

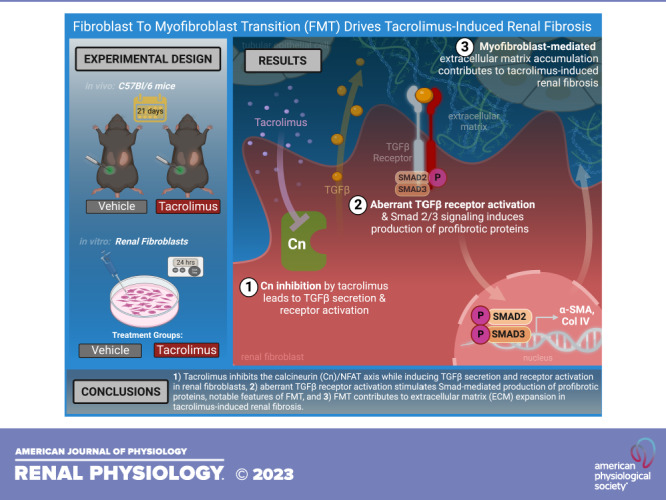
Keywords: calcineurin inhibitors, fibroblast activation, fibroblast-to-myofibroblast transition, renal fibrosis, tacrolimus
Abstract
Use of immunosuppressant calcineurin inhibitors (CNIs) is limited by irreversible kidney damage, hallmarked by renal fibrosis. CNIs directly damage many renal cell types. Given the diverse renal cell populations, additional targeted cell types and signaling mechanisms warrant further investigation. We hypothesized that fibroblasts contribute to CNI-induced renal fibrosis and propagate profibrotic effects via the transforming growth factor-β (TGF-β)/Smad signaling axis. To test this, kidney damage-resistant mice (C57BL/6) received tacrolimus (10 mg/kg) or vehicle for 21 days. Renal damage markers and signaling mediators were assessed. To investigate their role in renal damage, mouse renal fibroblasts were exposed to tacrolimus (1 nM) or vehicle for 24 h. Morphological and functional changes in addition to downstream signaling events were assessed. Tacrolimus-treated kidneys displayed evidence of renal fibrosis. Moreover, α-smooth muscle actin expression was significantly increased, suggesting the presence of fibroblast activation. TGF-β receptor activation and downstream Smad2/3 signaling were also upregulated. Consistent with in vivo findings, tacrolimus-treated renal fibroblasts displayed a phenotypic switch known as fibroblast-to-myofibroblast transition (FMT), as α-smooth muscle actin, actin stress fibers, cell motility, and collagen type IV expression were significantly increased. These findings were accompanied by concomitant induction of TGF-β signaling. Pharmacological inhibition of the downstream TGF-β effector Smad3 attenuated tacrolimus-induced phenotypic changes. Collectively, these findings suggest that 1) tacrolimus inhibits the calcineurin/nuclear factor of activated T cells axis while inducing TGF-β1 ligand secretion and receptor activation in renal fibroblasts; 2) aberrant TGF-β receptor activation stimulates Smad-mediated production of myofibroblast markers, notable features of FMT; and 3) FMT contributes to extracellular matrix expansion in tacrolimus-induced renal fibrosis. These results incorporate renal fibroblasts into the growing list of CNI-targeted cell types and identify renal FMT as a process mediated via a TGF-β-dependent mechanism.
NEW & NOTEWORTHY Renal fibrosis, a detrimental feature of irreversible kidney damage, remains a sinister consequence of long-term calcineurin inhibitor (CNI) immunosuppressive therapy. Our study not only incorporates renal fibroblasts into the growing list of cell types negatively impacted by CNIs but also identifies renal fibroblast-to-myofibroblast transition as a process mediated via a TGF-β-dependent mechanism. This insight will direct future studies investigating the feasibility of inhibiting TGF-β signaling to maintain CNI-mediated immunosuppression while ultimately preserving kidney health.
INTRODUCTION
The influence of calcineurin inhibitors (CNIs) such as cyclosporine (CsA) and tacrolimus has bolstered the field of organ transplantation with their immunosuppressive effects (1–3). However, the overall success of these immunosuppressant drugs is limited by nephropathy during chronic use. Long-term CNI therapy causes irreversible kidney damage, hallmarked by interstitial fibrosis and glomerulosclerosis (4–8). CNI-induced renal fibrosis is a pathological process characterized by increased extracellular matrix (ECM) protein deposition coupled with reduced matrix turnover, thereby promoting ECM expansion. These matrix disturbances contribute to the disruption of renal architecture and functional capacity. Consequently, ∼15% of patients on chronic CNI therapy experience an eventual progression to end-stage renal disease and organ failure (9, 10). The mechanisms underlying CNI-induced nephropathy remain a poorly understood dilemma in the field, and, to date, there are no specific therapeutic strategies to mitigate this damage. Kidneys of patients on chronic CNI therapy show increased expression of the profibrotic cytokine transforming growth factor-β (TGF-β). However, the precise role of TGF-β signaling in mediating renal damage induced by CNIs remains to be defined. These existing gaps in knowledge along with the detrimental outcomes present a critical need to identify the driving mechanisms by which CNIs induce renal damage. This insight will direct development of newer-generation CNI immunosuppressants exhibiting renopreservative potential.
Clinicians use CNIs because of their profound immunosuppressive effects. Upon immune cell stimulation, calcineurin subunits associate with Ca2+/calmodulin to form an active enzyme complex (11). Activation of this complex leads to the dephosphorylation of well-recognized downstream calcineurin targets, such as nuclear factor of activated T cells (NFAT) proteins (11–14). Once dephosphorylated, NFAT proteins translocate into the nucleus to promote transcription of proinflammatory cytokines (15). CNIs exert an immunosuppressive effect by complexing with cytosolic immunophilins [CsA: cyclophilin and tacrolimus: FK506-binding protein 12 (FKBP12)] to noncompetitively inhibit calcineurin (14, 16, 17). Thus, CNIs severely limit calcineurin-mediated dephosphorylation of target proteins. However, because of the ubiquitous expression of calcineurin in various tissues (18, 19), calcineurin inhibition in immune cells is often accompanied by the induction of collateral damage in other organ systems, particularly the kidneys (1, 10, 20). CNIs directly induce damage to many renal cell types including epithelial, endothelial, and mesangial cells (21–23). However, given the diverse cell populations in the kidney (24), additional targeted cell types and signaling mechanisms behind CNI-induced renal damage remain to be uncovered.
In the present study, we aimed to identify renal fibroblasts as a contributor to CNI-induced fibrosis through a phenotypic switch known as fibroblast-to-myofibroblast transition (FMT). FMT occurs after injury, where the quiescent fibroblast state is halted in favor of proliferation into an activated fibroblast (myofibroblast) state (25). Myofibroblasts have enhanced motility via upregulation of contractile markers [i.e., α-smooth muscle actin (α-SMA), etc.] in addition to forming collagen-rich ECM components that fill the interstitium (25). Progression of these events contributes to nephron loss and declining kidney function. Therefore, an abundance of myofibroblasts is recognized as a predictor of fibrosis in both human and animal models of renal disease (26). The myofibroblast cell type arises from circulating progenitors, tubular epithelial cells, and resident fibroblasts (27). Although the role of myofibroblasts in tacrolimus-induced renal damage is widely accepted (28), their origin is still a matter of debate.
Given the role of fibroblasts as the primary matrix-producing cells in the kidney, we investigated renal FMT as a molecular mechanism inducing renal fibrosis with CNI use. Specifically, using both in vivo and in vitro models of CNI-induced nephropathy, we demonstrated that 1) tacrolimus inhibits the calcineurin/NFAT axis while inducing TGF-β1 ligand secretion and receptor activation in renal fibroblasts; 2) aberrant TGF-β receptor activation stimulates Smad-mediated transcription of myofibroblast markers, notable features of FMT; and 3) FMT contributes to ECM expansion in tacrolimus-induced renal fibrosis. Taken together, these results not only incorporate renal fibroblasts into the growing list of cell types targeted by CNIs but also identify renal FMT as a process mediated via a TGF-β-dependent mechanism.
EXPERIMENTAL DESIGN
CNI Models
In vivo.
Although sex is not considered a risk factor for CNI-induced nephropathy in humans (29), male rats exhibited greater CNI-induced nephropathy than female rats (30). In light of these findings, male WT C57BL/6 mice (n = 16 total, n = 8/group, 8 wk old; Jackson Laboratory) were randomly assigned to receive intraperitoneal injections of FK-506 (tacrolimus, 10 mg/kg/day; Enzo) or vehicle (ethanol/DMSO/saline) for 21 days. Factoring in the drug pharmacokinetics in mice (31), this animal dose is consistent with the human equivalent tacrolimus dosing of organ transplant recipients (0.15–3 mg/kg/day) (32–34). Furthermore, similar doses (35) and time points (32, 35) have been shown to induce upregulation of profibrotic markers in experimental animal models. The order of daily injections was alternated between treatment groups to minimize potential confounding variables. Notably, no signs of distress were observed; therefore, no animals were excluded from the study (see Supplemental Table S1). For this study, mice were socially housed in standard cages with a regular 12:12-h light-dark cycle and fed standard rodent chow. Furthermore, all animal protocols and procedures were approved by the Animal Care and Use Committee of Wright State University and were conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
In vitro.
