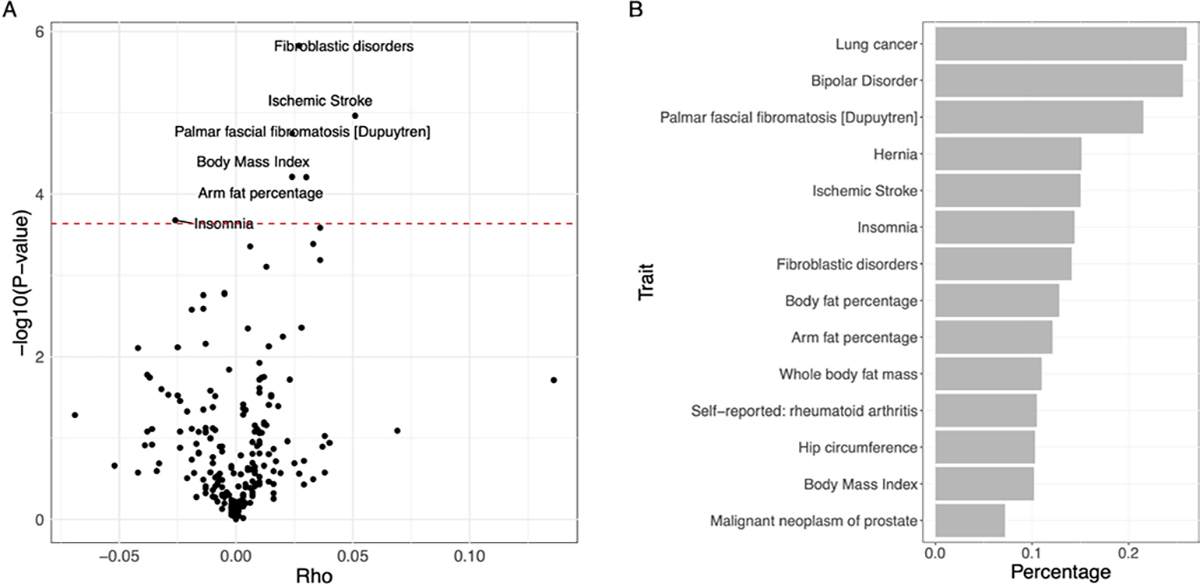
(A) Volcano plot of global genetic correlation between IPF and UK Biobank phenotypes. The red dashed line corresponds to the Bonferroni cut-off (0.05/216). Significantly associated traits were highlighted. (B) Bar plot of the proportions of correlated local regions between IPF and its genetically correlated phenotypes. IPF, idiopathic pulmonary fibrosis.
Figure 3.
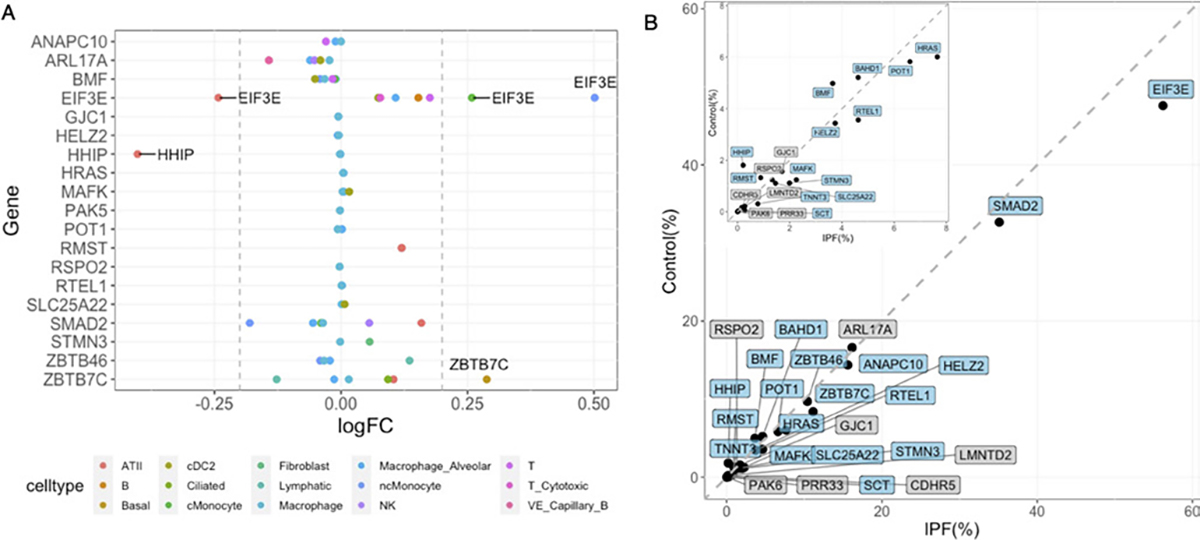
Single-cell expression patterns of 25 IPF candidate genes. (A) Dot plot of cell-type-specific differential expression. Genes with absolute logFC>0.2 and passed Bonferroni p-value cut-off (0.05/360) are labelled. (B) Proportions of cells expressing candidate genes in IPF and healthy lung samples. Genes with significantly different proportions are highlighted in blue (two-proportions z-test with Bonferroni cut-off as 0.05/25). The grey dashed line represents y=x. The panel in the left upper corner zooms in the axis between 0 to 8. IPF, idiopathic pulmonary fibrosis.
