

Abstract
Background
The diagnosis of soft tissue tumors is challenging, especially when the evaluable material procured is limited. As a result, diagnostic ancillary testing is frequently needed. Moreover, there is a trend in soft tissue pathology toward increasing use of molecular results for tumor classification and prognostication. Hence, diagnosing newer tumor entities such as CIC‐rearranged sarcoma explicitly requires molecular testing. Molecular testing can be accomplished by in situ hybridization, polymerase chain reaction, as well as next generation sequencing, and more recently such testing can even be accomplished leveraging an immunohistochemical proxy.
Conclusion
This review evaluates the role of different molecular tests in characterizing soft tissue tumors belonging to various cytomorphologic categories that have been sampled by small biopsy and cytologic techniques.
Keywords: cytopathology, fluorescent in situ hybridization, molecular, next‐generation sequencing, polymerase chain reaction, sarcoma, soft tissue
1. INTRODUCTION
Soft tissue tumors, whether benign or malignant, often pose a diagnostic challenge for pathologists. Many entities in this category have similar demographic prevalence, clinical history, sites of predilection, and morphology, making differentiation difficult. Immunohistochemistry (IHC) can be diagnostically helpful; however, staining may be patchy or negative in a subset of tumors (e.g., S100 and SOX10 in malignant peripheral nerve sheath tumors) and the immunoprofile of many soft tissue tumors often also show overlap. Molecular testing has accordingly emerged as an invaluable adjunct for diagnosing several soft tissue tumor types. Moreover, the WHO is beginning to introduce molecularly defined soft tissue tumors (e.g., CIC‐rearranged sarcoma) into their classification system alongside morphologically defined tumors. 1
In soft tissue pathology, there are several molecular modalities used to evaluate tumors, including IHC, in situ hybridization (ISH), polymerase chain reaction (PCR), and next‐generation sequencing (NGS). IHC is the most indirect (surrogate) method, but also the most familiar to practicing pathologists (e.g., MDM2 and STAT6). IHC can be used to evaluate for the presence or absence of a mutated protein, expression of a protein in the same pathway as the altered gene, or even a protein resulting from a gene fusion. ISH, most commonly using fluorescent probes (hence fluorescent in situ hybridization [FISH]), is used to directly observe DNA or RNA, highlighting a specific fusion (fusion probes), gene amplification, or disruption of a gene of interest (break‐apart probes). PCR, with or without sequencing, can also test for a point mutation, a specific fusion or, depending on the assay, identify a fusion between a known gene and an unknown partner gene. NGS, or massively parallel sequencing, refers to contemporary sequencing technologies of both DNA and RNA. NGS is being increasingly applied to interrogate sarcomas. 2 For example, the Archer FusionPlex sarcoma panel currently covers key fusions and variants in over 60 genes (e.g., BCOR, CAMTA1, CIC, EWSR1, FOSB, FOXO1, MDM2, SS18, STAT6, USP6, YAP1, etc.) relevant to sarcomas. 3 Given that cytology soft tissue tumor specimens may have limited procured material, making their morphologic examination challenging and restricting the use of large IHC panels, the need to conserve cellular material and employ ancillary molecular tests is becoming more desirable.
The aim of this review is to evaluate the role of molecular tests in characterizing soft tissue tumors sampled by cytologic techniques.
2. MOLECULAR ALTERATIONS IN SOFT TISSUE TUMORS
Although there are many ways by which oncogenes can become activated and tumor suppressor genes inactivated, the three main mechanisms that are particularly important in sarcomas are mutations, chromosomal imbalances (copy number changes), and chromosomal rearrangements/fusions.
2.1. Mutations in sarcomas
In general, mutations affecting a single nucleotide (single‐nucleotide variant [SNV]) or insertion/deletion (indel) are much less common in sarcomas than in carcinomas. This difference reflects the fact that many carcinomas develop through a gradual transformation from a normal cell, whereas most sarcomas seem to start out as malignancies without first arising from dysplastic or benign precursor lesions. Hence, mesenchymal tumors have fewer and infrequent recurrent mutations. However, important exceptions include gastrointestinal stromal tumors (GIST) which are characterized by specific oncogenic mutations that affect the two tyrosine kinase receptors KIT or platelet‐derived growth factor receptor‐alpha (PDGFRA), in approximately 80% and 5%–10% of cases, respectively. 4 Mutations that prevent beta (β)‐catenin degradation and activate Wnt signaling occur in a high proportion of desmoid tumors. 5 Activating mutations of the GNAS1 gene, encoding the stimulatory G protein, are present in most cases of intramuscular myxoma, 6 and inactivating INI1 (SMARCB1) mutations are present in approximately 50% of malignant rhabdoid tumors. 7
2.2. Chromosomal imbalances in sarcomas
Chromosomal imbalances or copy number changes may range from gain or loss of a single nucleotide to whole chromosomes, which has been reported in soft tissue tumors, especially in sarcomas. Amplifications of specific chromosomal regions may lead to the formation of ring chromosomes, which are particularly common in atypical lipomatous tumor/well‐differentiated liposarcoma (ALT/WDLPS) and dedifferentiated liposarcoma (DDLPS) as well as dermatofibrosarcoma protuberans (DFSP), where they occur in 80% and 50% of cases respectively, and often as the sole cytogenetic aberration. 8 The murine double minute‐2 (MDM2) gene located on chromosome 12, an oncogene important for cell cycle control, is highly amplified in a significant number of WDLPS and DDLPS cases. 9
2.3. Gene fusions/rearrangements in sarcomas
Unlike many epithelial neoplasms, where diverse genetic alterations usually underlie the stepwise progression of precursor lesions leading ultimately to the emergence of malignant clones, soft tissue malignancies have no identifiable precursor lesions and usually have a single genetic alteration typical of a particular type of sarcoma. In addition, chromosomal fusions in soft tissue sarcomas do not seem to represent a form of generalized genomic instability, as occurs with germline TP53 mutations or microsatellite instability associated with colon cancer. Numerous fusion genes have been identified in malignant soft tissue tumors (e.g., EWSR1‐FLI1 and EWSR1‐ERG in Ewing sarcoma, SS18‐SSX1/2/4 in synovial sarcoma, NAB2‐STAT6 in solitary fibrous tumor, and ETV6‐NTRK3 in infantile fibrosarcoma). 10 The majority of sarcoma translocations result in in‐frame fusion genes, resulting in abnormal chimeric transcription factors. 11 In a few cases, the gene fusion results in an aberrant tyrosine kinase. 12
3. CHARACTERISTIC GENETIC ALTERATIONS IN SOFT TISSUE TUMORS
Soft tissue tumors can be broadly grouped into seven categories based on their predominant cytomorphology, including: spindle cell tumors, myxoid tumors, small round cell tumors, epithelioid tumors, lipomatous tumors, pleomorphic tumors, and giant cell‐rich tumors. We review below the molecular findings in tumors from each of these categories.
3.1. Spindle cell tumors
Spindle cell soft tissue tumors comprise a large proportion of soft tissue diagnoses and include a wide spectrum of benign and malignant entities. Reactive conditions that mimic sarcoma (pseudosarcoma) and non‐mesenchymal tumors with spindle cells (e.g., sarcomatoid carcinoma, melanoma) should be excluded. Hence, when working up these cases it is recommended to begin by narrowing down the differential diagnoses using IHC as needed, before molecular testing. Nonetheless, given the advances in our understanding of spindle cell sarcomas molecular testing is becoming increasingly essential for these cases.
3.1.1. Fibroblastic/myofibroblastic tumors
Desmoid fibromatosis is characterized by bland spindle cells arranged in sweeping fascicles. Lesional cells harbor alterations in the Wnt signaling pathway, commonly a point mutation in either CTNNB1 (beta‐catenin gene) for sporadic cases or APC in hereditary cases with Gardner syndrome, 13 , 14 resulting in nuclear accumulation of beta‐catenin. 15 , 16 Other downstream targets of beta‐catenin, such as c‐Myc and cyclin D1, may also be positive but there are fewer supporting data. 17 , 18 , 19 Beta‐catenin expression is higher in familial adenomatous polyposis (FAP)‐associated tumors as well. 20 As these alterations are point mutations, FISH is not helpful. PCR is rarely used due to the relative increased expense compared to H&E staining and IHC. Although desmoid tumors may be harbingers of FAP, there are no widely adopted guidelines on when genetic testing for germline alterations should be performed. Large meta‐analyses suggest that the CTNNB1 S45F mutation portends a higher risk of recurrence. 21 , 22 A caveat of using beta‐catenin IHC to confirm the diagnosis is that a significant proportion of cases do not have nuclear beta‐catenin expression, and thus the negative predictive value is low. 23 The sensitivity and specificity of beta‐catenin immunoreactivity by IHC also depends on the clone used. 24
Nodular fasciitis is a benign (“transient”) neoplasm characterized by self‐limited growth and eventual resolution regardless of therapy. 25 The underlying molecular alteration is a fusion of the USP6 gene, often but not exclusively with myosin heavy chain 9 (MYH9), most commonly evaluated by USP6 break‐apart FISH when needed. 26 Not all cases of nodular fasciitis will be positive, as over time the percentage of cells within these tumors that harbor a fusion decreases drastically, and thus if performed on an “old” case (i.e., over 3 months), it may not meet criteria for positivity depending on the institutional cutoff and the area biopsied. 27 Reverse transcription (RT)‐PCR can identify alternate USP6 fusions, although there is currently no significance linked to these various fusions. Anti‐USP6 antibodies for IHC exist but have not been well evaluated in the literature. USP6‐rearranged tumors comprise an increasingly broad group that includes not only nodular fasciitis but also fibroma of tendon sheath, myositis ossificans, fibro‐osseous pseudotumor of digits, and aneurysmal bone cyst. 28
Solitary fibrous tumor (SFT) comprises a spectrum of benign to malignant tumors, and their prognosis of aggressive behavior is dependent on patient age, tumor size, mitotic rate, and extent of necrosis at resection. 29 They almost always harbor a NAB2‐STAT6 fusion, which is detectable via PCR, 30 or more commonly with nuclear expression of STAT6 IHC, which detects the resultant overexpressed STAT6 protein (Figure 1A). 31 , 32 STAT6 expression may be lost in cases with dedifferentiated SFT, 33 but the fusion remains detectable by PCR. 34 While any relationship between the exact fusion transcript and prognosis is uncertain, alternate prognostic molecular factors such as TP53 or TERT promoter mutations, evaluable by PCR, have been associated with worse outcome. 35 , 36 , 37 , 38 , 39 , 40
Dermatofibrosarcoma protuberans (DFSP) is a locally aggressive sarcoma of the dermis that frequently harbors fusions of COL1A1 and either PDGFB (most common, >90%) or PDGFD (<5%). ISH, either by FISH 41 , 42 or RNA ISH (Figure 2), 43 may be used to detect the more common COL1A1‐PDGFB fusion, with PCR testing to support alternate fusions when needed. 44 It should be noted that DFSP may undergo transformation and take on the appearance of another sarcoma (typically fibrosarcoma), portending a worse prognosis. 45 Despite their malignant change in appearance, these tumors retain their original fusion and commonly demonstrate other molecular alterations, most often in the TP53 gene. 46 , 47
Inflammatory myofibroblastic tumor (IMT) is a tumor of borderline malignant potential composed of atypical myofibroblastic and fibroblastic spindle cells, often intermixed with a lymphoplasmacytic infiltrate and/or eosinophils. They occur in the lung as well as several extrapulmonary sites (e.g., abdominal and somatic soft tissue). The majority (50–60%) of IMTs have a clonal rearrangement involving chromosome band 2p23 (ALK gene) with multiple fusion partners. 48 This, in turn, results in activation and overexpression of ALK. Immunoreactivty for ALK is accordingly detectable in those cases with an ALK gene rearrangement. 49 Depending on the ALK fusion partner, ALK staining may have a nuclear membranous pattern (e.g., RANBP2‐ALK), perinuclear cytoplasmic pattern (e.g., RRBP1‐ALK), granular cytoplasmic pattern (e.g., CLTC‐ALK), or show a diffuse cytoplasmic pattern (Figure 3). 50
NTRK‐rearranged spindle cell neoplasms with NTRK fusions, involving any of the NTRK1/2/3 genes, are found in infantile fibrosarcoma and an emerging group of molecularly defined soft tissue tumors such as lipofibromatosis‐like neural tumor. IHC stains for pan‐NTRK (which detects any NTRK protein) or a protein‐specific IHC are available to evaluate for overexpression. A recent large study examining 38,095 solid tumors compared IHC, DNA‐based NGS, and RNA‐based sequencing and found that overall, IHC demonstrated excellent sensitivity for tumors with fusions of NTRK1 (96%) or NTRK2 (100%), but not NTRK3 (79%), and DNA‐based sequencing was 81.1% sensitive and 99.9% specific. 51 However, for sarcomas (of which only 49 cases were included), IHC was only 80% sensitive and 74.4% specific. A sarcoma‐specific study evaluated 494 cases and found pan‐NTRK immunostaining in 16 (3.2%), of which five had NTRK fusions by NGS and 11 did not. 52 The NTRK‐fused tumors showed diffuse staining, while those without NTRK fusions had only focal (<50%) staining, indicating that this may be a useful adjunct if appropriately interpreted. Nonetheless, positive staining in a small biopsy sample may be misleading. As the Food and Drug Administration (FDA) in the United States has approved the NTRK inhibitor Larotrectinib, identifying this fusion may have therapeutic impact for NTRK sarcomas. 53
FIGURE 1.
