

Abstract
Background
Since 2009, Dutch patients with a confirmed diagnosis/suspicion of systemic sclerosis (SSc) can be referred to the Leiden Combined Care in Systemic Sclerosis (CCISS) cohort. This study evaluated whether early recognition of SSc has improved over time and whether disease characteristics and survival has changed over time.
Methods
643 SSc patients fulfilling American College of Rheumatology/European Alliance of Associations for Rheumatology 2013 SSc criteria were included and categorised into three groups based on cohort-entry year: (1) 2010–2013 (n=229 (36%)), (2) 2014–2017 (n=207 (32%)) and (3) 2018–2021 (n=207 (32%)). Variables including disease duration, interstitial lung disease (ILD), digital ulcers (DU), diffuse cutaneous SSc (dcSSc), antitopoisomerase (ATA) and anticentromere (ACA) antibodies, and survival from disease onset were compared between cohort-entry groups, including analyses stratified for sex and autoantibodies.
Results
Over time, duration between onset of disease symptoms and cohort entry decreased in males and females, but was always longer in females than in males.
The proportion of patients presenting with DU decreased, especially in ACA+SSc patients. Almost no ACA+ patients presented with ILD, while in ATA+ patients this proportion was 25% in 2010–2013 and decreased to 19% in 2018–2021. A reduction in patients presenting with clinically meaningful ILD and dcSSc was observed.
Overall 8-year survival for males was 59% (95% CI 40% to 73%) and for females 89% (95% CI 82% to 93%). Eight-year survival showed a trend for improvement over time, and was always worse in males.
Conclusion
We observed a decrease in disease duration in Leiden CCISS cohort at cohort entry, possibly indicating more timely diagnosis of SSc. This could provide opportunities for early interventions. While symptom duration at presentation is longer in females, mortality is consistently higher in males, underlining the urge for sex-specific treatment and follow-up.
Keywords: systemic sclerosis, patient reported outcome measures, autoimmunity
WHAT IS ALREADY KNOWN ON THIS TOPIC
In the past decade, advances in understanding, diagnosis and management of systemic sclerosis (SSc) have been made.
Identifying SSc patients early in the disease course is crucial as it might provide a window of opportunity for early risk stratification and early intervention, before irreversible organ damage occurs.
WHAT THIS STUDY ADDS
Over the past decade, the time elapsed between first symptoms and referral of SSc patients to an SSc expert centre has decreased.
The 8-year survival rate was 59% in SSc males and 89% in females and a trend for improvement over time was observed.
While symptom duration at presentation was longer in SSc females, mortality is consistently higher in males.
HOW THIS STUDY MIGHT AFFECT RESEARCH, PRACTICE OR POLICY
This study might indicate more timely diagnosis of SSc and provides opportunities for early interventions.
This study underlines the urge for sex-specific treatment and follow-up of male and female SSc patients.
Introduction
Systemic sclerosis (SSc) is a heterogeneous disease in which multiple organ systems including the skin, gastrointestinal tract, kidneys, heart and lungs can be affected.1 The clinical course of the disease can vary from rapidly progressive, to a milder course that develops over an extended period.2 For both physicians and patients, SSc represents a major challenge, as the prediction of the disease course for individual patients remains difficult.
Both SSc patients and physicians advocate standardised and regular screening for organ involvement in SSc.3 At Leiden University Medical Center, this is instituted in the prospective Combined Care in Systemic Sclerosis (CCISS) cohort, to which patients with Raynaud’s phenomenon (RP), a suspicion of SSc or other connective tissue diseases are referred to. This cohort was initiated in 2009 and continues to include new patients. As of 1 January 2022, 708 SSc patients fulfilling the American College of Rheumatology/European Alliance of Associations for Rheumatology (ACR/EULAR) 2013 SSc criteria4 have been included, and follow-up data have been gathered prospectively for over a decade.
In the past decade, advances in understanding and management of the disease have been made. During this cohort, the classification criteria for SSc have been updated (ACR/EULAR 2013 SSc criteria), leading to a higher sensitivity for diagnosis of SSc earlier in the disease course, and the mild and limited SSc subtypes.4 5 Moreover, clinical trials in SSc have been numerous, and new therapies have been approved.6 Despite increased SSc awareness, a recent Dutch study still found a diagnostic delay in SSc patients, especially in females.7 In the current study, we evaluated whether early recognition of SSc has improved over time and how this affects disease outcome.
To this end, we investigated disease characteristics and survival of SSc patients in the Leiden CCISS cohort and their change over time, using the cohort-entry year as an instrumental variable. This was evaluated for all patients, but also stratified for sex, and for anticentromere (ACA) or antitopoisomerase antibodies (ATA) positivity.
Methods
Study design and patients
Patients from the Leiden CCISS cohort were included in the study if they fulfilled the ACR/EULAR 2013 SSc criteria4 and had their cohort-entry visit between 1 January 2010 and 1 January 2022. SSc patients with a cohort-entry in 2009 (n=65) were excluded as these patients were often not newly referred. To structure the tables, patients were categorised into three groups based on the year they entered the CCISS cohort: (1) 2010–2013, (2) 2014–2017 and (3) 2018–2021. For these categories, we took into account the year 2013 in which the ACR/EULAR criteria for SSc were published, as well as similar sample sizes and cohort-entry years between the groups. To minimise the risk that our results are a coincidence, we performed a sensitivity analysis with two groups: (1A) 2010–2015 and (2A) 2016–2021.
