

Abstract
Hyper‐IgE syndromes (HIES) are a heterogeneous group of rare primary immunodeficiency diseases classically characterized by the triad of atopic dermatitis, and recurrent cutaneous and pulmonary infections. Autosomal dominant, loss‐of‐function STAT3 pathogenic variants are the most common genetic cause, which lead to deficiency of Th17 lymphocytes, impaired interferon gamma production, and IL‐10 signal transduction, and an unbalanced IL‐4 state. Dupilumab, a monoclonal antibody to the IL‐4a receptor, inhibits both IL‐4 and IL‐13, and has been shown to improve atopic dermatitis and other manifestations of HIES including asthma and allergic bronchopulmonary aspergillosis. We present a pediatric patient with HIES who presented predominantly with eosinophilic folliculitis, recurrent cutaneous infections, and other non‐eczematous findings and achieved sustained clearance with dupilumab.
Keywords: atopic dermatitis, dupilumab, eczema, genetic syndromes, hyper‐IgE syndrome
1. CASE
A 14‐month‐old girl with coarse facies, high arched palate, and a history of recurrent folliculocentric edematous papules and pustules complicated by methicillin‐resistant Staphylococcus aureus (MRSA) infection and pulmonary infections presented to dermatology clinic (Figure 1). She had been previously treated with five courses of trimethoprim‐sulfamethoxazole for culture‐confirmed MRSA and at least 12 courses of other oral antibiotics, in addition to topical corticosteroids. Prior workup for immunodeficiency showed persistent leukocytosis with peripheral eosinophilia (1700/uL, normal 0–700/uL), normal IgG (431 mg/dL, normal 206–676 mg/dL), IgM (96 mg/dL, normal 33–97 mg/dL), with mildly elevated IgE (65 IU/mL, normal 0–59 mg/dL), and IgA (83 mg/dL, normal 8–67 mg/dL). Biopsy in infancy demonstrated a superficial and deep perivascular, periadnexal interstitial infiltrate with eosinophils (Figure 2). Household empirical treatment for scabies led to no improvement.
FIGURE 1.
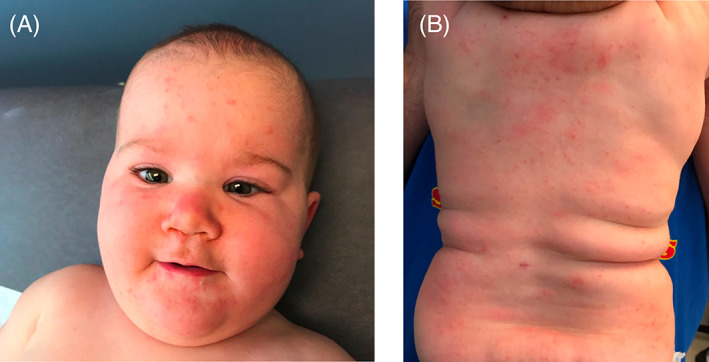
Skin findings at time of initial presentation
FIGURE 2.
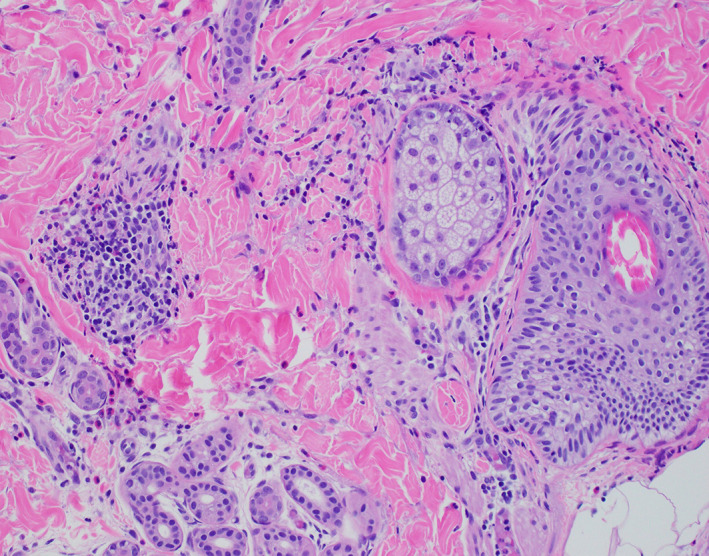
Biopsy demonstrating a superficial and deep perivascular, periadnexal interstitial infiltrate with eosinophils
Despite only mildly elevated IgE levels, STAT3‐associated HIES was considered, given the clinical and histopathologic evidence for eosinophilic folliculitis, and other dysmorphic features including coarse facial features. Testing was positive for a known pathogenic STAT3 variant (1144 C>T, Arg382Trp), confirming the diagnosis of autosomal dominant HIES. The patient was started on immune globulin replacement therapy to prevent life‐threatening infection, but this did not improve her skin findings. She was also treated with topical steroids, topical tacrolimus, emollients, long‐acting antihistamines, and anti‐staphylococcal therapeutics including topical mupirocin, bleach baths, and a host of oral antibiotics including oral cephalexin, erythromycin, and trimethoprim‐sulfamethoxazole. Despite these therapeutic efforts, her skin continued to flare every 2–3 weeks, and she subsequently developed crusting of the eyes, nose, and external auditory canal, with significant impairment in quality of life due to itch and pain (Figure 3). Multiple cultures from these sites were negative for microorganisms. At this time, dupilumab was considered due to case reports demonstrating improvement in atopic manifestations of hyper‐IgE syndrome, likely mechanistic efficacy based on the known pathologic disruption of IL‐4, and failure of all other trialed therapies. 1 Dupilumab was initiated with a loading dose of 600 mg followed by 300 mg every 4 weeks based on a weight 22 kg. At 2 months, she had excellent response with great improvement both clinically and symptomatically, with complete resolution of her recalcitrant itch and pain. Now, at 6 months after initiation of treatment at age 3 years, she continues to have durable response with no adverse effects, and no additional infections (Figure 4).
