

Abstract
Sex chromosomes vary greatly in their age and levels of differentiation across the tree of life. This variation is largely due to the rates of sex chromosome turnover in different lineages; however, we still lack an explanation for why sex chromosomes are so conserved in some lineages (e.g. mammals, birds) but so labile in others (e.g. teleosts, amphibians). To identify general mechanisms driving transitions in sex determination systems or forces which favour their conservation, we first require empirical data on sex chromosome systems from multiple lineages. Stickleback fishes are a valuable model lineage for the study of sex chromosome evolution due to variation in sex chromosome systems between closely‐related species. Here, we identify the sex chromosome and a strong candidate for the master sex determination gene in the brook stickleback, Culaea inconstans. Using whole‐genome sequencing of wild‐caught samples and a lab cross, we identify AmhY, a male specific duplication of the gene Amh, as the candidate master sex determination gene. AmhY resides on Chromosome 20 in C. inconstans and is likely a recent duplication, as both AmhY and the sex‐linked region of Chromosome 20 show little sequence divergence. Importantly, this duplicate AmhY represents the second independent duplication and recruitment of Amh as the sex determination gene in stickleback and the eighth example known across teleosts. We discuss this convergence in the context of sex chromosome turnovers and the role that the Amh/AmhrII pathway, which is crucial for sex determination, may play in the evolution of sex chromosomes in teleosts.
Keywords: Amh, sex chromosome evolution, sex chromosome turnover, stickleback, teleosts
Sex chromosomes vary greatly in their age and levels of differentiation across the tree of life, largely due to the rates of sex chromosome turnover in different lineages. Here, we identify a novel, and likely young, turnover in stickleback fishes. Using whole‐genome sequencing we identify AmhY, a male specific duplication of the autosomal gene Amh, as the candidate master sex determination gene in brook stickleback, Culaea inconstans. Importantly, this duplicate AmhY represents the second independent duplication and recruitment of Amh as the sex determination gene in stickleback and the eighth example known across teleosts. This convergence may offer important clues as to the mechanisms and forces shaping sex chromosome evolution in teleosts.

1. INTRODUCTION
Upon acquiring a master sex determination gene (MSD), recombination is often reduced or suppressed entirely in its vicinity, which opens the door to the accumulation of deleterious mutations due to Hill‐Robertson interference and Muller's ratchet. Given enough time, this process can lead to loss of gene function on the sex chromosomes and eventually to heteromorphic sex chromosomes (Charlesworth et al., 2005), typified by those of mammals and birds (Bachtrog et al., 2014). While there are a few cases in which recombination has been retained (or recently regained) around MSDs thought to be several millions of years old (Ieda et al., 2018; Kamiya et al., 2012; Rodrigues et al., 2018; Stöck et al., 2011), loss of recombination and the subsequent differentiation of sex chromosomes has been observed in the vast majority of old sex chromosome systems studied to date.
In some lineages, however, sex chromosomes escape this process via sex chromosome turnovers, the swapping of the chromosome used for sex determination. When this occurs, sex chromosome differentiation is reset (Vicoso, 2019) and, as such, lineages with labile sex determining systems often have homomorphic, undifferentiated sex chromosomes (Jeffries et al., 2018). Sex chromosome turnovers can, thus, be seen as one of the most impactful processes in the evolution of sex chromosomes and indeed the genome. However, we still lack a good understanding of what drives turnovers. One theory is that the accumulation of deleterious mutations in a sex‐linked, non‐recombining region should favour a transition to a new sex determination gene as a means of purging the genome of mutation load (Blaser et al., 2014). Alternatively, a transition to a new sex chromosome may be favoured if it harbours a sexually antagonistic gene which, when linked to a new MSD, provides a fitness increase to both sexes (van Doorn & Kirkpatrick, 2007, 2010). Finally, turnovers may occur via drift (Saunders et al., 2018). Understanding the relative importance of these drivers and other factors that may contribute to sex chromosome evolution and transitions is essential to explain the diversity and distribution of sex determining systems in nature. However, to achieve this understanding, we first need empirical evidence from multiple lineages to identify general evolutionary forces and mechanisms that drive transitions in sex determination systems or that favour their conservation.
Sticklebacks (Teleostei: Gasterosteidae) are a valuable lineage for addressing such questions, owing to their diversity of sex chromosomes (Figure 1; Ross et al., 2009; Dixon et al., 2018; Natri et al., 2019; Peichel et al., 2020; Sardell et al., 2021). Species of the Gasterosteus genus share a relatively well conserved XY sex determining system located on Chromosome 19 (Peichel et al., 2004), which harbours a strong candidate for sex determination in Gasterosteus, AmhY, a Y‐specific duplicate of the autosomal gene Amh (Peichel et al., 2020). This sex chromosome is estimated to have evolved approximately 22 million years ago (Peichel et al., 2020) and is heteromorphic (Ross & Peichel, 2008), with considerable loss of genes on the Y (Peichel et al., 2020; Sardell et al., 2021). However, in two Gasterosteus species, G. nipponicus and G. wheatlandi, independent Y‐autosome fusion events have created neo sex chromosomes which became sex linked within the past one to ten million years, respectively (Kitano et al., 2009; Ross et al., 2009; Sardell et al., 2021). In the genus Pungitius, Chromosome 19 is not involved in sex determination; instead, Chromosome 12 determines sex in P. pungitius (Natri et al., 2019; Rastas et al., 2015; Ross et al., 2009; Shapiro et al., 2009). However, sex chromosomes in several stickleback species have proven elusive. Despite interrogation of high‐quality genomic data sets, no sex chromosome could be identified for either P. sinensis or P. tymensis (Dixon et al., 2018), although genetic mapping results suggest that P. sinensis has a ZW system on Chromosome 7 (Natri et al., 2019). Similarly, the sex chromosome could not be identified in either Apeltes quadracus or Culaea inconstans using either cytogenetic techniques or genetic mapping with a small number of markers (Ross et al., 2009). The lack of detectable sex‐linked regions in these species could be explained by recent sex chromosome turnovers, resulting in young and undifferentiated sex chromosomes different to those in Gasterosteus or P. pungitius. However, it is also possible that they possess the same sex chromosome system as found in Gasterosteus or P. pungitius, but with much reduced divergence between gametologs, which would imply ongoing recombination and differing evolutionary trajectories of sex chromosomes across species. Identifying the sex chromosomes in additional species of the stickleback family will allow us to study the origin of the known systems in Gasterosteus and Pungitius, the rate of transition between systems, and the rate and dynamics of subsequent sex chromosome degradation after they arise.
FIGURE 1.

Phylogeny of known sex determination systems in Gasterosteidae. Identity of the sex chromosomes are in parentheses, with “+” indicating a Y‐autosome fusion. The candidate sex determination gene of G. nipponicus (AmhY) is inferred here based on the shared ancestry of the sex determination regions of all Gasterosteus species. Tree topology and node dates (given above nodes) are taken from: (a) Varadharajan et al. (2019); (b) Betancur‐R et al. (2015), Friedman et al. (2013), Near et al. (2013), Sanciangco et al. (2016); (c) Ravinet et al. (2018); (d) timetree.org; (e) Guo et al. (2019). Image credits: G. wheatlandi and G. nipponicus by Rene P. Martin, CC BY‐SA 4.0; G. aculeatus and C. inconstans by Ellen Edmonson, used with permission from the New York State Department of Environmental Conservation (NYSDEC); P. pungitius, P. sinensis, and P. tymensis by Nilo Sinnatamby, CC BY‐SA 4.0; A. quadracus by Hugh Chrisp, used with permission from NYSDEC; S. spinachia by Krüger ‐ Bibliothèque nationale de France, Public Domain
Here, we take a first step towards addressing these questions by identifying the sex chromosome and a strong candidate for the sex determination gene in the brook stickleback, Culaea inconstans. The genus Culaea is sister to Pungitius, and its phylogenetic placement in a clade with Pungitius and Apeltes (Kawahara et al., 2009) means that it has the potential to provide valuable insight into the origin and diversity of sex determination mechanisms across the Gasterosteidae family (Figure 1). We find convincing evidence that C. inconstans has undergone a sex chromosome turnover to a chromosome not previously known to be used in stickleback. This turnover seems to have been driven by a duplication and translocation of the well‐known sex determination gene, Amh, which is independent of the duplication of this gene in the Gasterosteus lineage. Thus, although the turnover involves a novel chromosome pair, the sex determination gene itself represents an example of convergence both in the identity of the gene and the mode of turnover and may provide clues as to why sex chromosomes in some lineages are so dynamic.