Study outcomes
A detailed explanation of the Leiden CCISS cohort is provided in online supplemental file. Briefly, patients included in the CCISS cohort undergo annual screening for organ involvement. Patients can be referred by general practitioners, rheumatologists or other medical specialists from other departments or hospitals. For the current evaluation, we included the following sociodemographic and disease characteristics. Sociodemographic data included age, ethnicity, sex, smoking habits (never, current or former), body mass index (BMI, kg/m2) and mortality. Disease duration was evaluated using three definitions: months between cohort entry and (1) date of diagnosis confirmed by a physician, (2) date of onset of RP and (3) date of first non-RP symptom or sign (=non-RP; including one or more of the following: puffy fingers, sclerodactyly, skin fibrosis, telangiectasias, digital ulcers (DU), pitting scars, calcinosis, interstitial lung disease (ILD), gastrointestinal involvement, myositis, cardiac involvement). Data on disease duration are evaluated at baseline in all patients following a standardised intake questionnaire in the consultation of the rheumatologist at the Care Pathway at the LUMC). Additionally, we calculated which patients fulfilled American Rheumatism Association (ARA) 1980 criteria for SSc.8
rmdopen-2022-002971supp001.pdf (279.7KB, pdf)
Disease characteristics included abnormalities on the nailfold capillaroscopy (scored according to Cutolo et al9) and presence of autoantibodies. The extent and severity of skin involvement was assessed based on the modified Rodnan Skin Score10 and patients were categorised as non-cutaneous, limited cutaneous or diffuse cutaneous.11 Cardiac involvement was based on a combined definition including two of the following: arrhythmias (>2% ventricular or supraventricular complexes, arrhythmia, atrial fibrillation), conduction problems, decreased LVEF <54%,12 diastolic or systolic dysfunction, pericarditis or pericardial effusion. Pulmonary arterial hypertension was defined as a mean pulmonary arterial pressure ≥25 mm Hg at rest, assessed by right heart catheterisation; presence of precapillary PH, defined by a pulmonary capillary wedge pressure ≤15 mm Hg; and a pulmonary vascular resistance >3 Wood units on right heart catheterisation.13 Diagnosis of ILD was determined based on presence of interstitial fibrosis or ground glass opacities on high-resolution CT (HRCT) of the thorax reported by a radiologist. A forced vital capacity (FVC) <80% on the pulmonary function test was used to determine whether radiologic ILD was clinically meaningful. Gastrointestinal involvement was defined by endoscopic evidence of gastric antral vascular ectasia, need of parenteral nutrition or by the presence of anaemia associated with BMI <20 kg/m2 plus one of the following symptoms: reflux, bloating, distension, diarrhoea and faecal incontinence. Musculoskeletal involvement was defined as the presence of one of the following symptoms: synovitis, myositis or tendon friction rubs.
Serum samples were collected in a dedicated biobank. At baseline, extensive autoantibody testing was performed. First, an ANA (detected by indirect immunofluorescence on HEP-2000 cells), and ENA screening (measured by fluorescence ELISA, using a Phadia250 system from Thermo Fisher Scientific, Nieuwegein, The Netherlands) were performed in all patients. ENA screening included screening for ACA, ATA, anti-U1RNP and anti-RNP70. If a patient was ANA-positive, but no SSc-related autoantibody was detected, further testing using Phadia250 for anti-RNA polymerase III, anti-fibrillarin (anti-U3RNP) and anti-Pm/Scl antibodies was performed. If these were also negative, antibodies to Th/To RNP (anti-Th/To) and anti-Ku antibodies were determined by a research chemiluminescence immuno-assay using the INOVA BioFlash (Werfen/INOVA, San Diego, USA).
Statistical analysis
Means and SDs were used for normally distributed data and median and IQRs ranges for non-normally distributed data. All descriptive data were evaluated for the total group and stratified for sex and autoantibody status. Data on disease duration are given per cohort-entry year, and data on organ involvement and survival are given per cohort-entry group as specified above. The cohort-entry groups were compared using independent t-tests, one-way analysis of variance tests or χ2 tests.
To evaluate study outcomes longitudinally, specifically mortality, we acknowledge that the 2018–2021 group has the shortest follow-up and these analyses were conducted exploratively. To minimise this limitation, we calculated the median follow-up time between the last available visit and the date of first non-RP symptom for the complete cohort, which was 8 years, and we chose this as the analysis period for survival. Eight-year survival was evaluated from the date of disease onset (time since first non-Raynaud’s symptom) instead of cohort entry to minimise survival bias in the analyses. Patients were censored at the time of last visit or after 8 years of disease duration. Kaplan-Meier methods were used to construct survival curves, which were calculated for the cohort-entry groups and stratified for sex and autoantibody status. Additionally, to explore if survival improved over the cohort-entry years, we performed a Cox proportional hazard model with cohort-entry year as a continuous variable and sex and age as covariates.
