


Abstract
Prolonged pausing of the transcription machinery may lead to the formation of three-stranded nucleic acid structures, called R-loops, typically resulting from the annealing of the nascent RNA with the template DNA. Unscheduled persistence of R-loops and RNA polymerases may interfere with transcription itself and other essential processes such as DNA replication and repair. Senataxin (SETX) is a putative helicase, mutated in two neurodegenerative disorders, which has been implicated in the control of R-loop accumulation and in transcription termination. However, understanding the precise role of SETX in these processes has been precluded by the absence of a direct characterisation of SETX biochemical activities. Here, we purify and characterise the helicase domain of SETX in parallel with its yeast orthologue, Sen1. Importantly, we show that SETX is a bona fide helicase with the ability to resolve R-loops. Furthermore, SETX has retained the transcription termination activity of Sen1 but functions in a species-specific manner. Finally, subsequent characterisation of two SETX variants harbouring disease-associated mutations shed light into the effect of such mutations on SETX folding and biochemical properties. Altogether, these results broaden our understanding of SETX function in gene expression and the maintenance of genome integrity and provide clues to elucidate the molecular basis of SETX-associated neurodegenerative diseases.
Graphical Abstract
Graphical Abstract.
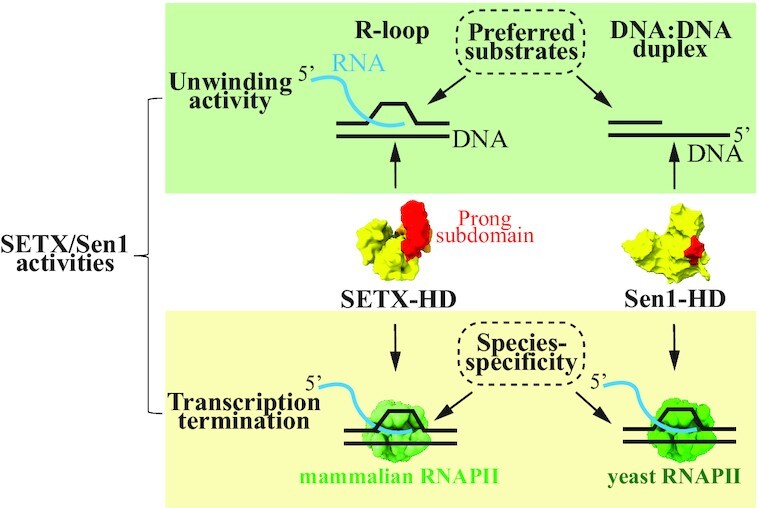
Comparison of the enzymatic activities of senataxin and Sen1 with respect to DNA unwinding and transcription termination.
INTRODUCTION
Essential cellular processes such as replication, recombination, and DNA repair occur alongside transcription on DNA. During transcription, RNA polymerase II (RNAPII) pauses at discrete sites, which in some instances is required for proper regulation of gene expression (1,2). Prolonged pausing and/or stalling of the transcribing RNAPII occurring either for regulatory purposes or under pathological conditions may impinge upon genomic functions by creating an obstacle for DNA-associated machineries (1,3). RNAPII pausing favours the appearance of R-loops, which are nucleic-acid structures formed when the nascent RNA anneals back to the complementary template DNA strand, thereby leaving the antiparallel non-template DNA strand unpaired. If they remain unprocessed, R-loops may compromise genome stability, either directly or by promoting particular histone modifications (4). On one side, the exposed single-strand DNA (ssDNA) is vulnerable to cleavage by endonucleases and oxidative damage (5,6). On the other side, R-loops may represent a block for DNA replication and repair (7–12). R-loops are indeed important contributors of transcription-replication conflicts (TRCs), which occur when the replication fork encounters a transcriptional block. Cells possess multiple factors at the interface of the replication and transcription machineries, to overcome TRCs, some of which are involved in the direct removal of R-loops and/or dislodging of RNAPIIs to enable replication fork restart (1,13–24).
Not only R-loops, but also stalled RNAPIIs themselves may act as a barrier for transcription, DNA replication, and DNA repair (1,25). Timely termination of transcription and removal of RNAPIIs and R-loops is, therefore, critical to maintain genome stability, successfully transmit genetic information across generations, and to timely and accurately execute the transcriptional programme.
Aside from their potentially harmful nature, R-loops also play a role in various important physiological processes (22,26) such as the regulation of gene expression (27–29), the regulation of homologous recombination during DNA repair and immunoglobulin class-switch, and the maintenance of telomeric and centromeric regions (8–10,12,30–34). Therefore, R-loop levels need to be tightly regulated to ensure a proper balance between its physiological and pathological roles.
Accumulation of detrimental R-loops may be prevented by mechanisms that avoid the formation of R-loops, for instance, by proper processing of pre-mRNAs, handling of topological changes, and chromatin remodelling during transcription (6,35–39). Efficient transcription termination, which leads to the release of RNAPII and the RNA transcript from DNA, may also decrease the likelihood of formation of R-loops by reducing the residency time of the RNA in the proximity to the template DNA (40,41). Once R-loops are formed, additional factors, including nucleases such as RNase H and RNA:DNA helicases, operate to directly remove them (5,8,14,32,40–49).
A key actor in the control of R-loop accumulation in humans is the putative RNA:DNA helicase, senataxin (SETX), which belongs to the superfamily 1B (SF1B) class of helicases. SETX is homologous to the well-characterised yeast protein Sen1, which plays a major role in transcription termination and the maintenance of genome stability. Detailed biochemical characterisation of Sen1 demonstrated that it is a helicase with the ability to translocate along single-strand DNA and RNA in the 5′ to 3′ direction and to unwind various nucleic acid duplexes (50–52). When Sen1 translocates on the nascent RNA and encounters the transcribing RNAPII, it induces the dissociation of the elongation complex, thereby releasing the RNAPII and the transcript from the DNA (50,53). During S-phase, Sen1 associates with the replication fork (54,55) and uses its transcription termination activity to remove RNAPIIs and associated R-loops from TRC sites to aid fork progression (56–58).
Akin to Sen1, SETX has been shown to prevent the accumulation of R-loops at sites of TRCs and/or DNA damage (6,7,59–61). Based on the strong conservation between SETX and Sen1 and the demonstrated ability of the yeast counterpart to unwind RNA:DNA hybrids in vitro, the prevailing model posits that SETX uses its helicase activity to directly resolve R-loops. However, it remains possible that SETX rather controls R-loop formation by acting on RNAPII genomic occupancy. Indeed, SETX has been implicated in RNAPII transcription termination (40,62–64), although it is unclear whether SETX has retained the ability of its yeast orthologue to induce termination or whether it rather plays an accessory role in this process.
The biological relevance of SETX is further underscored by the link between SETX mutations and two neurodegenerative diseases: ataxia with oculomotor apraxia (AOA2) and a juvenile form of amyotrophic lateral sclerosis dubbed ALS4 (65–71). While AOA2 is an autosomal recessive disease associated with SETX loss of function, ALS4 is an autosomal dominant disease provoked by toxic gain-of-function mutations in SETX (72). Several studies have reported decreased levels of R-loops in cells from ALS4 patients (27,73–75), however it remains unclear whether ALS4 mutations alter the catalytic activity of SETX and how such alteration could eventually have an impact on R-loop levels.
Because to date there is no direct biochemical characterisation of SETX activities, much uncertainty exists about the molecular function of SETX. Here, we wished to elucidate the precise role of SETX in the control of R-loop accumulation and in transcription termination. We have purified the helicase domain of SETX (SETX-HD) and characterised its catalytic activities in vitro. We have included in our experiments the helicase domain of Sen1 (Sen1-HD) to assess the conservation of the different activities, as well as potential divergences that may have appeared during evolution. We demonstrate that in vitro, SETX is an efficient nucleic acid-dependent ATPase, and that it unwinds duplex substrates with 5′-3′ polarity, similarly to Sen1. Interestingly, we have also uncovered several differences between SETX and Sen1. Notably, SETX-HD, unlike Sen1-HD, hydrolyses ATP more efficiently in the presence of RNA than DNA, likely due to a more dynamic nature of the interaction of SETX with the RNA compared to the DNA. Moreover, SETX-HD, but not Sen1-HD, seems to have evolved a clear preference for R-loops over other types of nucleic acid substrates. Importantly, we show that SETX has retained the ability of Sen1 to induce transcription termination and that this function requires specific properties or features of mammalian RNAPII.
Finally, we have taken advantage of our biochemical tools to assess the effect of disease-associated mutations on SETX properties and catalytic activities. We have found that the AOA2-associated substitution (L1976R), when introduced in the helicase domain of SETX, impinges upon protein folding, which is in line with the SETX loss-of-function phenotype observed in this disease. In addition, we have found that the R2136H substitution, linked to ALS4, reduces the thermostability of SETX-HD, with no discernible effect on its biochemical activities.
Taken together, our findings represent a major progress in the understanding of the function of SETX in the control of R-loop abundance and transcription termination and provide a framework to approach the molecular origin of neurodegenerative diseases linked to SETX mutations.
MATERIALS AND METHODS
Artificial intelligence-based modelling and data visualisation
AlphaFold 2.2 was used to generate a three-dimensional model of the helicase domain of SETX (amino acids 1699-2454) (76). Molecular graphic images were produced using the UCSF ChimeraX (77) package from the Resource for Biocomputing, Visualization, and Informatics at the University of California, San Francisco (supported by NIH P41 RR-01081).
Plasmid construction
A fragment of DNA containing the coding sequence of the putative helicase domain of human SETX (amino acids 1674-2677; SETX-HD), codon-optimised for expression in insect cells, was first cloned into plasmid 2BcT (pET His6 LIC cloning vector; addgene #37236) to generate a C-terminally tagged SETX-HD-(His)6. Subsequently, SETX-HD-(His)6 was subcloned into plasmid 438A (pFastBac cloning vector with BioBrick PolyPromoter LIC Subcloning, addgene #55218) for purification of SETX-HD-(His)6 from insect cells. Plasmids 2BcT and 438A were purchased directly from QB3 Macrolab (UC Berkeley). Point mutations resulting in the following amino acid substitutions: K1969R; L1976R; DE2181, 2182AA; V1884T; R2136H were introduced by site-directed mutagenesis. Oligonucleotides used are listed in Supplementary Table S1. The sequence integrity of all constructs was verified by sequencing.
Protein expression in insect cells
To generate viruses enabling the production of proteins in insect cells, the coding sequences and the necessary regulatory sequences of the aforementioned constructs were transposed into bacmids using the E. coli strain DH10bac. Viral particles were obtained by transfection of the corresponding bacmids into the Sf9 cells using the FuGENE Transfection Reagent and further amplification. Proteins were expressed in 600 ml of Hi5 cells (infected at 1 × 106 cells/ml) with the corresponding P1 virus at multiplicity of infection (MOI) >1. Cells were harvested 48 hours post-infection, washed with 1× PBS, and stored at –80°C.
Protein purification
Thawed insect cells expressing SETX-HD variants were resuspended in lysis buffer (50 mM Tris–HCl, pH 8; 0.5 M NaCl; 10% (v/v) glycerol; 1 mM DTT; 0.4% (v/v) Triton-X; 10 mM imidazole) containing protease inhibitors (0.66 μg/ml pepstatin, 5 μg/ml benzamidine, 4.75 μg/ml leupeptin, 2 μg/ml aprotinin) and 25 U benzonase per ml of lysate. The cleared lysate was passed through 2 ml of Ni-NTA beads (Qiagen), equilibrated with 50 mM Tris–HCl, pH 8; 500 mM NaCl; 10 mM imidazole; and 1 mM DTT. SETX-HD was eluted with an elution buffer containing 50 mM Tris–HCl, pH 8; 350 mM NaCl; 1 mM DTT and 400 mM imidazole. The elution fractions containing SETX-HD were diluted 2-fold with 50 mM Tris–HCl, pH 8; 1 mM DTT and loaded onto a 1 ml HiTrap Heparin HP column (Cytiva). Proteins were eluted with 50 mM Tris–HCl, pH 7.5; 1 mM DDT and a 30-column-volume (CV) gradient of 200 to 650 mM NaCl. Fractions containing homogeneous SETX-HD were concentrated, and glycerol was added to a final concentration of 10% before they were snap-frozen in liquid nitrogen and stored at −80°C. The identity of the purified variants of SETX-HD was verified by mass spectrometry. The results of the analyses are summarised in Supplementary Table S2.
Recombinant Sen1-HD was expressed from the T7 promoter in E. coli and purified by consecutive Ni‐affinity chromatography, heparin-affinity chromatography, and gel filtration as described in (52).
The ten-subunit RNAPII core enzyme was purified from S. cerevisiae strain BJ5464 (78) by Ni-affinity chromatography followed by anion exchange on a Mono Q column essentially as previously described (79). Recombinant (His)6‐tagged Rpb4/7 heterodimer was purified from E. coli by Ni-affinity chromatography followed by gel filtration as described in (79).
The mammalian, twelve-subunit RNAPII complex was purified from the calf (Bos taurus) thymus. Briefly, chromatin-bound RNAPII was purified to homogeneity by successive rounds of ion-exchange, immuno-affinity, ion-exchange, and size-exclusion chromatography as described in (80).
