

Abstract
Although the mechanisms underlying dystonia are largely unknown, dystonia is often associated with abnormal dopamine neurotransmission. DOPA-responsive dystonia (DRD) is a prototype disorder for understanding dopamine dysfunction in dystonia because it is caused by mutationsin genes necessary for the synthesis of dopamine and alleviated by the indirect-acting dopamine agonist l-DOPA. Although adaptations in striatal dopamine receptor-mediated intracellular signaling have been studied extensively in models of Parkinson’s disease, another movement disorders associated with dopamine deficiency, little is known about dopaminergic adaptations in dystonia. To identify the dopamine receptor-mediated intracellular signaling associated with dystonia, we used immunohistochemistry to quantify striatal protein kinase A activity and extracellular signal-related kinase (ERK) phosphorylation after dopaminergic challenges in a knockin mouse model of DRD. l-DOPA treatment induced the phosphorylation of both protein kinase A substrates and ERK largely in D1 dopamine receptor-expressing striatal neurons. As expected, this response was blocked by pretreatment with the D1 dopamine receptor antagonist SCH23390. The D2 dopamine receptor antagonist raclopride also significantly reduced the phosphorylation of ERK; this contrasts with models of parkinsonism in which l-DOPA-induced ERK phosphorylation is not mediated by D2 dopamine receptors. Further, the dysregulated signaling was dependent on striatal subdomains whereby ERK phosphorylation was largely confined to dorsomedial (associative) striatum while the dorsolateral (sensorimotor) striatum was unresponsive. This complex interaction between striatal functional domains and dysregulated dopamine-receptor mediated responses has not been observed in other models of dopamine deficiency, such as parkinsonism, suggesting that regional variation in dopamine-mediated neurotransmission may be a hallmark of dystonia.
Keywords: protein kinase A, extracellular signal-related kinase, dystonia, dopamine, striatum
Dystonia is characterized by involuntary muscle contractions that cause twisting movements and postures. Although the mechanisms underlying dystonia are largely unknown, dystonia is often associated with abnormal dopamine neurotransmission. Dystonia is a prominent feature of inherited disorders that disrupt dopamine neurotransmission such as DOPA-responsive dystonia (DRD), vesicular monoamine transporter 2 deficiency and dopamine transporter deficiency. Dystonia may occur in degenerative disorders that affect dopamine neurons, such as Parkinson’s disease or in response to therapy with dopamine receptor antagonists (Jinnah & Sun, 2019; Ribot et al., 2019). Further, dopamine neurotransmission is abnormal in idiopathic forms of dystonia that are not overtly associated with dopaminergic etiologies, such as writer’s cramp and spasmodic dysphonia (Berman et al., 2013; Simonyan et al., 2013).
DRD is a prototype disorder for understanding dopamine dysfunction in dystonia because it is caused by mutations in genes necessary for the synthesis of dopamine, including GTP cyclohydrolase 1 and tyrosine hydroxylase (Ichinose et al., 1994; Knappskog et al., 1995; Thony & Blau, 2006; van den Heuvel et al., 1998). Consequently, striatal dopamine concentrations in patients are reduced to <20% of normal (Furukawa et al., 1996; Ichinose et al., 1995; Ichinose et al., 1994), which is similar in magnitude to the dopamine deficiency associated with the onset of motor signs in Parkinson’s disease (Cheng et al., 2010), suggesting that dopamine concentrations alone are not predictive of the phenotypic outcome. It is instead likely that (mal)adaptive postsynaptic signaling contributes to the features of dystonia.
Adaptations in striatal dopamine receptor-mediated intracellular signaling in response to dopamine dysregulation have been studied extensively in rodent models of Parkinson’s disease such as unilateral 6-hydroxydopamine (6OHDA) lesions of nigrostriatal dopamine neurons. This work has largely focused on striatal spiny projection neurons (SPNs). Direct pathway SPNs (dSPNs) generally express D1 dopamine receptors (D1Rs) and project to internal pallidum/substantia nigra reticulata to promote movement, whereas indirect pathway SPNs (iSPNs) express D2 dopamine receptors (D2Rs) and project to the external pallidum to inhibit movement, although the functional and anatomical features are not strictly segregated (DeLong & Wichmann, 2007; Gerfen & Surmeier, 2011; Hjorth et al., 2020). After dopamine depletion in adult rodents, D1R activation abnormally induces phosphorylation of the MAP kinase extracellular signal-regulated kinase (ERK), a signaling molecule integral to activity-dependent neuronal plasticity (Gerfen et al., 2002; Pavon et al., 2006; Santini et al., 2009; Westin et al., 2007). D2R signaling cascades appear to be unaffected. These studies have been instrumental in the identification of dSPNs as targets for therapeutics in Parkinson’s disease.