FIGURE 3.
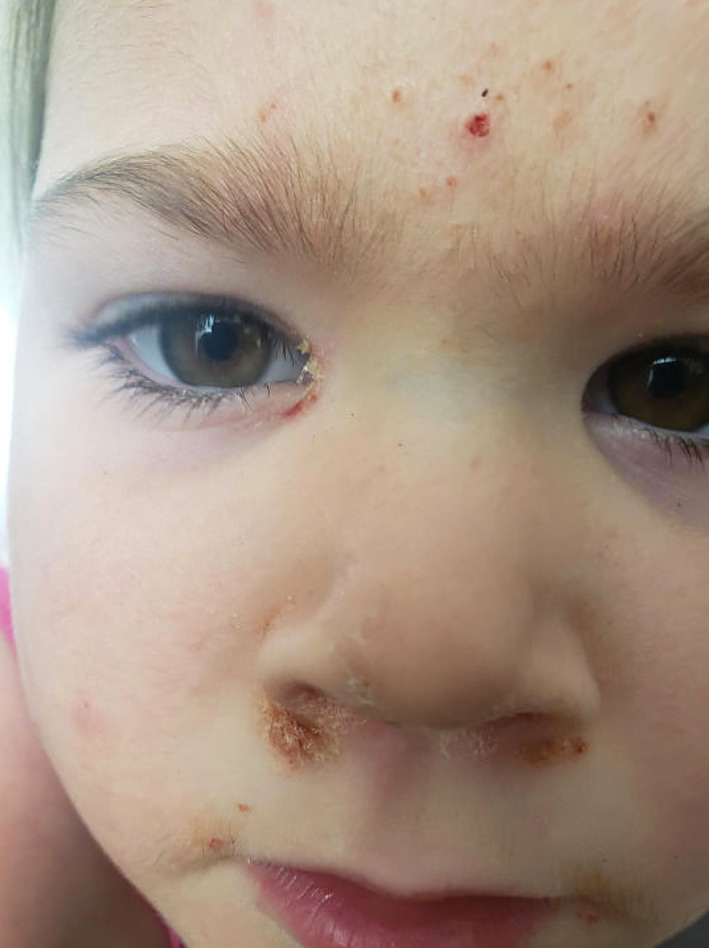
Painful yellow crusting of nasolabial folds and bilateral inner canthus of the eyes
FIGURE 4.