Synthetic nucleic acid substrates
Oligonucleotides for preparing synthetic fluorescently labelled (Cy3 or FITC) RNA/DNA substrates were purchased from Sigma (HPLC purified) and their sequences are available in Supplementary Table S1. The ssRNA oligonucleotides (pRM678 and pRMS740) were specifically designed to contain no secondary structures using the INFO RNA—sequence designer online tool (81–83). Duplex substrates were mixed in a 1:2 ratio of labelled versus unlabelled oligonucleotide and triplex substrates were prepared by mixing labelled molecules with a 5-fold excess of the unlabelled oligonucleotides in the annealing buffer (25 mM Tris–HCl, pH 7.5, 100 mM NaCl, 3 mM MgCl2), followed by initial denaturation at 75°C for 5 min and subsequent cooling to room temperature. The annealed substrates were purified from a polyacrylamide native gel. Schematic representation of the substrates used is shown in Supplementary Figure S1.
ATPase assays
The ATPase activity of SETX-HD and Sen1-HD proteins was assessed at 37°C in 10 μl reactions containing 10 mM Tris–HCl, pH 7.5, 75 mM NaCl, 1 mM MgCl2, 1 μM ZnCl2, 1 mM DTT, 10% (v/v) glycerol, 50 ng/μl of single-stranded DNA (ssDNA; oligo DL3701), or single-stranded RNA (ssRNA; oligo DL3316), 100 μM ATP-Mg, and 0.25 μM of 3000 Ci/mmol α32P‐ATP. After preincubation of the mixtures for 5 min at 37°C, the reactions were started with the addition of SETX-HD-WT (10 nM as the final concentration for reactions in the presence of ssDNA and 2.5 nM for reactions containing ssRNA), SETX-HD-DE/AA (25 nM as the final concentration), or Sen1-HD proteins (2 nM as the final concentration). Aliquots were taken at indicated time points, mixed with 2.5 volumes of quench buffer containing 10 mM EDTA and 0.5% (w/v) SDS, and then subjected to thin‐layer chromatography on PEI cellulose plates (Merck) in 0.35 M potassium phosphate pH 7.5. Plates were exposed to phosphorimager screens and scanned with a Typhoon scanner (GE Healthcare). The different radioactive species were analysed using the ImageQuant TL software (GE Healthcare).
Electrophoretic mobility shift assay (EMSA)
The indicated amounts of the different versions of SETX-HD and Sen1-HD proteins were incubated with fluorescently labelled nucleic acid substrates (at a final concentration of 10 nM) in buffer D (25 mM Tris–HCl, pH 7.5, 1 mM DTT, 0.1 mg/ml BSA, 5 mM MgCl2, and 100 mM NaCl and for nucleic acid substrates containing RNA, 2 U/μl of RNasin ribonuclease inhibitor (Promega)) for 20 min on ice. Loading buffer (60% (v/v) glycerol in Orange-G) was added to the reaction mixtures, the samples were loaded onto a 7.5% (w/v) polyacrylamide native gel in 0.5× TBE buffer, and run at 75 V for 1 h. The different nucleic acid substrates were visualised using a FLA-9000 Starion scanner and quantified with the MultiGauge software (Fujifilm). To calculate the relative amount of bound nucleic acid substrate the band intensity (using the band intensity—background option) of the control sample, without protein, was set as 0% of bound protein and the percent of bound protein for individual concentrations was calculated according to the control. Nucleic acid binding affinity graphs were generated with Prism-GraphPad 9 using the non-linear regression fit function.
Helicase assays
The indicated amounts of the different versions of SETX-HD and Sen1-HD were incubated with fluorescently labelled nucleic acid substrates (at a final concentration of 10 nM) in buffer H (25 mM Tris–HCl, pH 7.5, 1 mM DTT, 50 mM NaCl, 20 mM creatine phosphate, 20 μg/ml creatine kinase, 3.5 mM MgCl2, and 2 mM ATP (for nucleic acid substrates containing RNA, 2 U/μl of RNasin Ribonuclease Inhibitor (Promega)) for 30 min at 37°C. In reactions with duplex substrates a 5-molar excess of competitor oligonucleotide (pRMS680 for blunt-end, pRMS681 for 3′-overhang, and pRMS682 for 5′-overhang RNA:DNA hybrids) was added to trap the unwound, unlabelled oligonucleotide. The reactions were stopped by adding a mixture of 2.5% (w/v) SDS, and 0.5 mg/ml proteinase K and incubated for 3 min at 37°C. Loading buffer was added, and samples were loaded onto a 10–12% (w/v) polyacrylamide native gel in 1× TBE and run at 80 V for 1 h. Fluorescent nucleic acid species were visualised using a FLA-9000 Starion scanner and quantified by MultiGauge software (Fujifilm). Nucleic acid unwinding curves were generated by plotting the percentage of unwound product in Prism-GraphPad 9.
Biolayer-interferometry (BLI)
To determine the kinetics of SETX-HD and Sen1-HD association and dissociation to ssDNA and ssRNA, respectively, BLI experiments were performed using a single–channel BLItz instrument in Advanced Kinetics mode (ForteBio) at room temperature, with shaking at 2200 rpm. Prior to the measurements, the streptavidin biosensors (SAX, ForteBio) were pre–hydrated in BLI buffer (buffer D, supplemented with 0.05% (v/v) Tween 20) for 10 min. Biotinylated ssDNA (pRMS739) and ssRNA (pRMS740), respectively, were immobilised on the biosensor tip at a final concentration of 12 nM (oligonucleotide sequences can be found in Supplementary Table S1). The RNA substrate was diluted in BLI buffer supplemented with a RNase inhibitor at a final concentration of 2 U/μl (RNasin Ribonuclease inhibitor (Promega)). The output data are shown in a sensorgram as a change in optical thickness (in nm) induced by the binding of protein to the immobilised nucleic acid substrate over time. The protein association was measured at three different concentrations (12.5, 25, and 50 nM) for 180 s. Protein dissociation from the nucleic acid substrate was measured for 120 s by subsequent washing of the biosensor with BLI buffer. The association and dissociation constants were calculated in Prism GraphPad 9 software using the ‘Association then dissociation’ function of non-linear regression in global analysis mode (constraint HotNM—shared value for all datasets) from triplicate measurements of each concentration for each protein with a coefficient of determination (R2) indicating the appropriateness of the fit.
In vitro transcription termination (IVTT) assays
Termination assays were performed as previously described (78) with some modifications. Briefly, ternary elongation complexes (ECs) were assembled in a promoter‐independent manner by first annealing a fluorescently labelled RNA (oligonucleotide DL2492) with the template DNA (oligonucleotide DL3352) and subsequently incubating the RNA:DNA hybrid (typically 1 pmol per reaction) with purified RNAPII (typically 250 ng per reaction) for 10 min at 20°C. Next, a 2-fold excess of non‐template (NT) strand (oligonucleotide DL3353) was added to the mixture and incubated for an extra 10 min at 20°C. For reactions with yeast RNAPII we also added 2 pmol of recombinant Rpb4/7 and incubated the mixtures for additional 10 min. Ternary ECs were then immobilised on streptavidin beads (Dynabeads MyOne Streptavidin T1 from Invitrogen) and washed with transcription buffer (TB, 20 mM Tris–HCl, pH 7.5, 100 mM NaCl, 8 mM MgCl2, 10 μM ZnCl2, 10% (v/v) glycerol, and 2 mM DTT) containing 0.1% (v/v) Triton-X, TB containing 0.5 M NaCl, and finally TB. IVTT reactions were performed in TB in a final volume of 20 μl in the absence or the presence of 100 nM Sen1-HD or 600 nM SETX-HD. Transcription was initiated after addition of a mixture of ATP, UTP, and CTP (1 mM each as the final concentration in the reaction) and incubated for 10 min at 37°C. Reactions were then stopped by the addition of 1 μl of 0.5 M EDTA and the beads and supernatant fractions were separated. Beads fractions were directly resuspended in 8 μl of loading buffer (1× Tris–borate–EDTA, 8 M urea) whereas transcripts in the supernatant fractions were ethanol precipitated and resuspended in 8 μl of loading buffer. Samples were boiled for 5 min at 95°C and subjected to 10% (w/v) denaturing PAGE containing 8 M urea. Gels were scanned with a Typhoon scanner (GE Healthcare) and analysed with the ImageQuant TL software (GE Healthcare).
Differential scanning fluorimetry
The thermal stability of SETX-HD WT and the R2136H variant was measured using the Prometheus NT.38 instrument (NanoTemper Technologies). NanoDSF grade capillaries were filled with protein samples at 1 mg/ml in protein dilution buffer (25 mM Tris–HCl, pH 7.5, 150 mM NaCl, 0.01% (v/v) NP-40, and 1 mM DTT). Protein unfolding was detected at a temperature range of 20 to 95°C with a 1°C/min heating rate and 90% excitation power. Transition points (onset of protein unfolding (To) and protein melting temperature (Tm)) were determined from the ratio of tryptophan emission at 330 and 350 nm. The temperature of protein aggregation onset (Tagg) was determined by light scattering, which is the back-reflected light intensity passing through the sample.
Statistical analyses
Statistical analyses were performed using Prism GraphPad 9 (version 9.1.0). After assessing normal data distribution by Shapiro–Wilk normality test, a two-tailed unpaired Student's t-test was performed to compare individual datasets. In graphs showing nucleic acid-protein binding curves (EMSAs) and nucleic acid unwinding curves (helicase assays), the values for the last (highest) concentrations were compared. The resulting P-value (P) is depicted as: ns (non-significant): P > 0.05; *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001.
RESULTS
Expression and purification of the putative helicase domain of SETX
To investigate the mechanism of action of SETX we adopted an in vitro approach consisting in the purification and characterisation of the putative helicase domain of SETX (henceforth named SETX-HD), located at the C-terminal region of the protein comprising amino acids (aa) 1699–2454 (Figure 1A). AlfaFold2 predicts that SETX-HD adopts a fold similar to its yeast orthologue Sen1, with a notable difference in the length of the subdomain 1C (the ‘prong’) (Figure 1B), suggesting that it may confer different properties to SETX-HD (see discussion).
Figure 1.
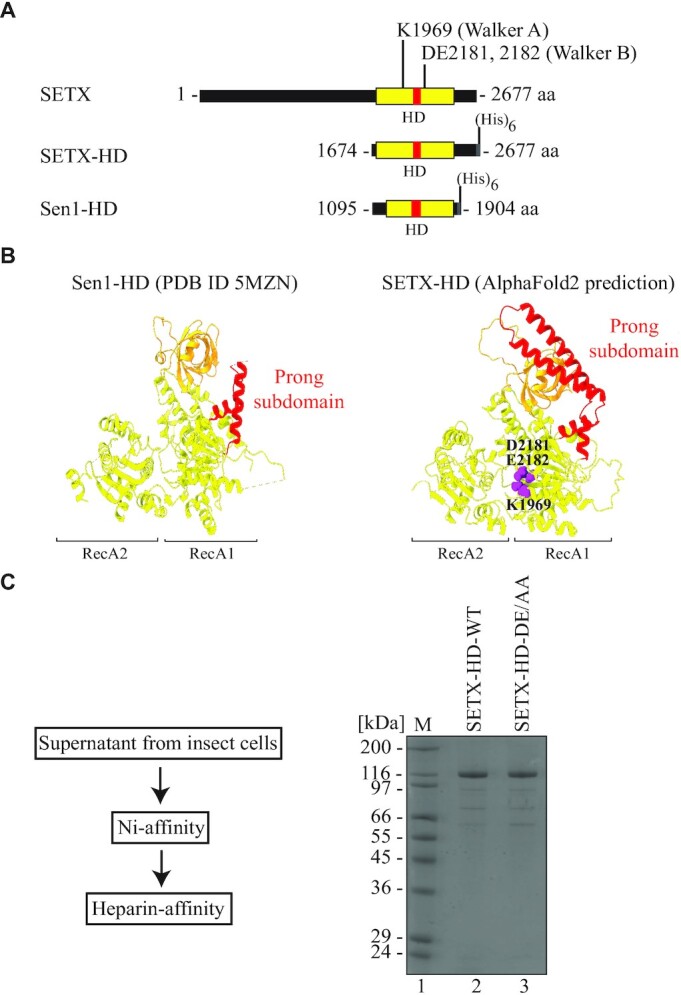
Design and purification of the helicase domain of senataxin. (A) Design of a variant of senataxin (SETX; amino acids (aa) 1674–2677), containing the putative helicase domain (HD, depicted in yellow), located between aa 1699 and 2454. A subdomain of the helicase domain, called the ‘prong’ is depicted in red. A (His)6-tag is fused to the C-terminus of the fragment. The positions of Walker A and Walker B motifs are indicated. A fragment of Sen1 (Sen1-HD, aa 1095–1904), containing its helicase domain, used as a reference, is depicted below. (B) Comparison of the structure of Sen1 helicase domain (PBD ID 5MZN -(52)) and the helicase domain of SETX, predicted by AI-mediated modelling using AlphaFold2. RecA1 and RecA2 domains are depicted in yellow, β-barrel in orange, and the ‘prong’ subdomain in red. In purple are highlighted the key residues responsible for ATP hydrolysis (K1969, within the Walker A motif), as well as for Mg2+ coordination (D2181, E2182, within the Walker B motif). (C) Procedure and representative results of purification of SETX-HD. Left: Cleared supernatant from insect cells was applied onto a Ni-affinity chromatography column and subsequently onto a heparin-affinity chromatography column. Right: Coomassie-stained SDS-PAGE gel showing purified SETX-HD variants (WT: wild-type and DE/AA: DE2181,2182AA). M: molecular weight marker.