The renal fibroblast cell line was kindly gifted to the Williams laboratory by Dr. Jennifer Gooch. These spontaneously immortalized cells were generated from the kidneys of wild-type male mice from a mixed genetic background as previously described (20, 36). Furthermore, cell lines were authenticated by IDEXX BioResearch. For in vitro experiments, renal fibroblasts were maintained in growth medium (DMEM, F-10 Ham’s, 10% FBS, penicillin-streptomycin, and Plasmocin) under standard conditions (37°C, 5% CO2). At 85% confluence, the culture medium was changed to serum-free medium for 24 h. Afterward, cells were exposed to serum-free medium containing either 1 nM tacrolimus or 0.01% vehicle (ethanol) for 24 h. Previous studies have shown that similar doses (37) and time points (12, 20, 38, 39) were sufficient to induce upregulation of profibrotic markers. In a separate set of experiments, both vehicle- and tacrolimus-treated cells were preincubated with 1 µM ionomycin [a Ca2+ ionophore known to induce calcineurin/NFAT signaling (40); catalog no. 73722, STEMCELL Technologies, Vancouver, BC, Canada] for 15 min. A final set of experiments consisted of fibroblasts treated with either 1 nM tacrolimus or 0.01% vehicle (ethanol) in the presence or absence of the Smad3 inhibitor SIS3 (500 nM; catalog no. HY-13013, MedChemExpress, Monmouth Junction, NJ). SIS3 acts as a selective inhibitor of Smad3 phosphorylation (activation).
Histology and Immunocytochemistry
Kidney samples were fixed in freshly prepared 4% paraformaldehyde, and routine histology was performed on 5-µm-thick sections. Alterations in kidney morphology were initially assessed by examination of hematoxylin and eosin-stained sections (conducted by AML Laboratories, St. Augustine, FL). Images of glomerular and tubular changes were captured with a Keyence BZ-X800 fluorescence microscope (×60 magnification, exposure time: 1/45 s). ECM accumulation (fibrosis) was assessed by collagen deposition with both Masson trichrome (catalog no. ab150686, Abcam, Cambridge, UK) and picrosirius red (catalog no. ab150681, Abcam) protocols. Images were acquired with a Keyence BZ-X800 fluorescence microscope (×60 magnification, exposure time: 1/80 s).
To assess myofibroblast cell abundance, fluorescent detection of the myofibroblast marker α-SMA was performed. Sections were blocked in 10% goat serum (catalog no. 16210-072, Life Technologies, Carlsbad, CA) in Tris-buffered saline-Tween for 30 min. Sections were then incubated with α-SMA-specific primary antibody (catalog no. 19245S, Cell Signaling Technology, Danvers, MA; 1:100 dilution). After incubation with Alexa Fluor 555 anti-rabbit secondary antibody (catalog no. A32794, Invitrogen, Waltham, MA; 1:1,000 dilution), images (5 or 6 sections from each treatment group) were captured with a Keyence BZ-X800 fluorescence microscope (×60 magnification, exposure time: 1/80 s). The Zen 3.1 software program was used to quantify α-SMA intensity.
To assess expression of the phosphorylated type I TGF-β receptor subunit (phospho-TGFβRI), chromogen staining was performed. Briefly, endogenous peroxidases were quenched in deparaffinized sections by incubation in 0.3% H2O2 (catalog no. ab64218, Abcam) for 15 min. Sections were then blocked in 10% goat serum (Life Technologies) in Tris-buffered saline-Tween for 30 min and incubated with phospho-TGFβRI-specific antibody (catalog no. PA5-40298, Invitrogen; 1:100 dilution). Sections were incubated with horseradish peroxidase-conjugated secondary antibody (catalog no. A32794, Invitrogen; 1:200 dilution). Bound antibody was identified by 3,3′-diaminobenzidine staining according to the manufacturer’s instructions (catalog no. SK-4100, Vector Laboratories, Burlingame, CA). In addition, sections were counterstained with hematoxylin according to the manufacturer’s instructions (catalog no. 220365, Abcam). Phospho-TGFβRI abundance was visualized with a Keyence BZ-X800 fluorescence microscope, and brightfield images at ×60 magnification and an exposure time of 1/45 s were captured.
Treated fibroblasts were fixed in 4% paraformaldehyde and permeabilized with 0.1% Triton-PBS before incubation with the following antibodies: α-SMA (Cell Signaling Technology; dilution 1:1,000), collagen type IV (Col IV; catalog no. sc-398655, Santa Cruz Biotechnology, Dallas, TX; 1:500 dilution), NFATc1 (catalog no. 5861S, Cell Signaling Technology; dilution 1:500), and phospho-TGFβRI (1:500 dilution; Invitrogen). Subsequently, cells were incubated with either Alexa Fluor 488 (catalog no. A32790, Invitrogen; 1:1,000 dilution) or Alexa Fluor 555 (Invitrogen; 1:1,000 dilution) secondary antibody and DAPI (catalog no. D1306, ThermoFisher; 1:1,000 dilution). Moreover, a subset of cells was also incubated with the Alexa Fluor 555-conjugated phalloidin probe (catalog no. A30106, Invitrogen; 1:1,000 dilution) and DAPI (1:1,000 dilution; ThermoFisher). Representative images were acquired with a Keyence BZ-X800 fluorescence microscope (at both ×20 and ×60 magnification; depending on antibody, exposure times ranged from 1/40 to 1/80 s). Relative intensities from five or six images (per treatment group) were quantified with both CellProfiler and ImageJ software programs. To quantify nuclear NFATc1 intensity, the following pipeline was used: the “IdentifyPrimaryObjects” module was used to identify DAPI-stained nuclei. The “MeasureObjectIntensity” module was then used to quantify the mean intensity of nuclear NFAT per cell. For quantification of α-SMA, Col IV, phalloidin, and phospho-TGFβRI intensity, the respective pipeline in CellProfiler was used. Initially, the “IdentifyPrimaryObjects” module was used for the identification of DAPI-stained nuclei. Next, the “IdentifySecondaryObjects” module propagation method was used to identify and select all cells present in the image. Finally, the “IdentifyTertiaryObjects” module was used to subtract the outlined nuclei from the cell selections, leaving only the cytoplasmic portion of each cell to be quantified. For a field of view of 282 × 182 µm, the total intensity for all fluorescent images was calculated, expressed as arbitrary light units, and displayed on the respective image. Finally, a heatmap of images was generated with ImageJ.
Motility Assessment
Scratch wound assays were used to assess fibroblast motility. Upon formation of an ∼100% confluent monolayer, a midline linear scratch (“wound”) was generated with a sterile pipette tip. After a sterile PBS wash to remove cellular debris, cells were treated with either 1 nM tacrolimus or 0.01% vehicle (ethanol) for 12 h. Images of initial scratches (time 0) and respective scratch closures (time = 12 h) were captured with an Invitrogen EVOS XL Core Imaging System (×10 magnification). Images of three focal areas per well were captured for each experiment. Differences in scratch closures between groups were quantified and averaged by a blinded reviewer using ImageJ software. The migration rate percentage was calculated with the following established formula (41):
Morphological Changes
To visualize morphological changes such as cell size and shape, brightfield images of treated cells were acquired with a Keyence BZ-X800 fluorescence microscope at ×60 magnification (exposure time: 1/50 s). Images of four focal areas per well were captured for each experiment. ImageJ software was also used to quantify both cell number and morphological changes by a blinded reviewer. Mean numbers of myofibroblasts were calculated based on cytoplasmic granule intensity compared with quiescent fibroblasts by an established method (27, 42).
Western Blot Analysis
Briefly, whole cell protein was extracted from both tissue and cells with RIPA lysis buffer, as previously described (20). In separate experiments, nuclear proteins were collected with a nuclear extraction kit (catalog no. 40410, Activ Motif, Carlsbad, CA), as described by the manufacturer. Both whole cell (40 μg) and nuclear (100 μg) protein were separated by 7.5% SDS-PAGE and transferred onto polyvinylidene difluoride (PVDF) membranes. PVDF membranes were then incubated in either 3% nonfat milk or 5% BSA in either PBS or Tris-buffered saline-Tween before incubation with primary antibodies specific for either α-SMA (Cell Signaling Technology; 1:1,000 dilution), total TGFβRI (catalog no. PA5-14959, Invitrogen; 1:1,000 dilution), phospho-TGFβRI (Invitrogen; 1:1,000 dilution), Col IV (Santa Cruz Biotechnology; 1:500 dilution), phospho-Smad2/3 (catalog no. Cell Signaling Technology; 1:500 dilution), β-actin (catalog no. 8457 L, Cell Signaling Technology; 1:10,000 dilution), or histone deacetylase 1 (catalog no. NB100, Novus Biologicals, Centennial, CO; 1:5,000 dilution). After incubation in either rabbit (catalog no. A0545, Sigma-Aldrich, St. Louis, MO; 1:5,000 dilution) or mouse (catalog no. 62-6520, Invitrogen; 1:5,000 dilution) secondary antibody, immunoreactive bands were detected with the Azure Biosystems c500 Infrared Imaging System and cSeries Capture Software. Densitometric values were obtained with AzureSpot Analysis Software. Values of proteins of interest were normalized to either β-actin (whole cell) or histone deacetylase 1 (nuclear extracts). Data are expressed as fold changes of vehicle.
ELISA
To examine NFAT activation, NFAT DNA-binding activity was assessed by a TransAM NFATc1 Transcription Factor Assay Kit (catalog no. 40796, Active Motif) as described by the manufacturer. This ELISA-based assay measures binding of active NFAT to its DNA recognition sequence. Briefly, nuclear cell extracts were incubated with an immobilized oligonucleotide containing a 5′- AGGAAA-3′ DNA motif. Bound NFAT was detected and quantified. Data are expressed as fold changes of vehicle.
To assess TGF-β1 ligand secretion, ELISAs were also conducted according to the manufacturer’s instructions (catalog no. DY167905, R&D Systems, Minneapolis, MN). With culture media collected from both vehicle- and tacrolimus-treated cells, active TGF-β1 concentrations were quantified by extrapolating the absorbance values of experimental samples from a standard curve of recombinant TGF-β1 proteins.