2. METHODS
2.1. Sample collection and sequencing
In this study, we examined two sample sets of the brook stickleback, C. inconstans. The first and largest is composed of wild‐caught individuals from a single population in Shunda Lake, Alberta, Canada (UTF‐8 encoded WGS84 coordinates: 52.453899 latitude, −116.146192 longitude). We collected a total of 84 samples in June of 2017 and 2019 using unbaited minnow traps (5 mm mesh). Samples were collected under a fisheries research licence issued by the Government of Alberta. Collection methods and the use of animals in research were approved by the Animal Care Committee at Mount Royal University (Animal Care Protocol ID 101029 and 101 795). We identified 46 males and 38 females at the site of capture by examining gonads, observing the extrusion of eggs, and by noting the presence of nuptial colouration in males. DNA was extracted using Qiagen DNEasy Blood and Tissue kits. DNA samples were sent to Genome Québec for shotgun DNA library preparation using an NEB Ultra II kit. Paired‐end sequencing (150 bp) was performed alongside other libraries; the samples in this study therefore received approximately one lane of Illumina HiSeqX (40 samples collected in 2017) and half of a NovaSeq6000 lane with an S4 flow cell (remaining 44 samples collected in 2019).
The second sample set consists of a single F1 lab cross between a female from Fox Holes Lake, Northwest Territories, Canada and a male from Pine Lake, Alberta, Canada; this cross was previously genotyped with a limited set of microsatellite markers (Ross et al., 2009). DNA was isolated from fin tissue using phenol‐chloroform extraction followed by ethanol precipitation. Four Nextera sequencing libraries were prepared: one using DNA from the mother, one using DNA from the father, one using DNA pooled from 16 daughters and one using DNA pooled from 14 sons. Paired‐end sequencing was performed on an Illumina NovaSeq SP flow cell for 300 cycles at the University of Bern Next Generation Sequencing Platform.
2.2. Data pre‐processing and SNP calling in wild‐caught C. inconstans
Sequencing of the 84 Shunda Lake stickleback yielded an average of 24.8 million read pairs per sample. Sequence quality was checked using FastQC, and an average of 0.59% (±0.13%) read pairs per sample were dropped during adapter and quality trimming using Trimmomatic v.0.36 (Bolger et al., 2014). Trimmed reads were then aligned using BWA‐mem v.0.7.17 (Li & Durbin, 2009) with default alignment parameters to a genome assembly of a P. pungitius male (Varadharajan et al., 2019) as it is the most closely related reference genome to C. inconstans (26.6 MYA) (Betancur‐R et al., 2015; Guo et al., 2019; Rabosky et al., 2013, 2018; Sanciangco et al., 2016). Alignment files were then processed with samtools v.1.10, and an average of 16.4% (± 7.0%) read pairs were then marked as PCR duplicates and removed using picard‐tools v.2.21.8. Remaining aligned reads resulted in an average read depth of 7.20 (± 2.14) for each sample; however, coverage was highly variable along the genome, with peaks of coverage of over 1500 reads in places, almost certainly driven by repeats.
Variant calling was performed using the Genome Analysis Toolkit (GATK) v.4.1.3.0 following the GATK best practices pipeline (DePristo et al., 2011) and resulted in 36 237 609 variants before filtering. Comparison of these variants to Hardy–Weinberg expectations revealed many variants with an excess of heterozygosity, likely because of repetitive regions. The full variant call sets for the wild‐caught samples were then filtered using VCFtools v0.1.15 (Danecek et al., 2011) to retain single nucleotide polymorphisms (SNPs) with the following attributes: within samples or pools, genotypes were retained if they had a minimum depth of 10 reads (‐‐minDP 10) and a minimum genotype quality score of 30 (‐‐minGQ 30). Across samples, loci were kept if they had a maximum mean depth across all samples of 200 or lower (max‐meanDP 200), less than 30% missing samples after genotype filters (‐‐max‐missing 0.3), and a minor allele frequency greater than 0.01 (‐‐maf 0.01). These filtering criteria reduced the call set to 249 485 variants, which were used for all analyses of the wild‐caught data set below. However, many loci showing excess heterozygosity persisted in the data set and were not filtered further, as this would likely remove signals of sex linkage (Figure S1, see Jupyter notebook JN_01, Appendix S1).
2.3. Identification of sex‐linked regions of the genome in wild‐caught C. inconstans
Sex‐linked genome regions typically exhibit two features that can be used to identify them using genomic data analysis. The first is that they often lose or gain segments of DNA on only one of the sex chromosomes. Such regions produce a read depth difference among the sexes in sequencing data reflecting their copy number in the genome. For instance, an X‐specific region will have roughly 2n coverage in XX females, and only 1n coverage in XY males, resulting in a ratio of male to female read depth of around 1:2. Secondly, sex‐linked regions accumulate sequence differences between the sex chromosomes. This often results in variants which are specific to the sex‐limited chromosome, which leads to an increase in SNP density and heterozygosity in sex‐linked regions in the heterogametic sex, relative to the homogametic sex. It is generally observed that small mutational differences accumulate on sex chromosomes in the early stages of their differentiation, and large loss or gain of DNA sequence is typically a sign of an older sex‐linked region. Here, we used both read depth and heterozygosity‐based approaches for assessing sex linkage across the genome in the wild‐caught data set from Shunda Lake.
For the read depth analysis, Deeptools v.2.5.4 was used to calculate coverage per sample across the genome in 1 kb windows, normalized by the average number of reads per kilobase mapped (RPKM). Normalized coverages for each window were then averaged for each sex and the mean male depth per window was then divided by that of females and plots were smoothed using a rolling average over 10 windows (JN_02, Appendix S1).
We then assessed genotypes for patterns of heterozygosity consistent for sex linkage. SNP calling resulted in many loci with excess heterozygosity, most likely due to reads from multiple repeat copies in C. inconstans aligning to a single (likely collapsed) repeat locus in the P. pungitius assembly (See above). However, repeats are common in sex‐linked genomic regions and therefore likely to contain signals of sex linkage, some of which can be salvaged (i.e., old repeat copies which are unique enough for robust assembly and alignment). Thus, we opted not to mask repeats in the P. pungitius genome prior to read alignment. Instead, we used a novel test for the association of heterozygosity at each locus with sex. For each locus, we calculated the probability of the observed pattern of heterozygotes across males and females occurring by chance using a non‐sequential random draw without replacement which takes into consideration the number of samples of each sex called at a given locus:
where N is the total number of samples called, N M is the number of males called, N F is the number of females called, H is the total number of heterozygotes, H M is the number of male heterozygotes and H F is the number of female heterozygotes. The resulting p‐values suffer from multiple testing. However, much like in genome wide association studies, the correction is not straightforward as genetic linkage between loci in close proximity to each other violates the multiple testing assumption that each test is independent. Here we avoided this issue by not invoking any threshold for “significant” sex linkage. Instead, we simply use our calculated p‐values as a relative measure of the extent of sex linkage. For reference, absolute sex linkage of a locus (i.e., heterozygous in all 46 males and homozygous in all 38 females) would yield p = 8.6 × 10−25 (log2(p) = −79.9), while, for a scenario with 20 heterozygotes evenly distributed across the sexes (11 male and 9 female heterozygotes), p = .2 (log2(p) = −2.3) (JN_03, Appendix S1).
2.4. Locating AmhY in the C. inconstans genome
Male coverage patterns suggested that there is an additional sex‐linked copy of Amh in the C. inconstans genome, implying that the duplicate must exist only on the Y chromosome (see Section 3). If a Y‐specific copy of Amh exists, then, as an artefact of the read alignment to a genome with only a single copy of Amh, any mutations that have arisen in the Y copy since the duplication will be observed as male‐specific SNPs within the Chromosome 08 copy of the gene. However, the Y‐linked alleles at these sites should show half the coverage (1n) of the autosomal alleles (2n). Thus, to test the hypothesis of a Y‐linked duplication of Amh, we compared allelic read depth ratios at the sex‐linked loci located in the Amy gene and compared them to the rest of the loci in the genome in the wild‐caught data set, using a slightly modified implementation of HDplot (McKinney et al., 2017) (JN_04, Appendix S1). We also performed a more explicit test of allele depth ratios using custom python scripts, specifying which allele is sex linked, and comparing its read depth to that of the putatively autosomal alleles (JN_04, Appendix S1).
