


Abstract
RecA-mediated homologous recombination (HR) is a key mechanism for genome maintenance and plasticity in bacteria. It proceeds through RecA assembly into a dynamic filament on ssDNA, the presynaptic filament, which mediates DNA homology search and ordered DNA strand exchange. Here, we combined structural, single molecule and biochemical approaches to characterize the ATP-dependent assembly mechanism of the presynaptic filament of RecA from Streptococcus pneumoniae (SpRecA), in comparison to the Escherichia coli RecA (EcRecA) paradigm. EcRecA polymerization on ssDNA is assisted by the Single-Stranded DNA Binding (SSB) protein, which unwinds ssDNA secondary structures that block EcRecA nucleofilament growth. We report by direct microscopic analysis of SpRecA filamentation on ssDNA that neither of the two paralogous pneumococcal SSBs could assist the extension of SpRecA nucleopolymers. Instead, we found that the conserved RadA helicase promotes SpRecA nucleofilamentation in an ATP-dependent manner. This allowed us to solve the atomic structure of such a long native SpRecA nucleopolymer by cryoEM stabilized with ATPγS. It was found to be equivalent to the crystal structure of the EcRecA filament with a marked difference in how RecA mediates nucleotide orientation in the stretched ssDNA. Then, our results show that SpRecA and EcRecA HR activities are different, in correlation with their distinct ATP-dependent ssDNA binding modes.
INTRODUCTION
Homologous recombination (HR) is a DNA strands exchange process essential for multiple pathways of genome maintenance and plasticity in all kingdoms of life (1,2). Defects in any of these pathways lead to deleterious consequences, such as cell death or various types of cancer (3). HR relies on the pairing of a single-stranded DNA (ssDNA) molecule with one complementary strand in a double-stranded DNA (dsDNA) to generate a three-stranded DNA structure, commonly referred to as a synaptic product or a D-loop structure (for displacement-loop) (4). This reaction is catalyzed by a widespread and conserved group of enzymes, defined hereafter as HR recombinases and named RecA in bacteria, Rad51/Dmc1 in eukaryotes, RadA in archaea (distinct from the widespread bacterial RadA recombination effector (also referred to as SMS; (5). They form the RecA/Rad51 protein family, unified by a conserved ATP binding and hydrolysis core domain and a common HR mechanism (2). They promote the pairing and exchange of homologous DNA molecules by polymerizing first on a ssDNA molecule to generate the so-called presynaptic nucleofilament (6). Once assembled, the presynaptic nucleofilament scans for an homologous sequence in dsDNA and promotes ssDNA pairing with a complementary DNA sequence to generate the D-loop (7). The assembly and disassembly of RecA/Rad51 nucleofilaments is finely tuned by the binding and hydrolysis of nucleotides at the interface between monomers in the polymer. As such, RecA/Rad51 nucleofilaments are dynamic and the regulation of the DNA-dependent NTP binding and hydrolysis cycle is at the heart of the HR process.
RecA from Escherichia coli (EcRecA) is a model protein for the bacterial HR recombinases (8). Crystal structures of a chimera made of 6 or 5 fused protomers of EcRecA truncated for N-terminal (1–30) and C-terminal (336–353) residues and bound to DNA in the presence of a non-hydrolyzable ATP derivative have revealed many key features about the organization of presynaptic filament assembly and the mechanism of its pairing with a complementary ssDNA molecule (9). First, ssDNA and ATP bind to RecA-RecA interfaces cooperatively, explaining the ATP dependency for RecA polymerization on ssDNA. Second, the γ-phosphate of ATP is sensed across the RecA-RecA interface by two lysine residues that stimulate ATP hydrolysis, providing a mechanism for DNA release. Third, the nucleoprotein filament adopts a right-handed helical shape with six EcRecA monomers per turn, which stretch the ssDNA about 1.6-fold in comparison with a B-form dsDNA. Remarkably, the ssDNA is organized in the filament in regularly separated B-form triplets of nucleotides, including two bases exposed externally that restricts the homology search to Watson–Crick-type base pairing within a recipient DNA. In addition, using a chimera of 9 fused EcRecA subunits, Yang et al. recently solved by high-resolution cryoEM the structure of a D-loop assembled by this chimeric EcRecA polymer in the presence of a non-hydrolysable ATP derivative (10). In parallel to these advances in the structural organization of EcRecA nucleofilaments HR intermediates, intensive biochemical ‘ensemble’ studies along with the development of single molecule (SM) techniques using fluorescently labelled EcRecA revealed important aspects in the dynamics of its ATP-dependent polymerization on DNA at the pre-synaptic and synaptic HR steps (2,10–13). These studies highlighted a biased growth of EcRecA filaments along ssDNA from 5’ to 3’ direction in presence of ATP, and a bidirectional growth in presence of the poorly hydrolysable ATPγS analogue that stabilizes the EcRecA polymer on DNA. In addition, SM analysis provided a direct imaging of the slow initial EcRecA interaction on ssDNA prior to the rapid growth of the nucleofilament, referred to as the nucleation and extension stages, respectively (14). These SM studies also unveiled key features of the DNA homology search and subsequent pairing stages mediated by EcRecA presynaptic filament, which proceeds through an inchworm mechanism and a 3-nucleotide stepping during DNA strand exchange, respectively (9). Similar structural and SM analysis conducted on Rad51/DMC1 pointed at the conservation of EcRecA properties in eukaryotic HR recombinases, pointing at their general character of these properties to all members of the RecA/Rad51 family (15).
Another common feature of HR recombinases is the assistance of accessory factors that modulate their polymerization/depolymerization from DNA templates and/or their DNA strands exchange activities (16). These HR modulators are differentially conserved, with some being found in a whole kingdom of life and others limited to few species. They compose distinct and partially overlapping subsets of HR effectors that define specific pathways of genome maintenance and plasticity. In bacteria, a widely conserved HR effector is the SSB protein (Single-Stranded DNA Binding). SSB is firstly known as being essential to cell growth via its action at the replication forks where it protects ssDNA and assists its replication by melting out secondary structures that could form of ssDNA and impede the progression of DNA polymerases. Reconstitution of E. coli HR in vitro has highlighted three distinct roles of its cognate SSB in counteracting or assisting its DNA interacting activities (17): one is to prevent RecA nucleation if bound first on the ssDNA; a second is to promote RecA polymerization along ssDNA, by removing the secondary structures that impede filament growth (providing the same assistance as to DNA polymerases); a third is to bind to the extruded parental strand during the DNA strand exchange reaction, stabilizing the recombination product and favoring the incorporation of ssDNA (a step referred to as a DNA branch migration). Another key and conserved bacterial effector acting in these postsynaptic stages of HR is RadA, which has been found to facilitate ssDNA recombination from D-loop stuctures via interaction with RecA and by driving DNA branch migration (5,18,19). Other known HR effectors act at one or more of these steps of the HR recombinase activity cycle through static or dynamic protein-protein interactions and/or DNA remodeling activities (3).
Not all bacterial RecAs exhibit the same intrinsic activities as EcRecA. Notable examples are RecA from Deinococcus radiodurans (Dr) and from Pseudomonas aeruginosa (Pa). These two bacterial species undergo high HR rate in their natural environment to sustain their growth under extreme radiative or oxidative conditions, respectively (20,21). DrRecA is less homologous to EcRecA than PaRecA, i.e. 61% and 71% of sequence identity, respectively. Their DNA binding affinity was found higher than that of EcRecA, along with a stronger ability to displace its cognate SSB from ssDNA for PaRecA and with a reverse recombination from dsDNA to ssDNA for DrRecA. Another reported deviation to the EcRecA paradigm is RecA from Streptococcus pneumoniae (Sp, the pneumococcus), which is well known to undergo high HR rate during the process of genetic transformation induced in response to multiple stress during the state of competence (22). Previous biochemical studies comparing SpRecA and EcRecA activities highlighted two marked differences between those two HR recombinases, which share 63% of identity. First, SpRecA was found intrinsically less efficient than EcRecA in directing DNA strand exchange between a long circular ssDNA with a complementary strand in a linear duplex DNA (23) a feature shared by RecA from Bacillus subtilis (BsRecA)(24); second, SpRecA interaction with ssDNA appears to be negatively challenged in the presynaptic stages of HR and positively assisted during synapsis by any of its two cognate paralogous SSB proteins, namely SsbA and SsbB (the former being essential and involved in DNA replication and genome maintenance processes and the latter being restrictively expressed during competence and involved in the mechanism of natural transformation; (24,25)); of note, the lack of assistance by SsbA from B. subtilis to its cognate BsRecA in presynaptic stages has also been reported, while the second SsbB also encoded by B. subtilis and specifically induced during competence assists BsRecA polymerization on ssDNA in early HR steps. Altogether, what precisely determine these functional deviations of SpRecA and BsRecA in comparison with EcRecA remains enigmatic.
Here, we combined classical biochemical techniques with single-molecule and structural approaches to closely examine the DNA interacting properties of SpRecA in comparison with EcRecA. Together, these experiments highlight significant variations in the ATP-dependent dynamics and structure of the SpRecA presynaptic filament, including the lack of assistance in its elongation by any SSB protein. Unexpectedly, however, we found that the SpRadA helicase could promote such an extension of the SpRecA presynaptic filament in an ATP-dependent manner. Altogether, this detailed analysis provides important molecular insights into the distinct efficiency of SpRecA in catalyzing HR in comparison with EcRecA.
MATERIAL AND METHODS
Proteins
SpRecA, SsbA, SsbB and E. coli SSB proteins and derivatives were purified following the same procedure described previously (19). Briefly, RecA or Ssb proteins were purified as soluble and native proteins, without any tag, as recombinant species following classical overexpressed in E. coli from pT7-directed plasmids. Protein expression was induced by adding 0.5 mM IPTG (37 °C for 2 h) to exponentially grown cells. Cells were resuspended in sucrose buffer (25 mM Tris–Cl, pH 8, 25% sucrose, 1 mM EDTA, 200 μg ml−1 lysozyme) before liquid nitrogen freezing. Cell pellets were next thawed overnight on ice and suspensions were clarified by ultracentrifugation. The clarified supernatant was transiently precipitated by addition of 0.1% Polymin P. After several salt washes, the protein fractions containing RecA or Ssb proteins were resolubilized in 20 mM Tris–Cl pH 7.6, 1 M NaCl, 1 mM DTT. The protein solution was then precipitated by ammonium sulfate. After centrifugation, the supernatants containing RecA and Ssb proteins were consecutively loaded on Phenyl, Heparin and Q columns and purified by FPLC.
EcRecA was purified following the same procedure as for SpRecA. Its ATPase and DNA strand exchange activities were found to be identical to those of commercial EcRecA from NEB. Thus, we used for this study either commercial or purified EcRecA.
