

Abstract
Introduction
Inflammatory myofibroblastic tumours are rare neoplasms which most commonly affect children and young adults. With an intermediate malignant potential, they are typically detected in the abdomen, lung, mediastinum, head and neck, gastrointestinal tract, and genitourinary tract.
Case description
We describe the case of a 33-year-old postpartum woman incidentally diagnosed with a pulmonary inflammatory myofibroblastic tumour following complaints of poorly controlled hypertension a week after caesarean section. She was ALK-negative and received an ALK inhibitor with complete resolution of the lesion. A ROS1–TFG fusion confirmed the diagnosis of an inflammatory myofibroblastic tumour after CT-guided fine needle aspiration.
Discussion
This case highlights an uncommon presentation posing a diagnostic and therapeutic challenge and the potential treatment option of crizotinib.
LEARNING POINTS
Inflammatory myofibroblastic tumour (IMT) is a rarely reported neoplasm arising in the abdominal soft tissues, the lung, mediastinum, head, neck, gastrointestinal tract, and genitourinary tract, with intermediate malignant potential.
IMT is definitively diagnosed only after histological examination following surgical biopsy, based on immunohistochemical markers and the molecular characteristics of the tumour, but small biopsies may have a role in a large lesion.
IMT did not cause any complications during gestation.
Therapeutic approaches include surgical resection and chemotherapy, including with crizotinib, an ALK tyrosine kinase inhibitor.
Keywords: Pulmonary inflammatory myofibroblastic tumor, hypertension
INTRODUCTION
An inflammatory myofibroblastic tumour (IMT) is a rarely metastasizing neoplasm composed of myofibroblastic and fibroblastic spindle cells accompanied by an inflammatory infiltrate of plasma cells, lymphocytes and/or eosinophils[1]. IMTs were first described in 1905 in orbital tissue. They were initially labelled ‘pseudo-tumours’ and have also been referred to as pseudo-sarcomatous myofibroblastic or fibromyxoid lesions, plasma cell granuloma benign myofibroblastoma, and inflammatory fibrosarcoma[2]. IMTs are rare, occur in less than one in 1 million people, and account for only 0.04–0.1% of all pulmonary neoplasms[3]. IMTs can occur in all age groups, but more than 50% of patients are below 40 years of age[4]. Half of IMTs have rearrangements involving the anaplastic lymphoma kinase (ALK) receptor, which are associated with localized tumours and better prognosis compared with ALK-negative IMTs. ROS1 is a receptor tyrosine kinase (RTK) structurally similar to ALK. ROS1 fusions, mainly YWHAE–ROS1 and TFG–ROS1, are present in about 10% of IMTs and half of ALK-negative IMT patients[5]. They are challenging to diagnose and can spread, invade or even reoccur before diagnosis. The prognosis is typically excellent if the tumour can be completely resected[3]. However, very little data is available on the prognosis of ALK-negative IMTs or on the role of ROS1 as a driver and potential therapeutic target[5]. We report the clinical case of a 33-year-old woman who was incidentally found to have an 8 cm mass postpartum with a ROS1 rearrangement and who had an extraordinary response to crizotinib.
CASE DESCRIPTION
A 33-year-old woman underwent a caesarean section for pre-labour rupture of membranes and non-reassuring fetal status at 37 weeks and 6 days; the fetus had lost variability and developed decelerations. One week later, the patient presented with complaints of hypertension, palpitations and headache, which had begun the day before. She had no significant past medical history, was a lifelong non-smoker and denied consumption of alcohol or drugs. She denied chest pain, chest pressure, cough, shortness of breath, fever, chills, abdominal pain, nausea, vomiting or diarrhoea. She was afebrile and had a heart rate of 66 bpm, a respiratory rate of 20, blood pressure of 158/91 mmHg, and oxygen saturation of 100% on room air. Her physical examination was unremarkable. Laboratory work-up revealed a haemoglobin of 9.3 g/dl, haematocrit 29, and platelets 689; her troponin was negative. A routine chest x-ray revealed a large right apical mass. CT of the chest without contrast showed a 7.6×7.8×7 cm right multilobulated and heterogeneous enhancing mass, which extended to the superior pleura from the right suprahilar region (Fig.1).
Figure 1.

Axial image of the right-sided 7.6×7.8×7 cm multilobulated mass
There was direct extension into the adjacent superior mediastinal fat on the right. There was cut-off of the right upper lobe bronchus and soft tissue thickening around the right middle and lower lobe bronchus suspicious for metastatic right hilar lymph node enlargement (Fig. 2).
Figure 2.

Coronal image of the right-sided 7.6×7.8×7 cm multilobulated mass
The patient had a positron emission tomography (PET) scan which showed uptake within the large right upper lobe mass with a maximum standardized uptake value of 15.7 with no hypermetabolic axillary, mediastinal, hilar or subcarinal lymph nodes (Fig. 3). No other hypermetabolic pulmonary nodules were seen.
Figure 3.

