

SUMMARY
Bats are distinctive among mammals due to their ability to fly, use laryngeal echolocation, and tolerate viruses. However, there are currently no reliable cellular models for studying bat biology or their response to viral infections. Here, we created induced pluripotent stem cells (iPSCs) from two species of bats: the wild greater horseshoe bat (Rhinolophus ferrumequinum) and the greater mouse-eared bat (Myotis myotis). The iPSCs from both bat species showed similar characteristics and had a gene expression profile resembling that of cells attacked by viruses. They also had a high number of endogenous viral sequences, particularly retroviruses. These results suggest that bats have evolved mechanisms to tolerate a large load of viral sequences and may have a more intertwined relationship with viruses than previously thought. Further study of bat iPSCs and their differentiated progeny will provide insights into bat biology, virus host relationships, and the molecular basis of bats’ special traits.
In brief
Generation of induced pluripotent stem cells from two diverse bat species opens the door to functional studies of bat cell biology, including the question of why they are distinctively able to harbor viruses of importance to human health.
Graphical Abstract
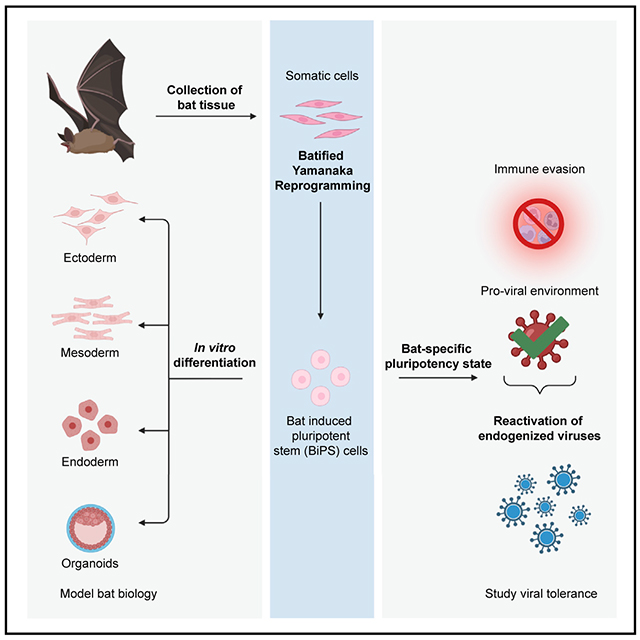
INTRODUCTION
It has been an age-old question: what makes bats so fascinating to humans? Bats account for one-fifth of all living mammalian species (n = 1,456),1 inhabiting diverse ecological niches, feeding on arthropods, fruit, nectar, leaves, fish, blood, and small vertebrates.2–5 They are the only mammals to have evolved true, self-powered flight and can use laryngeal echolocation to orient in complete darkness.4,6 They are found throughout the globe, absent only from extreme polar regions.4,6,7 Many bat species studied exhibit an extremely long lifespan relative to body size and a suspected low tumorigenesis rate.8,9 Still, what makes bats most distinctive is that many species (e.g., rhinolophids, hipposiderids, pteropodids) have been shown to tolerate and survive many viruses that have high mortality rates in humans,10–12 such as SARS-CoV, SARS-CoV-2, MERS-CoV, Marburg, and henipaviruses.12–18 This is potentially due to a modulation of their innate immune response rendering them as asymptomatic and tolerant viral hosts.19–22 Also, despite being among the smallest mammalian genomes, bat genomes contain the highest diversity of ancient and contemporary viral insertions of retroviral and non-retroviral origin,23–27 suggesting that bats have a long and tolerant evolutionary history with their viral pathogens. As some of the integrated retroviral sequences are full length and even of non-bat origin, sequencing bat genomes provides insights into the bat virosphere and the potential for zoonotic spillover15,28–30 but also uncovers mechanisms of viral persistence.26 To date, how bats deal with viruses is still poorly understood, with only a limited number of immune cells documented and characterized in a few bat species (e.g., Pteropus alecto, Eonycteris spelaea, Myotis lucifugus, Eptesicus fuscus).31–33 Further, developing cellular resources and assays is required to uncover and validate the molecular adaptations that have evolved in bats to tolerate these viral pathogens.25,31–33 The prevailing hypothesis supported by recent comparative genomic analyses of multiple bat families34 is that viral tolerance results from specific adaptations of their innate immune system. Accordingly, bats mount an inaugural antiviral reaction after viral inoculation—like all mammals—but then quickly “dampen” this very response before it becomes overly pathological.25,35,36 Critically, this unusual way to deal with viruses could be caused in part by molecular adaptations that stifle canonical virus sensing and the subsequent inflammatory response,18,19,37,38 such as the cGAS-STING, OAS-RNASE L, and NLPR3 systems.39–43
It is striking how closely the aforementioned genomic adaptations to the bat immune system mirror how viruses themselves typically dismantle the host response.44 Yet, viruses do not just use countermeasures against detection and quench inflammation. Viruses are also infinite masters of tweaking cell processes to their advantage to convert host cells into virus-producing factories. Hence, we wondered whether, in addition to the immune evasion strategies of viruses, bats also harbor the blueprints for productive viral replication, given the evolutionary maintenance of many intact and full-length viral elements in one of the smallest mammalian genomes and the potential evolutionary advantage gained from such a symbiosis.45,46
Here, we sought to test empirically the idea that bats genetically simulate the viral ploy for immune evasion and promote notably fertile ground for virus production. We conjectured that pluripotent stem cells would be an ideal experimental system for addressing this question. Given that pluripotent stem cells are the founding cells of the entire embryo, their cellular ground state provides an exclusive reference point for comparative studies, as all mammals must complete this stage in a similar manner.47 Importantly, the global epigenetic resetting that occurs as cells reprogram to pluripotency causes the transcriptional reactivation of endogenous viruses.48–51 As such, it would present a distinctive window into the abundant endogenized viral diversity within bat genomes, allowing the broad cataloging of active viruses and, in turn, the study of how viruses interface with host cell programs.
RESULTS
Bat Yamanaka reprogramming
Given the importance of bats as an emerging model system in multiple areas and the need to study their special biology and potential role in pandemics, we sought to develop an effective strategy to produce bat pluripotent stem cells (Figure 1A). Despite numerous attempts by different groups,36,52 and some initial success in partial reprograming,53 robust pluripotent stem cell lines from bat species with globally repeatable protocols have not been previously reported.
Figure 1. Derivation of pluripotent Rhinolophus ferrumequinum bat stem cells.
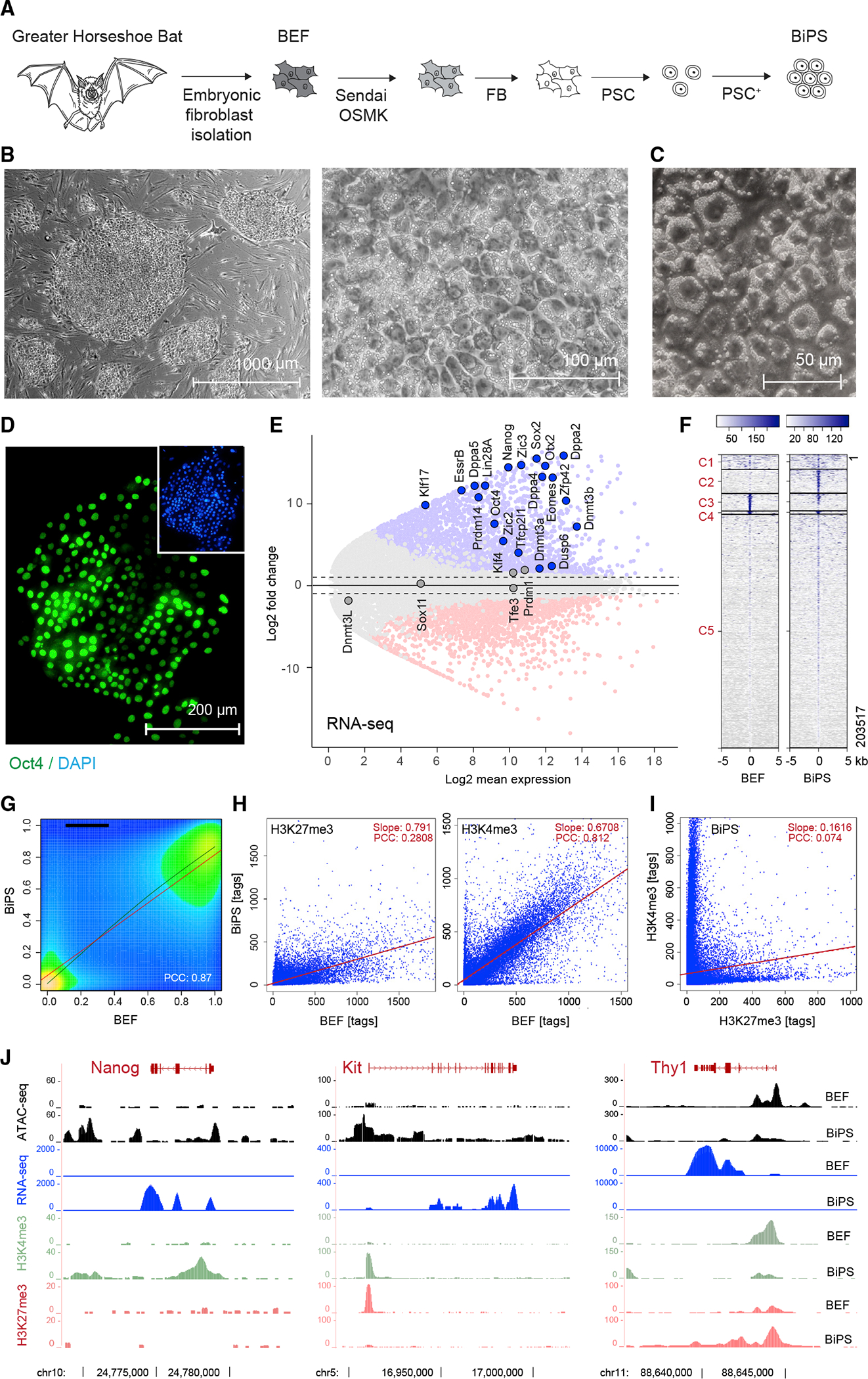
(A) Illustration of the bat pluripotent stem cell derivation strategy. BEF, embryonic fibroblasts; OSMK, Oct4, Sox2, cMyc, Klf4; FB, fibroblast medium; PSC, pluripotent stem cell medium; PSC+, PSC with additives.
(B) Microscopic images of bat pluripotent stem cells at different magnifications showing morphology of established BiPS cell colonies grown on mouse embryonic fibroblasts.
(C) Differential interference contrast microscopy image of BiPS cells highlighting prominent cytoplasmic vesicles.
(D) Immunofluorescent detection of Oct4 in BiPS cells.
(E) MA plot of RNA-seq data illustrating the transcriptional differences between bat embryonic fibroblast (BEF) and pluripotent stem cells (BiPS). Shown is the mean average of each gene from three replicates per cell type (n = 3). Selected genes with known functions in the establishment or maintenance of pluripotency are highlighted.
(F) Kmean cluster analysis of ATAC-seq signals obtained from BEF or BiPS cells. Shown is the representative result of one of two replicates per cell type. C, cluster.
(G) Density plot of RRBS results obtained from BEF and BiPS cells. Shown is the representative result of one of two replicates per cell type. PCC, Pearson correlation coefficient.
(H) Scatter plots of histone 3 methylation status at K4 (activating chromatin modification) or K27 (repressing chromatin modification) after ChIP-seq from BEF or BiPS cells, as indicated. Shown are the results of one sample for each chromatin mark.
(I) Scatter plot correlation of H3K4me3 and H3K27me3 in BiPS cells illustrating the occurrence of bivalent chromatin sites in BiPS cells.
(J) RNA-seq, ATAC-seq, and H3K4me3 or H3K27me3 ChIP-seq signals of selected genes with known roles in reprogramming that are activated (Nanog, Kit) or repressed (Thy1) in BiPS when compared with BEF cells. Shown are tracks of one representative sample.
We first focused on the original Yamanaka reprogramming paradigm, based on four reprogramming factors (Oct4, Sox2, Klf4, and cMyc), because it provides the most direct way to generate pluripotent stem cells in most species.54 As a starting point, we used bat embryonic fibroblast (BEF) cells isolated from wild-caught greater horseshoe bats (Rhinolophus ferrumequinum). Strikingly, the standard protocol55 that is highly effective in mice, and after adjustments in humans and other mammalian species (e.g., domestic dog [Canis familiaris], domestic pig [Sus scrofa], common marmoset [Callithrix jacchus]), failed in bats. Even though the standard reprogramming protocol failed, it provided us with a crucial insight: the Yamanaka factors triggered the formation of rudimentary stem cell-like colonies even though they ceased to expand. This observation prompted us to suspect that the core pluripotency network might be conserved in bats, whereas the signaling cascades that usually shield this network from differentiation cues may differ. We therefore empirically altered the ratios and amounts of the reprogramming factors and, through a combinatorial approach, activated and blocked various cellular signaling pathways to ascertain whether they enabled stem cell reprogramming in bats. To this end, we identified that a specific ratio of reprogramming factors, and the addition of Lif, Scf, the Pka activator forskolin,56 and Fgf2 to the culture medium, allowed for the uninterrupted growth of bat pluripotent stem cells (Figure 1A, detailed in the STAR methods section). Under these adjusted conditions, bat stem cell colonies typically appeared after 14–16 days of culture. These initial stem cell colonies were, however, not readily pickable and expandable using conventional EDTA (Versene)-, collagenase-, or trypsin-based methods that are normally used to passage pluripotent stem cells from other species. The only effective method seemed to be lightly flushing the cells off the feeder cell layer after gentle treatment with low concentrations of EDTA.
Bat pluripotent stem cells
Bat induced pluripotent stem cell (iPSC) colonies appeared tight and homogeneous. The cells had a large, apparent nucleus with one or two prominent nucleoli and were filled with tiny vesicles not seen in other mammalian pluripotent stem cells (Figures 1B, 1C, and S1A). Bat iPS cells expressed the pluripotency factor Oct4 as demonstrated by immunostaining (Figure 1D), and their proliferation rate was similar to human pluripotent cells. The cells retained a normal karyotype, with most cells containing 58 chromosomes (Figure S1B) and replicated in the absence of the exogenous reprogramming factors (Figure S1C), for now more than 100 passages without a change in morphology. RNA sequencing (RNA-seq) analyses (at passage 22) revealed the induced endogenous expression of canonical pluripotency-associated genes such as Oct4, Sox2 and Nanog (Figure 1E; Table S1A).
Next, we checked the effects of our reprogramming approach on the bat chromatin and epigenetic structures. A global epigenetic landscape survey using the assay for transposase-accessible chromatin with sequencing (ATAC-seq) revealed substantial chromatin configuration changes when bat fibroblasts transitioned into the pluripotent state (Figure 1F). Similarly, mapping the DNA methylome by reduced-representation bisulfite sequencing (RRBS) exposed major CpG methylation changes across the genome (Figures 1G, S1D; Tables S1C and S1D) after reprogramming. Finally, chromatin immunoprecipitation sequencing (ChIP-seq) for histone marks associated with active (H3K4me3) and developmentally repressed genes (H3K27me3) showed many changes (Figures 1H, 1I; Table S1E) which were also associated with pluripotency genes (Figures 1J and 2A). Approximately 18.2% of the bat stem cell genes were associated with a “bivalent” domain (H3K4me3 and H3K27me3; Figure S1E; Table S1F), a pluripotency chromatin hallmark initially found in human and mouse pluripotent cells.57 Interestingly, while there was an overlap between human58 and bat bivalency genes there were also some bat- or human-specific genes. Generally, there were strict correlations between an increase in gene expression and newly opened sites showing increased ATAC-seq signals and H3K4 trimethylation, along with decreased H3K27 trimethylation and DNA methylation in their promoters. Conversely, closed regions and gene shutdowns during the reprogramming process also corresponded to the absence of activating marks and presence of histone modifications, respectively (Figures 1J, 2A, and S1F–S1H). However, there were instances when we saw transcription and simultaneously active and repressive epigenetic marks, most likely as a result of spontaneous differentiation in our cultures (Figure 2A). Collectively, our results establish that our BiPS cells are reprogrammed, both transcriptionally and epigenetically. However, closer data inspection revealed that the expression and chromatin modification profiles (Figures 1J, 2A and 2B) did not fully match a single known pluripotency state. Instead, we saw factors indicative of the naive pluripotent state (e.g., Klf4, Klf17, Essrb, Tfcp2l1, Tfe3, Dppa, and Dusp6) expressed alongside genes typically found in the more advanced primed pluripotent cells (e.g., Otx2, Zic2), a phenomenon previously described for human cells under certain culture conditions, as it particularly pertains to lipid metabolism and the expression of Dusp6.59 Indeed, double immunostainings detecting four of the most commonly used primed/naive factors, Otx2/Tfe3 and Tfcp2l1/Zic2, respectively, show the co-expression of naive and primed markers in most cells (Figure 2C). In contrast, germ cell factors such as Dnmt3l and Dazl were absent (Table S1). Thus, while cellular heterogeneity might be at play, their uniform appearance makes it most likely that bat stem cells might correspond to the formative state of pluripotency60 or occupy a different, yet-to-be-characterized pluripotent default state.
Figure 2. Characteristics of pluripotency markers in pluripotent stem cells generated from Rhinolophus ferrumequinum fibroblasts.
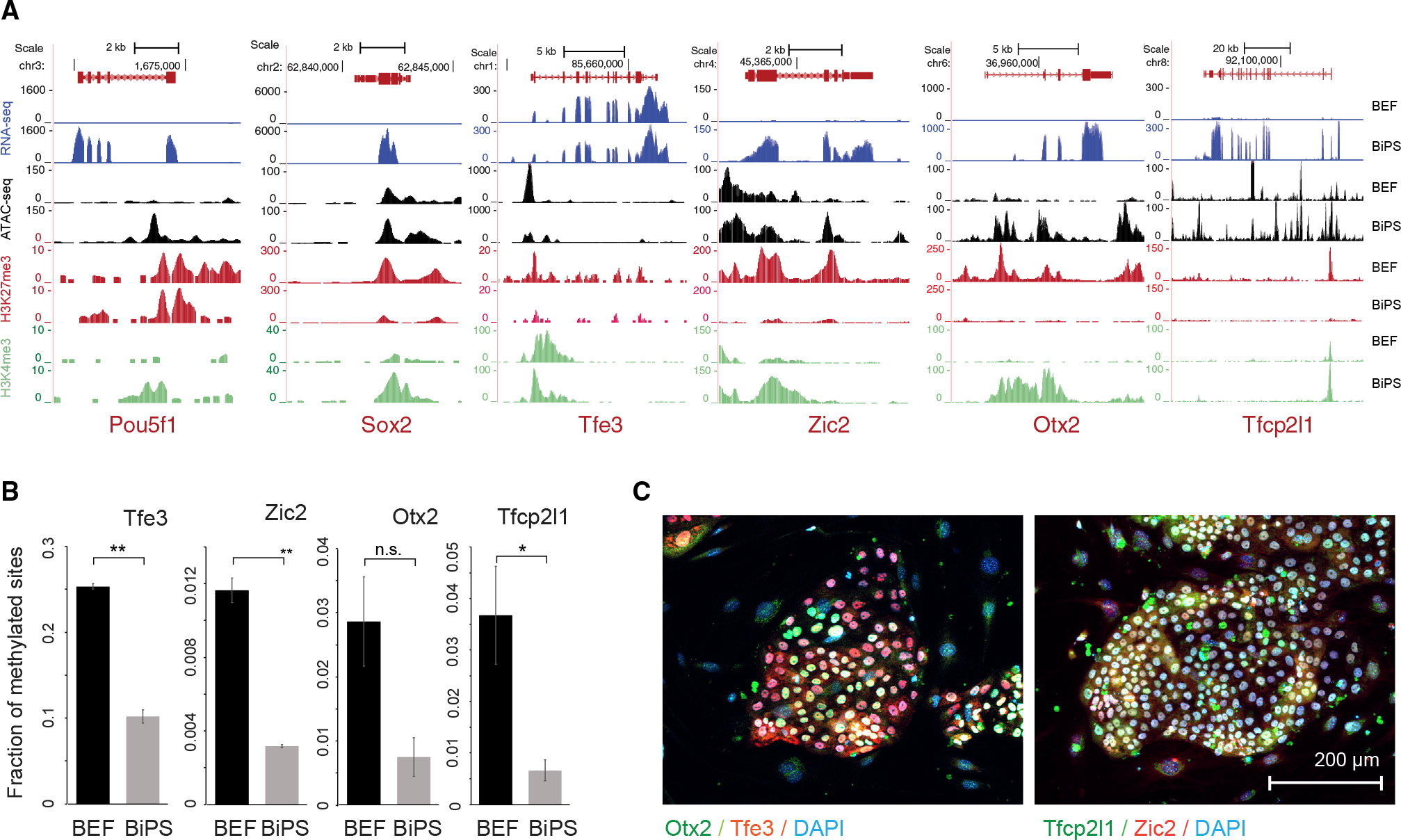
(A) Sequencing tracks showing expression, ATAC-seq signal, histone H3K27 trimethylation (H3K27me3), and histone H3K4 trimethylation (H3K4me3) status of pluripotency markers Oct4 and Sox2 in bat embryonic fibroblasts (BEF) or induced pluripotent stem cells (BiPS).
(B) Fraction of methylated sites in promoters of pluripotency genes that did show promoter methylation. Data are shown as mean ± SD of two replicates; p values were determined by t test: p = 0.0015, 0.0031, 0059, and 0.0481 from left to right. n.s., not significant. Note that we did not detect methylation in the promoters of Nanog, Pou5f1, or Sox2, which might be related to under-annotation of the R. ferrumequinum genome at present.
(C) Immunofluorescence images of bat pluripotent stem cells after staining of markers of naive (Tfe3 and Tfcp2l1) or primed pluripotency (Zic2 and Otx2). See also Table S1.
The transcriptional and epigenetic changes mentioned above suggest, but are not definitive proof of, developmental pluripotency. To obtain functional evidence, we subjected the bat stem cells to protocols optimized for directed differentiation into ectodermal, mesodermal, and endodermal fates (Figure S2A). In each case, the cells responded to the altered culture conditions by shifting their morphology profoundly and turned positive for Pax6 (a marker for ectoderm), T (mesoderm), or AFP (endoderm), respectively (Figure 3A). Because the cells used in this experiment were at an advanced passage (passage 37, an equivalent of about 6 months of continuous culture), the results also suggest that pluripotency can be maintained long term. We next probed the developmental plasticity of bat stem cells by subjecting them to embryoid body (EB) differentiation, another classical in vitro pluripotency assay.61 Again, the BiPS cells (referring to iPS cells derived from Rhinolophus ferrumequinum) differentiated and formed spherical arrangements typical for EBs (Figure S2B), which subsequently matured into elaborate three-dimensional structures positive for all three germ layer markers (Figure 3B). RNA-seq analyses of RNA isolated from the cells following monolayer differentiation and EB formation confirmed the respective cell fate changes (Figures 3C, and S2C and S2D; Table S2).
Figure 3. Differentiation potential of R. ferrumequinum bat pluripotent stem cells.