In contrast, less is known about dopamine receptor-mediated intracellular signaling pathways in dystonia. Therefore, to identify adaptations in striatal intracellular signaling caused by dopamine deficiency associated with dystonia, we examined dopamine receptor-mediated signal transduction in a mouse model of DRD. This knockin strain carries a human DRD-causing point mutation in tyrosine hydroxylase (ThDRD; DRD mice) that reduces striatal dopamine concentrations to <1% of normal (Knappskog et al., 1995; Rose et al., 2015). Like DRD patients, DRD mice exhibit dystonic movements that improve in response to l-DOPA. Further, the dystonia in DRD is also alleviated in response to D1R-selective agonists and D2R-selective agonists. Because both D1Rs and D2Rs mediate the dystonia (Rose et al., 2015), we assessed protein kinase A (PKA) activity, which is regulated by D1R- and D2R-mediated cAMP production, and ERK phosphorylation, which is abnormally regulated after dopamine depletion in adult rodents, in response to dopamine receptor agonists and antagonists. Further, because sensorimotor function, which is likely perturbed in dystonia, is mediated within the dorsolateral striatum (Balleine & O’Doherty, 2010; Yin & Knowlton, 2006) whereas dorsomedial striatum mediates associative functions (Devan & White, 1999; Yin et al., 2005) results were analyzed according to striatal subregions. Here, we demonstrate that dopamine-mediated intracellular signaling is differentially dysregulated within striatal subregions in a model of dystonia.
Experimental Procedures
Mice.
Male and female mice (3–5 month) homozygous for the c.1160C>A Th mutation (DRD mice) and control (+/+) littermates were generated as F1 hybrids of C57BL/6J +/ThDRD × DBA/2J +/ThDRD to circumvent the high perinatal lethality exhibited by inbred C57BL/6J DRD mice. For a subset of mice, the cell-type specific reporter transgene Drd2-EGFP (Heintz, Rockefeller University) inbred on C57BL/6J was bred onto the C57BL/6J +/ThDRD strain prior to the F1 hybrid cross (Chan et al., 2012). To maintain health, DRD and control mice received a daily subcutaneous injection of 10 mg/kg l-DOPA and 5 mg/kg benserazide (Sigma-Aldrich, St. Louis, MO) in 2.5 mg/ml ascorbic acid in saline (0.9% NaCl). l-DOPA was terminated >24 hours prior to all experiments. Mice were housed in standard mouse cages (2–5 mice/cage) in standard conditions (22°C, 40–50% relative humidity) with a 12 hour light cycle and ad libitum access to food and water. All procedures conformed to the NIH Guidelines for the Care and Use of Animals and were approved by the Emory University Animal Care and Use Committee.
Drug challenges.
Mice were injected subcutaneously (10 mL/kg) with either saline vehicle or drug dissolved in saline and euthanized 45 minutes after injection. The D1R-selective agonist SKF 81297 (0.2 mg/kg), the D2R-selective agonist quinpirole (0.1 mg/kg), and the indirect acting agonist l-DOPA (10 mg/kg) alleviate abnormal movements while the D1R-selective antagonist SCH 23390 (1 mg/kg) and the D2R-selective antagonist raclopride (1 mg/kg) worsen dystonia in DRD mice (Rose et al., 2015). To determine the role of D1Rs and D2Rs in l-DOPA-mediated signaling, mice were pretreated with SCH 23390 or raclopride 10 minutes prior to l-DOPA challenge. Because experiments tested multiple comparisons and large numbers of mice, vehicle and drug challenges in normal and DRD mice were distributed across experimental days to mitigate biases that could be inadvertently introduced by day-to-day environmental variation. SKF 81297 hydrobromide, (−)-quinpirole hydrochloride, SCH 23390 hydrochloride and raclopride were obtained from Tocris Biosciences (Minneapolis, MN). l-DOPA was obtained from Sigma-Aldrich (St. Louis, MO).
Immunohistochemistry.