As a sensitivity analysis, we also compared disease duration and data on organ involvement and survival between two groups using the cohort-entry years: 2010–2015 and 2016–2021. These results are provided in online supplemental file. In addition, in online supplemental file, we evaluated the use and start of immunosuppressive medication for patients who had a non-RP duration of less than 1 year at cohort entry to evaluate trends in treatment over time, and we compared rate of disease progression between the cohort-entry groups.
The descriptive analyses were conducted with SPSS V.25.0 software (SPSS), and the Kaplan-Meier survival curves with STATA SE V.16 (StataCorp).
Results
Patients
As of 1 January 2022, 1076 patients have been evaluated in the Care Pathway, with 708 fulfilling the ACR/EULAR 2013 criteria for SSc. Of these 708 SSc patients, 643 entered the cohort after 2010. The mean age at cohort entrance was 55 years (SD: 14). Eighty per cent of the 643 SSc patients were females, 95% had ANA, 43% ACA and 21% ATA. Four hundred and eighty-eight (76%) patients fulfilled ARA 1980 classification criteria for SSc.
Two hundred and twenty-nine (36%) patients entered the Leiden CCISS cohort between 2010 and 2013, 207 (32%) between 2014 and 2017 and 207 (32%) between 2018 and 2021. Characteristics of disease duration and organ involvement are shown in table 1.
Table 1.
Characteristics of SSc patients categorised on cohort-entry year
| 2010–2013 N=229 |
2014–2017 N=207 |
2018–2021 N=207 |
P value | |
| Age, mean (SD) | 53 (15) | 57 (14) | 55 (14) | 0.004 |
| Female | 195 (85) | 157 (76) | 157 (76) | 0.021 |
| RP duration, months, median (IQR) | 120 (44–240) | 93 (20–202) | 68 (21–210) | 0.010 |
| Non-RP duration, months, median (IQR) | 46 (16–127) | 20 (5–112) | 17 (6–55) | <0.001 |
| Time since diagnosis, months, median (IQR) | 22 (5–99) | 9 (1–51) | 4 (0–18) | <0.001 |
| Fulfilling ARA 1980 criteria for SSc | 181 (79) | 152 (73) | 155 (75) | 0.360 |
| Anticentromere antibodies | 88 (38) | 87 (42) | 100 (49) | 0.091 |
| Antitopoisomerase antibodies | 53 (23) | 49 (24) | 36 (18) | 0.248 |
| Disease subset: | 0.160 | |||
| Non-cutaneous | 43 (19) | 50 (24) | 43 (21) | |
| Limited cutaneous | 144 (62) | 109 (53) | 130 (63) | |
| Diffuse cutaneous | 42 (18) | 48 (23) | 34 (16) | |
| Pulmonary arterial hypertension | 8 (4) | 5 (2) | 6 (3) | 0.801 |
| Cardiac involvement | 11 (5) | 12 (6) | 25 (12) | 0.008 |
| Interstitial lung disease on HRCT | 99 (43) | 65 (31) | 64 (31) | 0.033 |
| Interstitial lung disease on HRCT and FVC <80% | 27 (12) | 20 (10) | 12 (6) | 0.092 |
| Gastrointestinal involvement | 56 (25) | 55 (27) | 46 (22) | 0.588 |
| Renal crisis | 5 (2) | 3 (1) | 5 (3) | 0.570 |
| Musculoskeletal involvement | 40 (18) | 42 (20) | 28 (14) | 0.185 |
| Digital ulcers | 45 (20) | 26 (13) | 23 (12) | 0.036 |
All binary variables are presented as numbers with percentages.
ARA, American Rheumatism Association; FVC, forced vital capacity; HRCT, high-resolution CT; ILD, interstitial lung disease; N, number; RP, Raynaud’s phenomenon; SSc, systemic sclerosis.
Disease duration
Disease duration over time is shown in figure 1.
Figure 1.
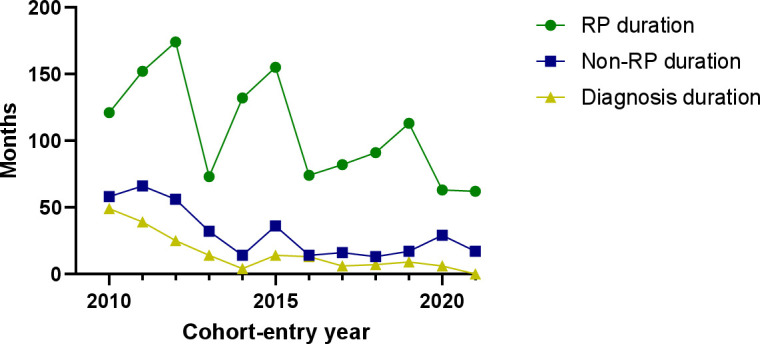
Disease duration at cohort entry of 643 SSc patients. Figure 1 shows the disease duration for patients included in the Leiden CCISS cohort per cohort-entry year. CCISS, Combined Care in Systemic Sclerosis; RP, Raynaud’s phenomenon; SSc, systemic sclerosis.