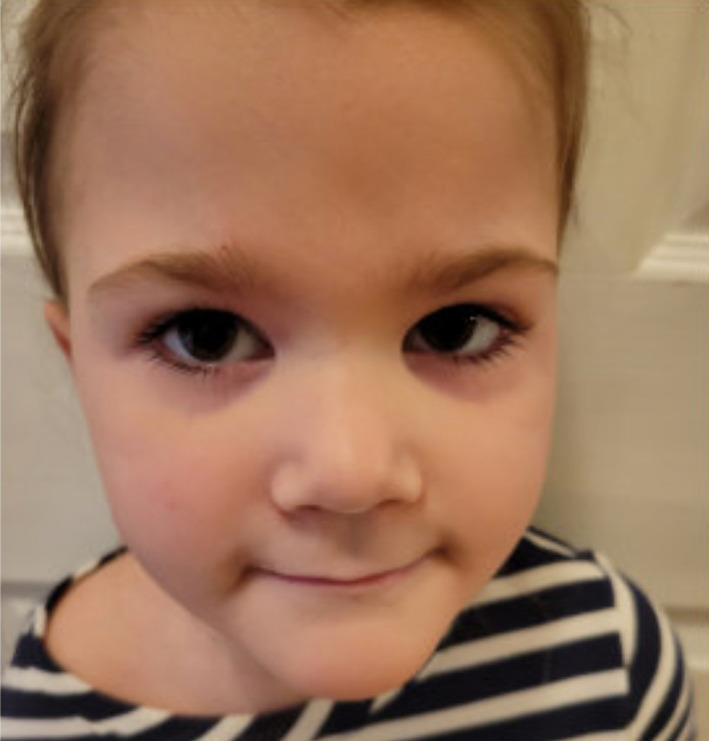
Skin improvement after 6 months of dupilumab treatment
2. DISCUSSION
The hyper‐IgE syndromes have historically been defined by the triad of elevated IgE, dermatitis, and recurrent cutaneous and pulmonary infections. 2 They represent a heterogeneous group of rare primary immunodeficiency diseases with an estimated annual incidence of 1:1,000,000. 3 Although autosomal dominant loss‐of‐function variants in STAT3 4 , 5 are the most common cause of HIES, a host of other genes including TYK2, 6 PGM3, 7 ZNF341, 8 CARD11, 9 and IL6ST 10 , 11 contribute to the disease‐causing spectrum. 12 The dermatologic manifestations of HIES typically manifest within the first few weeks of life and are most commonly eczematous in nature, involving the scalp and face. Eosinophilic folliculitis has been previously reported as a cutaneous manifestation of HIES. 13 Other common clinical findings include coarse or “leonine” facies, retained primary teeth, high palate, hyperextensibility, scoliosis, and minor trauma fractures. 2
In 1999, the NIH developed an HIES scoring system to aid in diagnosis. 14 This scoring system considers characteristic clinical findings and elevated serum IgE levels, which are often >2000 IU/mL. 15 However, IgE levels can be normal below 2 years of age and are therefore an unreliable diagnostic feature for HIES in this population. 16 Rather, identification of core clinical features should guide consideration of genetic testing in this population. This case demonstrates the importance of maintaining high clinical suspicion for HIES in infants with normal or only mild elevations in IgE levels, even in the absence of classic dermatologic findings such as atopic dermatitis. Further, this case underscores that recalcitrant eosinophilic folliculitis should raise suspicion for HIES in infants.
Management of HIES is challenging and involves topical skin care, as well as prevention and aggressive treatment of infections. Cutaneous manifestations sometimes respond to topical corticosteroids, regular use of emollients, and gentle skin care. Prophylactic antibiotics targeted against S. aureus, bleach baths, chlorhexidine washes, and immune globulin replacement are efficacious for prevention of infection. 17 Off‐label use of dupilumab, an IL‐4a receptor antagonist that disrupts the unbalanced IL‐4 signaling and the allergic type 2 cytokine signature in HIES, has demonstrated efficacy for atopic dermatitis in HIES, as well as other HIES manifestations including asthma and allergic bronchopulmonary aspergillosis. 1 , 18 This case report adds to the existing literature in demonstrating sustained clearance of atypical skin manifestations, including eosinophilic folliculitis and recurrent cutaneous infections. Though further studies are warranted, dupilumab demonstrates potential as an option for long‐term management of the cutaneous spectrum of HIES due to its targeted mechanism of action.
CONFLICTS OF INTEREST
Kristen E. Holland served as an investigator for Pfizer, Amgen, Incyte, Abbvie, and Sanofi. Abbvie also provides spouse salary. Anne Marie Singh has consulted for Abbvie and currently serves on the data monitoring board for Siolta Therapeutics. She receives research funding from the NIH. Lisa M. Arkin has consulted for Abbvie and Regeneron. The remaining authors declare no conflicts of interest.
ACKNOWLEDGMENT
The authors would like to thank Dr Karolyn Wanat for assisting with pathology.