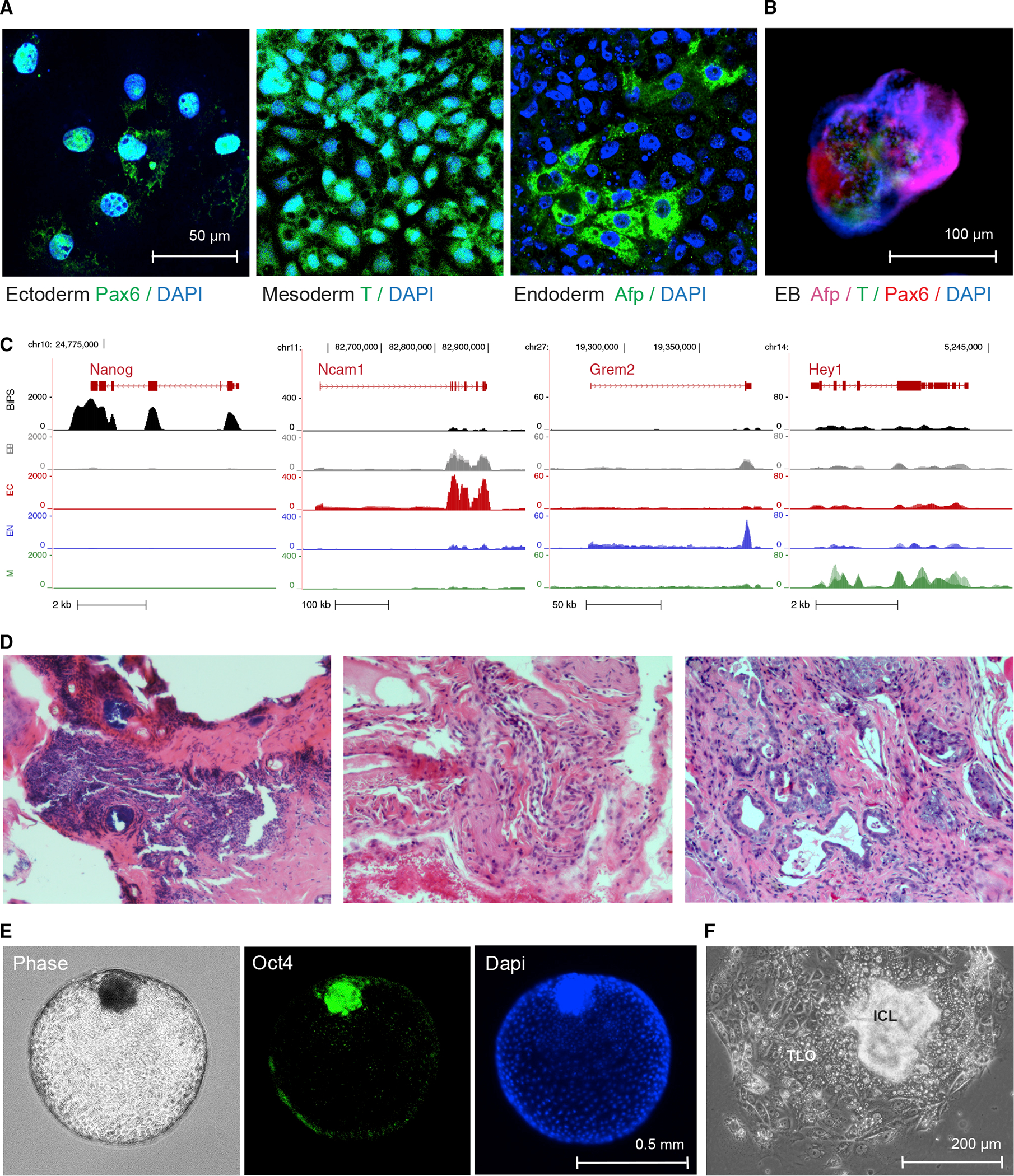
(A) Immunofluorescence microscopy images after staining with antibodies detecting the expression of lineage-specific markers Pax6, Afp, or brachyury (T) following specific directed differentiation into ectoderm, endoderm, or mesoderm, respectively.
(B) Immunofluorescence images of embryonic bodies (EB) that formed after 3D-differentiation of BiPS cells and were stained with antibodies to detect markers specific to all three germ layers as in (A).
(C) RNA-seq signals of selected lineage-specific marker genes in BiPS cells that underwent monolayer differentiation as in (A) or embryonic body differentiation as in (B). Shown is one representative sequencing track (n = 3) per condition. EB, embryonic body differentiation, EC, human ectoderm differentiation protocol; EN, human endoderm differentiation protocol; M, human mesoderm differentiation protocol.
(D) Microscopic images of hematoxylin-eosin-stained sections of tumor tissue after injection of BiPS cells into immunocompromised mice exhibiting ectodermal (left), mesodermal (middle), and endodermal (right) features.
(E) Images of floating blastoids that were obtained from BiPS cells after exposure to Bmp4 to capture their morphology by phase-contrast microscopy (left) and to detect Oct4 expression in inner-cell mass-like cell clusters after immunofluorescence staining (middle, right).
(F) Phase-contrast microscopy image of a typical blastocyst-outgrowth-like cell cluster that formed after the attachment of blastoids to the cell culture vessel surface during Bmp4-induced differentiation as in (E). ICL, inner cell mass-like; TLO, trophoblast-like outgrowth.
Next, we injected the R. ferrumequinum BiPS cells into immunocompromised mice because pluripotent stem cells typically form a particular tumor (teratoma) at the injection site,62 composed of ectodermal, mesodermal, and endodermal cells arranged in a semi-chaotic fashion. Although it took substantially longer than usual for pluripotent cells derived from other species, BiPS cells eventually formed a similar type of tumor after four to five months, albeit infrequently (33%) and relatively small (2–4 mm). The tumors consisted of immature tissue with epithelial, neural, and stromal characteristics (Figure 3D). The transcriptional profile of pivotal genes (Table S1A) previously reported critical for teratoma formation63,64 revealed that while some genes are downregulated in bat iPS cells in comparison with mouse iPS cells (like Eras), other genes like the hyaluronidases (HAS) and ADP ribosylation factors (ARFs) are indistinguishable between the experimental groups, making it likely that the antitumor effect seen in the rudimentary teratomas is a complex phenomenon. Although the host mice were severely immunocompromised and we had no access to immune-related tissues, the immaturity and delay in growth may suggest a yet-to-be-characterized anti-tumorigenic property of bat stem cells similar to, for instance, naked mole rats (Heterocephalus glaber),63,64 and which could also underlie the extended health span and cancer resistance reported in many bats.8,9 Finally, we created embryo-like structures from bat stem cells using a modified blastoid protocol.65 These bat blastoids recapitulated critical aspects of preimplantation embryos, including an Oct4-positive inner cell mass, the cystic cavity, and a bilayered epithelium (Figure 3E) consisting of a trophoblast that stained positive for Cdx2 (Figure S2E) and yolk sac cells. Replating these embryo structures resulted in their attachment with a flattened trophoblastic epithelium outgrowth and an expansion of the inner cell mass (Figure 3F). Our differentiation studies exemplify the potential of pluripotent bat stem cells to study important developmental events and serve as a powerful model to study the exceptional physiological adaptations of bats, including their reduced cancer phenotype. Finally, to see whether our protocol is broadly applicable to bats, we created primary Myotis fibroblast cells from 3 mm uropatagium (tail) biopsies of wild-caught adult bats. These fibroblasts were readily reprogrammable using our “batified” Yamanaka protocol and yielded bat iPS cells with a similar morphology that were Oct4 positive in immunostaining (Figures 4A and 4B). Importantly, they also showed co-expression of naive and primed pluripotency markers (Figure 4C) in most cells and differentiated into all three germ layers (Figure 4D). This suggests that our protocol is applicable across the deepest basal divergencies in bats.
Figure 4. Characterization of induced pluripotent stem cells derived Myotis myotis uropatagium fibroblasts.
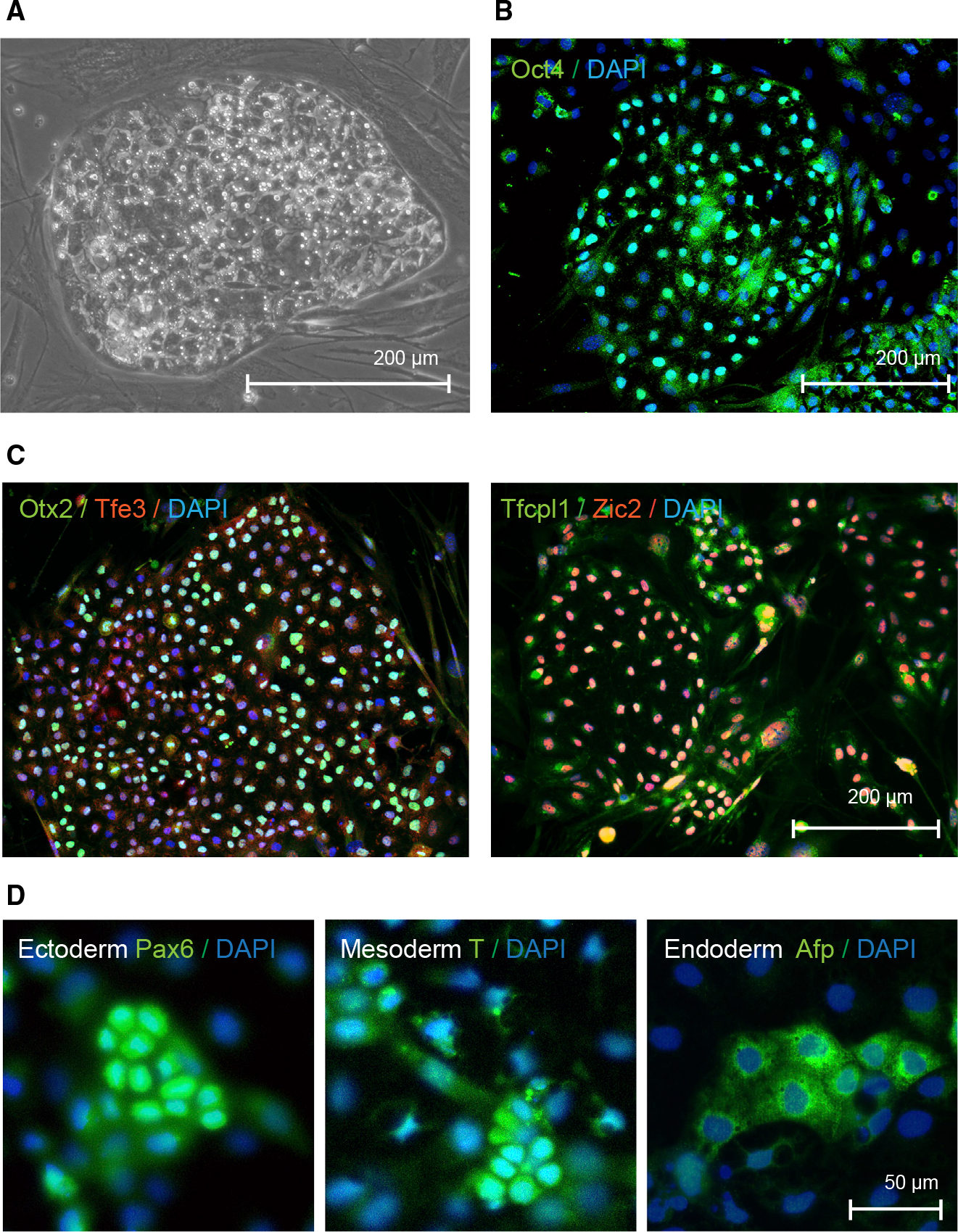
(A) Phase contrast image of Myotis myotis iPS cells.
(B) Microscopic image of Myotis myotis iPS cells after immunostaining to detect pluripotency marker Oct4.
(C) Immunofluorescence images of Myotis myotis pluripotent stem cells after staining of markers of naive (Tfe3 and Tfcp2l1) or primed pluripotency (Zic2 and Otx2)
(D) Microscopic images of Myotis myotis iPS cells that underwent differentiation and immunostaining to detect Pax6, brachyury (T) and Afp as markers for ectoderm, mesoderm, and endoderm, respectively.
Comparative transcriptomics
Next, we investigated whether our stem cell model could be used to gain insights into the distinctive evolutionary adaptations of bats. Phenotypic differences among species can be driven by evolutionary changes in gene expression.66 Therefore, given the special adaptations of bats, we should detect bat-specific gene expression patterns in bat stem cells, allowing us to compare the transcriptomic ground state across species, which is one of the earliest ontogenic comparisons possible. We collected transcriptome profiles of pluripotent stem cells from phylogenetically divergent mammal species (Mus musculus [mouse], Homo sapiens [human], Canis familiaris [dog], Sus scrofa [pig], and Callithrix jacchus [marmoset]) to compare them with our greater horseshoe bat (Rhinolophus ferrumequinum) data. Principal component analyses were performed to obtain a high-level overview of the number of commonalities and differences between bats and other mammals (Figure 5A). Remarkably, and supportive of our suspicion that bats are different, all other mammals grouped together in the PCA plot, while our bat stem cells formed a separate distinctive group, despite including other related laurasiatherian mammals.25
Figure 5. Distinct characteristics of bat pluripotent stem cells.
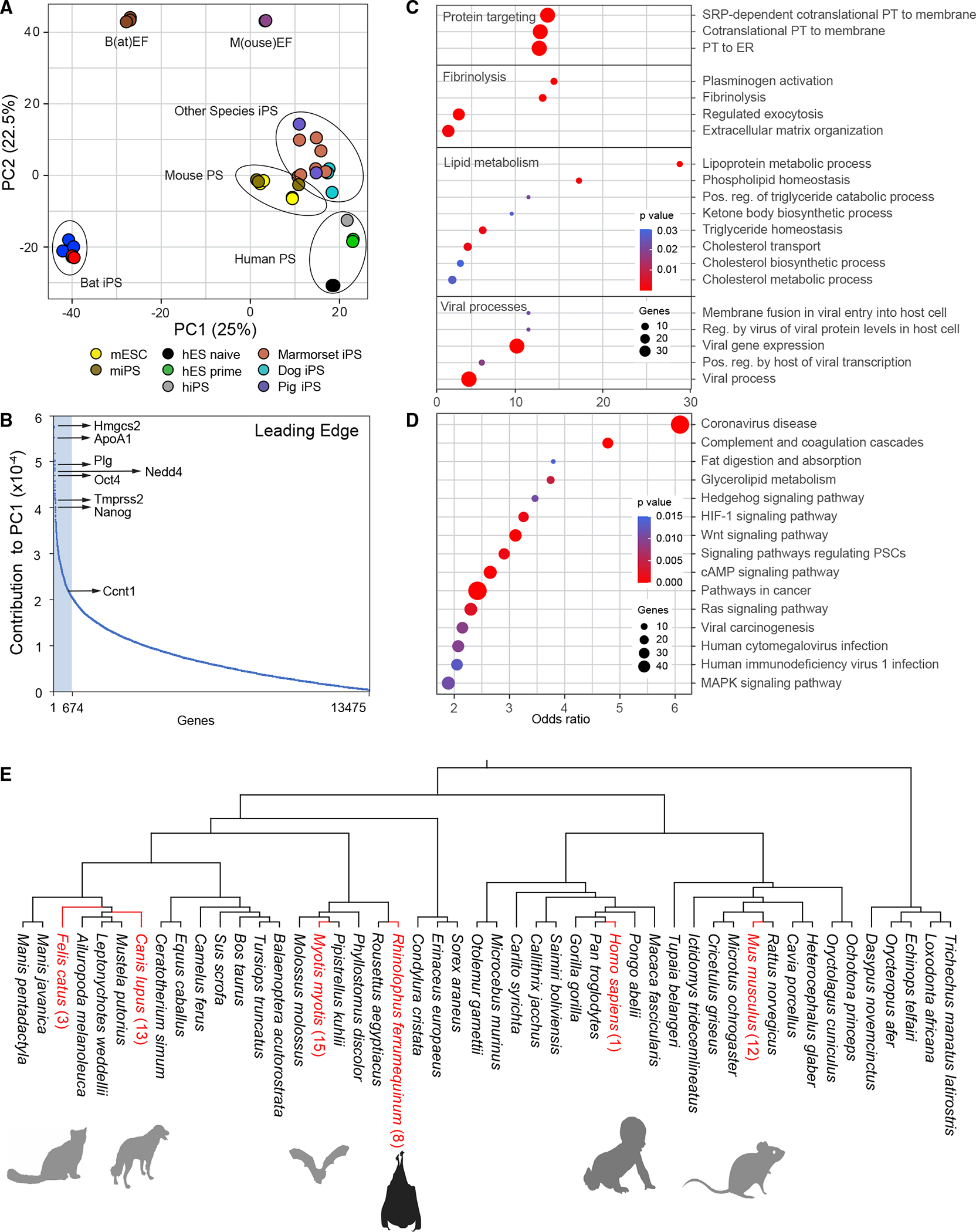
(A) Principal component analysis of R. ferrumequinum induced pluripotent bat stem cells (BiPS) in comparison to those derived from other species. h, human; m, mouse. PS, pluripotent stem cells; iPS, induced pluripotent stem cells; ES, embryonic stem cells; EF, embryonic fibroblasts. Each dot represents one dataset.
(B) Plot of genes that contribute to the differences of pluripotent bat and mouse stem cells as part of principal component 1 (PC1). Highlighted in light blue is the “leading edge” comprised of the top 5% of PC1-contributing genes.
(C) Selected GO and (D) KEGG pathways identified to be significantly enriched among the top 5% of PC1-contributing genes/leading-edge genes defined in (B) were plotted by their odds ratio, with the color of each circle indicating the enrichment p value and the size indicating the number of genes present in the respective category (see Data Tables S3B and S3C for a full list of enriched gene sets). ER, endoplasmic reticulum; PT, protein targeting; Pos, positive; Reg, regulation.
(D) Selection analyses of leading-edge genes by comparative genomics of the R. ferrumequinum lineage identified only eight genes (AARD, COL3A1, FAM111A, LAMB3, MUC1*, NES*, RGS5, RSPH1*) with significant evidence of positive selection, five of which showed at least one highly probable BEB site with no visual issues in the alignment region, while three genes (designated with *) did not (see Table S5E). Additional lineages and the number of leading-edge genes with significant evidence of positive selection found in them are highlighted in red.
See also Table S3.
We then determined the gene signature that contributed the most to the bat-specific gene expression profile. Specifically, we extracted the “leading edge,” corresponding to the top 5% of the genes that fortified the difference in principal component 1 (Figure 5B; Table S3A) when comparing bat with mouse pluripotent stem cells. The list covered genes belonging to a broad spectrum of transcription factors, kinases, and metabolic and homeostatic enzymes. For instance, it included Hmg-CoA synthase Hmgcs2, apolipoprotein Apoa1, cyclin Ccnt1, plasminogen PLG, pluripotency factors Oct4 and Nanog, Tmprss2 (required for SARS-CoV-2 entry in humans), and the ubiquitin ligase Nedd4, among many other genes. Given the broad spectrum of categories, we next asked whether the leading-edge genes were enriched for any particular biological pathway in gene ontology analyses (Figure 5C; Table S3B). We expected the genes to primarily encode developmental controllers and, indeed, some of the genes belonged to this class, but the vast majority fell into rather unexpected categories. Among the enriched functional families were proteins targeting membranes, including the endoplasmic reticulum, lipid and cholesterol biosynthesis, and fibrinogen production. However, the most prominent groups were viral gene expression, viral transcription, and many sets activated or suppressed after viral infection (Figure 5D; Table S3B). “Coronavirus disease” was by far the most significantly enriched category in any KEGG pathway (Figure S5D; Table S3C). These results suggest that bat stem cells execute a program that in other mammalian cells is activated only after viral infection.
Interestingly, out of the set of leading-edge genes, only a total of eight genes showed significant evidence of positive selection in R. ferrumequinum (Figure 5E; Tables S3D and S3E). Two of these genes, Col3a1, and Muc1, have roles in collagen formation in connective tissues,67 protect against pathogen infections,67 and show evidence of selection in another bat species, suggesting discrete, bat-specific adaptations in these genes. Our results might indicate that the distinctive bat signature is likely the consequence of the presence of viral sequences triggering the expression of antiviral cellular programs and that most of the coding leading-edge genes are not under positive selection pressure.
Endogenous viruses in bat stem cells
Throughout their evolution, bats have integrated diverse viral sequences into their genomes.21,27 This is in line with our findings that multiple virus-infection-related gene categories are highly enriched in bat stem cells (Figure 5; Table S3). Because endogenized viral sequences are often awakened in the developmental tabula rasa state of pluripotency in humans and mice,48 we hypothesized that our bat pluripotent stem cells would display a particularly rich set of expressed endogenized viral sequences and antigens compared with other mammals.
To advance the hypothesis, which that a particularly broad array of endogenized viral sequences would be re-activated in bat pluripotent stem cells to test, we looked first at endogenous retroviruses, which are abundant and diverse in bat genomes.24,25,27,68 As a starting point, we picked anchor points of retroviral sequences, and mapping our RNA-seq from both R. ferrumequinum and M. myotis bat iPS cells revealed the expression of a markedly diverse set of retroviral families in bat pluripotent stem cells when compared with fibroblasts (Figures 6A and S3; Tables S4A and S4B). Focusing on R. ferrumequinum iPS cells, we detected not only previously characterized full-length bat retroviruses (Figure S4A) but also thus-far-unknown ones (e.g., RFe-V-MD1) that are transcriptionally activated during reprogramming (Figure 6B; Table S4C). Importantly, chromatin in the vicinity of the expressed endogenous retroviruses opened up epigenetically during reprogramming, confirming our suspicion that the reprogramming process was responsible for revealing the diverse set of endogenous retroviruses (ERVs) sequences in our bat stem cells (Figure S4B).
Figure 6. Reactivation of endogenized retroviral elements in bat induced pluripotent stem cells.
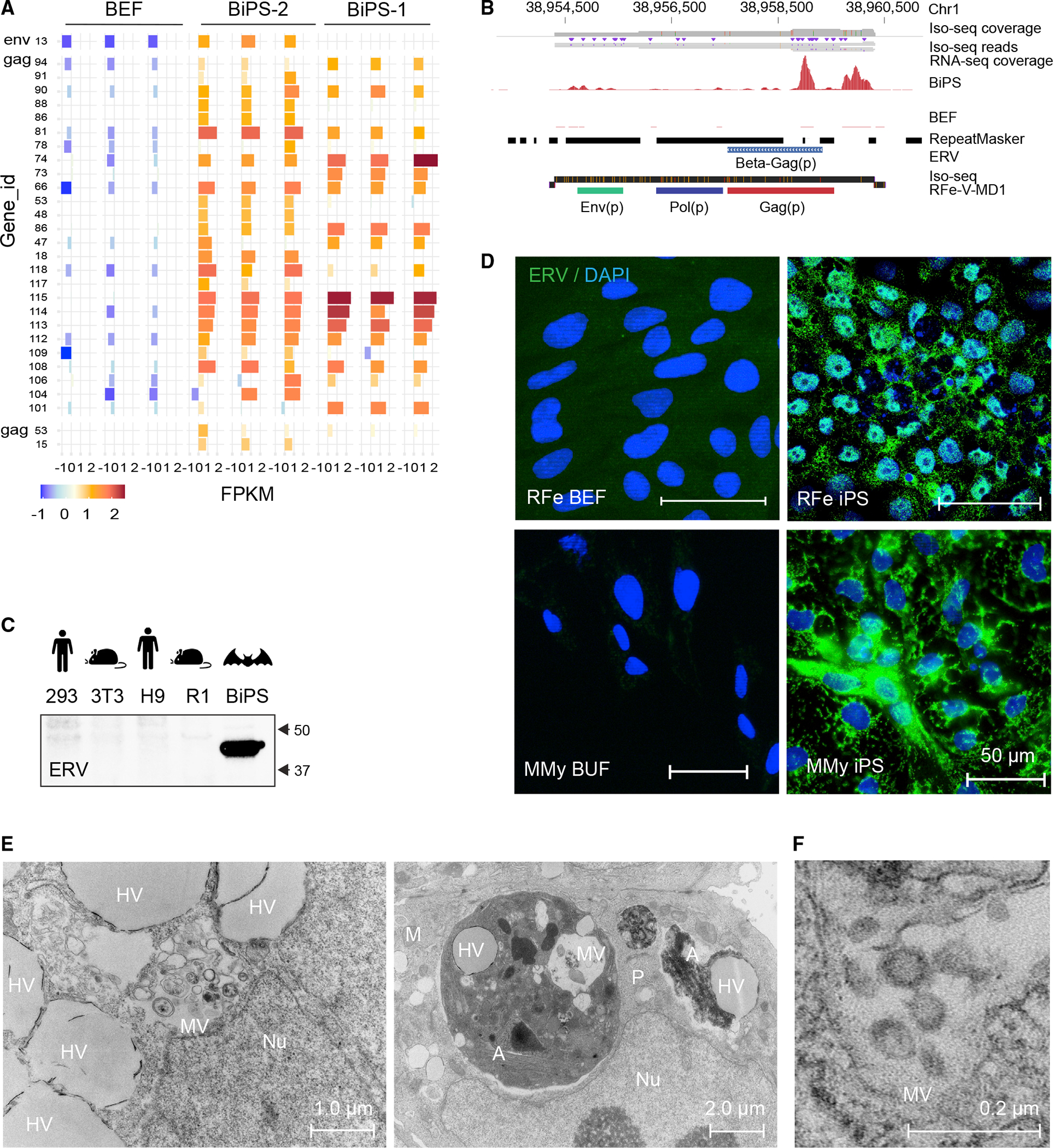
(A) Expression of indicated ERV elements in R. ferrumequinum bat embryonic fibroblasts (BEF) and iPS cells (BiPS), as determined by extracting the overlap between RNA-seq reads mapped to the R. ferrumequinum genome and known mapped ERV elements. Shown are the elements with the most evident differences (see Data Table S4A for a full list of expression data). All replicates (n = 3) per cell type are shown.
(B) RNA and Iso-seq sequencing tracks for an identified full-length retrovirus sequence, RFe-V-MD1, aligned to the R. ferrumequinum genome. The Iso-seq fragment represents a 6,088 bp-long transcript (ID: 39584940).
(C) Western blotting of protein lysates from human 293FT (kidney tumor cells) and human embryonic stem cells (H9), mouse 3T3 (fibroblasts) and mouse embryonic stem cells (R1), and R. ferrumequinum bat induced pluripotent stem cells (BiPS) with the endogenous retrovirus (ERV)-specific HERV K Cap antibody.
