

Abstract
We developed an IGFBP2-mimetic peptide fragment, JB2, and showed that it promotes basal synaptic structural and functional plasticity in cultured neurons and mice. We demonstrate that JB2 directly binds to dendrites and synapses, and its biological activity involves NMDA receptor activation, gene transcription and translation, and IGF2 receptors. It is not IGF1 receptor-dependent. In neurons, JB2 induced extensive remodeling of the membrane phosphoproteome. Synapse and cytoskeletal regulation, autism spectrum disorder (ASD) risk factors, and a Shank3-associated protein network were significantly enriched among phosphorylated and dephosphorylated proteins. Haploinsufficiency of the SHANK3 gene on chromosome 22q13.3 often causes Phelan-McDermid Syndrome (PMS), a genetically defined form of autism with profound deficits in motor behavior, sensory processing, language, and cognitive function. We identified multiple disease-relevant phenotypes in a Shank3 heterozygous mouse and showed that JB2 rescued deficits in synaptic function and plasticity, learning and memory, ultrasonic vocalizations, and motor function; it also normalized neuronal excitability and seizure susceptibility. Notably, JB2 rescued deficits in the auditory evoked response latency, alpha peak frequency, and steady-state electroencephalography response, measures with direct translational value to human subjects. These data demonstrate that JB2 is a potent modulator of neuroplasticity with therapeutic potential for the treatment of PMS and ASD.
INTRODUCTION
Neurodevelopmental disorders (NDDs), including autism spectrum disorder (ASD), pose a serious burden on society, and no effective disease-modifying pharmacological treatments exist. Converging evidence from large-scale genomics, postmortem neuroanatomy, brain imaging, and animal models have demonstrated that abnormalities in synaptic connections and circuits are key substrates of NDDs, including ASD [1, 2]. Therapeutically targeting synaptic plasticity may yield novel treatments for NDDs. However, our toolbox for modulating synaptic plasticity is limited, and there is a great unmet need to develop novel classes of modulators of synaptic plasticity with therapeutic potential.
According to the CDC, one in 44 children in the U.S. is diagnosed with ASD (https://www.cdc.gov/ncbddd/autism/data.html). With the high prevalence of ASD diagnoses among the general population, and with over 80% co-occurrence with other neurologic, genetic, and psychiatric diagnoses, the economic and social burden of the disease is enormous [3, 4]. Children and adolescents with ASD have median annual medical expenditures exceeding those of typically developing peers by a factor of 8 to 10 times. Diverse clinical and developmental symptoms of ASD are managed through a host of expensive behavioral interventions with varying degrees of success. Due to the immense etiological heterogeneity, the development of therapeutics for ASD has been extremely challenging. However, drug development for etiologically defined subtypes of ASD could be achievable and could pave the way for the treatment of other forms of autism [5–7].
Phelan-McDermid Syndrome (PMS) is an etiologically defined form of ASD. PMS is characterized by global developmental delay, variable degrees of ID, absent or delayed speech, ASD, epilepsy, sensory processing, attention, and motor deficits, hypotonia, regression, brain abnormalities, mild dysmorphic features, feeding and gastrointestinal problems, and a range of other comorbid clinical conditions [4, 8, 9]. The great majority of diagnosed cases of PMS are caused by terminal deletions in the 22q13.3 region, leading to SHANK3 haploinsufficiency [8]. PMS can also result from point mutations in one copy of the SHANK3 gene [10–13]. The development of an effective pharmacological treatment for PMS would have a substantial impact on the management of PMS and, potentially, ASD.
Insulin-like growth factor-binding proteins form a family of six proteins (IGFBP1 to 6), which play a broad array of functions within and outside of the central nervous system (CNS) [14, 15]. One major function of IGFBPs is to regulate the bioavailability and function of insulin-like growth factors (IGF-1 and IGF-2). In general, the functions of the IGF system are exerted through the IGF receptor 1 (IGF-1R). IGFBPs also function independently of binding to IGFs as autocrine and/or paracrine growth factors. Among IGFBPs, IGFBP2 is the most abundant in the cerebrospinal fluid (CSF) and is highly expressed during brain development [14]. Within the brain, IGFBP2 is highly expressed in the cortex, hippocampus, and cerebellum, structures affected in ASD. Several recent studies suggest that in addition to a role in neurodevelopment, IGFBP2 also modulates neuronal plasticity and cognitive functions [14, 16]. IGFBP2 has been shown to enhance excitatory synaptic transmission and promote long-term potentiation (LTP) by enhancing N-methyl-d-aspartate (NMDA) receptor-dependent excitatory postsynaptic currents [17].
Here we report the development of an IGFBP2-derived peptide fragment, JB2, and its effects on neuroplasticity, behavioral impairments, and cellular processes.
MATERIALS AND METHODS
Animals
Shank3 heterozygous (Shank3+/−) mice were generously provided by Joseph Buxbaum at the Icahn School of Medicine at Mount Sinai. The heterozygous mice were mated with wild-type (WT) mice to maintain the colonies. Littermates and Shank3+/− mice and WT mice were used for primary neuronal culture, electroencephalography (EEG)/electromyography (EMG), and behavioral tests. The mice were housed together in mixed-genotype home cages until testing. Within a specific-pathogen-free barrier area, mice were maintained on a 12:12 h light/dark schedule in an air-conditioned room under constant conditions of temperature and humidity. Food and tap water (membrane filter purified and autoclaved water) were provided ad libitum. All experiments were performed in accordance with the Institutional Animal Care and Use Committee at Northwestern University.
Neuronal cell cultures
Dissociated primary cortical neurons were prepared from E18 Sprague-Dawley rat embryos or P0 Shank3 WT × HT-breed pups. Neurons were plated on poly-D-lysine coated glass coverslips, fed with Neurobasal medium supplemented with 2% B27 Supplement, penicillin/streptomycin (100 U/mL and 100 mg/mL, respectively), and 2 mM GlutaMAX-I, as detailed in our previous publications [18, 19]. For immunocytochemistry or multi-electrode array (MEA) recordings, 0.85 × 105 or 0.74 × 105 cells/cm2 of cells were plated, respectively. Separately, 0.21 × 105 cells/cm2 were used for the analysis of ERK and AKT phosphorylation by immunocytochemistry. Neurons were then transfected with a plasmid expressing enhanced Green Fluorescent Protein (eGFP) at DIV 21; after 2 days, they were treated with 1 μM of JB2 for 1 h in media. Treated neurons were fixed for 10 min at 4 °C by using 4% paraformaldehyde in PBS and then stained with an antibody for GFP (Abcam; ab13970), followed by a fluorescent Alexa Fluor 488 secondary antibody (Invitrogen). The detailed information for immunocytochemistry was described previously [19]. With MEA plates, we performed all experiments at DIV 28.
Phosphoproteomic analysis
Primary cultured rat neurons were harvested after treatment with either the vehicle or 1 μM of JB2 (five 10-cm dishes per group) for 1 h at DIV 21. Cells were homogenized in sucrose buffer (20 mM HEPES pH 7.4, 320 mM sucrose, 5 mM EDTA with a protease inhibitor (Roche) and a phosphatase inhibitor cocktail 3 (Sigma)) and centrifuged at 3000 × g for 20 min at 4 °C. The protein concentration of the collected supernatant (cytosol and membrane) was measured by BCA; then, 5 mg of total protein was used from each group for the subsequent processes. The proteins were reduced and alkylated before trypsin digestion. Phosphopeptides from the digests were enriched using TiO2 beads, then each was fractionated by high pH reverse phase into four fractions. Each fraction was desalted prior to the mass spec analysis. We searched mass spectrometry data against a Rattus norvegicus database using the MaxQuant search engine, where MS1 peptide intensity was measured. We calculated the Log2 fold change value (Log2FC) between 2 groups. Among the phosphopeptides above 75% of localization probability (recommended cutoff), we defined a Log2FC ≥ 1 (2 Folds) as phosphorylated (in red) and ≤ −1 as dephosphorylated (in blue) in the JB2 sample. Biological process or cellular component enrichment was examined using the DAVID Bioinformatics database (v6.8) (https://david.ncifcrf.gov) or SynGO (https://www.syngoportal.org). More detailed information was described previously [20].
Imaging the binding of JB2 to dendrites in brain slices
A 3-month-old, Thy1-YFP (#003782; Jackson Laboratory) homozygous male mouse received a lethal intraperitoneal injection of pentobarbital (Euthasol, 150 mg/kg). The mouse was then transcardially perfused with ice-cold PFA 4% in PBS (pH = 7.2) and post-fixed overnight in the same fixative solution. The brain was incubated in PBS 1 x, sucrose 30%, for 24 h, and 40 μm brain sections were performed in a Cryostat (Leica). Slices were then rinsed in PBS, blocked and permeabilized in PBS, BSA 5%, and Triton 0.1% for 1 h, followed by overnight incubation with 1 μg/μl of JB2-cy3 at 4 C. Sections were then mounted in Prolong Gold between glass microscope slides (Colorfrost, Fisher Scientific) and #1.5 glass coverslips (Warner Instrument). Confocal images were acquired by a Nikon C2 confocal microscope using a 63x oil immersion objective (numerical aperture = 1.4). JB2-Cy3 was excited with a laser set at 561 nm and YFP at 488 nm. Images were acquired at a resolution of 100 um in the X-Y axes and 200 nm in the z-axis. Finally, 3D images were reconstructed using Imaris software.