When stratifying the groups in line with the tables, we observe a clear decrease in disease duration at cohort entry over time, independent of the definition for disease duration. RP duration decreased from 120 (IQR: 44–240) months in the 2010–2013 group to 93 months (IQR: 20–202) in the 2014–2017 group and 68 months (IQR: 21–210) in the 2018–2021 group. Non-RP duration decreased from 46 months (IQR: 16–127) in the 2010–2013 group to 20 months (IQR: 5–112) in the 2014–2017 group and 17 months (IQR: 6–55) in the 2018–2021 group. Diagnosis duration decreased from 22 months (IQR: 5–99) in the 2010–2013 group to 9 months (IQR: 1–51) in the 2014–2017 group and 4 months (IQR: 0–18) in the 2018–2021 group.
When also stratifying for sex, RP duration, non-RP duration and diagnosis duration at cohort entry decreased over time in both males and females, but was always longer in females than in males (figure 2A and table 2).
Figure 2.

(A) Disease duration for SSc patients, stratified for sex. (A) Shows the disease duration for patients included in the Leiden CCISS cohort, stratified for sex, per cohort-entry year. (B) Disease duration for SSc patients, stratified for autoantibody status. (B) shows the disease duration for patients included in the Leiden CCISS cohort, stratified for autoantibodies (ATA and ACA positive), per cohort-entry year. ACA, anticentromere autoantibody; ATA, antitopoisomerase autoantibody; CCISS, Combined Care in Systemic Sclerosis; RP, Raynaud’s phenomenon; SSc, systemic sclerosis.
Table 2.
Organ involvement at cohort entry stratified for sex
| 2010–2013 | 2014–2017 | 2018–2021 | ||||
| Female N=195 |
Male N=34 |
Female N=157 |
Male N=50 |
Female N=157 |
Male N=50 |
|
| RP duration, months | 121 (43–244) | 106 (44–231) | 118 (26–238) | 34 (7–168) | 96 (29–239) | 33 (9–81) |
| Non-RP duration, months | 46 (17–127) | 40 (9–143) | 21 (7–115) | 13 (3–88) | 18 (6–73) | 13 (5–36) |
| Diagnosis duration, months | 22 (7–99) | 14 (3–108) | 11 (2–65) | 3 (0–32) | 5 (0–21) | 3 (0–10) |
| Anticentromere antibodies | 82 (42) | 6 (18) | 76 (49) | 11 (22) | 89 (57) | 11 (22) |
| Antitopoisomerase antibodies | 42 (22) | 11 (32) | 28 (18) | 21 (42) | 20 (13) | 16 (33) |
| Disease subset: | ||||||
| Non-cutaneous | 39 (20) | 4 (12) | 44 (28) | 6 (12) | 40 (25) | 3 (6) |
| Limited cutaneous | 125 (64) | 19 (56) | 85 (54) | 24 (48) | 100 (64) | 30 (60) |
| Diffuse cutaneous | 31 (16) | 11 (32) | 28 (18) | 20 (40) | 17 (11) | 17 (34) |
| Pulmonary arterial hypertension | 7 (4) | 1 (3) | 4 (3) | 1 (2) | 5 (3) | 1 (2) |
| Cardiac involvement | 7 (4) | 4 (12) | 6 (4) | 6 (12) | 17 (11) | 8 (16) |
| Interstitial lung disease on HRCT | 83 (43) | 16 (47) | 45 (29) | 20 (40) | 43 (27) | 21 (42) |
| Interstitial lung disease on HRCT and FVC <80% | 18 (9) | 9 (27) | 15 (10) | 5 (10) | 5 (3) | 7 (14) |
| Gastrointestinal involvement | 41 (21) | 15 (44) | 37 (24) | 18 (36) | 26 (17) | 20 (40) |
| Renal crisis | 4 (2) | 1 (3) | 2 (1) | 1 (2) | 5 (4) | – |
| Musculoskeletal involvement | 32 (16) | 8 (24) | 25 (16) | 17 (34) | 21 (13) | 7 (14) |
| Digital ulcers | 40 (21) | 5 (15) | 20 (13) | 6 (12) | 18 (12) | 5 (10) |
Continuous variables are represented with medians and IQRs, and categorical variables with numbers and percentages.
For females: the following characteristics were significantly different between the cohort-entry groups: age (p=0.006), non-RP duration (p<0.001), diagnosis duration (p<0.001), presence of ACA (p=0.024), cardiac involvement (p=0.007), interstitial lung disease on HRCT (p=0.014) and interstitial lung disease on HRCT and FVC <80% (p=0.049).
For males: the following characteristics were significantly different between the cohort-entry groups: non-RP duration (p=0.012) and diagnosis duration (p=0.023).
ACA, anticentromere autoantibody; FVC, forced vital capacity; HRCT, high-resolution CT; RP, Raynaud’s phenomenon.
Stratification for autoantibody showed that for both ACA and ATA-positive SSc patients, RP duration, non-RP duration and diagnosis duration decreased especially in the first period. For ACA-positive SSc patients, RP and non-RP durations were always longer than in ATA-positive SSc patients. Duration since the first diagnosis was comparable between ACA and ATA-positive SSc patients (figure 2B and online supplemental table S1).
Patient characteristics at cohort entry
The proportion of female patients decreased from 85% in the 2010–2013 group to 76% in the 2014–2017 group and the 2018–2021 group (table 1). ACA positivity increased in the cohort-entry groups from 38% in the 2010–2013 group to 42% in 2014–2017 and 49% in 2018–2021.