The N-terminal boundary of the investigated variant was set at aa 1674, thereby including amino acids corresponding to a key structural feature of the helicase domain of Sen1 (the yeast orthologue of SETX; henceforth named Sen1-HD) termed the ‘brace’, which fastens three additional structural features within the helicase core: RecA1, the ‘stalk’, and the ‘barrel’ and is likely critical for proper protein folding (52). A hexa-histidine tag was placed on the C-terminus of SETX-HD to avoid possible interference with the folding of this domain, as well as to enrich the full-length protein during the purification. To gain an evolutionary perspective of the activities of SETX-HD, we included in our experiments Sen1-HD (aa 1095–1904), which has been extensively characterised previously (Supplementary Figure S2A (51,52)).
Two SETX-HD variants, mutated either in the Walker A motif, responsible for ATP hydrolysis (K1969R), or in the Walker B motif, responsible for Mg2+ binding (DE2181, 2182AA), were generated to determine the role of ATP hydrolysis in the potential activity(ies) of SETX-HD.
The wild-type (WT) version of SETX-HD (SETX-HD-WT), as well as the mutants in Walker A and Walker B motif, respectively, were expressed in insect cells, but only SETX-HD-WT and the mutant in the Walker B motif (hereafter SETX-HD-DE/AA) were soluble enough to be purified to near homogeneity (Figure 1C; Supplementary Figure S2B), which was necessary for subsequent analyses.
The helicase domain of SETX exhibits a preference for RNA over DNA
Initially, we aimed to characterise the ATPase activity of purified SETX-HD. As helicases strictly require the interaction with nucleic acids to hydrolyse ATP, we first investigated the binding of SETX-HD and Sen1-HD to single-stranded nucleic acid substrates (61 nt, see Supplementary Table S1) using electrophoretic mobility-shift assay (EMSA). Sen1-HD bound both ssDNA and ssRNA with similar affinity, in agreement with the existing literature (Figure 2A, Supplementary Figure S3A) (50,51). In contrast, SETX-HD-WT bound more readily ssDNA than ssRNA (at 12.5 nM SETX-HD bound 33.6% of the ssDNA substrate, whilst 19.4% of the ssRNA substrate; at 25 nM SETX-HD bound 71.5% ssDNA, whilst 35.9% ssRNA) (Figure 2A, Supplementary Figure S3A). The SETX-HD DE/AA variant had the same nucleic acid binding pattern as SETX-HD-WT (Supplementary Figure S3A, B).
Figure 2.

SETX, in contrast to Sen1, hydrolyses ATP more efficiently in the presence of single-stranded RNA than DNA. (A) Quantification of electrophoretic mobility-shift assays (EMSA) with SETX-HD and Sen1-HD, respectively, and single-stranded DNA (pRMS679, in black, 61 nt) or single-stranded RNA (pRMS678, in blue, 61 nt). The sequences of the different nucleic acid substrates are indicated in Supplementary Table S1. The data points in each graph represent the mean of three independent experiments (except for SETX-HD + ssDNA: n = 4), error bars represent the standard deviation (SD). The values obtained for the highest concentration were compared using a two-tailed unpaired Student′s t-test. The resulting P-values (P) are represented by ns (non-significant) P > 0.05 and ** P < 0.01. A scan of a gel of a representative experiment is shown in Supplementary Figure S3A. (B) Quantification of the amount of ATP hydrolysed by SETX-HD-WT, SETX-HD-DE/AA, and Sen1-HD, respectively, in the presence of single-stranded DNA (DL3701, in black, 44 nt) or RNA (DL3316, in blue, 44 nt), respectively, plotted as a function of time. The data points represent the mean of three independent experiments, error bars represent the standard deviation (SD). The values obtained for the highest concentration were compared using a two-tailed unpaired Student′s t-test. The resulting P-values (P) are represented by ns (non-significant) P > 0.05 and * P < 0.05. A scan of a representative experiment is included in Supplementary Figure S3C. (C) SETX-HD dissociates from RNA more rapidly than from DNA. Sensorgrams from bio-layer interferometry (BLI) assay represent the mean of 3 measurements for each condition. Twelve fmoles of biotinylated ssDNA (pRMS739, 41 nt) and ssRNA (pRMS740, 41 nt), respectively, were bound to a SAX biosensor. SETX-HD (50 nM) and Sen1-HD (50 nM), respectively, were then submerged into the biosensor, to measure protein association for 180 s. Protein dissociation from nucleic acids was measured in BLI buffer for 120 s. Sensorgrams depict the comparison between association and dissociation rates of SETX-HD (left) and Sen1-HD (right), respectively, to DNA (in black) and RNA (in blue). Binding curves were normalised in Prism GraphPad 9. (D) Quantification of BLI assays measuring the rate of association and dissociation of SETX-HD and Sen1-HD, respectively, to immobilised biotinylated single-stranded DNA and RNA. Graphs represent the association (Kon), dissociation (Koff), and dissociation (KD) constants, respectively, for SETX-HD and Sen1-HD with DNA (in black) and RNA (in blue). The data points represent the values calculated in Prism GraphPad 9 using the ‘Association then dissociation’ function under global analysis mode from triplicate measures of three concentrations (12.5, 25, and 50 nM) for each condition. Sensorgrams for individual concentrations can be found in Supplementary Figure S3D.
In agreement with the EMSA and the existing literature (50), both ssDNA and ssRNA (both 44 nt, see Supplementary Table S1) stimulated ATP hydrolysis by Sen1-HD to a similar extent (Figure 2B; Supplementary Figure S3C). Intriguingly, ssRNA stimulated the ATPase activity of SETX-HD nearly 1.7-fold more efficiently than ssDNA. As expected, SETX-HD-DE/AA did not hydrolyse ATP in the presence of neither ssDNA nor ssRNA, indicating that the observed ATPase activity is inherent to SETX-HD and not to any potential contaminating activity (Figure 2B, Supplementary Figure S3C).
To gain mechanistic insight into the observed different behaviour of SETX-HD and Sen1-HD, we employed biolayer-interferometry (BLI), which assesses the interaction between proteins and nucleic acids in real-time, enabling us to determine the kinetic parameters of the binding of SETX-HD and Sen1-HD to different nucleic acid substrates. We analysed the binding of SETX-HD and Sen1-HD to ssDNA and ssRNA (both 41 nt, see Supplementary Table S1) immobilised on a biosensor (Figure 2C, Supplementary Figure S3D). SETX-HD associated with ssRNA 3-times faster than with ssDNA, whilst Sen1-HD associated only slightly faster with ssRNA than ssDNA (SETX: KonRNA = 8.9 × 105 M−1 s−1; KonDNA = 3.3 × 105 M−1 s−1; Sen1: KonRNA = 1 × 106 M−1 s−1; KonDNA = 5.6 × 105 M−1 s−1). SETX-HD dissociated 4-times faster from ssRNA than ssDNA (KoffRNA = 4.2 × 10−3 s−1; KoffDNA = 9.4 × 10−4 s−1), whilst Sen1-HD dissociated twice faster from ssRNA than from ssDNA (KoffRNA = 3.8 × 10−3 s−1; KoffDNA = 1.9 × 10−3 s−1) (Figure 2D). Taken together, Sen1-HD has comparable affinity towards ssRNA and ssDNA (KDRNA = 3.7 × 10−9 M; KDDNA = 3.4 × 10−9 M). In contrast, SETX-HD exhibits 2-fold higher affinity towards ssDNA than ssRNA (KDRNA = 4.6 × 10−9 M; KDDNA = 2.8 × 10−9 M) (Figure 2D). These data are consistent with the EMSA experiments and provide a mechanistic understanding of the observed differences in the rates at which ssDNA and ssRNA stimulate the ATPase activity of SETX-HD and Sen1-HD, respectively.
We speculated that the faster dissociation rate of SETX-HD from ssRNA than ssDNA, compared to Sen1-HD, is due to accumulated mutations on the RNA-binding surface of SETX that appeared over the course of the evolution. We modelled the threading of the RNA substrate through Sen1 and SETX to rationalise their different nucleic acid binding properties (Supplementary Figure S4A) by using the available structure of the helicase domain of Upf1, a close homologue of Sen1/SETX, in complex with the RNA (PDB: 2XZO) (84) and taking advantage of the high conservation of the residues forming the helicase channel in the three proteins. Interestingly, residues contacting the sugar moieties are conserved between Sen1 and SETX, except for one at the entry channel, in which the T1289 in Sen1 is replaced by V1884 in SETX (Supplementary Figure S4B, C). We hypothesised that the threonine in Sen1 may contact both DNA and RNA equally well, as the threonine residue can switch between polar and hydrophobic contacts to the ribose and deoxyribose, respectively. In contrast, SETX has a valine at this position, which disfavours the interaction with the ribose moiety of RNA, thereby possibly explaining the less stable binding of SETX to RNA compared to DNA (Figure 2A, C and Supplementary Figure S3A, D). To test our hypothesis, we generated and purified the SETX-HD V1884T variant (Supplementary Figure S4D). To investigate the possible alterations in nucleic acid binding affinity of the SETX-HD-V1884T variant, we performed EMSA experiments alongside SETX-HD-WT (Supplementary Figure S4E, F). Whilst we did not observe a difference in ssRNA binding of the V1884T variant compared to the wild-type protein, the binding of the mutant to the ssDNA was slightly decreased compared to wild-type. This resulted in a very similar binding of SETX-HD-V1884T to both ssDNA and ssRNA, as observed for Sen1-HD (compare Figure 2A and Supplementary Figure S3A). To verify this observation, we performed BLI assays on immobilised ssDNA and ssRNA substrates, as before, with the SETX-HD-V1884T variant (Supplementary Figure S4G). We did not observe differences in association and dissociation of the V1884T variant to ssRNA, compared to SETX-HD-WT. However, we did observe faster dissociation of SETX-HD-V1884T from ssDNA, compared to SETX-HD-WT. The observed kinetics of protein association and dissociation to ssDNA of the V1884T variant were very similar to that of Sen1-HD. In summary, the presence of valine 1884 in SETX-HD instead of a threonine strengthens SETX binding to ssDNA, therefore leading to a slower protein turnover on ssDNA compared to ssRNA.
Collectively our data suggest that SETX-HD is indeed a potent ATPase, which, over the course of evolution, has adapted to respond more efficiently to ssRNA than to ssDNA.
SETX unwinds nucleic acid duplexes with a 5′-to-3′ polarity
Once we have established that the helicase domain of SETX has robust nucleic acid-dependent ATPase activity, we aimed to determine whether and with what polarity SETX-HD unwinds duplex substrates. To this end, we used a series of RNA:DNA hybrid substrates harbouring a 5′ ssRNA overhang, a 3′ ssRNA overhang, or no overhang (see Supplementary Figure S1), which enabled us to unambiguously determine the polarity of the translocation of SETX-HD on nucleic acids. First, we analysed the ability of SETX-HD-WT to bind such substrates and determined that SETX-HD displayed similar affinity for hybrids containing a 5′ ssRNA or a 3′ ssRNA overhang, whilst it did not bind a duplex, in agreement with the notion that SETX-HD only binds single-stranded nucleic acids (Supplementary Figure S5A, B). Subsequently, we performed a series of helicase assays with the same substrates, and we found that SETX-HD unwound only the substrate containing a 5′ ssRNA overhang, albeit less efficiently than Sen1-HD. This unwinding activity required the presence of ATP, as well as the ability of SETX-HD to hydrolyse ATP, since the SETX catalytic mutant DE/AA was virtually inactive (Figure 3A, B).
Figure 3.

SETX, similarly to Sen1, unwinds RNA:DNA hybrids and DNA:DNA duplexes with a 5′-to-3′ polarity. (A) SETX-HD unwinds RNA:DNA duplexes with a 5'-overhang in an ATP dependent manner. Helicase assays were performed in the presence of the indicated amounts of SETX-HD-WT, SETX-HD-DE/AA, and Sen1-HD, respectively, and 10 nM fluorescently labelled RNA:DNA duplexes (61 nt) containing either a 5' (lanes 1–15) or a 3′ (lanes 16–30) 41 nt single-stranded overhang (oh), or blunt ends (lanes 31–45). Where indicated, ATP was omitted. (B) Quantification of the experiments shown in (A). The data points in the graphs represent the mean of three independent experiments (for 5′-oh RNA:DNA duplex, n = 4) and the error bars represent the standard deviation (SD). The values obtained for the highest concentration were compared using a two-tailed unpaired Student′s t-test. The resulting P-values (P) are represented by * P < 0.05, and **** P < 0.0001. (C) SETX-HD unwinds DNA:DNA duplexes with a 5’-overhang in an ATP dependent manner. Helicase assays were performed in the presence of the indicated amounts of SETX-HD-WT, SETX-HD-DE/AA, and Sen1-HD, respectively, and 10 nM fluorescently labelled DNA:DNA duplexes (61 nt) containing either a 5' (lanes 1–15) or a 3′ (lanes 16–30) 41 nt single-stranded overhang (oh), or blunt ends (lanes 31–45). Where indicated, ATP was omitted. (D) Quantification of the experiments shown in (C). The data points in the graphs represent the mean of three independent experiments (for 5′-oh DNA:DNA duplex, n = 5) and the error bars represent the SD. The values obtained for the highest concentration were compared using a two-tailed unpaired Student′s t-test. The resulting P-values (P) are represented by **** P < 0.0001.
Whilst the helicase activity of SETX-HD on RNA:DNA hybrid substrates has potentially biological relevance, it has been shown that Sen1 is a potent DNA helicase (50,51). Therefore, we performed the aforementioned EMSA and helicase assays using DNA:DNA duplexes harbouring a 5’, a 3’ ssDNA overhang, or no overhang as substrates (Figure 3C, D and Supplementary Figure S5C, D). Similarly, to Sen1, we found that SETX-HD unwound DNA duplexes in an ATP-dependent manner, but only in the presence of a 5’ ssDNA overhang. In addition, both helicases are substantially more efficient at unwinding DNA:DNA duplexes than RNA:DNA hybrids, likely due to the lower stability of DNA:DNA hybrids.