Figure 4.
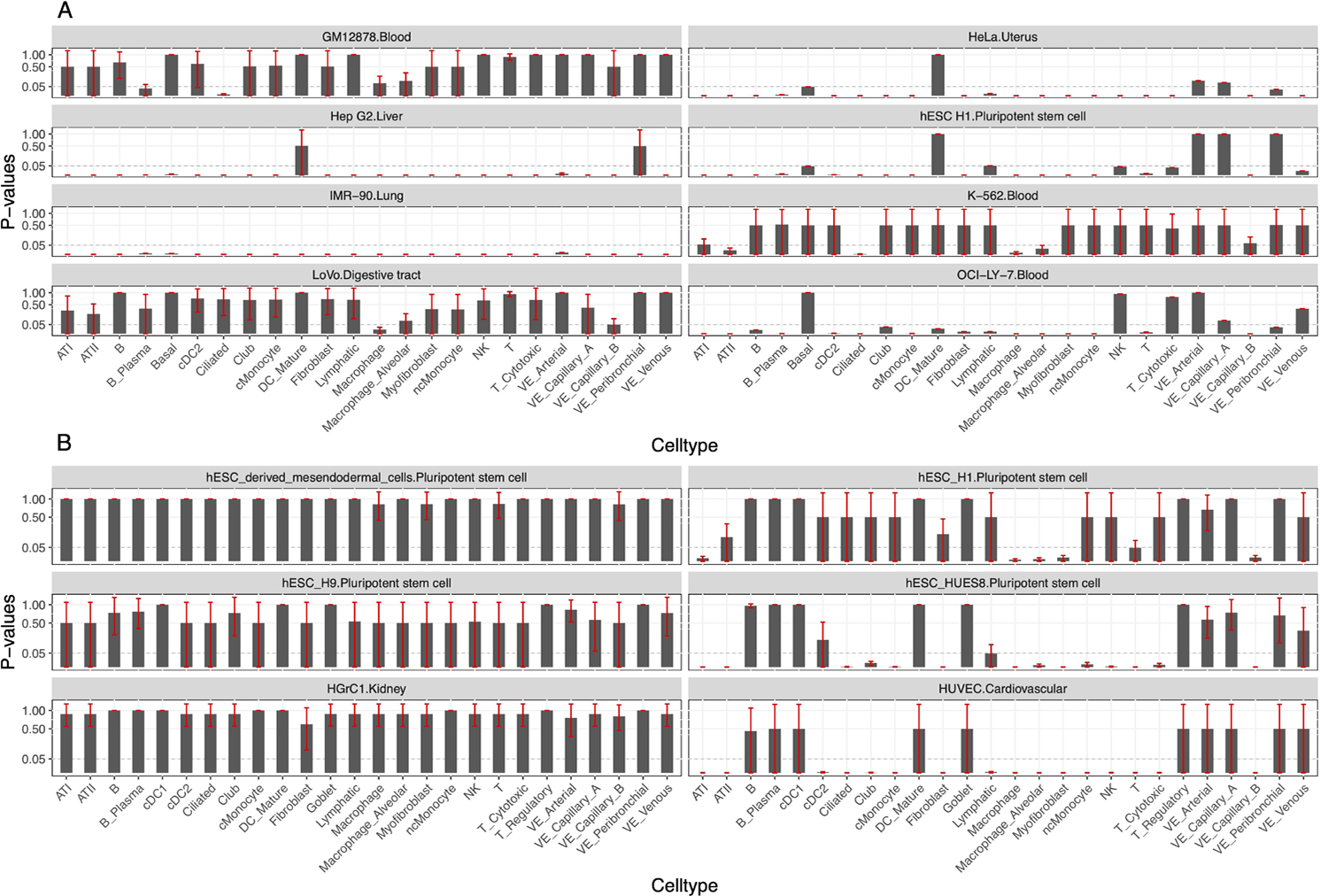
Bar plot for the enrichment analysis of MAFK and SMAD2 regulated genes among cell-type-specific DEGs between IPF and healthy lung samples. (A) Each panel represents results using MAFK target gene datasets from one cell line. For each panel, the bar plot represents hypergeometric test p-values after Bonferroni correction (0.05/448). (B) Each panel represents results using SMAD2 target gene datasets from one cell line. For each panel, the bar plot represents hypergeometric test p-values after Bonferroni correction (0.05/1209). The error bar for p-values is plotted in red. The dashed line corresponds to y=0.05. Only cell lines with at least one significant result were plotted. DEGs, differentially expressed genes.