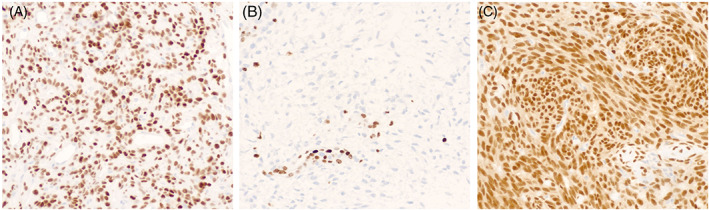
Immunohistochemical stains may act as a proxy for molecular alterations. The alteration may lead to overexpression of a protein, as with STAT6 in solitary fibrous tumor (A, STAT6 immunostain, 20×), or there may be loss of expression as is seen in malignant peripheral nerve sheath tumor, which loses the ability to methylate certain amino acids in histone proteins such as histone 3 lysine 27 (B, H3K27me3 immunostain, 20×). Note that the non‐neoplastic endothelial cells retain H3K27me3 expression. In rare cases, the chimeric fusion protein can be detected, as in synovial sarcoma (C, SS18‐SSX immunostain, 20×).
FIGURE 2.
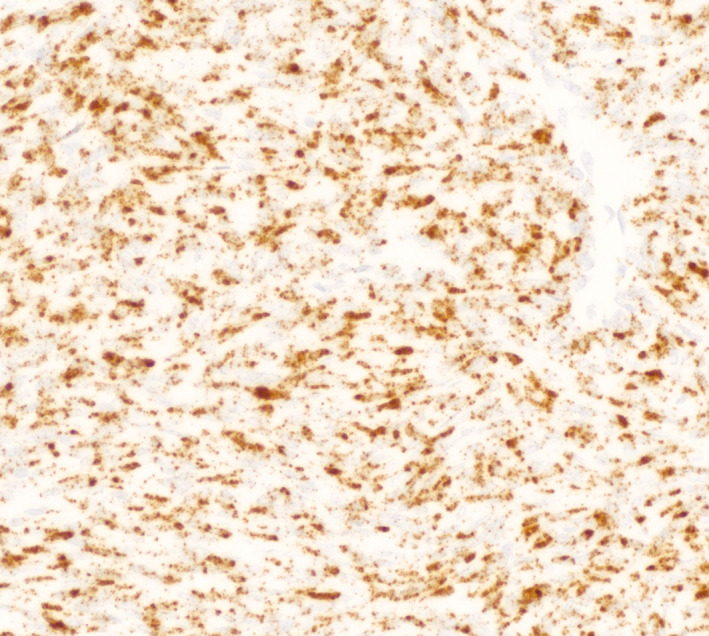
In dermatofibrosarcoma protuberans, RNA in situ hybridization for PDGFB allows for detection of a COL1A1‐PDGFB fusion without the need for FISH or PCR by demonstrating upregulation of PDGFB RNA expression. A tumor is considered positive if there are either five puncta or one aggregate in greater than 25% of tumor cells.
FIGURE 3.
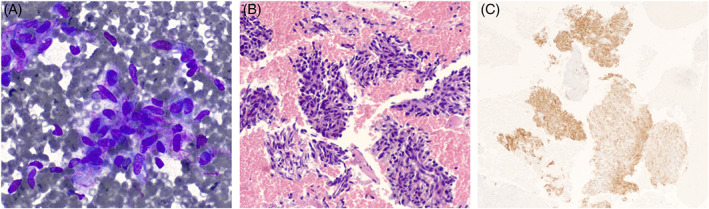
An inflammatory myofibroblastic tumor (IMT) seen on fine needle aspiration. The Diff‐Quik stain (A, 40×) and cell block (B, H&E, 20×) show clusters of mildly pleomorphic spindle cells within a background of chronic inflammation. An ALK immunostain (C, 10×) is positive in the tumor cells, suggesting the presence of an ALK rearrangement and confirming the diagnosis of IMT.
3.1.2. Nerve sheath tumors
Functional inactivation of genes on chromosome 22 is critical for development of schwannomas, regardless of whether they are sporadic lesions or part of a syndrome such as neurofibromatosis type 2 (NF2). 54 , 55 Neurofibroma development depends on inactivation of NF1 tumor suppressor gene on the long arm of chromosome 17. 56 , 57 Soft tissue perineuriomas harbor chromosomal losses, most commonly of chromosomes 22, 17, 2, or 6, 58 while intraneural perineuriomas often have TRAF7 mutations. 59 The much rarer hybrid nerve sheath tumors often have VGLL3 fusions instead, suggesting a different developmental pathway from simple nerve sheath tumors. 60 However, because the aforementioned molecular alterations are not pathognomonic currently the diagnosis of these benign nerve sheath tumors does not require molecular testing.
Malignant peripheral nerve sheath tumor (MPNST) may arise from a pre‐existing nerve sheath tumor, such as a neurofibroma, or de novo. In either case, they are aggressive sarcomas that have a strong tendency to recur and often metastasize. While genetically complex, alterations in the Polycomb repressor complex 2 (PRC2) are common. 61 , 62 These result in decreased histone methylation and thus loss of H3K27me3 (the trimethylated form of lysine 27 of the histone 3 protein), which is detectable by IHC (Figure 1B). 63 The loss of H3K27me2, the demethylated form, may be more specific for PRC2 loss. 64 Loss of H3K27me3 immunostaining is more frequent in high‐grade than low‐grade tumors. 65 FISH and PCR are not used for these tumors due to the variability of genetic changes.
3.1.3. Other spindle cell tumors
Synovial sarcoma is characterized by a unique t(X;18) translocation resulting in a fusion between the SS18 gene and a member of the SSX gene family on the X chromosome, most commonly SSX1 or SSX2 66 and rarely SSX4. 67 These sarcomas are commonly monophasic comprised of only spindle cells, but a small proportion are biphasic containing both fascicles of spindle cells and epithelial cells forming glands or cribriform structures. When poorly differentiated, synovial sarcoma may take on the appearance of a small round blue cell tumor. Traditionally, FISH for the SS18 gene or PCR were used to demonstrate the presence of one of the above fusions. 68 , 69 , 70 Recently, a new antibody that directly targets the fusion protein (SS18‐SSX) has been developed. 71 It may be paired with an antibody against the C‐terminus of SSX. 72 These antibodies are exquisitely sensitive and specific and detect fusions of SSX1, SSX2, or SSX4 due to sequence homology around the fusion point. IHC is positive in the vast majority of cases of synovial sarcoma (Figure 1C); only very rare fusions may result in a negative SS18‐SSX. 73 Indeed, a recent study found the IHC antibody to be more specific for the diagnosis of synovial sarcoma compared with FISH. 74
Gastrointestinal stromal tumors (GISTs) can harbor a range of alterations, most commonly substitution or insertion/deletion mutations, which is unusual for a mesenchymal neoplasm. IHC for DOG‐1 and CD117 are helpful in the diagnosis of GIST, with the DOG‐1 antibody being equivalent 75 or slightly superior to CD117. 76 Molecular testing increasingly guides therapy in these tumors, and thus molecular results are often requested. Alterations in KIT, PDGFRA, and SDH complex genes, and rarely NF1, KRAS, BRAF, NTRK, and others have been described. 77 While KIT and PDGFRA mutations are mutually exclusive, they are rarely found in conjunction with other mutations that may confer resistance to the tyrosine kinase inhibitor (TKI) imatinib despite the presence of a “susceptible” mutation. 78 At our institution, we test all GISTs for molecular alterations to guide treatment and follow a simple algorithm (see Table 1). KIT mutations are best detected by sequencing, as the exact mutation determines susceptibility to TKIs, but for some other alterations an IHC proxy can be used. Succinate dehydrogenase (SDH) deficient GISTs, which comprise a significant minority of gastric epithelioid GISTs, can be identified using an immunostain for SDHB, 79 and a PDGFRA immunostain can detect PDGFRA‐mutant GISTs, 80 although it was only 81% specific in a recent large study. 81 BRAF alterations are typically due to the common p.V600E mutation and thus the VE1 IHC antibody, sometimes used in melanoma, will be positive in these rare cases. 82 For NTRK‐rearranged GISTs, however, FISH is preferable due to the aforementioned low sensitivity of IHC. 83
TABLE 1.
Our in‐house GIST molecular testing algorithm.
| Gastric GIST | Non‐gastric/unknown site GIST | |
|---|---|---|
| Initial order | SDHB IHC | KIT mutation testing |
| If above fails | KIT mutation testing | PDGFRA mutation testing |
| If above fails | PDGFRA mutation testing | SDHB IHC |
Abbreviations: GIST, gastrointestinal stromal tumor; IHC, immunohistochemistry; PDGFRA, platelet‐derived growth factor receptor alpha; SDHB, succinate dehydrogenase, subunit B.
3.2. Myxoid tumors
Myxoid soft tissue tumors are among the most difficult to evaluate by cytopathology, as they are frequently markedly hypocellular and exhibit overlapping morphology. Furthermore, many “non‐myxoid” tumor types can develop myxoid change focally or globally (e.g., SFT with myxoid change). Myxoid tumors with available molecular testing include myxoid liposarcoma, low‐grade fibromyxoid sarcoma, and extraskeletal myxoid chondrosarcoma.
