

Significance
Gain-of-function mutations in voltage-gated sodium channel NaV1.7 cause the severe inherited pain syndrome inherited erythromelalgia (IEM). We introduced three IEM mutations into the ancestral bacterial sodium channel NaVAb, which substitutes threonine residues in the S4-S5 linker connecting the voltage sensor to the pore. These mutants recapitulated the pathogenic negative shift in voltage dependence of activation. Structural analysis revealed a common mechanism, in which new hydrogen bonds are formed between the S4-S5 linker and the pore. Because the S4-S5 linkers couple voltage sensor movements to pore opening, these newly formed hydrogen bonds would stabilize the activated state and promote hyperexcitability. These results provide key structural insights into how IEM mutations in the S4-S5 linkers may cause severe pain in IEM.
Keywords: sodium channel, action potential, channelopathies, pain, sensory nerves
Abstract
Gain-of-function mutations in voltage-gated sodium channel NaV1.7 cause severe inherited pain syndromes, including inherited erythromelalgia (IEM). The structural basis of these disease mutations, however, remains elusive. Here, we focused on three mutations that all substitute threonine residues in the alpha-helical S4-S5 intracellular linker that connects the voltage sensor to the pore: NaV1.7/I234T, NaV1.7/I848T, and NaV1.7/S241T in order of their positions in the amino acid sequence within the S4-S5 linkers. Introduction of these IEM mutations into the ancestral bacterial sodium channel NaVAb recapitulated the pathogenic gain-of-function of these mutants by inducing a negative shift in the voltage dependence of activation and slowing the kinetics of inactivation. Remarkably, our structural analysis reveals a common mechanism of action among the three mutations, in which the mutant threonine residues create new hydrogen bonds between the S4-S5 linker and the pore-lining S5 or S6 segment in the pore module. Because the S4-S5 linkers couple voltage sensor movements to pore opening, these newly formed hydrogen bonds would stabilize the activated state substantially and thereby promote the 8 to 18 mV negative shift in the voltage dependence of activation that is characteristic of the NaV1.7 IEM mutants. Our results provide key structural insights into how IEM mutations in the S4-S5 linkers may cause hyperexcitability of NaV1.7 and lead to severe pain in this debilitating disease.
Voltage-gated sodium (NaV) channels initiate and propagate action potentials in nerves and muscles (1, 2), and mutations in NaV channels lead to many diseases of hyperexcitability (3, 4). NaV1.7 is expressed mainly in peripheral somatic and visceral sensory neurons in dorsal root ganglia and in sympathetic ganglion neurons, where it serves as a threshold channel that sets the gain of nociceptors for generation of pain signals (4, 5). Its slow rate of closed-state inactivation allows smaller and slower depolarizations to activate the channel and thereby enhances responses to small nociceptive stimuli (4, 6).
Neuropathic pain syndromes affect millions of people and cost billions of dollars in health care expenditure (7, 8). They arise from damage in peripheral nerves from injury, diabetes, autoimmune disease, and cancer chemotherapy. However, rare inherited or sporadic pain syndromes are caused by mutations in NaV1.7. Inherited erythromelalgia (IEM), paroxysmal extreme pain disorder, and small fiber neuropathy are caused by heterozygous gain-of-function mutations in NaV1.7 (4). Patients with these syndromes have episodes of intense pain, redness, and swelling in different parts of the body. No targeted treatment for these pain syndromes is available, but some pharmacological interventions are effective in selected individuals (9). On the other hand, homozygous loss-of-function mutations caused by gene truncation or deletion of NaV1.7 produce Congenital Insensitivity to Pain (CIP), a rare disorder in which patients do not feel pain despite having injuries (10). As a result, NaV1.7 is an attractive drug target for novel pain treatments (8), but the development of effective analgesics targeting NaV1.7 has been fraught with difficulties (11).
NaV1.7 belongs to the family of nine human NaV channel subtypes (NaV1.1-NaV1.9), which are expressed in different excitable tissues (12, 13). NaV channels contain 24 transmembrane (TM) segments that form four homologous, but not identical, 6-TM domains (DI to DIV). The first four TM segments in each homologous 6-TM domain (S1 to S4) form the voltage-sensing module (VS), which is connected via the alpha-helical S4-S5 intracellular linker to the pore module (PM) formed by the S5 and S6 segments and the P loop between them. Structural studies of the homotetrameric bacterial sodium channel NaVAb showed that these four homologous domains come together to form a functional channel with an ion-conducting pore at its center (13, 14). Depolarization activates NaV channels according to the “sliding-helix model”, in which Arg or Lys gating charges in the S4 segment move outward toward the extracellular side of the membrane upon depolarization (15, 16). Outward S4 movement triggers a major conformational change in the S4-S5 linkers on the intracellular side of the membrane and induces the opening of the activation gate of the pore by bending and rotation of the intracellular ends of the S6 segments (17). Similar voltage-gating mechanisms have been proposed based on structural studies of the bacterial sodium channel NaChBac (16), the voltage-sensitive phosphatase CiVSP from the ancestral eukaryote Ciona (18), the two-pore-domain channel TPC from plants and mammals (19), the sea urchin hyperpolarization- and cyclic nucleotide-gated channel (20), the human proton channel (21), the human ether-a-go-go channel (22), and the mammalian sodium channel NaV1.7 (23–25).
IEM mutations shift the voltage dependence of activation to more negative membrane potentials, allowing smaller depolarizations to trigger an action potential (4). These disease mutations cluster in DI and DII. However, the molecular and structural basis for the pathogenic effects of mutations that result in pain phenotypes in patients remains unclear. Here, we have uncovered the structural basis for the gain-of-function phenotypes caused by three IEM mutations that substitute threonine (Thr) residues in the S4-S5 linkers of NaV1.7. Although the structure of NaV1.7 has been determined by cryo-EM (26, 27), progress toward resolving the structure with disease mutations at high resolution has been limited by challenges in obtaining sufficient quantity of NaV1.7 protein (28), intrinsic mobility of the VSs, and the limited resolution of the VS afforded by cryo-EM. To overcome these limitations, we introduced these IEM mutations into NaVAb, a bacterial ancestor of mammalian NaV channels, confirmed their functional effects in negatively shifting the voltage dependence of channel activation, and determined their structures at high resolution by X-ray crystallography (14). The backbone structures of NaVAb and NaV1.7 are similar within <3 Å rmsd in their core transmembrane domains, which is identical within the limit of resolution of the cryo-EM structures (14, 26, 27). Key structural elements involved in the activation of human NaV1.7 are transferable to other sodium channels (14, 23, 24, 29). Each mutation is represented four times in the homotetrameric structure of NaVAb, facilitating structure determination at high resolution by X-ray crystallography. This approach has revealed a common pathogenic mechanism in which the hydroxyl groups of substituted Thr residues form hydrogen bonds that stabilize the activated state of the voltage sensor and thereby negatively shift the voltage dependence of activation. Our results unveil an unexpected structural and physiological basis for NaV1.7 channelopathy caused by these three IEM mutations in the S4-S5 linker at near-atomic resolution.
Results
Function of NaVAb with NaV1.7 IEM Mutations in the S4-S5 Linker.
We surveyed IEM mutations in human NaV1.7 in the literature and selected three mutations that all substitute Thr residues in the S4-S5 linkers. These three IEM mutations are located in the alpha-helical S4-S5 linkers that connect the VS and PM: I234T and S241T in NaV1.7 DI and I848T in DII, which are equivalent to NaVAb I119T, V126T, and L123T, respectively (Fig. 1A). The positions of these mutations in the S4-S5 linkers are illustrated in the side views of NaV1.7 and NaVAb in Fig. 1 B and C, respectively. As viewed from the cytosolic side, the S4-S5 linkers of NaV1.7 surround the activation gate formed by the intracellular ends of the S6 segments (Fig. 1D). They are located in analogous positions in each of the four S4-S5 linkers in NaVAb (Fig. 1E). The genetic background, phenotypic properties and biophysical effects as determined in previous studies of these mutations in NaV1.7 are summarized in SI Appendix, Tables S1 and S2.