Preparation and immunostaining of acute brain slices for analysis of JB2 binding to synapses
Four P28 C57BL/6J mice (#000664; Jackson Laboratory) were deeply anesthetized with 5% isoflurane and euthanized. Brains were rapidly extracted and placed in ice-cold control artificial cerebrospinal fluid (ACSF), aerated with 95% O2/5% CO2, pH = 7.3 (ACSF compounds concentrations in mM: NaCl 124 mM, KCl 2.5 mM, NaH2PO4 1.2 mM, NaHCO3 24 mM, Hepes 5 mM, Glucose 12.5 mM, MgSO4•7H2O 2 mM, CaCl2 2 mM). Brains were glued on a vibratome stage (Leica Vt1000 S, Germany) to be cut at 200 μm in ice-cold ACSF. After a recovery phase of 1 hr at 32–33 °C, slices were incubated for 1 h with JB2-Cy3 or a control anti-human IgG-Cy3 (#C2571; Sigma-Aldrich), both compounds at 2 μg/ml. Stained slices were then washed three times in control ACSF for 10 minutes and are fixed for 30 min in 4% PFA in PBS and finally rinsed in PBS. Slices were then blocked and permeabilized with a solution of PBS, Triton 0.1%, BSA 5% for 1 h to be then incubated overnight with antibodies directed against PSD95 (1:500, K28/43; NeuroMab) and VGLUT1 (1:500, #135 304; Synaptic System). Slices were then rinsed three times in PBS 1 × and incubated with secondary antibodies against mouse IgG (1:500, A11029, conjugated with Alexa488; ThermoFisher) and guinea pig IgG (1:500, A21450, conjugated with Alexa 643; ThermoFisher). After three times washing in PBS, slices were mounted on glass slides in prolong gold mounting media. For analyzing the binding of JB2 on dendrites, three P120 male Thy1-YFP (#003782; Jackson Laboratory) mice were perfused transcardially with PFA 4%, PBS, and 30 microns slices were performed on a cryostat (Leica). Thy1-YFP slices were blocked in BSA for 30 min and stained with JB2-Cy3 for 1 h to be then mounted in glass slides in Prolong gold mounting media.
Confocal imaging and automatic 3D colocalization analyses of synaptic markers and JB2
All images were acquired on the same day on a C2 Nikon confocal microscope to limit laser intensity variations with a 60x oil objective, NA = 1.4. Stacks of 200 nm Z-steps were acquired from the surface of the slice to 15–20 um depth on the layer 2/3 of the somatosensory cortex, with a zoom 2, X-Y resolution: 100 nm. Images were deconvoluted using NIS element (Nikon) automatic deconvolution. Post-processed images were 3D-segmented using the toolbox Iterative Thresholding in ImageJ using the following parameters (min threshold: 20, Criteria method: Elongation, Segment Result: best, Threshold method STEP, Value method: 20). 3D colocalizations were analyzed in Fiji (ImageJ) using the Distance Analysis toolbox: DiAna to determine the percentage of JB2-Cy3 puncta or IgG-puncta colocalizing with either presynaptic marker VGLUT1 or postsynaptic marker PSD95 [21]. For the localization on dendritic compartments on Thy1-YFP slices, stacks were acquired and deconvoluted the same way as the immunostaining images. A 3D mask surrounding the YFP dendritic signal is used to easily see the puncta near the dendrites. Then puncta were assigned to one of the three defined compartments: head spines, spine neck, and dendritic shaft, and normalized in percentage for each neuron.
Auditory LTP
Noninvasive methods for measuring synaptic plasticity via LTP in mice were performed. Mice were anesthetized using isoflurane, and a cortical EEG was monitored via implanted skull screws (Pinnacle). EEG signals were captured via a tethered system (Pinnacle); auditory evoked potentials were recorded from frontal cortex skull screws using a cerebellum skull screw as a ground/reference. Data were acquired at 1000 samples per second using an A&M amplifier with high (0.1 Hz) and low pass (100 Hz) filters. Data was recorded using Data Wave acquisition software and analyzed using Brain Products Analyzer 2 software. LTP was induced by auditory tetanus (6-kHz, 50 ms in duration), presented 10 times per second for 5 min (total of 3000 tones) using a similar paradigm as Clapp et al. [22]. Mismatch negativity testing occurred immediately before tetanus (pre-tetanus) and 1 h after tetanus (post-tetanus) following a specific protocol. Post-pre-tetanus difference waves were generated to determine the range (in milliseconds) in which LTP occurred. Secondary endpoints included mismatch negativity and quantitative EEG.
fEPSP and LTP induction in mPFC neurons
Freely behaving rats implanted with medial prefrontal cortex bipolar stimulation electrodes (layer2–3) and monopolar recording electrodes (layer 5), and single shock (0.7 ms) induced field excitatory postsynaptic currents (fEPSPs) were recorded once every 30 seconds for 24 h. 1 h after the start of the study animals were either given an injection of vehicle (1 ml/kg sterile saline s.c.), and injection of NBQX (10 mg/kg sc), or JB2 (1 mg/kg sc). An additional LTP control group received 3 ×30-second train of theta (6 Hz) stimulation with three 0.07 ms (10 ms inter stimulus interval) stimulations given every 167 ms, with 30 sno stimulation period occurring after the first and second theta trains. The initial fEPSP slope was used for synaptic strength measurements, and the average of the last 15 min of stimulation before dosing or LTP was used as the baseline.
Multielectrode array (MEA) recordings
Primary cortical cultures (E18 rat or P0 mouse) were plated onto 48-well MEA plates (Axion Biosystems, USA). Multiunit action potential rates were determined by filtering the data using an IIR filter (low cutoff 300 Hz; high cutoff 3000 Hz) and applying a 2 × RMS threshold (Neuroexplore, USA). To determine the dose of JB2, 21 DIV primary cortical neurons were analyzed 1 hr after JB2 administration for field potential recordings by 48-well MEAs (Maestro, Axion biosystems). Dendritic growth in 96-well plates was measured by high content confocal microscopy (ImageXpress, molecular devices). Stimulating a central electrode(100 μA, 100 μs) of each array produced single shock-induced field potentials, and potentials were recorded from the remaining 15 electrodes per well. The initial field potential slope after the stimulation artifact was used for quantification. Previously it has been shown that the initial slope of the field potential primarily reflects post-synaptic AMPAR activation in primary neurons plated onto an MEA device, with approximately 80% of this effect being blocked by the AMPAR antagonist NBQX [23]. In our preparation, we found that >90% of this initial field potential slope was NBQX sensitive (Supplementary Fig. 1f). To measure the mean firing rate, electrodes with a response of more than 0.5 Hz were considered for the analysis. The dendritic length was quantified by MX6 software (molecular devices).
Measurement of JB2 concentration in cerebrospinal fluid (CSF)
In order to assess the blood-brain barrier permeability, we dosed JB2 at the therapeutic dose level (1 mg/kg; intravenous) in rats. 1 h after dosing, we anesthetized the animals with isoflurane and obtained blood-free CSF samples from the cisterna magna. Samples were protein precipitated by the addition of 3 volumes of acetonitrile and clarified by certification. JB2 levels were analyzed via LC/MS/MS and all samples were within the linear range (R^2 > .99) of the standard curve.
Electroencephalogram (EEG) and electromyogram (EMG) tests
Adult male mice were implanted with EEG and EMG electrodes (Pinnacle) and allowed at least 1 week to recover before testing, as described previously [24]. Electrical stimulation (1 mA, 100 us; AMPI, Israel) was applied to the medial prefrontal cortex (mPFC) scull screw (neck EMG ground), and field excitatory postsynaptic potentials (fEPSPs) between the ipsilateral mPFC scull screw ground and a cerebellar skull screw (A-M instruments) were recorded at 10,000 samples per second. Data were acquired using a data wave interface (USA) and analyzed using BrainVision software (Brain Products, Germany). For field potential studies, single-shock (10 μA, 100 μS) induced field potentials from the ipsilateral mPFC electrode were recorded and the initial slope after the stimulation artifact was quantified. For pre-ictal threshold studies, the electrical stimulation intensity (100 μS/pulse at 6 Hz) was increased until pre-ictal behaviors, such as head or limb twitching, were observed. At this point, the stimulation was immediately terminated, and the final stimulation intensity was noted by an experimenter blinded to drug conditions that were randomized using a Latin square design across test days.
Testing consisted of standard (6 kHz) or deviant (8 kHz) tone pips (50 ms duration) and an interstimulus interval of 350 ms, presented in a pseudorandom manner, with at least 2 standards occurring in a row. Tones were presented at 85 dB using a speaker (Avisoft, Germany) placed on the floor of the cage. One trial consisted of 1800 standard tones and 200 deviant tones (total, 2000 tones). Auditory LTP was induced by a high-frequency train (3 × 10 Hz for 5 min; 3000 tones) of auditory stimuli (6 kHz, 50-ms duration, 85 dB), and auditory LTP was measured by comparing the N1B amplitude to the standard tone of the mismatch negativity task pre vs. 1 h post-high-frequency stimulation. Auditory steady-state was induced by 40 Hz tone pips (0.1 ms in duration, 20 pips per 500 ms) with a 2-second inter-trial interval (total of 50 tones). Lastly, qEEG was measured for 3 min while the animal was alone in the test cage (resting, 3 min), as during subsequent social interaction with a conspecific (active, 3 min).
In vivo studies in mice
JB2 or a 0.09% sterile saline vehicle was administered to 2-to-8-month-old mice 1 h before testing, via subcutaneous injection at a volume of 10 ml/kg, for behavioral investigations. Observers were blind to which mice had been treated with which drug.
Motor tests.
Grip strength: force (grams) required to remove animals from the cage grid top while the experimenter gripped the animals at the base of the tall using the thumb and finger of their dominant hand, using a scale placed below the cage.
Paw slip assay: animals were placed on a grid, which was moved to a vertical position (~90 degrees to the floor). Animals were observed for paw slips while traversing the grid for 30 seconds.
Motor learning assay: Animals were placed on the grid, and the grid was rapidly inverted. The latency (seconds) for the animal to climb to the top of the grid was noted (i.e. righting reflex).