We then asked: where in the genome does the sex‐linked duplicate of Amh (and thus the Y chromosome) reside? Patterns of sex‐biased heterozygosity identified two regions of the P. pungitius reference genome with signs of sex linkage: Amh on Chromosome 08 and Chromosome 20 (see Section 3). However, it is not possible to have two strongly sex‐linked regions in a genome. Polygenic sex determination systems exist, but complete sex linkage signal would not be observed at either locus, which rules out this possibility in the current study. We therefore hypothesized that the two regions showing sex linkage are an artefact of the alignment to the P. pungitius genome assembly, and that, in C. inconstans, these sex‐linked loci lie in a single region. The most parsimonious scenario is that the duplicated Amh copy resides in the region of Chromosome 20 showing sex linkage. However, there were also regions that showed signs of Y‐specific duplications in this region of Chromosome 20 (see Section 3), raising the possibility that this region might also have duplicated, and that the sex determining region in C. inconstans may lie on a different chromosome altogether.
To test these competing theories, we first called structural variants in every wild‐caught sample using DELLY v0.7.8 (Rausch et al., 2012) and screened these variants for any translocations between the regions of sex linkage on Chromosomes 08 and 20, and for any variants showing patterns of sex linkage. Second, we manually screened the sequencing data for the presence of read pairs in which one read aligned near to Amh on Chromosome 08 and the other aligned within the sex‐linked region of Chromosome 20, which would occur if a read pair spanned the translocation break point (JN_05, Appendix S1). Third, we used Abyss v2.0.2 (with default parameters) to produce a de novo contig assembly of a pool of raw reads from the three highest coverage C. inconstans males, equating to approximately 100x coverage of the genome. The aim of this approach was to produce contigs that included either the autosomal Chromosome 08 copy of Amh or the sex‐linked AmhY flanked by regions syntenic to the sex‐linked region of Chromosome 20.
Lastly, we analysed the whole‐genome resequencing of the lab cross consisting of individually sequenced parents, a female offspring pool, and a male offspring pool (see “Sample collection and sequencing”). As there are only 30 offspring in this cross, it represents only 30 separate meiotic events that occurred in the father. Thus, there should have only been on the order of 30 crossover events between the X and the Y chromosome. This design should thus result in large linkage blocks along the genome, making it much easier to identify regions of the genome which are inherited in a sex‐linked fashion.
These sequence data were processed using the same procedures as for the wild‐caught data: data were quality checked and trimmed using fastQC and trimmomatic, resulting in 177.6 million retained reads for the father, 168 million for the mother, 160.8 million for the male offspring pool, and 75.7 million for the female offspring pool. These reads were aligned to the P. pungitius reference assembly, and duplicates were marked using picard‐tools v.2.21.8. We called variants in the parents using bcftools v1.10, which resulted in 19.7 million unfiltered variants in the male and 19.4 million in the female. To call variants in the pooled sequencing data, we used samtools v.1.10 to create an mpileup file which was then converted to allele frequencies using MAPGD v0.4.40 (Lynch et al., 2014). Variants were retained (using a custom python script JN_06, Appendix S1) only if they were present in the father, mother, male pool, and female pool, and if read depth in the parents was greater than 10 and parental genotype quality was greater than 30. To visualize the data, we plotted female ‐ male allele frequencies along the genome. Sex‐linked regions in which females are homozygous and males are heterozygous should show a female–male frequency of close to 0.5, compared to the autosomal expectation of close to 0. We then identified putatively sex‐linked variants that were heterozygous in the father, homozygous in the mother and where the X‐specific allele has a frequency between 0.4–0.8 in the male sequencing pool, and >0.98 in the female pool. These thresholds were chosen based on plotting male vs female pool frequencies (Figure S2, JN_06, Appendix S1).
2.5. Identifying the origin of AmhY in C. inconstans
To identify the origin of Amh duplication in brook stickleback, we compared the C. inconstans copies of this gene to each other and to its orthologs from other stickleback species. It was first necessary to identify Amh sequences for available closely related species. To do this we capitalized on already published whole‐genome sequencing data sets for 7 other stickleback species (G. aculeatus: 4 males, 4 females (White et al., 2015), G. nipponicus: 5 males, 5 females (Yoshida et al., 2014), G. wheatlandi: 4 males and 4 females (Liu et al., 2022), P. pungitius: 15 males, 15 females (Dixon et al., 2018), P. tymensis: 15 males, 11 females (Dixon et al., 2018), P. sinensis: 13 males, 9 females (Dixon et al., 2018) and Apeltes quadracus: 4 males, 4 females (Liu et al., 2022)).
We aligned adapter and quality trimmed reads from each species to the latest G. aculeatus genome assembly, which includes the Y chromosome (Peichel et al., 2020). We chose this reference over the P. pungitius assembly as it is already known that an Amh duplicate exists on the assembled Y chromosome of G. aculeatus (Peichel et al., 2020). Before aligning raw reads, we removed the Y chromosome scaffold from this assembly to ensure that reads from any and all copies of Amh in each species would align to the ancestral Amh copy on Chromosome 08 in the G. aculeatus assembly.
Alignments were again performed using BWA‐mem v0.7.17. Reads aligning to Amh on G. aculeatus Chromosome 08 were subsetted, and bcftools v1.10 was used to call variants. Variants were filtered using VCFtools to ensure that each genotype was based on a minimum read depth of 5, had a minimum genotype quality score of 30 and that data for each locus was present in at least 70% of samples within a species data set. The bcftools consensus tool was then used to produce a consensus sequence for each species using the major (highest frequency) allele at any polymorphic positions. Finally, in species where a sex‐linked copy of Amh exists, the consensus for the Y copy of Amh was output using a custom python script to phase SNPs based on their sex linkage (JN_07, Appendix S1). The resulting consensus sequences of Amh and AmhY were then aligned using the ClustalW algorithm implemented in MEGA v10.1 (Kumar et al., 2018), and a maximum likelihood tree was constructed using a Tamura‐Nei nucleotide substitution model with 500 bootstrap replicates, again implemented in MEGA.
Lastly, we predicted the effect of mutations between Amh paralogs in Gasterosteus and C. inconstans using Provean v1.1 (Choi, 2012; Choi et al., 2012; Choi & Chan, 2015). Provean compares a query protein to dozens of sequences from other taxa (in this case 66) taken from the NCBI protein database and computes a score which quantifies the conservation of each amino acid. As highly conserved amino acids are expected to be of high functional importance, this “Provean score” can be used to classify amino acid changes between two sequences of interest as putatively neutral (Provean score > −2.5) or putatively deleterious (Provean score ≤ −2.5). We compared Amh08 and AmhY in C. inconstans. For reference, we also compared the ancestral Amh08 and AmhY sequences for all Gasterosteus species, which were reconstructed using GRASP‐suite v2020.05.05.
3. RESULTS
3.1. Identification of sex‐linked regions in C. inconstans
Comparison of sequencing read depth between wild‐caught males and females from Shunda Lake failed to identify any large region of the genome with a reduction of read depth in one sex that would be indicative of well‐differentiated sex chromosomes. This analysis did, however, identify several very narrow regions throughout the genome with either male or female coverage bias (Figure 2). Two such regions were of particular interest. First, on Chromosome 08, there is a clear peak of high male versus female coverage (mean male/female coverage = 1.47 ± 0.09) centred at position 16.8325–16.839 Mb, which exactly matches the position of the gene Amh (Figure S3). Second, three peaks of high male vs. female coverage co‐localize within a ~5 Mb region on Chromosome 20 between position 2‐6 Mb (Figure 3). All three of these regions showed male coverage approximately 1.5 times that of female coverage, similar to the peak on Chromosome 08. The genome average for the male vs. female coverage ratio was 1.00 ± 0.07. Thus, the above‐mentioned peaks of high male coverage are consistent with a 3:2 ratio of male: female copy number in these regions and therefore suggest Y chromosome‐specific duplications of these loci.
FIGURE 2.
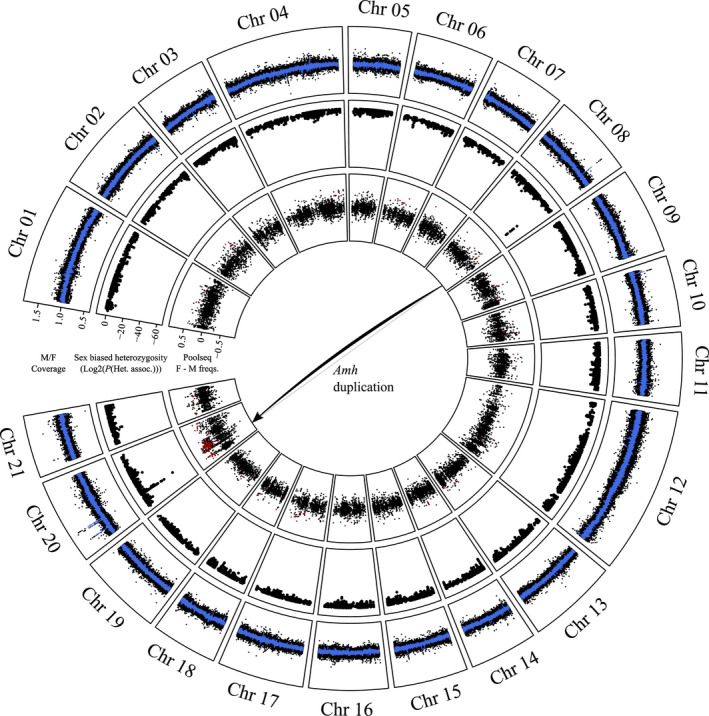
Sex linkage in C. inconstans, visualized on the P. pungitius genome assembly. Outer track: male/female coverage in 1 kb windows. The blue line represents the rolling average across 10 windows. Middle track: results of the test for association of heterozygosity patterns with sex. Inner track: female pool – male pool allele frequencies from the lab cross. The red points represent loci with parental genotypes and pool frequencies that fit expectations of sex linkage (see Section 2)
FIGURE 3.