(D) Immunofluorescence images of R. ferrumequinum (RFe) bat embryonic fibroblasts (BEFs) and iPS cells from (top) and M. myotis (MMy) bat uropatagium fibroblasts (BUF) and iPS cells (bottom) after detection of the endogenous retrovirus (ERV) HERV K Cap protein (green); DAPI (blue).
(E) Overview of transmission electron microscopy of R. ferrumequinum bat pluripotent stem cells. MV, vesicles filled with multimembrane structures; HV, other vesicle structures filled with homogenous content; Nu, Nucleus; A, autophagosome; M, mitochondria; P, phagosome.
(F) Higher magnification of electron microscopy images as in (E) showing the presence of aggregates that are morphologically compatible with the appearance of endogenous retrovirus-like particles.
To determine whether the transcript-based findings indeed hold true at the protein level, we then determined whether the ERV antigen was present in our bat stem cells. Indeed, western blotting and immunostaining revealed high levels of ERV antigen in the bat stem cells of both species, which were not detected in human or mouse stem cells or fibroblasts (Figures 6C and 6D). Additionally, transmission electron microscopy (TEM) revealed electron-dense particles with lucent cores, ranging in size from 50 to 100 nm, which in some instances resembled previously reported viral-like particles (VLPs) (Figure 6E).48 Although the exact nature of these particles needs to be explored further, virus-like activity was detectable in the supernatant (1.21 * 1010 viral particles per ml, as determined in a retroviral assay and 0.3 ng/well in a direct reverse transcriptase assay). However, when we inoculated Vero cells with the supernatant of our bat iPS cells in plaque assays, we did not detect any measurable cytotoxic effects, in contrast to the acute infectious virus particles that served as positive controls (Figure S4C). These findings imply that bat cells produce ERV antigen and, in some instances, possibly active endogenous viral-like assemblies at an unusual scale compared with other mammals.
In addition to ERVs, bat genomes have also assimilated a substantial number and diversity of endogenous viral elements (EVE).25 Hence, we attempted to obtain a broader portrait of integrated and expressed viral sequences. We developed pipelines based on the metagenomic classification of the stem cell RNA-seq data, using either Kraken2 or Microsoft Premonition (Figure S5; Tables S5 and S6), which included a series of strict classification (using the k-mer-based Kraken2 and the alignment-based Microsoft Premonition) and curation steps (de novo assembly of putative viral contigs and genome mapping) to identify true viral reads (Figures S6A and S6B; STAR methods). The analyses revealed that bat pluripotent stem cells display a variety of virus-associated endogenized sequences. For instance, Blast analysis of selected suspected viral Iso-seq reads, as identified by our metagenomics method, showed an unexpected region in the first intron of the Xpa gene (DNA damage and repair factor) on chromosome 12 (Figure S6C). The region showed homology to two human herpesvirus 4 isolates (HKD40 and HKNPC60), the human respiratory syncytial virus (Kilifi isolate), and a fragment of about 500 bp that was identified at the end of a SARS-CoV2 isolate from an infected patient (Figure S6D; Table S7A). We also found a nearly 50% identical sequence to either Scotophilus bat coronavirus 512 (Tables S7A and S7B) or RaTG13 coronavirus (one of the bat coronaviruses most closely resembling SARS-CoV-2), covering most of the spike encoding sequences (Tables S7A and S7C). Phylogenetic analysis revealed that these genomic sequences resembled the spike encoding genomic portion of viruses, such as the human coronavirus 229E and human coronavirus OC43, respectively (Figure S6E). Interestingly, these regions are flanked by LINE-1 sequences (Figure S6F). This suggests the possibility that LINE elements are directly involved in the homing of viral RNA, a possibility that was also explored recently in the context of SARS-CoV-2.69 Systematically scanning for expressed viral elements in our RNA we then assembled contigs and aligned them to viral and mammalian genomes using BLAST (Figure S5) and employing highly stringent parameters (Figures S6A and S6B; STAR methods). This procedure allowed us to identify previously unknown viral sequences and integration sites (Tables S6 and S7). Although many of the viral alignments were difficult to distinguish from cellular genes (e.g., oncogenes), especially in pipelines that did not include genome mapping steps, we detected what appeared to be bona fide purely viral-like elements that were integrated in the bat genome and induced in our pluripotent stem cells.
We identified numerous predicted and so-far-unknown retroviral integrations with homologies to the Koala retrovirus (Table S6), Mason-Pfizer monkey virus (Tables S7A and S7D), Jaagsiekte sheep retrovirus, or Ovine enzootic nasal tumor virus (Table S7A and S7E), to name a few. Translations of the extended regions covered by mapped RNA-seq reads using BLASTX revealed similarities to the Jaagsiekte sheep reverse transcriptase (Table S7E) and other known retroviral gag, pro, and pol proteins (Table S7). An example of an integrated DNA virus sequence is a region within scaffold_m29_p_1 of the R. ferrumequinum genome, which shows homologies to the Volepox, Variola, Squirrelpox, and Monkeypox viruses (Figure S7D). Here, translation of the extended region with mapped RNA-seq reads uncovered homologies with the cowpox protein CPXV051 and the monkeypox C10L protein (Tables S7A and S7G). Another region that is worth noting is located within scaffold_m29_p_20. We first identified this region through a short sequence homology with the SARS-related coronavirus isolate Rs7907, coding for an N-terminal fragment of a double-stranded RNA (dsRNA)-binding protein (Table S6N; Figure S7E). When extending this region to cover all mapped RNA-seq reads in the vicinity, a BLAST search identified several longer homologies with the White spot syndrome virus (Figure S7F; Tables S7A and S7H). Interestingly, we find mapped RNA-seq reads in fibroblasts and iPS cells and that the genomic location borders a “gap” region in the genome, indicating that even more of this non-mammalian virus might be present in the bat genome. Notably, a comparison of the metagenomic diversity, as determined by Kraken2 using RNA-seq data from BEFs and from human ES cells (Tables S5B and S5C), yielded some viral sequences, albeit to a much lesser degree (Figure S6A). In summary, we conclude that, while confounding effects including genomic contaminations can affect the metagenomic classification process, it is highly likely that a sizable body of proviral sequences and sequence fragments inhabit BiPS cells.
To support our transcriptomics-based findings, we next looked for antigen markers linked with the RNA virus lifestyle because bats have traditionally shown an extreme affinity for RNA viruses and, in some cases, coronaviruses.70 We first stained our bat iPS cells with an antibody detecting a corona-virus antigen. This was based on the fact that bats are known to host coronaviruses, and early during the SARS-CoV-2 pandemic Rhinolophidae sp. were considered as a host for SARS-CoV2, and our discovery of sequences that resemble corona-viruses (Figure S6; Table S7). Indeed, we found the BiPS cells to be positive in immunofluorescence and western blot analyses when compared with fibroblasts and pluripotent stem cells from other species (Figures 7A and 7B). Super-resolution microscopy showed clustered localization within the cytoplasm (Figure 7C). We then looked for the presence of dsRNA in immunostaining which is thought to be a sign of replicative genomes from both positive-strand dsRNA and DNA viruses (Figure 7D). Super-resolution imaging (Figure 7E) showed that the dsRNA was present in micron-order-sized aggregates throughout the cytoplasm but essentially absent from the nucleus. Further, ImageStream analysis revealed an overlap between the dsRNA signal and the distinctive intracellular vesicles found in bat stem cells (Figures 7F and 7G). However, the precise involvement of these vesicles in viral activities needs to be further investigated. Finally, in line with the pro-viral environment on a transcriptional level, we found that bat stem cells infected with an exogenous Metapneumovirus (MPV) revealed a particularly permissive environment for viral persistence when compared with mouse stem cells, further underscoring the supportive nature of bat stem cells for viruses (Figures S7G and S7H).
Figure 7. Reactivation of endogenized viral elements in bat pluripotent stem cells.
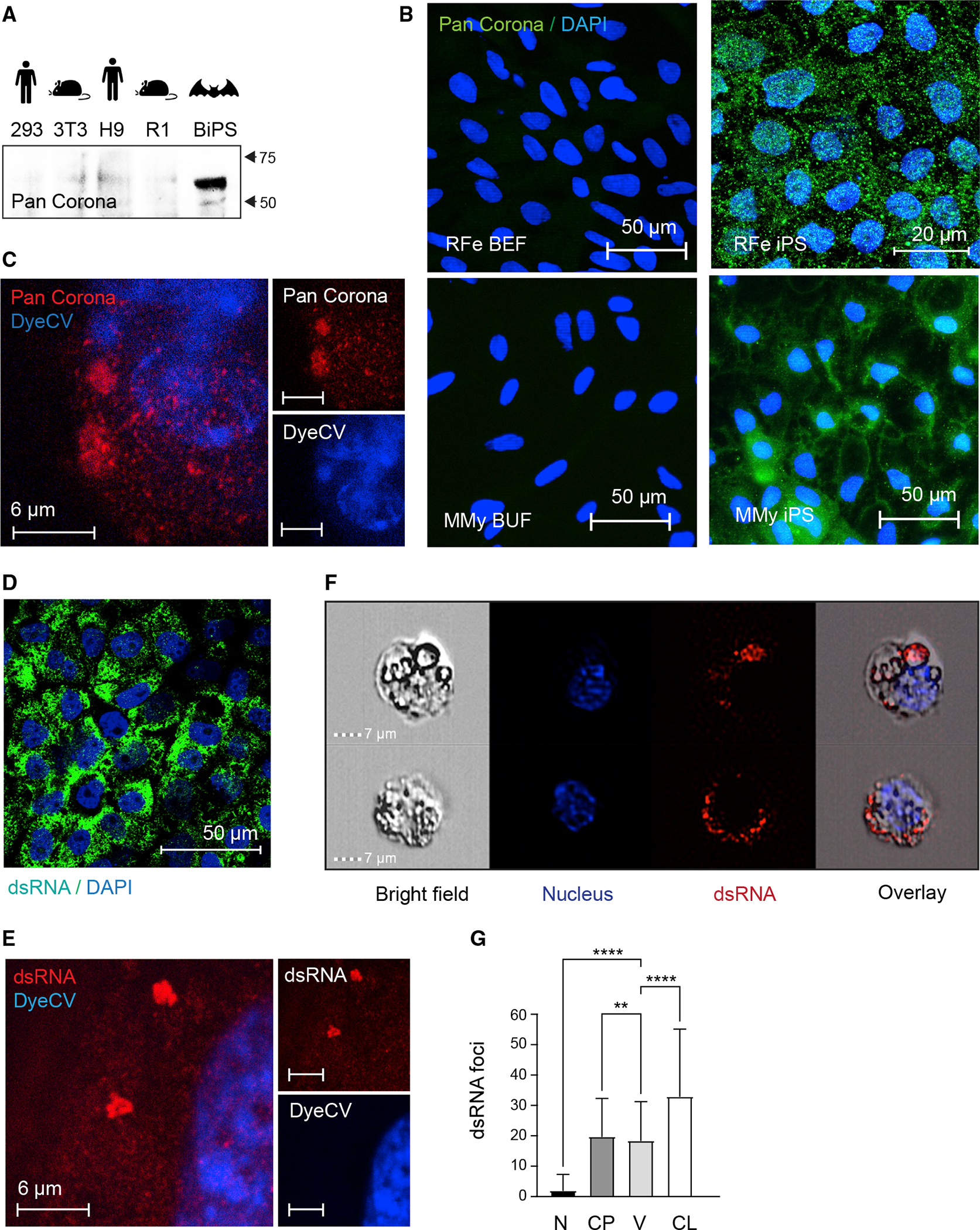
(A) Western blotting of protein lysates isolated from human 293FT and H9, mouse 3T3 and R1, and R. ferrumequinum BiPS cells with a pan coronavirus antibody known to be specific for the nucleocapsid; its reactivity includes, but might not be limited to, feline infectious peritonitis virus type 1 and 2, canine coronavirus (CCV), pig coronavirus transmissible gastroenteritis virus (TGEV), and ferret coronavirus.
(B) Immunofluorescence images of R. ferrumequinum (RFe) bat embryonic fibroblasts (BEFs) and iPS cells from (top) and M. myotis (MMy) bat uropatagium fibroblasts (BUF) and iPS cells (bottom) after detection of the pan coronavirus antigen (green); DAPI (blue).
(C) Representative STED microscopy image of R. ferrumequinum iPS cells after detecting the Corona antigen as in (B) and DyeCycle Violet (DyeCV) nuclear counter stain (blue).
(D) Immunofluorescence images of R. ferrumequinum BiPS cells after detection of double-stranded RNA (green) characteristic of RNA viruses; DAPI (blue).
(E) Representative STED microscopy image of R. ferrumequinum iPS cells after immunofluorescence staining of double-stranded RNA (dsRNA) as in (D).
(F) ImageStream analysis after immunofluorescence staining of BiPS cells as in (D and E). A brightfield image, DyeCycle Violet nuclear staining (blue), dsRNA staining (red), and an overlay is shown for each representative cell.
(G) Quantification of dsRNA foci by ImageStream in R. ferrumequinum iPS cells show in (F). Data are presented as mean +/− SD; n = 1,846 cells. **p < 0.01,****p < 0.0001 by one-way ANOVA with Bonferroni’s multiple comparisons test. RFe, Rhinolophus ferrumequinum; BEF, bat embryonic fibroblasts; CP, cytoplasm; CL, cell; iPS, induced pluripotent stem cells; MMy, Myotis myotis; N, nucleus; V, vesicle; BUF, bat uropatagium fibroblasts.
See also Figures S4–S7 and Tables S5, S6, and S7.
DISCUSSION
Bats have evolved an unusual lifestyle among mammals as they fly, use echolocation, and have a curious affinity for viruses. One possibility is that bats evolved a tolerance for viruses by evolving changes in their innate immunity resembling the virus evasion mechanisms of the mammalian immune response. Another possibility is that bats evolved mechanisms for a cellular program to support viral replication and persistence, comparable to how viruses manipulate the host cell. Our results support both perspectives.
Indeed, our results show that a potentially significant contingent of endogenous and exogenous viral products is present in bat pluripotent stem cells without severely compromising their ability to proliferate and grow and that this goes beyond how other pluripotent stem cells react to viruses. Viruses typically adapt their replication cycles to a particular cell type. Thus, one would not expect the pluripotent stem cell state to align with viruses’ often specialized requirements.49 Nevertheless, our data suggest that in bats the pluripotent state serves as an “umbrella” host for a highly divergent viral contingent. We propose that our culture model can help to carefully dissect the necessary balance for tolerance of viral infections. Our bat stem cell system will also provide insights into bats’ potential role as virus reservoirs and the relationship between bats and viruses. In vitro differentiation into immune cells and tissues, such as lung or gut epithelium, will illuminate emerging viruses, develop bats as new model study systems, provide new insights into how bats tolerate viral infections, and, in turn, allow us to better prepare for future pandemics.
Careful cataloging of acute exogenous bat viruses, tissue infection, persistent viruses, and endogenized viruses in geographically relevant regions71,72 will reveal members of diverse retroviral and non-retroviral sequences, potentially impacting the host and emerging viruses. It will also uncover rationales for virus persistence, including immune-modulatory strategies,3 symbiotic protection against other pathogens,73–75 biological warfare that bats use to deploy viruses,18 mammalian adaptive piRNA or CRISPR-like systems,76 and the augmentation of evolutionary processes.77 Although the events we specifically study in pluripotent stem cells do not directly impact those occurring in specific adult cells, we propose that pluripotent stem cells are, in their own right, an important cell system in the study of native immunity and viruses. They share a highly conserved78 and immunologically relevant common genetic program with somatic cells.79 Pluripotent stem cells in the embryo and the related trophoblastic tissue must establish their immunological barrier against the maternal tissue and are subject to viral infection, as also noted in bats.80 Furthermore, as some fundamental cell biological properties might be shared between stem cells,81,82 the finding may also extend to other stem cells often at risk of infection, such as hematopoietic stem cells,83 neural stem cells,84,85 and early human embryos where, for instance, the rubella virus can destroy the conceptus or cause severe congenital defects.86 Also, most basic native immunity systems with pathogen pattern sensing and inflammatory responses, including inflammasome, NFκB, and interferon, are basic tools largely present in their most ancient ancestors and broadly shared between species and cell types.87,88 Finally, viral RNA products present in bat cells might also represent a potential source for RNA recombination upon infection with exogenous homologous viruses, which could be a driver for viral evolution in bats.
Our bat stem cell system may also provide a much-needed experimental substrate to parse the tantalizing hypothesis that viruses and hosts are more entangled than previously thought and that viruses are fully competent agents and editors of host biology.46 Therefore, viruses must be rich sources of evolutionary instructions, especially given their extreme genetic adaptability and ability to transition between the living and chemical worlds.45,89 Allowing viral evolutionary processes to unfold in our bat cell lines and mapping out changes to the host transcriptome will be critical steps toward understanding host-virus editing interactions.
The results of our study provide proof-of-concept evidence that bat stem cells include a remarkable variety of sequences that are similar to viral genomic sequences. Additionally, our findings indicate that retroviruses, and parts of endogenous viruses other than retroviruses, are produced and active on a scale that is not generally seen in tumor or stem cell lines that originate from other animals or humans studied so far. We conclude that the transcriptionally permissive state of pluripotency can be exploited to discover bat viruses and derivative sequences that likely play an essential role in bat physiology and their ability to host viruses.
Bats are a critically needed model organism to better understand disease tolerance, but limited access to animal and cell models has hindered their study.36,90 Bat breeding colonies for some species are notoriously challenging to establish; most bat species are protected worldwide, and primary bat cell lines typically have a limited in vitro lifespan.36 Therefore, pluripotent stem cells offer a research tool that is a sine qua non for bat research. Once established, pluripotent stem cells divide indefinitely in culture and are very amenable to gene editing and molecular studies.91 Most importantly, pluripotent stem cells retain the ability to differentiate into any cell type in the body and are often used as a springboard for sophisticated tissue culture models92 and, more recently, virus studies.93,94 Future research on bat stem cells will directly impact every aspect of understanding bat biology, including bats’ amazing adaptations of flight, echolocation, extreme longevity, and unusual immunity. Although bat genomes are the natural starting point for studying such adaptations and are being generated by the Bat1K consortium,4,5 our pluripotent stem cell system will enable specific bat tissue studies, and organoids will let us test more complex relationships, gene editing, and specific evolutionary hypotheses. Bat stem cell lines and differentiated progeny will help address tantalizing physiology questions, provide the required tools for validation, and utilize the genomic basis of rare adaptations found in bats.
Limitations of the study
Currently, viral metagenomic classification of only recently assembled diverse non-model mammalian genomes and transcriptomes, such as those in bats, is still limited, given the lack of viral comparative studies and knowledge of these key taxa. These results will need further verification at the protein level and using future curated assemblies. Obtaining wild lethal and non-lethal bat samples can be a challenge, and the derived cell lines may not capture population-level diversity in ERV and other viruses. However, our results are a solid starting point to carefully revisit the plethora of genomic and expressed bat viral sequences and to test whether they are integrated sequences to defend against viruses and microbes and encode viral proteins in a self-vaccination scheme or are near full-length viruses to manipulate host physiology. While at this point it is not feasible to perform comprehensive quantitative comparative studies of viral-like sequences between species with any degree of certainty, mainly because the number of characterized human viruses vastly exceeds those of other species based on the bias toward human viral research, we hope that our study will serve as the foundation for such tool development. Also, the reactivation of endogenized virus fragments exposes only part of the host tissue response and needs to be investigated in other cell types and acute viruses. Furthermore, how closely our pluripotent stem cell lines resemble different inner cell mass (ICM) states, and the impact of our tissue culture protocol, will only be ascertained by comparing them to bat ICM cells and the generation of bat chimeras. This current proof-of-concept study establishes bat stem cells in particular, and bats in general, as tantalizing model systems, allowing us to both elucidate the diversity of viruses that bats can survive and the molecular adaptations that enable bats to asymptomatically tolerate these viruses, thus providing important insights into both disease surveillance and future therapeutics.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Thomas P. Zwaka (thomas.zwaka@mssm.edu).
Materials availability
All unique reagents in this study are available from the Lead Contact and will be provided upon request. There are restrictions to the availability of frozen cell vials due to the number of frozen stocks available.
Data and code availability
All high-throughput sequencing data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
This paper analyzes existing, publicly available data. These accession numbers for the datasets are listed in the key resources table.
This paper does not report any original code.
All software packages and their accessibility are described in the STAR Methods sections.