Myxoid liposarcoma (MLPS) is an adipocytic tumor characterized by prominent myxoid stroma and branching vessels. These tumors have a t(12;16) translocation and are accordingly molecularly defined by a fusion of the DDIT3 (formerly CHOP) gene, either with FUS or less commonly EWSR1. High‐grade MLPS (previously called round cell liposarcoma) has the same genetic abnormality. IHC for the DDIT3 chimeric oncoprotein is available, 84 , 85 although it has not been broadly tested on cytology specimens. FISH is more frequently used, employing a break‐apart probe targeted to the DDIT3 gene. 86 , 87 This has been tested in FNA samples yielding good results. 88 Care must be taken in interpretation, however, as the DDIT3 gene (12q13.3) may be co‐amplified with MDM2 (12q15) in dedifferentiated liposarcomas, especially in those with myxoid liposarcoma‐like areas, leading to a confusing FISH pattern when the DDIT3 telomeric tag is used. 89 , 90 Alternately, PCR for both FUS‐DDIT3 and EWSR1‐DDIT3 fusions can be performed. 91
Low‐grade fibromyxoid sarcoma (LGFMS) is a heterogeneous sarcoma characterized by bland fibrous and myxoid areas. They involve soft tissue locations, and infrequently deep body cavities or viscera. More than 90% of LGFMS cases harbor a t(7;16)(q33;p11), resulting in a FUS‐CREB3L2 fusion. The remaining cases have FUS‐CREB3L1 fusions. 92 If IHC (e.g., MUC4) is unhelpful, molecular testing by FISH or RT‐PCR to identify the FUS‐CREB3L2 or other rare fusions can be diagnostically helpful. 93
Extraskeletal myxoid chondrosarcoma (EMC) is a soft tissue tumor of uncertain differentiation that contains uniform interconnected spindle cells growing in cords or nests that are embedded in abundant myxoid matrix. These tumors are characterized by fusions involving the NR4A3 (previously CHN) gene. 94 While EWSR1 is the most common fusion partner, numerous others have been described (e.g., TAF15), and therefore for diagnostic tests the NR4A3 gene should be evaluated rather than a specific partner. IHC against the NR4A3 protein, although reported to be useful in salivary gland acinic cell carcinoma, 95 , 96 has not been found to be helpful in EMC in at least one study. 97 NR4A3 break‐apart FISH (Figure 4A) is the most useful test, given the variety of other genes that may fuse with it, 98 and this has also been shown to be effective in FNA specimens. 99 Not all cases will have detectable NR4A3 rearrangements, with sensitivity ranging from 61% to 100%. 100 , 101
FIGURE 4.
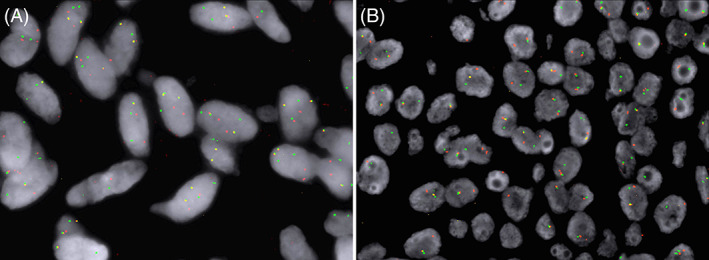
Representative photomicrographs of fluorescent in situ hybridization (FISH) of NR4A3 (A) and EWSR1 (B) dual‐color break‐apart probes showing separate red and green signals, indicating rearrangements of the NR4A3 and EWSR1 genes, respectively.
3.3. Small round cell tumors
Small round cell tumors (SRCTs) range from completely benign to extremely aggressive tumors. The differential diagnosis includes mesenchymal tumors that are differentiated (e.g., rhabdomyosarcoma) and undifferentiated (e.g., Ewing sarcoma), as well as non‐mesenchymal tumors (e.g., neuroblastoma, Wilms tumor). Rapid, accurate diagnosis is essential for planning therapy. In particular, malignant SRCTs tend to occur in younger patients, increasing the pressure for a quick turnaround on any biopsy. Molecular testing is most helpful to confirm the diagnosis for many of these tumors.
Ewing sarcoma may affect bone and extraskeletal sites. Classically these undifferentiated sarcomas have a fusion of the EWSR1 (previously EWS) gene, most commonly EWSR1‐FLI1 and EWSR1‐ERG (see Table 2). Previously, IHC for ERG and FLI1 have been used, but this is not reliable as many commercial anti‐FLI1 antibodies bind to the ETS domain, which is shared by other proteins (including ERG). 102 , 103 Some ERG antibodies, in particular clone EPR3864 (used for prostate cancer) may detect FLI‐1 as well. 104 Both are markers of vascular differentiation and are accordingly expressed in both normal blood vessels and neoplastic vascular tumors. 105 , 106 Both ERG and FLI1 have also been documented in other tumor types such as epithelioid sarcoma, which has a vastly different treatment approach. 107 Molecular testing is usually required to confirm the diagnosis of Ewing sarcoma. FISH for EWSR1 (Figure 4B) is of limited utility, both because this gene may be fused in a broad variety of sarcomas other than Ewing sarcoma (including desmoplastic small round cell tumor, clear cell sarcoma of soft tissue, extraskeletal myxoid chondrosarcoma, angiomatoid fibrous histiocytoma, myoepithelial tumor of soft tissue, and a subset of myxoid liposarcomas), and because a small subset of EWS have fusions of FUS rather than EWSR1. 108 PCR for the specific fusions found in Ewing sarcoma is the best and most accurate test to assist in making this diagnosis, 109 and multiplex PCR tests for multiple Ewing sarcoma fusions can be utilized if necessary. 110 NGS is particularly valuable because it provides details regarding both translocation partners.
Round cell sarcoma with EWSR1 – non‐ETS fusions are round and/or spindle cell sarcomas that affect bones more frequently than soft tissue. 111 They harbor EWSR1 or FUS fusions that involve partners unrelated to the ETS gene family, such as EWSR1‐NFATC2, FUS‐NFATC2, and EWSR1‐PATZ1. DNA methylation profiling reveals that these sarcomas are indeed distinct from Ewing sarcoma. 112 Rendering a definitive diagnosis requires identification of one of the characteristic fusions by means of molecular testing.
CIC‐rearranged sarcomas are a newly introduced, rare set of aggressive tumors that harbor CIC‐DUX4 or, less commonly CIC‐FOXO4 or CIC‐NUTM1 fusions. The former is much more common, accounting for approximately 57% of all reported CIC‐rearranged sarcomas in one large study. 113 While there is a DUX4 antibody with high specificity for tumors with this fusion, 114 other IHC stains are also helpful including WT1 (N and C terminal) and ETV4. 115 Alternately, FISH for the CIC gene may be used, which will catch either alteration; however, there are limitations to FISH testing as well, with a recent study showing that up to 26% of cases may have false negative FISH results. 116 PCR for CIC‐DUX4 would reveal this fusion in all cases. Rare, alternate DUX4‐fused sarcomas have been reported, but the data regarding DUX4 IHC in these cases are limited.
BCOR‐rearranged sarcomas are another emerging group of tumors, previously categorized as undifferentiated round cell sarcoma similar to Ewing sarcoma. 117 As is the case in high‐grade endometrial stromal sarcomas, BCOR IHC is an unreliable surrogate for BCOR fusion, with some studies showing that only 71% of cases are positive in BCOR‐CCNB3 fused cases. 118 FISH or PCR for BCOR may be more accurate.
Alveolar and embryonal rhabdomyosarcomas both have SRCT morphology. However, only alveolar rhabdomyosarcoma (ARMS) has recurrent genetic alterations, namely fusions between members of the paired homeobox family (PAX3 or PAX7) and FOXO1. FISH, PCR and NGS are available to test for these alterations. Absence of these fusions is suggestive of embryonal rhabdomyosarcoma. Recently, an immunostain was developed to evaluate for the epitope created by the fusion. 119 This antibody was quite successful in initial testing, with 91% sensitivity and 100% specificity, and it detected both fusions. However, in PAX7‐fused cases, only scattered cells were positive compared to the diffuse positivity of PAX3‐fused cases. Hence, small biopsies or FNA material may have only very rare positivity. Although it may not be needed for diagnosis, there is prognostic significance to the PAX gene involved in the fusion, with indications that PAX3‐FOXO1 portends a less favorable prognosis. 120 , 121
Desmoplastic small round cell tumor (DSRCT) is another EWSR1‐fused SRCT, in which nests of cytokeratin and desmin positive tumor cells are associated with prominent stromal desmoplasia. In DSRCT, the fusion partner is the Wilms tumor gene WT1. This results in increased expression of the WT1 protein, making WT1 IHC a potential surrogate, provided that the C‐terminus is targeted (as the N‐terminus is lost in the fusion). 122 , 123 Detection of EWSR1 and WT1 gene rearrangement is diagnostically helpful, as is PCR for the EWSR1‐WT1 chimeric transcript. There is a report of three tumors arising in the female genital tract that harbored EWSR1‐WT1 fusions but were inconsistent with DSRCT morphologically, of which one patient was progression free at 9 months following only resection. 124 Clearly these tumors need to be further evaluated, but this emphasizes the importance of morphology in addition to molecular testing.
Extraskeletal mesenchymal chondrosarcoma is a rare malignancy that displays a biphasic pattern, consisting of both primitive small round blue cells and well‐differentiated cartilage. This admixture of patterns is key to making the diagnosis but can be absent in a biopsy or FNA as the cartilaginous component may be scant and not readily aspirated. They typically harbor a HEY1‐NCOA2 fusion 125 or rarely an IRF2BP2‐CDX1 fusion, 126 both detectable by PCR. FISH has also been performed for HEY1‐NCOA2 with high sensitivity. 127
TABLE 2.
Chromosomal translocations and associated fusion transcripts identified in Ewing sarcoma. 109
| Chromosomal translocations | Fusion (genes involved) | Frequency (%) |
|---|---|---|
| t(11;22)(q24;q12) | EWSR1‐FLI1 | 85 |
| t(21;22)(q22;q12) | EWSR1‐ERG | 10 |
| t(2;22)(q33;q12) | EWSR1‐FEV | <1 |
| t(7;22)(p22;q12) | EWSR1‐ETV1 | <1 |
| t(17;22)(q21;q12) | EWSR1‐ETV4 | <1 |
| t(16;21)(p11;q22) | FUS‐ERG | <1 |
| t(2;16)(q35;p11) | FUS‐FEV | <1 |
3.4. Epithelioid tumors
Epithelioid tumors are the easiest to confuse with carcinomas, particularly without architecture to evaluate. Sheets of epithelioid cells without the formation of glands or nests is common in epithelioid sarcomas as opposed to carcinomas, although poorly differentiated carcinomas may also form large sheets of tumor cells. Epithelioid sarcoma (ES), malignant rhabdoid tumor (MRT), epithelioid hemangioendothelioma (EHE), clear cell sarcoma (CCS), alveolar soft part sarcoma (ASPS), and epithelioid MPNST all have recurrent genetic abnormalities that can assist in distinguishing them from carcinomas and from each other.
Epithelioid sarcoma (ES) and malignant rhabdoid tumor (MRT) both have alterations in the switch/sucrose non‐fermenting (SWI/SNF) pathway. In particular, both lose expression of the SMARCB1 protein (INI‐1), which is detectable by IHC. 128 , 129 This serves as a useful proxy for genetic testing, which demonstrates deletion of SMARCB1 in both entities. 130 , 131 ES and MRT can often be distinguished by their site of origin and other immunohistochemical features, such as the expression of ERG or SALL4. 132 Care must be taken when using INI‐1 staining alone, as many other tumors may show loss of this marker in a small subset of cases (e.g., epithelioid MPNST, renal medullary carcinoma, undifferentiated endometrial carcinoma, poorly differentiated chordoma), particularly those showing rhabdoid morphology. 133
Epithelioid hemangioendothelioma (EHE) is a malignant vascular neoplasm that was initially found to have WWTR1‐CAMTA1 fusions, the presence of which is testable using CAMTA1 IHC due to upregulation of CAMTA1 expression. 134 , 135 FISH for the WWTR1‐CAMTA1 fusion may be more specific, as some cases of epithelioid angiosarcoma, epithelioid sarcoma, and epithelioid hemangioma have been reported to be positive for CAMTA1 IHC, albeit with lower frequency than EHE. 136 , 137 A second, smaller group (<5%) of EHE with distinct morphologic features (e.g., more solid growth forming vascular spaces) were reported to harbor YAP1‐TFE3 fusions. 138 TFE3 expression or loss of the C‐terminus of YAP1 are helpful immunohistochemical markers for identifying this second fusion, 139 which can also be evaluated by FISH or PCR. Which of these two fusions is present may have prognostic significance, as a recent study found that patients with CAMTA1‐fused EHE fare worse than those with YAP1‐fused EHE. 140
Clear cell sarcoma (CCS) was previously termed “melanoma of soft parts” due to its unusual melanin production and expression of markers typical of melanocytic differentiation. 141 They harbor either EWSR1‐ATF1 (70%–90% of cases) or EWSR1‐CREB1 fusions, which are detectable by FISH or PCR, 142 although a thorough clinical history and imaging to rule out metastasis from a primary melanoma may be more useful in limited samples. Both gene fusions are also found in angiomatoid fibrous histiocytoma, clear cell sarcoma‐like tumor of the gastrointestinal tract, hyalinizing clear cell carcinoma of the salivary gland, and primary pulmonary myxoid sarcoma; however, these often have distinct clinical features. 143 While most CCS cases fail to demonstrate BRAF or NRAS mutations like melanoma, mutations in these genes have nevertheless been identified in a small subset of CCS. 144
Alveolar soft part sarcoma (ASPS) harbors a specific ASPSCR1‐TFE3 fusion. Similar to YAP1‐TFE3 fusion EHE, these tumors overexpress TFE3 by IHC. 145 PCR for the ASPSCR1‐TFE3 fusion can supplement immunostaining for diagnosis, although the immunomorphologic features of ASPS are typically sufficient. 146 The ASPSCR1‐TFE3 fusion may also be found in a unique subset of translocation‐associated renal cell carcinomas. 147
Epithelioid MPNST, in contrast to the typical spindle cell type, is driven in part by deletion or mutation of the SMARCB1 gene, which is adjacent to the NF2 gene on chromosome 22. 148 As a result, epithelioid MPNST frequently has loss of the INI‐1 protein by IHC, 149 while H3K27me3 expression is retained, as there is no disruption of the PRC2 complex. 148 No other recurrent molecular alterations have been found in epithelioid MPNST, limiting the utility of FISH and NGS testing.