Fig. 1.

IEM mutations in voltage sensors of NaV1.7. (A) Topology diagram of NaV1.7 with seven IEM mutations (red circles) in the VSDs and the S4-S5 linkers of DI and DII selected for this study. Equivalent IEM mutations were introduced into the homotetrameric bacterial channel NaVAb (cyan) for electrophysiological and structural characterization. (B) Structure of human NaV1.7 (PDB: 6J8I) with the locations of three IEM mutations mapped onto the model (red spheres). Each domain is colored as in A. (C) Structure of NaVAb (PDB: 3RVY) with the locations of three IEM mutations mapped onto the model (red spheres). For clarity, the IEM mutations are shown only in one subunit (teal). Due to the tetrameric nature of NaVAb, the mutations are also present in the remaining subunits (cyan). Residues are labeled with domain colors of their equivalent mutations in NaV1.7 as in B. (D) View of IEM mutations (red spheres) in NaV1.7 from the cytosolic side. (E) View of IEM mutations (red spheres) in NaVAb from the cytosolic side. The structure of NaV1.7 from panel D is superimposed (with the IEM mutations shown as pink spheres) with an rmsd of ~2.0 Å for Cα atoms in the S4-S5 linkers and PM.
We introduced each of these S4-S5 linker IEM mutations into the C-terminal-truncated construct NaVAbΔ28, which was then expressed and analyzed by whole-cell voltage clamp (30; Materials and Methods). All of these constructs conduct voltage-gated inward sodium currents that activate rapidly and inactivate more slowly than wild-type (WT) NaVAbΔ28 (Fig. 2 A–C). Their rates of activation are equal to or more rapid than NaVAb/WT in the critical voltage range from −50 mV to +10 mV, as illustrated in the plots of time-to-peak current vs. voltage in Fig. 2B. Similarly, these mutations increase the time constants for inactivation in the physiologically important voltage range of −60 to +10 mV (Fig. 2C), consistent with their hyperexcitability phenotype. In addition, the voltage dependence of activation (Va) for these mutants is negatively shifted to different extents as expected for IEM mutations (Fig. 2D). Surprisingly, the conductance/voltage (G/V) curves for mutants I119T and V126T are biphasic (Fig. 2D). Peak conductance in these G/V experiments is reached in 1 ms or less, raising the possibility that the voltage-dependent activation process of NaVAb has not reached steady state in that short time. Therefore, we repeated steady-state activation experiments for these two mutants by depolarizing to a series of test voltages for 10 ms, hyperpolarizing to a fixed negative voltage (−180 mV), and measuring the peaks of the resulting inward tail currents as a measure of pore opening (Fig. 2E). The resulting tail current G/V curves are monophasic and show a clear negative shift of Va, consistent with the hypothesis that these mutants do indeed require 1 ms to 10 ms of depolarization to reach steady-state activation. We propose a structural basis for these unexpected findings in the Discussion. Overall, the rapid activation, slowed inactivation, and negatively shifted Va of these mutants are consistent with their hyperexcitability phenotype in native NaV1.7 in vitro in transfected cells and in vivo in affected individuals.
Fig. 2.

Functional properties of NaVAb with IEM mutations. (A) Families of sodium currents. NaVAb/Δ28 WT and mutants were expressed in Hi5 insect cells and studied by whole-cell voltage clamp recording as described in Materials and Methods. (B) Time to reach peak current is plotted as a function of stimulus voltage as calculated from the results of panel A. (C) Mean time constants for inactivation calculated from the results of experiments similar to those in panel A as described in Materials and Methods. (D) Voltage dependence of activation of WT and the indicated IEM mutants in the S4-S5 linker. Current/voltage (I/V) relationships of the peak sodium currents were recorded in response to steps to voltages ranging from −160 mV to +60 mV in 10-mV increments from a holding potential of −160 mV or −180 mV. Conductance/voltage (G/V) relationships were calculated as described in Materials and Methods. (E) Voltage-dependent activation curves determined from peak tail current amplitudes measured at −180 mV. Short depolarizing pulses, 10 ms, were applied from a holding potential of −180 mV in 10-mV steps. Tail currents were normalized to the highest amplitudes. (Inset) Representative traces of inward sodium currents and tail currents for WT, I119T, and V126T.
Structures of NaVAb with IEM Mutations in S4-S5 Linkers.
We solubilized, purified, and crystallized the mutant NaVAb proteins. All structures were determined for channels embedded in lipid bicelles by X-ray crystallography at a resolution ranging from 2.7 Å to 3.1 Å, which yielded near atomic clarity in these cases (SI Appendix, Table S3). All of these structures have their VSs in the activated state, as determined by the outward position of the gating charges in the S4 segment, while the PM is closed at the activation gate formed by the intracellular ends of the S6 segments. Because the IEM mutations primarily affect the activation process, we also generated structural models of the NaVAb IEM mutants in the resting state, using the disulfide-crosslinked resting-state structure of NaVAb as a template (17). This gave insight into the conformational transitions that are altered by the IEM mutations during activation of the VS. Notably, all three of these amino acid substitutions contain hydroxyl side chains of Thr residues that form new hydrogen bonds in our structures, which would stabilize the voltage sensor in its activated conformation. In the sections below, we consider these three IEM mutations in their order in the amino acid sequence of the S4-S5 linker of NaVAb: I119T, L123T, and V126T (Fig. 1C).
NaVAb/I119T (NaV1.7/I234T).
NaVAb/Ile119 is analogous to Ile234 in the S4-S5 linker in DI of NaV1.7, which is moderately conserved by replacement with an amino acid residue with a hydrophobic side chain of medium size in all four domains (SI Appendix, Fig. S1). NaVAb/I119T shows a surprisingly large shift of −37.5 mV in Va (Fig. 2 D and E and SI Appendix, Table S2). This shift of Va is greater than the shift of −17.9 mV for NaV1.7/I234T, which was the largest negative shift among all IEM mutations in NaV1.7 (31).
The structure of NaVAb/I119T at 2.75-Å resolution (Fig. 3A) clearly indicates that the side chain of I119T forms a hydrogen bond with Ser132 on S5 of a neighboring subunit (referred to as Ser132′) (Fig. 3 A and B). Although the overall structure is similar to WT (Cα rmsd = 0.7 Å), the conformations of the hinge region between the S4-S5 linker and the S5 segment where S132′ is located, as well as the activation gate receptor region of the pore-lining S6 segment, differ from the WT structure significantly (Fig. 3C). In the S4-S5 linker/S5 hinge region, the Cα atoms of Ile119 and Ser132′ are ~8 Å apart in the WT structure (Fig. 3C). This distance is shortened to ~7 Å in the I119T mutant structure due to the formation of a strong hydrogen bond (~2.5 Å) between the oxygen atoms of the I119T and Ser132′ side chains (Fig. 3C). Notably, the methyl group of the Thr side chain retains the hydrophobic interaction of Ile119 with Val133 in S5, further strengthening the interaction of these two side chains (Fig. 3B).
Fig. 3.