Any additional information required to reanalyze the data reported in this paper (including the BLAST results related to Figures S5–S7) is available from the Lead Contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| anti-Afp | R&D Systems | AF1369; RRID:AB_354758 |
| anti-Pax6 | BioLegend | 901301; RRID:AB_2565003 |
| J2 anti-dsRNA | Scicons | 10010200; RRID:AB_2651015 |
| anti-(gag/pol)HERVK | Austrial Biological | HERM18315; RRID:AB_10890594 |
| anti-Pan Corona FIPV3-70 | Life Technologies | MA1-82189; RRID:AB_930177 |
| anti-Oct3/4-AF488 | Santa Cruz | sc-5279-AF488; RRID:AB_628051 |
| anti-Brachyury | R&D Systems | IC2085G;RRID:AB_2933975 |
| anti-Otx2 | R&D Systems | AF1979; RRID:AB_2157172 |
| anti-Zic2 | Abcam | ab150404; RRID:AB_2868423 |
| anti-Tfe3 | Sigma Aldrich | HPA023881; RID:AB_1857931 |
| anti-Tfcp2l1 | R&D Systems | AF5726; RRID:AB_2202564 |
| anti-Cdx2 | Invitrogen | PA5-20891; RRID:AB_11155394 |
| Donkey anti-chicken-Cy3 | Millipore | AP194C; RRID:AB_92679 |
| Goat anti-chicken-AF488 | Invitrogen | A-11039; RRID:AB_2534096 |
| Donkey anti-rabbit-AF647 | Invitrogen | A-31573; RRID:AB_2536183 |
| Goat anti-rabbit-AF488 | Invitrogen | A-10034; RRID:AB_2576217 |
| Goat anti-mouse-AF488 | Invitrogen | A-11029; RRID:AB_2534088 |
| Abberior Star 635P | Abberior | ST635P; RRID:AB_2893232 |
| H3K4me3 | Active Motif | 39159; RRID:AB_2615077 |
| H3K27me3 | Active Motif | 39155; RRID:AB_2561020 |
| anti-mouse-HRP | Promega | W4021; RRID:AB_430834 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| SARS-CoV-2, isolate USA-WA1/2020 | BEI Resources | NR-52281 |
| Human Metapneumovirus MPV-GFP1 | ViraTree | M121 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| DPBS (Life Technologies, 14190144), | Gibco | 14190144 |
| DPBS | Biowest | L0615-500 |
| 0.05% Trypsin | Gibco | 25300054 |
| DMEM | Gibco | 10569010 |
| DMEM | Gibco | 11995065 |
| Fetal bovine serum, FBS | Sigma-Aldrich | F4135 |
| Fetal bovine serum, FBS | Gibco | 10500064 |
| MEM Non-essential amino | Gibco | 11140050 |
| GlutaMax Supplement | Gibco | 35050061 |
| Penicillin-Streptomycin solution | Gibco | 15140122 |
| Gelatin | Millipore | ES006B |
| Antibiotic-Antimycotic (100X) | Gibco | 15240096 |
| Collagenase type II | Gibco | 17101015 |
| Gentle Cell Dissociation Reagent | StemCell Technologies | 07174 |
| DMEM/F-12 | Gibco | 11330057 |
| Knockout serum replacement | Gibco | 10828-028 |
| DMSO | Sigma-Aldrich | D8418-100ML |
| 2-mercaptoethanol | Fluka | 63689 |
| FGF2 | R&D Systems | 233-FB |
| Leukemia inhibitory factor | Millipore | ESG1107 |
| SCF | R&D Systems | PHC2111 |
| Forskolin | Sigma-Aldrich | F6886 |
| KaryoMAX Colcemid Solution in HBSS | Gibco | 15210040 |
| Sodium Pyruvate, 100 mM | Gibco | 11360070 |
| Potassium chloride | Sigma-Aldrich | P9327 |
| Glacial acetic | Fisher Scientific | A38-212 |
| Methanol | Sigma-Aldrich | A412-4 |
| KaryoMax Giemsa staining solution | Gibco | 10092013 |
| Gurr buffer tablets | Gibcco | 10582013 |
| Cytoseal 60 | Thermo Scientific | 23-244257 |
| SuperScript IV VILO™ Master Mix | Invitrogen | 11756050 |
| GoTaq Green Polymerase | Promega | M7123 |
| Cytofix/Cytoperm solution | Becton Dickinson | BDB554714 |
| NucBlue Dapi stain | Invitrogen | R37606 |
| Prolong Dimond antifade mounting medium | Invitrogen | P36965 |
| DyeCycle Violet stain | Invitrogen | V35003 |
| DNase | Qiagen | 79254 |
| Formaldehyde | Sigma-Aldrich | F8775 |
| Sodium chloride solution, 5M | Sigma-Aldrich | S5150 |
| EDTA (0.5 M), pH 8.0, RNase-free | Invitrogen | AM9260G |
| HEPES, 1M | Invitrogen | 15630080 |
| Glycine | Sigma-Aldrich | G8898 |
| Igepal | Sigma-Aldrich | I3021 |
| PMSF | Sigma-Aldrich | P7626 |
| mTeSR medium | StemCell Technologies | 85850 |
| Vitronectin | StemCell Technologies | 07180 |
| BMP4 | R&D Systems | 314-BP-010 |
| Versene | Gibco | 15040066 |
| 10% Formalin | Fisher Scientific | SF1004 |
| RIPA lysis and extraction buffer | Fisher Scientific | 89900 |
| Proteinase Inhibitor | Roche | 45582400 |
| BCA Protein assay kit | Pierce | 23252 |
| 12% TGX Precast gel | Bio-Rad | 4561044 |
| TGS buffer | Bio-Rad | 1610772 |
| the Precision Plus Kaleidoscope Protein standard | Bio-Rad | 161-3075 |
| EveryBlot blocking buffer | Bio-Rad | 12010020 |
| Tris-buffered saline | Fisher Scientific | BP2471500 |
| Tween 20 | Bio-Rad | 161-0781 |
| Clarity Western ECL Substrate | Bio-Rad | 170-5060 |
| Paraformaldehyde | Electron Microscopy Sciences | 19200 |
| Glutaraldehyde | Electron Microscopy Sciences | 16220 |
| Sodium Cacodylate Buffer | Electron Microscopy Sciences | 11652 |
| Aqueous osmium tetroxide | Sigma-Aldrich | 75633-2ML |
| 2% aqueous uranyl acetate | Electron Microscopy Sciences | 22400-2 |
| Ethanol | Electron Microscopy Sciences | 15055 |
| Epoxy resin EMbed 812 | Electron Microscopy Sciences | 14900 |
| Toluidine Blue | Electron Microscopy Sciences | 26074-15 |
| Lead citrate | Electron Microscopy Sciences | 22410 |
| Minimum Essential Medium | Gibco | 11935046 |
| 4 mM L-glutamine | Gibco | 25030081 |
| 0.7% agar | Fisher Scientific | AAJ1090722 |
| TrueBlue substrate | Seracare | 5510-0030 |
| Trizol reagent | Invitrogen | 15596018 |
| Bovine Serum Albumin | Sigma-Aldrich | A2153-50G |
|
| ||
| Critical commercial assays | ||
|
| ||
| CytoTune iPS2.0, Invitrogen, A16517 | Life Technologies | A16517 |
| STEMdiff Trilineage differentiation kit | StemCell Technologies | 05230 |
| QuickTiter Retrovirus Quantitation Kit | Cell Biolabs | VPK-120 |
| Colorimetric reverse transcriptase kit | Roche | 11468120910 |
| SMART-Seq v4 Ultra Low Input kit | Takara Bio | 634888 |
| Total RNA with Ribo-Zero Plus kit | Illumina | 20040529 |
| AllPrep DNA/RNA Mini Kit | Qiagen | 80204 |
| SMRTbell Express Template Preparation Kit v2.0 | PacBio | 100-938-900 |
| NEBNext Single Cell/Low Input cDNA Synthesis & Amplification Module | New England Biolabs | E6421L |
| SMRTbell Enzyme Clean Up Kit | PacBio | 101-746-400 |
| RNeasy Mini Kit | Qiagen | 74104 |
|
| ||
| Deposited data | ||
|
| ||
| Rhinolophus ferrumequinum assembled and annotated by the Vertebrate Genomes Project | Vertebrate Genome Project | GCF_004115265.1 |
| Rhinolophus ferrumequinum genome assembled and annotated by the Bat1K project | Bat1K | GCA_014108255.1 |
| Rhinolophus ferrumequinum genome, Ensembl annotation version 102 | Ensembl | GCA_004115265.2 |
| Bat1K annotation of known endogenous retroviruses (ERVs) for R. ferrumequinum | https://genome.senckenberg.de/) | N/A |
| Bat1K annotation of known endogenous retroviruses (ERVs) for Myotis myotis | https://genome.senckenberg.de/) | N/A |
| RFe_BEF-R1_RNA: R. ferrumequinum fibroblasts RNA-seq replicate 1 | This paper | GEO: GSE221965 |
| RFe_BEF-R2_RNA: R. ferrumequinum fibroblasts RNA-seq replicate 2 | This paper | GEO: GSE221965 |
| RFe_BEF-R3_RNA: R. ferrumequinum fibroblasts RNA-seq replicate 3 | This paper | GEO: GSE221965 |
| RFe_BiPS1-R1_RNA: R. ferrumequinum iPS#1 RNA-seq replicate 1 | This paper | GEO: GSE221965 |
| RFe_BiPS1-R2_RNA: R. ferrumequinum iPS#1 RNA-seq replicate 2 | This paper | GEO: GSE221965 |
| RFe_BiPS1-R3_RNA: R. ferrumequinum iPS#1 RNA-seq replicate 3 | This paper | GEO: GSE221965 |
| RFe_BiPS2-R1_RNA: R. ferrumequinum iPS#2 RNA-seq replicate 1 | This paper | GEO: GSE221965 |
| RFe_BiPS2-R2_RNA: R. ferrumequinum iPS#2 RNA-seq replicate 2 | This paper | GEO: GSE221965 |
| RFe_BiPS2-R3_RNA: R. ferrumequinum iPS#2 RNA-seq replicate 3 | This paper | GEO: GSE221965 |
| RFe_BiPS2-EB-R1_RNA: R. ferrumequinum EB RNA-seq replicate 1 | This paper | GEO: GSE221965 |
| RFe_BiPS2-EB-R2_RNA: R. ferrumequinum EB RNA-seq replicate 2 | This paper | GEO: GSE221965 |
| RFe_BiPS2-EB-R3_RNA: R. ferrumequinum EB RNA-seq replicate 3 | This paper | GEO: GSE221965 |
| RFe_BiPS2-EN-R1_RNA: R. ferrumequinum Endoderm RNA-seq replicate 1 | This paper | GEO: GSE221965 |
| RFe_BiPS2-EN-R2_RNA: R. ferrumequinum Endoderm RNA-seq replicate 2 | This paper | GEO: GSE221965 |
| RFe_BiPS2-EN-R3_RNA: R. ferrumequinum Endoderm RNA-seq replicate 3 | This paper | GEO: GSE221965 |
| RFe_BiPS2-ME-R1_RNA: R. ferrumequinum Mesoderm RNA-seq replicate 1 | This paper | GEO: GSE221965 |
| RFe_BiPS2-MS-R2_RNA: R. ferrumequinum Mesoderm RNA-seq replicate 2 | This paper | GEO: GSE221965 |
| RFe_BiPS2-MS-R3_RNA: R. ferrumequinum Mesoderm RNA-seq replicate 3 | This paper | GEO: GSE221965 |
| RFe_BiPS2-EC-R1_RNA: R. ferrumequinum Ectoderm RNA-seq replicate 1 | This paper | GEO: GSE221965 |
| RFe_BiPS2-EC-R2_RNA: R. ferrumequinum Ectoderm RNA-seq replicate 2 | This paper | GEO: GSE221965 |
| RFe_BiPS2-EC-R3_RNA: R. ferrumequinum Ectoderm RNA-seq replicate 3 | This paper | GEO: GSE221965 |
| RFe_BEF-R1_ATAC: R. ferrumequinum fibroblasts ATAC-seq replicate 1 | This paper | GEO: GSE221965 |
| RFe_BEF-R2_ATAC: R. ferrumequinum fibroblasts ATAC-seq replicate 2 | This paper | GEO: GSE221965 |
| RFe_BiPS2-R1_ATAC: R. ferrumequinum iPS#2 ATAC-seq replicate 1 | This paper | GEO: GSE221965 |
| RFe_BiPS2-R2_ATAC: R. ferrumequinum iPS #2 ATAC-seq replicate 2 | This paper | GEO: GSE221965 |
| RFe_BEF_H3K4: R. ferrumequinum fibroblasts H3K4me3 ChIP-seq | This paper | GEO: GSE221965 |
| RFe_BiPS2_H3K4: R. ferrumequinum iPS H3K4me3 ChIP-seq | This paper | GEO: GSE221965 |
| RFe_BEF_H3K27: R. ferrumequinum fibroblasts H3K4me3 ChIP-seq | This paper | GEO: GSE221965 |
| RFe_BiPS2_H3K27: R. ferrumequinum iPS#2 H3K4me3 ChIP-seq | This paper | GEO: GSE221965 |
| RFe_BEF-R1: RRBS: R. ferrumequinum fibroblasts RRBS replicate 1 | This paper | GEO: GSE221965 |
| RFe_BEF-R2: RRBS: R. ferrumequinum fibroblasts RRBS replicate 2 | This paper | GEO: GSE221965 |
| RFe_BiPS2-R1_RRBS: R. ferrumequinum iPS#2 RRBS replicate 1 | This paper | GEO: GSE221965 |
| RFe_BiPS2-R2_RRBS: R. ferrumequinum iPS#2 RRBS replicate 2 | This paper | GEO: GSE221965 |
| Dog iPS RNAseq: K9_1_iPS_P9 | GSE15249395 | GEO: GSM4616525 |
| Dog iPS RNAseq: K9_2_iPS_P8 | GSE15249395 | GEO: GSM4616526 |
| Dog iPS RNAseq: K9_3_iPS_P8 | GSE15249395 | GEO: GSM4616527 |
| Marmoset iPS RNAseq: CM421F_B0_12_iPS_P11 | GSE15249395 | GEO: GSM4617887 |
| Marmoset iPS RNAseq: CTXNS1_B1_iPS_P7 | GSE15249395 | GEO: GSM4617889 |
| Marmoset iPS RNAseq: CTXNS2_B1_iPS_P5 | GSE15249395 | GEO: GSM4617890 |
| Marmoset iPS RNAseq: E01F_A2_2_iPS_P10 | GSE15249395 | GEO: GSM4617891 |
| Marmoset iPS RNAseq: E02M_B0_7_iPS_P13 | GSE15249395 | GEO: GSM4617895 |
| Marmoset iPS RNAseq: I5061F_B3_15_iPS_P10 | GSE15249395 | GEO: GSM4617900 |
| Marmoset iPS RNAseq: I5061F_B3_3_iPS_P11 | GSE15249395 | GEO: GSM4617901 |
| Human iPS RNA-seq: BjiPS_P3 | GSE15249395 | GEO: GSM4616532 |
| Pig iPS cell RNA-seq: N01F_N1_iPS_P3 | GSE15249395 | GEO: GSM4616535 |
| Pig iPS cell RNA-seq: N01F_N2_iPS_P3 | GSE15249395 | GEO: GSM4616536 |
| Mouse ES cell RNA-seq: WT ESC -1 | GSE5321296 | GEO: GSM1287734 |
| Mouse ES cell RNA-seq: WT ESC -2 | GSE5321296 | GEO: GSM1287735 |
| Mouse ES cell RNA-seq: WT ESC -3 | GSE5321296 | GEO: GSM1287745 |
| Mouse ES cell RNA-seq: WT ESC -4 | GSE5321296 | GEO: GSM1287746 |
| Mouse ES cell RNA-seq: iPSC -1 | GSE5321296 | GEO: GSM1287736 |
| Mouse iPS cell RNA-seq: WT iPSC -2 | GSE5321296 | GEO: GSM1287747 |
| Mouse iPS cell RNA-seq: WT iPSC -3 | GSE5321296 | GEO: GSM1287748 |
| Mouse fibroblasts RNA-seq: WT MEFs -1 | GSE5321296 | GEO: GSM1287749 |
| Mouse fibroblasts RNA-seq: WT MEFs -2 | GSE5321296 | GEO: GSM1287750 |
| Human naive ES cell RNA-seq: Naive_Ex1 | GSE14499497 | GEO: GSM4303994 |
| Human naive ES cell RNA-seq: Naive_Ex2 | GSE14499497 | GEO: GSM4303995 |
| Human primed ES cell RNA-seq: Primed_Ex1 | GSE14499497 | GEO: GSM4304018 |
| Human primed ES cell RNA-seq: Primed_Ex2 | GSE14499497 | GEO: GSM4304019 |
| Human H9 ES cells RNA-seq: H9.WT.1.1 _RNA-seq | GSE14099798 | GEO: GSM4192140 |
| Human H9 ES cells RNA-seq: H9.WT.1.2_RNA-seq | GSE14099798 | GEO: GSM4192141 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| Rhinolophus ferrumequinum embryonic fibroblasts | This paper | BEF |
| Rhinolophus ferrumequinum induced | This paper | RFe.iPS#1(BiPS1) |
| pluripotent stem cells clone 1 | ||
| Rhinolophus ferrumequinum induced | This paper | RFe.iPS2 (BiPS#2) |
| pluripotent stem cells clone 2 | ||
| Myotis myotis uropatagium fibroblasts | This paper | MMy.BUF |
| Myotis myotis induced pluripotent stem cells | This paper | MMy.iPS |
| 293FT cells | Life Technologies | R700-07 |
| NIH/3T3 cells | ATCC | CRL-1658 |
| R1 mouse ES cells | ATCC | SCRC-1011 |
| H9 human ES cells | WiCell | WA09 |
| Vero-E6 | ATCC | CRL-1587 |
| Irradiated CF1 mouse fibroblasts | Gibco | A34181 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| 8-week-old male Fox Chase SCID Beige Mice | Charles River | 250 |
|
| ||
| Oligonucleotides | ||
|
| ||
| SeV-F: GGATCACTAGGTGATATCGAGC | CytoTune 2.0 | N/A |
| SeV-R: | CytoTune 2.0 | N/A |
| ACCAGACAAGAGTTTAAGAGATATGTATC | ||
| KOS-F: ATGCACCGCTACGACGTGAGCGC | CytoTune 2.0 | N/A |
| KOS-R: ACCTTGACAATCCTGATGTGG | CytoTune 2.0 | N/A |
| Klf4-F TTCCTGCATGCCAGAGGAGCCC | CytoTune 2.0 | N/A |
| Klf4-R: AATGTATCGAAGGTGCTCAA | CytoTune 2.0 | N/A |
| cMyc-F: TAACTGACTAGCAGGCTTGTCG | CytoTune 2.0 | N/A |
| cMyc-R: TCCACATACAGTCCTGGATGATGATG | CytoTune 2.0 | N/A |
| GAPDH_F1_GHB: TGGTGAAGGTCGGAGTGAAC | This paper | Z25-132 |
| GAPDH_R1_GHB: GAAGGGGTCATTGATGGCGA | This paper | Z25-133 |
|
| ||
| Software and algorithms | ||
|
| ||
| FastQC vO.11.9 | Andrews et al.99 | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| Trimmomatic v0.39 | Bolger et al.100 | http://www.usadellab.org/cms/?page=trimmomatic |
| HISAT2 v2.2.1 | Kim et al.101 | http://daehwankimlab.github.io/hisat2/ |
| SAMtools v1.10 | Li et al.102 | https://github.com/samtools/samtools/releases/ |
| featureCounts v2.0.1 | Liao et al.103 | https://subread.sourceforge.net/ |
| deepTools | Rami’rez et al.104 | https://deeptools.readthedocs.io/en/develop/content/tools/bamCoverage.html |
| DESeq2 v1.10.1 | Love et al.105 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| ClustVis (Beta) | Metsalu and Vilo106 | https://biit.cs.ut.ee/clustvis/ |
| R package ggpubr | Wickham and Grolemund107 | https://cran.r-project.org/web/packages/ggpubr/index.html |
| ggplot2 | Wickham and Grolemund107 | cran.r-project.org/web/packages/ggplot2/index.html |
| ggmaplot | Wickham and Grolemund107 | https://rpkgs.datanovia.com/ggpubr/reference/ggmaplot.html |
| ggscatter | Wickham and Grolemund107 | https://rpkgs.datanovia.com/ggpubr/reference/ggscatter.html |
| codeml package of the PAML suite software | Yang et al.108 | http://abacus.gene.ucl.ac.uk/software/paml.html |
| bedGraphToBigWig v4 | Kent et al.109 | https://www.encodeproject.org/software/bedgraphtobigwig/ |
| Enrichr | Xie et al.110 | maayanlab.cloud/Enrichr/ |
| Cytoscape Version 3.8.2 | Shannon et al.111 | https://cytoscape.org |
| SMRTLink version 10.1 | PacBio | https://www.pacb.com/support/software-downloads/ |
| Kraken2 v2.1.2 | Wood et al.112 | https://github.com/DerrickWood/kraken2 |
| gmap/gsnap | Wu et al.113 | http://research-pub.gene.com/gmap |
| Trimmgalore v.0.6.6 | Github, Open Source | https://github.com/FelixKrueger/TrimGalore |
| Cutadapt | Krueger n.d. Andrews114 | https://github.com/marcelm/cut adapt |
| stringTie v2.2.1 | Pertea et al.115 | http://ccb.jhu.edu/software/stringtie/index.shtml?t=manual |
| Seqtk v1.3 | Github, Open Source | https://github.com/lh3/seqtk |
| Trinity v2.12 | Grabbherr et al.116 | https://github.com/trinityrnaseq/trinityrnaseq/releases/tag/v2.12.0 |
| BLAST | Altschul et al.117 Camacho118 | https://blast.ncbi.nlm.nih.gov/Blast.cgi |
| SQUID | Eddy Lab, Harvard University | http://eddylab.org/software.html |
| INSPIRE 2.0 | Luminex | https://www.luminexcorp.com |
| IDEAS 6.2 | Luminex | https://www.luminexcorp.com |
|
| ||
| Other | ||
|
| ||
| ATAC-seq service | Active Motif | 25079 |
| RRBS Service, Active Motif, | Active Motif | 25069 |
| HistoPath ChIP-seq service, | Active Motif | 25001 |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Bats
Bat embryonic fibroblast isolation
A pregnant female bat of the species Rhinolophus ferrumequinum was captured with a hand-net in a roost near Jerez de la Frontera, Cádiz, Southern Spain in May 2020. Following the guidelines of the American Society of Mammalogists (2016) and with the approval of the Spanish bio-ethical authority and Andalusian regional government, the bat was euthanized by cervical dislocation and different tissues and the fetus were preserved in RPMI medium at 4°C till the processing of the samples 25 h later. In the lab, the female embryo (approximately developmental stage 20) acquired from a Spanish Rhinolophus ferrumequinum (RFe) was cut into several pieces while removing the head and as much of the inner organ tissue as possible. The pieces were then flushed with DPBS (Life Technologies, 14190144), and processed separately. The tissue was covered with 0.05% trypsin-EDTA (Life Technologies, 25300–120), minced with a scalpel, and incubated in a cell culture incubator at 37°C and 5% CO2 for 45 minutes. The trypsin was deactivated with fibroblast medium consisting of DMEM (Life Technologies, 10569–010), 10% fetal bovine serum (Sigma, F4135), 0.1 mM MEM Non-essential amino acids (Life Technologies, 11140–050), 2 mM GlutaMax supplement (Life Technologies, 35050061) and Penicillin-Streptomycin (10 U/ml and 10 μg/ml, respectively: Life Technologies, 15140122). The RFe cells were broken up by pipetting up and down 20 times, collected by centrifugation, transferred to a gelatin-coated (Sigma-Aldrich, G1890) T75 cell culture-treated flasks (Falcon, 353136) in 15 ml of fibroblast medium, and cultured at 37°C and 5% CO2. After 3 days, when reaching ~80% confluency, the attached RFe cells were washed with DPBS (Life Technologies, 14190144), treated with 0.05% trypsin-EDTA (Life Technologies, 25300120) to obtain a single cell solution and either split at a ratio of 1:4 or used directly in a reprogramming experiment.
Myotis myotis sampling and isolation of fibroblasts from tail biopsies
M. myotis (MMy) were sampled in Morbihan, Brittany in North-West France, July 2021 in accordance with the permits and ethical guidelines issued by ‘Arrêté’ by the Préfet du Morbihan and the University College Dublin ethics committee. This population has been transponded and followed since 2010 as part of on-going mark-recapture studies by Bretagne Vivante and the Teeling laboratory.119 Once captured, all bats were placed in individual cloth bags before processing. A single 3 mm biopsy was taken from the outstretched uropatagium of each bat using a sterile biopsy punch and immediately submerged in a cryotube (Sarstedt, 72.379) with 2 ml of DMEM cell culture medium (Gibco, 11995–065) supplemented with 20% FBS (Gibco, 10500064), 1% NEAA (Gibco, 11140050) and 1% Antibiotic-Antimycotic containing Streptomycin, Amphotericin B and Penicillin (Gibco, 15240096), maintaining as sterile conditions as possible. All bats were offered food and water and rapidly released after processing. Biopsies were then stored at 4°C and transported to the laboratory for processing within 6 days. Samples were further processed through a cell extraction methodology similar to a previously established protocol35 with a few modifications. The samples were rinsed with DPBS (Biowest, L0615-500) and cut finely within a minimal amount of cell culture medium using sterile blades (Swann-Morton, 0208) to result in six 0.5 mm pieces. These pieces were then transferred aseptically to a cryotube containing cell culture medium and incubated for 18 hours with collagenase type II (Life Technologies, 17101–015) at 37°C with 5% CO2 to allow for digestion. The pieces were collected by centrifugation for 5 minutes at 300 rcf, resuspended in 2 ml of fresh cell culture medium and transferred to a 35 mm cell culture treated plate (Corning, CLS430165) for initial P1 expansion. MMy cells were then fed every 2–3 days with cell culture medium as stated but a reduced 0.2% concentration of Antibiotic-Antimycotic. For the first feeding a ⅔ media change is performed to avoid sudden changes in antibiotic-antimycotic concentration from 1% to 0.2%. When the MMy cells reached 70% confluency, they were transferred to a T25 (Cellstar, 690175) in cell culture medium after treatment with 0.05% Trypsin (Gibco, 25300054) and were fed every 2–3 days as necessary. At 85% confluency, the MMy cells were trypsinized as before and 1×106 cells were frozen in 1 ml cell culture medium containing 10% DMSO (Sigma, D8418-100ML) within a cryotube through the use of a cell freezer (Thermo Scientific, 5100–0036) placed overnight in a −80°C freezer with subsequent transfer to long term storage in liquid nitrogen at −150°C. The particular MMy fibroblasts used in this study were isolated from one female bat.
Cell lines
293FT cells (Invitrogen, R700-07) embryonal kidney cells were cultured in DMEM (Gibco, 10569010) supplemented with 10% FBS (Sigma, F4135), 0.1 mM MEM Non-essential amino acids (Gibco, 11140–050), 2 mM GlutaMax supplement (Gibco, 35050–061), 1 mM Sodium Pyruvate (Gibco, 11360070) and Penicillin-Streptomycin (10 U/ml and 10 μg/ml, respectively; Gibco, 15140122). NIH/3T3 cells (ATCC, CRL-1658) cells were cultured in DMEM (Gibco, 10569010) supplemented with 10% FBS (Sigma, F4135). R1 mouse ES cells were cultured in DMEM (Gibco, 10569010) supplemented with 10% FBS (Sigma, F4135), 0.1 mM MEM Non-essential amino acids (Gibco, 11140–050), 2 mM GlutaMax supplement (Gibco, 35050–061), Penicillin-Streptomycin (10 U/ml and 10 μg/ml, respectively; Gibco, 15140122), 100 μM 2-mercaptoethanol (Fluka, 63689) and 10^4 U/ml Leukemia inhibitory factor (Millipore, ESG1107) on cell-culture dishes that were pretreated with 0.01% gelatin (Millipore, ES006B). Cell lines were split with 0.05% Trypsin-EDTA (Gibco) if not indicated otherwise. Bat induced pluripotent stem cells were cultured in DMEM/F-12 (Gibco, 11330–057) supplemented with 20% knockout serum replacement (Gibco, 10828–028), 0.1 mM MEM Non-essential amino acids (Gibco, 11140–050), 2 mM GlutaMax supplement (Gibco, 35050–061), Penicillin-Streptomycin (10 U/ml and 10 μg/ml, respectively; Gibco, 15140122), 100 μM 2-mercaptoethanol (Fluka, 63689), 100 ng/ml FGF2 (R&D Systems, 233-FB), 10^4 U/ml Leukemia inhibitory factor (Millipore, ESG1107), 100 ng/ml SCF (R&D Systems, PHC2111) and 20 nM Forskolin (Sigma, F6886). RFe and MMy iPSCs were cultured on a feeder layer of irradiated mouse embryonic fibroblasts (CF1, Gibco) and passaged with Gentle Cell Dissociation Reagent (GCDR, StemCell Technologies, 07174). After that, cells were passaged approximately every 5 days, or when they were confluent at a ratio of 1:6 to 1:12 on irradiated MEFs. H9 human embryonic stem cells were cultured in mTeSR medium (StemCell Technologies, 85850) on Vitronectin-coated (StemCell Technologies, 07180) cell culture dishes. H9 cells were split with Versene (Gibc, 15040066) at a ratio of 1:16.