Statistical Analysis
Graph creation and statistical analyses for all experiments were performed with GraphPad software (Prism; San Diego, CA). Statistical tests were conducted by using either a t test or two-way ANOVA to detect differences between the means of the experimental groups. A P value of <0.05 was considered statistically significant.
RESULTS
Tacrolimus Induces Kidney Damage in the Form of Renal Fibrosis in Mice
To establish that our in vivo model recapitulates the clinical morphological features of CNI-induced nephropathy, wild-type mice were treated with either tacrolimus (10 mg/kg) or vehicle (ethanol/DMSO) for 21 days (Fig. 1A). Tacrolimus- and vehicle-treated mouse kidneys were assessed for histological lesions via hematoxylin and eosin staining (Fig. 1B). Vehicle-treated kidneys retained a normal renal architecture that consisted of healthy glomeruli and tubules. However, tacrolimus-treated kidneys showed notable glomerular and tubular changes consistent with CNI nephrotoxicity (43). In particular, glomeruli from tacrolimus-treated kidneys displayed thrombotic microangiopathy, characterized by the presence of hyaline thrombi in glomerular capillaries (box in Fig. 1B). Additionally, the reduction of interstitial spacing in tacrolimus-treated kidneys was indicative of ECM expansion, another characteristic of CNI nephropathy (6). To confirm histological characteristics of chronic CNI use, kidneys were further assessed for collagen (ECM) accumulation via Masson trichrome staining (Fig. 1C). In tacrolimus-treated kidneys, ECM deposition (blue) was present in periglomerular and interstitial areas (arrows). These findings were confirmed with picrosirius red staining (Fig. 1D), an additional method to detect collagen abundance. Collectively, these findings establish that tacrolimus induces kidney damage in the form of renal fibrosis.
Figure 1.

Tacrolimus induces kidney damage in the form of renal fibrosis in mice. To establish that our in vivo model recapitulates the morphological features of calcineurin inhibitor-induced nephropathy, wild-type mice were treated with either 10 mg/kg tacrolimus or vehicle (ethanol/DMSO/saline) for 21 days (A). Tacrolimus-induced histological lesions were assessed by hematoxylin and eosin (H&E) staining (B, box). Extracellular matrix accumulation (collagen) was assessed by both Masson trichrome staining (blue; C, arrows) and picrosirius red staining (D). Representative images shown of 8 animals per group.
Tacrolimus-Induced Renal Fibrosis Is Accompanied by Fibroblast Activation in Mice
Although our findings establish that tacrolimus induces kidney fibrosis, the cellular source of ECM deposition was unknown. To examine whether fibroblast activation accompanies tacrolimus-induced renal fibrosis, the myofibroblast marker α-SMA (red) was assessed via immunofluorescence and quantified (Fig. 2A). Vehicle-treated kidneys localized α-SMA expression exclusively in the renal vasculature, with low glomerular expression. However, α-SMA expression in tacrolimus-treated kidneys was not limited only to vascular cells but was also present in the interstitium, lining nearby tubular and glomerular cells. These findings were confirmed via Western blot analysis (Fig. 2B), with tacrolimus-treated kidneys showing increased α-SMA expression (2.2 ± 0.42 arbitrary units) compared with vehicle-treated kidneys (1.0 ± 0.11 arbitrary units). Collectively, these findings suggest that fibroblasts, the primary matrix-producing cells in the kidney, serve as a culprit of CNI-induced renal fibrosis.
Figure 2.

Tacrolimus-induced renal fibrosis is accompanied by fibroblast activation in mice. To investigate whether fibroblast activation accompanied renal fibrosis, wild-type mice were treated with either 10 mg/kg tacrolimus or vehicle (ethanol/DMSO/saline) for 21 days. The myofibroblast marker α-smooth muscle actin (α-SMA) was also visualized via immunofluorescence (A, red) and quantified with Zen 3.1 software [expressed as arbitrary light units (alu)]. Western blot analysis (B) further quantified α-SMA expression. Representative images are shown. Values are means ± SE for 8 animals per group. Statistical tests were conducted by using a t test to detect differences between the means of experimental groups. *P < 0.05 and **P < 0.005 vs. vehicle.
Tacrolimus Inhibits Calcineurin Activity/NFAT Activation in Renal Fibroblasts
The kidney contains a diverse population of cell types (epithelial tubular cells, mesangial cells, immune cells, etc.), some of which have been identified as contributors to renal fibrosis (12, 32, 38, 44–47). Given their role as the primary matrix-producing cells in the kidney, we investigated whether renal fibroblasts were a cellular source of tacrolimus-induced renal fibrosis. In this endeavor, we initially assessed whether calcineurin activity/NFAT activation was inhibited in fibroblasts treated with tacrolimus. To this end, resting kidney fibroblasts (quiescent cells that have not transitioned into motile ECM-producing myofibroblasts) were treated with either tacrolimus (1 nM) or vehicle (0.01% ethanol) for 24 h (Fig. 3A). We then assessed activation of the well-known calcineurin substrate NFAT1. Whereas basal levels of nuclear NFAT (green) were present in vehicle-treated cells (Fig. 3Bi), tacrolimus-treated cells (Fig. 3Bii) showed decreased nuclear NFAT localization compared with vehicle treatment. In contrast, a surge of nuclear NFAT was induced in cells exposed to ionomycin (Fig. 3Biii), a Ca2+ ionophore known to induce calcineurin activation and subsequent NFAT nuclear translocation. However, tacrolimus treatment attenuated the surge in nuclear NFAT induced by ionomycin (Fig. 3Biv). Confirming these qualitative findings, NFAT activation assays (Fig. 3C) showed a statistically significant reduction in NFAT DNA binding in cells treated with tacrolimus compared with vehicle (tacrolimus: 0.74 ± 0.07 arbitrary units vs. vehicle: 1.0 ± 0.00 arbitrary units). In contrast, NFAT binding significantly increased with vehicle and ionomycin treatment (1.4 ± 0.13 arbitrary unit), an effect attenuated in tacrolimus-treated fibroblasts (0.87 ± 0.06 arbitrary units). Collectively, these findings demonstrate that tacrolimus effectively inhibits the calcineurin/NFAT axis in renal fibroblasts.
Figure 3.

Tacrolimus inhibits calcineurin (Cn) activity/nuclear factor of activated T cells (NFAT) activation in renal fibroblasts. To investigate whether tacrolimus inhibits Cn signaling in vitro, renal fibroblasts were treated with either 1 nM tacrolimus or vehicle for 24 h (A). Activation of a well-known Cn substrate, NFAT, was assessed. To confirm NFAT activation, both vehicle- and tacrolimus-treated cells were incubated with 1 µM ionomycin (a Ca2+ ionophore known to induce Cn/NFAT signaling) for 15 min. Nuclear NFAT abundance was visualized by immunofluorescence and quantified by CellProfiler software (B) [expressed as arbitrary light units (alu)], whereas NFAT activation was assessed via NFAT transcription factor assays (C). Representative images are shown. Values are means ± SE from 3 or 4 independent studies. Statistical tests were conducted using two-way ANOVA to detect variances between experimental groups. *P < 0.05, **P < 0.005, and ***P < 0.0005 vs. vehicle; ##P < 0.005 and ###P < 0.0005 vs. vehicle + ionomycin.
Tacrolimus Induces TGF-β Receptor Activation and Downstream Smad2/3 Phosphorylation in Mouse Kidneys
Next, we aimed to identify the signaling mediators driving tacrolimus-induced FMT. Kidneys of patients on long-term CNI therapy show increased expression of TGF-β1 ligand. However, the precise role of TGF-β signaling in mediating renal damage induced by CNIs remains to be defined. We then investigated whether tacrolimus induces TGF-β receptor activation. Western blot analysis (Fig. 4A) showed a decrease in total TGFβRI expression in tacrolimus-treated kidneys compared with vehicle (tacrolimus: 0.66 ± 0.09 arbitrary units vs. vehicle: 1.0 ± 0.05 arbitrary units). TGFβRI activation was then examined by assessing phospho-TGFβRI expression, also via Western blot analysis (Fig. 4B). Although tacrolimus treatment reduced total TGFβRI expression, phosphorylation of the present receptors was increased (tacrolimus: 1.4 ± 0.15 arbitrary units vs. vehicle: 1.0 ± 0.12 arbitrary units). These findings were confirmed by immunohistochemistry (Fig. 4C), as tacrolimus-treated kidneys showed greater phospho-TGFβRI expression (dark brown) in selected tubular epithelial cells. To assess the activation of downstream TGF-β receptor signaling mediators, phospho-Smad2/3 expression was quantified via Western blot analysis. Compared with vehicle, phospho-Smad2/3 abundance was significantly increased with tacrolimus treatment (tacrolimus: 1.17 ± 0.05 vs. vehicle: 1.0 ± 0.05 arbitrary units; Fig. 4D). Collectively, these findings demonstrate that tacrolimus activates the TGF-β/Smad signaling axis.
Figure 4.

Tacrolimus induces transforming growth factor-β (TGF-β) receptor activation and downstream Smad2/3 phosphorylation in mouse kidneys. To investigate whether tacrolimus induces TGF-β receptor activation, wild-type C57BL/6 mice were treated with either 10 mg/kg tacrolimus or vehicle (ethanol/DMSO/saline) for 21 days. Mouse kidneys were assessed for total type I TGF-β receptor subunit (TGFβRI) expression via Western blot (A). TGFβRI activation was examined by assessing phospho-TGFβRI expression via both Western blot (B) and immunohistochemistry (C). To assess whether tacrolimus induces downstream TGF-β receptor signaling, phospho-Smad2/3 abundance (D) was assessed via Western blot. Representative images are shown. Values are means ± SE for 7 or 8 animals per group. Statistical tests were conducted using a t test to detect differences between the means of experimental groups. *P < 0.05 and **P < 0.005 vs. vehicle.