Y-maze.
Y-maze spontaneous alternations tasks were performed similarly to a previous description [25]. Briefly, 1-hr post-dosing, mice were placed in the Y-maze and allowed to enter any of the three arms for 7 min. The order of arm entries was recorded and analyzed for spontaneous alternation. An alternation was scored for each set of three consecutive choices when no repeated entries occurred. Dividing the number of successive three-arm alternations by the maximum number of arm entries within 7 min yielded the percentage of spontaneous alternations. The chance level of alternation is 50%. This task requires working memory, as animals must maintain and update a mental log of recent arm entries.
NOR test.
NOR testing was conducted as described in a previous work [26].
Ultrasonic vocalization.
Ultrasonic vocalizations were quantified from 30 min home cage recordings of male and female mouse pairs using Avisoft (Germany) recording and analysis equipment as described in our previous paper [25].
Blinding and statistical analysis
Data from spine morphology and density analysis in pyramidal neurons (in vitro) were examined under blinded conditions (coverslip identity hidden and raw data pooled during quantification). MEA and MS experiments we not blinded but were analyzed using an experimenter-independent automatic computer algorithm. Whole animal drug studies were conducted blindly with the data analysis occurring before the blind code was broken. Visibly healthy cells were analyzed as previously described [27]. No animals were excluded from the analysis and no method of animal randomization was employed. All statistical tests were performed with GraphPad Prism9. A two-sample comparison was performed using Student’s t-test or two-tailed non-parametric Spearman correlation, and multiple comparisons were made using one-way ANOVA followed by a Bonferroni test. Bar graphs were displayed as mean ± SEM.
RESULTS
Generation and characterization of IGFBP2-derived JB2 peptide
IGFBP2 has been shown to promote dendritic spine growth and to improve depression and anxiety-related phenotypes in rats [16]. However, the relatively large size of the protein (~36 kDa) makes its direct use as a therapeutic agent difficult. Hence, we generated a series of peptide fragments to mimic its biological activity. The peptide fragment JB2, 6 amino acids (molecular weight of ~700 daltons), showed the greatest potency and activity in vivo using the rat-forced swim test [16]. We, therefore, reasoned based on our previously published paper showing the IGFBP2 produced and antidepressant-like effect in the Porsolt test as at the same doses that induced structural plasticity [16], that JB2 might also show might have general pro-plasticity activity.
To characterize the modulation of neuronal functional properties by JB2, we treated rat primary cortical neurons cultured on MEA plates with JB2 or the vehicle for 1 h. We then analyzed single shock-induced field potentials. Incubation with JB2 highly increased the initial field potential slope, indicating robust excitatory field potentiation (Fig. 1a). To determine the mechanisms underlying this potentiation, we used a set of pharmacological inhibitors. The IGF-1R antagonist, GSK1838705A, did not alter the JB2-dependent field potential slope, indicating independence of IGF1R. However, an anti-IGF2R antibody, but not IgG, fully inhibited the field potential slope increase, indicating dependence on IGF2R. JB2-dependent potentiation was also inhibited by mitogen-activated protein kinase (MEK) and extracellular signal-regulated kinase (ERK), but not the mammalian target of rapamycin (mTOR) inhibitors, indicating that JB2 signaled through the MAP kinase pathway. We examined the signal intensity of phosphor-ERK or phosphor-AKT, which is downstream of the mTOR signaling pathway, in rat primary cultured cortical neurons using immunocytochemistry. As a result, the intensity of phosphor-ERK was significantly increased by JB2 treatment, but not phosphor-AKT (Supplementary Fig. 1a–d). Finally, JB2-dependent potentiation was inhibited by transcription and translation inhibitors, indicating the requirement for new protein synthesis. JB2-dependent field potentiation was dose-dependent and was inhibited by the AMPA receptor antagonist, NBQX, indicating dependence on AMPA receptors (supplementary Fig. 1e). On the contrary, NMDA receptor antagonist, AP5, did not inhibit JB2-dependent field potentiation (supplementary Fig. 1f).
Fig. 1. JB2-induced field potential slope increase depends on IGF2R, MAPK/ERK pathway, and transcription and translation.
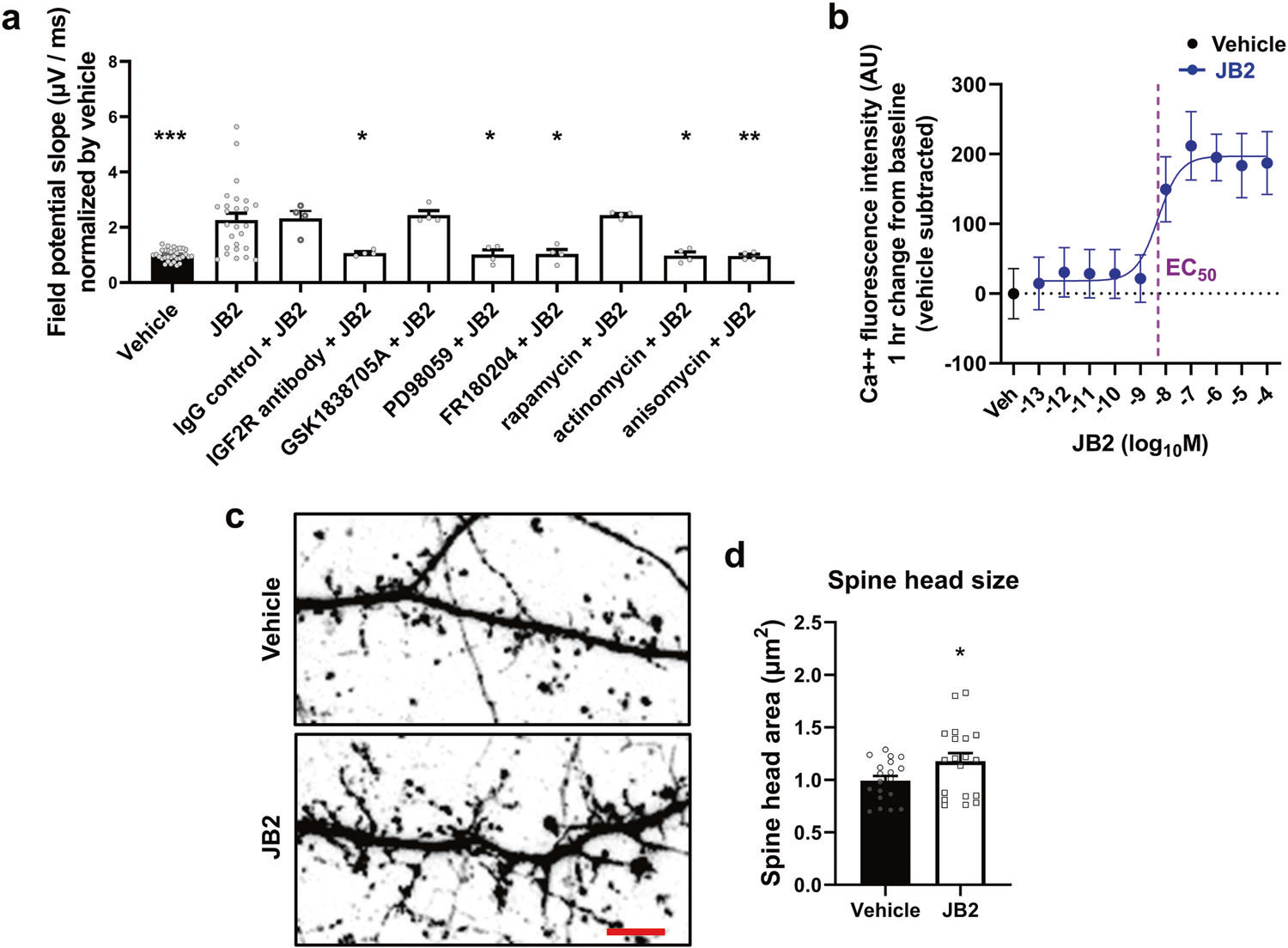
a Rat cortical culture (21 DIV) was examined using MEAs, and single shock-induced field potentials were measured. JB2 (1 μM) produces an enhancement in the excitatory field potential 1 h after dosing. Co-treatment with the IGF2R blocking antibody (1 μg/ml) was shown to reduce IGF2-induced spine growth blocked excitatory field potentials, whereas the IgG control (1 μg/ml) and the IGF1R antagonist, GSK1838705A (10 μM), failed to alter field potentials. Field potential effects were blocked with both MEK (PD98059; 10 μM) and ERK (FR180204; 10 μM) inhibitors, but not an mTOR (rapamycin; 10 μM) inhibitor. All drugs were added 5 min before JB2. Lastly, the field potential effects were both transcription (Actinomycin; 10 μM; 15 min before JB2) and translation (anisomycin; 10 μM; 15 min before JB2) dependent. Individual primary cultures were performed at least 4 times for each condition. F(9, 81) = 8.199; one-way analysis of variance was followed by Bonferroni post hoc tests. *p < 0.05; **p < 0.01; ***p < 0.001 vs. the JB2-alone group. b JB2 enhanced intracellular calcium levels in a dose-dependent manner in primary cultured rat cortical neurons. c Confocal images of neurons transfected with eGFP to observe the spine head size and morphologies between vehicle- and JB2-treated cultured mouse cortical neurons. Scale bar, 5 μm. d Spine head size from mushroom spines was analyzed from eGFP transfected neurons. t(36) = 2.058. *p = 0.047 vs vehicle; two-tailed Student’s t-test was performed. See also Supplementary Fig. 1.