3.5. Lipomatous tumors
Lipomatous tumors, despite being the most common soft tissue neoplasms, represent another difficult group to evaluate on a small biopsy. 150 In some cases, the challenge is to determine whether the tumor is benign or malignant, as atypical cells can be scant even in a large resection. In others, the difficulty lies in classifying the tumor as lipomatous, as adipocytes and lipoblasts may be rare or even absent. In this category, the diagnosis of spindle cell/pleomorphic lipoma, atypical spindle cell/pleomorphic lipomatous tumor, atypical lipomatous tumor/well‐differentiated liposarcomas (ALT/WDLPS), and dedifferentiated liposarcoma (DDLPS) can be aided by molecular testing. Myxoid liposarcoma was discussed above under myxoid neoplasms.
Spindle cell lipoma and pleomorphic lipoma are tumors of the same spectrum, along with atypical spindle cell/pleomorphic lipomatous tumors. These entities share not only histologic features, but also the loss of 13q14, a region which contains the RB1 gene that encodes the retinoblastoma (Rb) protein. Loss or a marked decrease in this protein by IHC is seen in the vast majority of these tumors as a result, with the caveat that inflammatory cells and endothelial cells will retain expression. 151 Atypical spindle cell/pleomorphic lipomatous tumor, a new entity in the 2020 WHO classification, also shows genetic losses in the same area with resultant Rb protein loss. 152
Atypical lipomatous tumor/well‐differentiated liposarcoma (ALT/WDLPS) have been combined in the new edition of the WHO. This combined entity, as well as most cases of DDLPS, is driven in part by the formation of supranumerary ring and giant marker chromosomes containing the 12q13‐15 locus. 153 At this locus are the MDM2, CDK4, and HMGA2 genes, which as a result are amplified. IHC with nuclear staining for MDM2 and/or CDK4 is often used to distinguish these tumors from mimics, 154 although with notable pitfalls – CDK4 is often cytoplasmic and may be positive in background benign cells, making evaluation of overexpression difficult, while MDM2 can be positive in activated histiocytes, as seen in fat necrosis. 155 A recent study found that even in cases with negative MDM2 and CDK4 IHC, there may be underlying amplification. 156 The limited tissue available on a core needle biopsy or FNA often renders IHC unreliable in making a diagnosis of ALT/WDLPS when no staining is seen. Rarely, MDM2 immunoreactivity may be noted in hibernomas. 157 MDM2 FISH is a much more reliable test in these situations, as it can directly evaluate for amplification. 155 , 158 , 159 FISH for MDM2 has been found to be 100% sensitive and specific on core needle biopsies of lipomatous tumors. 160 However, when performed on Pap‐stained slides, the background staining may be too high to allow for interpretation. 161 Additional modalities including brightfield chromogenic in situ hybridization (CISH), 162 RT‐PCR for MDM2 and CDK4 RNA, 163 and multiplex ligation‐dependent probe amplification (MLPA) 164 have comparable efficacy but are much less commonly used. This utility is restricted to adipocytic tumors only, as both IHC and FISH for MDM2 can also be seen in a variety of non‐fatty tumors (e.g., MPNST, myxofibrosarcoma, sclerosing rhabdomyosarcoma, gliomas, low‐grade osteosarcoma, intimal sarcoma, ependymoma, and some carcinomas). 165 , 166
3.6. Pleomorphic tumors
Many sarcomas, and rarely even benign tumors, may have a pleomorphic variant (e.g., rhabdomyosarcoma, leiomyosarcoma, liposarcoma). Undifferentiated pleomorphic sarcoma (UPS) and myxofibrosarcoma (MFS) are pleomorphic tumors without a corresponding non‐pleomorphic variant. UPS, formerly called malignant fibrous histiocytoma (MFH), demonstrates extensive genetic complexity. However, only pleomorphic liposarcomas have distinctive molecular alterations that can be leveraged in their diagnosis, although such testing is rarely necessary even on a limited sample.
Pleomorphic liposarcoma is a high‐grade sarcoma containing pleomorphic lipoblasts. Similar to pleomorphic lipomas and atypical pleomorphic lipomatous tumors, pleomorphic liposarcomas may have loss of the RB1 gene in superficial tumors, which is best assessed via Rb IHC. 167 They do not harbor MDM2 amplification, but TP53 is often disrupted, resulting in aberrant expression by IHC. 168
3.7. Giant cell‐rich tumors
Numerous tumors may contain giant cells, both reactive and neoplastic. However, the only tumors with a characteristic giant cell component that can be molecularly distinguished are tenosynovial giant cell tumor (TSGCT) and giant cell tumor of soft tissue‐like tumors.
Tenosynovial giant cell tumor (TSGCT) may present as either one tumor (localized type) or supplant the entire synovium of a joint (diffuse type). Both may have fusions of the colony stimulating factor 1 (CSF1) gene, most commonly with COL6A3, 169 although other fusion partners have been identified including S100A1, 170 VCAM1, FN1, CDH1, 171 and CD96. 172 Rare cases with non‐CSF1 fusions have been reported within the last few years, including NIPBL‐ERG, FN1‐ROS1, YAP1‐MAML2, 173 and a t(1;1)(p13;p34) translocation. 174 A significant number of cases do not have an identifiable fusion, and finding a fusion is not necessary for making a diagnosis. However, it may provide a potential target for therapy with CSFR1 inhibitors such as pexidartinib, which was recently approved by the Food and Drug Administration for symptomatic TSGCT. 175
Giant cell tumor of soft tissue (GCT‐ST) is a rare tumor that is morphologically identical to giant cell tumor of bone (GCT‐B). Unlike GCT‐B, however, GCT‐ST does not have H3F3A p.G34 mutations. 176 , 177 In fact, no molecular signature has been identified in GCT‐ST to date. Recently, a set of keratin‐positive giant cell‐containing tumors with similar morphologic appearance, but different immunophenotype, have been identified harboring HMGA2‐NCOR2 fusions, which are only detectable by PCR. 178
4. CONCLUSION
Soft tissue pathology is a challenging field, even more so when the evaluable material is limited. Molecular testing, whether by FISH/CISH, PCR, NGS, or now even an immunohistochemical proxy, is an invaluable adjunct to clinical presentation and morphology not only for rendering a definitive diagnosis, but on occasion for prognostication of these soft tissue tumors. 179 As discussed in this review article, there are a plethora of molecular tests now available for many soft tissue tumors to help distinguish them apart and from their mimics. However, it may not be practical to bring on every test for pathology laboratories, and the data are often limited or even non‐existent, on their utility in cytologic or core needle biopsy samples. Moreover, there are some cases as with MDM2 FISH, where the cytologic preparation of procured cellular material can hinder interpretation, and thus care must be taken with any new assay. Nevertheless, given the trend in soft tissue pathology toward increasing use of molecular results for tumor classification and diagnosis (e.g., the introduction of categories such as CIC‐rearranged sarcoma, which explicitly requires molecular testing to diagnose), concomitant with the demand for less invasive procedures, the use of these techniques is rapidly becoming unavoidable, even in the practice of cytopathology. 180 Not only must the correct molecular test be ordered, but the material must be triaged appropriately to ensure the best and most accurate diagnosis is achieved for patient care.
CONFLICT OF INTEREST
The authors declare no conflicts of interest.
Rottmann D, Abdulfatah E, Pantanowitz L. Molecular testing of soft tissue tumors. Diagnostic Cytopathology. 2023;51(1):12‐25. doi: 10.1002/dc.25013
DATA AVAILABILITY STATEMENT
Research data are not shared.