Tacrolimus Promotes FMT and ECM Production
Next, we investigated whether renal fibroblasts were a cellular source of tacrolimus-induced renal fibrosis (Fig. 5A). As assessed by immunofluorescence, heatmap analysis (Fig. 5B) showed enhanced α-SMA expression in tacrolimus-treated cells and revealed high luminescence around the cell perimeter (red). Consistently, Western blot analysis showed an increase in α-SMA protein abundance (tacrolimus: 1.6 ± 0.08 arbitrary units vs. vehicle: 1.0 ± 0.00 arbitrary units; Fig. 5C). To further confirm the myofibroblast phenotype, as indicated by the presence of actin stress fibers, phalloidin abundance was assessed by immunofluorescence. Heatmap analysis revealed that tacrolimus-treated cells showed increased phalloidin staining compared with vehicle (Fig. 5D). These findings establish the presence of activated renal myofibroblasts with tacrolimus treatment. To provide evidence of enhanced motility of these cells, the rate of fibroblast migration (in %) was assessed via scratch test assays (Fig. 5E). Compared with vehicle, tacrolimus-treated cells showed a 17% increase in migration rate after 12 h (tacrolimus: 75.5 ± 5.3% vs. vehicle: 58.2 ± 4.4%). Taken together, these findings suggest that tacrolimus directly stimulates renal fibroblast differentiation into myofibroblasts with enhanced motility. Finally, ECM production was examined in these activated myofibroblasts. Heatmap analysis revealed increased Col IV protein expression in tacrolimus-treated cells compared with control (Fig. 5F). Consistently, Western blot quantification confirmed elevated Col IV expression (tacrolimus: 1.2 ± 0.02 arbitrary units vs. vehicle: 1.0 ± 0.00 arbitrary units; Fig. 5G). Collectively, these findings demonstrate tacrolimus as an inducer of FMT and ECM production in the kidney.
Figure 5.
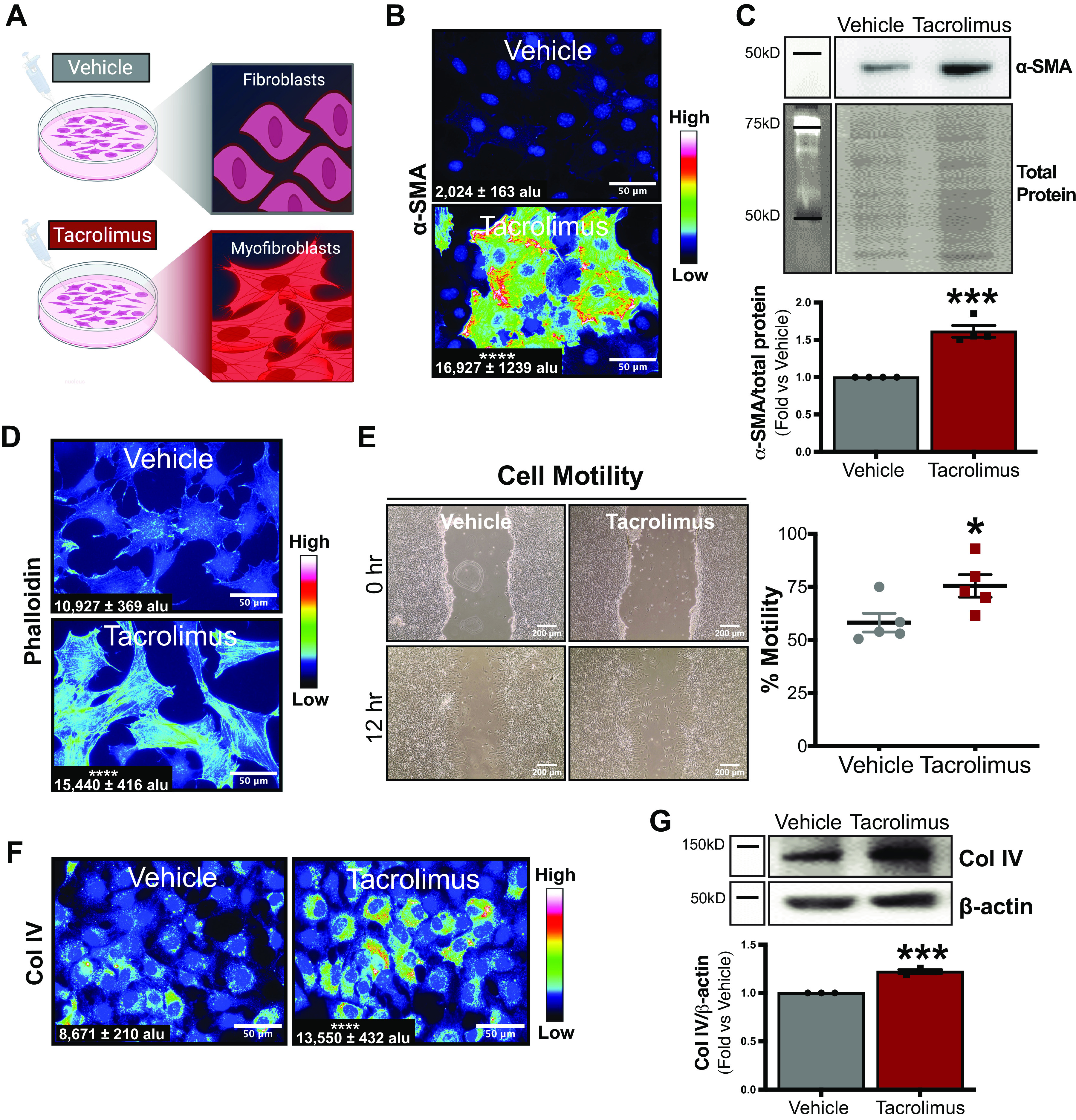
Tacrolimus promotes fibroblast-to-myofibroblast transition (FMT) and extracellular matrix production. To investigate fibroblasts as a source of tacrolimus-induced renal fibrosis, renal fibroblasts were treated with either tacrolimus (1 nM) or vehicle (0.01% ethanol) for 24 h (A). Myofibroblast marker α-smooth muscle actin (α-SMA) protein was visualized via immunofluorescence (B) and quantified with CellProfiler software [expressed as arbitrary light units (alu)]. Western blot (C) also quantified α-SMA expression. To further assess FMT, actin stress fiber content was visualized by immunofluorescent staining of phalloidin (D) and quantified with CellProfiler software (expressed as alu). To evaluate the effect of tacrolimus on fibroblast motility, scratch test assays were conducted and cell motility rates (in %) were calculated (E). Extracellular matrix production was assessed by both immunofluorescent detection and quantification of collagen type IV (Col IV) protein (with CellProfiler software, expressed as alu) (F). Western blot analysis further quantified Col IV expression (G). Representative images of at least 3 or 4 independent studies are shown. Values are means ± SE. Statistical tests were conducted using a t test to detect differences between experimental groups. *P < 0.05, ***P < 0.0005, and ****P < 0.00005 vs. vehicle.
The TGF-β Signaling Mediator Smad3 Drives Tacrolimus-Induced FMT and ECM Production in Renal Fibroblasts
Finally, we examined the role of TGFβRI signaling in tacrolimus-induced FMT (Fig. 6A). As assessed by ELISA (Fig. 6B), increased TGF-β1 ligand abundance was observed in media from tacrolimus-treated cells compared with control (tacrolimus: 421.1 ± 27.3 vs. vehicle: 107.9 ± 23.2). TGF-β receptor activation was then examined by visualizing phospho-TGFβRI expression via immunofluorescence (Fig. 6C). Compared with vehicle, tacrolimus-treated cells showed increased luminescent expression of phospho-TGFβRI, which was also confirmed by Western blot analysis (tacrolimus: 1.31 ± 0.11 arbitrary units vs. vehicle: 1.0 ± 0.00 arbitrary units; Fig. 6D). Western blot quantification also showed enhanced nuclear abundance of the TGF-β signaling mediator Smad2/3 in tacrolimus-treated cells compared with control (tacrolimus: 1.3 ± 0.10 arbitrary units vs. vehicle: 1.0 ± 0.00 arbitrary units; Fig. 6E). To assess the role of TGF-β receptor signaling in tacrolimus-induced FMT, cellular morphological changes were visualized in fibroblasts treated with either tacrolimus (1 nM) or vehicle (0.01% ethanol) in the presence or absence of the selective Smad3 inhibitor SIS3 (500 nM) for 24 h (Fig. 6F). Whereas fibroblasts maintained their normal spindlelike resting morphology with vehicle treatment (Fig. 6F, i and iii), tacrolimus treatment induced resting fibroblasts to adopt a more myofibroblast-like phenotype (Fig. 6Fii), which were greater in size and contained ruffled membranes and an enlarged endoplasmic reticulum. Interestingly, pretreatment of fibroblasts with SIS3 significantly attenuated the tacrolimus-induced myofibroblast phenotype (Fig. 6Fiv). Western blot quantification confirmed these morphological findings, as α-SMA (tacrolimus: 2.3 ± 0.13 arbitrary units vs. vehicle: 1.0 ± 0.00 arbitrary units) and Col IV (tacrolimus: 1.5 ± 0.11 arbitrary units vs. vehicle: 1.0 ± 0.00 arbitrary units) expression both increased with tacrolimus treatment (Fig. 6G). Notably, SIS3 cotreatment attenuated these tacrolimus-induced profibrotic effects (α-SMA, tacrolimus + SIS3: 1.4 ± 0.09 arbitrary units vs. tacrolimus: 2.3 ± 0.13 arbitrary units; Col IV, tacrolimus + SIS3: 1.1 ± 0.03 arbitrary units vs. tacrolimus: 1.5 ± 0.11 arbitrary units). Taken together, our results indicate that 1) fibroblasts are a source of tacrolimus-induced TGF-β secretion and 2) the TGFβRI signaling mediator Smad2/3 drives tacrolimus-induced FMT and ECM production.