To directly assess the activity of JB2 through the IGF2R, we treated primary cultured cortical rat neurons with JB2 (1 μM, 1 h post-dosing) and measured intracellular calcium using Flow8 as a readout for IGF2R activation (Fig. 1b). JB2 increased intracellular calcium in cultures and we calculated an EC50 of 5 nM for the cellular response to JB2 by fitting the data to a log (agonist) vs. response (three parameters) function. This result was confirmed by overexpressing IGF2R in HEK293 cells. The IGF2R-expressing HEK293 cells exhibited 11.3 pmol of IGF2R/mg as measured by Elisa, whereas the mock-transfected HEK293 cells had IGF2R levels that were below the threshold of quantification of the assay (~3 fmol/mg). JB2 increased intracellular calcium in expressing IGFR2 but not in mock-transfected HEK293 cells suggesting that the IGF2R was activated by JB2 (Supplementary Fig. 1g).
Because IGFBP2 promoted the enlargement of dendritic spines [16], we compared dendritic spine morphology in vehicle or JB2-treated rat cultured cortical neurons expressing eGFP (Fig. 1c). Following confocal imaging, we performed morphological classification of dendritic spines based on geometrical characteristics of spines [19]. JB2 treatment resulted in a significant enlargement in spine head size (Fig. 1d) and dendrite growth (Supplementary Fig. 1h), but did not alter total spine density (Supplementary Fig. 1i).
JB2 promotes the remodeling of the neuronal synaptic membrane phosphoproteome
To uncover the molecular cascades downstream of JB2 in neurons, we treated rat primary cortical cultured neurons with JB2 and isolated the P2 fraction containing synaptosomes and membrane-associated proteins. To analyze the relative changes of phosphorylation by the treatment of JB2, metal oxide affinity chromatography with TiO2 beads was performed. A phosphoproteome analysis using mass spectrometry (MS) (Fig. 2a) identified a total of 4,055 phosphoproteins (Supplementary Table 1), of which 358 had increased phosphorylation and 252 had reduced phosphorylation as compared to the vehicle control group (Fig. 2b). We then performed functional enrichment analysis on the significantly phosphorylated- or dephosphorylated-protein datasets using Gene Ontology (GO) tests. We found that the phosphorylated protein dataset was significantly enriched in cellular components including postsynaptic density (PSD), cytoplasm, neuron projection, microtubules, and axons (Fig. 2c). Biological processes enriched in the phosphorylated dataset included cytoskeleton organization, neuron projection, GTPase regulation, and axon extension, while biological processes enriched in the dephosphorylated dataset similarly included cytoskeleton organization, neuron projection, cell adhesion, GTPase activity, and axon extension (Supplementary Fig. 2a). In another GO database, SynGO, process and organization of synapse and synaptic vesicle cycle were confirmed in both groups (Supplementary Fig. 2b). We performed Western blotting on phosphor-CaMKII (tyrosine 231), which was phosphorylated by JB2 treatment, and phosphor-β-Catenin (Ser675), which was dephosphorylated (supplementary Fig. 3b). The results showed the same trend as phosphoproteomic outcomes by mass spectrometry.
Fig. 2. JB2 regulates the phosphorylation of postsynaptic autism risk factors in primary cortical cultured rat neurons.
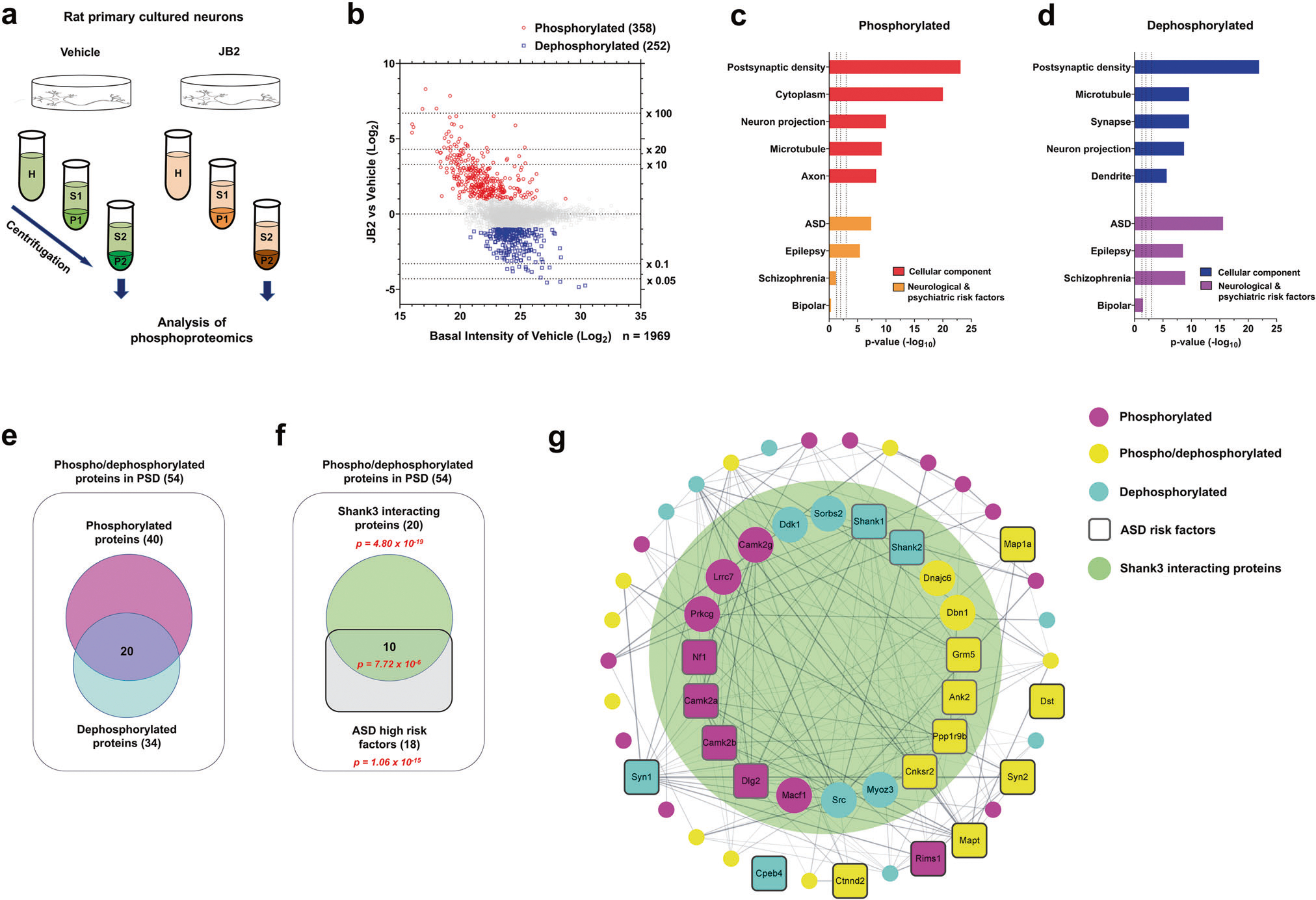
a Scheme of the experimental workflow of the phosphoproteomic analysis. b Scatter plot showing the fold change of individual phosphorylated or dephosphorylated protein levels, after JB2 treatment, and the vehicle. Phosphorylated proteins are in red (fold change ≥ 2), dephosphorylated proteins are in blue (fold change ≤0.5), and all other proteins are in gray (0.5 < fold change < 2). c, d Gene ontology (GO) analysis of statistically overrepresented biological processes among the differentially phosphorylated (in red) or dephosphorylated (in blue) proteins. The analysis was performed from data in b by DAVID. Enrichment of ASD, schizophrenia (SZ), epilepsy, and bipolar disorder (BD) risk factors (identified through SFARI gene archive, GWAS, and de novo studies) among phosphorylated (in orange) and dephosphorylated (in purple) proteins. Hypergeometry test. e Diagram showing the number of either phosphorylated or dephosphorylated proteins present in the postsynaptic density. f Diagram showing the number of either Shank3 interacting proteins or ASD high-risk factors (right panel). Significance (p-value) was demonstrated by hypergeometric tests. g Cytoscape analysis of PSD proteins with phosphor-/dephosphorylated proteins coded by color. Rounded rectangle nodes indicate ASD risk factors. Edges indicate predicted protein-protein interaction, including experimental data from the STRING database. The range of the big green circle indicates the Shank3 interacting proteins. See also supplementary Fig. 2–4.
To investigate the links between NDDs and phosphoproteome alterations induced by JB2 treatment, we assessed the enrichment of phospho- or dephosphoproteome datasets in disorder-specific gene sets (Fig. 2c, d). Intriguingly, we found that both datasets were enriched in NDD risk factors, with the most significant enrichment in ASD risk factors, followed by epilepsy, and schizophrenia. However, the phosphorylated data shows little to no enrichment in bipolar disorder gene sets. Using DAVID, we found a significant enrichment of pre- and postsynaptic proteins in both the phosphorylated protein dataset (n = 40/358) and dephosphorylated protein dataset (n = 34/252) (Fig. 2e; Supplementary Fig. 2a).
Notably, among the 54 phosphorylated/dephosphorylated PSD proteins, there was a statistically significant enrichment in Shank3-interacting proteins and ASD risk factors, as well as Shank3-interacting ASD risk factors (Fig. 2f). This indicates that JB2 treatment induces the remodeling of the phosphorylation state of a protein-protein interaction network associated with both Shank3 and ASD (Fig. 2g). Intriguingly, dephosphorylated serine 1287 in human Shank1 (matched with serine 1291 of rat Shank1) is close to Gly1316Asp reported as a missense variant in ASD patients [28]. Dephosphorylated serine 1104 in human Shank2 (matched with Ser729 of rat Shank2) is also close to the Thr1127Ile missense variant observed in an ASD and mental retardation patient [29] (Supplementary Fig. 4).