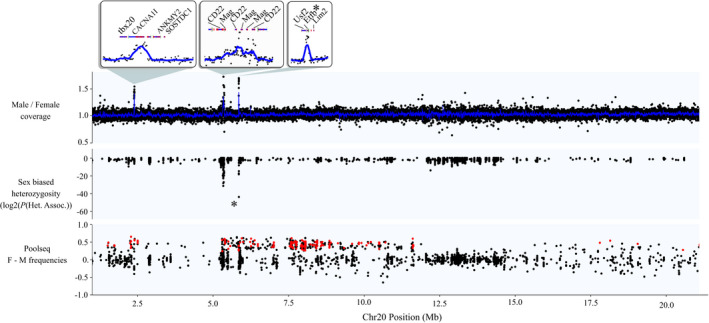
Signals of sex linkage along Chromosome 20, the putative sex chromosome in C. inconstans. For the coverage panel, each point represents male/female coverage in a 1 kb window. The blue line represents the rolling average across 10 windows. The asterisk in the heterozygosity panel and the zoomed box above represents the single completely sex‐linked variant that aligned to Chromosome 20 (in the gene Eftb). For the pooled sequencing panel, red points represent those with parental genotypes and pool frequencies that fit expectations of sex linkage (see Section 2)
The test for the association of heterozygosity patterns with sex was effective at identifying regions of sex linkage and overcoming the excess heterozygosity in the data set. The vast majority of loci showed high p‐values indicative of no sex linkage (Figure S4). Patterns of sex linkage were localized to two specific regions of the genome (Figure 2). Of the 10 loci that showed complete sex linkage (i.e., homozygous in all females and heterozygous in all, or all but a few, males), nine of them aligned to a narrow region on Chromosome 08. This region exactly coincides with a peak of male vs. female read depth mentioned above at the location of Amh (Figure S3). The vast majority of the remaining loci showing sex linkage (including one completely sex‐linked locus) aligned to a ~0.6 Mb region on Chromosome 20 (5.3–5.9 Mb), again coinciding with two peaks of increased male vs. female read depth (Figure 3). The loci showing sex linkage in these regions constitute the lowest probability values of the entire data set.
Together, the higher male coverage and the male‐biased heterozygosity suggest that a male specific (i.e., Y‐specific) Amh copy exists in the C. inconstans genome (henceforth referred to as AmhY), in addition to the ancestral Chromosome 08 copy (henceforth Amh08). If this is true, then the coverage of the male specific alleles identified by our heterozygosity analysis, which must have arisen on AmhY since the duplication, should be close to half that of Amh08 alleles. Read depth ratios for the nine completely sex‐linked SNPs fit this prediction, showing a clear departure from a 1:1 ratio, and were close to the 1:2 expected ratio, supporting the hypothesis that a Y‐specific Amh duplicate exists and harbours sex linked variation (Figure S5). This is the only gene in the genome to show complete sex linkage and is thus a strong candidate for the master sex determination gene in C. inconstans.
3.2. Chromosome 20 is the candidate sex chromosome in C. inconstans
While Amh08 was the only gene in the entire genome to show complete sex linkage, a ~0.6 Mb region on Chromosome 20 also showed partial sex linkage in our heterozygosity analysis, suggesting that AmhY resides somewhere in or near to this region (Figure 3). This raises the hypothesis that Chromosome 20 is the sex chromosome in C. inconstans. Additional support for this hypothesis comes from the fact that one of the ten loci showing complete sex linkage aligned to this region of Chromosome 20 (specifically within an intron of the gene Etfb, which also shows male biased coverage indicative of a Y‐specific duplication event).
To further determine whether Chromosome 20 is the sex chromosome, we first conducted three different analyses in the Shunda Lake genomic data set. First, we performed a structural variant analysis. However, we failed to find any evidence of the Amh duplication to this region, and further, there was not a single structural variant that showed the patterns of sex linkage expected for a Y‐specific Amh duplication (Figure S6). Second, we performed a more detailed search for split read pairs, but this analysis was also inconclusive. While many split read pairs were found with one read aligning close to Amh on Chromosome 08, the mates of these reads aligned to all other chromosomes, with no enrichment of mates mapping to Chromosome 20. We did find one read pair in which one read aligned immediately adjacent to the expected breakpoint location around Amh08 and the other aligned approximately 3 kb away from Etfb in the sex‐linked region of Chr20 (Figure S7), but a single read pair alone is not enough to confirm the insertion. This analysis did, however, reveal the extremely patchy nature of the read alignments within the Amh region on Chromosome 08, and indeed outside of coding regions across the entire genome (Figure S7), likely due to the evolutionary divergence time between C. inconstans and the P. pungitius reference genome. It is thus not surprising that we were unable to find stronger evidence based on either the structural variant analyses or the paired read alignments for the insertion of Amh on Chromosome 20. Third, we performed a de novo assembly of raw sequence reads from 10 males. This assembly yielded only a single contig containing sequence homologous to Amh, and this contig also contained regions homologous to the sequence flanking Amh08. Contigs aligning to the sex‐linked region of chromosome 20 were fragmented and showed no sign of containing AmhY. We were thus unable to show direct evidence of the theorized translocation event.
However, our analysis of the pooled sequencing data from a laboratory cross yielded 242 putatively sex‐linked markers, 158 of which aligned around the previously identified sex‐linked region of Chromosome 20 (see red points in Figure 3). In addition, there are many more markers (plotted in black) in this region with differences in female and male allele frequencies close to 0.5, as expected for sex‐linked loci, but which were not heterozygous in the male sample. These are likely also sex linked but lack heterozygous calls in the father due to allele dropout in low coverage regions. The high LD in this cross was therefore useful in that it allowed us to identify a region of sex‐linked inheritance approximately 11 Mb long, contrasting with the ~0.6 Mb region of sex linkage in the Shunda Lake data set. Importantly, these results provide unambiguous evidence that the sex‐determination locus is linked to Chromosome 20.
3.3. Convergent duplication and recruitment of Amh as the sex determination gene
Consistent with the presence of only four SNPs between Amh08 and AmhY in C. inconstans, the phylogenetic analyses of Amh sequences from all sticklebacks confidently clustered Amh08 and AmhY from C. inconstans together (Figure 4). Similarly, the Gasterosteus AmhY sequences clustered together as an outgroup of the Gasterosteus Amh08 sequences. These data, therefore, support an independent duplication of Amh in C. inconstans.
FIGURE 4.
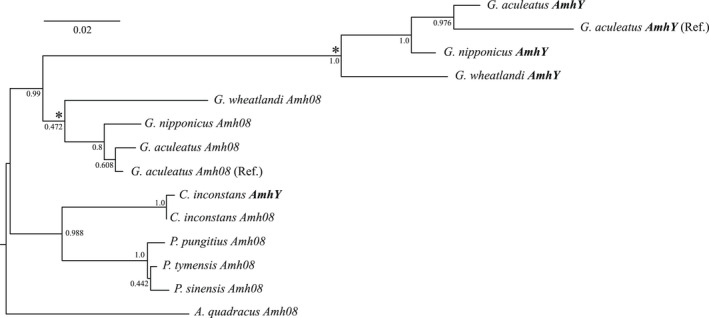
Maximum likelihood phylogeny of Amh consensus sequences for eight stickleback species. Node values represent confidence based on 500 bootstraps. Also included are the phased reference Amh and AmhY sequences from the G. aculeatus genome assembly (Peichel et al., 2020). Asterisks denote the nodes for which we reconstructed ancestral Amh and AmhY sequences for our mutation function predictions (see Section 2)
Provean analyses of amino acid conservation within Amh predicted that of the 93 inferred amino acid changes between the ancestral Amh08 and AmhY sequences in Gasterosteus, seven of them are likely to cause deleterious functional changes (Table S1). In contrast, all three of the amino acid changes between Amh08 and AmhY in C. inconstans were predicted to be neutral (Table S1).