Positron emission tomography (PET) scan of the right upper lobe mass with a maximum standardized uptake value of 15.7
CT-guided fine needle aspiration (FNA) biopsy of the lung lesion showed low-grade spindle cell proliferation and dense plasmacytic infiltrate of uncertain significance (Fig. 4).
Figure 4.

High-power view showing mixed inflammatory cells and low-grade spindle cells (H&E 400×)
This lesion comprised low-grade spindle and admixed inflammatory cells with prominent plasmacytic components. Mitoses were not identified, and necrosis was not seen. The lesion was positive for smooth muscle actin (SMA), and negative for ALK, desmin, pancytokeratins, p40, thyroid transcription factor 1 (TTF-1), and CD34 (Fig. 5).
Figure 5.

Immunochemistry for smooth muscle actin (SMA) shows the tumour cells are positive (SMA 200×)
On initial review, the lesion was thought to be an IMT. However, as ALK was negative, the diagnosis was uncertain, and a second opinion was sought. Next-generation sequencing (NGS) showed ROS1–TFG fusion, helping to confirm the diagnosis of an IMT. Due to the size and extension of the tumour, surgery would require a pneumonectomy, so NGS was submitted, and neoadjuvant chemotherapy with six cycles of doxorubicin and ifosfamide was planned. Due to the ROS1 oncogene and the stability of a repeat CT of the chest, crizotinib was started, and the patient was referred to cardiothoracic surgery for surgical resection after completion of induction therapy. The 3-month follow-up CT scan showed a significantly smaller tumour, and therapy was continued. Three months later, during a restaging examination, scans revealed a stable apical tumour and bilateral lung nodules. Re-biopsy was advised after cardiothoracic surgery. The transbronchial biopsy results, brushings and bronchoalveolar lavage were cancer-free (Fig. 6). A second PET scan revealed a complete response to treatment with resolution of the right upper lobe mass and lingering right upper lobe atelectasis. The patient continued to take crizotinib while under constant observation.
Figure 6.
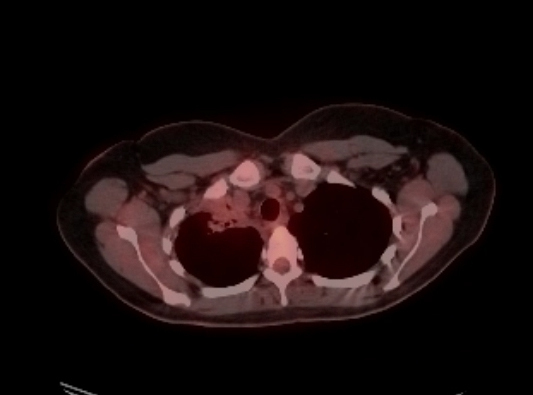
Complete response to therapy. Resolution of the right upper lobe mass with residual total right upper lobe atelectasis
DISCUSSION
IMTs of the lung consist of myofibroblastic spindle cells with inflammatory infiltration of plasma cells, lymphocytes and eosinophils[2]. The pathogenesis of IMTs is unknown, but they are associated with pulmonary tuberculosis, pseudomonas lung infection, Moraxella catarrhalis, actinomycetes, mycoplasma, mycobacteria, Epstein–Barr virus, and human herpesvirus 8 (HHV-8) as well as autoimmune diseases like Sjogren’s and IGG4[2]. ALK is apparent in approximately 40–50% of IMTs, and mutations in ROS1 RTK can be present in cases of ALK-negative ttumours. ROS1 rearrangements occur in approximately 1% of patients with NSCLC[1]. Of the estimated 1.5 million new cases of NSCLC worldwide each year, approximately 15,000 may be driven by oncogenic ROS1 fusions. As with ALK rearrangements, ROS1 rearrangements are more commonly found in patients who have never smoked or have a history of light smoking and who have histological features of adenocarcinoma. However, at the genetic level, ALK and ROS1 rearrangements rarely occur in the same tumour[6].
Clinical features of IMT are mainly determined by the site of origin[1]. Pulmonary IMT is sometimes associated with pleuritic pain, dyspnoea, cough, haemoptysis, or recurrent pneumonia. However, up to one-third of IMT patients exhibit a clinical condition characterized by fever, malaise, weight loss, microcytic hypochromic anaemia, thrombocytosis, polyclonal hypergammaglobulinaemia, high ESR, and raised C-reactive protein probably driven by cytokines[7,8]. Our patient complained of hypertension, palpitations and headache, most likely due to postpartum hypertension. Pulmonary IMTs occur more frequently in the lower lobes and peripheral lungs as a solitary, circumscribed, lobulated lesion. However, radiological features are variable and can include direct infiltration of airways, calcification, cavitation, and even pleural effusions. Our case was unique as the IMT was central and affected the upper lobe[4]. It is also the first case of IMT in a recently postpartum patient, signifying a relatively benign clinical course for the fetus and the mother during gestation.
Diagnosis of IMTs depends on surgical specimens because of the difficulty in distinguishing them from malignant tumours in small tissue samples obtained from bronchoscopic aspiration or FNA. The diagnosis of IMT can produce false positive and false negative results, and pathology is often reviewed at centres more experienced in diagnosis[4]. We were able to diagnose IMT with CT-guided FNA of the apical lung lesion, possibly indicating a more significant role for small biopsies in large IMTs, especially with the assistance of NGS. There have been rare cases of ALK and ROS-1 negative tumours[2]. NGS may be more appealing in this scenario because it can also detect other gene fusions characteristic for IMT, such as PDGFR, RET, FN1–IGF1R, and other rarer mutations targetable in ALK/ROS1-negative IMTs. Therefore, the molecular assessment of IMTs is crucial in order to choose the best target treatment[5].