SCID Beige Mice
8-week-old male Fox Chase SCID Beige mice (250) were retrieved from Charles River. All procedures were performed in accordance with protocol IACUC-2013-1433.
METHOD DETAILS
Reprogramming of bat fibroblasts
150,000 embryonic Rhinolophus ferrumequinum fibroblasts at passage 2, adult Myotis myotis at passage 3 were resuspended in 1 ml of fibroblast medium and mixed with Sendai-virus particles containing the reprogramming factors Oct4, Sox2, cMyc and Klf4 (CytoTune iPS 2.0, Invitrogen, A16517) with a final multiplicity of infection (MOI) of 10, 10, 10, 20, respectively. The RFe and MMy cells were plated on one gelatin-coated well of a 6-well plate (10 cm2, Corning, 353046) and cultured at 37°C with 5% CO2. The medium was replaced every 24 hours. 6 days after transduction, the RFe and MMy cells of each well were collected by treatment with 0.05% trypsin-EDTA, seeded at a density of 50,000 cells per 60 cm2 on irradiated CF1 mouse embryonic fibroblasts (MEFs; Gibco, A34180) in fibroblast medium. After 24 hours, the medium was switched to 50 % fibroblast medium and 50% pluripotent stem cell (PSC) medium consisting of DMEM/F-12 (Life Technologies, 11330–057), 20% knockout serum replacement (Life Technologies, 10828–028), 0.1 mM MEM Non-essential amino acids (Life Technologies, 11140–050), 2 mM GlutaMax supplement (Life Technologies, 35050–061), Penicillin-Streptomycin (10 U/ml and 10 μg/ml, respectively; Life Technologies, 15140122), 100 μM 2-mercaptoethanol (Fluka, 63689), and 40 ng/ml FGF2 (R&D Systems, 233-FB). From then on, the medium was replaced every day with PSC medium until day 14 when the FGF concentration was increased to 100 ng/ml and the medium was supplemented with 104 U/ml Leukemia inhibitory factor (Millipore, ESG1107), 100 ng/ml SCF (R&D Systems, PHC2111) and 20 nM Forskolin (Sigma, F6886). Colonies appeared 14 to 16 days after transduction, were picked on day 20 and expanded on irradiated MEFs with Gentle Cell Dissociation Reagent (GCDR, StemCell Technologies, 07174). After that, the RFe and MMy cells were passaged approximately every 5 days, or when they were confluent at a ratio of 1:6 to 1:12 on irradiated MEFs. Cell and colony morphology were recorded with an EVOS digital inverted microscope (Invitrogen).
Karyotyping
RFe iPSCs were treated with 100 ng/ml Colcemid solution (Life Technologies, 15210040) for 16 hours, then treated with 0.05% trypsin-EDTA for 15 minutes and filtered through a 40 μm cell strainer to remove clumps. RFe cells were collected by centrifugation, resuspended in 1 ml 0.075 M potassium chloride (Sigma-Aldrich, P9327) and incubated for 20 minutes at room temperature. 0.5 ml fixative [1 part glacial acetic (Fisher Scientific, A38-212) mixed with 3 parts methanol (Sigma-Aldrich, A412-4)] were added, RFe cells were collected as before, resuspended in 4 ml fixative, and incubated for 20 minutes at room temperature. The fixation step was repeated, the RFe cells collected as before and all but about 200 μl of the fixative was removed. The cells were resuspended in the remaining fixative and dropped onto slides that were precooled at −20°C. The slides were airdried and the RFe cells stained for 10 minutes with Giemsa Staining solution consisting of 1 part Giemsa solution (Life Technologies, 10092013), and 3 parts Gurr buffer (Invitrogen, 10582013). The slides were washed with water, dried, and mounted in Cytoseal 60 (Thermo Scientific, 23-244257). High-resolution pictures of chromosome spreads were acquired with an AxioObserver microscope (Zeiss) using the 100x oil objective.
RT-PCR
mRNA was extracted with the RNeasy Mini Kit (Qiagen, 74104). 500 ng of each sample were used to generate cDNA by reverse transcription using the SuperScript™ IV VILO™ Master Mix (Invitrogen, 11756050). 2 μl of the cDNA were used to detect the presence of Sendai virus transcripts using GoTaq Green Polymerase (Promega, M7123), and the oligos as recommended in the CytoTune iPS 2.0 kit (Invitrogen, A16517). Gapdh was amplified as loading control using oligos with the following sequence: Z25-132:GAPDH_F1_GHB: TGGTGAAGGTCGGAGTGAAC and Z25-133:GAPDH_R1_GHB: GAAGGGGTCA TTGATGGCGA). The PCR products were analyzed on a 2% agarose gel containing ethidium bromide.
Immunofluorescence staining
RFe and MMy cells were plated on μ-slides (Ibidi, 80286). After 4 days, cells were washed once with DPBS and fixed with Cytofix/Cytoperm solution (Becton Dickinson, BDB554714) for 20 minutes at 4°C. Cells were rinsed with Perm/Wash buffer (Becton Dickinson, BDB554714) and then incubated overnight at 4°C in Perm/Wash buffer containing primary anti-Afp (R&D Systems, AF1369), anti-Pax6 (BioLegend, 901301), J2 anti-dsRNA (Scicons, 10010200), anti-(gag/pol)HERVK (Austrial Biological, HERM18315), FIPV3-70 anti-Pan Corona (Life Technologies, MA1-82189) or directly conjugated anti-Oct3/4-AF488 (Santa Cruz, sc-5279-AF488), anti-Brachyury (R&D Systems, IC2085G), anti-Otx2 (R&D Systems, AF1979), anti-Zic2 (Abcam, ab150404), anti-Tfe3 (Sigma Aldrich, HPA023881), anti-Tfcp2l1 (R&D Systems, AF5726) or anti-Cdx2 (Life Technologies, PA5-20891) in a 1:50 (anti-Oct3/4) or 1:100 dilution (all others). RFe and MMy cells were rinsed and washed 3 times for 2 minutes with Perm/Wash solution at room temperature followed by a 1-hour incubation with a 1:200 dilution of the corresponding secondary antibodies (Donkey anti-chicken-Cy3, Millipore, AP194C; Goat anti-chicken-AF488, Life Technologies, A-11039; Donkey anti-rabbit-AF647, Life Technologies, A-31573; Goat anti-rabbit-AF488, Life Technologies, A-10034; Goat anti-mouse-AF488, Life Technologies, A-11029) in Perm/Wash buffer. RFe and MMy cells were rinsed, washed twice for 2 minutes with Perm/Wash Buffer and then incubated for 5 minutes with Perm/Wash buffer containing 2 drops per ml NucBlue Dapi stain (Invitrogen, R37606). The buffer was removed, and the cells were cover-slipped in Prolong Dimond antifade mounting medium (Invitrogen, P36965). Images were acquired with an AxioObserver fluorescence microscope with Apotome (Zeiss). For the simulated emission depletion (STED) microscopy (super-resolution), the RFe cells were plated on coverslips that were placed in wells of 6-well plates. The staining was performed as described above but with a 1:200 dilution of the Abberior Star 635P (Abberior, ST635P) secondary antibody in Perm/Wash buffer. The RFe cells were rinsed, washed twice for 2 minutes with Perm/Wash Buffer and then incubated for 5 minutes with Perm/Wash buffer containing 2 drops per ml DyeCycle Violet stain (Invitrogen, V35003). The coverslips were mounted face down on glass slides with Prolong Dimond antifade mounting medium (Invitrogen, P36965). Images were acquired with a TCS SP8 confocal microscope with STED 3x and White Light Laser (Leica) with a 100x oil objective. 405 nm and 594 nm lasers were used for excitation and 775 nm laser for depletion. Image resolution obtained was 19.8 μm by 19.8 μm using a zoom factor of 6x.
RNA-seq, differential expression analyses and visualization
For RNA-seq, RNA was extracted from RFe iPSCs cells at passage 22, RFe BEFs at passage 3, MMy iPSCs at passage 7 and MMy fibroblasts at passage 4. Total RNA was extracted with the RNeasy kit (Qiagen, 74104) following the manufacturer’s recommendations including the DNase (Qiagen, 79254) digest, and eluted in 50 μl RNase/DNase free H2O. RNA-seq libraries were prepared with the SMART-Seq v4 Ultra Low Input kit (Takara Bio, undifferentiated cells) or the Stranded Total RNA with Ribo-Zero Plus kit (Illumina, differentiated cells) and 100 bp paired-end sequencing reads (PE100) were generated by Illumina sequencing (NovaSeq 6000 S1) to a depth of at least 50 million reads (100 million total reads). The quality of the reads from the RNA sequencing was analysed with FastQC v0.11.9,99 and visualized using MultiQC v1.9.120 The mean phred score was around Q35 across each base position in the RFe and BEF and BiPS samples and no filtering or processing was necessary. In the differentiated samples (EB, endoderm, mesoderm, ectoderm differentiation), the quality of the first nucleotide was less than Q20 in many cases and the reads were processed with Trimmomatic v0.39.100 To carry out the differential expression analysis, the genome of Rhinolophus ferrumequinum was used as reference genome (RefSeq accession GCF_004115265.1 assembled and annotated by the Vertebrate Genomes Project (https://vertebrategenomesproject.org) or GenBank accession GCA_014108255.1 assembled and annotated by the Bat1K project (https://bat1k.com) as indicated. The reads were mapped with HISAT2 v2.2.1,101 the .sam files resulting from each mapping were converted into .bam files and indexed using SAMtools v1.10102 and the reads mapped against each gene were counted using featureCounts v2.0.1.103 The differential expression analysis was performed with DESeq2 v1.10.1.105 To visualize the RNA-seq data in the UCSC genome browser, bigwig files were generated using the bamCoverage command from deepTools (https://deeptools.readthedocs.io/en/develop/content/tools/bamCoverage.html).104
MA plot
The MA plots were generated based on the DESeq2 (see above) results with the ggmaplot function (https://rpkgs.datanovia.com/ggpubr/reference/ggmaplot.html) from the R package ggpubr (https://rpkgs.datanovia.com/ggpubr/).107 Genes are indicated by dots, plotted by their log2 fold change between RFe bat fibroblast and RFe pluripotent stem cells and the log2 mean of normalized counts (ratio of means). Blue dots indicate genes with an adjusted p value of (or FDR) of <0.05 and a fold change of 2 (log2 fold change of 1), red dots indicate genes with an adjusted p value (or FDR) of <0.05 and fold change of −2 (log2 fold change of −1). Dotted lines are drawn at fold change of 2/−2 (log2 fold change of 1/−1).
Heatmap
The heatmap of pluritest genes, with ClustVis (Beta), a web tool for visualizing clustering of multivariate data (https://biit.cs.ut.ee/clustvis/)).106 Rows are centered; unit variance scaling is applied to rows. Both rows and columns are clustered using correlation distance and average linkage.
ATAC-seq
ATAC-seq and bioinformatics analysis to detect open chromatin in RFe bat fibroblasts and RFe bat pluripotent stem cells was performed by Active Motif from 100,000 cryopreserved cells (ATAC-seq service, 25079). In brief, nuclei were isolated, and libraries of open chromatin were prepared with the Nextera Library Prep Kit (Illumina) by Tn5 tagmentation. The tagmented DNA was purified using the MinElute PCR purification kit (Qiagen), amplified with 10 cycles of PCR, and purified using Agencourt AMPure SPRI beads (Beckman Coulter). 42 bp paired-end sequencing reads (PE42) were generated by Illumina sequencing (using NextSeq 500) to a depth of at least 83 million total reads and mapped to the GCA_004115265.2 genome (Ensembl, annotation version 102) using the BWA algorithm with default settings (“bwa mem”). Alignment information for each read was stored as BAM file. Only reads that passed the Illumina’s purity filter, aligned with no more than 2 mismatches, and mapped uniquely to the genome were used in the subsequent analysis. Duplicate reads (“PCR duplicates”) are removed. Genomic regions with high levels of transposition/tagging events were then determined using the MACS2 peak calling algorithm.121 To identify the density of transposition events along the genome, the genome was divided into 32 bp bins and the number of fragments in each bin was determined. The data were then normalized by reducing the tag number of all samples by random sampling to the number of tags present in the smallest sample. Peak metrics between samples were compared by grouping overlapping Intervals into “Merged Regions”, which are defined by the start coordinate of the most upstream Interval and the end coordinate of the most downstream Interval (= union of overlapping Intervals; “merged peaks”). In locations where only one sample has an Interval, this Interval defines the Merged Region. Intervals and Merged Regions, their genomic locations along with their proximities to gene annotations and other genomic features were determined and average and peak (i.e., at “summit”) fragment densities were compiled. The sequencing tracks (number of fragments in each 32 bp bin stored as “.bigWig” file) were visualized with the UCSC genome browser.
Reduced representation bisulfite sequencing (RRBS)
Reduced representation bisulfite sequencing of RFe bat fibroblasts and pluripotent stem cells was performed by Active Motif (RRBS Service, Active Motif, 25069). Briefly, 500,000 cells were provided as a frozen pellet. Genomic DNA was isolated, and 100 ng were digested with TaqaI (NEB R0149) at 65°C for 2 hours followed by MspI (NEB R0106) at 37°C overnight. Following enzymatic digestion, samples were used for library generation with the Ovation RRBS Methyl-Seq System (Tecan, 0353–32) following the manufacturer’s instructions. In brief, digested DNA was randomly ligated, and, following fragment end repair, bisulfite converted using the EpiTect Fast DNA Bisulfite Kit (Qiagen, 59824) following the Qiagen protocol. After conversion and clean-up, samples were amplified resuming the Ovation RRBS Methyl-Seq System protocol for library amplification and purification. 75 bp single-end sequencing reads (SE75) were generated by Illumina sequencing (using NextSeq 500) to a depth of at least 27 million reads (total of 54 million reads), with at least 2.9 million covered CpGs. The reads were mapped to the GCA_004115265.2 genome (Ensembl, annotation version 102) and the percentage of methylation at CpG sites across the genome was calculated. To visualize the methylation ratios aligned to the gnome with the UCSC genome browser, the methylation ratio files containing the methylation ratio for each chromosomal position were first converted to bed files, that were then used to generate bigwig files with the “bedGraphToBigWig v4 tool” (https://www.encodeproject.org/software/bedgraphtobigwig/).109 Correlation scatter plots were generated to show the level of methylation at common CpG sites.
Chromatin immunoprecipitation sequencing (ChIP-seq)
5 million RFe iPS cells were fixed cells in 1% formaldehyde by adding 1/10 volume of freshly prepared formaldehyde solution (11% formaldehyde, 0.1 M NaCl, 1 mM EDTA, pH 8.0, 50 mM HEPES, pH 7.9) to the existing medium. The RFe cells were agitated for 15 minutes at room temperature and the fixation was stopped by addition of 1/20 volume of 2.5 M glycine solution (final concentration of 0.125 M) to the existing medium and incubation at room temperature for 5 minutes. The RFe cells were scraped off the wells, collected by centrifugation at 800 g and washed with 10 ml chilled 0.5 % Igepal in PBS per tube by pipetting up and down. RFe cells were pelleted by centrifugation as before and resuspended in 10 ml chilled PBS-Igepal containing 1 mM PMSF. RFe cells were collected as before, and the cell pellet was snap-frozen in liquid nitrogen. Further processing, chromatin immunoprecipitation and bioinformatics analysis to detect H3K4me3 and H3K27me3 was performed by Active Motif (HistoPath ChIP-seq service, 25001). In brief, chromatin was isolated by adding lysis buffer, followed by disruption with a dounce homogenizer. Lysates were sonicated and the DNA sheared to an average length of 300–500 bp with an EpiShear probe sonicator (Active Motif, 53051). Genomic DNA (Input) was prepared by treating aliquots of chromatin with RNase, proteinase K and heat for de-crosslinking, followed by SPRI beads clean up (Beckman Coulter) and quantitation with Clariostar (BMG Labtech). An aliquot of chromatin (20 μg) was precleared with protein A agarose beads (Life Technologies). Genomic DNA regions of interest were isolated using 4 μg of antibody against H3K4me3 (Active Motif, 39159) or H3K27me3 (Active Motif, 39155). Complexes were washed, eluted from the beads with SDS buffer, and subjected to RNase and proteinase K treatment. Crosslinks were reversed by incubation overnight at 65°C, and ChIP DNA was purified by phenol-chloroform extraction and ethanol precipitation. Illumina sequencing libraries were generated from the ChIP and Input DNAs with the standard consecutive enzymatic steps of end-polishing, dA-addition, and adaptor ligation. After a final PCR amplification step, 75-nt single-end (SE75) sequence reads were generated by Illumina sequencing (using NextSeq 500) to a depth of at least 36 million reads per sample and mapped to the GCA_004115265.2 genome (Ensembl, annotation version 102) using the BWA algorithm with default settings. Duplicate reads were removed, and only uniquely mapped reads (mapping quality >= 25) were used for further analysis. Alignments were extended in silico at their 3’-ends to a length of 200 bp, which is the average genomic fragment length in the size-selected library and assigned to 32-nt bins along the genome. The resulting histograms (genomic “signal maps”) were stored in “bigwig” files. To find peaks, the generic term “Interval” was used to describe genomic regions with local enrichments in tag numbers. Intervals were defined by the chromosome number and a start and end coordinate. Peak locations were determined using the MACS algorithm (v2.1.0) with a cutoff of p-value = 1e-734 Signal maps and peak locations were used as input data to Active Motifs proprietary analysis program, which creates Excel tables containing detailed information on sample comparison, peak metrics, peak locations and gene annotations. No normalization was performed on the H3K27me3 data, while standard normalization was applied to the H3K4me3 data. The tag number of all samples (within a comparison group) was reduced by random sampling to the number of tags present in the smallest sample. To compare peak metrics between 2 or more samples, overlapping Intervals were grouped into “Merged Regions”, which are defined by the start coordinate of the most upstream Interval and the end coordinate of the most downstream Interval (= union of overlapping Intervals; “merged peaks”). In locations where only one sample has an Interval, this Interval defines the Merged Region. The sequencing tracks (number of fragments in each 32 bp bin stored as a “bigwig” file) were visualized with the UCSC genome browser.
Three germ layer differentiation
The differentiation of RFe and MMy bat pluripotent stem cells was carried out with the STEMdiff Trilineage differentiation kit (StemCell Technologies, 05230) following the manufacturer’s protocol. RFe and MMy cells were plated at the desired densities in mTeSR medium (StemCell Technologies, 85850), and plated on Vitronectin-coated (StemCell Technologies, 07180) cell culture plates. After 5 days (endoderm or mesoderm) or 7 days (ectoderm) in culture as directed by the manufacturer. For the ectoderm differentiation, the floating three-dimensional structures were then replated and grown for 4 additional days in fibroblast medium. The RFe and MMy cells were stained with antibodies detecting the appropriate lineage markers as described above or cells were collected (surface area of 10 cm2 per replicate) for RNA isolation and RNA-seq after addition of 600 μl lysis buffer RTL (part of the RNeasy kit; Qiagen, 74104).
Embryonic body differentiation
RFe bat pluripotent stem cells grown on irradiated mouse embryonic fibroblasts from a total area of 60 cm2 were washed with PBS, treated for 10 minutes with Gentle Cell Dissociation Reagent (StemCell Technologies, 07174), collected by centrifugation and resuspended in 12 ml differentiation medium consisting of DMEM/F-12 (Life Technologies, 11330–057), 10% fetal bovine serum (Sigma, F4135), 0.1 mM MEM Non-essential amino acids (Life Technologies, 11140–050), 2 mM GlutaMax supplement (Life Technologies, 35050–061), Penicillin-Streptomycin (10 U/ml and 10 μg/ml, respectively; Life Technologies, 15140122) and 100 μM 2-mercaptoethanol (Fluka, 63689). The RFe cells were then transferred to one uncoated 60 cm2 petri dish (Corning, 351029). After 3 days in culture, as much as possible of the medium (about 2/3) was carefully exchanged without disturbing and removing the floating EBs that had formed. The floating EBs were collected after 3 more days (total of 6 days) in culture, fixed in Cytofix/Cytoperm fixation buffer (Becton Dickinson, BDB554714) overnight, and then stained with antibodies against as described above to detect differentiation markers of all three germ-layers by immunofluorescence. For RNA isolation and RNA-seq, EBs were formed as described, collected, resuspended in 6 ml differentiation medium, and distributed into three wells of cell-culture treated 6-well plates (10 cm2 each). After 2 more days in culture, the cells were washed with PBS, lysed with 600 μl buffer RTL (part of the RNeasy kit; Qiagen, 74104) and RNA was isolated as described above.
Blastoid differentiation
RFe iPSCs were harvested and plated as described for the embryonic body formation above. After 3 days in culture, 100 ng/ml BMP4 (R&D Systems, 314-BP-010) were added to the medium. 24 hours later the supernatant was diluted with 2/3 of fresh medium and transferred to two fresh uncoated petri dishes. The medium was exchanged after 3 more days in culture and floating blastoids were harvested 4 days later (total of 12 days of differentiation). The blastoids were fixed in Cytofix/Cytoperm fixation buffer (Becton Dickinson, BDB554714) overnight, and stained as described above to detect the expression of Oct4 and Cdx2 by immunofluorescence microscopy.
Teratoma formation
Two 6-well plates (12 wells) of RFe pluripotent stem cells grown on irradiated mouse embryonic fibroblasts were scraped off in 2 ml DMEM/F-12 medium (Life Technologies, 11330–057), collected by centrifugation and resuspended in 500 μl DMEM/F-12 medium. 100 μl of the cell suspension were injected into the hindleg muscle of 8-week-old male Fox Chase SCID Beige Mice (Charles River, 250). Tumor tissue that had formed after 16 weeks was harvested, fixed in 10% Formalin (Fisher Scientific, SF1004) overnight and then transferred to 70% ethanol. The tissue was submitted to the Pathology Core at the Icahn School of Medicine at Mount Sinai for paraffin embedding and hematoxylin and eosin staining of 5 μm sections. Images were acquired with an AxioObserver microscope (Zeiss).
Western blot analyses
RFe iPSCs were lysed with RIPA lysis and extraction buffer (Fisher Scientific, 89900) containing Proteinase Inhibitor (Roche, 45582400) for 30 minutes on ice, debris was removed by centrifugation and the supernatant was transferred to a new tube. Protein concentration was determined using the BCA Protein assay kit (Pierce, 23252) following the manufacturers recommendation in 96 well format. 20 μg of protein isolated from each cell line were separated on a 12% TGX Precast gel (Bio-Rad, 4561044) for 70 minutes at 200 V in TGS buffer (Bio-Rad, 1610772), the Precision Plus Kaleidoscope Protein standard (Bio-Rad, 161–3075) was used as size marker. The protein was transferred to a PVDF membrane (activated with methanol) with 1x TGS Buffer (Bio-Rad, 161–0374) supplemented with 20% methanol using the Turbo blot system (Bio-Rad, 1704150) for 30 minutes at 25 V (1 mA). The membrane was blocked with EveryBlot blocking buffer (Bio-Rad, 12010020) for 30 minutes, and then incubated with the anti-Pan Corona (Invitrogen, MA1-82189) or anti-HERVK(cap)(Austral Biologicals, HERM18315) antibodies in a 1:1000 dilution in EveryBlot blocking buffer for 1-hour. The membrane was washed four times with Tris-buffered saline (Fisher Scientific, BP2471500) supplemented with 0.05% Tween 20 (Bio-Rad, 161–0781) for 5 minutes, incubated with the secondary anti-mouse-HRP antibody in a 1:1000 dilution (Promega, PR-W4021) for 1 hour, and then washed as before. Signals were developed with the Clarity Western ECL Substrate (Bio-Rad, 170–5060) and detected with the ChemiDoc MP Imaging System (Bio-Rad, 170–01402).
Principal component analysis (PCA)
The DESeq2 output files of the RNA-seq analyses described above were subjected to a Variance Stabilizing Transformation (VST) using within-group-variability122 to compare the RFe bat pluripotent stem cell transcriptional profile with that of other species. The first two principal components of this result were plotted using the ggscatter function (https://rpkgs.datanovia.com/ggpubr/reference/ggscatter.html) from the R package ggpubr (https://cran.r-project.org/web/packages/ggpubr/index.html),107 and the weight of each gene’s contribution to the principal components were extracted for PC1 and PC2. The datasets used in the PCA were: GSM4616525, GSM4616526, GSM4616527 (dog iPS), GSM4617887, GSM4617889, GSM4617890, GSM4617891, GSM46 17895, GSM4617900, GSM4617901 (marmoset iPS), GSM4616532 (human iPS), GSM4616535, GSM4616536 (pigiPS) from study GSE152493,95 GSM1287734, GSM1287735, GSM1287745, GSM1287746 (mouse ESC), GSM1287736, GSM1287747, GSM128 7748 (mouse iPS), GSM1287749, GSM1287750 (mouse fibroblasts, MEF) from GSE53212,96 and GSM4303994, GSM4303995 (human naïve ES), GSM4304018, GSM4304019 (human primed ES) from GSE144994.97 Note, that each analysis only induced genes that were annotated in all of the respective species.