Altogether, our data indicate that SETX-HD can indeed translocate, in an ATP-dependent manner, on single-stranded nucleic acids with a 5′-to-3′ polarity.
SETX-HD preferentially unwinds R-loop structures with a 5′ overhang
SETX has been implicated in the suppression of R-loop structures that form in cells during transcription (7,40,59–61). However, a direct, mechanistic evidence that SETX resolves R-loops is still missing. As SETX harbours a helicase domain, capable of unwinding RNA:DNA duplexes, we next decided to investigate whether SETX-HD can dismantle R-loop structures.
Initially, we performed EMSA experiments on R-loop structures with a 5′ ssRNA overhang (5′-oh R-loop), a 3′ ssRNA overhang (3′-oh R-loop), and with no overhang (R-loop) and found that SETX-HD bound the three types of molecules with similar affinity, probably because they all harbour at least one region of single-stranded nucleic acid (Supplementary Figure S6A, B). However, SETX-HD dismantled preferentially 5′-oh R-loops in an ATP-dependent manner (Figure 4A–C; Supplementary Figure S6C, D), most likely by loading on the RNA and translocating from 5′ to 3′, in agreement with the results above (Figure 3). We observed a residual activity on the 3′-oh R-loop substrate (Supplementary Figure S6C, compare lane 4 and lane 9), which is likely caused by partial, spontaneous opening of duplex regions allowing SETX-HD loading, rather than by a 3′-to-5′ translocation of SETX-HD on the substrate (see section above).
Figure 4.

SETX-HD preferentially unwinds R-loop structures with a 5′-overhang. (A) SETX-HD unwinds the RNA:DNA duplex region of a 5'-oh R-loop in an ATP-dependent manner. Helicase assays with 70 nM SETX-HD WT and DE/AA, respectively, and 10 nM fluorescently labelled 5′-oh R-loop in the presence (lane 3 and 5) or absence (lane 2 and 4) of ATP. Lane 1 represents a control reaction with no protein. Samples containing all possible intermediates, resulting from either cleavage of ssRNA or SETX-mediated unwinding, were run on the gel for comparison. (B) SETX-HD unwinds 5'-oh R-loops in a concentration-dependent manner. Increasing concentration of SETX-HD-WT and SETX-HD-DE/AA (17.5, 35, and 70 nM) were mixed with 10 nM fluorescently labelled 5′-oh R-loop in the presence of ATP. Lanes 5 and 10 represent control experiments without ATP. (C) Quantification of the experiments shown in (B). The data points in the graph represent the mean of 5 (for WT + 5′-oh R-loop) and 3 (for DE/AA + 5′-oh R-loop) independent experiments and the error bars represent the SD. The values obtained for the highest concentration were compared using a two-tailed unpaired Student′s t-test. The resulting P-values (P) are represented by **** P < 0.0001. (D) SETX-HD preferentially unwinds 5′-oh R-loop structures, whilst Sen1-HD preferentially unwinds 5′-oh D-loop structures. Increasing concentrations of the indicated proteins were mixed with 10 nM fluorescently labelled 5′-oh R-loop (lanes 1–9 SETX-HD; 19–27 Sen1-HD) or 5′-oh D-loop (lanes 10–18 SETX-HD; 28–36 Sen1-HD). Where indicated, ATP was omitted. (E) Quantification of (D). Data points in graph represent the mean of three independent experiments and error bars represent the SD. The values obtained for the highest concentration were compared using a two-tailed unpaired Student′s t-test. The resulting P-values (P) are represented by ** P < 0.01.
Next, we analysed the helicase activity of SETX-HD and Sen1-HD on both 5′-oh D-loop and 5′-oh R-loop structures. Strikingly, we found that SETX-HD unwound 5′-oh R-loops significantly better than 5′-oh D-loops (Figure 4D, E), even though SETX-HD unwinding activity was substantially higher on DNA:DNA duplexes than on RNA:DNA hybrids (Figure 3). On the contrary, Sen1-HD unwound 5′-oh D-loops more efficiently than 5′-oh R-loops, which is in agreement with the above experiments conducted on RNA:DNA and DNA:DNA duplexes (50). Intriguingly, SETX-HD, unlike Sen1-HD, unwound triplex 5′-oh R-loops 4-fold more efficiently than duplex 5′-oh RNA:DNA hybrids (Supplementary Figure S6G, H), strengthening the notion that R-loops are the preferred substrate for SETX to dismantle.
In summary, our data suggest that SETX-HD, unlike Sen1-HD, dismantles R-loops more readily than D-loops if a 5′ overhang is present.
SETX terminates transcription in vitro in a species-specific manner
In addition to the function in the resolution of R-loops, several studies assigned to SETX a role in transcription termination (40,62,85). It has been shown that in vitro Sen1 efficiently terminates transcription by translocating along the nascent RNA to dislodge RNAPII from the DNA (50,53). Furthermore, Sen1-HD is sufficient to terminate transcription in vitro (50,52). However, it remains unclear whether SETX has retained the ability of its yeast orthologue to induce termination or whether it rather plays an indirect role in this process.
To address this question, we used an already established assay, initially developed to investigate the mechanism by which Sen1 terminates RNAPII-mediated transcription (50). In this assay, promoter-independent assembly of the elongation complex (EC) is performed using a 9 nt RNA:DNA hybrid, which occupies the catalytic centre of RNAPII (Figure 5A). Ternary ECs (yeast or mammalian) are attached to streptavidin beads via the 5′ biotin of the non-template strand, enabling subsequent separation of the bead-associated (B) and the supernatant (S) fractions. The RNA molecule is fluorescently labelled to allow its detection. The transcription template contains a G-less cassette followed by a G-stretch in the non-template strand. After adding a mixture of ATP, UTP, and CTP, RNAPII transcribes until it encounters the G-rich sequence. The ability of Sen1-HD and SETX-HD, to provoke dissociation of ECs paused at the G-rich stretch is then monitored by comparing the amount of nascent RNA released to the supernatant in the absence and the presence of these proteins in the reactions.
Figure 5.
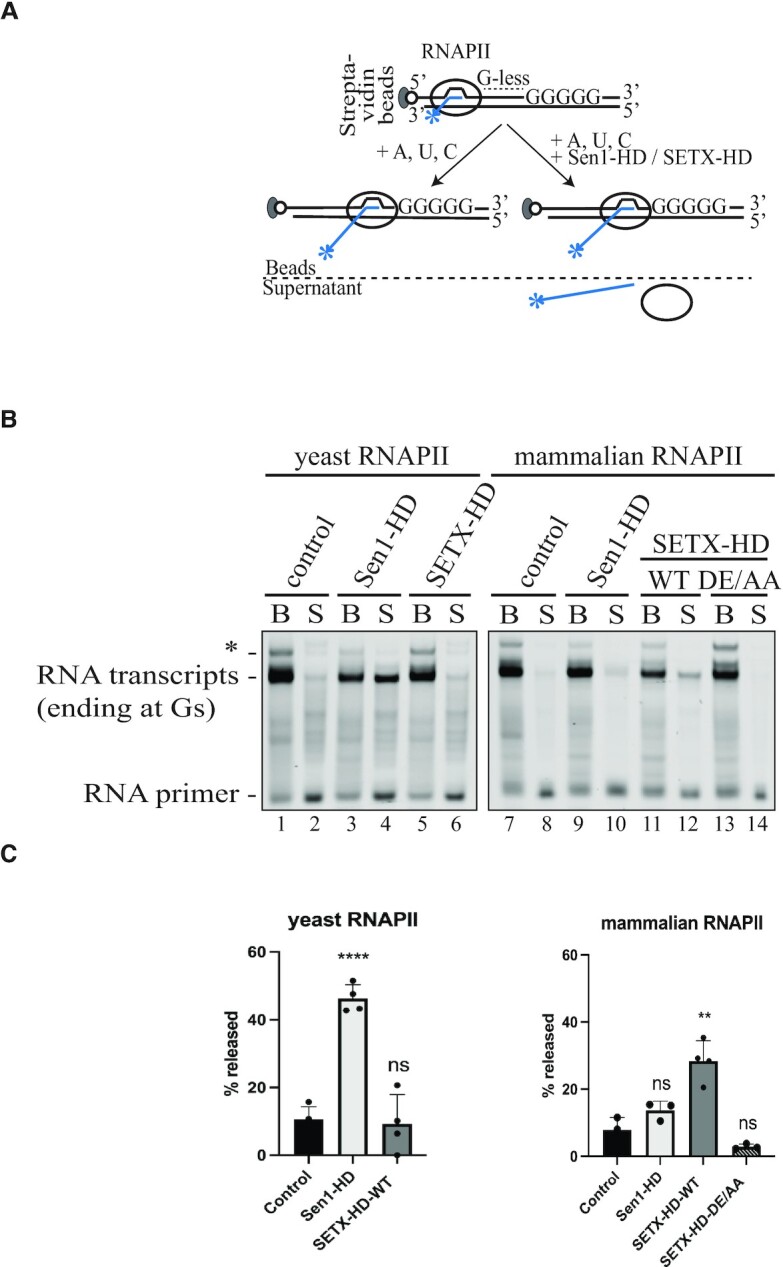
SETX-HD-WT terminates transcription in vitro in a species-specific manner. (A) Schematic representation of the in vitro transcription-termination (IVTT) assay (50). Promoter-independent assembly of the elongation complex (EC) is performed using a 9 nt RNA:DNA hybrid, which occupies the catalytic centre of RNAPII. Ternary ECs are attached to streptavidin beads via the 5′ biotin of the non-template strand, enabling subsequent separation of bead-associated (B) and supernatant (S) fractions. The RNA is fluorescently labelled with FAM at the 5′-end. The transcription template contains a G-less cassette followed by a G-stretch in the non-template strand. After adding an ATP, UTP, CTP mix, the RNAPII transcribes until it encounters the G-rich sequence. Subsequently, Sen1-HD/SETX-HD may provoke dissociation of ECs paused at the G-rich stretch and therefore the release of RNAPII and associated transcripts to the supernatant. (B) IVTT assay using either yeast RNAPII (lanes 1–6) or mammalian RNAPII (lanes 7–14). Transcript release from the beads fraction (B) to the supernatant fraction (S) was monitored upon addition of Sen1-HD, SETX-HD-WT or SETX-HD-DE/AA, as indicated. Lanes 1–2 and 7–8 represent control reactions with no protein. RNA species were analysed by denaturing PAGE. The RNA transcripts associated with RNAPIIs paused at the G-stretch are indicated. Asterisk denotes transcripts associated with RNAPIIs that over-read the G-stretch due to small contamination of the nucleotide solutions with GTP. (C) Quantification of (B). Quantification of RNA transcript in the supernatant fraction as an estimate of the efficiency of termination. Values represent the mean of three independent experiments (for Sen1-HD with yeast RNAPII: n = 4) and error bars denote the SD. The values obtained for each protein were compared to the control using a two-tailed unpaired Student′s t-test. The resulting P-values (P) are represented by ns (non-significant) P > 0.05; ** P < 0.01, and **** P < 0.0001.
First, we found that Sen1-HD efficiently dislodged yeast RNAPII from the DNA since ∼50% of the nascent RNA was present in the supernatant (Figure 5B, lane 4; Figure 5C), as previously shown (50). Under the same experimental set-up, however, SETX-HD did not promote the release of the polymerase and the nascent RNA from the DNA (Figure 5B, lane 6), indicating that either SETX has not retained the same ability of its yeast counterpart or that it displays a species-specific termination activity. To distinguish between these possibilities, we repeated the assay with mammalian RNAPII instead of yeast RNAPII. Interestingly, the addition of SETX-HD-WT, but not SETX-HD-DE/AA resulted in the release of the nascent RNA into the supernatant (Figure 5B, compare lanes 12 and 14; Figure 5C), indicating that SETX may mediate the dissociation of mammalian ECs in an ATP-dependent manner. Strikingly, Sen1-HD could not promote the efficient release of the mammalian RNAPII and the associated RNA into the supernatant (Figure 5B, compare lanes 4 and 10), indicating a specialisation for yeast RNAPII.
Collectively, these data support the notion that SETX-HD induces transcription termination, albeit less efficiently than Sen1-HD, and that this activity likely requires specific features of mammalian RNAPII.
Biochemical characterisation of variants of SETX associated with neurodegenerative diseases
Our thorough biochemical characterisation of the SETX-HD enabled us to assess the effect of mutations within the helicase domain that are associated with neurodegenerative diseases. Two different diseases are associated with mutations in SETX: (i) Ataxia with oculomotor apraxia type 2 (AOA2) and (ii) amyotrophic lateral sclerosis type 4 (ALS4). Whilst mutations causing AOA2 resemble SETX null phenotypes in vivo (65,67–70,74,86), mutations associated with ALS4 cause milder phenotypes (27,66,75,87). We constructed and attempted to purify two mutant versions of SETX: SETX-HD-L1976R (SETX-HD-L/R), associated with AOA2 (69) and SETX-HD-R2136H (SETX-HD-R/H), associated with ALS4 (66) (Figure 6A). Leucine 1976 is located within the RecA1 subdomain, suggesting that the L1976R substitution may impinge upon the ATPase activity and/or the overall structural integrity of the domain, whereas arginine 2136 is located at the bottom of the ‘prong’ subdomain, suggesting a milder effect of the R2136H substitution on the overall activity and/or folding of the domain (Figure 6B).