Figure 6.
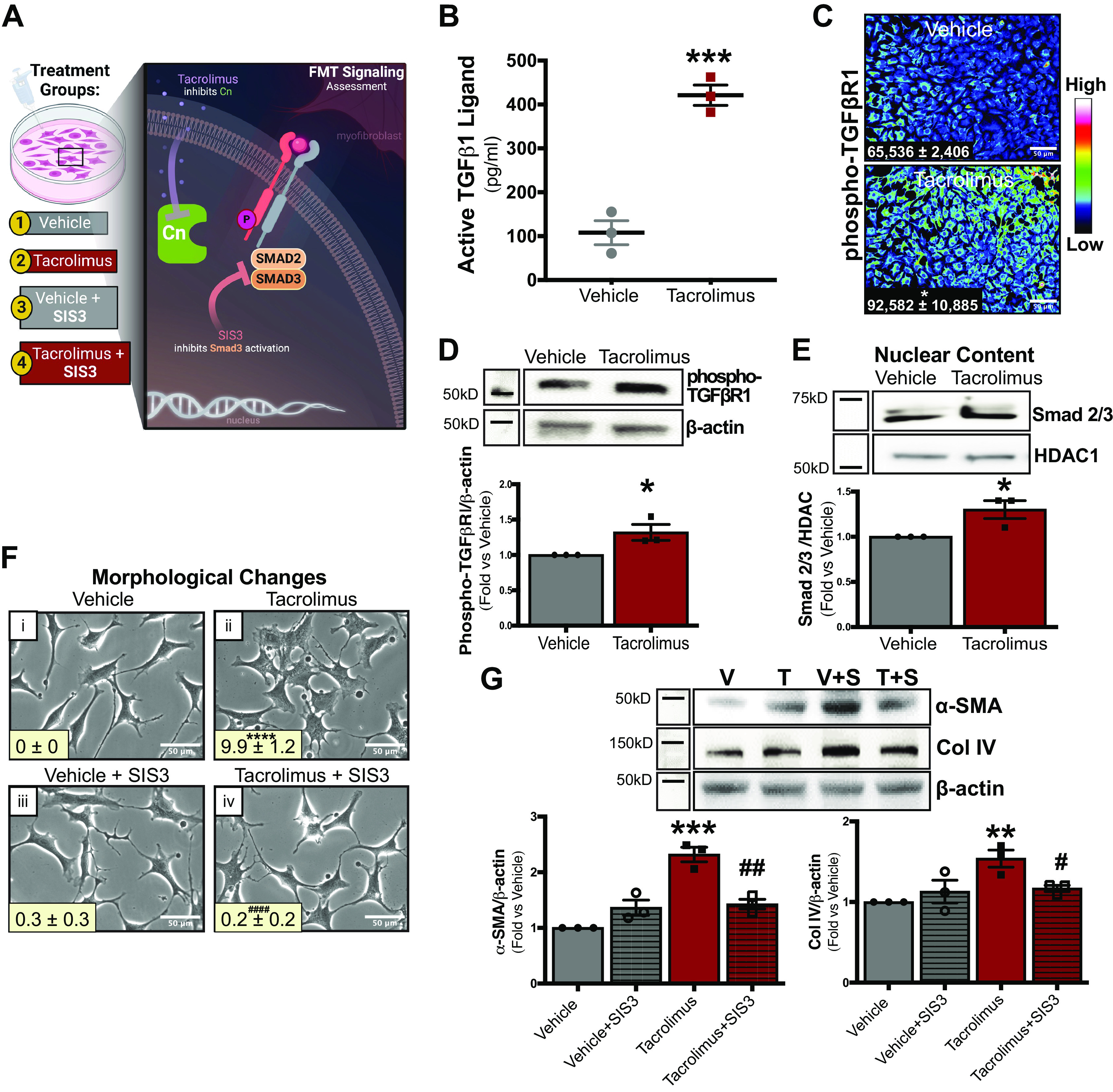
The transforming growth factor-β (TGF-β) signaling mediator Smad2/3 drives tacrolimus (T)-induced fibroblast-to-myofibroblast transition (FMT) and extracellular matrix production in renal fibroblasts. To examine the role of TGF-β receptor signaling in tacrolimus-induced FMT, renal fibroblasts were treated with either tacrolimus (1 nM) or vehicle (V; 0.01% ethanol) for 24 h (A). Type I TGF-β receptor subunit (TGFβRI) ligand secretion was quantified in media collected from both tacrolimus- and vehicle-treated cells by ELISA (B). TGFβRI activation was examined by visualizing phospho-TGFβRI expression via immunofluorescence (C) and quantified with CellProfiler software [expressed as arbitrary light units (alu)]. Western blot analysis (D) further quantified phospho-TGFβRI expression. Nuclear abundance of the TGF-β signaling mediator Smad2/3 was assessed by Western blot (E). To investigate the relevance of the TGF-β/Smad signaling pathway in tacrolimus-induced FMT and extracellular matrix accumulation, cellular morphological changes were visualized and quantified with ImageJ software in fibroblasts treated with either tacrolimus (1 nM) or vehicle (0.01% ethanol) in the presence or absence of the Smad3 inhibitor SIS3 (S; 500 nM; F). To confirm these morphological findings, α-smooth muscle actin (α-SMA) and collagen type IV (Col IV) abundance was assessed via Western blot (G). Representative images of at least 3 independent studies are shown. Values are means ± SE. Statistical tests were conducted using either two-way ANOVA or a t test to detect differences between the experimental groups. *P < 0.05, **P < 0.005, ***P < 0.0005, and ****P < 0.00005 vs. vehicle; #P < 0.05, ##P < 0.005, and ####P < 0.00005 vs. tacrolimus. Cn, calcineurin; HDAC, histone deacetylase 1.
DISCUSSION
It is well established that chronic CNI use invokes irreversible kidney damage in the form of renal fibrosis (10, 36, 48). CNIs are directly toxic to various renal cell types, including vascular (21), mesangial, glomerular, and tubular epithelial (22, 23) cells. However, given the diverse population of cells in the kidney (24), the additional targeted cell types and signaling mechanisms behind this progressive disease state continue to be defined. In this study, we identified renal fibroblasts as a contributor to CNI-induced fibrosis. The overall findings of our study revealed that the TGF-β/Smad pathway is a pivotal signaling event underlying FMT and subsequent CNI-induced renal fibrosis. Specifically, using both in vivo and in vitro models of CNI-induced nephropathy, we demonstrated that 1) tacrolimus inhibits the calcineurin/NFAT axis while inducing TGF-β1 ligand secretion and receptor activation; 2) aberrant TGF-β receptor activation stimulates Smad-mediated production of myofibroblast markers, notable features of FMT; and 3) FMT contributes to ECM expansion in tacrolimus-induced renal fibrosis. Taken together, these results not only incorporate renal fibroblasts into the growing list of cell types impacted by CNIs but also identify renal FMT as a process mediated via a TGF-β-dependent mechanism (Fig. 7).
Figure 7.

Proposed schema. Tacrolimus induced fibroblast-to-myofibroblast transition (FMT) via a transforming growth factor-β (TGF-β)-dependent mechanism to contribute to renal fibrosis: 1) tacrolimus inhibits the calcineurin (Cn)/nuclear factor of activated T cells (NFAT) axis while inducing TGF-β1 ligand secretion and receptor activation in renal fibroblasts; 2) aberrant TGF-β receptor activation stimulates Smad-mediated production of myofibroblast markers, notable features of FMT; and 3) FMT contributes to extracellular matrix (ECM) expansion in tacrolimus-induced renal fibrosis. Col IV, collagen type IV; α-SMA, α-smooth muscle actin.
Previous experimental studies have characterized features of CNI-induced renal damage using animal models susceptible to kidney damage (including CD-1, BALB/c, etc.) (22, 49). However, in the present study we provide additional evidence of CNI nephropathy in C57BL/6 mice, one of the most commonly used mouse strains in kidney injury research (50, 51). Given that C57BL/6 mice are resistant to chronic kidney disease-associated pathologies (50, 51), we demonstrated the profuse capacity of tacrolimus to induce histological signs of chronic renal damage, particularly interstitial fibrosis. Fibrosis was characterized by increased collagen deposition, accompanied by the appearance of α-SMA-positive myofibroblasts in damaged areas (Fig. 2A). Given our findings, the next step became the identification of the cellular source of tacrolimus-induced renal damage in addition to the signaling mechanism that drives this chronic (and irreversible) process.