REFERENCES
- 1. The WHO . Classification of Tumours Editorial Board, ed. Soft Tissue and Bone Tumours. 5th ed. International Agency for Research on Cancer (IARC); 2020. [Google Scholar]
- 2. Szurian K, Kashofer K, Liegl‐Atzwanger B. Role of next‐generation sequencing as a diagnostic tool for the evaluation of bone and soft‐tissue tumors. Pathobiology. 2017;84(6):323‐338. [DOI] [PubMed] [Google Scholar]
- 3. Racanelli D, Brenca M, Baldazzi D, et al. Next‐generation sequencing approaches for the identification of pathognomonic fusion transcripts in sarcomas: the experience of the Italian ACC sarcoma working group. Front Oncol. 2020;10:489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Corless CL, Barnett CM, Heinrich MC. Gastrointestinal stromal tumours: origin and molecular oncology. Nat Rev Cancer. 2011;11(12):865‐878. [DOI] [PubMed] [Google Scholar]
- 5. Kotiligam D, Lazar AJ, Pollock RE, Lev D. Desmoid tumor: a disease opportune for molecular insights. Histol Histopathol. 2008;23(1):117‐126. [DOI] [PubMed] [Google Scholar]
- 6. Delaney D, Diss TC, Presneau N, et al. GNAS1 mutations occur more commonly than previously thought in intramuscular myxoma. Mod Pathol. 2009;22(5):718‐724. [DOI] [PubMed] [Google Scholar]
- 7. Jackson EM, Sievert AJ, Gai X, et al. Genomic analysis using high‐density single nucleotide polymorphism‐based oligonucleotide arrays and multiplex ligation‐dependent probe amplification provides a comprehensive analysis of INI1/SMARCB1 in malignant rhabdoid tumors. Clin Cancer Res. 2009;15(6):1923‐1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fletcher CD, Akerman M, Dal Cin P, et al. Correlation between clinicopathological features and karyotype in lipomatous tumors. A report of 178 cases from the chromosomes and morphology (CHAMP) collaborative study group. Am J Pathol. 1996;148(2):623‐630. [PMC free article] [PubMed] [Google Scholar]
- 9. Thway K. Well‐differentiated liposarcoma and dedifferentiated liposarcoma: An updated review. Semin Diagn Pathol. 2019;36(2):112‐121. [DOI] [PubMed] [Google Scholar]
- 10. Mertens F, Antonescu CR, Mitelman F. Gene fusions in soft tissue tumors: recurrent and overlapping pathogenetic themes. Genes Chromosomes Cancer. 2016;55(4):291‐310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Bennicelli JL, Barr FG. Chromosomal translocations and sarcomas. Curr Opin Oncol. 2002;14(4):412‐419. [DOI] [PubMed] [Google Scholar]
- 12. Greco A, Roccato E, Miranda C, Cleris L, Formelli F, Pierotti MA. Growth‐inhibitory effect of STI571 on cells transformed by the COL1A1/PDGFB rearrangement. Int J Cancer. 2001;92(3):354‐360. [DOI] [PubMed] [Google Scholar]
- 13. Tejpar S, Nollet F, Li C, et al. Predominance of beta‐catenin mutations and beta‐catenin dysregulation in sporadic aggressive fibromatosis (desmoid tumor). Oncogene. 1999;18(47):6615‐6620. [DOI] [PubMed] [Google Scholar]
- 14. Trautmann M, Rehkämper J, Gevensleben H, et al. Novel pathogenic alterations in pediatric and adult desmoid‐type fibromatosis ‐ a systematic analysis of 204 cases. Sci Rep. 2020;10(1):3368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bhattacharya B, Dilworth HP, Iacobuzio‐Donahue C, et al. Nuclear beta‐catenin expression distinguishes deep fibromatosis from other benign and malignant fibroblastic and myofibroblastic lesions. Am J Surg Pathol. 2005;29(5):653‐659. [DOI] [PubMed] [Google Scholar]
- 16. An J, Woo HY, Lee Y, Kim HS, Jeong J, Kim SK. Clinicopathological features of 70 desmoid‐type fibromatoses confirmed by β‐catenin immunohistochemical staining and CTNNB1 mutation analysis. PLoS One. 2021;16(4):e0250619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Saito T, Oda Y, Tanaka K, et al. Beta‐catenin nuclear expression correlates with cyclin D1 overexpression in sporadic desmoid tumours. J Pathol. 2001;195(2):222‐228. [DOI] [PubMed] [Google Scholar]
- 18. Andino L, Cagle PT, Murer B, et al. Pleuropulmonary desmoid tumors: immunohistochemical comparison with solitary fibrous tumors and assessment of beta‐catenin and cyclin D1 expression. Arch Pathol Lab Med. 2006;130(10):1503‐1509. [DOI] [PubMed] [Google Scholar]
- 19. Jilong Y, Jian W, Xiaoyan Z, Xiaoqiu L, Xiongzeng Z. Analysis of APC/beta‐catenin genes mutations and Wnt signalling pathway in desmoid‐type fibromatosis. Pathology. 2007;39(3):319‐325. [DOI] [PubMed] [Google Scholar]
- 20. Colombo C, Foo WC, Whiting D, et al. FAP‐related desmoid tumors: a series of 44 patients evaluated in a cancer referral center. Histol Histopathol. 2012;27(5):641‐649. [DOI] [PubMed] [Google Scholar]
- 21. Timbergen MJM, Colombo C, Renckens M, et al. The prognostic role of β‐catenin mutations in desmoid‐type fibromatosis undergoing resection only: a meta‐analysis of individual patient data. Ann Surg. 2021;273(6):1094‐1101. [DOI] [PubMed] [Google Scholar]
- 22. Guo L, Wang X, Xu B, Lang R, Hu B. Prognostic significance of CTNNB1 mutation in recurrence of sporadic desmoid tumors. Future Oncol. 2021;17(4):435‐442. [DOI] [PubMed] [Google Scholar]
- 23. Koike H, Nishida Y, Kohno K, et al. Is immunohistochemical staining for β‐catenin the definitive pathological diagnostic tool for desmoid‐type fibromatosis? A multi‐institutional study. Hum Pathol. 2019;84:155‐163. [DOI] [PubMed] [Google Scholar]
- 24. Yamada Y, Hirata M, Sakamoto A, et al. A comparison of the usefulness of nuclear beta‐catenin in the diagnosis of desmoid‐type fibromatosis among commonly used anti‐beta‐catenin antibodies. Pathol Int. 2021;71(6):392‐399. [DOI] [PubMed] [Google Scholar]
- 25. Erickson‐Johnson MR, Chou MM, Evers BR, et al. Nodular fasciitis: a novel model of transient neoplasia induced by MYH9‐USP6 gene fusion. Lab Invest. 2011;91(10):1427‐1433. [DOI] [PubMed] [Google Scholar]
- 26. Shin C, Low I, Ng D, Oei P, Miles C, Symmans P. USP6 gene rearrangement in nodular fasciitis and histological mimics. Histopathology. 2016;69(5):784‐791. [DOI] [PubMed] [Google Scholar]
- 27. Sápi Z, Lippai Z, Papp G, et al. Nodular fasciitis: a comprehensive, time‐correlated investigation of 17 cases. Mod Pathol. 2021;34(12):2192‐2199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hiemcke‐Jiwa LS, van Gorp JM, Fisher C, Creytens D, van Diest PJ, Flucke U. USP6‐associated neoplasms: a rapidly expanding family of lesions. Int J Surg Pathol. 2020;28(8):816‐825. [DOI] [PubMed] [Google Scholar]
- 29. Demicco EG, Wagner MJ, Maki RG, et al. Risk assessment in solitary fibrous tumors: validation and refinement of a risk stratification model. Mod Pathol. 2017;30(10):1433‐1442. [DOI] [PubMed] [Google Scholar]
- 30. Guseva NV, Tanas MR, Stence AA, et al. The NAB2‐STAT6 gene fusion in solitary fibrous tumor can be reliably detected by anchored multiplexed PCR for targeted next‐generation sequencing. Cancer Genet. 2016;209(7–8):303‐312. [DOI] [PubMed] [Google Scholar]
- 31. Kao YC, Lin PC, Yen SL, et al. Clinicopathological and genetic heterogeneity of the head and neck solitary fibrous tumours: a comparative histological, immunohistochemical and molecular study of 36 cases. Histopathology. 2016;68(4):492‐501. [DOI] [PubMed] [Google Scholar]
- 32. Wakely PE, Rekhi B. Cytopathology of solitary fibrous tumor: a series of 34 cases. J Am Soc Cytopathol. 2021;10(4):382‐390. [DOI] [PubMed] [Google Scholar]
- 33. Olson NJ, Linos K. Dedifferentiated solitary fibrous tumor: a concise review. Arch Pathol Lab Med. 2018;142(6):761‐766. [DOI] [PubMed] [Google Scholar]
- 34. Dagrada GP, Spagnuolo RD, Mauro V, et al. Solitary fibrous tumors: loss of chimeric protein expression and genomic instability mark dedifferentiation. Mod Pathol. 2015;28(8):1074‐1083. [DOI] [PubMed] [Google Scholar]
- 35. Akaike K, Kurisaki‐Arakawa A, Hara K, et al. Distinct clinicopathological features of NAB2‐STAT6 fusion gene variants in solitary fibrous tumor with emphasis on the acquisition of highly malignant potential. Hum Pathol. 2015;46(3):347‐356. [DOI] [PubMed] [Google Scholar]
- 36. Tai HC, Chuang IC, Chen TC, et al. NAB2‐STAT6 fusion types account for clinicopathological variations in solitary fibrous tumors. Mod Pathol. 2015;28(10):1324‐1335. [DOI] [PubMed] [Google Scholar]
- 37. Huang SC, Li CF, Kao YC, et al. The clinicopathological significance of NAB2‐STAT6 gene fusions in 52 cases of intrathoracic solitary fibrous tumors. Cancer Med. 2016;5(2):159‐168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Chuang IC, Liao KC, Huang HY, et al. NAB2‐STAT6 gene fusion and STAT6 immunoexpression in extrathoracic solitary fibrous tumors: the association between fusion variants and locations. Pathol Int. 2016;66(5):288‐296. [DOI] [PubMed] [Google Scholar]
- 39. Machado I, Morales GN, Cruz J, et al. Solitary fibrous tumor: a case series identifying pathological adverse factors‐implications for risk stratification and classification. Virchows Arch. 2020;476(4):597‐607. [DOI] [PubMed] [Google Scholar]
- 40. Salguero‐Aranda C, Martínez‐Reguera P, Marcilla D, de Álava E, Díaz‐Martín J. Evaluation of NAB2‐STAT6 fusion variants and other molecular alterations as prognostic biomarkers in a case series of 83 solitary fibrous tumors. Cancers (Basel). 2021;13(20):5237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Segura S, Salgado R, Toll A, et al. Identification of t(17;22)(q22;q13) (COL1A1/PDGFB) in dermatofibrosarcoma protuberans by fluorescence in situ hybridization in paraffin‐embedded tissue microarrays. Hum Pathol. 2011;42(2):176‐184. [DOI] [PubMed] [Google Scholar]
- 42. Zhang Z, Chen H, Chen M, He X, Wang Y, Zhang H. Application of COL1A1‐PDGFB fusion gene detection by fluorescence in situ hybridization in biopsy tissue of dermatofibrosarcoma protuberans. J Dermatol. 2017;44(7):798‐802. [DOI] [PubMed] [Google Scholar]
- 43. Cloutier JM, Allard G, Bean GR, Hornick JL, Charville GW. PDGFB RNA in situ hybridization for the diagnosis of dermatofibrosarcoma protuberans. Mod Pathol. 2021;34(8):1521‐1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Dadone‐Montaudié B, Alberti L, Duc A, et al. Alternative PDGFD rearrangements in dermatofibrosarcomas protuberans without PDGFB fusions. Mod Pathol. 2018;31(11):1683‐1693. [DOI] [PubMed] [Google Scholar]
- 45. Liang CA, Jambusaria‐Pahlajani A, Karia PS, Elenitsas R, Zhang PD, Schmults CD. A systematic review of outcome data for dermatofibrosarcoma protuberans with and without fibrosarcomatous change. J Am Acad Dermatol. 2014;71(4):781‐786. [DOI] [PubMed] [Google Scholar]
- 46. Hisaoka M, Okamoto S, Morimitsu Y, Tsuji S, Hashimoto H. Dermatofibrosarcoma protuberans with fibrosarcomatous areas. Molecular abnormalities of the p53 pathway in fibrosarcomatous transformation of dermatofibrosarcoma protuberans. Virchows Arch. 1998;433(4):323‐329. [DOI] [PubMed] [Google Scholar]