Figure 6.
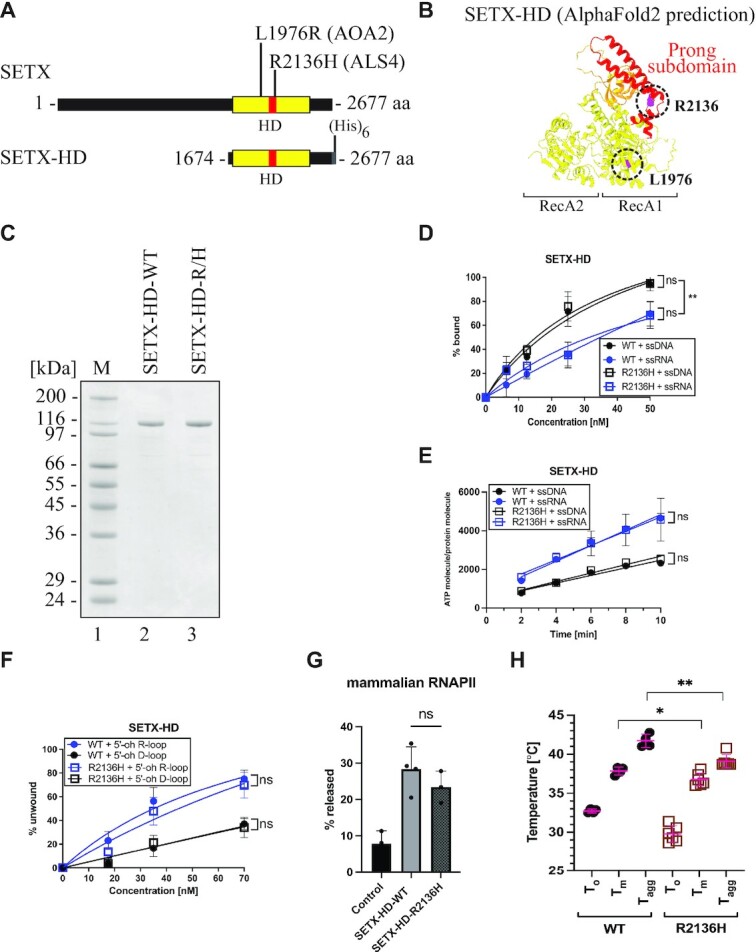
Purification and biochemical characterisation of SETX-HD variants associated with neurodegenerative diseases. (A) Schematic representation of SETX and position of the investigated mutations associated with AOA2 (the SETX-HD L1976R (L/R) variant) and ALS4 (the SETX-HD R2136H (R/H) variant). (B) Structure of the helicase domain of SETX predicted by AI-mediated modelling using AlphaFold2. RecA1 and RecA2 domains are depicted in yellow, β-barrel in orange, and the ‘prong’ subdomain in red. In purple are highlighted the positions of the amino acids mutated in AOA2 (L1976) and ALS4 (R2136). (C) SDS-PAGE gel showing the purified proteins SETX-HD-WT and SETX-HD-R/H. M: molecular weight marker. (D) SETX-HD-R/H binds nucleic acid substrates to the same extent as SETX-HD-WT. Quantification of EMSA with SETX-HD-WT and SETX-HD-R/H, respectively, in the presence of single-stranded DNA (pRMS679, in black, 61 nt) or single-stranded RNA (pRMS678, in blue, 61 nt). The data points in the graph represent the mean of several independent experiments (WT + ssDNA: n = 4; WT + ssRNA: n = 3; R/H + ssDNA: n = 3 and R/H + ssRNA: n = 4), while error bars represent the SD. The values obtained for the highest concentration were compared using a two-tailed unpaired Student′s t-test. The resulting P-values (P) are represented by ns (non-significant) P > 0.05. A scan of a representative experiment is included in Supplementary Figure S7C. (E) SETX-HD-R/H exhibits ATPase activity comparable to SETX-HD-WT. Quantification of ATP hydrolysis by SETX-HD-WT and SETX-HD-R/H in the presence of single-stranded DNA or RNA (both 44 nt) plotted as a function of time. The data points represent the mean of several independent experiments (WT/R/H + ssRNA: n = 4 and WT/R/H + ssDNA: n = 3), while error bars represent the SD. The values obtained for the highest concentration were compared using a two-tailed unpaired Student′s t-test. The resulting P-values (P) are represented by ns (non-significant) P > 0.05. A scan of a representative experiment is shown in Supplementary Figure S7D. (F) SETX-HD-R/H unwinds 5′-oh R-loop and 5′-oh D-loop structures similarly to SETX-HD-WT. Quantification of helicase assays performed in the presence of increasing concentrations of SETX-HD-WT and SETX-HD-R/H with 5′-oh R-loop (blue) or 5′-oh D-loop (black) as substrates. The data points represent the mean of 4 (for 5′-oh R-loop) or 3 (for 5′-oh D-loop) independent experiments and error bars represent the SD. The values obtained for the highest concentration were compared using a two-tailed unpaired Student′s t-test. The resulting P-values (P) are represented by ns (non-significant) P > 0.05. A scan of a representative experiment can be found in Supplementary Figure S7E. (G) SETX-HD-R/H is as efficient as SETX-HD-WT in terminating transcription in vitro. Quantification of RNA transcripts in the supernatant fraction. Values represent the mean of 3 or 4 (for SETX-HD-WT) independent experiments and error bars represent the SD. The values obtained for the mutant were compared to the WT using a two-tailed unpaired Student′s t-test. The resulting P-value (P) is indicated by ns (non-significant) P > 0.05 and ** P < 0.01. A representative scan of the experiment can be found in Supplementary Figure S7F. (H) SETX-HD-R/H variant is less thermostable than SETX-HD-WT. Quantification of the thermostability of SETX-HD-WT and SETX-HD-R/H measured by nanoDSF. Plots represent the mean of replicate measures (WT: n = 4; R/H: n = 6) indicating the temperatures corresponding to the onset of protein unfolding (To), the melting temperature (Tm), and the onset of protein aggregation (TAgg). Error bars represent the SD. The P-value (P) resulting from a two-tailed unpaired Student′s t-test is represented by * P < 0.05; ** P < 0.01. A representative thermograph can be found in Supplementary Figure S7G.
The SETX-HD-L/R variant could be expressed in insect cells, but it was insoluble (Supplementary Figure S7A), suggesting that the mutation affected the proper folding of the domain, thereby likely explaining the phenotype in vivo. On the other hand, SETX-HD-R/H was soluble and could be purified to near homogeneity (Figure 6C; Supplementary Figure S7B). We performed a complete set of biochemical experiments, comparing SETX-HD-R/H to the wild-type version. The behaviour of the SETX-HD-R/H variant was indistinguishable from that of the wild-type in terms of: (i) the binding to nucleic acid substrates (Figure 6D; Supplementary Figure S7C); (ii) ATP hydrolysis (Figure 6E; Supplementary Figure S7D); (iii) the helicase activity on both R-loop and D-loop substrates (Figure 6F; Supplementary Figure S7E); and (iv) the ability to induce transcription termination in vitro (Figure 6G; Supplementary Figure S7F). Finally, to monitor for possible differences in the thermal stability of SETX-HD-R/H, we conducted differential scanning fluorimetry. Indeed, we observed a statistically significant shift in the melting temperature (Tm) and the onset of protein aggregation (TAgg) for SETX-HD-R/H (Figure 6H; Supplementary Figure S7G), suggesting that this mutant variant may be less stable in vivo.
Taken together, our biochemical characterisation of the catalytic domain of SETX has enabled us to unveil that SETX is a bona fide helicase and transcription termination factor and has provided tools to directly address the impact of disease-associated mutations on the properties and activities of this relevant protein.
DISCUSSION
Over the recent years, the putative helicase SETX has emerged as a safeguard of gene expression and genome integrity. Studies conducted in a variety of cellular models, mainly based on diverse phenotypic characterisations upon SETX knock-down, have implicated SETX in critical processes such as transcription termination and the regulation of gene expression, the control of R-loop and DNA damage accumulation, and the regulation of transcription-replication conflicts. All functions attributed to SETX have been proposed to rely on its enzymatic properties as a helicase. However, despite the important role of SETX in several fundamental biological processes and the causal connection between SETX mutations and two devastating neurodegenerative diseases, the molecular function of SETX has remained elusive. In the present study, we fill this gap by providing the first purification and biochemical characterisation of the helicase domain of SETX, which is responsible for its catalytic activities.
Likely the most extensively studied aspect of SETX function is its involvement in R-loop homeostasis. As mentioned in the introduction, R-loops appear as a consequence of normal DNA metabolic activities, such as transcription, and participate in several physiological processes. Only when R-loops accumulate/persist, they become a threat for genome stability and transcription (22). One of the strategies to curb the accumulation of R-loops is their direct dismantling by helicases. Depletion of SETX most often results in an increase of R-loop levels (6,7,40,59–62,74,86,87). This has led many authors to propose that SETX directly resolves R-loops by unwinding the RNA:DNA hybrid portion of R-loops, therefore promoting the restoration of the natural double-stranded DNA. However, transcription, which is the major source of R-loops, is also substantially altered by SETX depletion (62–64,74). A couple of recent studies have shown that the perturbations of RNAPII genomic distribution provoked by SETX deletion/depletion are rather associated with R-loop losses at many loci (88,89). Therefore, the former data did not allow discriminating between a direct role for SETX in the resolution of R-loops from an indirect role via the regulation of transcription.
Here, using purified SETX helicase domain and synthetic R-loops we provide direct evidence that SETX can efficiently dismantle R-loops if they possess a 5′ ssRNA overhang (Figure 4 and Supplementary Figure S6C, D). Complementary experiments indicate that such R-loop resolving activity involves the loading of SETX to the exposed single-stranded RNA and its translocation in the 5′ to 3′ direction to unwind the RNA:DNA hybrid region of the R-loop. This type of R-loop (i.e. having a 5′ ssRNA overhang) is likely the most frequently formed during transcription when the nascent RNA invades the DNA behind the transcribing RNAPII. Additionally, R-loops may be formed at the front of RNAPII upon polymerase backtracking, for instance in response to damaged bases in the template DNA (90). According to our biochemical analyses, this kind of R-loop, lacking a ssRNA portion, would not be a good substrate for SETX, since we detected virtually no activity on such structures (Supplementary Figure S6C, lanes 11–15). It is possible that R-loops resulting from backtracking are removed by RNAPII itself once it resumes elongation, as it has been observed in vitro (90). Alternatively, other R-loop resolving RNA:DNA helicases, such as BLM, DDX5, DDX21, that are active on R-loops with no overhangs in vitro (91–93) may release the path for the polymerase. We also observed a weak SETX unwinding activity on R-loops with a 3′ ssRNA overhang (Supplementary Figure S6C, lanes 6–10). However, we believe that this activity is most likely the result of transient, spontaneous opening of the duplex DNA, followed by SETX translocation in 5′-3′ direction along the DNA strand annealed to the RNA. Such opening might be favoured by the 3′ ssRNA overhang. It is possible that this kind of R-loop structure (i.e. having a 3′ overhang) forms under certain circumstances. In that case, it may be resolved by other RNA helicases such as DHX9 or DDX17, which have the opposite 3′-5′ polarity (25,94–96). Altogether, our data allow us to propose that R-loops forming behind the transcribing RNAPII are the major targets of SETX in vivo (Figure 7).
Figure 7.

Schematic model of the mechanism by which senataxin may promote R-loop dissolution and transcription termination. RNAPIIs may pause/stall during transcription for instance upon reading specific DNA sequences or upon encountering other DNA-bound proteins or damaged template. SETX translocates on the single-stranded nascent RNA with a 5′-3′ polarity to dislodge RNAPII and its associated transcript. This process likely requires direct interactions between specific regions of SETX and RNAPII. Transcription termination by SETX may play a role in the regulation of gene expression and might also be important to prevent unscheduled persistence of RNAPIIs at TRC and DNA damage sites. R-loops may appear in various forms - with either a 5′, a 3′ RNA overhang, or possibly with no overhang. R-loops formed during transcription most often possess a 5′ RNA overhang. SETX can efficiently resolve this kind of structure by translocating in the 5′-3′ direction on single-stranded RNA to disrupt the RNA:DNA hybrid within the R-loop and release the RNA moiety.
Initial functional studies of SETX uncovered its involvement in several facets of RNA metabolism including transcription termination (85). Later reports showed a role for SETX in the regulation of the expression of several genes involved in the establishment of circadian clock and in the antiviral immune response (63,97). Specifically, SETX was proposed to promote premature transcription termination at those genes, in other words, to induce termination within the coding sequence leading to the production of truncated, non-functional mRNAs and, thus, gene downregulation. Additional studies also implicated SETX in transcription termination at the 3′ end of some genes (40,62,64). However, the function of SETX in transcription termination is often linked to R-loop metabolism since accumulation of both RNAPIIs and R-loops is observed at termination regions upon SETX depletion. Indeed, it has been proposed that SETX would not induce termination by itself but rather favour the action of XRN2, an exonuclease that plays a general role in transcription termination at protein-coding genes. By removing R-loops at termination regions, SETX would allow the access of XRN2 to the nascent transcript, since XRN2 is presumably unable to degrade the RNA hybridised to DNA; and thereby, XRN2 could ultimately reach the RNAPII and elicit its release from the DNA (40). Hence, the precise role of SETX in transcription termination has remained unclear.