Taken together, our data suggest that JB2 induces extensive remodeling of molecular pathways important for the organization of the synaptic cytoskeleton, and it could regulate phosphorylation or dephosphorylation of NDD-related proteins. Furthermore, our data support the potential utility of JB2 in altering protein networks and biological functions associated with Shank3 deficiency.
JB2 rescues neuroplasticity impairments in Shank3+/− mice
The Shank3 gene is extensively implicated in PMS and ASD and functions as a scaffolding protein in the postsynaptic density of excitatory neurons [30, 31]. Because our phosphoproteomic analysis of JB2-treated primary cortical cultured rat neurons showed a significant association to Shank3-related protein networks, and SHANK3 deletion or mutations identified in ASD lead to altered synaptic and circuit function, we hypothesized that JB2 might improve synaptic and circuit phenotypes relevant to PMS in mouse models.
First, to determine whether exogenous JB2 was able to target dendrites and synapses in the context of ex-vivo preparations, we applied Cy3-tagged JB2 to acute slices of cingulate cortex generated from mice sparsely expressing eGFP in a subset of pyramidal neurons. Confocal imaging showed that JB2-Cy3 was localized in clusters on the dendritic shaft and spine heads of the pyramidal neurons (Fig. 3a–c; supplementary Fig. 5a–c). We also applied Cy3-tagged JB2 or a control IgG-Cy3 to live acute coronal brain slices of the layer 2/3 somatosensory cortex in ACSF. After fixation and immunostaining for the presynaptic marker VGLUT1 and the postsynaptic marker PSD95, slices were imaged in confocal microscopy and stacks were deconvoluted to increase the precision of the colocalization analysis. Automatic 3D iterative thresholding was used to unbiasedly segment Cy3, PSD, and VGLUT1 elements and the 3D colocalization was performed using the DiAna Toolbox [21]. This analysis showed that JB2-Cy3 colocalizes significantly more with pre and postsynaptic markers than the control IgG-Cy3 (Fig. 3e).
Fig. 3. JB2 rescues synaptic circuit impairments in adult male Shank3+/− mice.

a Representative confocal image of the JB2-Cy3 binding to the dendritic compartment of a layer 2/3 pyramidal neurons from a Thy1-YFP mouse fixed slice and 3D rendering of JB2 on a spine head. b, c Quantification of the percentage of JB2-Cy3 puncta on the dendritic localization of 7 neurons from 3 mice per each group, 2–3 neurons per animal. F(2, 18) = 157.0. d Representative images of the JB2-Cy3 and control IgG-Cy3 (Red) staining and the immunolabeling of PSD95 (Green) and VGLUT1 (Gray) in acute cortical slices acquired in the layer 2/3 of the somatosensory cortex. e Quantification shows significantly higher percentage of JB2-Cy3 colocalizing with synaptic markers compared to the control protein, each dot represents an individual slice (n = 7–9 slices per condition from 4 mice). F(1, 28) = 5.743. Two-way ANOVA, post-hoc Bonferroni test: ***p < 0.001, *p < 0.05. Scale bars: 2 microns f Single shock-induced field potential slopes, which primarily reflects post-synaptic AMPA receptor activity. F(2, 17) = 15.46. g Single shock induced field potential slopes, which primarily reflect presynaptic excitatory glutamate receptor activity. F(2, 17) = 18.43. h Auditory LTP impaired in Shank3+/− mice, suggesting impaired NMDAR function. F(2, 24) = 19.75. i 40 Hz Auditory steady-state response of EEG F (2, 22) = 17.67. One-way analysis of variance was followed by Bonferroni post hoc tests. *p < 0.05; **p < 0.01; ***p < 0.001 vs wild-type vehicle and ##p < 0.01; ###p < 0.001 vs Shank3+/− vehicle. All data are represented as mean ± SEM. See also supplementary Fig. 5.
To determine the effect of JB2 treatment on the spine development of Shank3+/− neurons, the dendritic spines were observed after 1 h of JB2 treatment on mouse primary cultured cortical neurons. As a result, JB2 showed an increase in spine head size in both wild-type and Shank3+/− neurons, and no change in spine density (supplementary Fig. 5e, f). In addition, a significant increase in field potential was observed by JB2 treatment in both wild-type and Shank3+/− neurons (supplementary Fig. 5h).
Next, we analyzed the effect of JB2 on a set of functional phenotypes in PMS model mice. Drapeau et al. generated a mouse model in which all Shank3 isoforms are disrupted (Shank3Δ4-22) [9]. These mice recapitulate core symptoms of PMS, including deficits in learning and memory, sensory and motor functions, and stereotypic behaviors, thus have strong construct and face validity; so we used them in our analyses (referred to as Shank3+/− mice). To determine the CSF concentration of JB2, its level was analyzed through LC/MS/MS 1 h after treatment of 1 mg/kg JB2 by i.v. injection. This dose level produced CSF drug levels of 7 ± 0.6 nM (n = 3) 1 h after dosing. The EC50 for JB2 in vitro assays from primary cultured neurons was approximately 5 nM to induce an increase of Ca2+ fluorescence intensity. Hence the optimal in vivo dose used was 1 mg/kg.
Transcranial focal stimulation over the mPFC by means of a skull screw was used to create a shock-induced field potential in freely behaving animals. The slope of the initial field potential was measured after a single 100 μs shock. This slope decreased in shank3 mice, consistent with the previous literature [32], and it was rescued with JB2, suggesting a role of postsynaptic AMPAR function [33] (Fig. 3f). The ratio of the initial field potential slope after each of two 100 μs shocks separated by 50 ms was measured. This paired-pulse, which measures presynaptic glutamate release probability, increased in Shank3+/− mice, consistent with the data [32], although the literature is mixed; JB2 had no effect on this facilitation, suggesting that JB2 does not affect presynaptic release (Fig. 3g). In reduced preparations these potentials have been shown to primarily reflect post-synaptic AMPAR activity and presynaptic glutamate release respectively [7, 33, 34]. Given that transcranial focal electrical stimulation or single/paired-pulse transcranial magnetic stimulation can be used in a non-invasive way in humans, similar translational studies could be performed in PMS patients and normal volunteers.
Auditory LTP is a translational, noninvasive method for measuring synaptic plasticity via LTP in mice, measuring the difference in the N1B amplitude of the auditory evoked response before and 1 h after high-frequency auditory tetanus. We have previously shown that this response is increased by positive NMDAR modulation, and NMDAR agonists have been shown to eliminate this response [24, 35]. We found a deficit in auditory LTP in Shank3+/− mice which were rescued by JB2 (Fig. 3h).
The auditory steady-state click stimulation paradigm has been consistently shown to reduce entrainment as measured by electroencephalography (EEG) in patients [36]. This translational non-invasive method measures the entrainment of the brain at gamma frequency (~40 Hz) to a tone presented 40 times per second, and the response has been shown to be NMDAR dependent [37]. The 40-Hz Auditory Steady-State Response (ASSR) has been proposed as a biomarker of genetic defects in the SHANK3 gene [38]. Notably, this paradigm can also be used in mice. We found a deficit in auditory steady-state response in Shank3+/− mice which were rescued by JB2 (Fig. 3i).
JB2 rescues behavioral impairments in Shank3+/− mice
Because JB2 improved synaptic and circuit dysfunction in Shank3+/− mice, we hypothesized that it may also improve motor and behavioral deficits in these mice. Motor impairments are often present in PMS and ASD [31, 39–41]. Consistent with these clinical data, Shank3+/− mice showed an abnormal motor phenotype as measured by an increased number of paw slips while traversing a wire grid (Fig. 4a), increased righting latency (Fig. 4b), and reduced grip strength (Fig. 4c). JB2 (1 mg/kg, SC) reversed each of these phenotypes.
Fig. 4. JB2 rescues behavioral impairments in adult male Shank3+/− mice.