Genetic correlation
Global genetic correlation calculates the correlation of genetic effects of SNP on two traits. We estimated the genetic correlation between IPF and other phenotypes using the software GNOVA.20 GWAS summary statistics were from (1) UKBB and (2) non-UKBB. For UKBB, we obtained the second-round GWAS results. For non-UKBB data, we downloaded summary statistics for 31 traits from publicly available GWAS results. These 31 traits included GWAS well-studied traits with large sample sizes from different disease types (neuropsychiatric, immune, cancer, metabolic). Details of the non-UKBB GWASs are summarised in online supplemental table 9. All the GWASs were performed on samples majorly from European ancestry. We removed traits whose estimated heritability is less than 0.01 to reduce the uncertainty of correlation estimation. Together we calculated the genetic correlation between IPF and 216 phenotypes.
Local genetic correlation and prioritisation of candidate genes
Local genetic correlation provided additional information besides global genetic correlation as local regions can have heterogeneous correlation patterns. To ensure that the approximation was valid, we used the region partition provided by SUPERGNOVA and prefiltered regions with <250 SNPs shared between traits. To ensure the power and robustness of estimation, we selected 14 IPF genetically correlated traits with absolute genetic covariance>0.01 and FDR-adjusted p-value<0.05. We used FDR instead of Bonferroni correction to select more candidate traits. We applied SUPERGNOVA 14 to estimate and test the local genetic correlation for each candidate trait. We quantified the degree of local correlation and prioritised genes using the R package ashr23 and PLINK software. Details for gene prioritisation and sensitivity analyses can be found in online supplemental notes.
Transcriptome-wide association study
We used a joint-tissue TWAS method called UTMOST (URLs)19 to identify IPF-associated genes with its built-in gene expression imputation model. UTMOST provided imputation models of 44 tissues from the GTEx24 database and a cross-tissue joint test to improve the power. Details of UTMOST are found in online supplemental notes. Details of the tissue source are found in the GTEx portal. We used the joint test results to identify the associated genes. P value cut-off is 0.05/390625 using Bonferroni correction. We applied conditional analysis to prioritise candidate genes located in the same genomic region within 1Mb pairs.
Multitrait analysis and partitioned heritability
To explore TFs among candidate genes, we applied LDSC 25 to test the enrichment of the partitioned IPF heritability in each TF’s binding site.2 Annotations of TF binding sites were cell line-specific and were obtained from the IMPACT26 study. We conditioned the analysis on the 52 baseline annotations in LDSC. To improve the power of IPF GWAS, the above procedure was also performed on the IPF GWAS summary statistics adjusted by MTAG.11 MTAG is a multitrait framework leveraging the correlation of genetic effects to boost the power of the original GWAS. To ensure the accuracy and robustness of estimation, in addition to the criteria, we used to select candidate traits for local genetic correlation analysis, we selected traits that had estimated heritability >0.2 for MTAG adjustment. As the results, four traits, including whole-body fat mass, body fat percentage, arm fat percentage and hip circumference, were used for pairwise MTAG with IPF. As the result, we obtained updated IPF GWAS adjusted by MTAG with each of the four traits and conducted the same heritability enrichment analyses as above.
Single-cell expression analysis
We used the scRNA-Seq data of 2 39 707 cells and 38 cell types from 32 patients with IPF and 28 healthy distal lung parenchyma samples for single-cell expression analysis. Tissue procurement, sample processing and data quality control were performed.21 We obtained cell-type-specific DEGs using the R package Seurat 27 and the MAST28 hurdle test in the R package. To test the enrichment of TF target genes in DEGs for each cell type, we applied the hypergeometric test using cell-type-specific DEGs and cell-line-specific TF target gene sets (online supplemental table 6) predicted from the ChIP-Atlas (URLs).29 30 We also evaluated the difference in the proportions of cells that express candidate genes in IPF and healthy samples using the two-proportions z-test. Bonferroni correction is applied to obtain the p value cut-off (0.05/25). Details are found in the online supplemental notes.
RESULTS
Genetic correlation between IPF and UKBB traits
The workflow of our study is found in figure 1. To understand the genetic similarities between IPF and other traits, we identified six traits having significant genetic correlations with IPF (figure 2A; online supplemental table 1). Top correlated traits are fibroblastic disorders (ρ=0.027, p value=1.50E-6), ischaemic stroke (ρ=0.051, p value=1.09E-5) and body fat-related traits, for example, body mass index (ρ=0.024, p value=6.14E-5).