In the present study, we have used a highly purified in vitro system that has enabled us to assess the ability of the helicase domain of SETX to induce transcription termination directly and separately from its activity on R-loops because the DNA templates, which we use do not form any R-loops (53). Importantly, we have demonstrated that, akin to its yeast homologue Sen1, SETX behaves as a genuine transcription termination factor since it can directly induce the release of paused RNAPIIs and associated transcripts from the DNA (Figures 5 and 7). We therefore propose that SETX uses its ability to dislodge RNAPII from the DNA to regulate gene expression and possibly to remove RNAPIIs from TRC sites, as recently showed for its yeast counterpart (58). It also remains possible that SETX employs its transcription termination activity on RNAPIIs blocked by R-loops to promote genome stability.
Interestingly, we have observed that the helicase domain of SETX displays a clear transcription termination activity on bovine RNAPII, which is 99.9% identical to human RNAPII (80) but not on the more divergent budding yeast RNAPII (Figure 5). Similarly, Sen1-HD works much more efficiently on its cognate RNAPII, indicating that these proteins have coevolved with RNAPII to remain transcription termination factors in their respective organisms. The species-specific character of SETX/Sen1 transcription termination activity may explain the inability of SETX to complement Sen1 deficiency in S. cerevisiae (98) and provides potentially relevant information about the precise mechanisms of termination. As outlined above, to promote termination, Sen1 and likely SETX translocates along the nascent transcript until it collides with RNAPII. However, it is unclear how such a collision results in the dislodging of RNAPII from the DNA. The fact that both proteins require species-specific features of RNAPII suggests that specific interactions between Sen1/SETX and RNAPII might play an important role in termination, adding an extra layer of complexity to this process. We speculate that such protein-protein interactions might induce destabilising conformational changes in the transcription elongation complex. We hypothesise that one of the involved interaction surfaces could be located at the prong because: (i) this subdomain plays an essential role in Sen1-mediated transcription termination both in vitro and in vivo (52); (ii) its location predicts that it would be in direct contact with the polymerase upon SETX-RNAPII collision; and (iii) remarkable differences can be appreciated between Sen1 and SETX prongs. Indeed, structure-guided sequence alignment shows that the prong domain of SETX is shorter than the one of Sen1 (86 aa versus 104 aa) and there is poor sequence conservation (Supplementary Figure S8A). Whilst the central portion of the Sen1 prong (amino acids 1471–1538) is predicted to be disordered (52), the SETX structural model predicted by AplhaFold2 showed an additional large coiled-coil-like loop in the central region of the prong domain (Supplementary Figure S8B). Understanding what are the structural determinants of SETX/Sen1 species-specific transcription termination activity is an interesting matter for future investigation.
Our side-by-side comparison of the biochemical properties of SETX and Sen1 has enabled us to look at the activities of these proteins from an evolutionary perspective. We have uncovered several notable differences between the helicase domains of SETX and Sen1. Firstly, we have found that ssRNA stimulates the ATPase activity of SETX significantly more efficiently than ssDNA, which is not the case for Sen1 (Figure 2B and Supplementary Figure S3C). This is most likely due to a more dynamic interaction of SETX-HD with ssRNA than with ssDNA (Figure 2C, D), which may favour a higher exchange of SETX on the RNA substrate and, therefore, support a higher ATP hydrolysis rate. The nature of this more dynamic interaction likely depends on the type of nucleic acid contact with a non-conserved residue at the RNA entry channel of Sen1/SETX. As shown in Supplementary Figure 4C, the presence of a valine in position 1884 in SETX instead of a threonine in the equivalent site in Sen1 (T1289) promotes a more stable binding of SETX to ssDNA compared to ssRNA. Indeed, the SETX-V1884T-HD mutant exhibited nucleic acid binding kinetics that more closely (84) resembled the kinetics of Sen1-HD, which binds similarly to ssDNA and ssRNA.
In addition, our data indicate that, unlike Sen1, SETX is particularly avid for R-loops since it unwinds triplex substrates (i.e. R-loops) more efficiently than duplex substrates (i.e. RNA:DNA hybrids) and is more active on R-loops than on D-loops (Figure 4D, E and Supplementary Figure S6G, H). It is therefore possible to envisage that SETX has evolved to play a more prominent role in R-loop resolution than Sen1. Our present data do not allow us to ascertain the molecular basis of the preference of SETX for R-loop structures. It is possible that during the process of translocation this type of structure accommodates better in the helicase domain of SETX, which, together with the more efficient translocation of SETX along ssRNA, would enable robust R-loop dissolution. Structural analyses of intermediate stages of the reaction of R-loop resolution by SETX will be required to address this important question.
Although our biochemical data constitute a major progress in the comprehension of SETX molecular function, it is important to bear in mind that the properties revealed here concern the helicase domain of SETX and it remains possible that additional regions in the N-terminal domain of SETX play a role in modulating the catalytic activities of SETX through intra-molecular interactions. A side-by-side comparison of the behaviour of full-length SETX and SETX-HD will allow understanding this point.
Defects in transcription termination and R-loop metabolism have been implicated in the onset of several neurodegenerative diseases (27,85,87,89,99,100), highlighting the importance of understanding these processes. Mutations in SETX are associated with ataxia with oculomotor apraxia (AOA2) and a juvenile form of amyotrophic lateral sclerosis dubbed ALS4 (65–71). AOA2 mutations lead to SETX loss of function and are associated with transcriptional stress, R-loop accumulation, increased oxidative stress, elevated levels of double-strand breaks and genome instability, similarly to the phenotypes observed upon SETX depletion (6,71,74,85,101–103). A previous study used Sen1 as a surrogate for SETX to assess the effect of a subset of AOA2-associated mutations on SETX biochemical activities and found that the mutations analysed are associated with improper protein folding, impaired RNA binding, or reduced ATPase activity (52). However, a number of SETX disease-associated mutations do not fall into positions conserved in the yeast homologue, notably those linked to ALS4, and the properties of Sen1 and SETX present some clear divergencies, which are important limitations for using Sen1 as a model to study SETX-associated diseases.
In the present study, we report a series of methods to purify the helicase domain of SETX and assess its relevant biochemical activities, which provides an ideal system to investigate the molecular effect of SETX disease-associated mutations located at this region. To validate these tools, we have characterised one AOA2 and one ALS4 mutation (Figure 6). Specifically, we have found that the L1976R substitution (69), causing AOA2, results in protein insolubility (Supplementary Figure S7A), which explains the loss-of-function phenotypes. In contrast, we have found that the ALS4-associated substitution R2136H (66) does not significantly affect protein solubility nor any of the measurable SETX activities (Figure 6). Expression of SETX mutant versions harbouring either the L389S or the R2136H ALS4 substitutions induce ALS-like phenotypes in mice models, supporting the idea that ALS4 is provoked by toxic SETX gain of function (72). The comparison of ALS4 patient cells with control cell lines has revealed a global reduction of cellular R-loop abundance in ALS4 cells (72,87). In addition, decreased R-loop levels at the promoter of certain genes has also been observed in ALS4 cells and has been linked to the deregulation of a subset of genes (27). Our biochemical data indicate that the ALS4 substitution R2136H does not alter per se SETX catalytic properties leading to an increased R-loop resolving activity (Figure 6F and Supplementary Figure S7E), which could have explained the reduced R-loop levels observed in cellulo. However, it has been recently shown that the SETX L389S substitution impairs the interaction of SETX with the zinc finger protein ZPR1, which has been proposed to function as a regulator of SETX activity on R-loops (87). It is therefore possible that the R2136H substitution also interferes with the interaction of SETX with ZPR1 or with ZPR1-mediated regulation of SETX activity.
Alternatively, or additionally, the R2136H substitution might impact on the recently reported function of SETX in stress granules (SGs) (104). SGs are membrane-less organelles that form in the cytoplasm upon exposure to diverse stress conditions and that are highly enriched in RNAs and RNA-binding proteins such as the ALS-associated protein TDP-43 (105). Abnormal SGs have been proposed to nucleate protein inclusions that are typically observed in ALS patients and likely play a role in ALS pathogenesis (106,107). Among them, TDP-43 aggregates are the most frequently found in all ALS forms, including ALS4 (72,107). Interestingly, SETX has recently been found to associate with SGs and partake in their disassembly (104). We have observed that the R2136H version of SETX helicase domain has decreased thermostability compared to the wild type (Figure 6H, Supplementary Figure S7G), which might reflect a higher tendency of the mutant protein to aggregate under certain physicochemical conditions. We therefore speculate that SETX-R2136H might not preserve its proper protein folding and aggregate in SGs naturally forming in motor neurons, thus priming TDP-43 aggregation. The resulting toxic protein inclusions might in turn sequester important cellular factors (i.e. mRNAs and RNA-binding proteins), leading to global alterations in transcription and RNA processing that could result in the observed reduction in R-loop levels.
In conclusion, in this study we have provided direct biochemical evidence that SETX is endowed with the ability to resolve R-loops and induce transcription termination, and we have generated valuable tools to approach the molecular basis of SETX-associated neurodegenerative diseases. We anticipate that our results will enable better interpretation of the existing in vivo data and will possibly orient future studies on the biological function of SETX.
DATA AVAILABILITY
All primary data are available in this manuscript, supplementary information, and source data.
Supplementary Material
ACKNOWLEDGEMENTS
We thank members of the lab for their support. We thank Olga Jasnovidova (Tallinn University of Technology) for critically reading the manuscript. We thank Mario Spirek and Jakub Cibulka (Masaryk University) for assistance with BLI assays.
Author contributions: Experimentation: Z.H., O.P.; Protein purification: V.K., O.P.; In silico modelling: R.S.; Conceptualisation: O.P., M.S.; Funding acquisition: O.P., R.S., M.S.; Project administration: O.P., R.S., M.S.; Supervision: M.S.; Writing – original draft: Z.H., O.P., M.S.; Writing – review and editing: Z.H., V.K., O.P., R.S., M.S.
Contributor Information
Zdenka Hasanova, CEITEC-Central European Institute of Technology, Masaryk University, Brno CZ-62500, Czechia.
Veronika Klapstova, CEITEC-Central European Institute of Technology, Masaryk University, Brno CZ-62500, Czechia; National Centre for Biomolecular Research, Faculty of Science, Masaryk University, Brno CZ-62500, Czechia.
Odil Porrua, Université Paris Cité, CNRS, Institut Jacques Monod, F-75013 Paris, France; Institut de Génétique Moléculaire de Montpellier, Univ Montpellier, CNRS, Montpellier, France.
Richard Stefl, CEITEC-Central European Institute of Technology, Masaryk University, Brno CZ-62500, Czechia; National Centre for Biomolecular Research, Faculty of Science, Masaryk University, Brno CZ-62500, Czechia.
Marek Sebesta, CEITEC-Central European Institute of Technology, Masaryk University, Brno CZ-62500, Czechia.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Junior Star Grant from the Grant Agency of the Czech Republic [21-10464M to M.S.]; Fondation pour la Recherche Medicale [MND202003011479 to O.P.]; European Research Council (ERC) under the European Union′s Horizon 2020 research and innovation programme [649030 to R.S.], which supported initial experiments; CIISB, Instruct-CZ Centre of Instruct-ERIC EU consortium funded by MEYS CR infrastructure project [LM2018127] and European Regional Development Fund-Project ‘UP CIISB’ [CZ.02.1.01/0.0/0.0/18_046/0015974], which supported measurements at CEITEC Proteomics Core Facility and Biomolecular Interactions and Crystallization Core Facility, respectively. O.P. was also supported by the Centre Nationale de la Recherche Scientifique. Funding for open access charge: Grant Agency of the Czech Republic.
Conflict of interest statement. None declared.