Forty-five minutes after drug injection, mice were anesthetized with isoflurane and perfused with ice-cold, 4% buffered paraformaldehyde (pH 7.2). Brains were incubated in perfusate overnight at 4°C, incubated in 20% buffered sucrose overnight, and stored at 4°C in 30% buffered sucrose. Brains were sectioned (30 μm) and floating sections were treated with 0.5% Triton-X in Tris-buffered saline (TBS) for 30 minutes and transferred to 5% normal goat serum, 5% bovine serum albumin, 0.1% bovine gelatin, 0.05% Tween-80, and 0.01% sodium azide in TBS for 2 h. Sections were incubated with primary antibody (phospho-(Thr202/Tyr204)-ERK1/2 rabbit monoclonal antibody or phospho-(Ser/Thr) PKA substrate rabbit polyclonal antibody, both at 1:5000 dilution; Cell Signaling Technology) for 16–24 hours at 4°C and then for 2 hours at 4°C with secondary antibody (goat anti-rabbit IgG antibody (H+L), biotinylated, 1:800, Vector Laboratories) in 5% normal goat serum, 0.1% Triton-X-100 in TBS. Sections were incubated with avidin-biotin complex (Vector Laboratories) for 1 hour, and developed using 3–3’-diaminobenzidine (Sigma-Aldrich). Staining was batch-processed with up to ten different experimental conditions immunostained simultaneously to mitigate biases that could be introduced by inter-procedure technical variability. For immunofluorescence, after incubation with primary antibody, sections were incubated for 1 hour at 4°C with goat anti-rabbit IgG (H+L) highly cross-adsorbed, Alexa Fluor 594 antibody (1:500, Thermofisher Scientific) in 5% normal goat serum, 0.1% Triton-X-100 in TBS.
Brightfield image acquisition and cell counts.
Brightfield slides were digitized using an Aperio AT2 Scanner (Leica Biosystems). QuPath, an open-source software for whole slide image analysis, was used to define the dorsal striatum and automate cell counts, as previously described (Briscione et al., 2021). Because it is known that p-ERK increases after l-DOPA treatment in unilateral 6-OHDA-lesioned mice (Gerfen et al., 2002; Santini et al., 2009; Westin et al., 2007), sections from l-DOPA-challenged unilateral 6-OHDA-lesioned mice were used to optimize the cell counting parameters to ensure the reliable identification of immuno-positive cells. Background equivalent to that of the corpus callosum was set to account for variation in staining intensities between the sections. Cell counting was automated using the ‘positive cell detection’ function in QuPath after optimization of the parameters for each primary antibody, including adjustment of the cell size parameter to approximate medium spiny neuron perikarya area to 30–100 μm2 (Gagnon et al., 2017) to exclude most large cholinergic interneurons. To ensure that cell counting was not affected by staining intensity, several different dilutions of primary antibody were tested using the optimized parameters. Sections from the intermediate striatum (+1.145 to 0.145 bregma) were assessed because this striatal region receives projections from all cortical areas (Hintiryan et al., 2016) and has unique functional subregions whereby dorsomedial striatum mediates associative functions (Devan & White, 1999; Yin et al., 2005), and dorsolateral striatum mediates sensorimotor function (Balleine & O’Doherty, 2010; Yin & Knowlton, 2006). Cell counts were assessed in medial and lateral dorsal striatum, which were defined using QuPath, to provide a reproducible method for assigning regions for analysis. The entire striatum was outlined for each hemisphere. The midpoint of each striatum was defined by QuPath by a center x and y coordinate. Then, a line was drawn to connect the midpoint of the left with the right striatum. A perpendicular line was drawn from the center x,y coordinate of each striata to define the boundary between medial and lateral striatum. The total number of positive cells was divided by total area of the region to determine the number of positive cells per square mm. The brightfield photomicrographs were acquired using an Olympus BX51 upright microscope; the composite image in Figure 3C was adjusted for contrast and brightness to improve the clarity of the immunostaining.
Figure 3.

l-DOPA-mediated phosphorylation of ERK in the dorsomedial and dorsolateral striatum of normal and DRD mice. A. Quantification of the density of p-ERK positive striatal cells after acute administration of vehicle or l-DOPA (10 mg/kg) in normal and DRD mice (n = 6–9 mice/group). In addition, DRD mice were pretreated with either the D1R antagonist SCH 23390 (1 mg/kg) or the D2R antagonist raclopride (1 mg/kg) before l-DOPA challenge (n=7 mice/group). Mice were euthanized 45 minutes after treatment and the density of striatal cells immunolabeled with p-ERK antibody was quantified in dorsomedial and dorsolateral striatum. There was no effect of l-DOPA on control mice (mixed effects model, main effect of treatment, F1,14 = 2.76, P = 0.1189) but there was a main effect of region (F1,13 = 30.75, P < 0.0001) with higher expression in the dorsomedial than dorsolateral striatum. For DRD mice there was a significant treatment × region interaction effect (mixed effects model with repeated measures for region, F5,34 = 17.60, P < 0.0001 with Holm-Sidak’s post hoc analysis) whereby l-DOPA treatment increased the density of p-ERK positive cells in dorsomedial (P < 0.0001) but not dorsolateral (P = 0.7662) striatum. The dorsomedial-specific increase was attenuated by SCH 23390 (P < 0.0001) and raclopride (P < 0.0001) pretreatment. One sample from dorsolateral striatum of normal mice treated with l-DOPA and one sample from dorsolateral striatum of DRD mice treated with l-DOPA were identified as outliers and not included in the analyses. Data represent means ± SEMs; ††††P < 0.0001 compared to vehicle, ****P < 0.0001 compared to l-DOPA treatment. B. Double-labeling experiments of D2R+ striatal cells (green) and p-ERK-positive cells (magenta) in the dorsomedial striatum of a DRD mouse treated with l-DOPA. p-ERK immunoreactivity was not observed in D2R+ cells (merged image). C. Brightfield photomicrographs p-ERK immunoreactivity in the striatum of saline- and l-DOPA-treated DRD mice.