Of the investigated organ complications, a decrease in patients presenting with DU was observed (2010–2013: 20%; 2014–2017: 13% and 2018–2021: 12%; table 1). The proportion of patients presenting with ILD on the HRCT was lowest in the 2014–2017 and 2018–2021 group with a proportion of 31%, whereas this was 43% in the 2010–2013 group. A downward trend of the proportion of patients presenting with clinically meaningful ILD (HRCT and FVC <80%) between the cohort-entry groups was observed (2010–2013: 12%; 2014–2017: 10% and 2018–2021: 6%; table 1).
Stratifying for sex, an increase in ACA positivity for both female and male patients is noted, but this increase was greater in females (females vs males: 2010–2013: 42% vs 18%; 2014–2017: 49% vs 22% and 2018–2021: 57% vs 22%; table 2). Female SSc patients in the 2010–2013 group presented more often with ILD and DU compared with the 2014–2017 and 2018–2021 groups (table 2). In male SSc patients, the proportion of clinically meaningful ILD is highest in the 2010–2013 group with 27% and decreased to 10% in 2014–2017 and 14% in 2018–2021 (table 2). Strikingly, male SSc patients more often had gastrointestinal involvement (table 2).
When stratifying for autoantibody status, the presence of DU at cohort entry decreased between the cohort-entry groups in ACA+ patients, whereas for ATA+ patients this remained stable (2010–2013: 28% vs 15%; 2014–2017: 16% vs 14% and 2018–2021: 10% vs 17%; online supplemental table S1). Almost no ACA+ patient presented with clinically meaningful ILD, while in ATA+ patients this proportion was 25% in 2010–2013 and decreased to 19% in 2018–2021 (online supplemental table S1). Additionally, the proportion of ATA+ patients with the non-cutaneous subset increased in the cohort-entry groups and remained stable after (2010–2013: 5%; 2014–2017: 12% and 2018–2021: 11%; online supplemental table S1).
Data on treatment and disease progression are shown in online supplemental file. The proportions of patients using immunosuppressive medication were similar between the cohort-entry groups with only patients with a non-RP duration of less than 1 year (online supplemental table S3). In addition, the proportions of patients who started with or changed immunosuppressive medication after first evaluation in the care pathway were comparable between the cohort-entry groups. Total disease progression, and progression of DU, musculoskeletal involvement was lower in the 2018–2021 group compared with the two other cohort-entry groups (online supplemental table S4).
Eight-year survival
The survival curves are shown in figure 3. Overall 8-year survival was 82% (95% CI 76% to 87%), and was highest in the 2018–2021 group with 85% (95% CI 52% to 96%). In both males and females, survival showed an upward trend in the cohort-entry groups. Mortality was always higher in males than in females. Stratification for autoantibodies showed that ACA+SSc patients have a better survival than ATA+SSc patients. With a Cox proportional hazard model, increased risk for mortality was observed for male and age (HR male 4.7 (95% CI 2.3 to 9.5), HR age 1.1 (95% CI 1.05 to 1.1)), while a downwards trend was observed per cohort-entry year (HR 0.93 (95% CI 0.81 to 1.1)).
Figure 3.
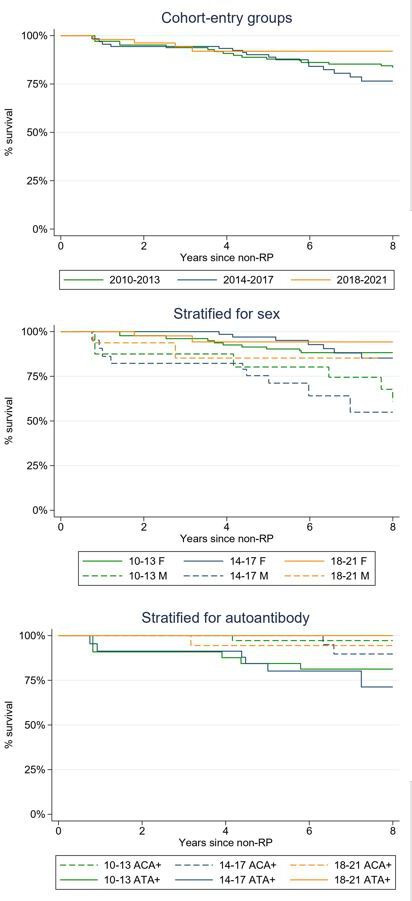
Kaplan-Meier curves for survival according to cohort-entry groups for the Leiden CCISS cohort. In each figure, the green lines represent the patients from 2010 to 2013 group, the blue lines from 2014 to 2017 and the orange lines from 2018 to 2021. The upper figure shows the Kaplan-Meier curves for 8-year survival for the cohort-entry groups, the middle for cohort-entry groups stratified for sex and the lower for the cohort-entry groups stratified for autoantibody status. ACA, anticentromere autoantibody; ATA, antitopoisomerase antibodies; CCISS, Combined Care in Systemic Sclerosis; F, female; M, male; RP, Raynaud’s phenomenon.
For the sensitivity analysis, the 2010–2015 group comprised 317 (49%) patients, and the 2016–2021 group 326 (51%). The same results were found when repeating these analyses with these two groups (online supplemental tables S5 and S6, online supplemental figure S1), strengthening our findings.