REFERENCES
- 1. Noe Gonzalez M., Blears D., Svejstrup J.Q.. Causes and consequences of RNA polymerase II stalling during transcript elongation. Nat. Rev. Mol. Cell Biol. 2021; 22:3–21. [DOI] [PubMed] [Google Scholar]
- 2. Core L., Adelman K.. Promoter-proximal pausing of RNA polymerase II: a nexus of gene regulation. Genes Dev. 2019; 33:960–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. García-Muse T., Aguilera A.. Transcription–replication conflicts: how they occur and how they are resolved. Nat. Rev. Mol. Cell Biol. 2016; 17:553–563. [DOI] [PubMed] [Google Scholar]
- 4. García-Pichardo D., Cañas J.C., García-Rubio M.L., Gómez-González B., Rondón A.G., Aguilera A.. Histone mutants separate R loop formation from genome instability induction. Mol. Cell. 2017; 66:597–609. [DOI] [PubMed] [Google Scholar]
- 5. Sollier J., Stork C.T., García-Rubio M.L., Paulsen R.D., Aguilera A., Cimprich K.A.. Transcription-coupled nucleotide excision repair factors promote R-loop-induced genome instability. Mol. Cell. 2014; 56:777–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Cristini A., Ricci G., Britton S., Salimbeni S., Huang S.yinN., Marinello J., Calsou P., Pommier Y., Favre G., Capranico G.et al.. Dual processing of R-loops and topoisomerase I induces transcription-dependent DNA double-strand breaks. Cell Rep. 2019; 28:3167–3181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Cohen S., Puget N., Lin Y.L., Clouaire T., Aguirrebengoa M., Rocher V., Pasero P., Canitrot Y., Legube G.. Senataxin resolves RNA:DNA hybrids forming at DNA double-strand breaks to prevent translocations. Nat. Commun. 2018; 9:533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Sessa G., En G Omez-Gonz Alez B., Silva S., P Erez-Calero C., Beaurepere R., Barroso S., Martineau S., Martin C., Ehl En A., Mart Inez J.S.et al.. BRCA2 promotes DNA-RNA hybrid resolution by DDX5 helicase at DNA breaks to facilitate their repair‡. EMBO J. 2021; 40:e106018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Ortega P., Mérida-Cerro J.A., Rondón A.G., Gómez-González B., Aguilera A.. Dna-rna hybrids at dsbs interfere with repair by homologous recombination. Elife. 2021; 10:e69881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jimeno S., Balestra F.R., Huertas P.. The emerging role of RNA modifications in DNA double-strand break repair. Front Mol Biosci. 2021; 8:309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Jimeno S., Prados-Carvajal R., Fernández-Ávila M.J., Silva S., Silvestris D.A., Endara-Coll M., Rodríguez-Real G., Domingo-Prim J., Mejías-Navarro F., Romero-Franco A.et al.. ADAR-mediated RNA editing of DNA:RNA hybrids is required for DNA double strand break repair. Nat. Commun. 2021; 12:5512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Marnef A., Legube G.. R-loops as Janus-faced modulators of DNA repair. Nat. Cell Biol. 2021; 23:305–313. [DOI] [PubMed] [Google Scholar]
- 13. Chappidi N., Nascakova Z., Boleslavska B., Zellweger R., Isik E., Andrs M., Menon S., Dobrovolna J., Balbo Pogliano C., Matos J.et al.. Fork cleavage-religation cycle and active transcription mediate replication restart after fork stalling at co-transcriptional R-loops. Mol. Cell. 2020; 77:528–541. [DOI] [PubMed] [Google Scholar]
- 14. Schwab R.A., Nieminuszczy J., Shah F., Langton J., Lopez Martinez D., Liang C.C., Cohn M.A., Gibbons R.J., Deans A.J., Niedzwiedz W.. The Fanconi anemia pathway maintains genome stability by coordinating replication and transcription. Mol. Cell. 2015; 60:351–361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Takedachi A., Despras E., Scaglione S., Guérois R., Guervilly J.H., Blin M., Audebert S., Camoin L., Hasanova Z., Schertzer M.et al.. SLX4 interacts with RTEL1 to prevent transcription-mediated DNA replication perturbations. Nat. Struct. Mol. Biol. 2020; 27:438–449. [DOI] [PubMed] [Google Scholar]
- 16. Saponaro M., Kantidakis T., Mitter R., Kelly G.P., Heron M., Williams H., Söding J., Stewart A., Svejstrup J.Q.. RECQL5 controls transcript elongation and suppresses genome instability associated with transcription stress. Cell. 2014; 157:1037–1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Helmrich A., Ballarino M., Nudler E., Tora L.. Transcription-replication encounters, consequences and genomic instability. Nat. Struct. Mol. Biol. 2013; 20:412–418. [DOI] [PubMed] [Google Scholar]
- 18. Aguilera A., Gómez-González B.. Genome instability: a mechanistic view of its causes and consequences. Nat. Rev. Genet. 2008; 9:204–217. [DOI] [PubMed] [Google Scholar]
- 19. Dutta D., Shatalin K., Epshtein V., Gottesman M.E., Nudler E.. Linking RNA polymerase backtracking to genome instability in E. coli. Cell. 2011; 146:533–543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Brickner J.R., Garzon J.L., Cimprich K.A.. Walking a tightrope: the complex balancing act of R-loops in genome stability. Mol. Cell. 2022; 82:2267–2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Petermann E., Lan L., Zou L.. Sources, resolution and physiological relevance of R-loops and RNA–DNA hybrids. Nat. Rev. Mol. Cell Biol. 2022; 23:521–549. [DOI] [PubMed] [Google Scholar]
- 22. Crossley M.P., Bocek M., Cimprich K.A.. R-Loops as cellular regulators and genomic threats. Mol. Cell. 2019; 73:398–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Wu W., Bhowmick R., Vogel I., Özer Ö., Ghisays F., Thakur R.S., Sanchez de Leon E., Richter P.H., Ren L., Petrini J.H.et al.. RTEL1 suppresses G-quadruplex-associated R-loops at difficult-to-replicate loci in the human genome. Nat. Struct. Mol. Biol. 2020; 27:424–437. [DOI] [PubMed] [Google Scholar]
- 24. Li M., Xu X., Chang C.W., Zheng L., Shen B., Liu Y.. SUMO2 conjugation of PCNA facilitates chromatin remodeling to resolve transcription-replication conflicts. Nat. Commun. 2018; 9:2706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chakraborty P., Huang J.T.J., Hiom K.. DHX9 helicase promotes R-loop formation in cells with impaired RNA splicing. Nat. Commun. 2018; 9:4346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. García-Muse T., Aguilera A.. R Loops: from physiological to pathological roles. Cell. 2019; 179:604–618. [DOI] [PubMed] [Google Scholar]
- 27. Grunseich C., Wang I.X., Watts J.A., Burdick J.T., Guber R.D., Zhu Z., Bruzel A., Lanman T., Chen K., Schindler A.B.et al.. Senataxin mutation reveals how R-loops promote transcription by blocking DNA methylation at gene promoters. Mol. Cell. 2018; 69:426–437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Ginno P.A., Lott P.L., Christensen H.C., Korf I., Chédin F.. R-Loop formation is a distinctive characteristic of unmethylated human CpG island promoters. Mol. Cell. 2012; 45:814–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tan-Wong S.M., Dhir S., Proudfoot N.J.. R-Loops promote antisense transcription across the mammalian genome. Mol. Cell. 2019; 76:600–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Jimeno S., Prados-Carvajal R., Huertas P.. The role of RNA and RNA-related proteins in the regulation of DNA double strand break repair pathway choice. DNA Repair (Amst.). 2019; 81:102662. [DOI] [PubMed] [Google Scholar]
- 31. Ohle C., Tesorero R., Schermann G., Dobrev N., Sinning I., Fischer T.. Transient RNA-DNA hybrids are required for efficient double-strand break repair. Cell. 2016; 167:1001–1013. [DOI] [PubMed] [Google Scholar]
- 32. Ribeiro de Almeida C., Dhir S., Dhir A., Moghaddam A.E., Sattentau Q., Meinhart A., Proudfoot N.J.. RNA helicase DDX1 converts RNA G-quadruplex structures into R-loops to promote IgH class switch recombination. Mol. Cell. 2018; 70:650–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Niehrs C., Luke B.. Regulatory R-loops as facilitators of gene expression and genome stability. Nat. Rev. Mol. Cell Biol. 2020; 21:167–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Liu Q., Liu Y., Shi Q., Su H., Wang C., Birchler J.A., Han ·Fangpu. Emerging roles of centromeric RNAs in centromere formation and function. Genes Genomics. 2021; 43:217–226. [DOI] [PubMed] [Google Scholar]
- 35. Li X., Manley J.L.. Inactivation of the SR protein splicing factor ASF/SF2 results in genomic instability. Cell. 2005; 122:365–378. [DOI] [PubMed] [Google Scholar]
- 36. el Hage A., French S.L., Beyer A.L., Tollervey D.. Loss of topoisomerase I leads to R-loop-mediated transcriptional blocks during ribosomal RNA synthesis. Genes Dev. 2010; 24:1546–1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Tuduri S., Crabbé L., Conti C., Tourrière H., Holtgreve-Grez H., Jauch A., Pantesco V., de Vos J., Thomas A., Theillet C.et al.. Topoisomerase I suppresses genomic instability by preventing interference between replication and transcription. Nat. Cell Biol. 2009; 11:1315–1324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Bayona-Feliu A., Barroso S., Muñoz S., Aguilera A.. The SWI/SNF chromatin remodeling complex helps resolve R-loop-mediated transcription–replication conflicts. Nat. Genet. 2021; 53:1050–1063. [DOI] [PubMed] [Google Scholar]
- 39. Huertas P., Aguilera A.. Cotranscriptionally formed DNA:RNA hybrids mediate transcription elongation impairment and transcription-associated recombination. Mol. Cell. 2003; 12:711–721. [DOI] [PubMed] [Google Scholar]
- 40. Skourti-Stathaki K., Proudfoot N.J., Gromak N.. Human senataxin resolves RNA/DNA hybrids formed at transcriptional pause sites to promote Xrn2-dependent termination. Mol. Cell. 2011; 42:794–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Cristini A., Groh M., Kristiansen M.S., Gromak N.. RNA/DNA hybrid interactome identifies DXH9 as a molecular player in transcriptional termination and R-loop-associated DNA damage. Cell Rep. 2018; 23:1891–1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Wahba L., Amon J.D., Koshland D., Vuica-Ross M.. RNase H and multiple RNA biogenesis factors cooperate to prevent RNA:DNA hybrids from generating genome instability. Mol. Cell. 2011; 44:978–988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Hodroj D., Recolin B., Serhal K., Martinez S., Tsanov N., Abou Merhi R., Maiorano D.. An ATR -dependent function for the Ddx19 RNA helicase in nuclear R-loop metabolism. EMBO J. 2017; 36:1182–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Lafuente-Barquero J., Luke-Glaser S., Graf M., Silva S., Gómez-González B., Lockhart A., Lisby M., Aguilera A., Luke B.. The Smc5/6 complex regulates the yeast Mph1 helicase at RNA-DNA hybrid-mediated DNA damage. PLos Genet. 2017; 13:e1007136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Li L., Germain D.R., Poon H.-Y., Hildebrandt M.R., Monckton E.A., McDonald D., Hendzel M.J., Godbout R.. DEAD Box 1 facilitates removal of RNA and homologous recombination at DNA double-strand breaks. Mol. Cell. Biol. 2016; 36:2794–2810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Sridhara S.C., Carvalho S., Grosso A.R., Gallego-Paez L.M., Carmo-Fonseca M., de Almeida S.F.. Transcription dynamics prevent RNA-mediated genomic instability through SRPK2-dependent DDX23 phosphorylation. Cell Rep. 2017; 18:334–343. [DOI] [PubMed] [Google Scholar]
- 47. Tran P.L.T., Pohl T.J., Chen C.F., Chan A., Pott S., Zakian V.A.. PIF1 family DNA helicases suppress R-loop mediated genome instability at tRNA genes. Nat. Commun. 2017; 8:15025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lin W.-L., Chen J.-K., Wen X., He W., Zarceno G.A., Chen Y., Chen S., Paull T.T., Liu H.. DDX18 prevents R-loop-induced DNA damage and genome instability via PARP-1. Cell Rep. 2022; 40:111089. [DOI] [PubMed] [Google Scholar]
- 49. Drolet M., Phoenix P., Menzel R., Massé E., Liu L.F., Crouch R.J.. Overexpression of RNase H partially complements the growth defect of an Escherichia coli delta topA mutant: r-loop formation is a major problem in the absence of DNA topoisomerase I. Proc. Natl. Acad. Sci. U.S.A. 1995; 92:3526–3530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Han Z., Libri D., Porrua O.. Biochemical characterization of the helicase Sen1 provides new insights into the mechanisms of non-coding transcription termination. Nucleic Acids Res. 2017; 45:1355–1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Martin-Tumasz S., Brow D.A.. Saccharomyces cerevisiae sen1 helicase domain exhibits 5′- to 3′-helicase activity with a preference for translocation on DNA rather than RNA. J. Biol. Chem. 2015; 290:22880–22889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Leonaitė B., Han Z., Basquin J., Bonneau F., Libri D., Porrua O., Conti E.. Sen1 has unique structural features grafted on the architecture of the Upf1-like helicase family. EMBO J. 2017; 36:1590–1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Porrua O., Libri D.. A bacterial-like mechanism for transcription termination by the Sen1p helicase in budding yeast. Nat. Struct. Mol. Biol. 2013; 20:884–891. [DOI] [PubMed] [Google Scholar]
- 54. Alzu A., Bermejo R., Begnis M., Lucca C., Piccini D., Carotenuto W., Saponaro M., Brambati A., Cocito A., Foiani M.et al.. Senataxin associates with replication forks to protect fork integrity across RNA-polymerase-II-transcribed genes. Cell. 2012; 151:835–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Appanah R., Lones E.C., Aiello U., Libri D., de Piccoli G.. Sen1 is recruited to replication forks via Ctf4 and Mrc1 and promotes genome stability. Cell Rep. 2020; 30:2094–2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. San Martin-Alonso M., Soler-Oliva M.E., García-Rubio M., García-Muse T., Aguilera A.. Harmful R-loops are prevented via different cell cycle-specific mechanisms. Nat. Commun. 2021; 12:4451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Zardoni L., Nardini E., Brambati A., Lucca C., Choudhary R., Loperfido F., Sabbioneda S., Liberi G.. Elongating RNA polymerase II and RNA:DNA hybrids hinder fork progression and gene expression at sites of head-on replication-transcription collisions. Nucleic Acids Res. 2021; 49:12769–12784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Aiello U., Challal D., Wentzinger G., Lengronne A., Appanah R., Pasero P., Palancade B., Libri D.. Sen1 is a key regulator of transcription-driven conflicts. Mol. Cell. 2022; 82:2952–2966. [DOI] [PubMed] [Google Scholar]