Fluorescent image acquisition and cell counts.
Immunofluorescence was used to determine the cell type of p-PKA-substrate and p-ERK-positive cells (red channel) in DRD mice carrying the Drd2-EGFP (endogenous eGFP fluorescence; green channel). Images were collected using a Leica SP8 confocal microscope at a 40X objective and were merged with NIH ImageJ software (1.52a). QuPath was used to automate positive counts of immunofluorescence, as described above. Fluorescent images were not adjusted for quantification, but the brightness was adjusted for presentation purposes. Cells were counted independently on the red and green channels and the percent overlap was determined in the merged image. Because 95% of neurons in the striatum are SPNs and less than 2% of SPNs express both D1Rs and D2Rs (Gagnon et al., 2017), positive cells in the red channel that overlapped with D2R-expressing SPNs in the green channel were presumptive iSPNs. Positive cells in the red channel that did not overlap with the green channel were presumptive dSPNs.
Data analyses.
GraphPad Prism was used for all analyses. The ROUT method (Q = 0.2%) was used to remove outliers. Ninety-six mice were tested with two data points for each mouse (dorsomedial and dorsolateral striatum) for a total of 192 data points; of these, 9 outliers were identified. Outliers are noted in the figure legends. For l-DOPA experiments, data were analyzed within each genotype using either two-way repeated measures ANOVA or a mixed effects model to accommodate missing outlier values with the striatal region as the repeated measure. Comparisons between the genotypes were not performed because l-DOPA is essentially inert in dopamine-replete normal animals. For direct-acting agonist and antagonist challenges, data were analyzed using a 3-way repeated measures ANOVA or a mixed effects model (genotype × treatment × region) with region as the repeated measure. QQ plots (normality distribution plots) were nearly linear and approximated the line of identity. Holm-Sidak’s multiple comparisons tests were used for post hoc analyses when there was a significant (P < 0.05) interaction effect or there were >2 variables within a factor with a significant main effect. Details of the statistical analyses are presented in the figure legends.
Results
Basal activity