To further locate genomic regions contributing to genetic similarities, we calculated the local genetic correlation between IPF and top correlated traits (the Methods section). Among these traits, 10 showed significant local region correlations across 13 local regions (online supplemental table 2). Based on local genetic correlation analysis, we estimated the proportion of correlated regions over whole-genome regions for each trait pair (figure 2B). Although the global genetic correlation between IPF and lung cancer only ranks middle in terms of both the correlation strength and significance level (ρ=−0.028, p value=0.0085), it has the largest proportion (26%) of correlated regions. In addition, we found that similar phenotypes were more likely to be correlated with IPF at the same genomic regions (online supplemental table 2). For example, there was significant local genetic correlation at a region (chr8:108,646,968–110,761,074) between IPF and fibroblastic disorders (ρ=0.0015, p value=3.55E-8) and palmar fascial fibromatosis (ρ=0.0014, p value=2.77E-8).
Local genetic correlation and TWAS-identified candidate IPF risk factors
Local genetic correlation analysis helps to identify local genomic regions that contribute to both traits. We plotted Manhattan plots for both traits on each local region (online supplemental figures 1–9). Both IPF and the other trait had at least one nominal signal (p value <0.001) standing out from the local region’s background. These findings motivated us to further locate pleiotropic genes. Thus, we investigated the overlapped signals in the local regions from both GWASs and identified 12 candidate genes. Many of these genes have been reported to be either directly related to IPF or related pathogenic functions. For example, POT1 and RTEL1 are related to telomere maintenance. Many are related to fibrosis signalling pathways, such as RSPO2 is related to Wnt/β-catenin signalling, and EIF3E and SMAD2 are related to TGF-β signalling. The detailed information for these 12 genes is found in online supplemental table 3.
TWAS incorporates eQTL information to improve the statistical power and biological interpretability of GWAS results. Thirty-seven genes were identified as significant for IPF through the UTMOST TWAS test using 44 GTEx tissues, and 36 of these genes remained significant after conditional analysis (online supplemental table 4). Of these genes, 23 of them have been reported in IPF GWAS, TWAS or found to be differentially expressed in patients with IPF versus healthy individuals.4 8 31–35 For the 13 novel genes, 6 are in different risk loci from the other 23 genes (online supplemental figures 10–12). Many of these genes are related to human carcinogenesis such as HRAS or the metabolic reprogramming for the formation of fibroblast in IPF such as SLC25A22. The detailed information for these 13 genes is found in online supplemental table 5.
Altogether, we have 25 new candidate genes, 12 were from local genetic correlation and 13 were from TWAS. Next, we investigated their expression patterns using scRNA-Seq data from IPF and control lungs. Three genes, EIF3E, HHIP and ZBTB7C (figure 3A), were found to be differentially expressed in patients with IPF versus healthy controls (adjusted p value <5.63E-5) in alveolar epithelial type II (ATII), classic monocyte, non-classic monocyte and basal cell type. Seventeen out of 25 genes have a significantly higher proportion of cells expressing them in IPF than control individuals (figure 3B).
The regulatory role of candidate TFs in IPF
There are seven TFs among the 25 identified candidate genes, including BAHD1, EIF3E, HELZ2, MAFK, SMAD2, ZBTB7C and ZBTB46. MAFK was identified using TWAS, and the rest TFs were identified by local genetic correlation. Their regulatory roles in IPF are of particular interest. We screened their target genes using available genomic annotations. MAFK is the only gene that has predicted TFBSs in four cell lines in the IMPACT data set. For ChIP-Atlas, MAFK has 13 predicted target gene sets across eight cell lines, and SMAD2 has 39 predicted target gene sets across 12 cell lines. The regulatory information of the other TFs is unavailable. Therefore, we conducted partitioned heritability analysis for MAFK and enrichment analysis for MAFK and SMAD2 target genes.
The partitioned IPF heritability showed strong enrichment in the TFBSs of MAFK but failed to reach a significant level after adjusting multiple tests. We applied MTAG1 on IPF GWAS summary statistics with four top genetically correlated traits (the Methods section) to boost the statistical power. The partitioned heritability of MAFK TFBSs after MTAG joint analysis had significant enrichment on all four cell lines, including the stem cell, fibroblasts cell from lung, myeloid and B cell from the blood (online supplemental table 7A). It showed similar results regardless of the trait used for MTAG. Partitioned heritability of MAFK TFBS in the lung consistently has the most significant enrichments, indicating that the regulatory effect of MAFK is most significant in IPF disease-relevant tissue. For example, arm fat percentage, which has the highest heritability and genetic covariance with IPF, showed the most significant enrichment in the lung (enrichment=1.73, p value=2.59E-7). To ensure the change of the enrichment analysis, results were not artefacts because of the auxiliary traits used in MTAG, we conducted the same partitioned heritability analysis on the original GWAS summary data of the correlated traits used in MTAG (ie, whole-body fat mass, body fat percentage, arm fat percentage and hip circumference). None showed significant enrichment for the TFBSs of MAFK in any cell type (online supplemental table 7B).