- 47. Takahira T, Oda Y, Tamiya S, et al. Microsatellite instability and p53 mutation associated with tumor progression in dermatofibrosarcoma protuberans. Hum Pathol. 2004;35(2):240‐245. [DOI] [PubMed] [Google Scholar]
- 48. Griffin CA, Hawkins AL, Dvorak C, Henkle C, Ellingham T, Perlman EJ. Recurrent involvement of 2p23 in inflammatory myofibroblastic tumors. Cancer Res. 1999;59(12):2776‐2780. [PubMed] [Google Scholar]
- 49. Coffin CM, Hornick JL, Fletcher CD. Inflammatory myofibroblastic tumor: comparison of clinicopathologic, histologic, and immunohistochemical features including ALK expression in atypical and aggressive cases. Am J Surg Pathol. 2007;31(4):509‐520. [DOI] [PubMed] [Google Scholar]
- 50. Yamamoto H. Inflammatory myofibroblastic tumor. The WHO Classification of Tumours Editorial Board. Soft Tissue and Bone Tumours. 5th ed. International Agency for Research on Cancer (IARC); 2020:109‐111. [Google Scholar]
- 51. Solomon JP, Linkov I, Rosado A, et al. NTRK fusion detection across multiple assays and 33,997 cases: diagnostic implications and pitfalls. Mod Pathol. 2020;33(1):38‐46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Brčić I, Godschachner TM, Bergovec M, et al. Broadening the spectrum of NTRK rearranged mesenchymal tumors and usefulness of pan‐TRK immunohistochemistry for identification of NTRK fusions. Mod Pathol. 2021;34(2):396‐407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Federman N, McDermott R. Larotrectinib, a highly selective tropomyosin receptor kinase (TRK) inhibitor for the treatment of TRK fusion cancer. Expert Rev Clin Pharmacol. 2019;12(10):931‐939. [DOI] [PubMed] [Google Scholar]
- 54. Bian LG, Sun QF, Tirakotai W, et al. Loss of heterozygosity on chromosome 22 in sporadic schwannoma and its relation to the proliferation of tumor cells. Chin Med J (Engl). 2005;118(18):1517‐1524. [PubMed] [Google Scholar]
- 55. Hilton DA, Hanemann CO. Schwannomas and their pathogenesis. Brain Pathol. 2014;24(3):205‐220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Storlazzi CT, Von Steyern FV, Domanski HA, Mandahl N, Mertens F. Biallelic somatic inactivation of the NF1 gene through chromosomal translocations in a sporadic neurofibroma. Int J Cancer. 2005;117(6):1055‐1057. [DOI] [PubMed] [Google Scholar]
- 57. De Raedt T, Maertens O, Chmara M, et al. Somatic loss of wild type NF1 allele in neurofibromas: comparison of NF1 microdeletion and non‐microdeletion patients. Genes Chromosomes Cancer. 2006;45(10):893‐904. [DOI] [PubMed] [Google Scholar]
- 58. Carter JM, Wu Y, Blessing MM, et al. Recurrent genomic alterations in soft tissue perineuriomas. Am J Surg Pathol. 2018;42(12):1708‐1714. [DOI] [PubMed] [Google Scholar]
- 59. Klein CJ, Wu Y, Jentoft ME, et al. Genomic analysis reveals frequent TRAF7 mutations in intraneural perineuriomas. Ann Neurol. 2017;81(2):316‐321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Nihous H, Baud J, Azmani R, et al. Clinicopathologic and molecular study of hybrid nerve sheath tumors reveals their common association with fusions involving VGLL3. Am J Surg Pathol. 2022;46(5):591‐602. [DOI] [PubMed] [Google Scholar]
- 61. Lee W, Teckie S, Wiesner T, et al. PRC2 is recurrently inactivated through EED or SUZ12 loss in malignant peripheral nerve sheath tumors. Nat Genet. 2014;46(11):1227‐1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Zhang X, Murray B, Mo G, Shern JF. The role of polycomb repressive complex in malignant peripheral nerve sheath tumor. Genes (Basel). 2020;11(3):287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Otsuka H, Kohashi K, Yoshimoto M, et al. Immunohistochemical evaluation of H3K27 trimethylation in malignant peripheral nerve sheath tumors. Pathol Res Pract. 2018;214(3):417‐425. [DOI] [PubMed] [Google Scholar]
- 64. Marchione DM, Lisby A, Viaene AN, et al. Histone H3K27 dimethyl loss is highly specific for malignant peripheral nerve sheath tumor and distinguishes true PRC2 loss from isolated H3K27 trimethyl loss. Mod Pathol. 2019;32(10):1434‐1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Röhrich M, Koelsche C, Schrimpf D, et al. Methylation‐based classification of benign and malignant peripheral nerve sheath tumors. Acta Neuropathol. 2016;131(6):877‐887. [DOI] [PubMed] [Google Scholar]
- 66. Crew AJ, Clark J, Fisher C, et al. Fusion of SYT to two genes, SSX1 and SSX2, encoding proteins with homology to the Kruppel‐associated box in human synovial sarcoma. EMBO J. 1995;14(10):2333‐2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Skytting B, Nilsson G, Brodin B, et al. A novel fusion gene, SYT‐SSX4, in synovial sarcoma. J Natl Cancer Inst. 1999;91(11):974‐975. [DOI] [PubMed] [Google Scholar]
- 68. Surace C, Panagopoulos I, Pålsson E, Rocchi M, Mandahl N, Mertens F. A novel FISH assay for SS18‐SSX fusion type in synovial sarcoma. Lab Invest. 2004;84(9):1185‐1192. [DOI] [PubMed] [Google Scholar]
- 69. Srinivasan R, Gautam U, Gupta R, Rajwanshi A, Vasistha RK. Synovial sarcoma: diagnosis on fine‐needle aspiration by morphology and molecular analysis. Cancer. 2009;117(2):128‐136. [DOI] [PubMed] [Google Scholar]
- 70. Zhang Y, Wessman S, Wejde J, Tani E, Haglund F. Diagnosing synovial sarcoma by fine‐needle aspiration cytology and molecular techniques. Cytopathology. 2019;30(5):504‐509. [DOI] [PubMed] [Google Scholar]
- 71. Baranov E, McBride MJ, Bellizzi AM, et al. A novel SS18‐SSX fusion‐specific antibody for the diagnosis of synovial sarcoma. Am J Surg Pathol. 2020;44(7):922‐933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Zaborowski M, Vargas AC, Pulvers J, et al. When used together SS18‐SSX fusion‐specific and SSX C‐terminus immunohistochemistry are highly specific and sensitive for the diagnosis of synovial sarcoma and can replace FISH or molecular testing in most cases. Histopathology. 2020;77(4):588‐600. [DOI] [PubMed] [Google Scholar]
- 73. Tahara S, Kohara M, Honma K, Morii E. Detection of synovial sarcoma with an atypical fusion transcript by using SS18‐SSX and SSX antibodies. Pathol Int. 2020;70(9):689‐691. [DOI] [PubMed] [Google Scholar]
- 74. Tay TKY, Sukma NB, Lim TH, Kuick CH, Goh JY, Chang KTE. Correlating SS18‐SSX immunohistochemistry (IHC) with SS18 fluorescent in situ hybridization (FISH) in synovial sarcomas: a study of 36 cases. Virchows Arch. 2021;479(4):785‐793. [DOI] [PubMed] [Google Scholar]
- 75. Miettinen M, Wang ZF, Lasota J. DOG1 antibody in the differential diagnosis of gastrointestinal stromal tumors: a study of 1840 cases. Am J Surg Pathol. 2009;33(9):1401‐1408. [DOI] [PubMed] [Google Scholar]
- 76. Fatima N, Cohen C, Siddiqui MT. DOG1 utility in diagnosing gastrointestinal stromal tumors on fine‐needle aspiration. Cancer Cytopathol. 2011;119(3):202‐208. [DOI] [PubMed] [Google Scholar]
- 77. Brčić I, Argyropoulos A, Liegl‐Atzwanger B. Update on molecular genetics of gastrointestinal stromal tumors. Diagnostics (Basel). 2021;11(2):194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Miranda C, Nucifora M, Molinari F, et al. KRAS and BRAF mutations predict primary resistance to imatinib in gastrointestinal stromal tumors. Clin Cancer Res. 2012;18(6):1769‐1776. [DOI] [PubMed] [Google Scholar]
- 79. Miettinen M, Wang ZF, Sarlomo‐Rikala M, Osuch C, Rutkowski P, Lasota J. Succinate dehydrogenase‐deficient GISTs: a clinicopathologic, immunohistochemical, and molecular genetic study of 66 gastric GISTs with predilection to young age. Am J Surg Pathol. 2011;35(11):1712‐1721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Agaimy A, Otto C, Braun A, Geddert H, Schaefer IM, Haller F. Value of epithelioid morphology and PDGFRA immunostaining pattern for prediction of PDGFRA mutated genotype in gastrointestinal stromal tumors (GISTs). Int J Clin Exp Pathol. 2013;6(9):1839‐1846. [PMC free article] [PubMed] [Google Scholar]
- 81. Papke DJ, Forgó E, Charville GW, Hornick JL. PDGFRA immunohistochemistry predicts PDGFRA mutations in gastrointestinal stromal tumors. Am J Surg Pathol. 2022;46(1):3‐10. [DOI] [PubMed] [Google Scholar]
- 82. Rossi S, Sbaraglia M, Dell'Orto MC, et al. Concomitant KIT/BRAF and PDGFRA/BRAF mutations are rare events in gastrointestinal stromal tumors. Oncotarget. 2016;7(21):30109‐30118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Castillon M, Kammerer‐Jacquet SF, Cariou M, et al. Fluorescent in situ hybridization must be preferred to pan‐TRK immunohistochemistry to diagnose NTRK3‐rearranged gastrointestinal stromal tumors (GIST). Appl Immunohistochem Mol Morphol. 2021;29(8):626‐634. [DOI] [PubMed] [Google Scholar]
- 84. Baranov E, Black MA, Fletcher CDM, Charville GW, Hornick JL. Nuclear expression of DDIT3 distinguishes high‐grade myxoid liposarcoma from other round cell sarcomas. Mod Pathol. 2021;34(7):1367‐1372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Scapa JV, Cloutier JM, Raghavan SS, Peters‐Schulze G, Varma S, Charville GW. DDIT3 immunohistochemistry is a useful tool for the diagnosis of myxoid liposarcoma. Am J Surg Pathol. 2021;45(2):230‐239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Downs‐Kelly E, Goldblum JR, Patel RM, et al. The utility of fluorescence in situ hybridization (FISH) in the diagnosis of myxoid soft tissue neoplasms. Am J Surg Pathol. 2008;32(1):8‐13. [DOI] [PubMed] [Google Scholar]
- 87. Narendra S, Valente A, Tull J, Zhang S. DDIT3 gene break‐apart as a molecular marker for diagnosis of myxoid liposarcoma–assay validation and clinical experience. Diagn Mol Pathol. 2011;20(4):218‐224. [DOI] [PubMed] [Google Scholar]
- 88. Wakely PE, Jin M. Myxoid liposarcoma: fine‐needle aspiration cytopathology in the molecular era. A report of 24 cases. J Am Soc Cytopathol. 2016;5(3):162‐169. [DOI] [PubMed] [Google Scholar]
- 89. Mantilla JG, Ricciotti RW, Chen EY, Liu YJ, Hoch BL. Amplification of DNA damage‐inducible transcript 3 (DDIT3) is associated with myxoid liposarcoma‐like morphology and homologous lipoblastic differentiation in dedifferentiated liposarcoma. Mod Pathol. 2019;32(4):585‐592. [DOI] [PubMed] [Google Scholar]
- 90. Kuczkiewicz‐Siemion O, Wiśniewski P, Dansonka‐Mieszkowska A, et al. The utility of fluorescence in situ hybridization (FISH) in determining DNA damage‐inducible transcript 3 (DDIT3) amplification in dedifferentiated liposarcomas ‐ an important diagnostic pitfall. Pathol Res Pract. 2021;225:153555. [DOI] [PubMed] [Google Scholar]
- 91. Powers MP, Wang WL, Hernandez VS, et al. Detection of myxoid liposarcoma‐associated FUS‐DDIT3 rearrangement variants including a newly identified breakpoint using an optimized RT‐PCR assay. Mod Pathol. 2010;23(10):1307‐1315. [DOI] [PubMed] [Google Scholar]