D-loop assay
The basic reaction solution contained 10 mM Tris–Cl (pH 7.5), 0,1 mg/ml BSA, 8% glycerol, 0,5 mM DTT or TCEP, 50 mM NaCl, 10 mM MgAc, 2 mM ATP, 10 nM of 5' Cy3 100-mer oligonucleotide (5′-TGCTTCCGGCTCGTATGTTGTGTGGAATTGTGAGCGGATAACAATTTCACACAGGAAACAGCTATGACCATGATTACGAATTCGAGCTCGGTACCCGGGG-3′) homologous to pUC18 sequence, and RecA (150–600 nM). After incubation of RecA with the ssDNA (oligonucleotide) for 10 min at 37°C, we added 5 nM of pUC18 vector into the reaction and further incubated 10 min at 37°C to allow oligonucleotide-pUC18 pairing (D-loop). The reaction was then kept on ice. The reaction was quenched (or deproteinized) with 1% SDS/10 mM EDTA (final concentrations). 0,5 μl of loading buffer (xylene cyanol in 30% glycerol) was added and reactions analysed by electrophoresis on a 1.2% agarose gel in a Tris-acetate–EDTA buffer at room temperature, 6 V/cm for 1 h in order to identify and estimate properly the amount of D-loop created. We detected the free and the bound Cy3 labelled oligonucleotides by a Fluor imager (Typhoon trio-Fuji-GE-healthcare) with an Abs/Em of 532/580 nm. Quantification of the proportion of D-loop created in this assay was performed with the Multigauge and Excel softwares.
TEM analysis
For transmission electron microscopy studies, a fraction of the reactions containing DNA-protein complexes was diluted and handled as previously described (26). For the reaction of RecA filament formation, 15 μM (nucleotides) ΦX ssDNA were first incubated with 5 μM SpRecA or EcRecA 15 min at 37°C in a buffer containing 10 mM Tris–HCl pH 7.5, 50 mM KCl, 5 mM MgCl2, 1 mM DTT and either 1.5 mM ATP or 1 mM ATPγS or 1.5 mM ATP plus 1.5 mM BeF3. In the experiments to test the assistance of RecA filament formation by SSB proteins, 0.25 μM (non-saturating amount) of either EcSSB or streptococcal SsbA or SsbB were added to the filament formation reaction 5 min after RecA. In some cases, saturating amount of EcSSB or SsbA or SsbB (1 μM) was added in the reaction. For statistical analysis of the length of filaments, 30–50 molecules were analyzed for each reaction.
Fluorescence labeling
SpRecAA488 was made by covalently modifying primary amines (lysines or N-ter) of the protein with Alexa 488-succinidimyl ester (Molecular Probes, ThermoFisher), in presence of an excess of ssDNA (M13 mp18, NEB) and ATPγS (Roche) in order to preserve both ATP and ssDNA binding surfaces of the purified recombinant protein. The free fluorescent probe was removed by a step of gel filtration chromatography (Superdex 200 Increase 10/300 GL; GE Healthcare) in the reaction buffer 50 mM Tris (pH7.5), 300mM NaCl, 1mM DTT. To remove any ATP or DNA contaminant, a final step of Anion Exchange chromatography was performed (MonoQ column GE Healthcare). RecAf was prepared as previously described (20). The ssDNA binding activity of RecA was determined by monitoring the ATP hydrolysis rate of RecA at increasing concentrations of ATP.
Production of DNA substrates
Gapped DNA substrates were prepared as described previously (27). The short fluorescent ssDNA substrate used in FCS experiments was prepared with synthetic oligonucleotides (Eurogentec) labeled either with Biotin or Alexa-488 in 5’ in order to generate a Biotin-labeled DNA strand and a fluorescently-labeled DNA strand (Sequence : Biotin-5’GCTTGCATGCCTGCAGGTCG3’; Alexa488-5’GCGGATAACAATTTCACACAGG3’) by PCR-amplification using the pUC18 plasmid as template. After PCR amplification (volume = 2 ml), the PCR reactions were loaded on Hi-Trap Streptavidin column (GE Healthcare). By addition of 60 mM NaOH, the fluorescent DNA strand is eluted, while the Biotin DNA strand retains on the column. The fluorescent ssDNA is then precipitated by Chloroform/Isopropyl alcohol, resuspended in 10 mM Tris–HCl pH 7.5; 50 mM NaCl and qunatified using a Nanodrop spectrophotometer.
Direct imaging of RecA assembly on single molecules of ssDNA
A gapped ssDNA substrate was prepared and biotinylated as described in (27). The gapped ssDNA molecules were injected into a flow cell and tethered to the surface of a coverslip via biotin-streptavidin interactions. Flow cells (4 mm × 0.4 mm × 0.07 mm) were assembled using a glass slide, a coverslip, and double-sided tape (3M Adhesive Transfer Tape 9437). Ports were drilled into the glass microscope slide, and flow was controlled using a motor-driven syringe pump (28,29). The surface of the coverslip was cleaned by the subsequent injection of 1 M NaOH for 10 min, rinsed with water and equilibrated in buffer containing 20 mM TrisOAc (pH 8.0), 20% sucrose and 50 mM DTT. The surface was then functionalized by injecting the above buffer containing 2 mg/ml biotin–BSA (Pierce) and incubated for 10 min, rinsed with buffer, equilibrated with 0.2 mg/ml streptavidin (Promega) for 10 min and then blocked with 1.5 mg/ml Roche Blocking Reagent (Roche) for 10 min. For imaging, the gapped DNA were allowed to incubate in the flow cell in the absence of flow for ∼5–15 min.
FCS measurements
Fluorescence correlation spectroscopy was performed on a custom-built setup with Pulse Interleaved Excitation (PIE) and Time Correlated Single Photon Counting (TCSPC) detection as described elsewhere (30). The FCS measurements were performed in the presence of the indicated amount of RecA proteins, 5 nM fluorescently labeled ssDNA (fluorescent probe: Alexa-488; size: 100 bases), in a buffer containing10 mM Tris–HCl, pH 7.5; BSA 0.5 mg/ml; 4 mM MgCl2; 50 mM NaCl; 0.5 mM DTT.
Equilibrium anisotropy fluorescent binding assays
Titrations to monitor the binding of RecA to ssDNA were performed by monitoring the anisotropy of fluorescence enhancement at 25°C, using a Horiba fluorescence spectrophotometer set at an excitation wavelength of 495 nm and an emission wavelength of 520 nm. Excitation and emission slits were set to a bandwidth of 10 nm. Titrations were performed in 25 mM Tris–HCl (pH 7.5), 1 mM DTT, 25mM NaCl, 2.5% glycerol, 10 mM MgCl2 and the indicated concentration of nucleotide. The Anisotropy of fluorescence values were corrected for dilution. An increased amount of RecA was added to the reaction solution containing the 25 nM of polydT of 65-mers. Data fitting using One-site-specific binding model was performed using GraphPad Prism. All equilibrium titrations were performed 3 times and the curves shown are the average of three with SEM represented.
Presynaptic and postsynaptic complex assembling
All the reaction steps were carried out at 37°C. For assembling presynaptic filaments, ΦX174 Virion single strand DNA (New England BioLabs) at 10 ATPγS g ml−1 was incubated with RadA at 50 μg ml−1 for 1 min in the reaction buffer comprising of 10 mM HEPES pH 7.5, 100 mM NaCl, 50 mM KCl, 0.5 mM DTT and 1.5 mM ATPγS; 50 μM Mg Cl2. Then, SpRecAwas added at final concentration of 200 μg ml−1 for 3.5 h at 37°C. For assembling postsynaptic filaments, Lambda double strand DNA (New England BioLabs) at 10 μg ml−1 was incubated with SpRecA at 200 μg ml−1 for 3.5 h at 37°C in the same reaction buffer. Complex formation was checked by negative stain on a CM120 electron microscope (FEI/Thermo Ficher).
Cryo-EM specimen preparation and electron microscopy data acquisition
For cryo-EM analyses, 3.5 μl of sample were deposited on glow-discharged Lacey carbon grids, blotted with filter paper to remove excess sample for 4 s, and plunge-frozen in liquid ethane using a FEI Vitrobot Mark IV (FEI/Thermo Fischer) with a blotting force of 0 in an environment with 100% humidity and 4°C temperature. Cryo-EM images were acquired on a Falcon 3 direct detector in counting mode for the presynaptic complex and in linear mode for the postsynaptic complex on a FEI Talos Arctica at 200 kV. For the presynaptic complex, a magnification of 190 000× was applied to record 40 movie frames with an exposure time of 0.8 s using a dose rate of 0.9 electrons per Å2 per frame for a total accumulated dose of 36 electrons per Å2 at a pixel size of 0.76 Å. For the postsynaptic complex, a magnification of 120,000 x was applied to record 20 movie frames with an exposure time of 1 s using a dose rate of 3 electrons per Å2 per frame, resulting in a total accumulated dose of 60 electrons per Å2 at a pixel size of 1.24 Å. The final datasets were composed of 2896 (for the presynaptic complex) and 2364 (for the postsynaptic complex) micrographs with defocus values ranging from –0.8 to –2.5 μm.
Helical reconstruction
Similar procedures were applied to the presynaptic complex and the postsynaptic complex datasets using helical reconstruction methods in RELION 2.1 (31). All frames were corrected for gain reference, binned by a factor of 2 only for the presynaptic complex, motion-corrected and dose-weighted using MOTIONCOR2 (32). Contrast transfer function (CTF) parameters were estimated by CTFind-4.1 (33).
Particles on micrographs of the presynaptic complex were picked manually in box sizes of 180 pixels and with an inter-box distance of 100 Å. Then, picked particles were classified into two-dimensional class averages to identify homogeneous subsets using a regularization value of T = 2. Selected classes were used as references for autopicking in RELION 2.1 (31). The total number of initial extracted segments (25653) was reduced to 7254 by subsequent rounds of two-dimensional classifications. After the best two-dimensional classes were selected, a first 3D reconstruction was done using featureless cylinder of 125 Å in diameter as an initial model. This was achieved by refining without imposing any helical symmetry and. This yielded a map at 7.6 Å in which helical symmetry was already apparent. The processing workflow is described in supplemental Figure 3.
Then, this map was used as reference for new autopicking on the micrographs of both complexes. The total number of extracted particles (363 828 segments for the presynaptic and 1 109 194 segments for the postsynaptic) was reduced to 188 475 and 715 954 by subsequent rounds of two-dimensional classifications. High-resolution refinements were performed in RELION’s 3D auto-refinement using the non-symmetrized map as a reference, optimizing both the helical twist (58.46° and 58.62° respectively) and rise (15.38 and 14.97 Å, respectively). The final resolution was 3.9 Å for the presynaptic complex and 3.8 Å for the postsynaptic complex, calculated with two masked half-maps refined independently, according to the gold standard Fourier shell correlation (FSC) 0.143 criterion using RELION. Local resolution, calculated with RELION with a B-factor applied of −141.9 and −153.07 respectively, retrieved a range between 3.7 and 7.7 Å. All of the densities obtained were subjected to Auto-sharpening in the Phenix software package (34).