Our case was challenging because the patient was negative for ALK. Fortunately, in ALK-negative cases, immunohistochemistry for ROS1 and molecular tests for non-ALK gene fusions are helpful and can confirm the diagnosis. Surgical resection is the recommended treatment, although there have been studies showing response with steroids and metformin for unresectable cases. Crizotinib, an ALK tyrosine kinase inhibitor, is a potential therapy that shows a good response in unresectable tumours with ALK rearrangement and has also been used in ALK-negative ROS-positive patients[2]. In our case, it was initially intended that our patient would undergo neoadjuvant therapy with crizotinib with plans for surgical resection; however, following her excellent response to treatment, it was decided to maintain therapy and continue to monitor. Repeat biopsies were negative, and a repeat PET scan showed complete resolution of the lung mass.
For ALK-negative IMTs, treatment is controversial, with few treatment options. Prognosis is excellent, especially after radical surgical excision, with 5-year survival rates above 91%. In some rare cases, spontaneous resolution has also been observed. However, recurrence has been reported to be 60% in those receiving incomplete resection, and unresectable cases pose a significant challenge[2]. Due to the lack of literature data because of the rarity of the disease, we know very little about the therapeutic response to targeted therapy in negative ALK and ROS1-positive patients. And unfortunately, we have little data on disease-free intervals with ROS1 inhibitors in this population. However, more excellent therapeutic options are becoming available, and other ROS1 inhibitors such as lorlatinib, entrectinib and repotrectinib may be a possible future therapeutic option in ROS1-positive disease.
CONCLUSION
In conclusion, pulmonary IMT is a rare entity that can be extremely challenging to diagnose, with very few options for treatment. We describe the case of a large IMT treated solely with crizotinib, demonstrating that it can be a good option for significant and non-resectable cases. Although prognosis is excellent with early resection, the non-specific course can lead to late diagnosis. We describe the first pulmonary IMT in a young woman who recently underwent a caesarean section.
Footnotes
Conflicts of Interests: The Authors declare that there are no competing interests.
Patient Consent: Not required by the institution.
REFERENCES
- 1.WHO Classification of Tumours Editorial Board. WHO classification of tumours. 5th ed. Vol. 3. Lyon, France: International Agency for Research on Cancer; 2020. Soft tissue and bone tumours. https://publications.iarc.fr/588 . [Google Scholar]
- 2.Khatri A, Agrawal A, Sikachi RR, Mehta D, Sahni S, Meena N. Inflammatory myofibroblastic tumor of the lung. Adv Resp Med. 2018;86:27–35. doi: 10.5603/ARM.2018.0007. [DOI] [PubMed] [Google Scholar]
- 3.Lee YJ, Jeong JS, Kim SR, Lee YC. Inflammatory myofibroblastic tumor of the lung with multifocal metastases. JTO Clin Res Rep. 2020;1:100007. doi: 10.1016/j.jtocrr.2020.100007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang F, Zhang W, Han C, Jiang H. A case of pulmonary inflammatory myofibroblastic tumor treated with bronchoscopic therapy plus lobectomy. J Cardiothorac Surg. 2021;16:144. doi: 10.1186/s13019-021-01528-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Comandini D, Catalano F, Grassi M, Pesola G, Bertulli R, Guadagno A, et al. Outstanding response in a patient with ROS1-rearranged inflammatory myofibroblastic tumor of soft tissues treated with crizotinib: case report. Front Oncol. 2021;11:658327. doi: 10.3389/fonc.2021.658327.ISSN=2234-943X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shaw AT, Ou SH, Bang YJ, Camidge DR, Solomon BJ, Salgia R, et al. Crizotinib in ROS1-rearranged non-small-cell lung cancer. N Engl J Med. 2014;371:1963–1971. doi: 10.1056/NEJMoa1406766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pettinato G, Manivel JC, De Rosa N, Dehner LP. Inflammatory myofibroblastic tumor (plasma cell granuloma). Clinicopathologic study of 20 cases with immunohistochemical and ultrastructural observations. Am J Clin Pathol. 1990;94:538–546. doi: 10.1093/ajcp/94.5.538. [DOI] [PubMed] [Google Scholar]
- 8.Coffin C, Watterson J, Priest J, Dehner L. Extrapulmonary inflammatory myofibroblastic tumor (inflammatory pseudotumor). A clinicopathologic and immunohistochemical study of 84 cases. Am J Surg Pathol. 1995;19:859–872. doi: 10.1097/00000478-199508000-00001. [DOI] [PubMed] [Google Scholar]