4. DISCUSSION
The study of taxa with dynamic sex chromosome systems is key to understanding which forces and mechanisms shape the diversity of sex chromosomes and sex determination throughout nature. In this study we have identified the sex chromosome in C. inconstans, a species for which there was previously no information. Thus, there are now six stickleback species across three genera with known sex chromosome systems, further solidifying sticklebacks as a valuable model for the study of sex chromosome evolution. Importantly, several attributes of the C. inconstans sex chromosome system allow us to speculate on the evolutionary processes at work in stickleback, which we discuss in detail below.
4.1. A duplicate of Amh is the candidate master sex determination gene in C. inconstans
Our genomic analyses in C. inconstans strongly suggest that the autosomal gene Amh has duplicated and that this duplicate (AmhY) has been recruited as the master sex determining gene in this species. Formally confirming the sex determination role for this gene would require knock‐in and knock‐out transgenic experiments, which are beyond the scope of this paper. However, the fact that AmhY is the only completely sex‐linked gene in the genome is very strong evidence for its role as the MSD. Our finding of an additional region of (partial) sex linkage on Chromosome 20 suggests that this is the location of AmhY. We were unable to directly confirm this with the available sequencing data as our structural variant analyses, split read pair analysis and genome assembly approach all failed to find evidence of the duplication of Amh from Chromosome 08 to Chromosome 20. However, this is not surprising given the available genomic resources. All three of the approaches above rely on reliable sequencing data across the break point of the inserted region on Chromosome 20. Unfortunately, sequence alignments throughout the non‐coding regions of the genome are patchy, likely due to the evolutionary distance from the reference genome and repetitive regions. Indeed, structural variants like the one hypothesized here are often mediated by repeat elements. Thus, it is very possible that the breakpoints fall within regions of the reference genome to which reads cannot be aligned. Construction of a high‐quality genome assembly based on long‐read sequencing data from a C. inconstans male is needed to resolve the structure of the AmhY locus on Chromosome 20. However, even in the absence of such data, our analysis of an independent genetic cross provides clear evidence that the sex‐determination locus in C. inconstans is linked to Chromosome 20.
Duplications of Amh have previously been implicated in sex determination in many fish species, including the pejerreys Odontesthes hatcheri and O. bonariensis (Hattori et al., 2012; Yamamoto et al., 2014), Nile tilapia Oreochromis niloticus (Eshel et al., 2014; Li et al., 2015), lingcod Ophiodon elongatus (Rondeau et al., 2016), the cobaltcap silverside Hypoatherina tsurugae (Bej et al., 2017), northern pike Esox lucius (Pan et al., 2019), Sebastes rockfish Sebastes schlegelii (Song et al., 2021) and the Gasterosteus clade of stickleback (Peichel et al., 2020; Sardell et al., 2021). Amh is also likely used for sex determination in Monotremes (Cortez et al., 2014), though in this case, both X and Y Amh homologues exist, and no duplication is apparent. Thus, Amh is clearly predisposed to becoming a master sex determination gene in teleosts, as exemplified by its independent recruitment in two stickleback lineages within the last 25–30 My.
It is interesting to note that, of the eight teleost examples now known, it is seemingly always a duplicate of Amh that determines sex, not the ancestral autosomal copy. This level of convergence implies that duplication is an important process in the recruitment of this gene as the master sex determination gene. While the results presented here do not allow us to investigate this observation directly, below we speculate on several possible explanations for this convergence. One hypothesis is that the ancestral, autosomal copies of Amh in these species play some vital role that cannot be altered but can be circumvented via its duplication and subsequent sub‐ or neo‐functionalization. A second hypothesis is that the duplication itself is the sex determining mutation, i.e. simply increasing the dose of this gene is enough to initiate male development. We speculate that the second of these hypotheses is more likely, based on two lines of reasoning. The first relies on the observation that, in all of the cases above (which are all XX/XY systems), the duplication is Y‐specific and is absent from the X. If the duplicate does not determine sex when it first arises (i.e. due to increased dose alone), then it is free to segregate like any autosomal gene on both homologues of its resident chromosome pair and, at least in some cases, it might be fixed. If one allele of the duplicate later acquires the male determination role, an X copy would still exist. Thus, to match the observation that no X homologue exists in any of the eight species discussed here, we would need to invoke multiple losses of the X homologues, which is unlikely. Alternatively, if, from the moment it arose, the duplicate could determine sex, homozygosity would be unlikely, as that would require males mating with males. This is theoretically possible in teleosts, as sex reversals induced by environmental conditions can allow for XY × XY matings (Imiuwa, 2020), though it is not clear how common this is in nature. Nonetheless, the lack of an X homologue is as we see here fits a scenario where the duplicate Amh determines sex from the moment of duplication.
The second line of reasoning rests on the prediction that the three amino acid changes between Amh08 and AmhY in C. inconstans have no effect on the function of the AmhY protein. If true, this would imply that no functional change was necessary for AmhY to assume the role of sex determination in C. inconstans and which would leave increased gene dose as the likely sex determination mechanism. However, such predictions (based on the evolutionary conservation of each base) come with uncertainty and more work would be needed to confirm that no major functional changes have occurred between Amh08 and AmhY. In contrast to the Amh duplication in C. inconstans, the duplication events in northern pike, Gasterosteus stickleback, rockfish, pejerreys, lingcod, the cobaltcap silverside, and Nile tilapia, are seemingly old and have accumulated substantial protein sequence divergence between the ancestral and duplicated Amh copies. It is, therefore, not possible to infer whether these mutations were important in the recruitment of the Amh duplicates for sex determination in these species, or whether they have arisen since. It would be interesting to follow up on this question in future studies, for example, by creating transgenic XX individuals with an additional Amh copy to test for the effect of increased Amh dose on sex determination, in the absence of any amino acid changes.
Another interesting observation highlighted by our results in C. inconstans is that all eight of the species known to have independently recruited Amh duplicates for sex determination belong to the clade Teleostei. This is unlikely to be a coincidence. In most vertebrates, Amh (Anti‐Müllerian hormone) is responsible for inhibiting the development of the Müllerian ducts in the female reproductive tract during embryogenesis (Capel, 2017) and thus promoting male development. However, teleosts lack Müllerian ducts (Adolfi et al., 2019) raising the interesting question of the role of Amh in this clade. Data from Nile tilapia (Li et al., 2015) and pejerreys (Hattori et al., 2012; Yamamoto et al., 2014) shows expression of the Amh duplicate genes occurs just prior to gonadal differentiation, suggesting that they likely play an important role in the proliferation and differentiation of germ cells during male gonad development. More work is still needed to identify the exact mechanisms by which Amh can determine sex in teleosts, but one could speculate that the loss of Müllerian ducts in teleosts has freed Amh of its primary role of Müllerian duct suppression and this allows it to be more easily repurposed for sex determination.
More broadly, while several theoretical studies have considered the evolutionary forces that might drive a new MSD to fixation, one understudied component of sex chromosome turnovers is the rate at which alternative MSDs arise. Given that the genetic architecture of the sex determination pathways differs drastically among taxa (Capel, 2017), it is not unreasonable to expect that taxa also differ in the number of possible alternative MSDs that exist. Thus, it is possible that in some lineages, the rate of sex chromosome turnovers is limited by the constraints of their existing sex determination pathway and the frequency with which alternative MSDs can arise, while, in others, there may be numerous potential MSDs, which can readily evolve via simple mutations (e.g., gene duplication). The Amh/AmhrII pathway in teleosts may be an example of such a scenario and may in turn help explain their rapid turnover rate. Indeed, AmhrII, the receptor of Amh has also been found to be the sex determiner in pufferfish (Ieda et al., 2018; Kamiya et al., 2012), the ayu Plecoglossus altivelis (Nakamoto et al., 2021) and the yellow perch Perca flavescens (Feron et al., 2020).
4.2. Sex chromosome turnover in stickleback
With the results of this study, there are now four independently evolved sex chromosome systems known in sticklebacks, Chromosome 19 (AmhY) in Gasterosteus (with a further two independent Y‐autosome fusion events within this clade), Chromosome 12 in Pungitius pungitius, Chromosome 7 in P. sinensis and now Chromosome 20 in C. inconstans. There must therefore be a minimum of three sex chromosome turnover events among these species. However, the inability to find sex linkage signal on any of these chromosomes in P. tymensis (Dixon et al., 2018) or in A. quadracus (Ross et al., 2009) might be suggestive of more turnovers.