We then evaluated the enrichment of MAFK and SMAD2 target genes in the cell-type-specific DEGs between patients with IPF and healthy individuals using single-cell data. For MAFK, all its target gene sets across eight cell lines showed significant enrichment in cell-type-specific DEGs (figure 4A; online supplemental table 8A). The top enrichments were in the differential expressed genes in myofibroblast, macrophage and alveolar macrophage for the target genes in the IMR-90 cell line from the lung. For SMAD2, target gene sets from 6 out of 12 cell lines had significant enrichment in cell-type-specific DEGs (figure 4B; online supplemental table 8B). The top enrichments were in the DEGs in cell types of vascular endothelial capillary B cell, myofibroblast and alveolar macrophage cell for target genes in the human umbilical vein endothelial cells (HUVEC) cell line, a cardiovascular cell type obtained from the umbilical cord.
DISCUSSION
This is a comprehensive study to understand the genetic similarities between IPF and other traits and use integrative analysis to improve the power of GWAS. Using the large-scale biobank data, transcriptome data and genomic regulatory information, we have conducted detailed analyses on the between-trait relationship of IPF to understand the degree of pleiotropy. We employed local genetic correlation and TWAS to identify novel IPF risk genes. TWAS connects the phenotype of interest to the predicted expression level of the genes by combining GWAS and eQTL signals. Genetic correlation combines the results of two GWASs and then connects the identified SNPs to their closest genes. Finally, we investigated the biological implications of identified genes to help with future disease mechanisms and drug development research.
First, the correlation analyses deepened our understanding of the relationship between IPF and other phenotypes. Especially, we found significant genetic correlations between IPF and many body fat-related traits. Different studies have found that obesity is a common comorbidity of IPF.31 32 Lower body mass index and body weight loss seemed to be related to poor outcomes.33 Altogether, the results suggested that metabolic dysregulation is a critical contributor to the pathogenesis of IPF on a genetic basis.34
We used the latest and largest IPF GWAS study and successfully identified 25 novel genes not identified in previous GWASs. We improved the power of GWAS by leveraging local genetic correlation and mapping genetic information to gene expression. First, applying local genetic correlation provided a new angle to find disease genes. For example, malignant neoplasm of the prostate was identified to be correlated with IPF in region chr18:45,314,528–46,208,355 harbouring ZBTB7C and SMAD2. ZBTB7C is related to cell proliferation through glutamine metabolism.35 Glutamate is required for TGF-β-induced collagen protein production in lung fibroblasts,36 and increased glutamate abundance was observed in IPF lung tissue in the previous study.37 It was also found to differ in prostate cancer.38 These findings suggested that glutamine metabolism involving ZBTB7C is a shared mechanism between prostate cancer and IPF.
SMAD2 plays an important role in TGF-β-induced apoptosis of prostate epithelial cells and tumour suppression.39 40 We found that SMAD2 targets have significant enrichment in IPF cell-type-specific DEGs. There are no significant differential expression results for SMAD2, indicating that SMAD2 is more likely to influence the phenotype by regulating other genes other than changing its expression. Previous research reported that the phosphorylation of SMAD2 is closely related to IPF through TGF-β and SMAD signalling to promote extracellular matrix gene expression and fibrosis.41 42 It will be interesting to study the role of SMAD2 in the TGF-β signalling to understand the shared mechanisms of IPF and cancers.
Furthermore, RSPO2 and EIFE3E were shared between IPF, palmar fascial fibromatosis and fibroblastic disorders. Both genes are related to fibrosis, suggesting basic fibrosis signalling pathways like Wnt/β-catenin signalling and TGF-β signalling shared by IPF and other fibrosis-related diseases. HHIP was found to be shared between IPF and hip circumference. A recent paper reported HHIP as the newly identified putative myofibroblast markers using single-cell data in mouse pulmonary fibrosis.43 As a gene closely related to COPD and lung function, HHIP suggests the important role of lung development or homeostasis in the developing lung diseases.
We investigated the regulatory role of the identified TFs. MAFK was identified through the TWAS joint test of 44 tissues. The GWAS p value of the leading SNP in MAFK is only 0.0048. However, another gene, MAD1L1, is only 0.3 Mb away from MAFK (online supplemental figure 10), harbouring a strong signal with the lead SNP rs12699415 (p value=7.15E-13). Despite that the signals from standard GWAS did not identify MAFK, with the help of eQTL, TWAS identified MAFK as the associated gene at this locus mainly through the eQTL effect of SNPs located in MAD1L1 on MAFK. For TWAS single-tissue-based results, MAFK did not show significant association in the lung (p value=0.5). MAFK expressed significantly in a higher proportion of cells in patients with IPF but was not found to be differentially expressed between IPF and health samples. We found significant heritability enrichment on MAFK TFBSs of multiple tissues, especially in the lung. There are significant enrichments of MAFK target genes, especially the target genes of the lung cell line, in IPF cell-type-specific DEGs among most cell types. We believe MAFK has a regulatory effect on IPF but is less likely to change its expression from the above results. MAFK can form a heterodimer to regulate antioxidant and xenobiotic-metabolising enzyme genes.44 Studies have identified that MAFK can modulate NF-kB activity45 and can be induced by TGF-β to regulate downstream genes.46 HMOX1, regulated by MAFK, was found to play a central role in the defence against oxidative and inflammatory insults in the lung47 and is related to many pulmonary diseases. Another downstream gene, GPNMB, is related to fibrosis by inducing epithelial-mesenchymal transition. 48 49 These findings suggest that MAFK may participate in the pathogenesis of IPF through its relationship with both fibrosis and inflammatory-related processes.