Nihal A, Comstock JR, Holland KE, Singh AM, Seroogy CM, Arkin LM. Clearance of atypical cutaneous manifestations of hyper‐IgE syndrome with dupilumab. Pediatr Dermatol. 2022;39(6):940‐942. doi: 10.1111/pde.15072
DATA AVAILABILITY STATEMENT
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
REFERENCES
- 1. Igelman S, Kurta AO, Sheikh U, et al. Off‐label use of dupilumab for pediatric patients with atopic dermatitis: a multicenter retrospective review. J Am Acad Dermatol. 2020;82(2):407‐411. doi: 10.1016/j.jaad.2019.10.010 [DOI] [PubMed] [Google Scholar]
- 2. Al‐Shaikhly T, Ochs HD. Hyper IgE syndromes: clinical and molecular characteristics. Immunol Cell Biol. 2018;97(4):368‐379. doi: 10.1111/imcb.12209 [DOI] [PubMed] [Google Scholar]
- 3. Hafsi W, Yarrarapu SNS. Job Syndrome. StatPearls Publishing; 2021. [PubMed] [Google Scholar]
- 4. Minegishi Y, Saito M, Tsuchiya S, et al. Dominant‐negative mutations in the DNA‐binding domain of STAT3 cause hyper‐IgE syndrome. Nature. 2007;448(7157):1058‐1062. doi: 10.1038/nature06096 [DOI] [PubMed] [Google Scholar]
- 5. Holland SM, DeLeo FR, Elloumi HZ, et al. STAT3 mutations in the hyper‐IGE syndrome. N Engl J Med. 2007;357(16):1608‐1619. doi: 10.1056/nejmoa073687 [DOI] [PubMed] [Google Scholar]
- 6. Minegishi Y, Saito M, Morio T, et al. Human tyrosine kinase 2 deficiency reveals its requisite roles in multiple cytokine signals involved in innate and acquired immunity. Immunity. 2006;25(5):745‐755. doi: 10.1016/j.immuni.2006.09.009 [DOI] [PubMed] [Google Scholar]
- 7. Sassi A, Lazaroski S, Wu G, et al. Hypomorphic homozygous mutations in phosphoglucomutase 3 (PGM3) impair immunity and increase serum IGE levels. J Allergy Clin Immunol. 2014;133(5):1410‐1419. doi: 10.1016/j.jaci.2014.02.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Béziat V, Li J, Lin J‐X, et al. A recessive form of hyper‐IGE syndrome by disruption of ZNF341‐dependent STAT3 transcription and activity. Sci Immunol. 2018;3(24):eaat4956. doi: 10.1126/sciimmunol.aat4956 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ma CA, Stinson JR, Zhang Y, et al. Germline hypomorphic CARD11 mutations in severe atopic disease. Nat Genet. 2017;49(8):1192‐1201. doi: 10.1038/ng.3898 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Schwerd T, Twigg SRF, Aschenbrenner D, et al. A biallelic mutation in IL6ST encoding the gp130 co‐receptor causes immunodeficiency and craniosynostosis. J Exp Med. 2017;214(9):2547‐2562. doi: 10.1084/jem.20161810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Shahin T, Aschenbrenner D, Cagdas D, et al. Selective loss of function variants in IL6ST cause hyper‐IgE syndrome with distinct impairments of T‐cell phenotype and function. Haematologica. 2018;104(3):609‐621. doi: 10.3324/haematol.2018.194233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bousfiha A, Jeddane L, Picard C, et al. Human inborn errors of immunity: 2019 update of the IUIS phenotypical classification. J Clin Immunol. 2020;40(1):66‐81. doi: 10.1007/s10875-020-00758-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Eberting CL, Davis J, Puck JM, Holland SM, Turner ML. Dermatitis and the newborn rash of hyper IgE syndrome. Arch Dermatol. 2004;140(9):1119‐1125. doi: 10.1001/archderm.140.9.1119 [DOI] [PubMed] [Google Scholar]
- 14. Grimbacher B, Schaffer AA, Holland SM, et al. Genetic linkage of hyper‐IgE syndrome to chromosome 4. Am J Hum Genet. 1999;65(3):735‐755. doi: 10.1086/302547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Grimbacher B, Holland SM, Puck JM. Hyper‐IgE syndromes. Immunol Rev. 2005;203(1):244‐250. doi: 10.1111/j.0105-2896.2005.00228.x [DOI] [PubMed] [Google Scholar]
- 16. Lindberg R, Arroyave C. Levels of IgE in serum from normal children and allergic children as measured by an enzyme immunoassay. J Allergy Clin Immunol. 1986;78(4):614‐618. doi: 10.1016/0091-6749(86)90078-3 [DOI] [PubMed] [Google Scholar]
- 17. Sowerwine KJ, Holland SM, Freeman AF. Hyper‐IgE syndrome update. Ann N Y Acad Sci. 2012;1250(1):25‐32. doi: 10.1111/j.1749-6632.2011.06387.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. James AE, West L, Schloss K, et al. Treatment of STAT3‐deficient hyper–immunoglobulin E syndrome with monoclonal antibodies targeting allergic inflammation. J Allergy Clin Immunol Pract. 2022;10(5):1367‐1370. doi: 10.1016/j.jaip.2022.01.011 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.