Discussion
Summary
Over the last decade, the Leiden CCISS cohort has provided tailored and standardised approaches for SSc. As many advances have been made in the research field of SSc, we investigated if disease duration and outcome as reflected by mortality have improved since initiation of the CCISS cohort. Indeed, symptom duration before cohort entry decreased over time, reflected by a decrease in duration of RP, duration since first non-RP symptom and duration since diagnosis. This observation might indicate that physicians recognise SSc earlier in the disease course. In line with this, the percentage of patients presenting with DU and clinically meaningful ILD decreased over time. Finally, we observed a trend for improved survival over time. Importantly, while time between first symptoms and first referral is longer in females than in males, survival is clearly and consistently worse in males.
Window of opportunity in SSc
We observe a decrease in the time elapsed between first symptoms and referral of SSc patients to an SSc expert centre. More awareness of SSc in the past decade is probably a major factor for this. Another contributing factor would be that the awareness of the existence of the expert centre of Leiden CCISS has increased. Moreover, in 2014, a screening programme for patients with RP was introduced, which could possibly have led to more referrals of patients with solely RP. Screening patients with RP is important to identify patients at risk for SSc resulting in early diagnosis of the disease.
Identifying SSc patients early in the disease course is crucial as it might provide a window of opportunity for early risk stratification and early intervention, before irreversible organ damage occurs. Whether this window of opportunity exists in SSc, and at which point of the disease course one should intervene to prevent irreversible damage, is currently unknown. In our study, we did not observe clear differences in the proportions of early SSc patients (<1 year since first non-RP) being treated with immunosuppressive treatment at cohort entry. Few studies suggest possible benefits of early intervention. Nihtyanova et al showed improved 5-year survival in diffuse cutaneous SSc patients over time and attributed this to improved recognition of pulmonary complications as a result of systematic annual screening.14 Recently, a study from Yomono and Kuwana showed that SSc patients in whom immunosuppressive treatment was started early (≤18 months disease duration at treatment introduction) more frequently showed improvement than those in whom treatment was started more than 18 months after disease onset.15 However, cumulative survival did not differ between the groups.15 In line with this, a paper on Swiss SSc patients in the EUropean Scleroderma Trials And Research group (EUSTAR) database concluded that survival of the Swiss SSc patients was not different from the EUSTAR patients although the Swiss patients had a 1-year shorter disease duration than the other SSc patients in the EUSTAR database.16 This might indicate that the optimal timing of intervention might even be earlier in the disease course. Indeed, also in our cohort, many patients present with severe organ involvement, for instance, nearly 30% of our SSc patients have signs of ILD on the HRCT at baseline, which is in line with findings from the Swiss and Norwegian national SSc cohorts.16 17 Moreover, duration since the first diagnosis was comparable between ACA+ and ATA+SSc patients, indicating either a delay in recognition of ACA+SSc and/or a significant difference in preclinical disease development stage. To foster insights in early disease and sequelae resulting in organ damage, more efforts are needed to identify patients before development of irreversible organ damage and recognise a possible ‘window of opportunity’. In addition, effective treatment strategies that can prevent this progression are urgently needed to employ this possible window of opportunity.
Sex prevalence and severity paradox in SSc
Many studies have demonstrated sex and gender disparities in healthcare use. In this study, we found that female SSc patients have a longer disease duration before cohort entry to a specialised SSc centre. A possible explanation could be that males more often have organ involvement at baseline than females, which suggests a more rapid disease course in male patients. Despite this diagnostic delay and inverse disease severity, this advocates for extra attention and awareness to diagnose SSc timely for female patients. Studies have shown that the burden of symptoms before the diagnosis is substantial.18 Moreover, earlier diagnosis seems to result in fewer DU (table 1), and hopefully in the future more options become available to prevent organ involvement.
Despite earlier referral, male SSc patients still show a more severe disease course.19 The reasons for these sex-based differences in SSc remain unclear. Sex does seem to play a role in the pathophysiology of SSc.20 This might affect treatment response. Recently, in a special issue on ‘sex and gender in rheumatology’ in Lancet Rheumatology, Volkmann et al performed a post hoc analysis stratified for sex of two randomized controlled trials (Scleroderma Lung Study I and II). This study showed that male SSc patients with ILD had a less favourable course of ILD both with and without active treatment, as well as worse long-term survival. The female participants had a more proinflammatory immune response, whereas males had a more profibrotic immune signature, which could account for the differences in treatment response.21 An improved understanding of the role of sex in the pathophysiology of SSc could contribute to personalised medicine. As treatment response in male and female SSc patients seems to differ, stratification based on sex should be taken into account in the design and analysis of future trials.