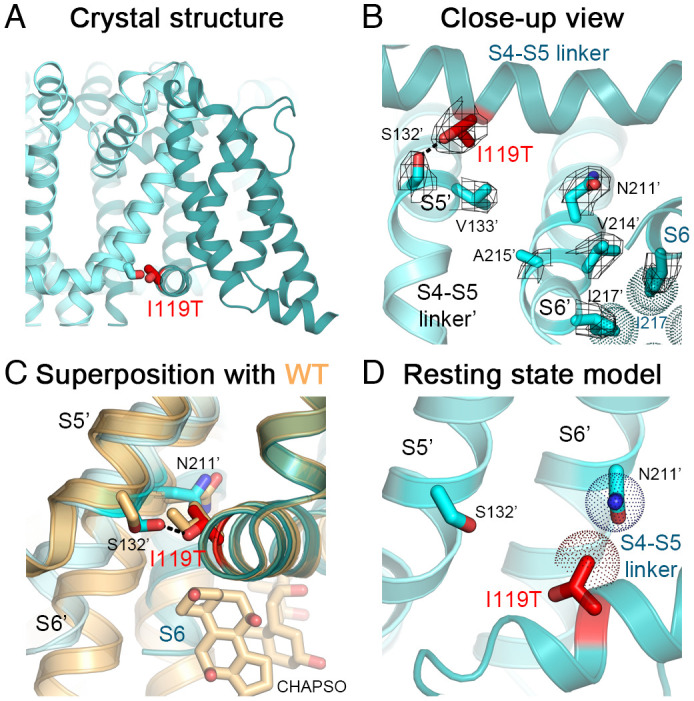
Structure of NaVAb/I119T. (A) Crystal structure of NaVAb/I119T with the mutation shown. (B) Close-up view of the NaVAb/I119T structure with the VS in the activated state (teal and cyan for different subunits). Side chains of residues I119T (red), I217 (teal) and key side chains in S5 and S6 of a neighboring subunit (cyan) including Ser132′ and Asn211′ are shown as sticks with the σA-weighted 2FO-FC electron density map contoured at 1.0σ level overlaid (mesh). (C) Comparison of NaVAb/I119T structure with the structure of NaVAb WT (light orange). Side chain of I119T forms a hydrogen bond (black dashes) with side chain of Ser132′. Conformational changes in the C-terminal end of the S6 segments starting from Asn211′ in the mutant structure abolish the CHAPSO binding site present in the WT structure. (D) Homology model of NaVAb/I119T structure in the resting state. The van der Waals hemispheres are shown as dots. NaVAbΔ28 (PDB: 6MWA) was used for the comparison.
In human NaV1.7, Asn857 in DII S5 is the equivalent residue to NaVAb Ser132′; therefore, the I234T mutation in the S4-S5 linker of DI of NaV1.7 is well positioned to form a hydrogen bond with the side chain of Asn857 in DII-S5 in the activated state, similar to NaVAb/Ser132′. This newly formed hydrogen bond would stabilize the conformation of the activated state and thereby contribute to the negative shift in Va.
The S6 segment structure also exhibits a conformational change as it approaches the activation gate in NaVAb/I119T, which starts with a kink around the conserved Asn211 residue and propagates by a rotation that causes a larger shift of ~4 Å at the last residue of S6 (Ala220) (Fig. 3C). This structural change may be induced by the formation of the hydrogen bond between I119T and Ser132′ and may contribute to its stabilization of the activated state and its unusually large negative shift of activation. The conformational change observed here disrupts two binding sites for CHAPSO detergent molecules from the bicelles that normally wedge between the S4-S5 linker and the pore-lining S6 segment in the WT structure, such that no electron density for the CHAPSO molecules was present in the mutant structure. Despite the conformational change in the activation gate, the pore structure of the mutant remains closed, with Ile217 forming a constriction site that seals the activation gate of the pore (Fig. 3B), similar to the WT structure.
The resting-state model of NaVAb/I119T suggests that the I119T side chain forms weaker van der Waals interactions with the outer surface of the pore module from the neighboring subunit. Notably, the I119T side chain makes van der Waals contacts with the conserved Asn211′ residue from S6 with a distance of ~4 Å (Fig. 3D), similar to that for the native Ile residue found in the resting state of NaVAb. Therefore, there is no evidence for a significant gain in stability by this mutation in the resting state. The gain of the hydrogen bond in the activated state from the I119T mutation, plus the conformational change induced in the S6 segment along with hydrogen bond formation, likely provides extra free energy that stabilizes the activated state and leads to channel hyperexcitability (Movie S1).
NaVAb/L123T (NaV1.7/I848T).
NaVAb/Leu123 is analogous to Ile848 in DII of NaV1.7, which is conservatively replaced by Leu in the other three domains of NaV1.7 (SI Appendix, Fig. S1). This conservation of Leu and Ile residues suggests an important role for a hydrophobic side chain at this position. Electrophysiological recordings of NaVAb/L123T show a shift of −15.5 mV in Va (Fig. 2D and SI Appendix, Table S2), similar to negative shifts in Va of −13.8 mV to −7.5 mV in previous studies of NaV1.7/I848T (32–34).
The structure of NaVAb/L123T in the activated state at 2.7-Å resolution indicates that the substituted Thr side chain forms a hydrogen bond with the highly conserved Asn211 side chain in the S6 segment of a neighboring subunit (referred to as Asn211′) (Fig. 4 A and B). Asn211 is a conserved residue that is important for electromechanical coupling and critical for the gating process by bridging interactions between the S4-S5 linker and the S6 activation gate (17). In the WT structure, Asn211′ forms part of the wall of the ion-conducting pore with its side chain pointing toward the lumen, while also interacting with Val126 in the S4-S5 linker (Fig. 4C). In the L123T structure, the Asn211′ side chain adopts a different rotamer that points away from the lumen of the pore toward the S4-S5 linker and forms a hydrogen bond with a distance of ~3.1 Å to the hydroxyl oxygen of the L123T side chain (Fig. 4 B and C). The hydrogen bond formed between these two residues from the S4-S5 linker and the S6′ segment would provide additional stabilization of the activated state and therefore significantly shift voltage-dependent activation to more negative membrane potentials.
Fig. 4.
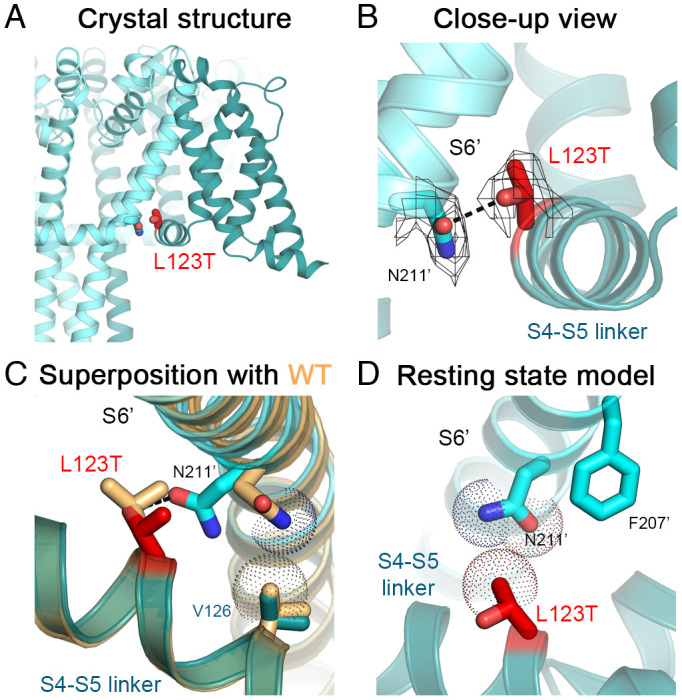
Structure of NaVAb/L123T. (A) Crystal structure of NaVAb/L123T with the mutation shown. (B) Close-up view of NaVAb/L123T structure with the VS in the activated state (teal and cyan for different subunits). Side chains of residues L123T (red) and Asn211′ (cyan) are shown as sticks with the σA-weighted 2FO-FC electron density map contoured at 1.0σ level overlaid (mesh). (C) Comparison of NaVAb/L123T structure with the structure of NaVAb WT (light orange). Side chain of N211′ makes a van der Waals contact with the side chain of V126 in the WT structure but rotates to form a hydrogen bond (black dashes) with side chain of L123T in the mutant structure. The van der Waals hemispheres are shown as dots. (D) Homology model of NaVAb/L123T structure in the resting state.