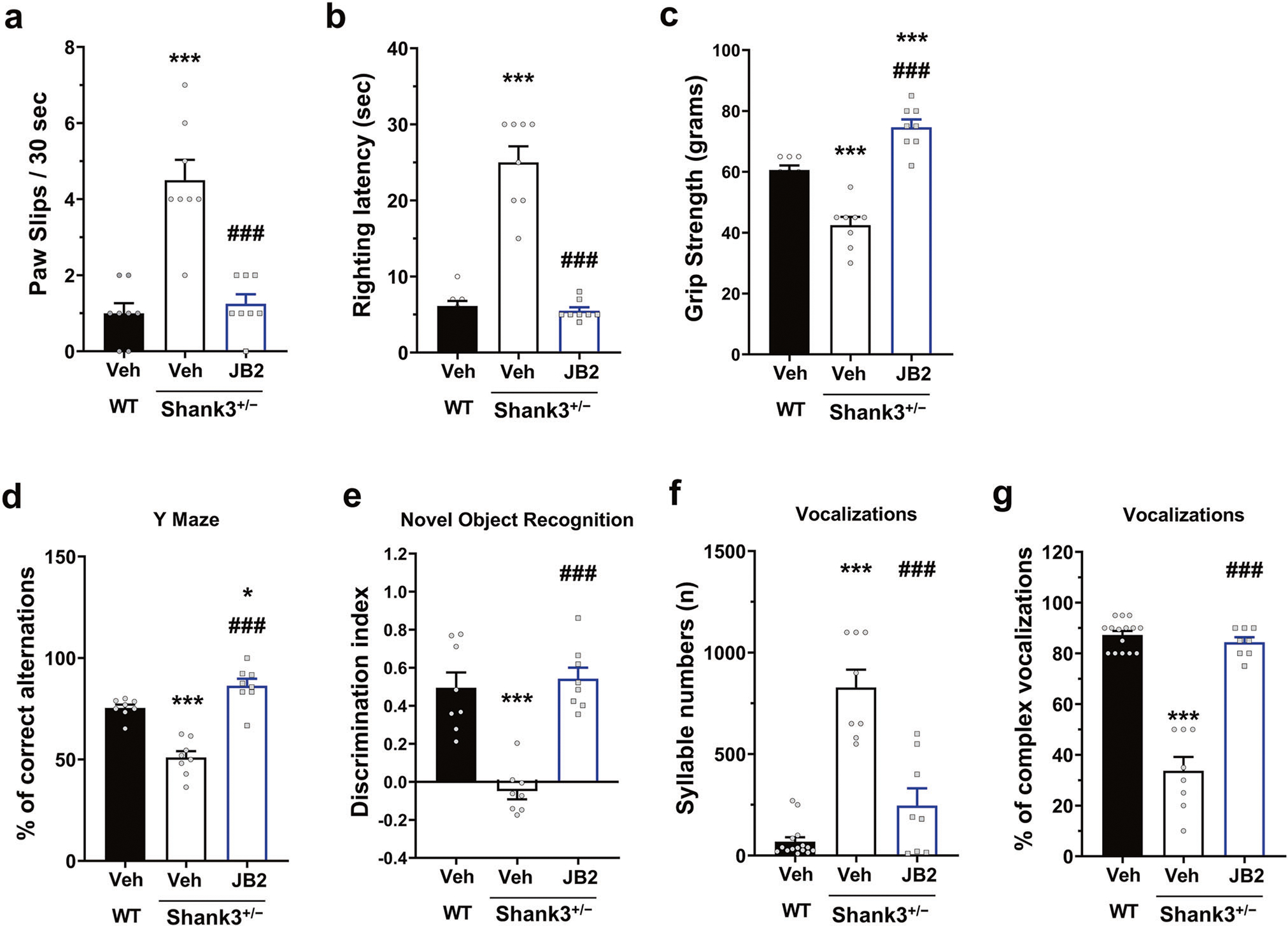
a–e JB2 rescued wide variety deficits in adult male Shank3+/− mice in the behavioral domains. (a) Paw slips F (2, 28) = 195.2, (b) latency to righting F (2, 28) = 490.3 on a wire grid, and c grip strength F (2, 28) = 30.67. d Y-maze F (2, 28) = 21.62 and (e) NOR learning F (2, 25) = 31.93. The discrimination index (DI) [(time spent exploring the novel object—time spent exploring the familiar object)/total exploration time] was then calculated for retention trials. f Syllable numbers of adult male mice. F (2, 28) = 46.01. g Complex ultrasonic vocalizations of adult male mice. F (2, 28) = 97.84. One-way analysis of variance was followed by Bonferroni post hoc tests. *p < 0.05; ***p < 0.001 vs wild-type vehicle and ###p < 0.001 vs Shank3+/− vehicle. All data are represented as mean ± SEM. See also supplementary Fig. 6.
ID is highly common among patients with PMS and with mutations in SHANK3 [31, 39–41]. Therefore, we tested the effect of JB2 on cognitive deficits in Shank3+/− mice. Cognitive deficits were observed in Shank3+/− mice during spontaneous, alternating Y-maze tasks (Fig. 4d) and novel object recognition (Fig. 4e) both of which were rescued with JB2. Importantly, we did not observe gender differences in the behavioral effects of JB2 in Shank3+/− mice, as female mice showed comparable deficits and drug rescue to male mice (Supplementary Fig. 7).
Abnormalities in vocal and communication abilities are common in both PMS and ASD [31, 39–41]. Consistent with this, the syllable numbers and bandwidth/spectral complexity of ultrasonic calls were impaired in Shank3+/− male mice (Fig. 4f and g), but no change of syllable duration and energy (data not shown). The monotonous nature of ultrasonic calls in Shank3+/− mice may model speech deficits in PMS. Notably, JB2 rescued the syllable numbers and complexity of vocalizations in these mice.
JB2 rescues translational endpoints in Shank3+/− mice
Seizures and epilepsy occur commonly in patients with PMS as well as ASD [31, 39–41]. Consequently, we examined the effects of JB2 on neuronal excitability and seizure susceptibility in Shank3+/− neurons and mice. A previous study reported that Shank3-deficient neurons were hyperexcitable due to impaired hyperpolarization-activated cation (Ih) currents [42]. Consistent with this, MEA recordings from mouse primary cortical cultures showed that Shank3+/− neurons exhibited a higher basal firing rate as compared to wild-type neurons (Fig. 5a). While JB2 slightly increased the firing rate of wild-type neurons, it attenuated the hyperexcitability of Shank3+/− neurons. This suggested that JB2 may improve seizure susceptibility in Shank3+/− mice. To test this, we surgically implanted mPFC skull screws on adult male wild-type and Shank3+/− mice, injected with either the vehicle or JB2, and tested them for theta train-induced seizure thresholds. We determined the effects of JB2 or LTP on fEPSP of wild-type mice after 1 hr or 24 h, and the results showed that the significant increase in fEPSP was maintained at both 1 hr and 24 h (supplementary Fig. 8). JB2 increased the pre-ictal threshold in both wild-type and Shank3+/− mice, rescuing the seizure susceptibility in mutant mice (Fig. 5b). This suggests that JB2 may have anti-seizure properties.
Fig. 5. JB2 produced therapeutic-like effects in Shank3+/− mice using direct mouse-to-human non-invasive translational measures.
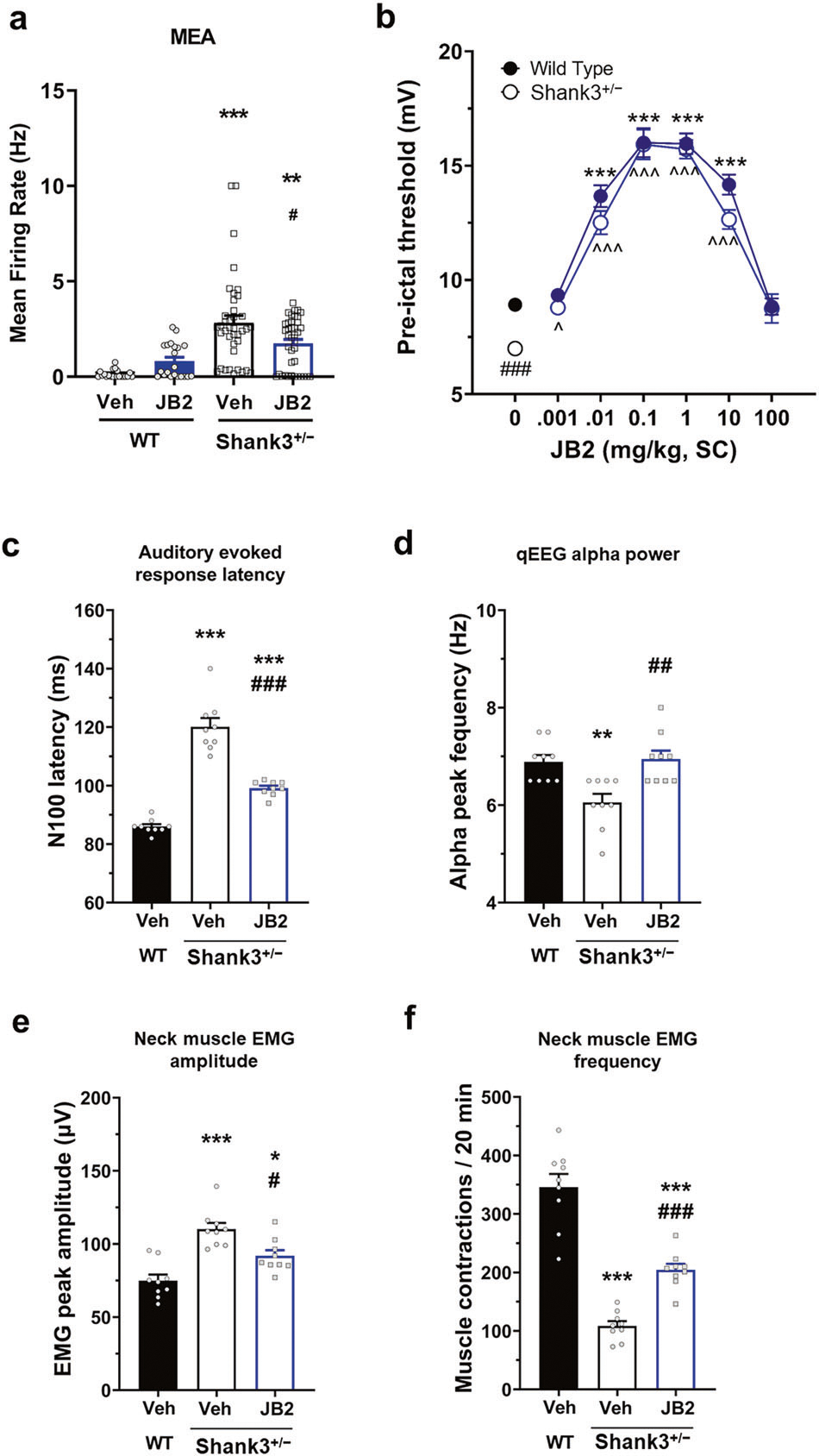
a Firing rate of cortical neurons at DIV28 from an MEA plate. F(3, 116) = 15.39. b Adult male mice with surgically implanted mPFC skull screws were tested for theta train-induced seizure thresholds F (1, 118) = 10.104. Two-way analysis of variance was followed by Bonferroni post hoc tests. c N100 latencies in response to an auditory tone F (2, 24) = 86.45. d Alpha peak frequency F (2, 24) = 9.181. e, f Adult male mice with surgically implanted neck EMG electrodes (cerebellar reference) were tested for theta train-induced seizure thresholds. Amplitude (e) F (2, 24) = 18.42 and frequency (f) F (2, 24) = 62.81 were measured. One-way analysis of variance was followed by Bonferroni post hoc tests. *p < 0.05; **p < 0.01; ***p < 0.001 vs wild-type vehicle and #p < 0.05; ##p < 0.01; ###p < 0.001 vs Shank3+/− vehicle. All data are represented as mean ± SEM.