After activation by cAMP, PKA phosphorylates numerous proteins including transcription factors, ion channels and other signaling molecules (Nagai et al., 2016; Surmeier et al., 1995; Walaas et al., 1983). Therefore, striatal PKA activity was assessed using the phospho-PKA substrate antibody (p-PKA-sub) which recognizes phospho-Ser/Thr residues preceded by arginine at the −3 or −2 positions. After vehicle treatment, the density of p-PKA-sub-positive cells was comparable in control and DRD mice. In both genotypes, the density of p-PKA-sub-positive cells in dorsolateral striatum was lower than in dorsomedial striatum (Figure 1A).
Figure 1.
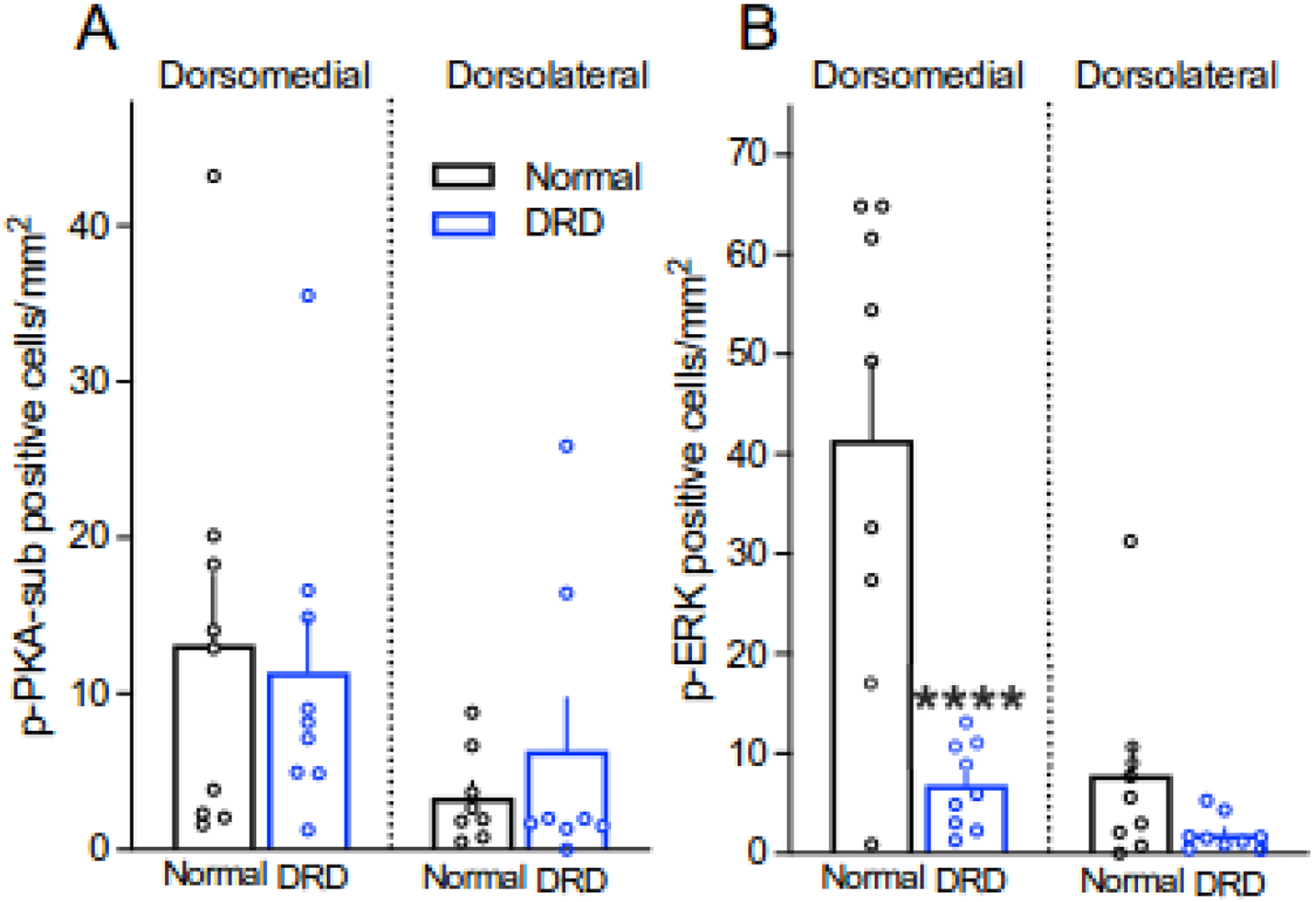
Baseline densities of p-PKA-sub-positive cells or p-ERK-positive cells in the dorsomedial and dorsolateral striatum of normal and DRD mice (n = 8–9 mice/group). A. The density of p-PKA-sub-positive cells was overall lower in dorsolateral striatum compared to dorsomedial striatum but there was no difference between normal and DRD mice (linear mix model with region as the repeated measure, main effect of genotype, F1,16 = 0.071, P = 0.793; main effect of region, F1,14 = 6.205, P = 0.0259). One sample from dorsolateral striatum of normal mice and one sample from dorsolateral striatum of DRD mice were identified as outliers and not included in the analyses. B. The density of p-ERK-positive cells in the dorsomedial striatum was reduced in DRD mice compared to normal mice (two-way repeated measures ANOVA with region as the repeated measure, genotype × region interaction effect, F1,16 = 20.31, P = 0.0004; Holms-Sidak’s post hoc analysis P < 0.0001). Data represent means ± SEMs; ****P < 0.0001 compared to normal mice.
The mitogen-activated protein kinase pathway consisting of Ras, MEK and the extracellular signal-regulated kinase ERK is expressed throughout the CNS. Once activated through phosphorylation by MEK, ERK exerts downstream effects to mediate neuronal plasticity, excitability and gene transcription (Thomas & Huganir, 2004). To quantify the density of phosphorylated ERK-positive cells, we used an antibody to phospho-(Thr202/Tyr204)-ERK1/2 (p-ERK), a MEK phosphorylation site. In normal mice, the density of p-ERK-positive cells after vehicle treatment was significantly higher in the dorsomedial striatum than the dorsolateral striatum. In contrast, the density of p-ERK-positive cells in DRD mice was comparable in dorsomedial and dorsolateral striatum because the density of p-ERK cells in the dorsomedial striatum of DRD mice was significantly lower than normal mice (Figure 1B).