Tacrolimus Inhibits the Calcineurin/NFAT Axis While Inducing TGF-β1 Ligand Secretion and Receptor Activation
In mesangial cells, TGF-β (partially via the calcineurin/NFAT axis) upregulates ECM production (52). To determine whether renal fibroblasts are also a cellular source of tacrolimus-induced renal fibrosis, we initially assessed whether calcineurin/NFAT activation was inhibited in tacrolimus-treated fibroblasts. Our findings demonstrated that tacrolimus inhibited the calcineurin/NFAT axis in renal fibroblasts (Fig. 3). This study, along with previous reports (52, 53), suggests that either activation or suppression of NFAT could mediate CNI-induced nephropathy. Given the capacity of TGF-β to induce ECM production in both mesangial cells (52) and tubular epithelia (12), this study aimed to investigate TGF-β signaling as a driver of tacrolimus-induced FMT. TGF-β1 has been reported as a lead signaling mediator of CsA-induced nephrotoxicity (36, 45, 54). We found that tacrolimus treatment also resulted in elevated renal fibroblast secretion of TGF-β1 ligand (Fig. 6B), which becomes available to bind receptors in both a paracrine and an autocrine fashion (55). Previous reports were consistent with our findings, as increased TGF-β1 expression was observed in renal biopsies obtained from transplant recipients with CNI nephropathy (32, 56) and in kidneys of tacrolimus-treated Sprague-Dawley rats (57).
In the presence of TGF-β1 ligand binding, membrane-bound type II TGF-β receptor subunits phosphorylate and thereby activate TGFβRI to initiate downstream TGF-β signaling (58). In addition to promoting extracellular TGF-β1 ligand release, we showed both in vivo and in vitro that TGFβRI activation increased with tacrolimus treatment (Fig. 4, B and C, and Fig. 6C). Our findings illustrate that tacrolimus reduced total TGFβRI protein expression in vivo (Fig. 4A); however, this observation could be attributed to a compensatory downregulation of the total TGF-β receptor pool, to prevent further activation of the receptor. It is worth noting that tacrolimus can potentially induce TGFβRI activation through association with its cytosolic immunophilin, FKBP12. TGFβRI activation is usually inhibited by FKBP12 binding at the cytoplasmic tail of the receptor, blocking access to activation signals (57). However, in the presence of tacrolimus (FK506), FKBP12 binds tacrolimus, prompting release from the receptor to cause downstream kinase activation and subsequent TGF-β receptor signaling (21).
Aberrant TGF-β Receptor Activation Stimulates Smad-Mediated Production of Myofibroblast Markers, Notable Features of FMT
TGF-β receptor activity prompts downstream recruitment and activation of the intracellular mediator Smad2/3, which translocate to the nucleus to regulate gene transcription (24). A previous in vitro study has reported that tacrolimus upregulated Smad2 expression in mesangial cells (38). However, we expanded these findings by demonstrating that tacrolimus treatment increased Smad2/3 activation in both kidneys and renal fibroblasts (Fig. 4D and Fig. 6E). We further demonstrated that Smad signaling drives tacrolimus-induced FMT (Fig. 6, F and G). Assessing morphological features of FMT, tacrolimus treatment induced phenotypic changes indicative of a transition into contractile myofibroblasts (Figs. 5 and 6, F and G) (27). Morphological changes were accompanied by upregulation of established myofibroblast markers (Fig. 6, F and G). In contrast, pharmacological inhibition of Smad3 activation attenuated these tacrolimus-induced phenotypic changes (Fig. 6, F and G). Our findings reveal that the TGF-β/Smad signaling axis mediates tacrolimus-induced FMT in kidneys, which is consistent with both observations from patient renal biopsies (56) and a study conducted with human kidney fibroblasts (44).
FMT Contributes to ECM Expansion in Tacrolimus-Induced Renal Fibrosis
The progression of kidney disease toward end-stage renal failure is facilitated by pathological changes such as ECM accumulation (47). In chronic kidney disease, the primary cell source responsible for ECM accumulation is the myofibroblast, a mesenchymal cell type possessing the phenotypic characteristics of both fibroblasts and smooth muscle cells (27). Myofibroblasts arise from circulating progenitors, tubular epithelial cells, and resident fibroblasts (27). Although the role of myofibroblasts in tacrolimus-induced renal damage is widely accepted, their origin is still a matter of debate (28). Human renal proximal tubular epithelial cells have been extensively used in the investigation of both CsA and tacrolimus nephropathy (32, 45, 54, 59). However, given their role as the primary matrix-producing cells in the kidney, we selected mouse renal fibroblasts to further investigate mechanisms underlying renal fibrosis with CNI use. Resting renal fibroblasts exposed to tacrolimus for 24 h exhibited profound morphological alterations consistent with FMT (27), which was confirmed by significant increases in de novo α-SMA and Col IV protein expression (Fig. 5, B, C, F, and G). The myofibroblast phenotype was further validated by detection of abundant stress fibers in tacrolimus-exposed cells, enabling the acquisition of enhanced cell motility and contractibility (Fig. 5, D and E). In line with our in vivo findings, these data indicate that tacrolimus treatment is sufficient to induce FMT, a process capable of causing sinister consequences in the kidney.
It is worth noting that epithelial-to-mesenchymal transition (EMT) is recognized as a contributor to the myofibroblast population in renal interstitial fibrosis and is well known to be present in CNI-treated kidneys (12, 45, 54). EMT is the phenomenon characterized by disruption of polarized tubular epithelial cell morphology into characteristic features of contractile fibroblast cells (27, 60). However, given its capacity to release the profibrotic factor TGF-β into the extracellular environment, our data suggest that activated fibroblasts may be initiators of tacrolimus-induced EMT (12, 44). This conclusion is based on our 1) in vivo data showing a lack of α-SMA-positive tubular epithelial cells and 2) in vitro data demonstrating that tacrolimus significantly increased α-SMA protein levels after only 24 h, whereas a previously published study showed tacrolimus-induced expression of α-SMA mRNA in tubular epithelial cells after 48 h (12). Moreover, because our in vivo findings also showed increased TGFβRI activation in select nephron segments, we suspect that this occurrence is due to fibroblast secretion of TGF-β1 ligand, binding and stimulating nearby epithelial cell receptors. Fibroblast activation preceding EMT is quite possible, as this phenomenon has been noted in the pathogenesis of tumor invasion (61), acute renal injury (62), and pulmonary fibrosis (63, 64). Fibroblast activation is associated with chronic renal disease, consisting of progressive interstitial scarring and tubular cell atrophy. Our findings indicate that tacrolimus triggers TGF-β1 signaling to induce FMT, initiating a cascade of downstream profibrotic events that cause further damage to the renal parenchyma (Fig. 7).
Perspectives and Significance
This study expands understanding of the many cell types and central signaling mediators driving CNI-induced renal fibrosis. Not only does our work incorporate renal fibroblasts into the growing list of cell types negatively impacted by CNIs, but it also identifies renal FMT as a process mediated via a TGF-β-dependent mechanism. Ultimately, our findings provide a more comprehensive picture of how calcineurin inhibition and profibrotic pathways intersect in the kidneys. Considering the impact of aberrant TGF-β signaling on multiple cell types in the kidney, it is now necessary to demonstrate the feasibility of inhibiting TGF-β signaling as a means of preventing renal fibrosis associated with chronic CNI use. Understanding the cellular and molecular mechanisms of tacrolimus-induced renal damage and their role in the initiation of fibrosis will inform the development of novel therapeutic strategies to ameliorate the nephropathy observed in patients requiring long-term CNI therapy.
DATA AVAILABILITY
Data will be made available upon reasonable request.
SUPPLEMENTAL DATA
Supplemental Table S1: https://doi.org/10.6084/m9.figshare.21770534.
GRANTS
This work was supported by the following National Institutes of Health Grants: F30DK130531 (to A.C.U.), R21DK119879 (to C.R.W.), R21DK119879-S (to A.C.U.), R01DK133698 (to C.R.W.), 5R01AR077574 (to H.R.), and F30NS124237 (to J.N.S.). This work was also supported by the William Townsend Porter Predoctoral Fellowship from the American Physiological Society (to A.C.U.), by a Cornerstone Grant from The Histochemical Society (to A.C.U.), by a Medical Student Research Grant from Wright State University Boonshoft School of Medicine (to A.C.U.), by American Heart Association Grant 16SDG27080009 (to C.R.W.), by Department of Defense Development Award W81XWH2110679 (to H.R.), and by an American Society of Nephrology KidneyCure Transition to Independence Grant (to C.R.W.).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
A.C.U., T.Y.W., and C.R.W. conceived and designed research; A.C.U. and T.Y.W. performed experiments; A.C.U., T.Y.W., C.P.O., J.N.S., and C.R.W. analyzed data; A.C.U., T.Y.W., D.N.A., J.N.S., and C.R.W. interpreted results of experiments; A.C.U., T.Y.W., C.P.O., J.N.S., A.M.W., and J.K. prepared figures; A.C.U. drafted manuscript; A.C.U., T.Y.W., J.N.S., C.P.O., D.N.A., K.S., E.S.B., H.R., and C.R.W. edited and revised manuscript; C.R.W. approved final version of manuscript.