Modeling NaVAb/L123T in the resting state suggests that the side chain of L123T makes van der Waals interactions with the conserved Asn211' side chain of an adjacent subunit (Fig. 4D). In contrast, the native Leu side chain in the NaVAb resting state makes an additional hydrophobic interaction with the conserved Phe207’ residue located one helical turn from Asn211′. This interaction is absent in the L123T structure as the length of the side chain is reduced by the mutation, suggesting destabilization of the resting state by this mutation.
In human NaV1.7, Phe1435 and Asn1439 in DIII S6 are found at the position equivalent to NaVAb/Phe207 and NaVAb/Asn211, respectively. Similarly, the I848T mutation likely abolishes the hydrophobic interaction with Phe1435 in the resting state, while creating a new hydrogen bond with Asn1439 in the activated state. Therefore, the loss of the hydrophobic interaction in the resting state and the gain of a hydrogen bond in the activated state both likely contribute to the large negative shift in Va (Movie S2).
NaVAb/V126T (NaV1.7/S241T).
NaVAb/Val126 is analogous to NaV1.7/Ser241. It is conserved in DI, DII, and DIV of NaV1.7 but is replaced by Ala in DIII (SI Appendix, Fig. S1). Electrophysiological recordings of NaVAb/V126T showed a shift of −12 mV in Va (Fig. 2 D and E and SI Appendix, Table S2), similar to the shift of −8.4 mV for NaV1.7/S241T (35). These results indicate that NaVAb/V126T is a valid structural model for this IEM mutation.
The crystal structure of NaVAb/V126T at 3.1-Å resolution (Fig. 5A) suggests that the V126T side chain forms a long-range hydrogen bond with the side chain of the Asn211′ residue in the S6 segment of a neighboring subunit (Fig. 5B). The V126T side chain rotates almost 180° from the Val side chain in the WT structure, twisting around the axis of the Cα–Cβ bond to reorient the oxygen atom toward the Asn211′ side chain with a distance of ~3.4 Å while maintaining van der Waals interaction with the side chain of Ile216 on S6 (Fig. 5C). In the WT structure, Val126 interacts with Asn211′ through van der Waals interactions with a distance of ~4 Å. The V126T mutation changes the chemistry of the interaction to a hydrogen bond while preserving the volume of the side chain and the hydrophobic interaction with Ile216. The hydrogen bond between Val126T and Asn211, which is absent in the resting-state homology model (Fig. 5D and (17)), likely enhances activation by providing additional stability to the activated state and thereby leads to channel hyperexcitability (Movie S3).
Fig. 5.
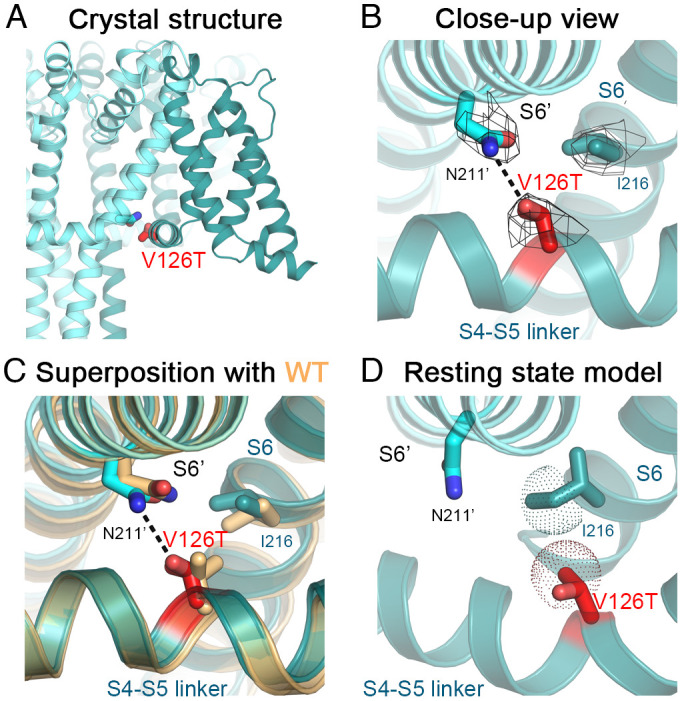
Structure of NaVAb/V126T. (A) Crystal structure of NaVAb/V126T with the mutation shown. (B) Close-up view of NaVAb/V126T structure with the VS in the activated state (teal and cyan for different subunits). Side chains of residues V126T (red), Ile216 (teal), and Asn211’ (cyan) are shown as sticks with the σA-weighted 2FO-FC electron density map contoured at 1.0σ level overlaid (mesh). (C) Comparison of NaVAb/V126T structure with the structure of NaVAb WT (light orange). Side chain of V126T rotates to form a hydrogen bond (black dashes) with side chain of Asn211’. (D) Homology model of NaVAb/L123T structure in the resting state. The van der Waals hemispheres are shown as dots.
Modeling the Mechanism of Action of IEM Mutations in NaV1.7 Channels.
NaV1.7 has four distinct domains rather than four identical subunits as in NaVAb. Similar mammalian NaV channels have complex voltage-dependent gating mechanisms, typically involving four resting states, four resting/inactivated states, one open state, one fast inactivated state, and one or more slow inactivated states (36). The available cryo-EM structures of NaV1.7 are most likely to be in slow inactivated states, considering that the protein has been in a depolarized environment for an extended time (25, 26). Nevertheless, it is of interest to explore the potential functional relevance of the mechanism of action of the IEM mutations revealed here by modeling their impact on the available NaV1.7 structures (26) (Fig. 6). Of the three substituted Thr residues, we have studied, NaV1.7/I234T in the DI S4-S5 linker fits our proposed mechanism most closely. As in NaVAb, the mutant Thr residue is predicted to form a high-energy hydrogen bond with nearby Asn857 in the DII S5 segment of NaV1.7, while maintaining van der Waals interaction with Leu858 (Fig. 6A). Similarly, the nearby residue NaV1.7/S241T is predicted to form a lower energy hydrogen bond with Asn950 in DII S6 of NaV1.7 (Fig. 6B). In contrast, NaV1.7/I848T in the S4-S5 linker in DII is rotated away from the potential hydrogen-bonding residue, Asn1439 in DIII S6 in the available NaV1.7 structure (Fig. 6C). However, it is only one helical turn away from making this contact if the S4-S5 linker were to rotate counterclockwise by 60° to 90° from its position in the NaV1.7 inactivated state structure. Therefore, it is entirely possible that these two residues in NaV1.7 move near each other as the S4-S5 linker rotates during the activation process, similar to the small rotation of the S4-S5 linker in our gating model of NaVAb (Movie S2 and (17)). This subtle movement would allow hydrogen bond formation to stabilize the activated state of NaV1.7.
Fig. 6.

Potential hydrogen bond formation between mutant IEM residues in the S4-S5 linker and the PM of NaV1.7. (A) NaV1.7/I234T. (B) NaV1.7/S241T. (C) NaV1.7/I848T.
Discussion
Our study provides the first structural characterization of IEM mutations of human NaV1.7 channels and gives insight into the potential molecular basis for hyperexcitability in three sets of affected IEM families (SI Appendix, Table S1). We focused on the S4-S5 linkers of NaV1.7 where IEM mutations are clustered and coupling of voltage-dependent activation to pore opening takes place (15). By expressing these IEM mutations in the context of the ancestral sodium channel NaVAb, we were able to isolate sufficient quantity of protein in the homogeneous form to allow crystallization and high-resolution structure determination of each mutation in its native state with an activated VS. In addition, because NaVAb is the only sodium channel whose resting-state structure is known (17), we were able to use molecular modeling methods to define the effects of IEM mutations on the structures of both resting and activated states. By comparing the structural effects of these mutations in these two distinct states of the VS, a unified mechanism emerged for these three IEM mutants, which suggests a plausible structural basis for their hyperexcitability.