Model building and refinement
The initial atomic model of SpRecA protomers in both presynaptic and post-synaptic complexes were generated from the crystal structure of E. coli RecA (PDB ID: 3cmw and 3cmx) by SWISS-MODEL (35). Rigid-bodies, comprising four molecules of ATPγS and the DNA, were docked into the autosharpened electron density map in UCSF-Chimera (36). The coordinates of the obtained single-chain model were modified manually using Coot and refined with repeated rounds of Phenix real-space refine function. The structure was further refined in real-space in PHENIX with secondary structure restraint (34). The atomic models were validated using the Cryo-EM validation tools of Phenix (37). Briefly, each model was firstly refined against the sharpened map (Supplemental data). To monitor the refinement of the model and avoid over-fitting, the final model was refined against one half map and tested against the other half map by calculating the Fourier Shell Correlation curves (not reported), which indicated that the refinement of the atomic coordinates did not suffer from over-fitting. The asymmetric unit is composed of three protomers. We included four protomers to show the contacts between two adjacent asymmetric units. Extending the PDB further can just be obtained by applying the symmetry operators.
RESULTS
SpRecA forms short presynaptic filaments
First, we purified SpRecA and analyzed by transmission electron microscopy (TEM) its ATP-dependent polymerizing activity on ssDNA in comparison with purified EcRecA, by using circular form of ΦX174 bacteriophage (5386 nucleotides long) as a template. SpRecA added in saturating concentration to fully cover all ssDNA molecules forms dense structures on ssDNA in the presence of ATP (Figure 1B). These structures result from SpRecA binding to ssDNA as they are not observed with naked ssDNA (Figure 1A). The same experiment performed with an ATP regenerating system did not change the result (Figure 1C). By contrast, short polymers could be detected in the presence of the poorly hydrolysable ATPγS derivative (Figure 1D). We also observed similar extended filaments in the presence of ATP and of BeF3, a Pi analogue known to notably inhibit ATP hydrolysis (Figure 1E and Supplemental Figure 1). The same TEM analysis performed with EcRecA showed that it also forms small dense structures on ssDNA (Figure 1F) in the presence of ATP. By contrast, EcRecA was able to form extended filaments in the presence of ATP together with an ATP regenerating system (Figure 1G), showing not only its binding but also its assembly along ssDNA. Furthermore, EcRecA appeared to polymerize extensively along ΦX174 ssDNA in the presence of ATPγS (Figure 1H), and for a 6-fold longer distance than SpRecA in the same conditions (mean value of 767 and 122 nm, respectively; see Figure 1I). Both recombinases could generate several filaments on the same ΦX174 ssDNA molecule in experiments performed with ATPγS, indicative of several nucleation events for both recombinases followed by their polymerization. The total length of SpRecA nucleofilaments per individual ssDNA molecule was ∼0.34 μm in the presence of ATPγS (and ∼0.47 μm in the presence of ATP and BeF3), contrasting with the ∼2.06 μm measured for EcRecA in the presence of ATPγS. Also, in these conditions, EcRecA does not fully cover the circular ssDNA template, indicating that its polymerization is blocked at some sites, most probably secondary structures that formed on ssDNA (27). Interestingly, SpRecA seems to be less efficient than EcRecA to unfold such structures, explaining why SpRecA generate more and shorter filaments on the long ssDNA template. Altogether, these results revealed a marked difference between SpRecA and EcRecA in their ability to extend their nucleofilamentation under these stabilizing conditions that block ATP hydrolysis and their release from ssDNA.
Figure 1.

TEM analysis of SpRecA and EcRecA polymerisation on ssDNA. From a to h, representative electron micrographs images of ΦX ssDNA alone (A) or incubated with SpRecA and ATP (B), SpRecA, ATP and an ATP regenerating system (C), SpRecA and ATPγS (D), SpRecA and ATP-BeF3 (E), with EcRecA and ATP (F), RecAEc, ATP and an ATP regenerating system (G), RecAEc and ATPγS (H). In (I): measured length of the filaments made with EcRecA or SpRecA in presence of ATPγS (50 molecules per experiments, n = 2). All scale bars represent 200 nm.
SpRecA polymerisation on ssDNA is not assisted by any SSB protein.
EcRecA presynaptic filamentation is well known to be assisted by its cognate SSB via its melting activity of secondary structures that form on ssDNA (38). This has been generalized to several bacterial RecA similarly studied in vitro, with the marked exceptions of SpRecA and BsRecA (17). Indeed, E. coli SSB appears to counteract the presynaptic HR step of SpRecA, as indirectly evaluated by measuring the rate of ssDNA induced SpRecA ATP hydrolysis, while still stimulating the subsequent DNA strand exchange step (25). This negative competitive effect of SSB on SpRecA interaction with ssDNA evaluated in similar ATPase assays was observed with any of the two paralogous pneumococcal SSB, SsbA and SsbB (39,40). Conversely, SsbA and SsbB behave similarly as EcSSB in stimulating EcRecA ATPase and HR activities. These earlier studies pointed at a distinct HR activity of SpRecA in comparison with EcRecA, which is differently challenged by SSB proteins. We studied by TEM this interplay between SpRecA and SSB proteins. Typical images obtained by TEM using saturating amounts of SsbA and SsbB (1 μM) incubated with ΦX174 ssDNA template are presented in Figure 2A and B, respectively, revealing an identical pattern of interaction with ssDNA. Based on what is commonly done to achieve EcRecA filament formation in vitro, a non-saturating concentration of SsbA/SsbB (0.25 μM) was added to SpRecA pre-bound to ssDNA to test their potential ability to assist SpRecA filament elongation. Addition of either of these two SSB proteins in the reaction in the presence of ATP alone or with an ATP regenerating system did not promote SpRecA polymerization but rather led to the same nucleocomplexes observed with the SSBs alone. In the same vein, addition of SsbA or SsbB to the short SpRecA filaments formed in the presence of ATPγS did not promote their elongation but led to the binding of either of these two SSBs to the ssDNA unoccupied by SpRecA (Figures 2C and E, respectively). By contrast, we confirmed that EcSSB added to EcRecA pre-incubated with ssDNA in the presence of ATPγS promotes a full coverage of the circular ssDNA molecules by EcRecA (Figure 2D). The same result has been obtained by adding EcSSB to EcRecA incubated with ssDNA in the presence of ATP and an ATP regenerating system. Furthermore, we also found that EcRecA polymerization along ssDNA could also be assisted by the two pneumococcal SSBs and, conversely, that EcSSB failed to assist SpRecA nucleofilamentation in any conditions. Altogether, these findings highlight that SpRecA ATP-dependent filamentation on ssDNA is not assisted by any SSB, highlighting a main functional deviation between SpRecA and the EcRecA paradigm and all other RecA proteins similarly studied so far.
Figure 2.

TEM analysis of SpRecA and EcRecA assembly on ssDNA in presence of SpSsbA and SpSsbB, or EcSSB. Representative electron micrographs images of ΦX ssDNA (15 μM) incubated either with saturating amount (1 μM) of SsbA (A) or SsbB (B), or pre-incubated with SpRecA and, next, incubated with non-saturating amount (0,25 μM) of SsbA (C) or SsbB (E), or pre-incubated with EcRecA and, next, incubated with non-saturating amount of EcSSB (D). All scale bars represent 200 nm.
The SpRadA HR helicase extends SpRecA polymerization on ssDNA.
We next wondered whether SpRecA nucleofilamentation on ssDNA could be assisted by another HR effector. An obvious candidate was the SpRadA protein, which we found to interact with RecA and to be a DnaB-type hexameric helicase that canonically translocates along ssDNA fueled by ATP hydrolysis in the 5’ to 3’ direction (19). We investigated by TEM analysis whether SpRadA could modulate SpRecA extension along ssDNA, by using the circular form of the M13 bacteriophage. While a few small polymers were formed in the presence of ATP in the absence of SpRadA, incubation of SpRecA with SpRadA in the presence of ATPγS promoted the formation of nucleofilaments 10-fold longer than those observed with SpRecA alone in those conditions (Figure 3A and C). Sub-stoichiometric amounts of SpRadA with respect to SpRecA concentration (1:4) were sufficient to generate these polymers. Observation by negative staining showed that these longer filaments formed by mixing SpRecA with SpRadA are comparable to the helical filaments generated by SpRecA alone, indicating that SpRadA promoted SpRecA polymerization extension along ssDNA. Next, we reproduced these experiments with SpRadAK101A point mutant, which was previously shown to be unable to hydrolyze ATP (19). This SpRadAK101A mutant, which still assembled into hexamers as wild-type protein (as visible on the EM grid; Figure 3B), was no longer able to promote the formation of long filaments when mixed with SpRecA. This result shows that ATPγS hydrolysis is necessary to extend SpRecA filamentation along ssDNA. The need for SpRadA ATPase activity supports that it might act by translocating on the ssDNA to unwind the secondary structures that impede SpRecA filament growth, but without physically blocking SpRecA assembly on ssDNA as SSB does. However, we could not exclude that SpRadA assists SpRecA filamentation by another ATP-dependent mechanism relying on their interaction. Finally, based on this TEM analysis, we could not conclude whether SpRadA is associated to these nucleofilaments.
Figure 3.

SpRadA extends SpRecA polymerization along ssDNA. (a and b) Negatively stained EM images of presynaptic filaments in presence of SpRadAFL (A) and of SpRadAK101A (B), the presynaptic filaments and SpRadA proteins are shown by black arrows and circles respectively. The insets on the right show a zoom of presynaptic filaments and SpRadAFL (A), and SpRadAK101A (B) self-assembled into ring-shaped hexamers. (C) Histogram of SpRecA presynaptic filaments average length in the presence of SpRadAFL and RadAK101A.
CryoEM analysis of SpRecA filaments assembled on ssDNA and dsDNA.