REFERENCES
- 1. Starzl TE, Fung J, Jordan M, Shapiro R, Tzakis A, McCauley J, Johnston J, Iwaki Y, Jain A, Alessiani M. Kidney transplantation under FK 506. JAMA 264: 63–67, 1990. doi: 10.1001/jama.1990.03450010067032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Kahan BD. Cyclosporine: a powerful addition to the immunosuppressive armamentarium. Am J Kidney Dis 3: 444–455, 1984. doi: 10.1016/S0272-6386(84)80009-8. [DOI] [PubMed] [Google Scholar]
- 3. Kahan BD. Cyclosporine. N Engl J Med 321: 1725–1738, 1989. doi: 10.1056/NEJM198912213212507. [DOI] [PubMed] [Google Scholar]
- 4. Legendre C, Thervet E, Skhiri H, Mamzer-Bruneel MF, Cantarovich F, Noël LH, Kreis H. Histologic features of chronic allograft nephropathy revealed by protocol biopsies in renal transplant recipients. Transplantation 65: 1506–1509, 1998. doi: 10.1097/00007890-199806150-00020. [DOI] [PubMed] [Google Scholar]
- 5. de Mattos AM, Olyaei AJ, Bennett WM. Nephrotoxicity of immunosuppressive drugs: long-term consequences and challenges for the future. Am J Kidney Dis 35: 333–346, 2000. doi: 10.1016/S0272-6386(00)70348-9. [DOI] [PubMed] [Google Scholar]
- 6. Naesens M, Kuypers DR, Sarwal M. In-depth review calcineurin inhibitor nephrotoxicity. Clin J Am Soc Nephrol 4: 481–508, 2009. doi: 10.2215/CJN.04800908. [DOI] [PubMed] [Google Scholar]
- 7. Martins L, Ventura A, Branco A, Carvalho MJ, Henriques AC, Dias L, Sarmento AM, Amil M. Cyclosporine versus tacrolimus in kidney transplantation: are there differences in nephrotoxicity? Transplant Proc 36: 877–879, 2004. doi: 10.1016/j.transproceed.2004.03.083. [DOI] [PubMed] [Google Scholar]
- 8. Randhawa PS, Shapiro R, Jordan ML, Starzl TE, Demetris AJ. The histopathological changes associated with allograft rejection and drug toxicity in renal transplant recipients maintained on FK506. Clinical significance and comparison with cyclosporine. Am J Surg Pathol 17: 60–68, 1993. doi: 10.1097/00000478-199301000-00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ojo AO, Held PJ, Port FK, Wolfe RA, Leichtman AB, Young EW, Arndorfer J, Christensen L, Merion RM. Chronic renal failure after transplantation of a nonrenal organ. N Engl J Med 349: 931–940, 2003. doi: 10.1056/NEJMoa021744. [DOI] [PubMed] [Google Scholar]
- 10. Ume AC, Wenegieme TY, Williams CR. Calcineurin inhibitors: a double-edged sword. Am J Physiol Renal Physiol 320: F336–F341, 2021. doi: 10.1152/ajprenal.00262.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Rusnak F, Mertz P. Calcineurin: form and function. Physiol Rev 80: 1483–1521, 2000. doi: 10.1152/physrev.2000.80.4.1483. [DOI] [PubMed] [Google Scholar]
- 12. Bennett J, Cassidy H, Slattery C, Ryan MP, McMorrow T. Tacrolimus modulates TGF-β signaling to induce epithelial-mesenchymal transition in human renal proximal tubule epithelial cells. J Clin Med 5: 50, 2016. doi: 10.3390/jcm5050050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Klee CB, Draetta GF, Hubbard MJ. Calcineurin. Adv Enzymol Relat Areas Mol Biol 61: 149–200. 1988. doi: 10.1002/9780470123072.ch4. [DOI] [PubMed] [Google Scholar]
- 14. Sieber M, Baumgrass R. Novel inhibitors of the calcineurin/NFATc hub—alternatives to CsA and FK506? Cell Commun Signal 7: 25, 2009. doi: 10.1186/1478-811X-7-25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. O’Keefe SJ, Tamura J, Kincaid RL, Tocci MJ, O’Neill EA. FK-506- and CsA-sensitive activation of the interleukin-2 promoter by calcineurin. Nature 357: 692–694, 1992. doi: 10.1038/357692a0. [DOI] [PubMed] [Google Scholar]
- 16. Perrino BA, Wilson AJ, Ellison P, Clapp LH. Substrate selectivity and sensitivity to inhibition by FK506 and cyclosporin A of calcineurin heterodimers composed of the α or β catalytic subunit. Eur J Biochem 269: 3540–3548, 2002. doi: 10.1046/j.1432-1033.2002.03040.x. [DOI] [PubMed] [Google Scholar]
- 17. Fruman DA, Klee CB, Bierer BE, Burakoff SJ. Calcineurin phosphatase activity in T lymphocytes is inhibited by FK 506 and cyclosporin A. Proc Natl Acad Sci USA 89: 3686–3690, 1992. doi: 10.1073/pnas.89.9.3686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kung L, Batiuk TD, Palomo-Pinon S, Noujaim J, Helms LM, Halloran PF. Tissue distribution of calcineurin and its sensitivity to inhibition by cyclosporine. Am J Transplant 1: 325–333, 2001. doi: 10.1034/j.1600-6143.2001.10407.x. [DOI] [PubMed] [Google Scholar]
- 19. Jiang H, Xiong F, Kong S, Ogawa T, Kobayashi M, Liu JO. Distinct tissue and cellular distribution of two major isoforms of calcineurin. Mol Immunol 34: 663–669, 1997. doi: 10.1016/S0161-5890(97)00054-0. [DOI] [PubMed] [Google Scholar]
- 20. Cheriyan AM, Ume AC, Francis CE, King KN, Linck VA, Bai Y, Cai H, Hoover RS, Ma HP, Gooch JL, Williams CR. Calcineurin A-α suppression drives nuclear factor-κB-mediated NADPH oxidase-2 upregulation. Am J Physiol Renal Physiol 320: F789–F798, 2021. doi: 10.1152/ajprenal.00254.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Giordano A, Romano S, Mallardo M, D’Angelillo A, Calì G, Corcione N, Ferraro P, Romano MF. FK506 can activate transforming growth factor-β signalling in vascular smooth muscle cells and promote proliferation. Cardiovasc Res 79: 519–526, 2008. doi: 10.1093/cvr/cvn079. [DOI] [PubMed] [Google Scholar]
- 22. Vandenbussche C, Van der Hauwaert C, Dewaeles E, Franczak J, Hennino MF, Gnemmi V, Savary G, Tavernier Q, Nottet N, Paquet A, Perrais M, Blum D, Mari B, Pottier N, Glowacki F, Cauffiez C. Tacrolimus-induced nephrotoxicity in mice is associated with microRNA deregulation. Arch Toxicol 92: 1539–1550, 2018. doi: 10.1007/s00204-018-2158-3. [DOI] [PubMed] [Google Scholar]
- 23. Lusco MA, Fogo AB, Najafian B, Alpers CE. AJKD Atlas of Renal Pathology: calcineurin inhibitor nephrotoxicity. Am J Kidney Dis 69: e21–e22, 2017. doi: 10.1053/j.ajkd.2017.02.003. [DOI] [PubMed] [Google Scholar]
- 24. Fuchs MA, Broeker KA, Schrankl J, Burzlaff N, Willam C, Wagner C, Kurtz A. Inhibition of transforming growth factor β1 signaling in resident interstitial cells attenuates profibrotic gene expression and preserves erythropoietin production during experimental kidney fibrosis in mice. Kidney Int 100: 122–137, 2021. doi: 10.1016/j.kint.2021.02.035. [DOI] [PubMed] [Google Scholar]
- 25. Grande MT, López-Novoa JM. Fibroblast activation and myofibroblast generation in obstructive nephropathy. Nat Rev Nephrol 5: 319–328, 2009. doi: 10.1038/nrneph.2009.74. [DOI] [PubMed] [Google Scholar]
- 26. Meran S, Steadman R. Fibroblasts and myofibroblasts in renal fibrosis. Int J Exp Pathol 92: 158–167, 2011. doi: 10.1111/j.1365-2613.2011.00764.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Strutz F, Zeisberg M. Renal fibroblasts and myofibroblasts in chronic kidney disease. J Am Soc Nephrol 17: 2992–2998, 2006. doi: 10.1681/ASN.2006050420. [DOI] [PubMed] [Google Scholar]
- 28. Forino M, Torregrossa R, Ceol M, Murer L, Della Vella M, Del Prete D, D’Angelo A, Anglani F. TGFβ1 induces epithelial-mesenchymal transition, but not myofibroblast transdifferentiation of human kidney tubular epithelial cells in primary culture. Int J Exp Pathol 87: 197–208, 2006. doi: 10.1111/j.1365-2613.2006.00479.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Xia T, Zhu S, Wen Y, Gao S, Li M, Tao X, Zhang F, Chen W. Risk factors for calcineurin inhibitor nephrotoxicity after renal transplantation: a systematic review and meta-analysis. Drug Des Devel Ther 12: 417–428, 2018. doi: 10.2147/DDDT.S149340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. El-Bassossy HM, Eid BG. Cyclosporine A exhibits gender-specific nephrotoxicity in rats: Effect on renal tissue inflammation. Biochem Biophys Res Commun 495: 468–472, 2018. doi: 10.1016/j.bbrc.2017.11.042. [DOI] [PubMed] [Google Scholar]
- 31. Nair AB, Jacob S. A simple practice guide for dose conversion between animals and human. J Basic Clin Pharm 7: 27–31, 2016. doi: 10.4103/0976-0105.177703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Khanna A, Plummer M, Bromberek C, Bresnahan B, Hariharan S. Expression of TGF-β and fibrogenic genes in transplant recipients with tacrolimus and cyclosporine nephrotoxicity. Kidney Int 62: 2257–2263, 2002. doi: 10.1046/j.1523-1755.2002.00668.x. [DOI] [PubMed] [Google Scholar]
- 33. Nishiyama M, Izumi S, Okuhara M. Discovery and development of FK506 (tacrolimus), a potent immunosuppressant of microbial origin. In: The Search for Anti-Inflammatory Drugs, edited by Merluzzi VJ, Adams J.. Boston, MA: Birkhäuser, 1995, p. 65–104. [Google Scholar]