To facilitate the development of novel therapeutics, it is important to assess mouse phenotypes that have direct translational value to human patients. Hence, we determined whether such phenotypes were present in the Shank3+/− mice and if they could be rescued with JB2. Patients with autism show increased latency of auditory evoked potentials and a slower alpha peak frequency when measured with EEG [43–45]. Consistent with this, we detected deficits in N100 latencies and alpha peak frequency in Shank3+/− mice (Fig. 5c, d). Shank3+/− mice also showed high amplitude and irregular spontaneous electromyographic activity (Fig. 5e, f) recorded from the neck muscle that may be similar to the myoclonic jerks reported in PMS patients [46]. JB2 reduced the impairment in Shank3+/− mice.
DISCUSSION
Here we show that the IGFBP2-derived peptide JB2 is a novel modulator of neuroplasticity with unique properties. While a major function of IGFBPs is to modulate IGF1 and -2 bioavailability and function through IGF1R, the effects of JB2 are distinct from IGF1, which acts through IGF1R. JB2 likely corresponds to one active site of IGFBP2. Indeed, our data support a mechanism of action through IGF2R. Our inhibitor studies indicate that JB2 activates the MEK/ERK signaling pathway, consistent with growth factor-like properties. The plasticity-promoting effects of JB2 on neurons are transcription and translation-dependent, which are also consistent with growth factor-like properties. More importantly, our findings indicate that JB2 required IGF2R activation in neurons and induced IGF2R-dependent calcium influx in cells. As such, JB2 can be employed as a tool to mimic the IGF1 and IGF1R-independent functions of IGFBP2.
In wild-type neurons, JB2 promotes dendritic spine head size increase, but not density changes, consistent with the spine-promoting effects of IGFBP2 [16]. In the wild-type rat neurons, JB2 induces large-scale, bidirectional global remodeling of the synaptic phosphoproteome. These changes in phosphorylation converge on synaptic and cytoskeletal proteins, many of which are encoded by ASD risk genes. Remarkably, JB2 binds directly to dendritic and synaptic regions in mouse cortical slices. Our global phosphoproteomic investigations also pointed to Shank3 deficits as being potentially rescuable by JB2.
So far, studies have been conducted on the mechanisms and treatment of various drugs to restore synaptic deficits and ASD-like behaviors in PMS models. IGF1 treatment has been reported to reverse not only a decrease in fEPSP in Shank3+/− mice [47], but also a decrease in excitatory synaptic transmission in PMS patient-derived neurons [48]. The IGF signaling pathway is known to be involved in intracellular protein synthesis and cell division and growth through phosphorylation of Akt, phosphoinositide 3-kinase, MEK, and ERK [49]. A decrease in Akt T308/S473 phosphorylation was observed in PMS iPSC-derived patient neurons, and it was due to enhanced activation of protein phosphatase 2 mediated by Cdc2-like kinase 2 (CLK2) [50]. CLK2 inhibitor, TG003, could recover not only structural and electro-physiological synaptic deficits, but also social motivation impairment in Shank3Δ21 mice. In Shank3Δ11 mice, another PMS model, the metabotropic glutamate receptor subtype 5 (mGluR5) was reduced in postsynapse of cortical neurons [51]. Treatment with mGluR5 activator, 3-Cyano-N-(1,3-diphenyl-1H-pyrazol-5-yl)benzamide, restored the decreased intracellular release of calcium in Shank3Δ21 neurons and rescued ASD-like behaviors. We may suggest that JB2 could be regarded as a novel treatment approach through the MEK and ERK pathway initiated from IGF2R, which is separated from the previously reported pathways for drugs.
Importantly, JB2 rescued a broad range of deficits in PMS model mice across multiple motor, cognitive and sensory domains that capture the core deficits of the disorder. We chose to investigate the Shank3Δ4-22 mouse line with a complete disruption of the Shank3 gene, because this model more closely replicates human PMS. Shank3Δ4-22 mice were reported to show stereotypies, sensory alterations, motor deficits, and some learning deficits, but no social deficits [9]. In addition to those, we discovered novel neurophysiological and translational phenotypes relevant for PMS and ASD, including alterations in postsynaptic dendritic strength, auditory LTP, auditory steady-state response, neuronal hyperexcitability, seizure susceptibility, N100 latencies, alpha peak frequency, as well as motor evoked potentials, which have not yet been shown in Shank3 mutant mice, but have direct relevance to autism patient clinical phenotypes [52]. These are particularly interesting and relevant for the pathogenesis of PMS, and provide a pathophysiological link between the genetic alterations and the behavioral phenotypes. Importantly, JB2 rescued synaptic and circuit function, motor behaviors, sensory processing, learning, and memory, as well as communication behavior. Synaptic dysfunction is extensively implicated in ASD [2], and deficits in synaptic strength are likely to underlie the deficits in Shank3+/− mice. JB2’s therapeutic effects are likely due to enhanced synaptic strength, which is a cellular mechanism. This is the first use of IGFBP-derived peptides as a potential therapeutic.
Currently, there are no pharmacological treatments for core neurodevelopmental symptoms of PMS or ASD. Oxytocin, AMO-01, balovaptan (a vasopressin 1a receptor antagonist), and IGF1 are now in clinical trials for PMS, but already have challenges in being used as long-term therapies due to their side effects and age range limitations. Of these, IGF1 has been extensively investigated in PMS [53, 54]. By contrast, JB2 acts through a different mechanism and improves a broad range of synaptic, circuit, motor, and behavioral phenotypes in mice, several of which are directly translatable to human subjects. For these reasons, it could serve as the basis for a disease-modifying pharmacological treatment by reversing core neurodevelopmental phenotypes of PMS.
Supplementary Material
ACKNOWLEDGEMENTS
This work was supported by R41MH121140 to PP. Proteomics services were performed by the Northwestern Proteomics Core Facility, generously supported by NCI CCSG P30 CA060553 awarded to the Robert H Lurie Comprehensive Cancer Center, instrumentation award (S10OD025194) from NIH Office of Director, and the National Resource for Translational and Developmental Proteomics supported by P41 GM108569. We also thank NU Nikon Cell Imaging Facility. We would also like to thank Joseph Buxbaum for generously providing the Shank3 mice and Paulina Rychenkova for her knowledge and insight into Phelan-McDermid Syndrome.
Footnotes
COMPETING INTERESTS
JSB is a full-time employee of Gate Neurosciences Inc., (Mount Carmel IN, USA). Northwestern University has the rights to the JB2 with pending patents.
ADDITIONAL INFORMATION
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41380-022-01904-0.
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
REFERENCES
- 1.Gao R, Penzes P. Common mechanisms of excitatory and inhibitory imbalance in schizophrenia and autism spectrum disorders. Curr Mol Med. 2015;15:146–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Forrest MP, Parnell E, Penzes P. Dendritic structural plasticity and neuropsychiatric disease. Nat Rev Neurosci. 2018;19:215–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Buescher AVS, Cidav Z, Knapp M, Mandell DS. Costs of autism spectrum disorders in the United Kingdom and the United States. JAMA Pediatr. 2014;168:721–8. [DOI] [PubMed] [Google Scholar]
- 4.Lavelle TA, Weinstein MC, Newhouse JP, Munir K, Kuhlthau KA, Prosser LA. Economic burden of childhood autism spectrum disorders. Pediatrics. 2014;133:e520–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Voineagu I, Wang XC, Johnston P, Lowe JK, Tian Y, Horvath S, et al. Transcriptomic analysis of autistic brain reveals convergent molecular pathology. Nature. 2011;474:380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Spooren W, Lindemann L, Ghosh A, Santarelli L. Synapse dysfunction in autism: a molecular medicine approach to drug discovery in neurodevelopmental disorders. Trends Pharm Sci. 2012;33:669–84. [DOI] [PubMed] [Google Scholar]
- 7.Bourgeron T From the genetic architecture to synaptic plasticity in autism spectrum disorder. Nat Rev Neurosci. 2015;16:551–63. [DOI] [PubMed] [Google Scholar]