- 92. Moller E, Hornick JL, Magnusson L, Veerla S, Domanski HA, Mertens F. FUS‐CREB3L2/L1‐positive sarcomas show a specific gene expression profile with upregulation of CD24 and FOXL1. Clin Cancer Res. 2011;17(9):2646‐2656. [DOI] [PubMed] [Google Scholar]
- 93. Patel RM, Downs‐Kelly E, Dandekar MN, et al. FUS (16p11) gene rearrangement as detected by fluorescence in‐situ hybridization in cutaneous low‐grade fibromyxoid sarcoma: a potential diagnostic tool. Am J Dermatopathol. 2011;33(2):140‐143. [DOI] [PubMed] [Google Scholar]
- 94. Antonescu CR, Argani P, Erlandson RA, Healey JH, Ladanyi M, Huvos AG. Skeletal and extraskeletal myxoid chondrosarcoma: a comparative clinicopathologic, ultrastructural, and molecular study. Cancer. 1998;83(8):1504‐1521. [DOI] [PubMed] [Google Scholar]
- 95. Haller F, Skálová A, Ihrler S, et al. Nuclear NR4A3 immunostaining is a specific and sensitive novel marker for Acinic cell carcinoma of the salivary glands. Am J Surg Pathol. 2019;43(9):1264‐1272. [DOI] [PubMed] [Google Scholar]
- 96. Skaugen JM, Seethala RR, Chiosea SI, Landau MS. Evaluation of NR4A3 immunohistochemistry (IHC) and fluorescence in situ hybridization and comparison with DOG1 IHC for FNA diagnosis of acinic cell carcinoma. Cancer Cytopathol. 2021;129(2):104‐113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Vargas AC, Maclean FM, Bonar F, Mahar A, Gill AJ. NR4A3 immunohistochemistry lacks sensitivity for the diagnosis of Extraskeletal Myxoid chondrosarcoma. Am J Surg Pathol. 2019;43(12):1726‐1728. [DOI] [PubMed] [Google Scholar]
- 98. Wang WL, Mayordomo E, Czerniak BA, et al. Fluorescence in situ hybridization is a useful ancillary diagnostic tool for extraskeletal myxoid chondrosarcoma. Mod Pathol. 2008;21(11):1303‐1310. [DOI] [PubMed] [Google Scholar]
- 99. Wakely PE. Extraskeletal myxoid chondrosarcoma: combining cytopathology with molecular testing to achieve diagnostic accuracy. J Am Soc Cytopathol. 2021;10(3):293‐299. [DOI] [PubMed] [Google Scholar]
- 100. Noguchi H, Mitsuhashi T, Seki K, et al. Fluorescence in situ hybridization analysis of extraskeletal myxoid chondrosarcomas using EWSR1 and NR4A3 probes. Hum Pathol. 2010;41(3):336‐342. [DOI] [PubMed] [Google Scholar]
- 101. Paioli A, Stacchiotti S, Campanacci D, et al. Extraskeletal myxoid chondrosarcoma with molecularly confirmed diagnosis: a multicenter retrospective study within the Italian sarcoma group. Ann Surg Oncol. 2021;28(2):1142‐1150. [DOI] [PubMed] [Google Scholar]
- 102. Folpe AL, Hill CE, Parham DM, O'Shea PA, Weiss SW. Immunohistochemical detection of FLI‐1 protein expression: a study of 132 round cell tumors with emphasis on CD99‐positive mimics of Ewing's sarcoma/primitive neuroectodermal tumor. Am J Surg Pathol. 2000;24(12):1657‐1662. [DOI] [PubMed] [Google Scholar]
- 103. Wang WL, Patel NR, Caragea M, et al. Expression of ERG, an Ets family transcription factor, identifies ERG‐rearranged Ewing sarcoma. Mod Pathol. 2012;25(10):1378‐1383. [DOI] [PubMed] [Google Scholar]
- 104. Tomlins SA, Palanisamy N, Brenner JC, et al. Usefulness of a monoclonal ERG/FLI1 antibody for immunohistochemical discrimination of Ewing family tumors. Am J Clin Pathol. 2013;139(6):771‐779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Folpe AL, Chand EM, Goldblum JR, Weiss SW. Expression of Fli‐1, a nuclear transcription factor, distinguishes vascular neoplasms from potential mimics. Am J Surg Pathol. 2001;25(8):1061‐1066. [DOI] [PubMed] [Google Scholar]
- 106. Miettinen M, Wang ZF, Paetau A, et al. ERG transcription factor as an immunohistochemical marker for vascular endothelial tumors and prostatic carcinoma. Am J Surg Pathol. 2011;35(3):432‐441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Stockman DL, Hornick JL, Deavers MT, Lev DC, Lazar AJ, Wang WL. ERG and FLI1 protein expression in epithelioid sarcoma. Mod Pathol. 2014;27(4):496‐501. [DOI] [PubMed] [Google Scholar]
- 108. Chen S, Deniz K, Sung YS, Zhang L, Dry S, Antonescu CR. Ewing sarcoma with ERG gene rearrangements: a molecular study focusing on the prevalence of FUS‐ERG and common pitfalls in detecting EWSR1‐ERG fusions by FISH. Genes Chromosomes Cancer. 2016;55(4):340‐349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Rodríguez‐Martín C, Alonso J. Molecular approaches to diagnosis in Ewing sarcoma: RT‐PCR. Methods Mol Biol. 2021;2226:85‐103. [DOI] [PubMed] [Google Scholar]
- 110. Ueno‐Yokohata H, Okita H, Nakasato K, et al. Establishment of multiplex RT‐PCR to detect fusion genes for the diagnosis of Ewing sarcoma. Diagn Pathol. 2021;16(1):102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. Le Loarer F, Szuhai K, Tirode F. Round cell sarcoma with EWSR1–non‐ETS fusions. Soft Tissue and Bone Tumours. 5th ed. International Agency for Research on Cancer (IARC); 2020:326‐329. [Google Scholar]
- 112. Koelsche C, Kriegsmann M, Kommoss FKF, et al. DNA methylation profiling distinguishes Ewing‐like sarcoma with EWSR1‐NFATc2 fusion from Ewing sarcoma. J Cancer Res Clin Oncol. 2019;145(5):1273‐1281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Antonescu CR, Owosho AA, Zhang L, et al. Sarcomas with CIC‐rearrangements are a distinct pathologic entity with aggressive outcome: a clinicopathologic and molecular study of 115 cases. Am J Surg Pathol. 2017;41(7):941‐949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Siegele B, Roberts J, Black JO, Rudzinski E, Vargas SO, Galambos C. DUX4 immunohistochemistry is a highly sensitive and specific marker for CIC‐DUX4 fusion‐positive round cell tumor. Am J Surg Pathol. 2017;41(3):423‐429. [DOI] [PubMed] [Google Scholar]
- 115. Le Guellec S, Velasco V, Pérot G, Watson S, Tirode F, Coindre JM. ETV4 is a useful marker for the diagnosis of CIC‐rearranged undifferentiated round‐cell sarcomas: a study of 127 cases including mimicking lesions. Mod Pathol. 2016;29(12):1523‐1531. [DOI] [PubMed] [Google Scholar]
- 116. Cocchi S, Gamberi G, Magagnoli G, et al. CIC rearranged sarcomas: a single institution experience of the potential pitfalls in interpreting CIC FISH results. Pathol Res Pract. 2022;231:153773. [DOI] [PubMed] [Google Scholar]
- 117. Kao YC, Owosho AA, Sung YS, et al. BCOR‐CCNB3 fusion positive sarcomas: a clinicopathologic and molecular analysis of 36 cases with comparison to morphologic spectrum and clinical behavior of other round cell sarcomas. Am J Surg Pathol. 2018;42(5):604‐615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Li L, Zhang M, Chen S, et al. Detection of BCOR gene rearrangement in Ewing‐like sarcoma: an important diagnostic tool. Diagn Pathol. 2021;16(1):50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Azorsa DO, Bode PK, Wachtel M, et al. Immunohistochemical detection of PAX‐FOXO1 fusion proteins in alveolar rhabdomyosarcoma using breakpoint specific monoclonal antibodies. Mod Pathol. 2021;34(4):748‐757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Missiaglia E, Williamson D, Chisholm J, et al. PAX3/FOXO1 fusion gene status is the key prognostic molecular marker in rhabdomyosarcoma and significantly improves current risk stratification. J Clin Oncol. 2012;30(14):1670‐1677. [DOI] [PubMed] [Google Scholar]
- 121. Kubo T, Shimose S, Fujimori J, Furuta T, Ochi M. Prognostic value of PAX3/7‐FOXO1 fusion status in alveolar rhabdomyosarcoma: systematic review and meta‐analysis. Crit Rev Oncol Hematol. 2015;96(1):46‐53. [DOI] [PubMed] [Google Scholar]
- 122. Hill DA, Pfeifer JD, Marley EF, et al. WT1 staining reliably differentiates desmoplastic small round cell tumor from Ewing sarcoma/primitive neuroectodermal tumor. An immunohistochemical and molecular diagnostic study. Am J Clin Pathol. 2000;114(3):345‐353. [DOI] [PubMed] [Google Scholar]
- 123. Barnoud R, Sabourin JC, Pasquier D, et al. Immunohistochemical expression of WT1 by desmoplastic small round cell tumor: a comparative study with other small round cell tumors. Am J Surg Pathol. 2000;24(6):830‐836. [DOI] [PubMed] [Google Scholar]
- 124. Schoolmeester JK, Folpe AL, Nair AA, et al. EWSR1‐WT1 gene fusions in neoplasms other than desmoplastic small round cell tumor: a report of three unusual tumors involving the female genital tract and review of the literature. Mod Pathol. 2021;34(10):1912‐1920. [DOI] [PubMed] [Google Scholar]
- 125. Wang L, Motoi T, Khanin R, et al. Identification of a novel, recurrent HEY1‐NCOA2 fusion in mesenchymal chondrosarcoma based on a genome‐wide screen of exon‐level expression data. Genes Chromosomes Cancer. 2012;51(2):127‐139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Nyquist KB, Panagopoulos I, Thorsen J, et al. Whole‐transcriptome sequencing identifies novel IRF2BP2‐CDX1 fusion gene brought about by translocation t(1;5)(q42;q32) in mesenchymal chondrosarcoma. PLoS One. 2012;7(11):e49705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127. Nakayama R, Miura Y, Ogino J, et al. Detection of HEY1‐NCOA2 fusion by fluorescence in‐situ hybridization in formalin‐fixed paraffin‐embedded tissues as a possible diagnostic tool for mesenchymal chondrosarcoma. Pathol Int. 2012;62(12):823‐826. [DOI] [PubMed] [Google Scholar]
- 128. Hornick JL, Dal Cin P, Fletcher CD. Loss of INI1 expression is characteristic of both conventional and proximal‐type epithelioid sarcoma. Am J Surg Pathol. 2009;33(4):542‐550. [DOI] [PubMed] [Google Scholar]
- 129. Pawel BR. SMARCB1‐deficient tumors of childhood: a practical guide. Pediatr Dev Pathol. 2018;21(1):6‐28. [DOI] [PubMed] [Google Scholar]
- 130. Sullivan LM, Folpe AL, Pawel BR, Judkins AR, Biegel JA. Epithelioid sarcoma is associated with a high percentage of SMARCB1 deletions. Mod Pathol. 2013;26(3):385‐392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Rousseau‐Merck MF, Versteege I, Legrand I, et al. hSNF5/INI1 inactivation is mainly associated with homozygous deletions and mitotic recombinations in rhabdoid tumors. Cancer Res. 1999;59(13):3152‐3156. [PubMed] [Google Scholar]
- 132. Kohashi K, Yamada Y, Hotokebuchi Y, et al. ERG and SALL4 expressions in SMARCB1/INI1‐deficient tumors: a useful tool for distinguishing epithelioid sarcoma from malignant rhabdoid tumor. Hum Pathol. 2015;46(2):225‐230. [DOI] [PubMed] [Google Scholar]
- 133. Hollmann TJ, Hornick JL. INI1‐deficient tumors: diagnostic features and molecular genetics. Am J Surg Pathol. 2011;35(10):e47‐e63. [DOI] [PubMed] [Google Scholar]
- 134. Shibuya R, Matsuyama A, Shiba E, Harada H, Yabuki K, Hisaoka M. CAMTA1 is a useful immunohistochemical marker for diagnosing epithelioid haemangioendothelioma. Histopathology. 2015;67(6):827‐835. [DOI] [PubMed] [Google Scholar]
- 135. Doyle LA, Fletcher CD, Hornick JL. Nuclear expression of CAMTA1 distinguishes epithelioid hemangioendothelioma from histologic mimics. Am J Surg Pathol. 2016;40(1):94‐102. [DOI] [PubMed] [Google Scholar]