Evolutionary selection analysis
To explore evidence of positive selection in R. ferrumequinum for the 674 genes identified as part of the “leading” edge in the PCA analysis described above, we extracted all gene alignments that were available for these transcripts (n = 491) and had previously been annotated,25 in addition to annotating 169 alignments that had been made available as part of BAT1K but were currently unannotated. These alignments contained a maximum of 48 species from all eutherian mammalian superorders, with the species tree published by Jebb et al.25 used for all selection analyses. A total of 660 of these alignments contained representative genes for R. ferrumequinum and were analysed for positive selection using the branch-site models in the codeml package of the PAML suite software.108 Positive selection was inferred using likelihood-derived dN/dS (ω) values under both a null (foreground and background ω constrained to be less than 1) and alternative (foreground ω can vary) model. The R. ferrumequinum lineage was designated as foreground branch to detect unique instances of taxon-specific positive selection. A likelihood ratio test (LRT, 2*lnLalt–lnL null) was used to compare the fit of both models, with a p-value calculated assuming chi-squared distributed LRTs. P-values were corrected for multiple testing using the Benjamin-Hochberg False Discovery Rate (FDR) method via ‘padjust’ implemented in R. Any significant gene showing a p-value greater than 0.05 with ω >1 was explored further. Significant sites showing positive selection were identified using Bayes Empirical Bayes (BEB) scores with a probability > 0.95. All significant genes were subject to a visual inspection of the alignment, to rule out potential false positive results having occurred due to misaligned sequences. In addition to R. ferrumequinum, the M. myotis (n=637 representative genes), Homo sapiens (n=652), Mus musculus (n=628), Canis lupus (n=593) and Felis catus (n=603) lineages were also independently designated as foreground branches for all genes containing a representative sequence shared with R. ferrumequinum. This served as a means of determining whether positive selection identified in R. ferrumequinum was truly unique to the species lineage or a consequence of bat-specific, Laurasiatherian-specific, or eutherian mammal-specific instances of sequence evolution.
Gene ontology and KEGG pathway analyses
Gene ontology and KEGG pathways that are enriched within a group of genes were identified with the Enrichr tool (maayanlab.cloud/Enrichr/).110 The odd ratios were then plotted with ggplot2 (cran.r-project.org/web/packages/ggplot2/index.html)107 with the odds ratio displayed on the x-axis, the dot size reflecting the gene count (number of genes present in the top 5% of PC1 contributing genes) and the dot color reflecting the p-value.
Protein interaction network analysis
The genes of the Corona virus disease related KEGG pathway were retrieved from the PathCards database (https://pathcards.genecards.org). The differential expression analysis was performed between RFe bat (this study) and mouse iPS cells (GSM1287736, GSM1287747 and GSM1287748 from Study GSE5321295 using DESeq2.105 The Corona virus disease-related genes were then illustrated with Cytoscape (Version 3.8.2)111 using the STRING protein query with a 0.8 confidence score cutoff. The nodes were colored based on the log2 Fold-change with a negative (blue) fold change indicating down-regulation and a positive (red) fold change indicating upregulation in bat pluripotent stem cells. Bold borders indicate proteins that were present in the top 5% of PC1 in the PCA analysis described above.
Electron microscopy
RFe iPSCs were grown in chambered Permanox slides (LabTek, 70390) on irradiated mouse embryonic fibroblasts as described above for 5 days and then further processed by the Biorepository and Pathology core at the Icahn School of Medicine at Mount Sinai. Briefly, the RFe iPSCs were rinsed once with DPBS and fixed overnight with 2% paraformaldehyde and 2% glutaraldehyde in 0.01 M sodium cacodylate buffer at 4°C. Sections were rinsed in 0.1 M sodium cacodylate buffer, followed by a quick rinse with ddH2O. RFe iPSCs were post fixed with 1% aqueous osmium tetroxide for 1 hour, followed with an En bloc stain of 2% aqueous uranyl acetate for 1 hour. Sections were washed again in ddH2O, dehydrated through graduated ethanol (25–100%), infiltrated through an ascending ethanol/epoxy resin mixture (Embed 812, Electron Microscopy Sciences), and then covered with pure resin overnight. Chambers were separated from the slides, and a modified #3 BEEM embedding capsule (Electron Microscopy Sciences) was placed over defined areas containing cells. Capsules were filled with pure resin and placed in vacuum oven to polymerize at 60°C for 72 hours. Immediately after polymerization, the capsules were snapped from the substrate to dislodge the cells from the slide. Semi-thin sections (0.5 – 1 μm) were obtained using a Leica UC7 ultramicrotome (Leica, Buffalo Grove, IL), counterstained with 1% Toluidine Blue, cover slipped and viewed under a light microscope to identify successful dislodging of cells. Ultra-thin sections (85 nm) were collected on 300 hexagonal mesh copper grids (Electron Microscopy Sciences) using a Coat-Quick adhesive pen (EMS). Sections were counter-stained with uranyl acetate and lead citrate and imaged with a Hitachi 7700 Electron Microscope (Hitachi High-Technologies) using an advantage CCD camera (Advanced Microscopy Techniques). Images were adjusted for brightness, contrast, and size using Adobe Photoshop CS4 11.0.1.
Image-based flow cytometry (ImageStream)
RFe iPSCs were seeded onto 6-well plates and separated from irradiated MEFs via two-stage trypsinization after four days. Wells were dosed and incubated with 0.25ml prewarmed (37°C) trypsin which was removed and discarded at 4 minutes. An additional 0.25ml trypsin was added and the plate was again incubated. After eight minutes cells were removed and pelleted via centrifugation. The cells were washed twice in PBS containing 0.5% BSA (Sigma), fixed and permeabilized with Cytofix/Cytoperm (Invitrogen, C10337). The Primary antibody was added at a dilution of 1:200 in wash buffer incubated overnight at 4°C. The RFe cells were washed twice with 0.5% BSA/PBS, resuspended in wash buffer containing the secondary goat anti-mouse AF568 antibody (Life Technologies, A-11029) in a 1:200 dilution and incubated for 1 hour at 4°C. The RFe cells were washed as before resuspended in 0.5% BSA/PBS containing two drops/ml DyeCycle Violet to stain the nuclei. Imaging was conducted with the ImageStream MkII, at 60x magnification with the extended depth of field mode for probe resolution. Images were acquired using the INSPIRE 2.0 software at the lowest flow speed. Fluorophores were excited by the 405 nm and 568 nm lasers at 60 mW and 100 mW, respectively. RFe cells in focus were gated via histogram of brightfield gradient R.M.S. values and an aspect ratio vs. area plot was used to select the population of single cells. 5000 individual images of focused single cells were taken. Gating was refined further post-acquisition via the IDEAS 6.2 software suite by the same methods and plots, yielding n=1846 (BiPS). This software was used also for image processing, in which a set of custom masks defined by logical operators were used to denote vesicles and sensitively assess probes. For vesicles, it was observed that they may be selected from other cell component by contrast (bright and dark) and also by aspect ratio, and therefore are defined here by “Dilate(Range(Dilate(Range(System(Peak. (Threshold(M01, BF, 70), BF, Bright, 1), BF, 20), 0–5000, 0.4–1), 1), 0–5000, 0.4–1), 1) Or Range (AdaptiveErode(Level Set(M01, BF, Dim, 5), BF, 75), 0–5000, 0.5–1).” BF and BF2 represent each brightfield image taken of a single cell from each of the two cameras, M01 and M09 represent the corresponding channel masks for each channel and the remaining terms represent mask modifiers and their associated values in the IDEAS software. For resolving immunofluorescence, “Peak(System(M05, Ch05, 3), Ch05, Bright, 1)” where Ch05 represents the staining of interest and M05 represents the corresponding channel mask. Modification was necessary to sensitively include all representative fluorescence, and to distinguish individual foci. The nuclear mask corresponding to DyeCycle Violet staining was defined “Object(M07, Ch07, Tight)” and the cytoplasm was defined through subtraction of the nuclear and vesicle masks from the cell mask through the logical operator available in the software (“Not”). Vesicle-nucleus overlap was determined in favor of vesicles by excluding them from the nuclear mask (“Not”). Probe localization was then defined according to these entities using the respective definitions and the operator “And.” Statistics for foci were generated using the Spot Count feature with a connectedness of 4. Prism 9 was used for graphs and statistics.
Retrovirus assay
2 ml of tissue culture medium were collected, and retroviral particle concentrations were determined using the QuickTiter Retrovirus Quantitation Kit (Cell Biolabs, VPK-120) according to the manufacturer’s instructions.
Reverse transcriptase assay
Reverse transcriptase enzyme levels were determined with the colorimetric reverse transcriptase kit (Roche) per the manufacturer protocol. Cells lines represented were lysed in RIPA buffer, frozen at −80°C, thawed on ice, collected, and resuspended in the kit lysis buffer (10 μL pellet in 40 μL lysis buffer per colorimetric well). Incubation duration (15 h at 37°C) was selected for maximal sensitivity to the limit of the kit (1–5 pg RT). Absorbance at 405 nm was measured by microtiter ELISA plate reader. Sample absorbance measurements were fitted to a linear regression of the measured HIV-1 RT standards (Y=2.549X) to obtain RT concentrations in units of ng/well.
Plaque assay
Supernatants were centrifuged at 10000 rpm for 5 min to remove cellular debris, and the cleared lysates transferred to new tubes. Lysates were then diluted in 10-fold dilutions 6 times. Quantification of infectious titer was then performed by plaque assays in comparison to SARS-CoV-2 infection as positive control. Briefly, Vero-E6 cells were plated as confluent monolayers in 12 well dishes. Media was removed, and wells washed in 1 ml of PBS. 200 μl of diluted lysates were then added per well and allowed to incubate for 1 hour at 37°C. After viral adsorption, lysates were removed from the well and cells were overlaid with Minimum Essential Media supplemented with 2% FBS, 4 mM L-glutamine, 0.2% BSA, 10 mM HEPES and 0.12% NaHCO3 and 0.7% agar. 72h post infection, agar plugs were fixed in 10% formalin for 24h before being removed. Plaques were visualized by staining with TrueBlue substrate (KPL-Seracare) and viral titers calculated and expressed as PFU/ml.
Metapneumovirus (MPV) infection of BiPS and mES cells
50,000 mouse ES cells (R1) or RFe iPSCs were plated per well of a 12-well plate on irradiated CF1 mouse embryonic fibroblasts using mouse and bat culture medium respectively. After 24 hours, culture medium containing human Metapneumovirus with GFP (MPV-GFP) (ViraTree, M121) with a final multiplicity of infection (MOI) of 3. Medium was changed daily, and samples were dissociated at 3 and 5 dpi using 0.05% Trypsin-EDTA and the infection rate was determined by fluorescence activated cell sorting (FACS).
Iso-Seq library preparation and sequencing
RFe iPSCs and BEFs at passage 27 and passage 2–4 were lyzed in 400 μl Trizol reagent (Life Technologies, 15596–018) and total RNA was extracted using the AllPrep DNA/RNA Mini Kit (Qiagen, #80204) including a DNase digest to remove any potential contamination from carryover of genomic DNA using RNase-free DNase (Qiagen, 79254) according to the manufacturer’s instructions. The extracted RNA was then purified using 1.8X RNAClean XP beads (Beckman Coulter) to remove any molecular impurities. Iso-Seq SMRTbell libraries were prepared as recommended by the manufacturer (Pacific Biosciences). Briefly, 300 nanograms of total RNA (RIN > 8) from each sample was used as input for cDNA synthesis using the NEBNext Single Cell/Low Input cDNA Synthesis & Amplification Module (NEB, E6421L), which employs a modified oligodT primer and template switching technology to reverse-transcribe full-length polyadenylated transcripts. Following double-stranded cDNA amplification and purification, the full-length cDNA was used as input into SMRTbell library preparation, using SMRTbell Express Template Preparation Kit v2.0. Briefly, a minimum of 100 ng of cDNA from each sample were treated with a DNA Damage Repair enzyme mix to repair nicked DNA, followed by an End Repair and A-tailing reaction to repair blunt ends and polyadenylate each template. Next, overhang SMRTbell adapters were ligated onto each template and purified using 0.6X AMPure PB beads to remove small fragments and excess reagents (Pacific Biosciences). The completed SMRTbell libraries were further treated with the SMRTbell Enzyme Clean Up Kit to remove unligated templates. The final libraries were then annealed to sequencing primer v4 and bound to sequencing polymerase 2.0 before being sequenced on one SMRTcell 8M on the Sequel II system with a 24-hour movie each. After data collection, the raw sequencing subreads were imported to the SMRTLink analysis suite, version 10.1 for processing. Intramolecular error correcting was performed using the circular consensus sequencing (CCS) algorithm to produce highly accurate (>Q10) CCS reads, each requiring a minimum of 3 polymerase passes. The polished CCS reads were then passed to the lima tool to remove Iso-Seq and template-switching oligo sequences and orient the isoforms into the correct 5’ to 3’ direction. The refine tool was then used to remove polyA tails and concatemers from the full-length reads to generate final full-length, non-concatamer (FLNC) isoforms. The FLNC isoforms were then clustered together using the cluster tool to generate final, polished consensus isoforms per sample.
Identification and illustration of viral sequences in the bat pluripotent stem cell transcriptome
The existence of viruses in the Rhinolophus ferrumequinum transcriptome was explored by analysing the RNA-seq and Iso-seq data based on a metagenomic approach using Kraken2 v2.1.2 112 First, the adaptors in the RNA-seq data were removed with Trimgalore v0.6.7114 and all replicates for corresponding datasets were joined in one file. The reference library “RefSeq complete viral genomes / proteins” was downloaded and a custom database was built to identify matches within the processed RNA-seq or Iso-seq. To eliminate false positive hits that could be due to matches with any cellular transcript such as oncogenes that are carried by some viruses, a second analysis was performed after eliminating all reads from the RNA-seq and Iso-seq datasets that matched any annotated Rhinolophus ferrumequinum transcript. To do this, the Iso-Seq FLNC isoforms or RNA-seq trimmed fastq sequences were first mapped to the “Rhinolophus ferrumequinum genomic rna exons RefSeq” file “GCF_004115265.1_mRhiFer1_v1.p_rna_from_genomic.fna” using gmap/gsnap.113 The sequences with no mappings were then used to identify viral sequences using Kraken2 as before.
In a first approximation, we mapped the sequencing reads against a virus database, using a metagenomic classification tool (Kraken2). The results revealed a taxonomically highly diverse “zoo” of assigned viruses belonging to several major viral families. We also classified viral sequences using RNA-seq data from BEFs (Table S5B) and from human ES cells (Table S5C) notably yielding some viral sequences albeit to a lesser degree (Figure S11A). This observation in BEFs was surprising as post-implantation tissues typically do not exhibit endogenous viral activity,50 underscoring the notion that bat cells harbor pro-viral environments. Further, the results support our hypothesis that the epigenetic reprogramming of bat fibroblasts reactivates a considerable number of additional dormant viral sequences.
Further, we investigated potential confounding effects that might impact interpretation of the metagenomic data. Three potential sources for distortion are (i) statistical stringency, (ii) cellular genes containing viral-like sequences (e.g., oncogenes), and (iii) potential xeno sequence pollution originating from the feeder cells. To address the first point, we utilized progressively higher statistical stringency, yielding an expected decrease in matches in our R. ferrumequinum RNA-seq data. However, even with the most stringent parameters tested, there remained a sizable number of hits. To exclude potential cellular genes misinterpreted by the classification algorithm as viruses, we depleted the RNA-seq and Iso-seq from all sequences that match exons, which only marginally affected the number of hits. When the Kraken2 database was expanded to include bacteria, archaea, and the whole human genome, the yield was only 0.65 percent, compared to 2.53 percent when the viral database was probed (Figure S11B). Finally, we checked if some of the classified sequences were of murine origin and found that this was the case for several retroviruses, and for example, the sequences classified as Diatom colony associated ssRNA virus 1. The latter matched a genomic region on mouse chromosome chr9: 65,969,137–65,969,198 (GRCmm39/mm39).
In addition to Kraken2, we also processed sequences through the Microsoft Premonition metagenomics pipeline (http://microsoft.com/premonition), which uses an alignment-based approach (cf Kraken2’s k-mer based method) to identify matches, in addition to employing a larger reference database, factors which can increase sensitivity/reduce false negatives. The Premonition pipeline also uses a statistical model to probabilistically assign reads to taxonomic levels, favoring matches with better coverage of the reference genome, increasing specificity/reducing false positives.
Mapping of RNA-seq reads to bat genomes and quantifying expression of ERVs
To trim adapters and generate quality metrics of the fastq files, we used Trimmgalore v.0.6.6 (https://github.com/FelixKrueger/Trim-Galore), a wrapper for Cutadapt (https://github.com/marcelm/cutadapt) and FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Then, reads were mapped to the genome of R. ferrumequinum and M. myotis (Bat1K assembly HLrhiFer5) using HISAT2 v.2.2101 suppressing unpaired alignments for paired reads (–no-mixed), suppressing discordant alignments for paired reads (–no-discordant), and setting a function for the maximum number of ambiguous characters per read (–n-ceil L,0,0.05). Output files were then filtered to remove any unmapped reads (-F 4), sorted and indexed using samtools.102 Aligned reads were then assembled into transcripts using stringTie v2.2.1115 in stranded mode (-rf). To generate a Ballgown readable expression output with normalized expression units of fragments per kilobase of transcript per million mapped fragments (FPKMs), we also used as input in strigTie the Bat1K annotation of known endogenous retroviruses (ERVs) for R. ferrumequinum and M. myotis (https://genome.senckenberg.de/).25 Output counts were post-process and plotted with a custom R script.
De novo assembly of potential virus-derived RNA-seq
The trimmed reads that were identified by Kraken2 v2.1.2 to map to viral sequences with a confidence score of 0 as described above were first grouped together based on their taxonomic ID assigned by Kraken and then classified as either mammalian or non-mammalian using the VIRION database.123 The data were converted to FASTA format using the Seqtk v1.3 program and the reads were assembled using the Trinity v2.12 software.116 To check and gather successful assemblies that had produced at least one contig, a custom BASH script was applied for both groups of mammalian and non-mammalian viruses.
Mapping transcripts to viral and mammalian databases
To determine if the assembled transcripts represented an expressed viral sequence, all transcripts were mapped to a database of viral genomes using BLAST.117,118 The viral database consisted of genomes whose host species contained either ‘human’ or ‘vertebrate’ as specified in the NCBI database. Initially this list contained over 17,000 genomes. However, this was reduced to 3,922 genomes by taking only unique virus/strain names. An additional non-mammalian virus database was generated by combining all genomic sequences of viruses identified by Kraken2 and classified as non-mammalian via VIRION. Transcripts were also mapped to a combined database of RFe bat, human and mouse genomes to both confirm their presence in the bat and to exclude the possibility of false positives through contamination. For each of these transcripts, expected values for both bat and viral genome BLAST results were combined into a single metric via the following formula: Log (bat-expected value+1 × virus-expected value+1). A threshold of less than 0.3, representing a combined e-value of less than 1e−50 for both viral and bat hits, was used to rule out potential false positives. In addition, we used SQUID (http://eddylab.org/software.html) to shuffle the 63 (bottom-up) and 82 (top-down) sequences while preserving the dinucleotide distribution (parameter -d) to obtain a conservative threshold to distinguish bona fide viral homology from matches by random chance. Shuffled sequences were mapped to both the bat genome and viral genome databases, with the same BLAST threshold applied. All transcripts passing this threshold were extended by 5000 bp flanks within the bat genome and these regions were subsequently mapped to the viral database to confirm their presence in a viral genome.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical details of experiments are described in the figures, figure legends, and methods. In general, statistical analyses are presented as mean ± SEM or SDEV. Comparisons between two groups were determined by Student’s t-test. Multiple comparisons were determined by one- or two-way ANOVA followed by post-hoc testing as indicated in the figure legends. Significance criterion was set at a P-value of ≥0.05.
Supplementary Material
Highlights.
Understanding of bat biology has been limited by lack of cellular models
Induced pluripotent stem cells are produced from two evolutionarily distant bat species
The core pluripotency expression profile resembles that of cells attacked by viruses
Bat stem cells accommodate a substantial load of endogenous viral sequences
ACKNOWLEDGMENTS
We thank Prof. Eloy Revilla Ebd-CSIC’s director, the EBD-CSIC’s Bio-Ethical Committee, the CABIMER, the Consejería de Medio-Ambiente of the Junta de Andalucía and the Subdirección General de Acuerdos Sanitarios y Control en Frontera of the Spanish Ministry of Agricultura, Pesca y Alimentación, for their support and help in obtaining the export permits in record time and many other people for making the shipment and delivery of the Rhinolophus samples from Seville, Spain, to New York, USA, under the COVID-19 pandemic’s most arduous conditions possible. We thank the members of Bretagne Vivante, local volunteers, and students from University College Dublin for their help in sample collection and the owners/local authorities for allowing access to the sites to collect Myotis samples in France. We also thank Michael Schotsaert and Carles Martinez for helping with the bat tissue import, Allison Sova and Bill Williams from the Microscopy Core and Advanced Bioimaging Center at the Icahn School of Medicine for preparing the cells for the electron microscopy and imaging, as well as Glenn Doherty and Nikolas Tzavaras for their assistance with the confocal microscopy. We also thank the Genomics and the Biorepository and Pathology Core Facility members at the Icahn School of Medicine at Mount Sinai for performing the RNA-seq and histology, respectively. This work was supported in part by the computational resources and staff expertise provided by Scientific Computing at the Icahn School of Medicine at Mount Sinai. E.C.T. is supported by Irish Research Council Laureate Award IRCLA/2017/58 and Science Foundation Ireland Future Frontiers 19/FFP/6790. G.M.H. is funded by a University College Dublin Ad Astra Fellowship. M.D. and T.P.Z. are supported by the Huffington Foundation. T.P.Z. is supported by NIH GM129329 and HD100518. A.G.S. is supported by HR0011-19-2-0020, awarded by DARPA, and grant No. W81XWH-20-1-0270, awarded by the Department of Defense(DoD). This work was also partially supported by NIAID grant U19AI135972 and by CRIPT (Center for Research on Influenza Pathogenesis and Response), a NIAID-supported Center of Excellence for Influenza Research and Response (CEIRR, contract #75N93019R00028) to A.G.S. and by grant 2021-244135(5384) from the Open Philanthropy Project Fund to A.G.S. and T.P.Z., while J.J. and C.I. are supported by the Spanish Ministerio de Ciencia e Innovación (SAF2017-89355-P). R.A.Y. is supported by NIH R35 GM144283. A.E.M. and M.H. are supported by the LOEWE-Centre for Translational Biodiversity Genomics (TBG). The graphical abstract was created with BioRender.com.
INCLUSION AND DIVERSITY
We support the inclusive, diverse, and equitable conduct of research.
Footnotes
DECLARATION OF INTERESTS
T.P.Z., M.D., and A.G.S. are inventors on patents and patent applications on the use of bat iPS cells, owned by the Icahn School of Medicine at Mount Sinai, New York. T.P.Z. and R.A.Y. are founders and shareholders of Paratus Sciences, and R.A.Y. is a founder and shareholder of Syros Pharmaceuticals, Camp4 Therapeutics, Omega Therapeutics, and Dewpoint Therapeutics. The A.G.S. laboratory has received research support from Pfizer, Senhwa Biosciences, Kenall Manufacturing, Avimex, Johnson & Johnson, Dynavax, 7Hills Pharma, Pharmamar, ImmunityBio, Accurius, Nanocomposix, Hexamer, N-Fold LLC, Model Medicines, and Merck, outside of the reported work. A.G.S. has consulting agreements for the following companies involving cash and/or stock: Vivaldi Biosciences, Contrafect, 7Hills Pharma, Avimex, Vaxalto, Pagoda, Accurius, Esperovax, Farmak, Applied Biological Laboratories, and Pfizer, outside of the reported work. A.G.S. is inventor on patents and patent applications on the use of antivirals and vaccines for the treatment and prevention of virus infections and cancer, owned by the Icahn School of Medicine at Mount Sinai, New York, outside of the reported work. A.M. is the creator of Omics Bioinformatics and owns all the stocks of this company. R.P.S. has a consulting agreement with Sema4 involving cash and is also a stockholder of this company. A.P. and S.D.W.F. are employees of Microsoft Corporation. S.D.W.F. is co-founder of DIOSynVax, Ltd. and an inventor on patent applications on the design of vaccine immunogens for the prevention of virus infections, outside of the reported work.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.cell.2023.01.011.