- 8.Kolevzon A, Angarita B, Bush L, Wang AT, Frank Y, Yang A, et al. Phelan-McDermid syndrome: a review of the literature and practice parameters for medical assessment and monitoring. J Neurodev Disord. 2014;6:39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Drapeau E, Riad M, Kajiwara Y, Buxbaum JD. Behavioral phenotyping of an improved mouse model of phelan-mcdermid syndrome with a complete deletion of the Shank3 Gene. eNeuro 2018;5:e0046–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Durand CM, Betancur C, Boeckers TM, Bockmann J, Chaste P, Fauchereau F, et al. Mutations in the gene encoding the synaptic scaffolding protein SHANK3 are associated with autism spectrum disorders. Nat Genet. 2007;39:25–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Soorya L, Kolevzon A, Zweifach J, Lim T, Dobry Y, Schwartz L, et al. Prospective investigation of autism and genotype-phenotype correlations in 22q13 deletion syndrome and SHANK3 deficiency. Mol Autism. 2013;4:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cochoy DM, Kolevzon A, Kajiwara Y, Schoen M, Pascual-Lucas M, Lurie S, et al. Phenotypic and functional analysis of SHANK3 stop mutations identified in individuals with ASD and/or ID. Mol Autism. 2015;6:23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.De Rubeis S, Siper PM, Durkin A, Weissman J, Muratet F, Halpern D, et al. Delineation of the genetic and clinical spectrum of Phelan-McDermid syndrome caused by SHANK3 point mutations. Mol Autism 2018;9:31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Khan S IGFBP-2 signaling in the brain: from brain development to higher order brain functions. Front Endocrinol. 2019;10:822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lewitt MS, Boyd GW. The role of insulin-like growth factors and insulin-like growth factor-binding proteins in the nervous system. Biochem Insights. 2019;12:1178626419842176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Burgdorf J, Colechio EM, Ghoreishi-Haack N, Gross AL, Rex CS, Zhang XL, et al. IGFBP2 produces rapid-acting and long-lasting effects in rat models of post-traumatic stress disorder via a novel mechanism associated with structural plasticity. Int J Neuropsychoph. 2017;20:476–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Khan S, Lu X, Huang Q, Tang J, Weng J, Yang Z, et al. IGFBP2 plays an essential role in cognitive development during early life. Adv Sci. 2019;6:1901152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Smith KR, Kopeikina KJ, Fawcett-Patel JM, Leaderbrand K, Gao RQ, Schurmann B, et al. Psychiatric risk factor ANK3/Ankyrin-G nanodomains regulate the structure and function of glutamatergic synapses. Neuron. 2014;84:399–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yoon S, Myczek K, Penzes P. cAMP signaling-mediated phosphorylation of diacylglycerol lipase alpha regulates interaction with Ankyrin-G and dendritic spine morphology. Biol Psychiatry. 2021;90:263–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yoon S, Piguel NH, Khalatyan N, Dionisio LE, Savas JN, Penzes P. Homer1 promotes dendritic spine growth through ankyrin-G and its loss reshapes the synaptic proteome. Mol Psychiatry. 2021;26:1775–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Gilles JF, Dos Santos M, Boudier T, Bolte S, Heck N. DiAna, an ImageJ tool for object-based 3D co-localization and distance analysis. Methods. 2017;115:55–64. [DOI] [PubMed] [Google Scholar]
- 22.Clapp WC, Kirk IJ, Hamm JP, Shepherd D, Teyler TJ. Induction of LTP in the human auditory cortex by sensory stimulation. Eur J Neurosci. 2005;22:1135–40. [DOI] [PubMed] [Google Scholar]
- 23.van Vliet E, Stoppini L, Balestrino M, Eskes C, Griesinger C, Sobanski T, et al. Electrophysiological recording of re-aggregating brain cell cultures on multi-electrode arrays to detect acute neurotoxic effects. Neurotoxicology. 2007;28:1136–46. [DOI] [PubMed] [Google Scholar]
- 24.Burgdorf JS, Christian EP, Sorensen L, Stanton PK, Leaderbrand K, Madsen TM, et al. A translational EEG-based approach to assess modulation of long-lasting NMDAR-dependent synaptic plasticity. Psychopharmacology. 2019;236:3687–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Srivastava DP, Jones KA, Woolfrey KM, Burgdorf J, Russell TA, Kalmbach A, et al. Social, communication, and cortical structural impairments in Epac2-deficient mice. J Neurosci. 2012;32:11864–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rajagopal L, Burgdorf JS, Moskal JR, Meltzer HY. GLYX-13 (rapastinel) ameliorates subchronic phencyclidine- and ketamine-induced declarative memory deficits in mice. Behav Brain Res. 2016;299:105–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yoon S, Penzes P. A fluorescence recovery after photobleaching protocol to measure surface diffusion of DAGLalpha in primary cultured cortical mouse neurons. STAR Protoc. 2022;3:101118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sato D, Lionel AC, Leblond CS, Prasad A, Pinto D, Walker S, et al. SHANK1 deletions in males with autism spectrum disorder. Am J Hum Genet. 2012;90:879–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Berkel S, Marshall CR, Weiss B, Howe J, Roeth R, Moog U, et al. Mutations in the SHANK2 synaptic scaffolding gene in autism spectrum disorder and mental retardation. Nat Genet. 2010;42:489–91. [DOI] [PubMed] [Google Scholar]
- 30.Durand CM, Perroy J, Loll F, Perrais D, Fagni L, Bourgeron T, et al. SHANK3 mutations identified in autism lead to modification of dendritic spine morphology via an actin-dependent mechanism. Mol Psychiatry. 2012;17:71–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Harony-Nicolas H, De Rubeis S, Kolevzon A, Buxbaum JD. Phelan McDermid Syndrome: from genetic discoveries to animal models and treatment. J Child Neurol. 2015;30:1861–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Arons MH, Thynne CJ, Grabrucker AM, Li D, Schoen M, Cheyne JE, et al. Autism-associated mutations in ProSAP2/Shank3 impair synaptic transmission and neurexin-neuroligin-mediated transsynaptic signaling. J Neurosci. 2012;32:14966–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Malenka RC, Bear MF. LTP and LTD: an embarrassment of riches. Neuron. 2004;44:5–21. [DOI] [PubMed] [Google Scholar]
- 34.Jackman SL, Regehr WG. The mechanisms and functions of synaptic facilitation. Neuron. 2017;94:447–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Clapp WC, Eckert MJ, Teyler TJ, Abraham WC. Rapid visual stimulation induces N-methyl-D-aspartate receptor-dependent sensory long-term potentiation in the rat cortex. Neuroreport. 2006;17:511–5. [DOI] [PubMed] [Google Scholar]
- 36.Sivarao DV, Chen P, Senapati A, Yang Y, Fernandes A, Benitex Y, et al. 40 Hz auditory steady-state response is a pharmacodynamic biomarker for cortical NMDA receptors. Neuropsychopharmacology. 2016;41:2232–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sivarao DV. The 40-Hz auditory steady-state response: a selective biomarker for cortical NMDA function. Ann N Y Acad Sci. 2015;1344:27–36. [DOI] [PubMed] [Google Scholar]
- 38.Neklyudova AK, Portnova GV, Rebreikina AB, Voinova VY, Vorsanova SG, Iourov IY, et al. 40-Hz Auditory steady-state response (ASSR) as a biomarker of genetic defects in the SHANK3 gene: a case report of 15-year-old girl with a rare partial SHANK3 Duplication. Int J Mol Sci. 2021;22:1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Frank Y The neurological manifestations of Phelan-McDermid Syndrome. Pediatr Neurol. 2021;122:59–64. [DOI] [PubMed] [Google Scholar]
- 40.Vogels A, Droogmans G, Vergaelen E, Van Buggenhout G, Swillen A. Recent developments in Phelan-McDermid syndrome research: an update on cognitive development, communication and psychiatric disorders. Curr Opin Psychiatry. 2021;34:118–22. [DOI] [PubMed] [Google Scholar]
- 41.Levy T, Foss-Feig JH, Betancur C, Siper PM, Trelles-Thorne MDP, Halpern D, et al. Strong evidence for genotype-phenotype correlations in Phelan-McDermid syndrome: results from the developmental synaptopathies consortium. Hum Mol Genet. 2022;31:625–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yi F, Danko T, Botelho SC, Patzke C, Pak C, Wernig M, et al. Autism-associated SHANK3 haploinsufficiency causes Ih channelopathy in human neurons. Science. 2016;352:aaf2669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bomba MD, Pang EW. Cortical auditory evoked potentials in autism: a review. Int J Psychophysiol. 2004;53:161–9. [DOI] [PubMed] [Google Scholar]
- 44.Dickinson A, DiStefano C, Senturk D, Jeste SS. Peak alpha frequency is a neural marker of cognitive function across the autism spectrum. Eur J Neurosci. 2018;47:643–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pillion JP, Boatman-Reich D, Gordon B. Auditory brainstem pathology in autism spectrum disorder: a review. Cogn Behav Neurol. 2018;31:53–78. [DOI] [PubMed] [Google Scholar]
- 46.Kurtas N, Arrigoni F, Errichiello E, Zucca C, Maghini C, D’Angelo MG, et al. Chromothripsis and ring chromosome 22: a paradigm of genomic complexity in the Phelan-McDermid syndrome (22q13 deletion syndrome). J Med Genet. 2018;55:269–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bozdagi O, Tavassoli T, Buxbaum JD. Insulin-like growth factor-1 rescues synaptic and motor deficits in a mouse model of autism and developmental delay. Mol Autism. 2013;4:9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Shcheglovitov A, Shcheglovitova O, Yazawa M, Portmann T, Shu R, Sebastiano V, et al. SHANK3 and IGF1 restore synaptic deficits in neurons from 22q13 deletion syndrome patients. Nature. 2013;503:267–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hakuno F, Takahashi SI. IGF1 receptor signaling pathways. J Mol Endocrinol. 2018;61:T69–T86. [DOI] [PubMed] [Google Scholar]
- 50.Bidinosti M, Botta P, Kruttner S, Proenca CC, Stoehr N, Bernhard M, et al. CLK2 inhibition ameliorates autistic features associated with SHANK3 deficiency. Science. 2016;351:1199–203. [DOI] [PubMed] [Google Scholar]
- 51.Vicidomini C, Ponzoni L, Lim D, Schmeisser MJ, Reim D, Morello N, et al. Pharmacological enhancement of mGlu5 receptors rescues behavioral deficits in SHANK3 knock-out mice. Mol Psychiatry. 2017;22:784. [DOI] [PubMed] [Google Scholar]
- 52.Wang J, Barstein J, Ethridge LE, Mosconi MW, Takarae Y, Sweeney JA. Resting state EEG abnormalities in autism spectrum disorders. J Neurodev Disord. 2013;5:24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Costales J, Kolevzon A. The therapeutic potential of insulin-like growth factor-1 in central nervous system disorders. Neurosci Biobehav Rev. 2016;63:207–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Vahdatpour C, Dyer AH, Tropea D. Insulin-like growth Factor 1 and related compounds in the treatment of childhood-onset neurodevelopmental disorders. Front Neurosci. 2016;10:450. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.