Despite the above findings, our study has several limitations. First, for genetic correlation analysis, we mainly used processed GWAS summary statistics from the UKBB. Furthermore, we did not manually select the phenotype to avoid potential selection bias. However, the definition of many phenotypes was vague or general, posing challenges to interpreting the results and comparisons with other studies. For example, some of the top correlated phenotypes are ‘diseases of the nervous system’ and ‘self-reported: rheumatoid arthritis’. Their definitions are vague and heterogeneous. Nevertheless, this is also an advantage of our analysis as biobank data simultaneously enable the investigation of a wide range of phenotypes. Second, although our results suggest that local regions may contain disease risk genes that failed to be identified in GWAS, these regions might be false positives because it is difficult to locate the right-correlated genes in the local genomic regions. Although we prefiltered regions with a small number of SNPs, the best practice is to replicate the results on an independent data set. Currently, no study meets the requirements. In the future, additional studies are needed to investigate whether these candidate genes are genuinely related to IPF. Third, due to the lack of the target gene database, we only verified two TFs, and our approaches were limited to computational verification. In the future, biological experiments, such as knocking out MAFK in mice, are needed to further explore and verify the mechanism of the identified TFs in IPF. In addition, we also noticed that some well-known IPF genes like MUC5B were not identified in TWAS. This is because the UTMOST joint test needs prediction models from 44 tissues. However, take MUC5B as an example. It had available prediction models in only 11 out of the 44 tissues due to the relatively low expression in the GTEx data. In lung tissue, its association is not significant (effect size=0.036, p value=0.27). Utilising other eQTL data may help to mitigate this problem in the future.
Taken together, through the investigation of the plethora of data sets, we identified seven traits with significant genetic correlation with IPF. By integrating GWAS data with pleiotropy information and transcriptome data, we discovered 25 novel genes from local multitrait and TWAS studies. Functional analyses showed the differential expression and gene expression regulatory function among these novel genes. These findings will provide new avenues for understanding the underlying biology and investigating potential therapeutics in this deadly disease.
Supplementary Material
WHAT IS ALREADY KNOWN ON THIS TOPIC
Although genome-wide association studies have identified some genomic regions associated with IPF, the causal genes and functions remain largely unknown.
WHAT THIS STUDY ADDS
We identified 25 novel genes associated with IPF and discussed their biological implications through integrated analysis of multiple phenotypes and gene expression data. We found evidence of the regulatory functions of two transcription factors, MAFK and SMAD2, in lung tissue and major cell types in the lung.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
Our study indicated shared genetic factors between IPF and other traits. The identified gene provided new insights into the disease mechanism.
Acknowledgements
We gratefully acknowledge all the studies and databases that made GWAS summary data, expression data, and annotation data available as listed in the supplemental file. This research was conducted by using the UK Biobank resource under application numbers 29900.
Funding
LVW holds a GSK/British Lung Foundation Chair in Respiratory Research. The research was partially supported by the NIHR Leicester Biomedical Research Centre; the views expressed are those of the author(s) and not necessarily those of the NHS, the NIHR or the Department of Health. MHC is supported by R01HL135142, R01 HL137927, R01 HL089856, R01 HL147148. NK is supported by R01HL127349, R01HL141852, U01HL145567, UH2HL123886, and a generous gift from Three Lakes Partners. The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. The funding body has no role in the design of the study and collection, analysis, and interpretation of data and in writing the manuscript. HZ is supported by NIH grant R01 GM134005 and NSF grant DMS 1902903.
Footnotes
Competing interests LVW has received funding from GSK and Orion, outside of the submitted work. MHC has received grant support from GSK and Bayer, consulting or speaking fees from Genentech, AstraZeneca, and Illumina. NK served as a consultant to Biogen Idec, Boehringer Ingelheim, Third Rock, Pliant, Samumed, NuMedii, Theravance, LifeMax, Three Lake Partners, Optikira, Astra Zeneca, Veracyte, Augmanity and CSL Behring, over the last 3 years, reports Equity in Pliant and a grant from Veracyte and non-financial support from MiRagen and Astra Zeneca. NK has IP on novel biomarkers and therapeutics in IPF licensed to Biotech.
Patient consent for publication Not applicable.
Ethics approval Not applicable.
Provenance and peer review Not commissioned; externally peer reviewed.