SpRecA nucleofilaments formed on long ssDNA molecules in the presence of ATPγS are too short to allow their structural analysis by cryoEM. This drawback has been overcome via the action of SpRadA (see above). Long nucleoprotein filaments formed on M13 ssDNA were deposited on cryoEM Lacey grids and visualized using a 200 keV Talos Arctica cryo-electron microscope (See Supplemental Figure 2A). We used helical reconstruction in Relion to obtain a 3.9 Å resolution map. Using a non-symmetrized 3D reconstruction, we could determine in real space that these filaments display a helical symmetry and estimate the helical parameters (see Supplemental Figure 3). These helical parameters were then imposed and refined during reconstruction to obtain the final map with 15.38 Å rise and 58.46 degrees of twist (corresponding to 6.16 subunits per turn of helix) (see methods and Table 1). The final cryoEM map displayed key structural features of SpRecA with the bulky side chains clearly visible (Supplemental Figure 2G). A homology structural model of the SpRecA was obtained using Swissmodel using EcRecA crystal structure as template. SpRecA and EcRecA proteins share 63% identity (sequence-based alignment) and we postulated that their structure should be similar in term of secondary structure elements and overall fold. This initial model was docked into the map and the SpRecA structure was entirely rebuilt in our cryoEM map using coot (41). Densities for the backbone and bases of ssDNA were clearly visible. Since the sequence of M13 ssDNA is different from one filament to another, a poly-dT (thymine) DNA molecule was built in these averaged densities. Finally, ATPγS molecules were built in the corresponding densities of the map. SpRecA model with ssDNA and ATPγS was refined against the cryoEM map using real-space refinement in Phenix (34).
Table 1.
Cryo-EM structure determination and model statistics for SpRecA-ssDNA and SpRecA-dsDNA complexes
| Data collection | SpRecA–ssDNA complex | SpRecA–dsDNA complex |
|---|---|---|
| Magnification | X190 000 | X120 000 |
| Defocus range (μm) | –0.8 to –2.5 | –0.8 to –2.5 |
| Voltage (kV) | 200 | 200 |
| Microscope | Talos | Talos |
| Camera | Falcon 3 | Falcon 3 |
| Frame exposure time (s) | 0.8 | 1 |
| # movie frames | 40 | 20 |
| Total electron dose (e−/Å−2) | 36 | 60 |
| Pixel size (Å) | 0.76 | 1.24 |
| Reconstruction | RecA–ssDNA complex | RecA–dsDNA complex |
| Boxe size (pixel) | 180 | 180 |
| Inter-box distance (Å) | 100 | 100 |
| # segments extracted | 363 828 | 1 109 194 |
| # segements after Class2D | 188 475 | 715 954 |
| Resolution (Å) | 3.9 | 3.8 |
| Map sharpening B-factor (Å-2) | –141.9 | –153.07 |
| Helical rise (Å) | 15.38 | 14.97 |
| Helical twist (°) | 58.46 | 58.62 |
| Atomic model | RecA–ssDNA complex | RecA–dsDNA complex |
| Chains | 9 | 10 |
| # unique non-hydrogen atoms | 10 381 | 10 515 |
| R.m.s.d. Length (Å) | 0.006 | 0.007 |
| R.m.s.d. Angles (°) | 0.993 | 0.963 |
| Molprobity score | 1.84 | 1.86 |
| Molprobity clashscore, all atoms | 5.10 | 6.90 |
| Ramachandran outliers (%) | 0 | 0 |
| Ramachandran allowed (%) | 10.73 | 7.85 |
| Ramachandran favored (%) | 89.27 | 92.15 |
| Rotamer outliers (%) | 0.77 | 0.77 |
| Cβ outliers (%) | 0 | 0 |
| Model versus map | RecA–ssDNA complex | RecA–dsDNA complex |
| Correlation coefficient (mask) | 0.80 | 0.84 |
| Correlation coefficient (box) | 0.51 | 0.55 |
| Correlation coefficient (peaks) | 0.25 | 0.25 |
| Correlation coefficient (volume) | 0.80 | 0.84 |
| Correlation coefficient CC for ligands | 0.80 | 0.87 |
The SpRecA nucleofilament structure assembled and stabilized on ssDNA with ATPγS was found to be globally superimposable with the crystal structure of the EcRecA-ssDNA nucleofilament (Figure 4A and C) obtained in presence of ADP–AlF4–Mg2+ (9). Each protomer binds to three nucleotides, organizing the ssDNA in triplets of a nearly B-form conformation that are separated from each other by 7.2 Å (Figure 4B and D).
Figure 4.
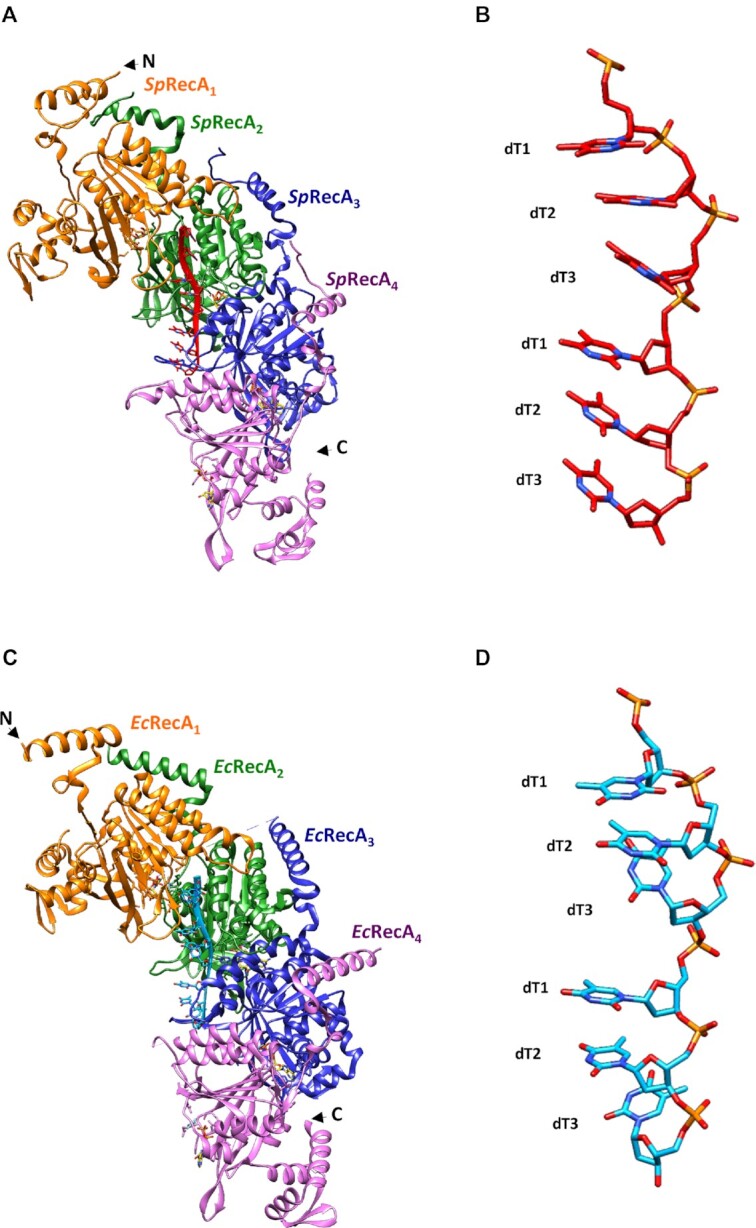
Structure comparison of the presynaptic nucleoprotein filaments from Sp and Ec. (A and C) Structure of the RecA–ATPγS–dT complex from SpRecA (A) and EcRecA (C). Four RecA protomers are numbered from the N-terminus of the first protomer to the C-terminus of the last protomer, coloured in orange, green, blue and purple respectively. A single stranded DNA (ssDNA) molecule composed of eight thymidine nucleotides bound to SpRecA and EcRecA are represented in red and blue respectively. Four ATPγS molecules are shown in gold. (B and D) Zoom on the single strand B-form DNA from S. pneumoniae (B) and E. coli (D) presynaptic filaments. The ssDNA is numbered starting with the 5′-most nucleotide in each nucleotide triplet. The ssDNA binds with a stoichiometry of exactly three nucleotides per RecA, and the repeating unit of the DNA structure is a group of three nucleotides with a 3.5–4.2 Å spacing.
The structure of each SpRecA protomer in the nucleofilament appears to be very similar to the crystal structure of the EcRecA protomer unbound to DNA (9). It is composed of a N-terminal extension (residues 9–55), a typical α/β ATPase core domain (residues 56–286) containing a canonical nucleotide binding motif and the conserved DNA interacting loops L1 and L2, and a globular C-terminal domain (residues 287–341). The charged C-terminal tail (residues 342–388) could not be resolved. In the SpRecA filament, we numbered consecutive SpRecA protomers along ssDNA in the 5’ end to the 3’ end direction. Within the nucleofilaments, the SpRecA protomers interact mostly through their ATP binding domains. In addition, the N-terminal extension of the SpRecAn protomer lies on the ATPase domain of the adjacent SpRecAn-1 protomer.
Within the SpRecA nucleofilament, the DNA binding pockets are delineated by three consecutive SpRecA protomers to accommodate a DNA triplet. In each pocket, the L1 and L2 loops of SpRecA n and SpRecAn+1 protomers play a crucial role in contacting the ssDNA. Together, they encircle the DNA backbone (Figure 5A). Residues from three consecutive SpRecA protomers contribute to ssDNA binding through hydrogen bonds. Within the phosphate backbone of each triplet of nucleotides, from 5’ to 3’ end, the first phosphate group interacts with the backbone amide groups of E210 in SpRecAn and R226 in SpRecAn+1, the second phosphate interacts with the backbone amide groups of G224 and G225 in SpRecAn+1 and the third phosphate interacts with side chains from R209 in SpRecAn+1 and S185 in SpRecAn+2 (Figure 5B). The V212 residue found in the L2 loop inserts between consecutive triplets compensating the lack of base stacking in the inter-triplet junction (Figure 5B). The ATPγS binding pocket is shared by two consecutive SpRecA protomers. In the SpRecAn protomer, the Walker A motif (residues 79–86) contacts ATPγS with conserved residues G84, K85 and T86 contacting the ATPγS phosphate groups (Supplemental Figure 5) The third ATPγS phosphate makes several hydrogen bonds with the residues K265 and K267 in the RecAn+1, stabilizing the SpRecAn/SpRecAn+1 interface. Finally, the highly conserved catalytic glutamate (E109) amongst RecA proteins is also found in the vicinity of the phosphate moieties.
Figure 5.

Interaction comparison between ssDNA and RecA protomers from from Sp and Ec. (A) Surface representation comparaison of the RecA-ATPyS-dT complex from S. pneumoniae (SpRecA) on the left and from E. coli (EcRecA) on the right. Four RecA protomers are numbered from the N-terminus of the first protomer to the C-terminus of the last protomer, coloured in different grey. A single stranded DNA (ssDNA) molecule composed of eight thimidine nucleotides bound to SpRecA and EcRecA are represented in red and blue respectively. (B and C) Zoom of the RecA-ssDNA contacts from S. pneumoniae (B) and E. coli (C) presynaptic filaments. Each nucleotide triplet is bound by three consecutive RecA protomers. RecA protomers and ssDNA are numbered and coloured as the Figure 4A. Residues V177, R182 and V212 from S. pneumoniae presynaptic filament (B) and residues M164, R169, and I199 from E. coli presynaptic filament (C) are coloured in red. (D) Superimposition of the nucleotide triplets bound to SpRecA (red) and EcRecA (blue). The two first nucleotides can be superimposed while the last nucleotide of each triplet shows a difference in orientation, represented by a dotted line of 6.5 Å long. (E) Top view of the last nucleotide of each triplet superimposed and numbered dT3. The superimposition shows a shift of 53°.