l-DOPA-mediated signal transduction
l-DOPA ameliorates the dystonia in DRD mice. Because l-DOPA increases overall brain dopamine concentrations, dopamine signaling is enhanced at both D1Rs and D2Rs. D1Rs promote the formation of cAMP, which activates PKA. Conversely, D2Rs normally inhibit the formation of cAMP, reducing PKA activity (Nagai et al., 2016). However, we have previously demonstrated that D2R agonist-mediated inhibition of adenylate cyclase activity is blunted in DRD mice (Rose et al., 2015), suggesting that PKA activation in DRD mice may not conform to typical signal transduction pathways. Control and DRD mice were challenged with vehicle or 10 mg/kg l-DOPA and p-PKA-sub immunostaining was assessed in dorsomedial and dorsolateral striatum (Figure 2A). Control mice did not respond to l-DOPA, as control mice are dopamine-replete and l-DOPA treatment does not increase striatal dopamine release in normal mice (Downs et al., 2019). In contrast, l-DOPA treatment induced a significant ~6-fold increase in the density of p-PKA-sub immunoreactive cells in both the dorsomedial and dorsolateral striatum of DRD mice compared to vehicle-treated DRD mice. To determine the SPN subtype in which PKA activity was induced, we performed double-label immunofluorescence to identify D2R-expressing iSPNs and p-PKA-sub-positive neurons (Figure 2B). p-PKA-sub immunoreactivity was not observed in iSPNs suggesting that the PKA-mediated response occurs largely in dSPNs in DRD mice, although PKA may be induced in other striatal cell types also.
Figure 2.

l-DOPA-mediated PKA signaling in the dorsomedial and dorsolateral striatum of normal and DRD mice. A. Quantification of the density of p-PKA-sub-positive striatal cells after acute challenge with vehicle or l-DOPA (10 mg/kg) in normal and DRD mice (n = 7–9 mice/group). In addition, DRD mice were pretreated with either the D1R antagonist SCH 23390 (1 mg/kg) or the D2R antagonist raclopride (1 mg/kg) before l-DOPA challenge (n = 6–7 mice/group). Mice were euthanized 45 minutes after treatment and the density of p-PKA-sub-positive striatal cells was quantified in dorsomedial and dorsolateral striatum. There was no effect of l-DOPA in control mice (mixed effects model, main effect of treatment, F1,14 = 0.316, P = 0.5829). There was a main effect of treatment for DRD mice (mixed effects model, main effect of treatment, F5,34 = 3.702, P = 0.0088 with Holm-Sidak’s post hoc analysis) whereby l-DOPA increased the density of p-PKA-sub-positive striatal cells in dorsomedial (P = 0.0207) and dorsolateral (P = 0.0077) striatum; SCH 23390 and raclopride attenuated the effects of l-DOPA in dorsomedial (P = 0.0111 and p= 0.0556) and dorsolateral striatum (P = 0.0061 and P = 0.0099). One l-DOPA + RAC DRD mouse was not included the analysis because both dorsomedial and dorsolateral points were identified as outliers. Data represent means ± SEMs; †P < 0.05 and ††P < 0.01 compared to vehicle, *P < 0.05 and **P < 0.01 compared to l-DOPA treatment. B. Double-labeling experiments of D2R+ striatal cells (green) and p-PKA-sub-positive cells (magenta) in the dorsomedial striatum of a DRD mouse treated with l-DOPA. p-PKA-sub immunoreactivity was not observed in D2R+ cells (merged image).
To determine whether the l-DOPA-induced increase in PKA substrate phosphorylation was attributable to activation of one or both dopamine receptor subtypes, DRD mice were treated with the subtype-selective dopamine receptor antagonists SCH 23390 or raclopride prior to administration of l-DOPA (Figure 2A). Pretreatment with the D1R-selective antagonist SCH 23390 (1 mg/kg) attenuated the l-DOPA-induced p-PKA-sub immunoreactivity compared to l-DOPA alone in both dorsomedial and dorsolateral striatum. Pretreatment with the D2R-selective antagonist raclopride (1 mg/kg) also reduced the l-DOPA-induced p-PKA-sub immunoreactivity in DRD mice in both dorsomedial and dorsolateral striatum. The reduction in l-DOPA-induced p-PKA-sub-positive striatal cells by a D2R antagonist was unexpected because l-DOPA-induced PKA substrate phosphorylation induction occurred specifically in D1R-expressing SPNs.