The sex chromosome of C. inconstans shows the lowest divergence of any of the sex chromosomes now described in stickleback when compared to the heteromorphism observed in Gasterosteus (Peichel et al., 2020; Ross & Peichel, 2008; Sardell et al., 2021) and the large region of differentiation found in P. pungitius (Dixon et al., 2018). Consistent with a lack of extensive degeneration on the Y, there is no evidence of a reduction in read coverage in males relative to females. Furthermore, there are very few loci that show evidence of differentiation between the X and the Y (i.e., differences in heterozygosity between males and females). In fact, if we are correct in our hypothesis that AmhY lies near to the gene Eftb on Chromosome 20, then the completely sex‐linked region of this chromosome may be on the order of 1 Mb in length. Given that, in general, recombination loss and sex chromosome differentiation expand outwards from the sex determination locus over time, this would suggest that the turnover event in C. inconstans was recent, and that the sex chromosomes in this species are young. However, several examples of old sex chromosomes with small non‐recombining regions do exist (Ieda et al., 2018; Kamiya et al., 2012; Rodrigues et al., 2018; Stöck et al., 2011), and thus sex chromosome differentiation may not be the best method for estimating the age of a sex determining system. Unfortunately, we cannot precisely estimate the age of the sex‐linked region without a good quality long read assembly of the X and Y chromosomes of C. inconstans. However, if it is recent, as our current data suggest, it may be possible to infer the identity of the ancestral sex chromosome pair by searching for signals left behind during its time in this role (e.g. reduced effective population size, increased repeat content [Vicoso & Bachtrog, 2013]). This would be an interesting topic of further study and could help to further characterize the transitions among sex chromosomes in stickleback.
Interestingly, our coverage analyses also identified several other genes in this sex‐linked region which seem to have male specific copies (see inlays in Figure 3). None of these genes have roles that have previously been associated with sex, though it is possible that their duplication and linkage with the sex determination gene may still be adaptive, for example, as a means of resolving genomic conflict at a sexually antagonistic locus (Bergero et al., 2019). The gene locations in this study are based on those in the P. pungitius assembly; however, it is likely that these genes are in approximately the same location in C. inconstans because they also show evidence of sex linkage. In addition, a manual examination of the G. aculeatus genome assembly places tbx20 and ANKMY2 next to Mag between positions 5–6 Mb on Chromosome 20 (CACNA1I, CD22, Eftb, and Lim2 were unfortunately not annotated in G. aculeatus). These locations are, therefore, likely ancestral. However, given the proximity of tbx20, ANKMY2 and Mag in G. aculeatus, there may, in fact, be an inversion around 2–5 Mb specific to P. pungitius, which would explain the distance between the sex linked tbx20 duplicate and the region of sex linkage identified in the wild‐caught data and the dearth of sex‐linked variants in the lab cross in this region. Again, a high‐quality reference assembly for the C. inconstans X and Y chromosomes is needed to resolve the speculation above.
In the context of studying young sex chromosomes, it is useful to highlight the utility of different data types in our analyses. Though pooled sequencing strategies lose individual haplotype information and the ability to examine heterozygosities, the fact that this data came from an F1 cross limited the number of recombination events in the data set. The resulting large blocks of linkage disequilibrium along the genome made this data set ideal for looking for a broad signal of sex linkage and, in this case, was essential for confidently identifying the sex chromosome. In fact, given how broad the sex linkage signal is in this cross, individual based sequencing would have been overkill, as increasing marker density far beyond the size of linkage blocks would add no more biological information. It should be noted, however, that such an approach will always overestimate the extent of true recombination suppression between sex chromosomes because the detection of rare recombination events is limited by the number of individuals in the cross. In contrast, the population‐level data set from Shunda Lake represents a large sampling and sequencing effort in terms of both money and time but provided the extremely fine resolution needed to distinguish between the complete sex linkage of a single gene, infer its duplication, and to infer partial sex linkage elsewhere in the genome. Importantly, this resolution comes not only from the high marker density and numerous samples but also from the large number of recombination events that have happened in the ancestors of all individuals sampled. Incorporating long coalescent times into a sample set captures linkage information from thousands of recombination events, and it is this that allowed us to map the sex‐linked regions of the genome so finely in this study. We highlight this point with the hope of aiding future researchers to design the most informative and cost‐effective data set for their purposes.
5. CONCLUSIONS
In the present study, we have identified a very strong candidate sex determination gene in C. inconstans, AmhY, which represents the eighth independent recruitment of this gene in teleosts, and the second within stickleback. The convergence on this gene and the potential role of gene duplication in this process offers promising clues as to the factors that allow for the lability of sex chromosomes seen in teleosts. However, studies identifying sex determination genes, sex chromosomes and transitions in many more taxa are needed before we have sufficient knowledge to gain more concrete insights into the general drivers of sex chromosome evolution.
CONFLICT OF INTEREST
The authors have no conflicts of interest to declare.
OPEN RESEARCH BADGES
This article has earned Open Data and Open Materials badges. Data and materials are available at the above NCBI SRA accessions, and in appendix S1.
PEER REVIEW
The peer review history for this article is available at https://publons.com/publon/10.1111/jeb.14034.
Supporting information
Appendix S1
Figure S1
Figure S2
Figure S3
Figure S4
Figure S5
Figure S6
Figure S7
Table S1
ACKNOWLEDGEMENTS
We would like to acknowledge and thank Melanie Hiltbrunner for making the sequencing libraries for the lab cross herein. We would also like to thank Rene Martin and Nilo Sinnatamby for their wonderful illustrations of several species in Figure 1. This work was funded by the Swiss National Science Foundation grant (number 31003A_176130) to C.L.P Open access funding provided by Universität Bern.
Jeffries, D. L. , Mee, J. A. , & Peichel, C. L. (2022). Identification of a candidate sex determination gene in Culaea inconstans suggests convergent recruitment of an Amh duplicate in two lineages of stickleback. Journal of Evolutionary Biology, 35, 1683–1695. 10.1111/jeb.14034
DATA AVAILABILITY STATEMENT
The individual‐level whole‐genome sequencing data from wild‐caught fish from Shunda Lake is available from the NCBI SRA under accession number PRJNA838194. The pooled (offspring) and individual‐level (parents) whole‐genome sequencing data from the lab cross is available under accession PRJNA843597. All custom python scripts can be found in Jupyter notebooks in the Appendix S1, along with relevant intermediate files from the evolutionary analyses.