The SpRecA filament structure is in a native conformation, without protomeric fusion. Indeed, the published crystal structure of the pre- and post-synaptic EcRecA filament was obtained using a chimera of six EcRecA protomers truncated for Nter (1–30) and Cter (336–353) residues and mutated to avoid oligomerization (C117M, S118V and Q119R). This chimera was bound to ssDNA and ADP–AlF4–Mg2+ (9). In SpRecA filament, the presence of Nter or Cter regions does not modify the overall organization. When superimposed, EcRecA and SpRecA protomers have a RMSD of 1.4 Å. Similar helical parameters are found both for EcRecA and SpRecA presynaptic filaments. Structure-based alignment shows 55.86% identity between EcRecA and SpRecA sequences. Conservation of the residues is distributed across the whole structure. The ssDNA binding pocket and the L1/L2 loops are also particularly conserved between EcRecA and SpRecA. All interactions found between ssDNA and EcRecA are also found in SpRecA. The only notable differences are the V177 and V212 residues in SpRecA, which correspond to the M164 and I199 residues in EcRecA, respectively (Figure 5C). They are located at the tip of the L2 and L1 loops, respectively. These residues belonging to two consecutive RecA protomers close the L1/L2 loops around the primary ssDNA in the pre-synaptic nucleofilament and intercalate between the DNA triplet bound to RecA. However, one marked distinction stands out. Both recombinases stretch the ssDNA molecule the B-form of DNA in a non-uniform manner, and remarkably, while the third base of each triplet was found turned towards the interior of the EcRecA protein filament, the three bases of the nucleotide triplet are all aligned toward the outer surface of the SpRecA protein filament.
In parallel, we analyzed SpRecA filamentation on dsDNA and, by contrast with the ssDNA matrix, we found that SpRecA could self-assemble on dsDNA into long and stable filaments in the presence of ATPγS. We successfully analyzed their structure by cryoEM by applying a similar procedure as with filaments obtained on ssDNA. A 3.8 Å resolution map of these filaments has been obtained, in which we built and refined the structure of SpRecA bound to dsDNA and ATPγS with a helical symmetry of 14.97 Å rise and 58.62 Å twist (corresponding to 6.14 subunits per turn). The overall structure of the individual SpRecA protomer, as well as the interactions between SpRecA protomers and with the ATPγS in this filament assembled on dsDNA are identical to those characterized for the filament assembled on ssDNA (Supplemental Figure 2B). Remarkably, however, the overall dsDNA B-form structure has been notably modified by polymerization of SpRecA protomers. These were found to interact with one DNA strand as in the filament built with ssDNA. The complementary strand makes very few contacts with SpRecA protomers (Supplemental Figure 4). Like in EcRecA filaments, we propose that this strand interacts with this primary DNA strand through Watson–Crick hydrogen bonds. Interestingly, this structural organization of the dsDNA generated by SpRecA polymerization appeared to be identical to the crystal structure of the dsDNA molecule resulting from the pairing of a ssDNA strand pre-bound by EcRecA with its complementary ssDNA strand (9).
Direct imaging of SpRecA assembly on single molecules of DNA by TIRFm.
Next, we undertook the analysis of nucleofilamentation dynamics of SpRecA. To this end, we performed real-time observation of its polymerization on a single DNA molecule by Total Internal Reflection Fluorescence microscopy (TIRFm), following the same procedure previously developed for the study of EcRecA nucleofilamentation (27). We used a DNA substrate composed of a central ssDNA gap of 8155 nucleotides flanked by biotinylated dsDNA ‘handles’ of 21 080 and 24 590 bp (Figure 6A) and a fluorescently labeled SpRecA (Alexa 488, named SpRecAA488 hereafter) characterized for several activities. The purified labeled SpRecAA488 was demonstrated to be active for ssDNA binding, D-loop formation and ssDNA-dependent ATP hydrolysis with a slight defect compared to the non-labeled protein (Supplemental Figure 6). DNA molecules were then attached to the surface of a streptavidin-coated glass coverslip in a microfluidic chamber and visualized by TIRFm. We detected interaction of SpRecAA488 with DNA in the presence of ATPγS, but not with ATP, reproducing our previous TEM experiments (Figure 1D). SpRecA filament formation first appeared as a single spot in the minute range.
Figure 6.
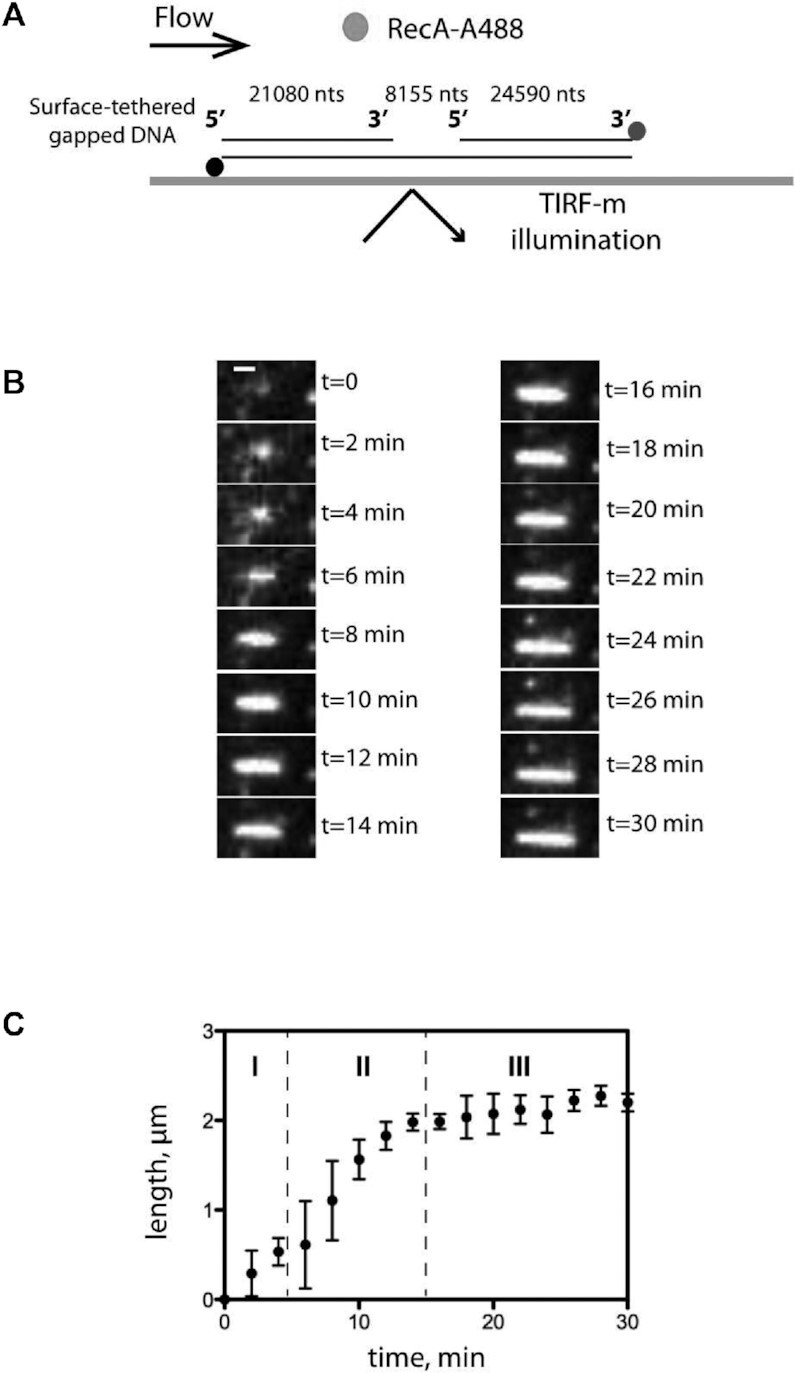
TIRFm analysis of SpRecA assembly on single molecules of ssDNA. (A) Schematic of the experimental set up combining TIRFm and microfluidics for direct imaging of SpRecAA488 filament assembly in presence of ATPγS on a single molecule of ssDNA tethered within a microfluidic flow chamber. (B) Sequential images of SpRecAA488 filament assembly in presence of ATPγS. Scale bars represent 1μm and the time interval in min (min) is indicated in the images. (C) The length of SpRecAA488 filament clusters increases linearly with time. The plots are the average of three experiments and the standard error of the mean (sem) is represented.
Then, the size of individual SpRecAA488 filaments gradually grew over time (Figure 6B) to reach a stable length after 15 min. The final filament occupies the place on the DNA molecule that is unbound by Sytox and, therefore, corresponds to the ssDNA portion. The averaged SpRecA filament growth rate measured on three individual DNA molecules showed a consistent and reproducible polymerization into three distinct stages, referred to as initiation, elongation, termination (Figure 6C). The elongation rate of SpRecA nucleofilament was 165 ± 18 nm min−1, which is similar to the elongation rate previously reported measured for EcRecA and measured on the same DNA molecule (50–500 nm min−1, Bell et al., 2012). The SpRecAA488 filament assembly eventually reached a stable and maximum length of 2.1 ± 0.1 μm, which is 10–20-fold longer than SpRecA nucleofilaments measured by TEM in the experimental conditions of TEM (compare Figure 2C and Figure 3). However, this measured length of the nucleofilament does not correspond to the maximum length of 4 μm expected for a saturating coverage of the ssDNA portion of the DNA substrate by one RecA molecule every 3 nucleotides (as demonstrated with the solved cryoEM structure of the SpRecA nucleofilament; Figure 4).
Altogether, this TIRFm analysis shows that SpRecA filament growth follows the same dynamics as that reported for EcRecA. The two recombinases appear to differ in their intrinsic capacity to overcome some secondary structures to extend along the ssDNA matrix, but not in their elongation rate along ssDNA.
SpRecA ATP-dependent ssDNA binding mode
As we were not able to detect any SpRecA assembly on DNA by TIRFm in the presence of ATP, we used Fluorescence Correlation Spectroscopy (FCS) and Fluorescence Anisotropy (FA) to measure the kinetics of formation of short SpRecA polymers on ssDNA that could not have been detected by TIRF microscopy.