IEM Mutations in the S4-S5 Linker Form Hydrogen Bonds with the Pore Module of NaVAb.
The three IEM mutations we studied cause little or no change in the conformation of the S4-S5 linker. However, our structures indicate that these IEM mutations provide extra stability to the activated state by forming a new hydrogen bond with a residue from the PM of the adjacent subunit when the channel is in the activated state (Ser132 on S5 for I119T; Asn211 on S6 for L123T and V126T). All three of these mutated residues have been proposed to be important for electromechanical coupling and pore gating of the channel, based on the structure of NaVAb in the resting and activated states (17). These hydrogen bonds are well-positioned to stabilize the conformation of the VS in the activated state. In contrast, these mutations do not form a hydrogen bond with any residue in the resting state, in which the S4-S5 linker protrudes as an elbow into the cytosol (17). The fact that mutations of these residues cause hyperexcitability that leads to severe pain in IEM further highlights the critical role of these residues in channel gating.
IEM Mutations Differentially Stabilize the Activated States of NaVAb and NaV1.7.
When we introduced these IEM mutations into NaVAb, they caused negative shifts in Va by −12 mV to −37.5 mV, with the mutation equivalent to I234T causing the largest change. For all three of these IEM mutations, the negative shift of Va in NaVAb is significantly greater than that for the same mutation in NaV1.7 (SI Appendix, Table S2). In the most prominent example, Va for NaVAb/I119T is −37.5 mV compared to −17.9 mV for Va in NaV1.7/I234T (SI Appendix, Table S2). In comparison, the shift of Va in NaVAb/L123T is −15.5 mV compared to −10.4 mV (mean of n = 3 published studies; SI Appendix, Table S2) for Va in NaV1.7/I848T, whereas the shift of Va in NaVAb/V126T is −12 mV compared to −8.4 mV for Va in NaV1.7/S241T (SI Appendix, Table S2). It is likely that two distinct factors contribute to these differences in Va. First, the energy of hydrogen bonds diminishes with their length, and the bond lengths for NaVAb/I119T, NaVAb/L123T, and NaVAb/V126T are approximately 2.5 Å, 3.1 Å, and 3.4 Å, respectively, providing a structural basis for their different shifts of Va based on their hydrogen bond length. Second, we consider it likely that the shifts of Va are larger in NaVAb because of conformational coupling among the four copies of the mutation in the tetramer, and this coupling energy may be greater for NaVAb/I119T than for the other two mutants.
IEM Mutations Are Well-Positioned to Form Hydrogen Bonds in NaV1.7.
The I234T, S241T, and I848T mutations are located in the S4-S5 linkers in DI and DII of NaV1.7. Models of these mutations in the structure of NaV1.7 directly support the formation of hydrogen bonds in the activated state for two of these three mutations, and the third mutation is in position to form a potential new hydrogen bond with modest structural change during the activation process (Fig. 6). These newly formed hydrogen bonds could contribute up to 6 kcal/mol to stabilize the activated state in NaV1.7, which would make a substantial contribution to the free energy required for the 8 mV to 18 mV negative shifts in Va of these mutants in NaV1.7 (SI Appendix, Table S2). Stabilization of the activated state would lead to voltage-sensor trapping, similar to the effects of neurotoxins that negatively shift Va or block fast inactivation by binding with high affinity to the activated state of NaV channels (37, 38). Voltage sensor trapping in the activated state leads to prolonged trains of action potentials, similar to those induced by IEM mutations (4, 31). Interestingly, recent studies of phosphorylation of NaV1.7/I848T also showed a negative shift of −9.8 mV in Va mediated by Protein Kinase C (34), which would further enhance the negative shift of Va caused by this mutation that we have observed in NaVAb (ΔV1/2 of −15.5 mV). Thus, our high-resolution information on these focused structural changes induced by IEM mutations gives unprecedented insights into IEM pathogenesis at near-atomic resolution and provides a potential molecular template for mutation-specific therapy of this debilitating disease.
Biphasic Kinetics of IEM Mutants Suggests Hydrogen Bond Formation During Activation.
Mutations NaVAb/I119T and NaVAb/V126T give two-component Boltzmann fits of their activation curves, as if the voltage sensor activates in two steps (Fig. 2D). These results raise the possibility that the formation of hydrogen bonds with Ser132' and Asn211' is incomplete, resulting in a major fraction of the channels that open with negatively shifted voltage dependence stabilized by hydrogen bond formation and a minor fraction of channels that do not form hydrogen bonds successfully during the brief stimulus pulses. Although our stimulus pulses were 50 ms in duration, actual measurements of peak sodium currents were made at stimulus times less than 1 ms at the most positive stimulus potentials (Fig. 2 A–D). To test whether these short stimulus durations are responsible for the biphasic activation curves, we measured G/V curves from tail currents. In this protocol, cells were depolarized to different membrane potentials during a prepulse of 10 ms, and inward sodium currents through the channels that have opened were measured during a hyperpolarizing pulse to −180 mV (Fig. 2E). During this protocol, the VS have up to 10 ms to activate at all stimulus potentials. Remarkably, the biphasic activation curves were resolved to approximately single Boltzmann fits with this protocol (Fig. 2E), confirming that biphasic activation curves were caused by the short times available to reach peak current in the standard G/V curve protocol. This striking feature of our data provides further support for the importance of hydrogen bond formation to stabilize the activated state of the VS and cause a negative shift in Va and further suggests that these amino acid side chains can align and hydrogen bonds can form completely in less than 10 ms.
Slow Inactivation of IEM Mutants in NaVAb.
Mutations that cause IEM have variable effects on slow inactivation of NaV1.7 channels, suggesting that these effects are not central to the IEM phenotype but may modify it in individuals with specific mutations (4, 7). Slow inactivation of mammalian sodium channels involves conformational movements of the outer pore region (39–41). Bacterial NaV channels have a slow inactivation process that causes pore closure, similar to the slow inactivation of mammalian NaV channels, and analogous to conformational changes in transmembrane closure of the pore in the slow inactivation of the bacterial potassium channel KcsA (42–45). NaVAb has a complex three-phase mode of slow inactivation involving movements of the selectivity filter and outer pore as well as asymmetric closure of the intracellular activation gate formed by the S6 segments (30, 44). Consistent with previous studies, we found that the IEM mutations we studied here primarily slowed the rate of inactivation in the physiologically important membrane potential range from −60 mV to 0 mV (Fig. 2C). These effects on slow inactivation of IEM mutations expressed in NaVAb are consistent with their hyperexcitable phenotype in vivo. However, individual IEM mutations have a range of effects from little or no change to substantial delay of slow inactivation during trains of action potentials (4, 7); therefore, they probably do not contribute in a major way to the disease phenotype in most individuals with mutations in human NaV1.7 (46). Negatively shifted Va, stabilized by the formation of new hydrogen bonds, is most likely to be the underlying mechanism for hyperexcitability for the IEM mutations studied here.
Materials and Methods
Materials Availability.
Further information and requests for resources and reagents should be directed to the corresponding author William A. Catterall (wcatt@uw.edu). All reagents generated in this study are available from the corresponding author with a completed Materials Transfer Agreement.
Microbe Strains.
Escherichia coli GC10 was cultured at 37 °C in an LB medium supplemented with 100 mg/mL of ampicillin for plasmid DNA extraction. E. coli DH10Bac was cultured at 37 °C in LB medium supplemented with 50 mg/mL kanamycin sulfate, 7 mg/mL gentamicin, and 10 mg/mL tetracycline for bacmid production.
Cell Lines.