REFERENCES
- Adolfi, M. C. , Nakajima, R. T. , Nóbrega, R. H. , & Schartl, M. (2019). Intersex, hermaphroditism, and gonadal plasticity in vertebrates: evolution of the Müllerian Duct and Amh/Amhr2 signaling. Annual Review of Animal Biosciences, 7, 149–172. [DOI] [PubMed] [Google Scholar]
- Bachtrog, D. , Mank, J. E. , Peichel, C. L. , Kirkpatrick, M. , Otto, S. P. , Ashman, T.‐L. , Hahn, M. W. , Kitano, J. , Mayrose, I. , Ming, R. , Perrin, N. , Ross, L. , Valenzuela, N. , Vamosi, J. C. , & The Tree of Sex Consortium . (2014). Sex determination: Why so many ways of doing it? PLoS Biology, 12, e1001899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bej, D. K. , Miyoshi, K. , Hattori, R. S. , Strüssmann, C. A. , & Yamamoto, Y. (2017). A duplicated, truncated amh gene is involved in male sex determination in an old world silverside. G3, 7, 2489–2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergero, R. , Gardner, J. , Bader, B. , Yong, L. , & Charlesworth, D. (2019). Exaggerated heterochiasmy in a fish with sex‐linked male coloration polymorphisms. Proceedings of the National Academy of Sciences of the United States of America, 116, 6924–6931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Betancur‐R, R. , Ortí, G. , & Pyron, R. A. (2015). Fossil‐based comparative analyses reveal ancient marine ancestry erased by extinction in ray‐finned fishes. Ecology Letters, 18, 441–450. [DOI] [PubMed] [Google Scholar]
- Blaser, O. , Neuenschwander, S. , & Perrin, N. (2014). Sex‐chromosome turnovers: The hot‐potato model. The American Naturalist, 183, 140–146. [DOI] [PubMed] [Google Scholar]
- Bolger, A. M. , Lohse, M. , & Usadel, B. (2014). Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics, 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capel, B. (2017). Vertebrate sex determination: evolutionary plasticity of a fundamental switch. Nature Reviews. Genetics, 18, 675–689. [DOI] [PubMed] [Google Scholar]
- Charlesworth, D. , Charlesworth, B. , & Marais, G. (2005). Steps in the evolution of heteromorphic sex chromosomes. Heredity, 95, 118–128. [DOI] [PubMed] [Google Scholar]
- Choi, Y. 2012. A fast computation of pairwise sequence alignment scores between a protein and a set of single‐locus variants of another protein. In: Proceedings of the ACM conference on bioinformatics, computational biology and biomedicine, pp. 414–417. Association for Computing Machinery. [Google Scholar]
- Choi, Y. , & Chan, A. P. (2015). PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics, 31, 2745–2747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi, Y. , Sims, G. E. , Murphy, S. , Miller, J. R. , & Chan, A. P. (2012). Predicting the functional effect of amino acid substitutions and indels. PLoS One, 7, e46688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortez, D. , Marin, R. , Toledo‐Flores, D. , & Froidevaux, L. (2014). Origins and functional evolution of Y chromosomes across mammals. Nature, 508, 488–493. [DOI] [PubMed] [Google Scholar]
- Danecek, P. , Auton, A. , Abecasis, G. , Albers, C. A. , Banks, E. , DePristo, M. A. , Handsaker, R. E. , Lunter, G. , Marth, G. T. , Sherry, S. T. , McVean, G. , Durbin, R. , & 1000 Genomes Project Analysis Group . (2011). The variant call format and VCFtools. Bioinformatics, 27, 2156–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DePristo, M. A. , Banks, E. , Poplin, R. , Garimella, K. V. , Maguire, J. R. , Hartl, C. , Philippakis, A. A. , del Angel, G. , Rivas, M. A. , Hanna, M. , McKenna, A. , Fennell, T. J. , Kernytsky, A. M. , Sivachenko, A. Y. , Cibulskis, K. , Gabriel, S. B. , Altshuler, D. , & Daly, M. J. (2011). A framework for variation discovery and genotyping using next‐generation DNA sequencing data. Nature Genetics, 43, 491–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dixon, G. , Kitano, J. , & Kirkpatrick, M. (2018). The origin of a new sex chromosome by introgression between two stickleback fishes. Molecular Biology and Evolution, 36, 28–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eshel, O. , Shirak, A. , Dor, L. , Band, M. , Zak, T. , Markovich‐Gordon, M. , Chalifa‐Caspi, V. , Feldmesser, E. , Weller, J. I. , Seroussi, E. , Hulata, G. , & Ron, M. (2014). Identification of male‐specific amh duplication, sexually differentially expressed genes and microRNAs at early embryonic development of Nile tilapia (Oreochromis niloticus). BMC Genomics, 15, 774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feron, R. , Zahm, M. , Cabau, C. , Klopp, C. , Roques, C. , Bouchez, O. , Eché, C. , Valière, S. , Donnadieu, C. , Haffray, P. , Bestin, A. , Morvezen, R. , Acloque, H. , Euclide, P. T. , Wen, M. , Jouano, E. , Schartl, M. , Postlethwait, J. H. , Schraidt, C. , … Guiguen, Y. (2020). Characterization of a Y‐specific duplication/insertion of the anti‐Mullerian hormone type II receptor gene based on a chromosome‐scale genome assembly of yellow perch, Perca flavescens . Molecular Ecology Resources, 20, 531–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friedman, M. , Keck, B. P. , Dornburg, A. , Eytan, R. I. , Martin, C. H. , Hulsey, C. D. , Wainwright, P. C. , & Near, T. J. (2013). Molecular and fossil evidence place the origin of cichlid fishes long after Gondwanan rifting. Proceedings of the Royal Society B: Biological Sciences, 280, 20131733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo, B. , Fang, B. , Shikano, T. , Momigliano, P. , Wang, C. , Kravchenko, A. , & Merilä, J. (2019). A phylogenomic perspective on diversity, hybridization and evolutionary affinities in the stickleback genus Pungitius. Molecular Ecology, 28, 4046–4064. [DOI] [PubMed] [Google Scholar]
- Hattori, R. S. , Murai, Y. , Oura, M. , Masuda, S. , Majhi, S. K. , Sakamoto, T. , Fernandino, J. I. , Somoza, G. M. , Yokota, M. , & Strüssmann, C. A. (2012). A Y‐linked anti‐Müllerian hormone duplication takes over a critical role in sex determination. Proceedings of the National Academy of Sciences of the United States of America, 109, 2955–2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ieda, R. , Hosoya, S. , Tajima, S. , Atsumi, K. , Kamiya, T. , Nozawa, A. , Aoki, Y. , Tasumi, S. , Koyama, T. , Nakamura, O. , Suzuki, Y. , & Kikuchi, K. (2018). Identification of the sex‐determining locus in grass puffer (Takifugu niphobles) provides evidence for sex‐chromosome turnover in a subset of Takifugu species. PLoS One, 13, e0190635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imiuwa, M. E. (2020). Induction of gonadal sex reversal in adult gonochorist teleost by chemical treatment: an examination of the changing paradigm. The Journal of Basic and Applied Zoology, 81, 1–7. [Google Scholar]
- Jeffries, D. L. , Lavanchy, G. , Sermier, R. , Sredl, M. J. , Miura, I. , Borzée, A. , Barrow, L. N. , Canestrelli, D. , Crochet, P. A. , Dufresnes, C. , Fu, J. , Ma, W. J. , Garcia, C. M. , Ghali, K. , Nicieza, A. G. , O’Donnell, R. P. , Rodrigues, N. , Romano, A. , Martínez‐Solano, Í. , … Perrin, N. (2018). A rapid rate of sex‐chromosome turnover and non‐random transitions in true frogs. Nature Communications, 9, 4088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamiya, T. , Kai, W. , Tasumi, S. , Oka, A. , Matsunaga, T. , Mizuno, N. , Fujita, M. , Suetake, H. , Suzuki, S. , Hosoya, S. , Tohari, S. , Brenner, S. , Miyadai, T. , Venkatesh, B. , Suzuki, Y. , & Kikuchi, K. (2012). A trans‐species missense SNP in Amhr2 is associated with sex determination in the tiger pufferfish, Takifugu rubripes (fugu). PLoS Genetics, 8, e1002798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara, R. , Miya, M. , Mabuchi, K. , Near, T. J. , & Nishida, M. (2009). Stickleback phylogenies resolved: evidence from mitochondrial genomes and 11 nuclear genes. Molecular Phylogenetics and Evolution, 50(2), 401–404. [DOI] [PubMed] [Google Scholar]
- Kitano, J. , Ross, J. A. , Mori, S. , Kume, M. , Jones, F. C. , Chan, Y. F. , Absher, D. M. , Grimwood, J. , Schmutz, J. , Myers, R. M. , Kingsley, D. M. , & Peichel, C. L. (2009). A role for a neo‐sex chromosome in stickleback speciation. Nature, 461, 1079–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar, S. , Stecher, G. , Li, M. , Knyaz, C. , & Tamura, K. (2018). MEGA X: Molecular evolutionary genetics analysis across computing platforms. Molecular Biology and Evolution, 35, 1547–1549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, H. , & Durbin, R. (2009). Fast and accurate short read alignment with Burrows‐Wheeler transform. Bioinformatics, 25, 1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, M. , Sun, Y. , Zhao, J. , Shi, H. , Zeng, S. , Ye, K. , Jiang, D. , Zhou, L. , Sun, L. , Tao, W. , Nagahama, Y. , Kocher, T. D. , & Wang, D. (2015). A tandem duplicate of anti‐Müllerian hormone with a missense SNP on the Y chromosome is essential for male sex determination in Nile tilapia, Oreochromis niloticus . PLoS Genetics, 11, e1005678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, Z. , Roesti, M. , Marques, D. , Hiltbrunner, M. , Saladin, V. , & Peichel, C. L. (2022). Chromosomal fusions facilitate adaptation to divergent environments in threespine stickleback. Molecular Biology and Evolution, 39, msab358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch, M. , Bost, D. , Wilson, S. , Maruki, T. , & Harrison, S. (2014). Population‐genetic inference from pooled‐sequencing data. Genome Biology and Evolution, 6, 1210–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKinney, G. J. , Waples, R. K. , Seeb, L. W. , & Seeb, J. E. (2017). Paralogs are revealed by proportion of heterozygotes and deviations in read ratios in genotyping‐by‐sequencing data from natural populations. Molecular Ecology Resources, 17, 656–669. [DOI] [PubMed] [Google Scholar]