FCS allows the detection of fluorescently labeled molecules that diffuse through a sub-femtoliter detection volume, giving rise to intensity fluctuations in real time and at the millisecond scale (Figure 7A) and allowing to calculate their diffusion time within the observation volume τD. We used ssDNA substrates with random sequences, labeled with Alexa 488 at the 5’ end. We tested several lengths of ssDNA substrates, i.e. 1000, 500 and 100 nucleotides long, and we were able to detect exploitable signal changes only for the small 100-mers. Upon addition of SpRecA or EcRecA and ATP, the diffusion time increased with time prior reaching a plateau that reports on the ssDNA assembly kinetics of the two recombinases (Figure 7B and C, for 250 and 400 nM of each RecA, respectively). Thus, in these conditions, we were able to detect SpRecA and EcRecA assembly on ssDNA in the presence of hydrolysable ATP and to compare their kinetics in those conditions. To this end, we measured the average half-time to reach the plateau value in each condition. This was slightly shorter for SpRecA in both conditions, i.e. 72 s for SpRecA and 112 s for EcRecA at 250 nM, and 69 s for SpRecA and 132 s for EcRecA at 400 nM. In addition, the kinetics of assembly on ssDNA appeared to be clearly different for the two recombinases. Indeed, in the very early stage of EcRecA assembly (Figure 7, blue curve, zoom), the curve showed a cooperative mode, whereas SpRecA assembly was faster and showed no cooperativity.
Figure 7.

FCS analysis of SpRecA and EcRecA assembly on single molecules of ssDNA. (A) Schematic of the experimental set up using FCS for the direct measurement of the change of diffusion time of fluorescently labeled (A488) ssDNA of 100 nucleotides length upon binding of SpRecA and EcRecA. (B and C) Averaged curve of three measurements of diffusion time at 250 nM of protein (B) showing a mean of half-time polymerization of 72.54 ± 11.85 s for SpRecA and 111.99 ± 18.24 s for EcRecA and at 400 nM of protein (C) showing a mean of half-time polymerization of 69 ± 6.0 s for SpRecA and 132, 35 ± 29.8 s for EcRecA. (D–H) Equilibrium binding of EcRecA (D) of SpRecA (E) of EcRecA in the presence of ATP. Fluorescence anisotropy (FA) variation with 1 mM ATP (black circles) and 0.1 mM ATP (red circles) with a Kd of 65 nM and 35 nM in presence of 1 mM and 0.1 mM ATP, respectively. The plots are the average of three experiments and the standard error of the mean (sem) is represented. In the presence of ATPγS (F); ATP-BeF3 (G); parameters table (Kd) obtained from the above measurements (H).
In these FCS experiments, ATP hydrolysis by both recombinases triggered by their binding to ssDNA does not impact the stability of their interaction on ssDNA during such short periods of time. Complementarily, we characterized ssDNA binding affinity of SpRecA and EcRecA protein at steady state. To this end, we measured by fluorescence anisotropy (FA) their apparent affinities constants for a short 65 nucleotides long fluorescent ssDNA molecule (T65). FA measurements were performed at 0.1 mM and 1 mM ATP, in large excess compared to the EcRecA and SpRecA concentrations used (Figure 7D and E, respectively). In those conditions, the measured apparent affinity for ssDNA (Kd) was 6- to 2-fold lower for SpRecA than for EcRecA at 0.1 mM ATP and 1 mM of ATP, respectively (Figure 7H). In addition, the maximum FA value reached –0.26 for EcRecA, whereas it was <0.2 for SpRecA, pointing at a different apparent molecular size or folding of the nucleoprotein complexes. Thus, while the FCS analysis demonstrates that the two recombinases present a nearly equivalent half-time of association on ssDNA in the presence of ATP, the FA analysis indicates that they display a different ssDNA binding mode. Notably, no difference in the binding of EcRecA to ssDNA was observed at the two ATP concentrations tested. In marked contrast, the plateau value reached for SpRecA was found to be lower at 0.1 mM than at 1 mM ATP, and this latter value was lower than the one measured for EcRecA. To test the impact of ATP hydrolysis in these differences, we reproduced these FA experiments in the presence of ATPγS or ATP-BeF3. Interestingly, in those conditions the ssDNA binding curves obtained for SpRecA were found identical whatever nucleotide concentration used, either 0.1 or 1 mM and the value of the plateau matched with EcRecA curves generated in the presence of ATP (Figures 7F and G to compare with 7D). This FA analysis showed that ATP hydrolysis modulates differently SpRecA and EcRecA interaction on ssDNA, despite both exhibit a similar ssDNA-induced ATP hydrolysis rate (Supplemental Figure 7). Altogether, these results show that SpRecA binding on ssDNA appears markedly less stable upon ATP hydrolysis, pointing at a distinct and more dynamic mode of interaction with ssDNA for SpRecA in comparison with that of EcRecA. Also, ssDNA binding activities of both RecA proteins measured by FCS and FA revealed that ATP hydrolysis impacts differently the stability of their interaction on ssDNA, while they display a similar rate of ssDNA-dependent ATP hydrolysis.
SpRecA is more efficient than EcRecA in a D-loop assay.
Then, we measured the intrinsic ATP-dependent DNA strand-exchange activity of SpRecA and EcRecA. To this end, we used the D-loop assay illustrated in Figure 8b. In this assay, a 100-nucleotides (nts) linear oligonucleotide, fluorescently labeled with Cy3 at its 5’end, was incubated with increasing amounts of SpRecA or EcRecA and mixed with a homologous supercoiled plasmid. Following protein denaturation, the fluorescent D-loop product was separated from free ssDNA by agarose gel electrophoresis and quantified. SpRecA was found to be up to three times more efficient than EcRecA (3.1% ± 1.15 versus 1.29% ± 0.57, respectively; Figure 1C). This result contrasts with a previous analysis reporting a less efficient HR activity of SpRecA in comparison with EcRecA when similarly tested alone in the presence of ATP (40). However, the HR assay used was markedly different. In this former assay depicted in Figure 8A, the HR reaction is initiated by DNA strand exchange at one end of a linear dsDNA molecule with its complementary sequence on a long circular homologous ssDNA molecule (>5000 nts) and is followed by DNA branch migration over a long distance to get the final product. By contrast, the D-loop product results from the invasive pairing between a short ssDNA molecule with its complementary sequence in a supercoiled dsDNA molecule. Thus, SpRecA and EcRecA appear to be oppositely and differently active in catalyzing the initial ssDNA pairing with a complementary sequence and in extending ssDNA recombination by DNA branch migration. Altogether, these functional divergences between these two bacterial RecA appear to stem from the different stability of their presynaptic filaments independent of the ATP hydrolysis rate.
Figure 8.

Comparison of SpRecA and EcRecA recombination activity in a D-loop assay. (A and B) Schematics of in vitro DNA strands exchange assays commonly used to measure the recombination activity of HR recombinases; the D-loop assay is depicted in (B). (C) Left: deproteinized agarose gel of the D-loop reaction performed in presence of 10 nM of cy3 oligonucleotide (100 mers) 5 nM (1 ml) of pUC18 vector, and increased amount (150–600 nM) of SpRecA or EcRecA as indicated above the gel (Sp and Ec, respectively). Right: quantification of the D-loop product generated at 600 nM of SpRecA and EcRecA concentration. The percentage of D-loop formed is given as mean values ± standard error of the mean (sem) of three reactions.
DISCUSSION
We report a comprehensive molecular study of SpRecA functional properties, which provides important insights into its DNA interaction properties in relation with its DNA strand exchange activities. This in vitro analysis shows that SpRecA markedly differs from the EcRecA paradigm in the early stages of HR. Our findings collectively concur to the conclusion that the main deviation between the two HR recombinases mostly stems from their ATP-dependent ssDNA binding mode and independently of their ATPase rates. Our studies also highlight the lack of synergy between SpRecA and its two cognate and paralogous SSB proteins in elongating its presynaptic filamentation. We found that SpRecA filamentation on ssDNA could be assisted by the conserved RadA helicase, previously known to act coordinately with RecA on postsynaptic HR intermediates (1,2). In addition, our findings support a model of HR mechanism in which the SpRecA presynaptic filament would be more efficient than EcRecA in homology search and ssDNA pairing within a recipient complementary dsDNA molecule.
Key variations in the ATP-dependent ssDNA interaction dynamics of SpRecA and EcRecA.
A central intermediate of the HR mechanism is the presynaptic filament, which is dynamically assembled and disassembled on ssDNA by ATP binding and hydrolysis between the protomers of the recombinase (3). This ssDNA-dependent ATP cycle is not uniformly conserved between bacterial RecA, leading to various lengths of presynaptic filaments (1,42). We report here that SpRecA interaction with ssDNA in the presence of ATP evaluated by FA analysis is more dynamic than that of EcRecA (Figure 7). These results are consistent with previous studies showing in SpRecA biochemical assays an enhanced ssDNA binding and DNA strand exchange activities in the presence of dATP or ATPγS instead of ATP (42,48). This finding indicates that SpRecA forms shorter nucleofilaments than EcRecA, as further supported by TEM analysis (Figure 1). However, this marked difference between the two recombinases is not due to a different affinity for ATP, nor to a different ssDNA-dependent ATP hydrolysis rate, nor to a different kinetic in ATP-dependent interaction with ssDNA (Supplemental figure 7) (48). Thus, a possible cause of the limited extension of the presynaptic filament of SpRecA would be a lower binding stability of its protomers on ssDNA. Within the HR presynaptic filament, each protomer interacting with ssDNA is further stabilized via interaction with two adjacent protomers through ATP binding (43). Upon ATP hydrolysis, protomers located at the tips of the filament are less stably bound to ssDNA, as they are engaged in only one interaction with an adjacent protomer. Thus, it has been shown by biochemical and SM analysis that the ATP-EcRecA filament mainly disassembles at the 5’ side and grows in the 3’ direction of the ssDNA (44). In direct line with such a polymerization dynamic, the 5’ terminal protomer of the SpRecA filament might be less stable on ssDNA upon ATP hydrolysis than in the case of the EcRecA filament. In addition, the release of Pi from the ADP-Pi product will change the interactions between the two protomers, which will alter their interaction with ssDNA. Thus, a possible source of difference between SpRecA and EcRecA impacting the length of their presynaptic filaments would be the ADP.Pi release and/or ATP turnover at the interface of two protomers bound to ssDNA.
Furthermore, even under stabilizing conditions that restrain ATP hydrolysis (with the use of ATPγS or by adding BeF3 to ATP), SpRecA appears less prone than EcRecA to elongate on ssDNA (Figure 1). We interpret this difference as a lower capability of SpRecA to melt ssDNA secondary structures in comparison with EcRecA, limiting differentially their filament growth. The longer SpRecA presynaptic filaments observed in TIRFm than in TEM experiments supports this proposal (Figures 2 and 3). SM observation of SpRecA filamentation by TIRFm is performed in real time in a microfluidic chamber on an attached DNA molecule and under flux, which will physically extend the ssDNA and limit its self-pairing into secondary structures. As a result, SpRecA could extend its polymerization, stabilized by limiting ATP hydrolysis, on a longer distance than on an unstretched ssDNA molecule as in the TEM experiments.