- 136. Yusıflı Z, Kösemehmetoğlu K. CAMTA1 immunostaining is not useful in differentiating epithelioid hemangioendothelioma from its potential mimickers. Turk Patoloji Derg. 2014;30(3):159‐165. [DOI] [PubMed] [Google Scholar]
- 137. Yang P, Zhang S, Yu C, et al. Fluorescence in situ hybridization for WWTR1‐CAMTA1 has higher sensitivity and specificity for epithelioid hemangioendothelioma diagnosis. Am J Transl Res. 2020;12(8):4561‐4568. [PMC free article] [PubMed] [Google Scholar]
- 138. Antonescu CR, Le Loarer F, Mosquera JM, et al. Novel YAP1‐TFE3 fusion defines a distinct subset of epithelioid hemangioendothelioma. Genes Chromosomes Cancer. 2013;52(8):775‐784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139. Anderson WJ, Fletcher CDM, Hornick JL. Loss of expression of YAP1 C‐terminus as an ancillary marker for epithelioid hemangioendothelioma variant with YAP1‐TFE3 fusion and other YAP1‐related vascular neoplasms. Mod Pathol. 2021;34(11):2036‐2042. [DOI] [PubMed] [Google Scholar]
- 140. Rosenbaum E, Jadeja B, Xu B, et al. Prognostic stratification of clinical and molecular epithelioid hemangioendothelioma subsets. Mod Pathol. 2020;33(4):591‐602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Chung EB, Enzinger FM. Malignant melanoma of soft parts. A reassessment of clear cell sarcoma. Am J Surg Pathol. 1983;7(5):405‐413. [DOI] [PubMed] [Google Scholar]
- 142. Wang WL, Mayordomo E, Zhang W, et al. Detection and characterization of EWSR1/ATF1 and EWSR1/CREB1 chimeric transcripts in clear cell sarcoma (melanoma of soft parts). Mod Pathol. 2009;22(9):1201‐1209. [DOI] [PubMed] [Google Scholar]
- 143. Thway K, Fisher C. Tumors with EWSR1‐CREB1 and EWSR1‐ATF1 fusions: the current status. Am J Surg Pathol. 2012;36(7):e1‐e11. [DOI] [PubMed] [Google Scholar]
- 144. Hocar O, Le Cesne A, Berissi S, et al. Clear cell sarcoma (malignant melanoma) of soft parts: a clinicopathologic study of 52 cases. Dermatol Res Pract. 2012;2012:984096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145. Rekhi B, Ingle A, Agarwal M, Puri A, Laskar S, Jambhekar NA. Alveolar soft part sarcoma 'revisited': clinicopathological review of 47 cases from a tertiary cancer referral centre, including immunohistochemical expression of TFE3 in 22 cases and 21 other tumours. Pathology. 2012;44(1):11‐17. [DOI] [PubMed] [Google Scholar]
- 146. Rekhi B, Rao V, Ramadwar M. Revisiting cytomorphology, including unusual features and clinical scenarios of 8 cases of alveolar soft part sarcoma with TFE3 immunohistochemical staining in 7 cases. Cytopathology. 2021;32(1):20‐28. [DOI] [PubMed] [Google Scholar]
- 147. Tretiakova MS, Wang W, Wu Y, Tykodi SS, True L, Liu YJ. Gene fusion analysis in renal cell carcinoma by FusionPlex RNA‐sequencing and correlations of molecular findings with clinicopathological features. Genes Chromosomes Cancer. 2020;59(1):40‐49. [DOI] [PubMed] [Google Scholar]
- 148. Schaefer IM, Dong F, Garcia EP, Fletcher CDM, Jo VY. Recurrent SMARCB1 inactivation in epithelioid malignant peripheral nerve sheath tumors. Am J Surg Pathol. 2019;43(6):835‐843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. Jo VY, Fletcher CD. Epithelioid malignant peripheral nerve sheath tumor: clinicopathologic analysis of 63 cases. Am J Surg Pathol. 2015;39(5):673‐682. [DOI] [PubMed] [Google Scholar]
- 150. Dodd LG. Update on liposarcoma: a review for cytopathologists. Diagn Cytopathol. 2012;40(12):1122‐1131. [DOI] [PubMed] [Google Scholar]
- 151. Chen BJ, Mariño‐Enríquez A, Fletcher CD, Hornick JL. Loss of retinoblastoma protein expression in spindle cell/pleomorphic lipomas and cytogenetically related tumors: an immunohistochemical study with diagnostic implications. Am J Surg Pathol. 2012;36(8):1119‐1128. [DOI] [PubMed] [Google Scholar]
- 152. Creytens D, Mentzel T, Ferdinande L, et al. "atypical" pleomorphic lipomatous tumor: a clinicopathologic, Immunohistochemical and molecular study of 21 cases, emphasizing its relationship to atypical spindle cell lipomatous tumor and suggesting a morphologic spectrum (atypical spindle cell/pleomorphic lipomatous tumor). Am J Surg Pathol. 2017;41(11):1443‐1455. [DOI] [PubMed] [Google Scholar]
- 153. Dei Tos AP, Doglioni C, Piccinin S, et al. Coordinated expression and amplification of the MDM2, CDK4, and HMGI‐C genes in atypical lipomatous tumours. J Pathol. 2000;190(5):531‐536. [DOI] [PubMed] [Google Scholar]
- 154. Sirvent N, Coindre JM, Maire G, et al. Detection of MDM2‐CDK4 amplification by fluorescence in situ hybridization in 200 paraffin‐embedded tumor samples: utility in diagnosing adipocytic lesions and comparison with immunohistochemistry and real‐time PCR. Am J Surg Pathol. 2007;31(10):1476‐1489. [DOI] [PubMed] [Google Scholar]
- 155. Clay MR, Martinez AP, Weiss SW, Edgar MA. MDM2 and CDK4 immunohistochemistry: should it be used in problematic differentiated lipomatous tumors?: a new perspective. Am J Surg Pathol. 2016;40(12):1647‐1652. [DOI] [PubMed] [Google Scholar]
- 156. Machado I, Vargas AC, Maclean F, Llombart‐Bosch A. Negative MDM2/CDK4 immunoreactivity does not fully exclude MDM2/CDK4 amplification in a subset of atypical lipomatous tumor/well differentiated liposarcoma. Pathol Res Pract. 2022;232:153839. [DOI] [PubMed] [Google Scholar]
- 157. Tsuda Y, Matsuyama A, Makihara K, et al. Nuclear expression of MDM2 in hibernoma: a potential diagnostic pitfall. Virchows Arch. 2021;478(3):527‐534. [DOI] [PubMed] [Google Scholar]
- 158. Weaver J, Downs‐Kelly E, Goldblum JR, et al. Fluorescence in situ hybridization for MDM2 gene amplification as a diagnostic tool in lipomatous neoplasms. Mod Pathol. 2008;21(8):943‐949. [DOI] [PubMed] [Google Scholar]
- 159. Kashima T, Halai D, Ye H, et al. Sensitivity of MDM2 amplification and unexpected multiple faint alphoid 12 (alpha 12 satellite sequences) signals in atypical lipomatous tumor. Mod Pathol. 2012;25(10):1384‐1396. [DOI] [PubMed] [Google Scholar]
- 160. Weaver J, Rao P, Goldblum JR, et al. Can MDM2 analytical tests performed on core needle biopsy be relied upon to diagnose well‐differentiated liposarcoma? Mod Pathol. 2010;23(10):1301‐1306. [DOI] [PubMed] [Google Scholar]
- 161. Sugiyama K, Washimi K, Sato S, et al. Differential diagnosis of lipoma and atypical lipomatous tumor/well‐differentiated liposarcoma by cytological analysis. Diagn Cytopathol. 2022;50(3):112‐122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162. Zhang W, McElhinny A, Nielsen A, et al. Automated brightfield dual‐color in situ hybridization for detection of mouse double minute 2 gene amplification in sarcomas. Appl Immunohistochem Mol Morphol. 2011;19(1):54‐61. [DOI] [PubMed] [Google Scholar]
- 163. Sasaki T, Ogose A, Kawashima H, et al. Real‐time polymerase chain reaction analysis of MDM2 and CDK4 expression using total RNA from core‐needle biopsies is useful for diagnosing adipocytic tumors. BMC Cancer. 2014;14:468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164. Creytens D, van Gorp J, Ferdinande L, Speel EJ, Libbrecht L. Detection of MDM2/CDK4 amplification in lipomatous soft tissue tumors from formalin‐fixed, paraffin‐embedded tissue: comparison of multiplex ligation‐dependent probe amplification (MLPA) and fluorescence in situ hybridization (FISH). Appl Immunohistochem Mol Morphol. 2015;23(2):126‐133. [DOI] [PubMed] [Google Scholar]
- 165. Kimura H, Dobashi Y, Nojima T, et al. Utility of fluorescence in situ hybridization to detect MDM2 amplification in liposarcomas and their morphological mimics. Int J Clin Exp Pathol. 2013;6(7):1306‐1316. [PMC free article] [PubMed] [Google Scholar]
- 166. Sciot R. MDM2 amplified sarcomas: a literature review. Diagnostics (Basel). 2021;11(3):496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167. Berg SH, Massoud CM, Jackson‐Cook C, Boikos SA, Smith SC, Mochel MC. A reappraisal of superficial pleomorphic liposarcoma. Am J Clin Pathol. 2020;154(3):353‐361. [DOI] [PubMed] [Google Scholar]
- 168. Anderson WJ, Jo VY. Pleomorphic liposarcoma: updates and current differential diagnosis. Semin Diagn Pathol. 2019;36(2):122‐128. [DOI] [PubMed] [Google Scholar]
- 169. West RB, Rubin BP, Miller MA, et al. A landscape effect in tenosynovial giant‐cell tumor from activation of CSF1 expression by a translocation in a minority of tumor cells. Proc Natl Acad Sci U S A. 2006;103(3):690‐695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170. Panagopoulos I, Brandal P, Gorunova L, Bjerkehagen B, Heim S. Novel CSF1‐S100A10 fusion gene and CSF1 transcript identified by RNA sequencing in tenosynovial giant cell tumors. Int J Oncol. 2014;44(5):1425‐1432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171. Tsuda Y, Hirata M, Katayama K, et al. Massively parallel sequencing of tenosynovial giant cell tumors reveals novel CSF1 fusion transcripts and novel somatic CBL mutations. Int J Cancer. 2019;145(12):3276‐3284. [DOI] [PubMed] [Google Scholar]
- 172. Mejbel H, Siegal GP, Wei S. Intramuscular tenosynovial giant cell tumor harboring a novel CSF1‐CD96 fusion. Int J Surg Pathol. 2022;30(3):335‐338. [DOI] [PubMed] [Google Scholar]
- 173. Vougiouklakis T, Shen G, Feng X, Hoda ST, Jour G. Molecular profiling of atypical tenosynovial giant cell tumors reveals novel non‐CSF1 fusions. Cancers (Basel). 2019;12(1):100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174. Nakayama S, Nishio J, Nakatani K, Nabeshima K, Yamamoto T. Giant cell tumor of tendon sheath with a t(1;1)(p13;p34) chromosomal translocation. Anticancer Res. 2020;40(8):4373‐4377. [DOI] [PubMed] [Google Scholar]
- 175. Baldi GG, Gronchi A, Stacchiotti S. Pexidartinib for the treatment of adult symptomatic patients with tenosynovial giant cell tumors. Expert Rev Clin Pharmacol. 2020;13(6):571‐576. [DOI] [PubMed] [Google Scholar]
- 176. Lee JC, Liang CW, Fletcher CD. Giant cell tumor of soft tissue is genetically distinct from its bone counterpart. Mod Pathol. 2017;30(5):728‐733. [DOI] [PubMed] [Google Scholar]
- 177. Mancini I, Righi A, Gambarotti M, et al. Phenotypic and molecular differences between giant‐cell tumour of soft tissue and its bone counterpart. Histopathology. 2017;71(3):453‐460. [DOI] [PubMed] [Google Scholar]
- 178. Agaimy A, Michal M, Stoehr R, et al. Recurrent novel HMGA2‐NCOR2 fusions characterize a subset of keratin‐positive giant cell‐rich soft tissue tumors. Mod Pathol. 2021;34(8):1507‐1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Anderson WJ, Hornick JL. Immunohistochemical correlates of recurrent genetic alterations in sarcomas. Genes Chromosomes Cancer. 2019;58(2):111‐123. [DOI] [PubMed] [Google Scholar]
- 180. Koelsche C, Schrimpf D, Stichel D, et al. Sarcoma classification by DNA methylation profiling. Nat Commun. 2021;12(1):498. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Research data are not shared.