- Nakamoto, M. , Uchino, T. , Koshimizu, E. , Kuchiishi, Y. , Sekiguchi, R. , Wang, L. , Sudo, R. , Endo, M. , Guiguen, Y. , Schartl, M. , Postlethwait, J. H. , & Sakamoto, T. (2021). A Y‐linked anti‐Müllerian hormone type‐II receptor is the sex‐determining gene in ayu, Plecoglossus altivelis . PLoS Genetics, 17, e1009705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Natri, H. M. , Merilä, J. , & Shikano, T. (2019). The evolution of sex determination associated with a chromosomal inversion. Nature Communications, 10, 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Near, T. J. , Dornburg, A. , Eytan, R. I. , Keck, B. P. , Smith, W. L. , Kuhn, K. L. , Moore, J. A. , Price, S. A. , Burbrink, F. T. , Friedman, M. , & Wainwright, P. C. (2013). Phylogeny and tempo of diversification in the superradiation of spiny‐rayed fishes. Proceedings of the National Academy of Sciences of the United States of America, 110, 12738–12743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan, Q. , Feron, R. , Yano, A. , Guyomard, R. , Jouanno, E. , Vigouroux, E. , Wen, M. , Busnel, J. M. , Bobe, J. , Concordet, J. P. , Parrinello, H. , Journot, L. , Klopp, C. , Lluch, J. , Roques, C. , Postlethwait, J. , Schartl, M. , Herpin, A. , & Guiguen, Y. (2019). Identification of the master sex determining gene in Northern pike (Esox lucius) reveals restricted sex chromosome differentiation. PLoS Genetics, 15, e1008013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peichel, C. L. , McCann, S. R. , Ross, J. A. , Naftaly, A. F. S. , Urton, J. R. , Cech, J. N. , Grimwood, J. , Schmutz, J. , Myers, R. M. , Kingsley, D. M. , & White, M. A. (2020). Assembly of the threespine stickleback Y chromosome reveals convergent signatures of sex chromosome evolution. Genome Biology, 21, 177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peichel, C. L. , Ross, J. A. , Matson, C. K. , Dickson, M. , Grimwood, J. , & Schmutz, J. (2004). The master sex‐determination locus in threespine sticklebacks is on a nascent Y chromosome. Current Biology, 14, 1416–1424. [DOI] [PubMed] [Google Scholar]
- Rabosky, D. L. , Chang, J. , Title, P. O. , Cowman, P. F. , Sallan, L. , Friedman, M. , Kaschner, K. , Garilao, C. , Near, T. J. , Coll, M. , & Alfaro, M. E. (2018). An inverse latitudinal gradient in speciation rate for marine fishes. Nature, 559, 392–395. [DOI] [PubMed] [Google Scholar]
- Rabosky, D. L. , Santini, F. , Eastman, J. , Smith, S. A. , Sidlauskas, B. , Chang, J. , & Alfaro, M. E. (2013). Rates of speciation and morphological evolution are correlated across the largest vertebrate radiation. Nature Communications, 4, 1958. [DOI] [PubMed] [Google Scholar]
- Rastas, P. , Calboli, F. C. F. , Guo, B. , Shikano, T. , & Merilä, J. (2015). Construction of ultradense linkage maps with Lep‐MAP2: stickleback F2 recombinant crosses as an example. Genome Biology and Evolution, 8, 78–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rausch, T. , Zichner, T. , Schlattl, A. , Stütz, A. M. , Benes, V. , & Korbel, J. O. (2012). DELLY: Structural variant discovery by integrated paired‐end and split‐read analysis. Bioinformatics, 28, i333–i339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravinet, M. , Yoshida, K. , Shigenobu, S. , Toyoda, A. , Fujiyama, A. , & Kitano, J. (2018). The genomic landscape at a late stage of stickleback speciation: High genomic divergence interspersed by small localized regions of introgression. PLoS Genetics, 14, e1007358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodrigues, N. , Studer, T. , Dufresnes, C. , & Perrin, N. (2018). Sex‐chromosome recombination in common frogs brings water to the Fountain‐of‐Youth. Molecular Biology and Evolution, 35, 942–948. [DOI] [PubMed] [Google Scholar]
- Rondeau, E. B. , Laurie, C. V. , Johnson, S. C. , & Koop, B. F. (2016). A PCR assay detects a male‐specific duplicated copy of Anti‐Müllerian hormone (amh) in the lingcod (Ophiodon elongatus). BMC Research Notes, 9, 230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross, J. A. , & Peichel, C. L. (2008). Molecular cytogenetic evidence of rearrangements on the Y chromosome of the threespine stickleback fish. Genetics, 179, 2173–2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross, J. A. , Urton, J. R. , Boland, J. , Shapiro, M. D. , & Peichel, C. L. (2009). Turnover of sex chromosomes in the stickleback fishes (Gasterosteidae). PLoS Genetics, 5, e1000391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanciangco, M. D. , Carpenter, K. E. , & Betancur‐R, R. (2016). Phylogenetic placement of enigmatic percomorph families (Teleostei: Percomorphaceae). Molecular Phylogenetics and Evolution, 94, 565–576. [DOI] [PubMed] [Google Scholar]
- Sardell, J. M. , Josephson, M. P. , Dalziel, A. C. , Peichel, C. L. , & Kirkpatrick, M. (2021). Heterogeneous histories of recombination suppression on stickleback sex chromosomes. Molecular Biology and Evolution, 38, 4403–4418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders, P. A. , Neuenschwander, S. , & Perrin, N. (2018). Sex chromosome turnovers and genetic drift: A simulation study. Journal of Evolutionary Biology, 31, 1413–1419. [DOI] [PubMed] [Google Scholar]
- Shapiro, M. D. , Summers, B. R. , Balabhadra, S. , Aldenhoven, J. T. , Miller, A. L. , Cunningham, C. B. , Bell, M. A. , & Kingsley, D. M. (2009). The genetic architecture of skeletal convergence and sex determination in ninespine sticklebacks. Current Biology, 19, 1140–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, W. , Xie, Y. , Sun, M. , Li, X. , Fitzpatrick, C. K. , Vaux, F. , O'Malley, K. G. , Zhang, Q. , Qi, J. , & He, Y. (2021). A duplicated amh is the master sex‐determining gene for Sebastes rockfish in the Northwest Pacific. Open Biology, 11, 210063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stöck, M. , Horn, A. , Grossen, C. , Lindtke, D. , Sermier, R. , Betto‐Colliard, C. , Dufresnes, C. , Bonjour, E. , Dumas, Z. , Luquet, E. , Maddalena, T. , Sousa, H. C. , & Martinez‐Solano, I. (2011). Ever‐young sex chromosomes in European tree frogs. PLoS Biology, 9, e1001062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Doorn, G. S. , & Kirkpatrick, M. (2007). Turnover of sex chromosomes induced by sexual conflict. Nature, 449, 909–912. [DOI] [PubMed] [Google Scholar]
- van Doorn, G. S. , & Kirkpatrick, M. (2010). Transitions between male and female heterogamety caused by sex‐antagonistic selection. Genetics, 186, 629–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varadharajan, S. , Rastas, P. , Löytynoja, A. , Matschiner, M. , Calboli, F. C. F. , Guo, B. , Nederbragt, A. J. , Jakobsen, K. S. , & Merilä, J. (2019). A high‐quality assembly of the nine‐spined stickleback (Pungitius pungitius) genome. Genome Biology and Evolution, 11, 3291–3308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicoso, B. (2019). Molecular and evolutionary dynamics of animal sex‐chromosome turnover. Nature Ecology & Evolution, 3, 1632–1641. [DOI] [PubMed] [Google Scholar]
- Vicoso, B. , & Bachtrog, D. (2013). Reversal of an ancient sex chromosome to an autosome in Drosophila. Nature, 499, 332–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White, M. A. , Kitano, J. , & Peichel, C. L. (2015). Purifying selection maintains dosage‐sensitive genes during degeneration of the threespine stickleback Y chromosome. Molecular Biology and Evolution, 32, 1981–1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto, Y. , Zhang, Y. , Sarida, M. , Hattori, R. S. , & Strüssmann, C. A. (2014). Coexistence of genotypic and temperature‐dependent sex determination in pejerrey Odontesthes bonariensis. PLoS One, 9, e102574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida, K. , Makino, T. , Yamaguchi, K. , Shigenobu, S. , Hasebe, M. , Kawata, M. , Kume, M. , Mori, S. , Peichel, C. L. , Toyoda, A. , Fujiyama, A. , & Kitano, J. (2014). Sex chromosome turnover contributes to genomic divergence between incipient stickleback species. PLoS Genetics, 10, e1004223. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1
Figure S1
Figure S2
Figure S3
Figure S4
Figure S5
Figure S6
Figure S7
Table S1
Data Availability Statement
The individual‐level whole‐genome sequencing data from wild‐caught fish from Shunda Lake is available from the NCBI SRA under accession number PRJNA838194. The pooled (offspring) and individual‐level (parents) whole‐genome sequencing data from the lab cross is available under accession PRJNA843597. All custom python scripts can be found in Jupyter notebooks in the Appendix S1, along with relevant intermediate files from the evolutionary analyses.