- 59. Zlem Yüce O., West S.C.. Senataxin, defective in the neurodegenerative disorder ataxia with oculomotor apraxia 2, lies at the interface of transcription and the DNA damage response. Mol. Cell. Biol. 2013; 33:406–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Hatchi E., Skourti-Stathaki K., Ventz S., Pinello L., Yen A., Kamieniarz-Gdula K., Dimitrov S., Pathania S., McKinney K.M., Eaton M.L.et al.. BRCA1 recruitment to transcriptional pause sites is required for R-loop-driven DNA damage repair. Mol. Cell. 2015; 57:636–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Makharashvili N., Arora S., Yin Y., Fu Q., Wen X., Lee J.-H., Kao C.-H., Leung J.W., Miller K.M., Paull T.T.. Sae2/CtIP prevents R-loop accumulation in eukaryotic cells. Elife. 2018; 7:e42733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Yanling Zhao D., Gish G., Braunschweig U., Li Y., Ni Z., Schmitges F.W., Zhong G., Liu K., Li W., Moffat J.et al.. SMN and symmetric arginine dimethylation of RNA polymerase II C-terminal domain control termination. Nature. 2016; 529:48–53. [DOI] [PubMed] [Google Scholar]
- 63. Miller M.S., Rialdi A., Ho J.S.Y., Tilove M., Martinez-Gil L., Moshkina N.P., Peralta Z., Noel J., Melegari C., Maestre A.M.et al.. Senataxin suppresses the antiviral transcriptional response and controls viral biogenesis. Nat. Immunol. 2015; 16:485–494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Gatti V., Fierro C., Compagnone M., Banca V.la, Mauriello A., Montanaro M., Scalera S., de Nicola F., Candi E., Ricci F.et al.. ΔNp63-Senataxin circuit controls keratinocyte differentiation by promoting the transcriptional termination of epidermal genes. Proc. Natl. Acad. Sci. U.S.A. 2022; 119:e2104718119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Moreira M.C., Klur S., Watanabe M., Németh A.H., le Ber I., Moniz J.C., Tranchant C., Aubourg P., Tazir M., Schöls L.et al.. Senataxin, the ortholog of a yeast RNA helicase, is mutant in ataxia-ocular apraxia 2. Nat. Genet. 2004; 36:225–227. [DOI] [PubMed] [Google Scholar]
- 66. Chen Y.Z., Bennett C.L., Huynh H.M., Blair I.P., Puls I., Irobi J., Dierick I., Abel A., Kennerson M.L., Rabin B.A.et al.. DNA/RNA helicase gene mutations in a form of juvenile amyotrophic lateral sclerosis (ALS4). Am. J. Hum. Genetics. 2004; 74:1128–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. le Ber I., Bouslam N., Rivaud-Péchoux S., Guimarães J., Benomar A., Chamayou C., Goizet C., Moreira M.C., Klur S., Yahyaoui M.et al.. Frequency and phenotypic spectrum of ataxia with oculomotor apraxia 2: a clinical and genetic study in 18 patients. Brain. 2004; 127:759–767. [DOI] [PubMed] [Google Scholar]
- 68. Nanetti L., Cavalieri S., Pensato V., Erbetta A., Pareyson D., Panzeri M., Zorzi G., Antozzi C., Moroni I., Gellera C.et al.. SETX mutations are a frequent genetic cause of juvenile and adult onset cerebellar ataxia with neuropathy and elevated serum alpha-fetoprotein. Orphanet. J. Rare. Dis. 2013; 8:123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Datta N., Hohler A.. A new SETX mutation producing AOA2 in two siblings. Int. J. Neurosci. 2013; 123:670–673. [DOI] [PubMed] [Google Scholar]
- 70. Asaka T., Yokoji H., Ito J., Yamaguchi K., Matsushima A.. Autosomal recessive ataxia with peripheral neuropathy and elevated AFP: novel mutations in SETX. Neurology. 2006; 66:1580–1581. [DOI] [PubMed] [Google Scholar]
- 71. Anheim M., Monga B., Fleury M., Charles P., Barbot C., Salih M., Delaunoy J.P., Fritsch M., Arning L., Synofzik M.et al.. Ataxia with oculomotor apraxia type 2: clinical, biological and genotype/phenotype correlation study of a cohort of 90 patients. Brain. 2009; 132:2688–2698. [DOI] [PubMed] [Google Scholar]
- 72. Bennett C.L., Dastidar S.G., Ling S.C., Malik B., Ashe T., Wadhwa M., Miller D.B., Lee C., Mitchell M.B., van Es M.A.et al.. Senataxin mutations elicit motor neuron degeneration phenotypes and yield TDP-43 mislocalization in ALS4 mice and human patients. Acta Neuropathol. 2018; 136:425–443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Grunseich C., Patankar A., Amaya J., Watts J.A., Li D., Ramirez P., Schindler A.B., Fischbeck K.H., Cheung V.G.. Clinical and Molecular Aspects of Senataxin Mutations in Amyotrophic Lateral Sclerosis 4. Ann. Neurol. 2020; 87:547–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Kanagaraj R., Mitter R., Kantidakis T., Edwards M.M., Benitez A., Chakravarty P., Fu B., Becherel O., Yang F., Lavin M.F.et al.. Integrated genome and transcriptome analyses reveal the mechanism of genome instability in ataxia with oculomotor apraxia 2. Proc. Natl. Acad. Sci. U.S.A. 2022; 119:e2114314119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Hirano M., Quinzii C.M., Mitsumoto H., Hays A.P., Roberts J.K., Richard P., Rowland L.P.. Senataxin mutations and amyotrophic lateral sclerosis. Amyotrophic Lateral Sclerosis. 2011; 12:223–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Jumper J., Evans R., Pritzel A., Green T., Figurnov M., Ronneberger O., Tunyasuvunakool K., Bates R., Žídek A., Potapenko A.et al.. Highly accurate protein structure prediction with AlphaFold. Nature. 2021; 596:583–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Pettersen E.F., Goddard T.D., Huang C.C., Meng E.C., Couch G.S., Croll T.I., Morris J.H., Ferrin T.E.. UCSF ChimeraX: structure visualization for researchers, educators, and developers. Protein Sci. 2021; 30:70–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Porrua O., Libri D.. Characterization of the mechanisms of transcription termination by the helicase Sen1. Methods Mol. Biol. 2015; 1259:313–331. [DOI] [PubMed] [Google Scholar]
- 79. Kireeva M.L., Lubkowska L., Komissarova N., Kashlev M.. Assays and affinity purification of biotinylated and nonbiotinylated forms of double-tagged core RNA polymerase II from Saccharomyces cerevisiae. Methods Enzymol. 2003; 370:138–155. [DOI] [PubMed] [Google Scholar]
- 80. Bernecky C., Herzog F., Baumeister W., Plitzko J.M., Cramer P.. Structure of transcribing mammalian RNA polymerase II. Nature. 2016; 529:551–554. [DOI] [PubMed] [Google Scholar]
- 81. Busch A., Backofen R.. INFO-RNA - a fast approach to inverse RNA folding. Bioinformatics. 2006; 22:1823–1831. [DOI] [PubMed] [Google Scholar]
- 82. Raden M., Ali S.M., Alkhnbashi O.S., Busch A., Costa F., Davis J.A., Eggenhofer F., Gelhausen R., Georg J., Heyne S.et al.. Freiburg RNA tools: a central online resource for RNA-focused research and teaching. Nucleic Acids Res. 2018; 46:W25–W29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Busch A., Backofen R.. INFO-RNA-a server for fast inverse RNA folding satisfying sequence constraints. Nucleic Acids Res. 2007; 35:W310–W313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Chakrabarti S., Jayachandran U., Bonneau F., Fiorini F., Basquin C., Domcke S., le Hir H., Conti E.. Molecular mechanisms for the RNA-dependent ATPase activity of Upf1 and its regulation by Upf2. Mol. Cell. 2011; 41:693–703. [DOI] [PubMed] [Google Scholar]
- 85. Suraweera A., Lim Y.C., Woods R., Birrell G.W., Nasim T., Becherel O.J., Lavin M.F.. Functional role for senataxin, defective in ataxia oculomotor apraxia type 2, in transcriptional regulation. Hum. Mol. Genet. 2009; 18:3384–3396. [DOI] [PubMed] [Google Scholar]
- 86. Richard P., Feng S., Manley J.L.. A SUMO-dependent interaction between Senataxin and the exosome, disrupted in the neurodegenerative disease AOA2, targets the exosome to sites of transcription-induced DNA damage. Genes Dev. 2013; 27:2227–2232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Kannan A., Cuartas J., Gangwani P., Branzei D., Gangwani L.. Mutation in senataxin alters the mechanism of R-loop resolution in amyotrophic lateral sclerosis 4. Brain. 2022; 145:3072–3094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Abraham K.J., Khosraviani N., Chan J.N.Y., Gorthi A., Samman A., Zhao D.Y., Wang M., Bokros M., Vidya E., Ostrowski L.A.et al.. Nucleolar RNA polymerase II drives ribosome biogenesis. Nature. 2020; 585:298–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Richard P., Feng S., Tsai Y.L., Li W., Rinchetti P., Muhith U., Irizarry-Cole J., Stolz K., Sanz L.A., Hartono S.et al.. SETX (senataxin), the helicase mutated in AOA2 and ALS4, functions in autophagy regulation. Autophagy. 2020; 17:1889–1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Zatreanu D., Han Z., Mitter R., Tumini E., Williams H., Gregersen L., Dirac-Svejstrup A.B., Roma S., Stewart A., Aguilera A.et al.. Elongation factor TFIIS prevents transcription stress and R-loop accumulation to maintain genome stability. Mol. Cell. 2019; 76:57–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Chang E.Y.C., Novoa C.A., Aristizabal M.J., Coulombe Y., Segovia R., Chaturvedi R., Shen Y., Keong C., Tam A.S., Jones S.J.M.et al.. RECQ-like helicases Sgs1 and BLM regulate R-loop- associated genome instabil. J. Cell Biol. 2017; 216:3991–4005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Song C., Hotz-Wagenblatt A., Voit R., Grummt I.. SIRT7 and the DEAD-box helicase DDX21 cooperate to resolve genomic R loops and safeguard genome stability. Genes Dev. 2017; 31:1370–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Mersaoui S.Y., Yu Z., Coulombe Y., Karam M., Busatto F.F., Masson J.-Y., Richard S.. Arginine methylation of the DDX5 helicase RGG/RG motif by PRMT5 regulates resolution of RNA:DNA hybrids. EMBO J. 2019; 38:e100986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Chakraborty P., Grosse F.. Human DHX9 helicase preferentially unwinds RNA-containing displacement loops (R-loops) and G-quadruplexes. DNA Repair (Amst.). 2011; 10:654–665. [DOI] [PubMed] [Google Scholar]
- 95. Dutta A., Kwon Y., Sung P.. Biochemical analysis of RNA–DNA hybrid and R-loop unwinding via motor proteins. Methods in Molecular Biology. 2022; 2528:Humana Press Inc; 305–316. [DOI] [PubMed] [Google Scholar]
- 96. Boleslavska B., Oravetzova A., Shukla K., Nascakova Z., Ibini O.N., Hasanova Z., Andrs M., Kanagaraj R., Dobrovolna J., Janscak P.. DDX17 helicase promotes resolution of R-loop-mediated transcription–replication conflicts in human cells. Nucleic Acids Res. 2022; 50:12274–12290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Padmanabhan K., Robles M.S., Westerling T., Weitz C.J.. Feedback regulation of transcriptional termination by the mammalian circadian clock PERIOD complex. Science (1979). 2012; 337:599–602. [DOI] [PubMed] [Google Scholar]
- 98. Chen X., Müller U., Sundling K.E., Brow D.A.. Saccharomyces cerevisiae Sen1 as a model for the study of mutations in human Senataxin that elicit cerebellar ataxia. Genetics. 2014; 198:577–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Giannini M., Bayona-Feliu A., Sproviero D., Barroso S.I., Cereda C., Aguilera A.. TDP-43 mutations link Amyotrophic Lateral Sclerosis with R-loop homeostasis and R loop-mediated DNA damage. PLoS Genet. 2020; 16:e1009260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Kannan A., Bhatia K., Branzei D., Gangwani L.. Combined deficiency of Senataxin and DNA-PKcs causes DNA damage accumulation and neurodegeneration in spinal muscular atrophy. Nucleic Acids Res. 2018; 46:8326–8346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Becherel O.J., Sun J., Yeo A.J., Nayler S., Fogel B.L., Gao F., Coppola G., Criscuolo C., de Michele G., Wolvetang E.et al.. A new model to study neurodegeneration in ataxia oculomotor apraxia type 2. Hum. Mol. Genet. 2015; 24:5759–5774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. de Amicis A., Piane M., Ferrari F., Fanciulli M., Delia D., Chessa L.. Role of senataxin in DNA damage and telomeric stability. DNA Repair (Amst.). 2011; 10:199–209. [DOI] [PubMed] [Google Scholar]
- 103. Roda R.H., Rinaldi C., Singh R., Schindler A.B., Blackstone C.. Ataxia with oculomotor apraxia type 2 fibroblasts exhibit increased susceptibility to oxidative DNA damage. J. Clin. Neurosci. 2014; 21:1627–1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Marmor-Kollet H., Siany A., Kedersha N., Knafo N., Rivkin N., Danino Y.M., Moens T.G., Olender T., Sheban D., Cohen N.et al.. Spatiotemporal proteomic analysis of stress granule disassembly using APEX reveals regulation by SUMOylation and links to ALS pathogenesis. Mol. Cell. 2020; 80:876–891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Riggs C.L., Kedersha N., Ivanov P., Anderson P.. Mammalian stress granules and P bodies at a glance. J. Cell Sci. 2020; 133:jcs242487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106. Wolozin B., Ivanov P.. Stress granules and neurodegeneration. Nat. Rev. Neurosci. 2019; 20:649–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Taylor J.P., Brown R.H., Cleveland D.W.. Decoding ALS: from genes to mechanism. Nature. 2016; 539:197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All primary data are available in this manuscript, supplementary information, and source data.