REFERENCES
- 1.Simmons N, and Cirranello A (2020). Bat species of the world: a taxonomic and geographic database. https://batnames.org/. [Google Scholar]
- 2.Allen G (1939). Bats (Harvard University Press; ). [Google Scholar]
- 3.Nagel T (1974). What is it like to be a bat. The. Philosophical Review 83, 159–168. [Google Scholar]
- 4.Teeling E, Dool S, and Springer M (2012). Phylogenies, fossils and functional genes: the evolution of echolocation in bats. Evol. Hist. Bats Fossils Mol. Morphol, 1–22. [Google Scholar]
- 5.Teeling EC, Vernes SC, Dávalos LM, Ray DA, Gilbert MTP, and Myers E; Bat1K Consortium (2018). Bat biology, genomes, and the Bat1K project: to generate chromosome-level genomes for all living bat species. Annu. Rev. Anim. Biosci. 6, 23–46. [DOI] [PubMed] [Google Scholar]
- 6.Teeling EC, Springer MS, Madsen O, Bates P, O’Brien SJ, and Murphy WJ (2005). A molecular phylogeny for bats illuminates biogeography and the fossil record. Science 307, 580–584. [DOI] [PubMed] [Google Scholar]
- 7.Altringham JD (2011). Bats: from evolution to conservation (Oxford University Press; ). [Google Scholar]
- 8.Munshi-South J, and Wilkinson GS (2010). Bats and birds: exceptional longevity despite high metabolic rates. Ageing Res. Rev. 9, 12–19. [DOI] [PubMed] [Google Scholar]
- 9.Wilkinson GS, and Adams DM (2019). Recurrent evolution of extreme longevity in bats. Biol. Lett. 15, 20180860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hermida Lorenzo RJ, Cadar D, Koundouno FR, Juste J, Bialonski A, Baum H, García-Mudarra JL, Hakamaki H, Bencsik A, Nelson EV, et al. (2021). Metagenomic snapshots of viral components in Guinean bats. Microorganisms 9, 599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Luis AD, Hayman DT, O’Shea TJ, Cryan PM, Gilbert AT, Pulliam JR, Mills JN, Timonin ME, Willis CK, Cunningham AA, et al. (2013). A comparison of bats and rodents as reservoirs of zoonotic viruses: are bats special? Proc. R. Soc. Lond. B 280, 20122753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Olival KJ, Hosseini PR, Zambrana-Torrelio C, Ross N, Bogich TL, and Daszak P (2017). Host and viral traits predict zoonotic spillover from mammals. Nature 546, 646–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Anthony SJ, Johnson CK, Greig DJ, Kramer S, Che X, Wells H, Hicks AL, Joly DO, Wolfe ND, Daszak P, et al. (2017). Global patterns in coronavirus diversity. Virus Evol. 3, vex012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brook CE, and Dobson AP (2015). Bats as ‘special’reservoirs for emerging zoonotic pathogens. Trends Microbiol. 23, 172–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Drexler JF, Corman VM, Müller MA, Maganga GD, Vallo P, Binger T, Gloza-Rausch F, Cottontail VM, Rasche A, Yordanov S, et al. (2012). Bats host major mammalian paramyxoviruses. Nat. Commun. 3, 796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.O’Shea TJ, Cryan PM, Cunningham AA, Fooks AR, Hayman DT, Luis AD, Peel AJ, Plowright RK, and Wood JL (2014). Bat flight and zoonotic viruses. Emerg. Infect. Dis. 20, 741–745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Van Brussel K, and Holmes EC (2022). Zoonotic disease and virome diversity in bats. Curr. Opin. Virol. 52, 192–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang LF, Walker PJ, and Poon LL (2011). Mass extinctions, biodiversity and mitochondrial function: are bats ‘special’ as reservoirs for emerging viruses? Curr. Opin. Virol. 1, 649–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Banerjee A, Baker ML, Kulcsar K, Misra V, Plowright R, and Mossman K (2020). Novel insights into immune systems of bats. Front. Immunol. 11, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Brook CE, Boots M, Chandran K, Dobson AP, Drosten C, Graham AL, Grenfell BT, Müller MA, Ng M, Wang LF, and Leeuwen A.v. (2020). Accelerated viral dynamics in bat cell lines, with implications for zoonotic emergence. eLife 9, e48401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pourrut X, Souris M, Towner JS, Rollin PE, Nichol ST, Gonzalez JP, and Leroy E (2009). Large serological survey showing cocirculation of Ebola and Marburg viruses in Gabonese bat populations, and a high seroprevalence of both viruses in Rousettus aegyptiacus. BMC Infect. Dis. 9, 159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Yob JM, Field H, Rashdi AM, Morrissy C, van der Heide B, Rota P, bin Adzhar A, White J, Daniels P, Jamaluddin A, and Ksiazek T (2001). Nipah virus infection in bats (order Chiroptera) in peninsular Malaysia. Emerging infectious diseases 7. 439–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cui J, Tachedjian M, Wang L, Tachedjian G, Wang LF, and Zhang S (2012). Discovery of retroviral homologs in bats: implications for the origin of mammalian gammaretroviruses. J. Virol. 86, 4288–4293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hayward JA, Tachedjian M, Cui J, Cheng AZ, Johnson A, Baker ML, Harris RS, Wang LF, and Tachedjian G (2018). Differential evolution of antiretroviral restriction factors in pteropid bats as revealed by APOBEC3 gene complexity. Mol. Biol. Evol. 35, 1626–1637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Jebb D, Huang Z, Pippel M, Hughes GM, Lavrichenko K, Devanna P, Winkler S, Jermiin LS, Skirmuntt EC, Katzourakis A, et al. (2020). Six reference-quality genomes reveal evolution of bat adaptations. Nature 583, 578–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Skirmuntt EC, Escalera-Zamudio M, Teeling EC, Smith A, and Katzourakis A (2020). The potential role of endogenous viral elements in the evolution of bats as reservoirs for zoonotic viruses. Annu. Rev. Virol. 7, 103–119. [DOI] [PubMed] [Google Scholar]
- 27.Skirmuntt EC, and Katzourakis A (2019). The evolution of endogenous retroviral envelope genes in bats and their potential contribution to host biology. Virus Res. 270, 197645. [DOI] [PubMed] [Google Scholar]
- 28.Cui J, Tachedjian G, and Wang LF (2015). Bats and rodents shape mammalian retroviral phylogeny. Sci. Rep. 5, 16561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Escalera-Zamudio M, Rojas-Anaya E, Kolokotronis SO, Taboada B, Loza-Rubio E, Méndez-Ojeda ML, Arias CF, Osterrieder N, and Greenwood AD (2016). Bats, primates, and the evolutionary origins and diversification of mammalian gammaherpesviruses. mBio 7. e01425–01416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hayman DT, Bowen RA, Cryan PM, McCracken GF, O’Shea TJ, Peel AJ, Gilbert A, Webb CT, and Wood JL (2013). Ecology of zoonotic infectious diseases in bats: current knowledge and future directions. Zoonoses Public Health 60, 2–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Middleton DJ, Morrissy CJ, Van Der Heide BM, Russell GM, Braun MA, Westbury HA, Halpin K, and Daniels PW (2007). Experimental Nipah virus infection in pteropid bats (Pteropus poliocephalus). J. Comp. Pathol. 136, 266–272. [DOI] [PubMed] [Google Scholar]
- 32.Swanepoel R, Leman PA, Burt FJ, Zachariades NA, Braack LE, Ksiazek TG, Rollin PE, Zaki SR, and Peters CJ (1996). Experimental inoculation of plants and animals with Ebola virus. Emerg. Inf. Dis. 2, 321–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Watanabe S, Masangkay JS, Nagata N, Morikawa S, Mizutani T, Fukushi S, Alviola P, Omatsu T, Ueda N, Iha K, et al. (2010). Bat coronaviruses and experimental infection of bats, the Philippines. Emerg. Inf. Dis. 16, 1217–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Moreno Santillán DD, Lama TM, Gutierrez Guerrero YT, Brown AM, Donat P, Zhao H, Rossiter SJ, Yohe LR, Potter JH, Teeling EC, et al. (2021). Large-scale genome sampling reveals unique immunity and metabolic adaptations in bats. Mol. Ecol. 30, 6449–6467. [DOI] [PubMed] [Google Scholar]
- 35.Kacprzyk J, Hughes GM, Palsson-McDermott EM, Quinn SR, Puechmaille SJ, O’Neill LAJ, and Teeling EC (2017). A potent anti-inflammatory response in bat macrophages may be linked to extended longevity and viral tolerance. Acta Chiropterol. 19, 219–228. [Google Scholar]
- 36.Wang LF, Gamage AM, Chan WOY, Hiller M, and Teeling EC (2021). Decoding bat immunity: the need for a coordinated research approach. Nat. Rev. Immunol. 21, 269–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Baker ML, Schountz T, and Wang LF (2013). Antiviral immune responses of bats: a review. Zoonoses Public Health 60, 104–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Irving AT, Ahn M, Goh G, Anderson DE, and Wang LF (2021). Lessons from the host defences of bats, a unique viral reservoir. Nature 589, 363–370. 10.1038/s41586-020-03128-0. [DOI] [PubMed] [Google Scholar]
- 39.Ahn M, Anderson DE, Zhang Q, Tan CW, Lim BL, Luko K, Wen M, Chia WN, Mani S, Wang LC, et al. (2019). Dampened NLRP3-mediated inflammation in bats and implications for a special viral reservoir host. Nat. Microbiol. 4, 789–799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Goh G, Ahn M, Zhu F, Lee LB, Luo D, Irving AT, and Wang LF (2020). Complementary regulation of caspase-1 and IL-1β reveals additional mechanisms of dampened inflammation in bats. Proc. Natl. Acad. Sci. USA 117, 28939–28949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mozzi A, Pontremoli C, Forni D, Clerici M, Pozzoli U, Bresolin N, Cagliani R, and Sironi M (2015). OASes and STING: adaptive evolution in concert. Genome Biol. Evol. 7, 1016–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Pavlovich SS, Lovett SP, Koroleva G, Guito JC, Arnold CE, Nagle ER, Kulcsar K, Lee A, Thibaud-Nissen F, Hume AJ, et al. (2018). The Egyptian rousette genome reveals unexpected features of bat antiviral immunity. Cell 173, 1098–1110.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Xie J, Li Y, Shen X, Goh G, Zhu Y, Cui J, Wang LF, Shi ZL, and Zhou P (2018). Dampened STING-dependent interferon activation in bats. Cell Host Microbe 23, 297–301.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.García-Sastre A (2017). Ten strategies of interferon evasion by viruses. Cell Host Microbe 22, 176–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Villarreal LP, Defilippis VR, and Gottlieb KA (2000). Acute and persistent viral life strategies and their relationship to emerging diseases. Virology 272, 1–6. [DOI] [PubMed] [Google Scholar]
- 46.Witzany G (2010). Biocommunication and natural genome editing. World J. Biol. Chem. 1, 348–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Wray J, Kalkan T, and Smith AG (2010). The ground state of pluripotency. Biochem. Soc. Trans. 38, 1027–1032. [DOI] [PubMed] [Google Scholar]
- 48.Grow EJ, Flynn RA, Chavez SL, Bayless NL, Wossidlo M, Wesche DJ, Martin L, Ware CB, Blish CA, Chang HY, et al. (2015). Intrinsic retroviral reactivation in human preimplantation embryos and pluripotent cells. Nature 522, 221–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Macfarlan TS, Gifford WD, Driscoll S, Lettieri K, Rowe HM, Bonanomi D, Firth A, Singer O, Trono D, and Pfaff SL (2012). Embryonic stem cell potency fluctuates with endogenous retrovirus activity. Nature 487, 57–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rowe HM, and Trono D (2011). Dynamic control of endogenous retroviruses during development. Virology 411, 273–287. [DOI] [PubMed] [Google Scholar]
- 51.Wang J, Xie G, Singh M, Ghanbarian AT, Raskó T, Szvetnik A, Cai H, Besser D, Prigione A, Fuchs NV, et al. (2014). Primate-specific endogenous retrovirus-driven transcription defines naive-like stem cells. Nature 516, 405–409. [DOI] [PubMed] [Google Scholar]
- 52.Mo X, Li N, and Wu S (2014). Generation and characterization of bat-induced pluripotent stem cells. Theriogenology 82, 283–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Aurine N, Baquerre C, Gaudino M, Jean C, Dumont C, Rival-Gervier S, Kress C, Horvat B, and Pain B (2021). Reprogrammed Pteropus Bat stem cells as A model to study Host-Pathogen Interaction during Henipavirus Infection. Microorganisms 9, 2567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Takahashi K, and Yamanaka S (2006). Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell 126, 663–676. [DOI] [PubMed] [Google Scholar]
- 55.Hochedlinger K, and Jaenisch R (2015). Induced pluripotency and epigenetic reprogramming. Cold Spring Harb. Perspect. Biol. 7, a019448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hanna J, Cheng AW, Saha K, Kim J, Lengner CJ, Soldner F, Cassady JP, Muffat J, Carey BW, and Jaenisch R (2010). Human embryonic stem cells with biological and epigenetic characteristics similar to those of mouse ESCs. Proc. Natl. Acad. Sci. USA 107, 9222–9227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bernstein BE, Mikkelsen TS, Xie X, Kamal M, Huebert DJ, Cuff J, Fry B, Meissner A, Wernig M, Plath K, et al. (2006). A bivalent chromatin structure marks key developmental genes in embryonic stem cells. Cell 125, 315–326. [DOI] [PubMed] [Google Scholar]
- 58.Court F, and Arnaud P (2017). An annotated list of bivalent chromatin regions in human ES cells: a new tool for cancer epigenetic research. Oncotarget 8, 4110–4124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Cornacchia D, Zhang C, Zimmer B, Chung SY, Fan Y, Soliman MA, Tchieu J, Chambers SM, Shah H, Paull D, et al. (2019). Lipid deprivation induces a stable, naive-to-primed intermediate state of pluripotency in human PSCs. Cell Stem Cell 25, 120–136.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Smith A (2017). Formative pluripotency: the executive phase in a developmental continuum. Development 144, 365–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wiles MV, and Keller G (1991). Multiple hematopoietic lineages develop from embryonic stem (ES) cells in culture. Development 111, 259–267. [DOI] [PubMed] [Google Scholar]
- 62.Damjanov I, and Andrews PW (2007). The terminology of teratocarcinomas and teratomas. Nat. Biotechnol. 25. 1212; discussion 1212. [DOI] [PubMed] [Google Scholar]
- 63.Miyawaki S, Kawamura Y, Oiwa Y, Shimizu A, Hachiya T, Bono H, Koya I, Okada Y, Kimura T, Tsuchiya Y, et al. (2016). Tumour resistance in induced pluripotent stem cells derived from naked mole-rats. Nat. Commun. 7, 11471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tan L, Ke Z, Tombline G, Macoretta N, Hayes K, Tian X, Lv R, Ablaeva J, Gilbert M, Bhanu NV, et al. (2017). Naked mole rat cells have a stable epigenome that resists iPSC reprogramming. Stem Cell Rep. 9, 1721–1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Yu L, Wei Y, Duan J, Schmitz DA, Sakurai M, Wang L, Wang K, Zhao S, Hon GC, and Wu J (2021). Blastocyst-like structures generated from human pluripotent stem cells. Nature 591, 620–626. [DOI] [PubMed] [Google Scholar]
- 66.Romero IG, Ruvinsky I, and Gilad Y (2012). Comparative studies of gene expression and the evolution of gene regulation. Nat. Rev. Genet. 13, 505–516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kuivaniemi H, and Tromp G (2019). Type III collagen (COL3A1): gene and protein structure, tissue distribution, and associated diseases. Gene 707, 151–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhuo X, Rho M, and Feschotte C (2013). Genome-wide characterization of endogenous retroviruses in the bat Myotis lucifugus reveals recent and diverse infections. J. Virol. 87, 8493–8501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhang L, Richards A, Barrasa MI, Hughes SH, Young RA, and Jaenisch R (2021). Reverse-transcribed SARS-CoV-2 RNA can integrate into the genome of cultured human cells and can be expressed in patient-derived tissues. Proc. Natl. Acad. Sci. USA 118. e2105968118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Villarreal LP (2009). Persistence pays: how viruses promote host group survival. Curr. Opin. Microbiol. 12, 467–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Wacharapluesadee S, Tan CW, Maneeorn P, Duengkae P, Zhu F, Joyjinda Y, Kaewpom T, Chia WN, Ampoot W, Lim BL, et al. (2021). Evidence for SARS-CoV-2 related coronaviruses circulating in bats and pangolins in Southeast Asia. Nat. Commun. 12, 972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhou H, Ji J, Chen X, Bi Y, Li J, Wang Q, Hu T, Song H, Zhao R, Chen Y, et al. (2021). Identification of novel bat coronaviruses sheds light on the evolutionary origins of SARS-CoV-2 and related viruses. Cell 184, 4380–4391.e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Barton ES, White DW, Cathelyn JS, Brett-McClellan KA, Engle M, Diamond MS, Miller VL, and Virgin HW (2007). Herpesvirus latency confers symbiotic protection from bacterial infection. Nature 447, 326–329. [DOI] [PubMed] [Google Scholar]
- 74.Eaton BT, Broder CC, Middleton D, and Wang LF (2006). Hendra and Nipah viruses: different and dangerous. Nat. Rev. Microbiol. 4, 23–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Roossinck MJ (2011). The good viruses: viral mutualistic symbioses. Nat. Rev. Microbiol. 9, 99–108. [DOI] [PubMed] [Google Scholar]
- 76.Ophinni Y, Palatini U, Hayashi Y, and Parrish NF (2019). piRNA-guided CRISPR-like immunity in eukaryotes. Trends Immunol. 40, 998–1010. [DOI] [PubMed] [Google Scholar]
- 77.Feschotte C, and Gilbert C (2012). Endogenous viruses: insights into viral evolution and impact on host biology. Nat. Rev. Genet. 13, 283–296. [DOI] [PubMed] [Google Scholar]
- 78.Endo Y, Kamei KI, and Inoue-Murayama M (2020). Genetic signatures of evolution of the pluripotency gene regulating network across mammals. Genome Biol. Evol. 12, 1806–1818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Wu X, Thi VLD, Huang Y, Billerbeck E, Saha D, Hoffmann HH, Wang Y, Silva LAV, Sarbanes S, Sun T, et al. (2018). Intrinsic immunity shapes viral resistance of stem cells. Cell 172, 423–438.e25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Aikawa H, Tamai M, Mitamura K, Itmainati F, Barber GN, and Tagawa Y. i. (2014). Innate immunity in an in vitro murine blastocyst model using embryonic and trophoblast stem cells. J. Biosci. Bioeng. 117, 358–365. [DOI] [PubMed] [Google Scholar]
- 81.Ivanova NB, Dimos JT, Schaniel C, Hackney JA, Moore KA, and Lemischka IR (2002). A stem cell molecular signature. Science 298, 601–604. [DOI] [PubMed] [Google Scholar]
- 82.Ramalho-Santos M, Yoon S, Matsuzaki Y, Mulligan RC, and Melton DA (2002). “ Stemness”: transcriptional profiling of embryonic and adult stem cells. Science 298, 597–600. [DOI] [PubMed] [Google Scholar]
- 83.Carter CC, McNamara LA, Onafuwa-Nuga A, Shackleton M, Riddell J, Bixby D, Savona MR, Morrison SJ, and Collins KL (2011). HIV-1 utilizes the CXCR4 chemokine receptor to infect multipotent hematopoietic stem and progenitor cells. Cell Host Microbe 9, 223–234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Janssens S, Schotsaert M, Karnik R, Balasubramaniam V, Dejosez M, Meissner A, García-Sastre A, and Zwaka TP (2018). Zika virus alters DNA methylation of neural genes in an organoid model of the developing human brain. mSystems 3. e00219–00217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Li H, Saucedo-Cuevas L, Regla-Nava JA, Chai G, Sheets N, Tang W, Terskikh AV, Shresta S, and Gleeson JG (2016). Zika virus infects neural progenitors in the adult mouse brain and alters proliferation. Cell Stem Cell 19, 593–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Naeye RL, and Blanc W (1965). Pathogenesis of congenital rubella. JAMA 194, 1277–1283. [PubMed] [Google Scholar]
- 87.Daugherty MD, and Malik HS (2012). Rules of engagement: molecular insights from host-virus arms races. Annu. Rev. Genet. 46, 677–700. [DOI] [PubMed] [Google Scholar]
- 88.Siddle KJ, and Quintana-Murci L (2014). The Red Queen’s long race: human adaptation to pathogen pressure. Curr. Opin. Genet. Dev. 29, 31–38. [DOI] [PubMed] [Google Scholar]
- 89.Villarreal LP (2007). Review article virus-host symbiosis mediated by persistence. Symbiosis. [Google Scholar]
- 90.Rasweiler JJ, Cretekos CJ, and Behringer RR (2009). Feeding short-tailed fruit bats (Carollia perspicillata). Cold Spring Harb. Protoc. 2009. pdb.prot5159. [DOI] [PubMed] [Google Scholar]
- 91.Takahashi K, and Yamanaka S (2016). A decade of transcription factor-mediated reprogramming to pluripotency. Nat. Rev. Mol. Cell Biol. 17, 183–193. [DOI] [PubMed] [Google Scholar]
- 92.Lancaster MA, Renner M, Martin CA, Wenzel D, Bicknell LS, Hurles ME, Homfray T, Penninger JM, Jackson AP, and Knoblich JA (2013). Cerebral organoids model human brain development and microcephaly. Nature 501, 373–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.White KM, Rosales R, Yildiz S, Kehrer T, Miorin L, Moreno E, Jangra S, Uccellini MB, Rathnasinghe R, Coughlan L, et al. (2021). Plitidepsin has potent preclinical efficacy against SARS-CoV-2 by targeting the host protein eEF1A. Science 371, 926–931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yang L, Han Y, Nilsson-Payant BE, Gupta V, Wang P, Duan X, Tang X, Zhu J, Zhao Z, Jaffré F, et al. (2020). A human pluripotent stem cell-based platform to study SARS-CoV-2 tropism and model virus infection in human cells and organoids. Cell Stem Cell 27, 125–136.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Yoshimatsu M, Ohnishi H, Zhao C, Hayashi Y, Kuwata F, Kaba S, Okuyama H, Kawai Y, Hiwatashi N, Kishimoto Y, et al. (2021). In vivo regeneration of rat laryngeal cartilage with mesenchymal stem cells derived from human induced pluripotent stem cells via neural crest cells. Stem Cell Res. 52, 102233. [DOI] [PubMed] [Google Scholar]
- 96.Carter AC, Davis-Dusenbery BN, Koszka K, Ichida JK, and Eggan K (2014). Nanog-independent reprogramming to iPSCs with canonical factors. Stem Cell Rep. 2, 119–126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Io S, Kabata S,M, Iemura Y, Semi K, Morone N, Minagawa A, Wang B, Okamoto I, Nakamura T, Kojima Y, et al. (2021). Capturing human trophoblast development with naive pluripotent stem cells in vitro. Cell Stem Cell 28, 1023–1039.e13. [DOI] [PubMed] [Google Scholar]
- 98.Selmi T, Hussain S, Dietmann S, Heiß M, Borland K, Flad S, Carter JM, Dennison R, Huang YL, Kellner S, et al. (2021). Sequence-and structure-specific cytosine-5 mRNA methylation by NSUN6. Nucleic Acids Res. 49, 1006–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Andrews S, Krueger F, Segonds-Pichon A, Biggins L, Krueger C, and Wingett S (2010). FastQC. A Quality Control Tool for High Throughput Sequence Data 370. [Google Scholar]
- 100.Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Kim D, Paggi JM, Park C, Bennett C, and Salzberg SL (2019). Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat. Biotechnol. 37, 907–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Subgroup (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- 104.Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Metsalu T, and Vilo J (2015). ClustVis: a web tool for visualizing clustering of multivariate data using principal component analysis and heatmap. Nucleic Acids Res. 43, W566–W570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Wickham H, and Grolemund G (2016). R for data science: import, tidy, transform, visualize, and model data (“ O’Reilly Media, Inc.”; ). [Google Scholar]
- 108.Yang Z (2007). PAML 4: phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 24, 1586–1591. [DOI] [PubMed] [Google Scholar]
- 109.Kent WJ, Zweig AS, Barber G, Hinrichs AS, and Karolchik D (2010). BigWig and BigBed: enabling browsing of large distributed datasets. Bioinformatics 26, 2204–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Xie Z, Bailey A, Kuleshov MV, Clarke DJB, Evangelista JE, Jenkins SL, Lachmann A, Wojciechowicz ML, Kropiwnicki E, Jagodnik KM, et al. (2021). Gene set knowledge discovery with enrichr. Curr. Protoc. 1, e90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, and Ideker T (2003). Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res. 13, 2498–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Wood DE, Lu J, and Langmead B (2019). Improved metagenomic analysis with Kraken 2. Genome Biol. 20, 257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Wu TD, and Nacu S (2010). Fast and SNP-tolerant detection of complex variants and splicing in short reads. Bioinformatics 26, 873–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Krueger F, and Andrews SR (2012). Quality Control, Trimming and Alignment of Bisulfite-Seq Data (Prot 57) (Department of Medicine, Hematology and Oncology). [Google Scholar]
- 115.Pertea M, Pertea GM, Antonescu CM, Chang TC, Mendell JT, and Salzberg SL (2015). StringTie enables improved reconstruction of a transcriptome from RNA-seq reads. Nat. Biotechnol. 33, 290–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Grabherr MG, Haas BJ, Yassour M, Levin JZ, Thompson DA, Amit I, Adiconis X, Fan L, Raychowdhury R, Zeng Q, et al. (2011). Trinity: reconstructing a full-length transcriptome without a genome from RNA-Seq data. Nat. Biotechnol. 29, 644–652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Altschul SF, Gish W, Miller W, Myers EW, and Lipman DJ (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403–410. [DOI] [PubMed] [Google Scholar]
- 118.Camacho C, Coulouris G, Avagyan V, Ma N, Papadopoulos J, Bealer K, and Madden TL (2009). Blast+: architecture and applications. BMC Bioinformatics 10, 421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Huang Z, Whelan CV, Foley NM, Jebb D, Touzalin F, Petit EJ, Puechmaille SJ, and Teeling EC (2019). Longitudinal comparative transcriptomics reveals unique mechanisms underlying extended healthspan in bats. Nat. Ecol. Evol. 3, 1110–1120. [DOI] [PubMed] [Google Scholar]
- 120.Ewels P, Magnusson M, Lundin S, and Käller M (2016). MultiQC: summarize analysis results for multiple tools and samples in a single report. Bioinformatics 32, 3047–3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Zhang Y, Lin YH, Johnson TD, Rozek LS, and Sartor MA (2014). PePr: a peak-calling prioritization pipeline to identify consistent or differential peaks from replicated ChIP-Seq data. Bioinformatics 30, 2568–2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Anders S, and Huber W (2010). Differential expression analysis for sequence count data. Genome Biol. 11, R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Carlson CJ, Gibb RJ, Albery GF, Brierley L, Connor RP, Dallas TA, Eskew EA, Fagre AC, Farrell MJ, Frank HK, et al. (2022). The Global Virome in One Network (virion): an atlas of vertebrate-virus associations. mBio 13. e02985–02921. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All high-throughput sequencing data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
This paper analyzes existing, publicly available data. These accession numbers for the datasets are listed in the key resources table.