Sf9 (Spodoptera frugiperda) insect cells were maintained in Grace’s Insect Medium and supplemented with 8 to 10% fetal bovine serum (FBS) and penicillin/streptomycin at 27 °C and passaged at 80 to 95% confluence for baculovirus production. Hi5 (Trichoplusia ni) insect cells were maintained and infected in Grace’s Insect Medium supplemented with 8 to 10% FBS and glutamine/penicillin/streptomycin at 27 °C for electrophysiology and protein expression.
Mutagenesis and Baculovirus Production of NaVAb with IEM Mutations.
The pFastBac-NaVAbΔ28 plasmid carrying a C-terminal 28-residue truncation (residues 1 to 239) of NaVAb gene (30) was used as a template for site-directed mutagenesis. To generate a desired plasmid of a NaVAb mutant, overlapping oligonucleotide primers with a codon changed to a specific mutation were synthesized (Integrated DNA Technologies) and used for site-directed mutagenesis PCR with PfuUltra II Fusion HotStart DNA Polymerase (Agilent). The PCR products were treated with DpnI (New England Biolabs) and transformed into E. coli GC10 competent cells (Genesee Scientific). Plasmid DNAs were isolated from transformed colonies using the QIAprep Spin Miniprep Kit (Qiagen) according to the manufacturer’s protocol and sequenced to confirm the mutation. Plasmids containing NaVAbΔ28 with the IEM mutation were used to transform E. coli DH10Bac competent cells for bacmid production and baculoviruses were prepared with Sf9 insect cells using the Bac-to-Bac protocol according to the manufacturer (Life Technologies).
Electrophysiology of NaVAb with IEM Mutations.
Hi5 insect cells (Trichoplusia ni) were infected with baculovirus containing NaVAbΔ28 with an IEM mutation (30). After 24 to 48 h, whole-cell sodium currents were recorded using an Axopatch 200 amplifier (Molecular Devices) with glass micropipettes (1.5 to 2.5 MΩ) (30). The intracellular pipette solution contained (in mM): 35 NaCl, 105 CsF, 10 EGTA, 10 HEPES, pH 7.4 (adjusted with CsOH). The extracellular solution contained (in mM): 140 NaCl, 2 CaCl2, 2 MgCl2, 10 HEPES, pH 7.4 (adjusted with NaOH). Linear capacitance was subtracted and 80 to 90% of series resistance was compensated using internal amplifier circuitry. I/V relationships of the peak currents were recorded in response to steps to voltages ranging from −160 mV to +160 mV in 10-mV increments from a holding potential of −160 mV or −180 mV. In the case of voltage-dependent activation curves determined from peak tail current amplitudes measured at −180 mV, 10-ms depolarizing pulses were applied from a holding potential of −180 mV in 10-mV steps. Tail currents were normalized to the highest amplitudes. Pulses were generated and currents were recorded using Pulse software controlling an InstruTECH ITC-18 interface (HEKA). Data were analyzed using Igor Pro 8 (WaveMetrics).
Analysis of Electrophysiological Data.
Voltage-clamp data were analyzed using IGOR Pro 8 (WaveMetrics). Peak current at each voltage of the current family was plotted as a function of the stimulus voltage to visualize the current vs. voltage (I/V) relationship. For WT and mutant L123T, normalized conductance/voltage (G/V) curves were calculated from the I/V curves and fit with a simple one-component Boltzmann equation: 1/(1+exp(Va−Vm)/k) in which Vm is the stimulus potential, Va is the half-activation voltage, and k is a slope factor. Similarly, for WT, I119T, and V126T, G/V curves were determined from peak tail current amplitudes by fit to this simple one-component Boltzmann relationship.
In contrast, biphasic G/V curves for I119T and V126T were calculated from I/V curves and fit with a two component Boltzmann equation:
where y0 and A are the minimum and the maximum values taken by y. p is the fraction of the curve comprising first sigmoidal curve, (1 − p) is the fraction of the curve comprising the second sigmoidal curve, and Va1, Va2, k1, and k2 are the voltage midpoints and slope factors of the two phases. The results for the I119T mutation fit well to this two-component Boltzmann equation with Va1 = −127.6 mV, p = 0.76, k1 = 5.34 mV, Va2 = −64 mV, 1-p = 0.24, and k2 = 8.3 mV. However, the double-sigmoidal curve did not fit well for the second phase of activation of the V126T mutation, so only the first component was fitted with Va = −102 mV, k = 5.6 mV.
The half-time (τ) for inactivation was measured from the peak of each current trace. A single exponential function was used to fit the inactivation kinetics of all three mutants.
The data were presented as mean and SEM. Statistical significance was evaluated with Student’s t test.
Protein Expression and Purification of NaVAb with IEM Mutations.
Third passage (P3) baculoviruses for NaVAbΔ28 with an IEM mutation were used to infect Hi5 cells in single-layer culture dishes (~12 dishes for each preparation), and the cells were incubated at 27 °C for ~72 h. The cells were harvested by centrifugation, and the pellets were resuspended in Buffer A (50 mM Tris HCl pH 7.5 and 200 mM NaCl) supplemented with 1 mM PMSF, 2× SigmaFast protease inhibitor cocktails (MilliporeSigma), benzamidine HCl, and DNase I. Cells were lysed by sonication and membranes were solubilized with 1% high-purity digitonin (MilliporeSigma) for 1 h at 4 °C with gentle mixing. The mixture was centrifuged at 15,000×g for 30 min at 4 °C, and the supernatant was incubated with Anti-FLAG-M2 affinity gel (MilliporeSigma) for 1 h at 4 °C with gentle mixing. The resins were washed with Buffer B (Buffer A supplemented with 0.12% digitonin) and bound protein was eluted with Buffer C (Buffer B supplemented with 200 μM FLAG peptide). Eluted protein was concentrated to 1 mL using Vivaspin20 100 kDa MWCO (Cytiva) and further purified with Superdex S200 size-exclusion chromatography (Cytiva) using 10 mM Tris HCl pH 7.5, 100 mM NaCl, and 0.12% digitonin as a column running buffer. Elution fractions were evaluated using SDS-PAGE, and peak fractions were combined and concentrated to final concentration of ~20 mg/ml. Protein concentrations were estimated using 1 A280 absorbance unit = 1 mg/mL on a NanoDrop spectrophotometer.
Crystallization of NaVAb with IEM Mutations.
NaVAb-bicelle complexes were prepared by mixing NaVAbΔ28 with an IEM mutation with 10% bicelle (7.5% w/v DMPC and 2.5% w/v CHAPSO) at 1:4 or 1:5 volume ratios to obtain final 3.0 to 3.5 mg/ml protein concentration. The complexes were screened for crystallization conditions under 1.70 to 1.95 M ammonium sulfate and 0.1 M sodium citrate tribasic, pH 4.6 to 6.0 (Hampton Research). Crystals were cryo-protected by stepwise transfers to a series of cryo-protectant solutions containing 6 to 30% glucose (6% increments) with the same concentration of ammonium sulfate and sodium citrate in which the crystals were grown.
X-ray Data Collection and Structure Determination of NaVAb with IEM Mutations.
Crystals were tested for diffraction, and data were collected at Advanced Light Source beamline 8.2.1 and 8.2.2 Howard Hughes Medical Institute. Diffraction data were processed using the HKL2000 program (47) with anisotropic scaling and truncation due to anisotropic diffraction. Structures were solved by molecular replacement with PHASER (48) using the previously determined NaVAb structure PDB: 3RVY (14) or PDB: 6MW (30) as a search model and refined with REFMAC (49) in the CCP4 program suite (50). Manual model building and local real space refinement were carried out in COOT (51), followed by structure refinement in REFMAC. Subsequently structures were refined with Phenix.refine module and the final structures were analyzed and validated using MolProbity in the Phenix program suite (52) (SI Appendix, Table S3).
Modeling of NaVAb Structure with IEM Mutations in the Resting State.