Limitations
Our research is not without limitations. This is a monocentric cohort study and it needs to be replicated in other settings. The CCISS cohort is a referral centre for SSc. We cannot exclude that patients with more severe disease tend to be referred more often from other hospitals, which might explain the somewhat higher proportion of male patients (proportion of female patients in Leiden CCISS: 79%, EUSTAR 86%,22 Spanish Scleroderma Registry (RESCLE) 89%,23 Australian cohort 86%,24 Oslo 84%17). Disease duration is evaluated at cohort entrance in all patients following a standardised intake questionnaire by physicians, which is prone to recall bias. However, the same method is applied to all patients and, therefore, it is unlikely that recall bias is different between the cohort-entry groups. Furthermore, we did not adjust for disease subset in the analyses, as this additional stratification would result in very low numbers of patients per group, in particular for male patients. Another limitation is that the 2018–2021 group had, in general, a shorter follow-up. Follow-up duration considered in this study is short for a chronic disease, especially to evaluate the survival rate. Though we performed the survival analyses from disease onset to minimise the risk of survival and lead time bias, it is important to realise that bias still could have occurred. Therefore, robust conclusions can not be drawn and we always refer to ‘trends’ in the result section. P values should be interpreted with caution as this is a single centre descriptive study. Finally, it is important to emphasise that external factors, unrelated to the standardised follow-up in the CCISS cohort, such as the change in classification criteria and the availability of effective treatment opportunities, also impact the observed trends.
Conclusions
To conclude, since the Leiden care pathway has been instituted, we observe a shorter disease duration at the time of referral, possibly indicating a more timely diagnosis but also providing opportunities for early interventions. Even though for both males and females disease duration and mortality decrease, our data still highlight a diagnostic delay in females, and considerable mortality, specifically in males. Whether and to what extent all patients benefit from a standardised care programme, and how mortality can be modified by tighter follow-up, is still an open question. More data from prospective cohorts would be needed to elucidate this point.
Acknowledgments
The authors would like to thank the patients for participating in the CCISS cohort, Jozé Krol-van Berkel, Cedric Kromme and Samantha Jurado Zapata for their assistance in data management and Jessica Vlot for her help in organising the clinical studies. The authors would like to thank Coen van der Meulen for his diligently proofreading of the manuscript.
Footnotes
Contributors: SIEL, JC, SA, TWJH and JDV-B conceived the research question. All authors contributed to data collection. SIEL, JC, NMvL, MB, SA and JDV-B had access to the raw data and verified it. SIEL and JDV-B wrote the statistical analysis plan, and performed the statistical analyses. TWJH, JC, NMvL, MB, SA and CFA helped with interpretation of the results. SIEL wrote the paper, with input from all authors for the final manuscript. All authors had final responsibility for the decision to submit for publication.
Funding: The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests: None declared.
Provenance and peer review: Not commissioned; externally peer reviewed.
Supplemental material: This content has been supplied by the author(s). It has not been vetted by BMJ Publishing Group Limited (BMJ) and may not have been peer-reviewed. Any opinions or recommendations discussed are solely those of the author(s) and are not endorsed by BMJ. BMJ disclaims all liability and responsibility arising from any reliance placed on the content. Where the content includes any translated material, BMJ does not warrant the accuracy and reliability of the translations (including but not limited to local regulations, clinical guidelines, terminology, drug names and drug dosages), and is not responsible for any error and/or omissions arising from translation and adaptation or otherwise.
Data availability statement
Data are available on reasonable request. Data from the authors are available on reasonable request, in accordance with the Dutch privacy laws.
Ethics statements
Patient consent for publication
Consent obtained directly from patient(s).
Ethics approval
All patients included in the cohort provided written informed consent prior to inclusion. The cohort was approved by the LUMC Ethics Committee (CME no. B16.037, REU 043/SH/sh, P09.003/SH/s).
References
- 1.Denton CP, Khanna D. Systemic sclerosis. Lancet 2017;390:1685–99. 10.1016/S0140-6736(17)30933-9 [DOI] [PubMed] [Google Scholar]
- 2.Allanore Y, Simms R, Distler O, et al. Systemic sclerosis. Nat Rev Dis Primers 2015;1:15002. 10.1038/nrdp.2015.2 [DOI] [PubMed] [Google Scholar]
- 3.Meijs J, Schouffoer AA, Ajmone Marsan N, et al. Therapeutic and diagnostic outcomes of a standardised, comprehensive care pathway for patients with systemic sclerosis. RMD Open 2016;2:e000159. 10.1136/rmdopen-2015-000159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.van den Hoogen F, Khanna D, Fransen J, et al. 2013 classification criteria for systemic sclerosis: an American College of Rheumatology/European League against rheumatism collaborative initiative. Ann Rheum Dis 2013;72:1747–55. 10.1136/annrheumdis-2013-204424 [DOI] [PubMed] [Google Scholar]