Supplemental material This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information. We gratefully acknowledge all the studies and databases that made GWAS summary data, expression data and annotation data available as listed in the supplemental file. This research was conducted by using the UK Biobank resource under application numbers 29900. All data relevant to the study are included in the article or uploaded as supplementary information.
REFERENCES
- 1.Kaur A, Mathai SK, Schwartz DA. Genetics in idiopathic pulmonary fibrosis pathogenesis, prognosis, and treatment. Front Med 2017;4:154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Allen RJ, Guillen-Guio B, Oldham JM, et al. Genome-Wide association study of susceptibility to idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2020;201:564–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Noth I, Zhang Y, Ma S-F, et al. Genetic variants associated with idiopathic pulmonary fibrosis susceptibility and mortality: a genome-wide association study. Lancet Respir Med 2013;1:309–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fingerlin TE, Murphy E, Zhang W, et al. Genome-Wide association study identifies multiple susceptibility loci for pulmonary fibrosis. Nat Genet 2013;45:613–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Watanabe K, Stringer S, Frei O, et al. A global overview of pleiotropy and genetic architecture in complex traits. Nat Genet 2019;51:1339–48. [DOI] [PubMed] [Google Scholar]
- 6.van Moorsel CHM. Trade-Offs in aging lung diseases: a review on shared but opposite genetic risk variants in idiopathic pulmonary fibrosis, lung cancer and chronic obstructive pulmonary disease. Curr Opin Pulm Med 2018;24:309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Nobakht M Gh BF, Aliannejad R, Rezaei-Tavirani M, et al. The metabolomics of airway diseases, including COPD, asthma and cystic fibrosis. Biomarkers 2015;20:5–16. [DOI] [PubMed] [Google Scholar]
- 8.Kusko RL, Brothers JF, Tedrow J, et al. Integrated genomics reveals convergent transcriptomic networks underlying chronic obstructive pulmonary disease and idiopathic pulmonary fibrosis. Am J Respir Crit Care Med 2016;194:948–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Burgess S, Butterworth A, Thompson SG. Mendelian randomization analysis with multiple genetic variants using summarized data. Genet Epidemiol 2013;37:658–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bulik-Sullivan B, Finucane HK, Anttila V, et al. An atlas of genetic correlations across human diseases and traits. Nat Genet 2015;47:1236–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Turley P, Walters RK, Maghzian O, et al. Multi-trait analysis of genome-wide association summary statistics using MTAG. Nat Genet 2018;50): :229–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Grotzinger AD, Rhemtulla M, de Vlaming R, et al. Genomic structural equation modelling provides insights into the multivariate genetic architecture of complex traits. Nat Hum Behav 2019;3:513–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shi H, Mancuso N, Spendlove S, et al. Local genetic correlation gives insights into the shared genetic architecture of complex traits. Am J Hum Genet 2017;101:737–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang Y, Lu Q, Ye Y, et al. SUPERGNOVA: local genetic correlation analysis reveals heterogeneous etiologic sharing of complex traits. Genome Biol 2021;22:262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Porter HF, O’Reilly PF. Multivariate simulation framework reveals performance of multitrait GWAS methods. Sci Rep 2017;7:38837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cichonska A, Rousu J, Marttinen P, et al. metaCCA: summary statistics-based multivariate meta-analysis of genome-wide association studies using canonical correlation analysis. Bioinformatics 2016;32:1981–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bhattacharjee S, Rajaraman P, Jacobs KB, et al. A subset-based approach improves power and interpretation for the combined analysis of genetic association studies of heterogeneous traits. Am J Hum Genet 2012;90:821–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gusev A, Ko A, Shi H, et al. Integrative approaches for large-scale transcriptome-wide association studies. Nat Genet 2016;48:245–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hu Y, Li M, Lu Q, et al. A statistical framework for cross-tissue transcriptome-wide association analysis. Nat Genet 2019;51:568–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lu Q, Li B, Ou D, et al. A powerful approach to estimating annotation-stratified genetic covariance via GWAS summary statistics. Am J Hum Genet 2017;101:939–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Adams TS, Schupp JC, Poli S, et al. Single-Cell RNA-seq reveals ectopic and aberrant lung-resident cell populations in idiopathic pulmonary fibrosis. Sci Adv 2020;6:eaba1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Villaseñor-Altamirano AB, Moretto M, Maldonado M, et al. PulmonDB: a curated lung disease gene expression database. Sci Rep 2020;10:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stephens M False discovery rates: a new deal. Biostatistics 2017;18:275–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ardlie K, GTEx Consortium. Human genomics. The Genotype-Tissue expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science 2015;348:648–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Finucane HK, Bulik-Sullivan B, Gusev A, et al. Partitioning heritability by functional annotation