Homology models of NaVAb tetramer structure with the IEM mutations in the resting state were generated with Modeller 10.2 (53) using the disulfide-locked NaVAb/KAV/G94C/Q150C cryo-EM structure (PDB: 6P6W) (17) as a template. Structure figures and morph movies were made with Pymol (Schrodinger).
Modeling of NaV1.7 Structure with IEM Mutations.
Cryo-EM structure of human NaV1.7 (PDB: 7W9K) was used for modeling in Coot (51). Residues with IEM mutations were mutated to threonine and the most probable side chain rotamer without clashes was selected. Different rotamers of residues that potentially form hydrogen bond pairs with the IEM mutations were tested and a rotamer within a hydrogen bond distance without clashes was selected.
Supplementary Material
Appendix 01 (PDF)
Transition of NaVAb/I119T from the resting state to the activated state from the intracellular view. I119T and Val126 are shown as red and orange sticks, respectively. Ser132’ and Asn211’ from a neighboring subunit are shown as yellow and cyan sticks, respectively. S4 is colored in magenta, the S4-S5 linker in orange, and the pore module S5 to S6 in cyan. Dashes indicate distance in Å.
Transition of NaVAb/L123T from the resting state to the activated state from the intracellular view. L123T, and Asn211’ from a neighboring subunit are shown as red and cyan sticks, respectively. Dashes indicate distance in Å.
Transition of NaVAb/V126T from the resting state to the activated state from the intracellular view. V126T, Ile216, and Asn211’ from a neighboring subunit are shown as red, teal, and cyan sticks, respectively. Dashes indicate distance in Å.
Acknowledgments
We thank the beamline staff at the Advanced Light Source (BL8.2.1 and BL8.2.2) for assistance during X-ray data collection and Dr. Jin Li (Department of Pharmacology, University of Washington) for the technical and editorial support. This research was supported by NIH research grants R01 NS015751 (W.A.C.), R35 NS111573 (W.A.C.), and R01 HL112808 (N.Z. and W.A.C.) and by the Howard Hughes Medical Institute (N.Z.). The Berkeley Center for Structural Biology is supported in part by the Howard Hughes Medical Institute. The Advanced Light Source is a Department of Energy, Office of Science User Facility under Contract No. DE-AC02-05CH11231. This research also used resources of the Advanced Photon Source, a U.S. Department of Energy Office of Science user facility operated by Argonne National Laboratory under Contract No. DE-AC02-06CH11357.
Author contributions
G.W., T.M.G.E.-D., N.Z., and W.A.C. designed research; G.W. and T.M.G.E.-D. performed research; G.W. and T.M.G.E.-D. analyzed data; and G.W., T.M.G.E.-D., N.Z., and W.A.C. wrote the paper.
Competing interests
The authors declare no competing interest.
Footnotes
Reviewers: E.P., The University of Chicago; P.C.R., Simon Fraser University; and V.Y.-Y., University of California Davis.
Contributor Information
Ning Zheng, Email: nzheng@uw.edu.
William A. Catterall, Email: wcatt@uw.edu.
Data, Materials, and Software Availability
The coordinates and the structure factors for the reported crystal structures have been deposited in the Protein Data Bank (PDB) under accession codes 8DIZ (54), 8DJ0 (55), and 8DJ1 (56) for NaVAbΔ28 I119T, L123T, and V126T, respectively. All study data are included in the article and/or SI Appendix.
Supporting Information
References
- 1.Hodgkin A. L., Huxley A. F., A quantitative description of membrane current and its application to conduction and excitation in nerve. J. Physiol. 117, 500–544 (1952). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Adrian R. H., Chandler W. K., Hodgkin A. L., Voltage clamp experiments in striated muscle fibres. J. Physiol. 208, 607–644 (1970). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Mantegazza M., Cestèle S., Catterall W. A., Sodium channelopathies of skeletal muscle and brain. Physiol. Rev. 101, 1633–1689 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.S. D. Dib-Hajj, Y. Yang, J. A. Black, S. G. Waxman, The Nav1.7 sodium channel from molecule to man. Nat. Rev. Neurosci. 14, 49–62 (2013). [DOI] [PubMed] [Google Scholar]
- 5.Klugbauer N., Lacinova L., Flockerzi V., Hofmann F., Structure and functional expression of a new member of the tetrodotoxin-sensitive voltage-activated sodium channel family from human neuroendocrine cells. EMBO J. 14, 1084–1090 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cummins T. R., Howe J. R., Waxman S. G., Slow closed-state inactivation: A novel mechanism underlying ramp currents in cells expressing the hNE/PN1 sodium channel. J. Neurosci. 18, 9607–9619 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Waxman S. G., et al. , Sodium channel genes in pain-related disorders: Phenotype-genotype associations and recommendations for clinical use. Lancet Neurol. 13, 1152–1160 (2014). [DOI] [PubMed] [Google Scholar]
- 8.Dib-Hajj S. D., Waxman S. G., Translational pain research: Lessons from genetics and genomics. Sci. Transl. Med. 6, 249sr244 (2014). [DOI] [PubMed] [Google Scholar]
- 9.Geha P., et al. , Pharmacotherapy for pain in a family with inherited erythromelalgia guided by genomic analysis and functional profiling. JAMA Neurol 73, 659–667 (2016). [DOI] [PubMed] [Google Scholar]
- 10.Cox J. J., et al. , An SCN9A channelopathy causes congenital inability to experience pain. Nature 444, 894–898 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mulcahy J. V., et al. , Challenges and opportunities for therapeutics targeting the voltage-gated sodium channel Isoform NaV1.7. J. Med. Chem. 62, 8695–8710 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Catterall W. A., From ionic currents to molecular mechanisms: The structure and function of voltage-gated sodium channels. Neuron 26, 13–25 (2000). [DOI] [PubMed] [Google Scholar]
- 13.Ahern C. A., Payandeh J., Bosmans F., Chanda B., The hitchhiker’s guide to the voltage-gated sodium channel galaxy. J. Gen. Physiol. 147, 1–24 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Payandeh J., Scheuer T., Zheng N., Catterall W. A., The crystal structure of a voltage-gated sodium channel. Nature 475, 353–358 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Catterall W. A., Wisedchaisri G., Zheng N., The chemical basis for electrical signaling. Nat. Chem. Biol. 13, 455–463 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yarov-Yarovoy V., et al. , Structural basis for gating charge movement in the voltage sensor of a sodium channel. Proc. Natl. Acad. Sci. U.S.A. 109, E93–E102 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wisedchaisri G., et al. , Resting-state structure and gating mechanism of a voltage-gated sodium channel. Cell 178, 993–1003.e1012 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li Q., et al. , Structural mechanism of voltage-dependent gating in an isolated voltage-sensing domain. Nat. Struct. Mol. Biol. 21, 244–252 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dickinson M. S., Myasnikov A., Eriksen J., Poweleit N., Stroud R. M., Resting state structure of the hyperdepolarization activated two-pore channel 3. Proc. Natl. Acad. Sci. U.S.A. 117, 1988–1993 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dai G., Aman T. K., DiMaio F., Zagotta W. N., The HCN channel voltage sensor undergoes a large downward motion during hyperpolarization. Nat. Struct. Mol. Biol. 26, 686–694 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li Q., et al. , Resting state of the human proton channel dimer in a lipid bilayer. Proc. Natl. Acad. Sci. U.S.A. 112, E5926–5935 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mandala V. S., MacKinnon R., Voltage-sensor movements in the Eag Kv channel under an applied electric field. Proc. Natl. Acad. Sci. U.S.A. 119, e2214151119 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wisedchaisri G., et al. , Structural basis for high-affinity trapping of the NaV1.7 channel in its resting state by a tarantula toxin. Mol. Cell 81, 38–48.e34 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Xu H., et al. , Structural basis of NaV1.7 inhibition by a gating-modifier spider toxin. Cell 176, 702–715.e14 (2019). [DOI] [PubMed] [Google Scholar]