Another distinct ssDNA interaction property between these two recombinases has been uncovered from the characterization by cryoEM of the SpRecA nucleofilament structure stabilized by ATPγS. This structure is superimposed to a large extent on the EcRecA filament resolved by crystallization (9). In both SpRecA and EcRecA nucleofilaments, the ssDNA molecule is bound into an identical helical and extended conformation organized in triplets of nucleotides. However, the 3 bases of each triplet of nucleotides are fully exposed toward the exterior in the SpRecA filament, contrasting with the EcRecA filament where the third base of each nucleotide triplet is flipped inward (9). These characteristics suggest that the ssDNA conformation in the SpRecA filament is potentially more favorable for the homology search.
Lack of SSB assistance in the extension of SpRecA presynaptic filamentation.
One of the key roles of SSB in the early HR steps is to assist presynaptic filament extension by melting out ssDNA secondary structures (17). This interplay originally characterized between RecA and SSB of E. coli has been generalized to RecA of many other species, with some exceptions such as for SpRecA or B. subtilis RecA (45).Indeed, either of the two pneumococcal SsbA and SsbB proteins, or EcSSB were found to inhibit SpRecA binding to ssDNA, as deduced from their inhibition of the ssDNA dependent SpRecA ATPase activity (25). Here, we directly observed by TEM analysis that any of these SSB outcompetes ATP-dependent SpRecA polymerization on long ssDNA molecules. Furthermore, their addition to the short SpRecA filaments stabilized by ATPγS simply conduct to their binding on ssDNA portions unbound by SpRecA,without promoting the extension of SpRecA nucleofilaments as in the case of EcRecA (Figure 2). This result firmly demonstrated the lack of assistance by any SSB in elongating SpRecA polymers on ssDNA. They also indicate that the inhibition by SSB proteins of the ATP-dependent SpRecA interaction on ssDNA is the result of a more stable binding of SSB in comparison to the highly dynamic binding of SpRecA, leading to a full occupancy of the ssDNA by SSB in these conditions. This also shows that SSB can bind to ssDNA parts that are inaccessible to SpRecA and inferred to be secondary structures.
An elegant genetic screen of EcRecA mutants more efficient in conjugational recombination resulted in the selection of several point mutants that were all found to exhibit in vitro a greater persistence on ssDNA and a more efficient displacement of SSB than wild type EcRecA (46). The EcRecA region randomly mutated in this screening corresponds to the large N-ter region involved in RecA subunit-subunit interaction. This shows that modulations in this interacting interface could impact the intrinsic ssDNA interacting and polymerizing property of RecA on ssDNA. However, comparison of this interaction surface between SpRecA and EcRecA could not highlight a particular difference that would explain the lower persistence of SpRecA on ssDNA that we report here (Supplemental Figure 8). In addition, other subtle variations between RecA proteins might influence their intrinsic stability on ssDNA. Indeed, another possible source of variation could be the residues engaged in direct interaction with ssDNA, as we found here in the structure of the presynaptic filaments of SpRecA in comparison with EcRecA (see above). However, further studies are needed to establish whether this different organization of the presynaptic filament modifies their dynamism.
An unprecedented role of the RadA HR effector in extending RecA presynaptic filamentation.
The less stable ssDNA binding in SpRecA filament leads to their limited extension, which is impeded by SSB proteins or ssDNA secondary structures and unfavorable for the branch migration step in HR reaction (25). SSB proteins are well known effectors that assist RecA dynamics and filament length (47). For E. coli, Pseudomonas aeruginosa, Neisseria gonorraheae, Herbaspirillum seropedicae or Bacillus subtilis (Bs) RecA proteins, SSB proteins remove structures in ssDNA to facilitate formation of EcRecA nucleoprotein filaments on ssDNA (48). In the experimental conditions tested here, SpSsbA or SpSsbB protein improve only very slightly or compete with the SpRecA filament extension. Like for DrRecA, SpRecA ATP hydrolysis is inhibited by SpSsbA or EcSSB. So, regarding SSB proteins, SpRecA showed a distinct behavior shared with DrRecA. In contrast, SpRadA helicase enhanced the SpRecA filament extension. It does so without co-polymerizing with it. The use of the ATP hydrolysis mutant of SpRadA (SpRadAK101A) showed that the ATP hydrolysis activity of SpRadA is required to enhance SpRecA filament extension. This strongly suggests that helicase activity of SpRadA could remove ssDNA secondary structures to help SpRecA extension. Interestingly, RecA filament growth is well known to proceeds from 5’ to 3’ on ssDNA, which is also the translocation directionality of SpRadA when acting as helicase (19). Thus, SpRadA not only acts in HR mechanism at the post-synaptic step by promoting DNA branch migration (5,18,19) but also at the presynaptic step, by relieving the stem-loop structures that form on ssDNA and that impede RecA polymerization. Interestingly, SpRecA is markedly inefficient in directing these two HR steps by itself (this study; (25)). By marked contrast, we found that SpRecA is intrinsically highly efficient in promoting homologous ssDNA pairing in dsDNA template, even more than the EcRecA paradigm (Figure 8C).
Altogether, this detailed structural and biochemical analysis of ATP-dependent DNA interacting properties of SpRecA points at their balanced intrinsic efficiency by comparison with the EcRecA paradigm. Also, these activities could be differently compensated by accessory effectors. These key variations on the conserved RecA-directed HR mechanism points at its adaptation amongst bacterial species, which could reflect specific needs and/or its particular integration with other processes at work on their genome.
DATA AVAILABILITY
The cryo-EM densities of RecA nucleofilaments on ssDNA and dsDNA have been deposited in the Electron Microscopy Data Bank under ID codes EMD-15524 and EMD-15525 respectively. The models for RecA nucleofilaments on ssDNA and dsDNA have been deposited in the PDB under ID codes PDB 8AMD and 8AMF respectively. Raw cryo-EM data are available on request.
Supplementary Material
Notes
Present address: Unité de biologie Moléculaire, Cellulaire et du Développement (UMR 5077) Centre de Biologie Intégrative; 169, avenue Marianne Grunberg-Manago, CNRS - Université Paul Sabatier - Bât 4R4, 118, route de Narbonne 31062, Toulouse cedex 09, France
Contributor Information
Maud Hertzog, Laboratoire de Microbiologie et de Génétique Moléculaire (UMR 5100). Centre de Biologie Intégrative; 169, avenue Marianne Grunberg-Manago; CNRS - Université Paul Sabatier - Bât 4R4, 118, route de Narbonne; 31062 Toulouse cedex 09, France.
Thomas Noé Perry, Structure and Function of Bacterial Nanomachines – Institut Européen de Chimie et Biologie, Microbiologie fondamentale et pathogénicité, UMR 5234, CNRS, University of Bordeaux, 2 rue Robert Escarpit, 33600 Pessac, France.
Pauline Dupaigne, Genome Maintenance and Molecular Microscopy UMR 9019 CNRS, Université Paris-Saclay, Gustave Roussy, F-94805 Villejuif Cedex, France.
Sandra Serres, Laboratoire de Microbiologie et de Génétique Moléculaire (UMR 5100). Centre de Biologie Intégrative; 169, avenue Marianne Grunberg-Manago; CNRS - Université Paul Sabatier - Bât 4R4, 118, route de Narbonne; 31062 Toulouse cedex 09, France.
Violette Morales, Laboratoire de Microbiologie et de Génétique Moléculaire (UMR 5100). Centre de Biologie Intégrative; 169, avenue Marianne Grunberg-Manago; CNRS - Université Paul Sabatier - Bât 4R4, 118, route de Narbonne; 31062 Toulouse cedex 09, France.
Anne-Lise Soulet, Laboratoire de Microbiologie et de Génétique Moléculaire (UMR 5100). Centre de Biologie Intégrative; 169, avenue Marianne Grunberg-Manago; CNRS - Université Paul Sabatier - Bât 4R4, 118, route de Narbonne; 31062 Toulouse cedex 09, France.
Jason C Bell, 10x Genomics, Inc., Pleasanton, CA, USA.
Emmanuel Margeat, CBS (Centre de Biologie Structurale), Univ Montpellier, CNRS, INSERM, Montpellier, France.
Stephen C Kowalczykowski, Department of Microbiology and Molecular Genetics and Department of Molecular and Cellular Biology, University of California, Davis, CA 95616, USA.
Eric Le Cam, Genome Maintenance and Molecular Microscopy UMR 9019 CNRS, Université Paris-Saclay, Gustave Roussy, F-94805 Villejuif Cedex, France.
Rémi Fronzes, Structure and Function of Bacterial Nanomachines – Institut Européen de Chimie et Biologie, Microbiologie fondamentale et pathogénicité, UMR 5234, CNRS, University of Bordeaux, 2 rue Robert Escarpit, 33600 Pessac, France.
Patrice Polard, Laboratoire de Microbiologie et de Génétique Moléculaire (UMR 5100). Centre de Biologie Intégrative; 169, avenue Marianne Grunberg-Manago; CNRS - Université Paul Sabatier - Bât 4R4, 118, route de Narbonne; 31062 Toulouse cedex 09, France.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Centre National de la Recherche Scientifique, University Paul Sabatier; Agence Nationale de la Recherche [ANR-10-BLAN-1331]; IdeX Toulouse funding for Emergence with the SMART project ‘Single Molecule Analysis of Homologous Recombination’ attributed to M.H. and for Equipment with the ‘Go ahead in life sciences in Toulouse’ project; Fondation pour la Recherche Médicale (FRM) [ING20150532556] for the salary of S.S.; European Research Council (ERC) consolidator grant TransfoPneumo (725554) attributed to R.F.; S.C.K. was supported by grants from NIH (GM62653, GM64745, and R35 GM131900) and DOD-CDMRP (BC171869); France Bio Imaging (FBI) funding attributed to E.M. & M.H.; We thank Chantal Prevost for helpful discussions. Funding for open access charge: ERC PneumoTransfo.
Conflict of interest statement. None declared.