After dopamine depletion in adult rodents, p-ERK is abnormally induced in response to both acute and chronic l-DOPA treatment (Alcacer et al., 2012; Pavon et al., 2006; Santini et al., 2009; Westin et al., 2007). To determine if ERK is similarly mediated in response to early-life dopamine deficiency, we challenged DRD mice with vehicle or l-DOPA and assessed the density of p-ERK-positive cells. In normal mice, p-ERK-positive cell density was not affected by l-DOPA treatment, consistent with previous studies (Kim et al., 2000; Westin et al., 2007). In DRD mice, l-DOPA treatment induced a robust increase in striatal p-ERK immunoreactivity (Figure 3A). Dual immunofluorescence revealed that l-DOPA-induced p-ERK immunoreactivity was not detected in iSPNs, suggesting that p-ERK was induced in dSPNs in DRD mice (Figure 3B). The effect was region specific whereby l-DOPA-induced a >100-fold increase in the density of p-ERK-labeled cells in the dorsomedial striatum compared to vehicle treatment, but not in the dorsolateral striatum (Figure 3A & C). To determine whether p-ERK activation was mediated by one or both dopamine receptor subtypes, DRD mice were pretreated with subtype-selective dopamine receptor antagonists and then administered l-DOPA (Figure 3A). Administration of the D1R antagonist SCH23390 prior to l-DOPA reduced the density of p-ERK positive cells compared to l-DOPA treatment alone. As expected, no effect was observed in dorsolateral striatum considering that l-DOPA treatment alone did not significantly increase the density of p-ERK positive cells in dorsolateral striatum. Administration of the D2R antagonist raclopride prior to l-DOPA treatment also reduced the density of p-ERK positive cells compared to l-DOPA treatment alone in the dorsomedial striatum but not in the dorsolateral striatum.
D1R agonist-mediated signal transduction
Because normal mice are dopamine replete, l-DOPA-stimulated intracellular signaling occurred only in DRD mice. Therefore, to reveal abnormalities in dopamine receptor-mediated signaling that distinguish DRD from control mice, we used the direct-acting agonist SKF 81297 (0.2 mg/kg) to activate D1Rs in both control and DRD mice. We have previously demonstrated that this low dose alleviates the dystonia in DRD mice, while higher doses cause seizures (Rose et al., 2015). SKF 81297 did not affect the density of striatal p-PKA-sub labeled cells in control mice. In contrast, SKF 81297 treatment increased the density of p-PKA-sub-positive neurons in DRD mice by >10-fold in dorsomedial and dorsolateral striatum (Figure 4A). In fact, the SKF 81297-induced increase in the density of p-PKA-sub-positive neurons was more robust than that of l-DOPA, despite the low dose of SKF 81297, suggesting that D1R signaling is supersensitive in DRD mice.
Figure 4.

Quantification of D1R and D2R agonist-mediated p-PKA-sub-positive or p-ERK-positive striatal cell density in normal and DRD mice. A & B. Mice were treated with vehicle or 0.2 mg/kg SKF 81297 (n = 6–9 mice/group) and euthanized 45 minutes after vehicle or drug challenge. A. SKF 81297 treatment had no effect on normal mice but significantly increased the density of p-PKA-sub-positive cells in DRD mice (three-way mixed effects model, treatment × genotype interaction effect, F1,29 = 6.930, P = 0.0134 with Holm-Sidak’s post hoc analysis) in both dorsomedial (P = 0.0087) and dorsolateral (P = 0.0070) striatum. One sample from the dorsolateral striatum of normal mice treated with SK81297 was identified as an outlier and not included in the analysis. B. The density of p-ERK-positive cells was not affected by SKF 81297 treatment in either normal or DRD mice (three-way repeated measures ANOVA, main effect of treatment, F1,28 = 0.1233, P = 0.7281). There was a genotype × region interaction effect (three-way RM-ANOVA, F1,28 = 7.609, P = 0.0101), reflecting the low basal p-ERK-positive cell density in the dorsomedial striatum of DRD mice. One DRD mouse treated with SKF 81297 was excluded from the analysis because both dorsomedial and dorsolateral datapoints were identified as outliers. C & D. Normal or DRD mice were treated with vehicle or 0.1 mg/kg quinpirole (n = 5–9 mice/group) and euthanized 45 minutes after vehicle or drug challenge. C. There was no effect of quinpirole treatment on the density of p-PKA-sub-positive cells in either normal or DRD mice (three-way mixed effects model, main effect of treatment, F1,22 = 1.310, P = 0.2647). D. There was no effect of quinpirole treatment on the density of p-ERK-positive cells in either normal or DRD mice (three-way repeated measures ANOVA, main effect of treatment, F1,26 = 0.0046, P = 0.9463). Data represent means ± SEMs. **P < 0.01 compared to vehicle.
Although previous studies have demonstrated that p-ERK is abnormally induced in response to D1R agonist treatment after chronic dopamine depletion in adults (Gerfen et al., 2002; Santini et al., 2009; Westin et al., 2007), SKF 81297 treatment did not significantly affect p-ERK immunoreactivity in either control or DRD mice (Figure 4B). The lack of an SKF 81297-induced ERK response is not likely attributable to the low dose of SKF 81297 because this same dose induced a significant increase in p-PKA-sub-positive neurons in the same mice.