This paper does not report any original code.
All software packages and their accessibility are described in the STAR Methods sections.
Any additional information required to reanalyze the data reported in this paper (including the BLAST results related to Figures S5–S7) is available from the Lead Contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| anti-Afp | R&D Systems | AF1369; RRID:AB_354758 |
| anti-Pax6 | BioLegend | 901301; RRID:AB_2565003 |
| J2 anti-dsRNA | Scicons | 10010200; RRID:AB_2651015 |
| anti-(gag/pol)HERVK | Austrial Biological | HERM18315; RRID:AB_10890594 |
| anti-Pan Corona FIPV3-70 | Life Technologies | MA1-82189; RRID:AB_930177 |
| anti-Oct3/4-AF488 | Santa Cruz | sc-5279-AF488; RRID:AB_628051 |
| anti-Brachyury | R&D Systems | IC2085G;RRID:AB_2933975 |
| anti-Otx2 | R&D Systems | AF1979; RRID:AB_2157172 |
| anti-Zic2 | Abcam | ab150404; RRID:AB_2868423 |
| anti-Tfe3 | Sigma Aldrich | HPA023881; RID:AB_1857931 |
| anti-Tfcp2l1 | R&D Systems | AF5726; RRID:AB_2202564 |
| anti-Cdx2 | Invitrogen | PA5-20891; RRID:AB_11155394 |
| Donkey anti-chicken-Cy3 | Millipore | AP194C; RRID:AB_92679 |
| Goat anti-chicken-AF488 | Invitrogen | A-11039; RRID:AB_2534096 |
| Donkey anti-rabbit-AF647 | Invitrogen | A-31573; RRID:AB_2536183 |
| Goat anti-rabbit-AF488 | Invitrogen | A-10034; RRID:AB_2576217 |
| Goat anti-mouse-AF488 | Invitrogen | A-11029; RRID:AB_2534088 |
| Abberior Star 635P | Abberior | ST635P; RRID:AB_2893232 |
| H3K4me3 | Active Motif | 39159; RRID:AB_2615077 |
| H3K27me3 | Active Motif | 39155; RRID:AB_2561020 |
| anti-mouse-HRP | Promega | W4021; RRID:AB_430834 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| SARS-CoV-2, isolate USA-WA1/2020 | BEI Resources | NR-52281 |
| Human Metapneumovirus MPV-GFP1 | ViraTree | M121 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| DPBS (Life Technologies, 14190144), | Gibco | 14190144 |
| DPBS | Biowest | L0615-500 |
| 0.05% Trypsin | Gibco | 25300054 |
| DMEM | Gibco | 10569010 |
| DMEM | Gibco | 11995065 |
| Fetal bovine serum, FBS | Sigma-Aldrich | F4135 |
| Fetal bovine serum, FBS | Gibco | 10500064 |
| MEM Non-essential amino | Gibco | 11140050 |
| GlutaMax Supplement | Gibco | 35050061 |
| Penicillin-Streptomycin solution | Gibco | 15140122 |
| Gelatin | Millipore | ES006B |
| Antibiotic-Antimycotic (100X) | Gibco | 15240096 |
| Collagenase type II | Gibco | 17101015 |
| Gentle Cell Dissociation Reagent | StemCell Technologies | 07174 |
| DMEM/F-12 | Gibco | 11330057 |
| Knockout serum replacement | Gibco | 10828-028 |
| DMSO | Sigma-Aldrich | D8418-100ML |
| 2-mercaptoethanol | Fluka | 63689 |
| FGF2 | R&D Systems | 233-FB |
| Leukemia inhibitory factor | Millipore | ESG1107 |
| SCF | R&D Systems | PHC2111 |
| Forskolin | Sigma-Aldrich | F6886 |
| KaryoMAX Colcemid Solution in HBSS | Gibco | 15210040 |
| Sodium Pyruvate, 100 mM | Gibco | 11360070 |
| Potassium chloride | Sigma-Aldrich | P9327 |
| Glacial acetic | Fisher Scientific | A38-212 |
| Methanol | Sigma-Aldrich | A412-4 |
| KaryoMax Giemsa staining solution | Gibco | 10092013 |
| Gurr buffer tablets | Gibcco | 10582013 |
| Cytoseal 60 | Thermo Scientific | 23-244257 |
| SuperScript IV VILO™ Master Mix | Invitrogen | 11756050 |
| GoTaq Green Polymerase | Promega | M7123 |
| Cytofix/Cytoperm solution | Becton Dickinson | BDB554714 |
| NucBlue Dapi stain | Invitrogen | R37606 |
| Prolong Dimond antifade mounting medium | Invitrogen | P36965 |
| DyeCycle Violet stain | Invitrogen | V35003 |
| DNase | Qiagen | 79254 |
| Formaldehyde | Sigma-Aldrich | F8775 |
| Sodium chloride solution, 5M | Sigma-Aldrich | S5150 |
| EDTA (0.5 M), pH 8.0, RNase-free | Invitrogen | AM9260G |
| HEPES, 1M | Invitrogen | 15630080 |
| Glycine | Sigma-Aldrich | G8898 |
| Igepal | Sigma-Aldrich | I3021 |
| PMSF | Sigma-Aldrich | P7626 |
| mTeSR medium | StemCell Technologies | 85850 |
| Vitronectin | StemCell Technologies | 07180 |
| BMP4 | R&D Systems | 314-BP-010 |
| Versene | Gibco | 15040066 |
| 10% Formalin | Fisher Scientific | SF1004 |
| RIPA lysis and extraction buffer | Fisher Scientific | 89900 |
| Proteinase Inhibitor | Roche | 45582400 |
| BCA Protein assay kit | Pierce | 23252 |
| 12% TGX Precast gel | Bio-Rad | 4561044 |
| TGS buffer | Bio-Rad | 1610772 |
| the Precision Plus Kaleidoscope Protein standard | Bio-Rad | 161-3075 |
| EveryBlot blocking buffer | Bio-Rad | 12010020 |
| Tris-buffered saline | Fisher Scientific | BP2471500 |
| Tween 20 | Bio-Rad | 161-0781 |
| Clarity Western ECL Substrate | Bio-Rad | 170-5060 |
| Paraformaldehyde | Electron Microscopy Sciences | 19200 |
| Glutaraldehyde | Electron Microscopy Sciences | 16220 |
| Sodium Cacodylate Buffer | Electron Microscopy Sciences | 11652 |
| Aqueous osmium tetroxide | Sigma-Aldrich | 75633-2ML |
| 2% aqueous uranyl acetate | Electron Microscopy Sciences | 22400-2 |
| Ethanol | Electron Microscopy Sciences | 15055 |
| Epoxy resin EMbed 812 | Electron Microscopy Sciences | 14900 |
| Toluidine Blue | Electron Microscopy Sciences | 26074-15 |
| Lead citrate | Electron Microscopy Sciences | 22410 |
| Minimum Essential Medium | Gibco | 11935046 |
| 4 mM L-glutamine | Gibco | 25030081 |
| 0.7% agar | Fisher Scientific | AAJ1090722 |
| TrueBlue substrate | Seracare | 5510-0030 |
| Trizol reagent | Invitrogen | 15596018 |
| Bovine Serum Albumin | Sigma-Aldrich | A2153-50G |
|
| ||
| Critical commercial assays | ||
|
| ||
| CytoTune iPS2.0, Invitrogen, A16517 | Life Technologies | A16517 |
| STEMdiff Trilineage differentiation kit | StemCell Technologies | 05230 |
| QuickTiter Retrovirus Quantitation Kit | Cell Biolabs | VPK-120 |
| Colorimetric reverse transcriptase kit | Roche | 11468120910 |
| SMART-Seq v4 Ultra Low Input kit | Takara Bio | 634888 |
| Total RNA with Ribo-Zero Plus kit | Illumina | 20040529 |
| AllPrep DNA/RNA Mini Kit | Qiagen | 80204 |
| SMRTbell Express Template Preparation Kit v2.0 | PacBio | 100-938-900 |
| NEBNext Single Cell/Low Input cDNA Synthesis & Amplification Module | New England Biolabs | E6421L |
| SMRTbell Enzyme Clean Up Kit | PacBio | 101-746-400 |
| RNeasy Mini Kit | Qiagen | 74104 |
|
| ||
| Deposited data | ||
|
| ||
| Rhinolophus ferrumequinum assembled and annotated by the Vertebrate Genomes Project | Vertebrate Genome Project | GCF_004115265.1 |
| Rhinolophus ferrumequinum genome assembled and annotated by the Bat1K project | Bat1K | GCA_014108255.1 |
| Rhinolophus ferrumequinum genome, Ensembl annotation version 102 | Ensembl | GCA_004115265.2 |
| Bat1K annotation of known endogenous retroviruses (ERVs) for R. ferrumequinum | https://genome.senckenberg.de/) | N/A |
| Bat1K annotation of known endogenous retroviruses (ERVs) for Myotis myotis | https://genome.senckenberg.de/) | N/A |
| RFe_BEF-R1_RNA: R. ferrumequinum fibroblasts RNA-seq replicate 1 | This paper | GEO: GSE221965 |
| RFe_BEF-R2_RNA: R. ferrumequinum fibroblasts RNA-seq replicate 2 | This paper | GEO: GSE221965 |
| RFe_BEF-R3_RNA: R. ferrumequinum fibroblasts RNA-seq replicate 3 | This paper | GEO: GSE221965 |
| RFe_BiPS1-R1_RNA: R. ferrumequinum iPS#1 RNA-seq replicate 1 | This paper | GEO: GSE221965 |
| RFe_BiPS1-R2_RNA: R. ferrumequinum iPS#1 RNA-seq replicate 2 | This paper | GEO: GSE221965 |
| RFe_BiPS1-R3_RNA: R. ferrumequinum iPS#1 RNA-seq replicate 3 | This paper | GEO: GSE221965 |
| RFe_BiPS2-R1_RNA: R. ferrumequinum iPS#2 RNA-seq replicate 1 | This paper | GEO: GSE221965 |
| RFe_BiPS2-R2_RNA: R. ferrumequinum iPS#2 RNA-seq replicate 2 | This paper | GEO: GSE221965 |
| RFe_BiPS2-R3_RNA: R. ferrumequinum iPS#2 RNA-seq replicate 3 | This paper | GEO: GSE221965 |
| RFe_BiPS2-EB-R1_RNA: R. ferrumequinum EB RNA-seq replicate 1 | This paper | GEO: GSE221965 |
| RFe_BiPS2-EB-R2_RNA: R. ferrumequinum EB RNA-seq replicate 2 | This paper | GEO: GSE221965 |
| RFe_BiPS2-EB-R3_RNA: R. ferrumequinum EB RNA-seq replicate 3 | This paper | GEO: GSE221965 |
| RFe_BiPS2-EN-R1_RNA: R. ferrumequinum Endoderm RNA-seq replicate 1 | This paper | GEO: GSE221965 |
| RFe_BiPS2-EN-R2_RNA: R. ferrumequinum Endoderm RNA-seq replicate 2 | This paper | GEO: GSE221965 |
| RFe_BiPS2-EN-R3_RNA: R. ferrumequinum Endoderm RNA-seq replicate 3 | This paper | GEO: GSE221965 |
| RFe_BiPS2-ME-R1_RNA: R. ferrumequinum Mesoderm RNA-seq replicate 1 | This paper | GEO: GSE221965 |
| RFe_BiPS2-MS-R2_RNA: R. ferrumequinum Mesoderm RNA-seq replicate 2 | This paper | GEO: GSE221965 |
| RFe_BiPS2-MS-R3_RNA: R. ferrumequinum Mesoderm RNA-seq replicate 3 | This paper | GEO: GSE221965 |
| RFe_BiPS2-EC-R1_RNA: R. ferrumequinum Ectoderm RNA-seq replicate 1 | This paper | GEO: GSE221965 |
| RFe_BiPS2-EC-R2_RNA: R. ferrumequinum Ectoderm RNA-seq replicate 2 | This paper | GEO: GSE221965 |
| RFe_BiPS2-EC-R3_RNA: R. ferrumequinum Ectoderm RNA-seq replicate 3 | This paper | GEO: GSE221965 |
| RFe_BEF-R1_ATAC: R. ferrumequinum fibroblasts ATAC-seq replicate 1 | This paper | GEO: GSE221965 |
| RFe_BEF-R2_ATAC: R. ferrumequinum fibroblasts ATAC-seq replicate 2 | This paper | GEO: GSE221965 |
| RFe_BiPS2-R1_ATAC: R. ferrumequinum iPS#2 ATAC-seq replicate 1 | This paper | GEO: GSE221965 |
| RFe_BiPS2-R2_ATAC: R. ferrumequinum iPS #2 ATAC-seq replicate 2 | This paper | GEO: GSE221965 |
| RFe_BEF_H3K4: R. ferrumequinum fibroblasts H3K4me3 ChIP-seq | This paper | GEO: GSE221965 |
| RFe_BiPS2_H3K4: R. ferrumequinum iPS H3K4me3 ChIP-seq | This paper | GEO: GSE221965 |
| RFe_BEF_H3K27: R. ferrumequinum fibroblasts H3K4me3 ChIP-seq | This paper | GEO: GSE221965 |
| RFe_BiPS2_H3K27: R. ferrumequinum iPS#2 H3K4me3 ChIP-seq | This paper | GEO: GSE221965 |
| RFe_BEF-R1: RRBS: R. ferrumequinum fibroblasts RRBS replicate 1 | This paper | GEO: GSE221965 |
| RFe_BEF-R2: RRBS: R. ferrumequinum fibroblasts RRBS replicate 2 | This paper | GEO: GSE221965 |
| RFe_BiPS2-R1_RRBS: R. ferrumequinum iPS#2 RRBS replicate 1 | This paper | GEO: GSE221965 |
| RFe_BiPS2-R2_RRBS: R. ferrumequinum iPS#2 RRBS replicate 2 | This paper | GEO: GSE221965 |
| Dog iPS RNAseq: K9_1_iPS_P9 | GSE15249395 | GEO: GSM4616525 |
| Dog iPS RNAseq: K9_2_iPS_P8 | GSE15249395 | GEO: GSM4616526 |
| Dog iPS RNAseq: K9_3_iPS_P8 | GSE15249395 | GEO: GSM4616527 |
| Marmoset iPS RNAseq: CM421F_B0_12_iPS_P11 | GSE15249395 | GEO: GSM4617887 |
| Marmoset iPS RNAseq: CTXNS1_B1_iPS_P7 | GSE15249395 | GEO: GSM4617889 |
| Marmoset iPS RNAseq: CTXNS2_B1_iPS_P5 | GSE15249395 | GEO: GSM4617890 |
| Marmoset iPS RNAseq: E01F_A2_2_iPS_P10 | GSE15249395 | GEO: GSM4617891 |
| Marmoset iPS RNAseq: E02M_B0_7_iPS_P13 | GSE15249395 | GEO: GSM4617895 |
| Marmoset iPS RNAseq: I5061F_B3_15_iPS_P10 | GSE15249395 | GEO: GSM4617900 |
| Marmoset iPS RNAseq: I5061F_B3_3_iPS_P11 | GSE15249395 | GEO: GSM4617901 |
| Human iPS RNA-seq: BjiPS_P3 | GSE15249395 | GEO: GSM4616532 |
| Pig iPS cell RNA-seq: N01F_N1_iPS_P3 | GSE15249395 | GEO: GSM4616535 |
| Pig iPS cell RNA-seq: N01F_N2_iPS_P3 | GSE15249395 | GEO: GSM4616536 |
| Mouse ES cell RNA-seq: WT ESC -1 | GSE5321296 | GEO: GSM1287734 |
| Mouse ES cell RNA-seq: WT ESC -2 | GSE5321296 | GEO: GSM1287735 |
| Mouse ES cell RNA-seq: WT ESC -3 | GSE5321296 | GEO: GSM1287745 |
| Mouse ES cell RNA-seq: WT ESC -4 | GSE5321296 | GEO: GSM1287746 |
| Mouse ES cell RNA-seq: iPSC -1 | GSE5321296 | GEO: GSM1287736 |
| Mouse iPS cell RNA-seq: WT iPSC -2 | GSE5321296 | GEO: GSM1287747 |
| Mouse iPS cell RNA-seq: WT iPSC -3 | GSE5321296 | GEO: GSM1287748 |
| Mouse fibroblasts RNA-seq: WT MEFs -1 | GSE5321296 | GEO: GSM1287749 |
| Mouse fibroblasts RNA-seq: WT MEFs -2 | GSE5321296 | GEO: GSM1287750 |
| Human naive ES cell RNA-seq: Naive_Ex1 | GSE14499497 | GEO: GSM4303994 |
| Human naive ES cell RNA-seq: Naive_Ex2 | GSE14499497 | GEO: GSM4303995 |
| Human primed ES cell RNA-seq: Primed_Ex1 | GSE14499497 | GEO: GSM4304018 |
| Human primed ES cell RNA-seq: Primed_Ex2 | GSE14499497 | GEO: GSM4304019 |
| Human H9 ES cells RNA-seq: H9.WT.1.1 _RNA-seq | GSE14099798 | GEO: GSM4192140 |
| Human H9 ES cells RNA-seq: H9.WT.1.2_RNA-seq | GSE14099798 | GEO: GSM4192141 |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| Rhinolophus ferrumequinum embryonic fibroblasts | This paper | BEF |
| Rhinolophus ferrumequinum induced | This paper | RFe.iPS#1(BiPS1) |
| pluripotent stem cells clone 1 | ||
| Rhinolophus ferrumequinum induced | This paper | RFe.iPS2 (BiPS#2) |
| pluripotent stem cells clone 2 | ||
| Myotis myotis uropatagium fibroblasts | This paper | MMy.BUF |
| Myotis myotis induced pluripotent stem cells | This paper | MMy.iPS |
| 293FT cells | Life Technologies | R700-07 |
| NIH/3T3 cells | ATCC | CRL-1658 |
| R1 mouse ES cells | ATCC | SCRC-1011 |
| H9 human ES cells | WiCell | WA09 |
| Vero-E6 | ATCC | CRL-1587 |
| Irradiated CF1 mouse fibroblasts | Gibco | A34181 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| 8-week-old male Fox Chase SCID Beige Mice | Charles River | 250 |
|
| ||
| Oligonucleotides | ||
|
| ||
| SeV-F: GGATCACTAGGTGATATCGAGC | CytoTune 2.0 | N/A |
| SeV-R: | CytoTune 2.0 | N/A |
| ACCAGACAAGAGTTTAAGAGATATGTATC | ||
| KOS-F: ATGCACCGCTACGACGTGAGCGC | CytoTune 2.0 | N/A |
| KOS-R: ACCTTGACAATCCTGATGTGG | CytoTune 2.0 | N/A |
| Klf4-F TTCCTGCATGCCAGAGGAGCCC | CytoTune 2.0 | N/A |
| Klf4-R: AATGTATCGAAGGTGCTCAA | CytoTune 2.0 | N/A |
| cMyc-F: TAACTGACTAGCAGGCTTGTCG | CytoTune 2.0 | N/A |
| cMyc-R: TCCACATACAGTCCTGGATGATGATG | CytoTune 2.0 | N/A |
| GAPDH_F1_GHB: TGGTGAAGGTCGGAGTGAAC | This paper | Z25-132 |
| GAPDH_R1_GHB: GAAGGGGTCATTGATGGCGA | This paper | Z25-133 |
|
| ||
| Software and algorithms | ||
|
| ||
| FastQC vO.11.9 | Andrews et al.99 | https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ |
| Trimmomatic v0.39 | Bolger et al.100 | http://www.usadellab.org/cms/?page=trimmomatic |
| HISAT2 v2.2.1 | Kim et al.101 | http://daehwankimlab.github.io/hisat2/ |
| SAMtools v1.10 | Li et al.102 | https://github.com/samtools/samtools/releases/ |
| featureCounts v2.0.1 | Liao et al.103 | https://subread.sourceforge.net/ |
| deepTools | Rami’rez et al.104 | https://deeptools.readthedocs.io/en/develop/content/tools/bamCoverage.html |
| DESeq2 v1.10.1 | Love et al.105 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| ClustVis (Beta) | Metsalu and Vilo106 | https://biit.cs.ut.ee/clustvis/ |
| R package ggpubr | Wickham and Grolemund107 | https://cran.r-project.org/web/packages/ggpubr/index.html |
| ggplot2 | Wickham and Grolemund107 | cran.r-project.org/web/packages/ggplot2/index.html |
| ggmaplot | Wickham and Grolemund107 | https://rpkgs.datanovia.com/ggpubr/reference/ggmaplot.html |
| ggscatter | Wickham and Grolemund107 | https://rpkgs.datanovia.com/ggpubr/reference/ggscatter.html |
| codeml package of the PAML suite software | Yang et al.108 | http://abacus.gene.ucl.ac.uk/software/paml.html |
| bedGraphToBigWig v4 | Kent et al.109 | https://www.encodeproject.org/software/bedgraphtobigwig/ |
| Enrichr | Xie et al.110 | maayanlab.cloud/Enrichr/ |
| Cytoscape Version 3.8.2 | Shannon et al.111 | https://cytoscape.org |
| SMRTLink version 10.1 | PacBio | https://www.pacb.com/support/software-downloads/ |
| Kraken2 v2.1.2 | Wood et al.112 | https://github.com/DerrickWood/kraken2 |
| gmap/gsnap | Wu et al.113 | http://research-pub.gene.com/gmap |
| Trimmgalore v.0.6.6 | Github, Open Source | https://github.com/FelixKrueger/TrimGalore |
| Cutadapt | Krueger n.d. Andrews114 | https://github.com/marcelm/cut adapt |
| stringTie v2.2.1 | Pertea et al.115 | http://ccb.jhu.edu/software/stringtie/index.shtml?t=manual |
| Seqtk v1.3 | Github, Open Source | https://github.com/lh3/seqtk |
| Trinity v2.12 | Grabbherr et al.116 | https://github.com/trinityrnaseq/trinityrnaseq/releases/tag/v2.12.0 |
| BLAST | Altschul et al.117 Camacho118 | https://blast.ncbi.nlm.nih.gov/Blast.cgi |
| SQUID | Eddy Lab, Harvard University | http://eddylab.org/software.html |
| INSPIRE 2.0 | Luminex | https://www.luminexcorp.com |
| IDEAS 6.2 | Luminex | https://www.luminexcorp.com |
|
| ||
| Other | ||
|
| ||
| ATAC-seq service | Active Motif | 25079 |
| RRBS Service, Active Motif, | Active Motif | 25069 |
| HistoPath ChIP-seq service, | Active Motif | 25001 |