- 34. Staatz CE, Tett SE. Clinical pharmacokinetics and pharmacodynamics of tacrolimus in solid organ transplantation. Clin Pharmacokinet 43: 623–653, 2004. doi: 10.2165/00003088-200443100-00001. [DOI] [PubMed] [Google Scholar]
- 35. Lim SW, Jin L, Luo K, Jin J, Shin YJ, Hong SY, Yang CW. Klotho enhances FoxO3-mediated manganese superoxide dismutase expression by negatively regulating PI3K/AKT pathway during tacrolimus-induced oxidative stress. Cell Death Dis 8: e2972, 2017. doi: 10.1038/cddis.2017.365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Gooch JL, Roberts BR, Cobbs SL, Tumlin JA. Loss of the α-isoform of calcineurin is sufficient to induce nephrotoxicity and altered expression of transforming growth factor-β. Transplantation 83: 439–447, 2007. doi: 10.1097/01.tp.0000251423.78124.51. [DOI] [PubMed] [Google Scholar]
- 37. Esposito C, Foschi A, Parrilla B, Cornacchia F, Fasoli G, Plati AR, De Mauri A, Mazzullo T, Scudellaro R, Dal Canton A. Effect of calcineurin inhibitors on extracellular matrix turnover in isolated human glomeruli. Transplant Proc 36: 695–697, 2004. doi: 10.1016/j.transproceed.2004.03.013. [DOI] [PubMed] [Google Scholar]
- 38. Qi R, Li W, Yu S. FK506 inhibits the mice glomerular mesangial cells proliferation by affecting the transforming growth factor-β and Smads signal pathways. Ren Fail 36: 589–592, 2014. doi: 10.3109/0886022X.2014.882713. [DOI] [PubMed] [Google Scholar]
- 39. Khanna AK. The immunosuppressive agent tacrolimus induces p21WAF/CIP1WAF1/CIP1 via TGF-β secretion. Biochem Biophys Res Commun 303: 266–272, 2003. doi: 10.1016/S0006-291X(03)00340-1. [DOI] [PubMed] [Google Scholar]
- 40. Gooch JL, Gorin Y, Zhang BX, Abboud HE. Involvement of calcineurin in transforming growth factor-beta-mediated regulation of extracellular matrix accumulation. J Biol Chem 279: 15561–15570, 2004. doi: 10.1074/jbc.M308759200. [DOI] [PubMed] [Google Scholar]
- 41. Kavachale A, Shrivastava T, Balekar N. Wound healing potential of grandiflorenic acid isolated from Wedelia trilobata Linn. J Drug Deliv Therapeut 7: 232–234, 2017. doi: 10.22270/jddt.v7i7.1648. [DOI] [Google Scholar]
- 42. Strutz F, Zeisberg M, Hemmerlein B, Sattler B, Hummel K, Becker V, Müller GA. Basic fibroblast growth factor expression is increased in human renal fibrogenesis and may mediate autocrine fibroblast proliferation. Kidney Int 57: 1521–1538, 2000. doi: 10.1046/j.1523-1755.2000.00997.x. [DOI] [PubMed] [Google Scholar]
- 43. Liptak P, Ivanyi B. Primer: histopathology of calcineurin-inhibitor toxicity in renal allografts. Nat Clin Pract Nephrol 2: 398–404, 2006. doi: 10.1038/ncpneph0225. [DOI] [PubMed] [Google Scholar]
- 44. Kern G, Mair SM, Noppert SJ, Jennings P, Schramek H, Rudnicki M, Mueller GA, Mayer G, Koppelstaetter C. Tacrolimus increases Nox4 expression in human renal fibroblasts and induces fibrosis-related genes by aberrant TGF-beta receptor signalling. PloS One 9: e96377, 2014. doi: 10.1371/journal.pone.0096377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. McMorrow T, Gaffney MM, Slattery C, Campbell E, Ryan MP. Cyclosporine A induced epithelial-mesenchymal transition in human renal proximal tubular epithelial cells. Nephrol Dial Transplant 20: 2215–2225, 2005. doi: 10.1093/ndt/gfh967. [DOI] [PubMed] [Google Scholar]
- 46. Wolf G, Killen PD, Neilson EG. Cyclosporin A stimulates transcription and procollagen secretion in tubulointerstitial fibroblasts and proximal tubular cells. J Am Soc Nephrol 1: 918–922, 1990. doi: 10.1681/ASN.V16918. [DOI] [PubMed] [Google Scholar]
- 47. Mackensen-Haen S, Bader R, Grund KE, Bohle A. Correlations between renal cortical interstitial fibrosis, atrophy of the proximal tubules and impairment of the glomerular filtration rate. Clin Nephrol 15: 167–171, 1981. [PubMed] [Google Scholar]
- 48. Wang D, Chen X, Fu M, Xu H, Li Z. Tacrolimus increases the expression level of the chemokine receptor CXCR2 to promote renal fibrosis progression. Int J Mol Med 44: 2181–2188, 2019. doi: 10.3892/ijmm.2019.4368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Lopes CT, Gallo AP, Palma PV, Cury PM, Bueno V. Skin allograft survival and analysis of renal parameters after FTY720 + tacrolimus treatment in mice. Transplant Proc 40: 856–860, 2008. doi: 10.1016/j.transproceed.2008.02.051. [DOI] [PubMed] [Google Scholar]
- 50. Sears SM, Sharp CN, Krueger A, Oropilla GB, Saforo D, Doll MA, Megyesi J, Beverly LJ, Siskind LJ. C57BL/6 mice require a higher dose of cisplatin to induce renal fibrosis and CCL2 correlates with cisplatin-induced kidney injury. Am J Physiol Renal Physiol 319: F674–F685, 2020. doi: 10.1152/ajprenal.00196.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Rabe M, Schaefer F. Non-transgenic mouse models of kidney disease. Nephron 133: 53–61, 2016. doi: 10.1159/000445171. [DOI] [PubMed] [Google Scholar]
- 52. Cobbs SL, Gooch JL. NFATc is required for TGFβ-mediated transcriptional regulation of fibronectin. Biochem Biophys Res Commun 362: 288–294, 2007. doi: 10.1016/j.bbrc.2007.07.186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Beljaars L, Daliri S, Dijkhuizen C, Poelstra K, Gosens R. WNT-5A regulates TGF-related activities in liver fibrosis. Am J Physiol Gastrointest Liver Physiol 312: G219–G227, 2017. doi: 10.1152/ajpgi.00160.2016. [DOI] [PubMed] [Google Scholar]
- 54. Slattery C, Campbell E, McMorrow T, Ryan MP. Cyclosporine A-induced renal fibrosis: a role for epithelial-mesenchymal transition. Am J Pathol 167: 395–407, 2005. doi: 10.1016/S0002-9440(10)62984-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Massagué J. TGF-β signal transduction. Annu Rev Biochem 67: 753–791, 1998. doi: 10.1146/annurev.biochem.67.1.753. [DOI] [PubMed] [Google Scholar]
- 56. Rivelli RF, Gonçalves RT, Leite M, Santos MA, Delgado AG, Cardoso LR, Takiya CM. Early withdrawal of calcineurin inhibitor from a sirolimus-based immunosuppression stabilizes fibrosis and the transforming growth factor-β signalling pathway in kidney transplant. Nephrology (Carlton) 20: 168–176, 2015. doi: 10.1111/nep.12368. [DOI] [PubMed] [Google Scholar]
- 57. Huse M, Chen YG, Massagué J, Kuriyan J. Crystal structure of the cytoplasmic domain of the type I TGF beta receptor in complex with FKBP12. Cell 96: 425–436, 1999. doi: 10.1016/S0092-8674(00)80555-3. [DOI] [PubMed] [Google Scholar]
- 58. Massagué J. TGFβ signalling in context. Nat Rev Mol Cell Biol 13: 616–630, 2012. doi: 10.1038/nrm3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Jiang H, Yamamoto S, Nishikawa K, Kato R. Anti-tumor-promoting action of FK506, a potent immunosuppressive agent. Carcinogenesis 14: 67–71, 1993. doi: 10.1093/carcin/14.1.67. [DOI] [PubMed] [Google Scholar]
- 60. Sato Y, Yanagita M. Resident fibroblasts in the kidney: a major driver of fibrosis and inflammation. Inflamm Regen 37: 17, 2017. doi: 10.1186/s41232-017-0048-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Ding W, You H, Dang H, LeBlanc F, Galicia V, Lu SC, Stiles B, Rountree CB. Epithelial-to-mesenchymal transition of murine liver tumor cells promotes invasion. Hepatology 52: 945–953, 2010. doi: 10.1002/hep.23748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Masola V, Zaza G, Gambaro G, Onisto M, Bellin G, Vischini G, Khamaysi I, Hassan A, Hamoud S, Nativ O, Hayman SN, Lupo A. Heparanase: a potential new factor involved in the renal epithelial mesenchymal transition (EMT) induced by ischemia/reperfusion (I/R) injury. PLoS One 11: e0160074, 2016. doi: 10.1371/journal.pone.0160074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Zhu L, Fu X, Chen X, Han X, Dong P. M2 macrophages induce EMT through the TGF-β/Smad2 signaling pathway. Cell Biol Int 41: 960–968, 2017. doi: 10.1002/cbin.10788. [DOI] [PubMed] [Google Scholar]
- 64. Chen T, Nie H, Gao X, Yang J, Pu J, Chen Z, Cui X, Wang Y, Wang H, Jia G. Epithelial–mesenchymal transition involved in pulmonary fibrosis induced by multi-walled carbon nanotubes via TGF-beta/Smad signaling pathway. Toxicol Lett 226: 150–162, 2014. doi: 10.1016/j.toxlet.2014.02.004. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table S1: https://doi.org/10.6084/m9.figshare.21770534.
Data Availability Statement
Data will be made available upon reasonable request.