REFERENCES
- 1. Cox M.M. Regulation of bacterial RecA protein function. Crit. Rev. Biochem. Mol. Biol. 2007; 42:41–63. [DOI] [PubMed] [Google Scholar]
- 2. Bell J.C., Kowalczykowski S.C.. Mechanics and single-molecule interrogation of DNA recombination. Annu. Rev. Biochem. 2016; 85:193–226. [DOI] [PubMed] [Google Scholar]
- 3. Liu J., Ehmsen K.T., Heyer W.-D., Morrical S.W.. Presynaptic filament dynamics in homologous recombination and DNA repair. Crit. Rev. Biochem. Mol. Biol. 2011; 46:240–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Michel B., Leach D.. Homologous recombination—enzymes and pathways. EcoSal Plus. 2012; 5: 10.1128/ecosalplus.7.2.7. [DOI] [PubMed] [Google Scholar]
- 5. Cooper D.L., Lovett S.T.. Recombinational branch migration by the RadA/Sms paralog of RecA in Escherichia coli. Elife. 2016; 5:e10807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kowalczykowski S.C. An overview of the molecular mechanisms of recombinational DNA repair. Cold Spring Harb. Perspect. Biol. 2015; 7:a016410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Renkawitz J., Lademann C.A., Jentsch S.. Mechanisms and principles of homology search during recombination. Nat. Rev. Mol. Cell Biol. 2014; 15:369–383. [DOI] [PubMed] [Google Scholar]
- 8. Del Val E., Nasser W., Abaibou H., Reverchon S.. RecA and DNA recombination: a review of molecular mechanisms. Biochem. Soc. Trans. 2019; 47:1511–1531. [DOI] [PubMed] [Google Scholar]
- 9. Chen Z., Yang H., Pavletich N.P.. Mechanism of homologous recombination from the RecA-ssDNA/dsDNA structures. Nature. 2008; 453:489–484. [DOI] [PubMed] [Google Scholar]
- 10. Yang H., Zhou C., Dhar A., Pavletich N.P.. Mechanism of strand exchange from RecA-DNA synaptic and D-loop structures. Nature. 2020; 586:801–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lin Y.H., Chu C.C., Fan H.F., Wang P.Y., Cox M.M., Li H.W.. A 5'-to-3' strand exchange polarity is intrinsic to RecA nucleoprotein filaments in the absence of ATP hydrolysis. Nucleic Acids Res. 2019; 47:5126–5140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Konforti B.B., Davis R.W.. The preference for a 3' homologous end is intrinsic to RecA-promoted strand exchange. J. Biol. Chem. 1990; 265:6916–6920. [PubMed] [Google Scholar]
- 13. Konforti B.B., Davis R.W.. ATP hydrolysis and the displaced strand are two factors that determine the polarity of RecA-promoted DNA strand exchange. J. Mol. Biol. 1992; 227:38–53. [DOI] [PubMed] [Google Scholar]
- 14. Joo C., McKinney S.A., Nakamura M., Rasnik I., Myong S., Ha T.. Real-time observation of RecA filament dynamics with single monomer resolution. Cell. 2006; 126:515–527. [DOI] [PubMed] [Google Scholar]
- 15. Lee J.Y., Terakawa T., Qi Z., Steinfeld J.B., Redding S., Kwon Y., Gaines W.A., Zhao W., Sung P., Greene E.C.. DNA recom. Base triplet stepping by the Rad51/RecA family of recombinases. Science. 2015; 349:977–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Antony E., Lohman T.M.. Dynamics of E. coli single stranded DNA binding (SSB) protein-DNA complexes. Semin. Cell Dev. Biol. 2019; 86:102–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Bianco P.R. The tale of SSB. Prog. Biophys. Mol. Biol. 2017; 127:111–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Torres R., Serrano E., Alonso J.C.. Bacillus subtilis RecA interacts with and loads RadA/Sms to unwind recombination intermediates during natural chromosomal transformation. Nucleic Acids Res. 2019; 47:9198–9215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Marie L., Rapisarda C., Morales V., Bergé M., Perry T., Soulet A.-L., Gruget C., Remaut H., Fronzes R., Polard P.. Bacterial RadA is a DnaB-type helicase interacting with RecA to promote bidirectional D-loop extension. Nat. Commun. 2017; 8:15638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Baitin D.M., Bakhlanova I.V., Kil Y.V., Cox M.M., Lanzov V.A.. Distinguishing characteristics of hyperrecombinogenic RecA protein from Pseudomonas aeruginosa acting in Escherichia coli. J. Bacteriol. 2006; 188:5812–5820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cox M.M., Battista J.R.. Deinococcus radiodurans - the consummate survivor. Nat. Rev. Microbiol. 2005; 3:882–892. [DOI] [PubMed] [Google Scholar]
- 22. Johnston C., Martin B., Fichant G., Polard P., Claverys J.-P.. Bacterial transformation: distribution, shared mechanisms and divergent control. Nat. Rev. Microbiol. 2014; 12:181–196. [DOI] [PubMed] [Google Scholar]
- 23. Steffen S.E., Bryant F.R.. Purification and characterization of the single-stranded DNA binding protein from Streptococcus pneumoniae. Arch. Biochem. Biophys. 2001; 388:165–170. [DOI] [PubMed] [Google Scholar]
- 24. Attaiech L., Olivier A., Mortier-Barrière I., Soulet A.-L., Granadel C., Martin B., Polard P., Claverys J.-P.. Role of the single-stranded DNA-binding protein SsbB in pneumococcal transformation: maintenance of a reservoir for genetic plasticity. PLoS Genet. 2011; 7:e1002156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Grove D.E., Bryant F.R.. Effect of Mg2+ on the DNA binding modes of the Streptococcus pneumoniae SsbA and SsbB proteins. J. Biol. Chem. 2006; 281:2087–2094. [DOI] [PubMed] [Google Scholar]
- 26. Dupaigne P., Le Breton C., Fabre F., Gangloff S., Le Cam E., Veaute X.. The Srs2 helicase activity is stimulated by Rad51 filaments on dsDNA: implications for crossover incidence during mitotic recombination. Mol. Cell. 2008; 29:243–254. [DOI] [PubMed] [Google Scholar]
- 27. Bell J.C., Plank J.L., Dombrowski C.C., Kowalczykowski S.C.. Direct imaging of RecA nucleation and growth on single molecules of SSB-coated ssDNA. Nature. 2012; 491:274–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Amitani I., Liu B., Dombrowski C.C., Baskin R.J., Kowalczykowski S.C.. Watching individual proteins acting on single molecules of DNA. Methods Enzymol. 2010; 472:261–291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Forget A.L., Kowalczykowski S.C.. Single-molecule imaging of DNA pairing by RecA reveals a 3-dimensional homology search. Nature. 2012; 482:423–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Olofsson L., Margeat E.. Pulsed interleaved excitation fluorescence spectroscopy with a supercontinuum source. Opt. Express. 2013; 21:3370–3378. [DOI] [PubMed] [Google Scholar]
- 31. Scheres S.H. RELION: implementation of a Bayesian approach to cryo-EM structure determination. J. Struct. Biol. 2012; 180:519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Zheng S.Q., Palovcak E., Armache J.-P., Verba K.A., Cheng Y., Agard D.A.. MotionCor2 - anisotropic correction of beam-induced motion for improved cryo-electron microscopy. Nat Methods. 2017; 14:331–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Rohou A., Grigorieff N.. CTFFIND4: fast and accurate defocus estimation from electron micrographs. J. Struct. Biol. 2015; 192:216–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Afonine P.V., Poon B.K., Read R.J., Sobolev O.V., Terwilliger T.C., Urzhumtsev A., Adams P.D.. Real-space refinement in PHENIX for cryo-EM and crystallography. Acta Cryst. D. 2018; 74:531–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schwede T., Kopp J., Guex N., Peitsch M.C.. SWISS-MODEL: an automated protein homology-modeling server. Nucleic Acids Res. 2003; 31:3381–3385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E.. UCSF chimera? A visualization system for exploratory research and analysis. J. Comput. Chem. 2004; 25:1605–1612. [DOI] [PubMed] [Google Scholar]
- 37. Adams P.D., Afonine P.V., Bunkóczi G., Chen V.B., Davis I.W., Echols N., Headd J.J., Hung L.-W., Kapral G.J., Grosse-Kunstleve R.W.et al.. PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 2010; 66:213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kowalczykowski S.C., Clow J., Somani R., Varghese A.. Effects of the Escherichia coli SSB protein on the binding of Escherichia coli RecA protein to single-stranded DNA. Demonstration of competitive binding and the lack of a specific protein-protein interaction. J. Mol. Biol. 1987; 193:81–95. [DOI] [PubMed] [Google Scholar]
- 39. Steffen S.E., Katz F.S., Bryant F.R.. Complete inhibition of Streptococcus pneumoniae RecA protein-catalyzed ATP hydrolysis by single-stranded DNA-binding protein (SSB protein): implications for the mechanism of SSB protein-stimulated DNA strand exchange. J. Biol. Chem. 2002; 277:14493–500. [DOI] [PubMed] [Google Scholar]
- 40. Grove D.E., Anne G., Hedayati M.A., Bryant F.R.. Stimulation of the Streptococcus pneumoniae RecA protein-promoted three-strand exchange reaction by the competence-specific SsbB protein. Biochem. Biophys Res. Commun. 2012; 424:40–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Emsley P., Lohkamp B., Scott W.G., Cowtan K.. Features and development of Coot. Acta Crystallogr. D Biol. Crystallogr. 2010; 66:486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Morrical S.W. DNA-pairing and annealing processes in homologous recombination and homology-directed repair. Cold Spring Harb. Perspect. Biol. 2015; 7:a016444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Nayak S., Bryant F.R.. Kinetics of the ATP and dATP-mediated formation of a functionally-active RecA-ssDNA complex. Biochem. Biophys Res. Commun. 2015; 463:1257–1261. [DOI] [PubMed] [Google Scholar]
- 44. Bell J.C., Kowalczykowski S.C.. RecA: regulation and mechanism of a molecular search engine. Trends Biochem. Sci. 2016; 41:491–507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Steffen S.E., Bryant F.R.. Reevaluation of the nucleotide cofactor specificity of the RecA protein from Bacillus subtilis. J. Biol. Chem. 1999; 274:25990–25994. [DOI] [PubMed] [Google Scholar]
- 46. Kim T., Chitteni-Pattu S., Cox B.L., Wood E.A., Sandler S.J., Cox M.M.. Directed evolution of RecA variants with enhanced capacity for conjugational recombination. PLoS Genet. 2015; 11:e1005278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Roy R., Kozlov A.G., Lohman T.M., Ha T.. SSB protein diffusion on single-stranded DNA stimulates RecA filament formation. Nature. 2009; 461:1092–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Gruenig M.C., Stohl E.A., Chitteni-Pattu S., Seifert H.S., Cox M.M.. Less is more: Neisseria gonorrhoeae RecX protein stimulates recombination by inhibiting RecA. J. Biol. Chem. 2010; 285:37188–37197. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The cryo-EM densities of RecA nucleofilaments on ssDNA and dsDNA have been deposited in the Electron Microscopy Data Bank under ID codes EMD-15524 and EMD-15525 respectively. The models for RecA nucleofilaments on ssDNA and dsDNA have been deposited in the PDB under ID codes PDB 8AMD and 8AMF respectively. Raw cryo-EM data are available on request.