using genome-wide association summary statistics. Nat Genet 2015;47:1228–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Amariuta T, Luo Y, Gazal S, et al. Impact: genomic annotation of cell-state-specific regulatory elements inferred from the epigenome of bound transcription factors. Am J Hum Genet 2019;104:879–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Stuart T, Butler A, Hoffman P, et al. Comprehensive integration of single-cell data. Cell 2019;177:1888–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Finak G, McDavid A, Yajima M, et al. Mast: a flexible statistical framework for assessing transcriptional changes and characterizing heterogeneity in single-cell RNA sequencing data. Genome Biol 2015;16:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Oki SO. T. ChIP-Atlas, 2015. Available: http://chip-atlas.org [Google Scholar]
- 30.Oki S, Ohta T, Shioi G, et al. ChIP-Atlas: a data-mining suite powered by full integration of public ChIP-Seq data. EMBO Rep 2018;19:e46255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Faverio P, De Giacomi F, Sardella L, et al. Management of acute respiratory failure in interstitial lung diseases: overview and clinical insights. BMC Pulm Med 2018;18:70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Guler SA, Hur SA, Lear SA, et al. Body composition, muscle function, and physical performance in fibrotic interstitial lung disease: a prospective cohort study. Respir Res 2019;20:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Faverio P, Bocchino M, Caminati A, et al. Nutrition in patients with idiopathic pulmonary fibrosis: critical issues analysis and future research directions. Nutrients 2020;12:1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Phan THG, Paliogiannis P, Nasrallah GK, et al. Emerging cellular and molecular determinants of idiopathic pulmonary fibrosis. Cell Mol Life Sci 2021;78:2031–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hur M-W, Yoon J-H, Kim M-Y, et al. Kr-POK (ZBTB7c) regulates cancer cell proliferation through glutamine metabolism. Biochim Biophys Acta Gene Regul Mech 2017;1860:829–38. [DOI] [PubMed] [Google Scholar]
- 36.Hamanaka RB, O’Leary EM, Witt LJ, et al. Glutamine metabolism is required for collagen protein synthesis in lung fibroblasts. Am J Respir Cell Mol Biol 2019;61:597–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kang YP, Lee SB, Lee J-M, et al. Metabolic profiling regarding pathogenesis of idiopathic pulmonary fibrosis. J Proteome Res 2016;15:1717–24. [DOI] [PubMed] [Google Scholar]
- 38.Zacharias NM, McCullough C, Shanmugavelandy S, et al. Metabolic differences in glutamine utilization lead to metabolic vulnerabilities in prostate cancer. Sci Rep 2017;7:16159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yang J, Wahdan-Alaswad R, Danielpour D. Critical role of Smad2 in tumor suppression and transforming growth factor-beta-induced apoptosis of prostate epithelial cells. Cancer Res 2009;69:2185–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brodin G, ten Dijke P, Funa K, et al. Increased Smad expression and activation are associated with apoptosis in normal and malignant prostate after castration. Cancer Res 1999;59:2731–8. [PubMed] [Google Scholar]
- 41.Kolosova I, Nethery D, Kern JA. Role of Smad2/3 and p38 MAP kinase in TGF-β1-induced epithelial-mesenchymal transition of pulmonary epithelial cells. J Cell Physiol 2011;226:1248–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Walton KL, Johnson KE, Harrison CA. Targeting TGF-β mediated Smad signaling for the prevention of fibrosis. Front Pharmacol 2017;8:461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xie T, Wang Y, Deng N, et al. Single-Cell deconvolution of fibroblast heterogeneity in mouse pulmonary fibrosis. Cell Rep 2018;22:3625–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Katsuoka F, Motohashi H, Ishii T, et al. Genetic evidence that small Maf proteins are essential for the activation of antioxidant response element-dependent genes. Mol Cell Biol 2005;25:8044–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Hwang Y-J, Lee E-W, Song J, et al. Mafk positively regulates NF-κB activity by enhancing CBP-mediated p65 acetylation. Sci Rep 2013;3:3242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Okita Y, Kamoshida A, Suzuki H, et al. Transforming growth factor-β induces transcription factors MafK and Bach1 to suppress expression of the heme oxygenase-1 gene. J Biol Chem 2013;288:20658–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Fredenburgh LE, Perrella MA, Mitsialis SA. The role of heme oxygenase-1 in pulmonary disease. Am J Respir Cell Mol Biol 2007;36:158–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bhattacharyya S, Feferman L, Sharma G, et al. Increased GPNMB, Phospho-Erk1/2, and MMP-9 in cystic fibrosis in association with reduced arylsulfatase B. Mol Genet Metab 2018;124:168–75. [DOI] [PubMed] [Google Scholar]
- 49.Katayama A, Nakatsuka A, Eguchi J, et al. Beneficial impact of Gpnmb and its significance as a biomarker in nonalcoholic steatohepatitis. Sci Rep 2015;5:16920. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data relevant to the study are included in the article or uploaded as supplementary information. We gratefully acknowledge all the studies and databases that made GWAS summary data, expression data and annotation data available as listed in the supplemental file. This research was conducted by using the UK Biobank resource under application numbers 29900. All data relevant to the study are included in the article or uploaded as supplementary information.