- 25.Huang G., et al. , Unwinding and spiral sliding of S4 and domain rotation of VSD during the electromechanical coupling in Nav1.7. Proc. Natl. Acad. Sci. U.S.A. 119, e2209164119 (2022). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shen H., Liu D., Wu K., Lei J., Yan N., Structures of human NaV1.7 channel in complex with auxiliary subunits and animal toxins. Science 363, 1303–1308 (2019). [DOI] [PubMed] [Google Scholar]
- 27.Huang G., et al. , High-resolution structures of human NaV1.7 reveal gating modulation through α-π helical transition of S6IV. Cell Rep. 39, 110735 (2022). [DOI] [PubMed] [Google Scholar]
- 28.Shen H., Yan N., Pan X., Structural determination of human NaV1.4 and NaV1.7 using single particle cryo-electron microscopy. Methods Enzymol. 653, 103–120 (2021). [DOI] [PubMed] [Google Scholar]
- 29.Ahuja S., et al. , Structural basis of NaV1.7 inhibition by an isoform-selective small-molecule antagonist. Science 350, aac5464 (2015). [DOI] [PubMed] [Google Scholar]
- 30.Gamal El-Din T. M., Lenaeus M. J., Ramanadane K., Zheng N., Catterall W. A., Molecular dissection of multiphase inactivation of the bacterial sodium channel NaVAb. J. Gen. Physiol. 151, 174–185 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ahn H. S., et al. , A new NaV1.7 sodium channel mutation I234T in a child with severe pain. Eur. J. Pain 14, 944–950 (2010). [DOI] [PubMed] [Google Scholar]
- 32.Cummins T. R., Dib-Hajj S. D., Waxman S. G., Electrophysiological properties of mutant NaV1.7 sodium channels in a painful inherited neuropathy. J. Neurosci. 24, 8232–8236 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wu M. T., Huang P. Y., Yen C. T., Chen C. C., Lee M. J., A novel SCN9A mutation responsible for primary erythromelalgia and is resistant to the treatment of sodium channel blockers. PLoS One 8, e55212 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kerth C. M., Hautvast P., Körner J., Lampert A., Meents J. E., Phosphorylation of a chronic pain mutation in the voltage-gated sodium channel NaV1.7 increases voltage sensitivity. J. Biol. Chem. 296, 100227 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lampert A., Dib-Hajj S. D., Tyrrell L., Waxman S. G., Size matters: Erythromelalgia mutation S241T in NaV1.7 alters channel gating. J. Biol. Chem. 281, 36029–36035 (2006). [DOI] [PubMed] [Google Scholar]
- 36.Fohlmeister J. F., Voltage gating by molecular subunits of Na+ and K+ ion channels: Higher-dimensional cubic kinetics, rate constants, and temperature. J. Neurophysiol. 113, 3759–3777 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cestèle S., et al. , Voltage sensor-trapping: Enhanced activation of sodium channels by beta-scorpion toxin bound to the S3–S4 loop in domain II. Neuron 21, 919–931 (1998). [DOI] [PubMed] [Google Scholar]
- 38.Jiang D., et al. , Structural basis for voltage-sensor trapping of the cardiac sodium channel by a deathstalker scorpion toxin. Nat. Commun. 12, 128 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vilin Y. Y., Makita N., George A. L., Ruben P. C., Structural determinants of slow inactivation in human cardiac and skeletal muscle sodium channels. Biophys. J. 77, 1384–1393 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Balser J. R., et al. , External pore residue mediates slow inactivation in μ1 rat skeletal muscle sodium channels. J. Physiol. 494, 431–442 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ong B. H., Tomaselli G. F., Balser J. R., A structural rearrangement in the sodium channel pore linked to slow inactivation and use dependence. J. Gen. Physiol. 116, 653–662 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pavlov E., et al. , The pore, not cytoplasmic domains, underlies inactivation in a prokaryotic sodium channel. Biophys. J. 89, 232–242 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vilin Y. Y., Ruben P. C., Slow inactivation in voltage-gated sodium channels: Molecular substrates and contributions to channelopathies. Cell Biochem. Biophys. 35, 171–190 (2001). [DOI] [PubMed] [Google Scholar]
- 44.Payandeh J., Gamal El-Din T. M., Scheuer T., Zheng N., Catterall W. A., Crystal structure of a voltage-gated sodium channel in two potentially inactivated states. Nature 486, 135–139 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li J., Ostmeyer J., Cuello L. G., Perozo E., Roux B., Rapid constriction of the selectivity filter underlies C-type inactivation in the KcsA potassium channel. J. Gen. Physiol. 150, 1408–1420 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lampert A., Eberhardt M., Waxman S. G., Altered sodium channel gating as molecular basis for pain: Contribution of activation, inactivation, and resurgent currents. Handb. Exp. Pharmacol. 221, 91–110 (2014). [DOI] [PubMed] [Google Scholar]
- 47.Otwinowski Z., Minor W., Processing of X-ray diffraction data collected in oscillation mode. Methods Enzymol. 276, 307–326 (1997). [DOI] [PubMed] [Google Scholar]
- 48.McCoy A. J., et al. , Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Murshudov G. N., et al. , REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr. D Biol. Crystallogr. 67, 355–367 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Winn M. D., et al. , Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Emsley P., Lohkamp B., Scott W. G., Cowtan K., Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 66, 486–501 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Liebschner D., et al. , Macromolecular structure determination using x-rays, neutrons, and electrons: Recent developments in Phenix. Acta Crystallogr. D Struct. Biol. 75, 861–877 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Eswar N., et al. , Comparative protein modeling using modeller. Curr. Protoc. Bioinformatics 54, 5.6.1–5.6.37 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Wisedchaisri G., Gamal El-Din T. M., Zheng N., Catterall W. A., Crystal structure of NaVAb I119T as a basis for the human NaV1.7 Inherited Erythromelalgia I234T mutation. Protein Data Bank. https://www.rcsb.org/structure/8DIZ. Deposited 29 June 2022. [Google Scholar]
- 55.Wisedchaisri G., Gamal El-Din T. M., Zheng N., Catterall W. A., Crystal structure of NaVAb L123T as a basis for the human NaV1.7 Inherited Erythromelalgia I848T mutation. Protein Data Bank. https://www.rcsb.org/structure/8DJ0. Deposited 29 June 2022. [Google Scholar]
- 56.Wisedchaisri G., Gamal El-Din T. M., Zheng N., Catterall W. A., Crystal structure of NaVAb V126T as a basis for the human NaV1.7 Inherited Erythromelalgia S241T mutation. Protein Data Bank. https://www.rcsb.org/structure/8DJ1. Deposited 29 June 2022. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix 01 (PDF)
Transition of NaVAb/I119T from the resting state to the activated state from the intracellular view. I119T and Val126 are shown as red and orange sticks, respectively. Ser132’ and Asn211’ from a neighboring subunit are shown as yellow and cyan sticks, respectively. S4 is colored in magenta, the S4-S5 linker in orange, and the pore module S5 to S6 in cyan. Dashes indicate distance in Å.
Transition of NaVAb/L123T from the resting state to the activated state from the intracellular view. L123T, and Asn211’ from a neighboring subunit are shown as red and cyan sticks, respectively. Dashes indicate distance in Å.
Transition of NaVAb/V126T from the resting state to the activated state from the intracellular view. V126T, Ile216, and Asn211’ from a neighboring subunit are shown as red, teal, and cyan sticks, respectively. Dashes indicate distance in Å.
Data Availability Statement
The coordinates and the structure factors for the reported crystal structures have been deposited in the Protein Data Bank (PDB) under accession codes 8DIZ (54), 8DJ0 (55), and 8DJ1 (56) for NaVAbΔ28 I119T, L123T, and V126T, respectively. All study data are included in the article and/or SI Appendix.