- 5.Boonstra M, Ninaber MK, Ajmone Marsan N, et al. Prognostic properties of anti-topoisomerase antibodies in patients identified by the ACR/EULAR 2013 systemic sclerosis criteria. Rheumatology (Oxford) 2019;58:730–2. 10.1093/rheumatology/kez005 [DOI] [PubMed] [Google Scholar]
- 6.Bukiri H, Volkmann ER. Current advances in the treatment of systemic sclerosis. Curr Opin Pharmacol 2022;64:102211. 10.1016/j.coph.2022.102211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Spierings J, van den Ende CHM, Schriemer RM, et al. How do patients with systemic sclerosis experience currently provided healthcare and how should we measure its quality? Rheumatology 2020;59:1226–32. 10.1093/rheumatology/kez417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Preliminary criteria for the classification of systemic sclerosis (scleroderma). Subcommittee for scleroderma criteria of the American Rheumatism Association Diagnostic and Therapeutic Criteria Committee. Arthritis Rheum 1980;23:581–90. 10.1002/art.1780230510 [DOI] [PubMed] [Google Scholar]
- 9.Cutolo M, Sulli A, Pizzorni C, et al. Nailfold videocapillaroscopy assessment of microvascular damage in systemic sclerosis. J Rheumatol 2000;27:155–60. [PubMed] [Google Scholar]
- 10.Valentini G, D’Angelo S, Della Rossa A, et al. European scleroderma study group to define disease activity criteria for systemic sclerosis. IV. Assessment of skin thickening by modified rodnan skin score. Ann Rheum Dis 2003;62:904–5. 10.1136/ard.62.9.904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.LeRoy EC, Medsger TA. Criteria for the classification of early systemic sclerosis. J Rheumatol 2001;28:1573–6. [PubMed] [Google Scholar]
- 12.Lang RM, Bierig M, Devereux RB, et al. Recommendations for chamber quantification: a report from the American Society of Echocardiography’s guidelines and standards Committee and the chamber quantification writing group, developed in conjunction with the European association of echocardiography, a branch of the European Society of cardiology. J Am Soc Echocardiogr 2005;18:1440–63. 10.1016/j.echo.2005.10.005 [DOI] [PubMed] [Google Scholar]
- 13.Humbert M, Kovacs G, Hoeper MM, et al. 2022 ESC/ERS guidelines for the diagnosis and treatment of pulmonary hypertension. Eur Heart J 2022;43:3618–731. 10.1093/eurheartj/ehac237 [DOI] [PubMed] [Google Scholar]
- 14.Nihtyanova SI, Tang EC, Coghlan JG, et al. Improved survival in systemic sclerosis is associated with better ascertainment of internal organ disease: a retrospective cohort study. QJM 2010;103:109–15. 10.1093/qjmed/hcp174 [DOI] [PubMed] [Google Scholar]
- 15.Yomono K, Kuwana M. Outcomes in patients with systemic sclerosis undergoing early vs delayed intervention with potential disease-modifying therapies. Rheumatology (Oxford) 2022;61:3677–85. 10.1093/rheumatology/keab931 [DOI] [PubMed] [Google Scholar]
- 16.Hernández J, Jordan S, Dobrota R, et al. The burden of systemic sclerosis in switzerland - the Swiss systemic sclerosis EUSTAR cohort. Swiss Med Wkly 2021;151:w20528. 10.4414/smw.2021.20528 [DOI] [PubMed] [Google Scholar]
- 17.Hoffmann-Vold A-M, Fretheim H, Halse A-K, et al. Tracking impact of interstitial lung disease in systemic sclerosis in a complete nationwide cohort. Am J Respir Crit Care Med 2019;200:1258–66. 10.1164/rccm.201903-0486OC [DOI] [PubMed] [Google Scholar]
- 18.Spierings J, van den Ende C, Schriemer R, et al. Optimal care for systemic sclerosis patients: recommendations from a patient-centered and multidisciplinary mixed-method study and working conference. Clin Rheumatol 2019;38:1007–15. 10.1007/s10067-018-4358-x [DOI] [PubMed] [Google Scholar]
- 19.Liem SIE, Boonstra M, le Cessie S, et al. Sex-Specific risk of anti-topoisomerase antibodies on mortality and disease severity in systemic sclerosis: 10-year analysis of the Leiden CCISS and EUSTAR cohorts. The Lancet Rheumatology 2022;4:e699–709. 10.1016/S2665-9913(22)00224-7 [DOI] [PubMed] [Google Scholar]
- 20.Hughes M, Pauling JD, Armstrong-James L, et al. Gender-related differences in systemic sclerosis. Autoimmun Rev 2020;19:102494. 10.1016/j.autrev.2020.102494 [DOI] [PubMed] [Google Scholar]
- 21.Volkmann ER, Tashkin DP, Silver R, et al. Sex differences in clinical outcomes and biological profiles in systemic sclerosis-associated interstitial lung disease: a post-hoc analysis of two randomised controlled trials. The Lancet Rheumatology 2022;4:e668–78. 10.1016/S2665-9913(22)00193-X [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sobanski V, Giovannelli J, Allanore Y, et al. Phenotypes determined by cluster analysis and their survival in the prospective European scleroderma trials and research cohort of patients with systemic sclerosis. Arthritis Rheumatol 2019;71:1553–70. 10.1002/art.40906 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Freire M, Rivera A, Sopeña B, et al. Clinical and epidemiological differences between men and women with systemic sclerosis: a study in a Spanish systemic sclerosis cohort and literature review. Clin Exp Rheumatol 2017;35 Suppl 106:89–97. [PubMed] [Google Scholar]
- 24.Fairley JL, Hansen D, Proudman S, et al. Clinical features of systemic sclerosis-mixed connective tissue disease and systemic sclerosis overlap syndromes. Arthritis Care Res (Hoboken) 2021;73:732–41. 10.1002/acr.24167 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
rmdopen-2022-002971supp001.pdf (279.7KB, pdf)
Data Availability Statement
Data are available on reasonable request. Data from the authors are available on reasonable request, in accordance with the Dutch privacy laws.