D2R agonist-mediated signalling
We have previously demonstrated that quinpirole (0.1 mg/kg) ameliorates dystonia in DRD mice (Rose et al., 2015). At this dose quinpirole inhibits adenylate cyclase activity in control mice but has little effect on adenylate cyclase activity in DRD mice. Quinpirole treatment did not significantly affect the density of p-PKA-sub labeled cells in either control or DRD mice (Figure 4C). Similarly, quinpirole treatment did not affect p-ERK immunoreactivity (Figure 4D).
D1R antagonist-mediated signalling
Because we have previously demonstrated that administration of SCH 23390 exacerbates the dystonia in DRD mice (Rose et al., 2015), we also assessed the effects of D1R antagonist treatment on PKA substrate phosphorylation and ERK phosphorylation in normal and DRD mice. There was no significant effect of SCH 23390 treatment on the density of p-PKA-sub positive striatal cells in either normal or DRD mice in either dorsomedial or dorsolateral striatum (Figure 5A). However, SCH 23390 treatment significantly reduced the density of p-ERK-positive cells in dorsomedial striatum of normal mice to the low levels observed in saline-treated DRD mice (Figure 5B). These results suggest that normal baseline extracellular dopamine concentrations at D1Rs are necessary for the low level of p-ERK-positive cells observed in normal mice. Indeed, a reduction in D1R stimulation by either blockade with SCH23390 in normal mice or the very low concentrations of dopamine in DRD mice (<1% of normal) attenuated ERK phosphorylation.
Figure 5.

Quantification of D1R and D2R antagonist-mediated p-PKA-sub-positive or p-ERK-positive striatal cell density in normal and DRD mice. A & B. Normal or DRD mice were treated with vehicle or 1 mg/kg SCH 23390 (n = 4–9 mice/group) and euthanized 45 minutes after vehicle or drug challenge. A. SCH 23390 treatment did not alter the density of p-PKA-sub-positive striatal cells in either normal or DRD mice (3-way mixed effects analysis, main effect of treatment, F1,22 = 1.310, P = 0.2647). B. SCH 23390 treatment significantly reduced the density of p-ERK-positive cells in dorsomedial striatum of normal mice (P < 0.0001) but the effect was not significant (P < 0.9767) in DRD mice (three-way repeated measures ANOVA, genotype × region × treatment interaction effect F1,22 = 6.549, P = 0.0179 with Holm-Sidak’s post hoc analysis). C & D. Normal or DRD mice were treated with vehicle or 1 mg/kg raclopride (n = 7–9 mice/group) and euthanized 45 minutes after vehicle or drug challenge. C. Raclopride treatment significantly increased the density of p-PKA-sub-positive cells in the dorsolateral striatum of normal mice (three-way mixed effects analysis, treatment × genotype interaction effect F1,29 = 4.615, P = 0.0402; Holm-Sidak’s post hoc analysis, P = 0.0464). D. There was not a significant effect of raclopride on the density of p-ERK-positive striatal cells (three-way repeated measures ANOVA, main effect of treatment, F1,29 = 2.109, P = 0.1571), but there was a significant region × genotype interaction effect (three-way repeated measures ANOVA, F1,29 = 32.48, P < 0.0001; Holm-Sidak’s post hoc analysis, P < 0.0001 vehicle-treated dorsomedial normal mice versus vehicle-treated dorsomedial DRD mice). Data represent means ± SEMs. *P < 0.05 and ****P < 0.0001 compared to vehicle.
D2R antagonist-mediated signalling
Previous work has demonstrated that blocking the inhibitory effects of D2Rs is an effective strategy for unmasking abnormal signaling pathways in the striatum (Gerfen et al., 2002). Further, we have previously demonstrated that in DRD mice, blockade of D2Rs exacerbates dystonia (Rose et al., 2015). Raclopride treatment increased the density of p-PKA-sub labeled cells in normal mice but not in DRD mice (Figure 5C), but there was no effect of raclopride on p-ERK immunoreactivity (Figure 5D).
Discussion
Here, we demonstrate that intracellular signaling is dysregulated in DRD mice. Both dopamine receptor subtype-mediated responses and striatal subregion-specific responses were abnormal. At baseline, when DRD mice are dystonic, the density of p-ERK-positive cells was significantly lower in DRD mice than normal mice, while the PKA baseline was comparable in DRD and normal mice. These observations suggest that PKA maintains homeostatic levels despite the dopamine deficit, while ERK fails to adapt. The low p-ERK baseline in DRD mice may result from the deficit in dopamine-induced D1R stimulation, but it is not solely attributable to the deficit in dopamine, as baseline p-ERK levels are not reduced after 6OHDA-lesion in adult mice (Sugiyama et al., 2021). These observations suggest that the ERK dysregulation is instigated by early-life dopamine loss. Abnormal p-ERK regulation has been observed in mouse models of TOR1A, THAP1 and GNAL dystonia (Melis et al., 2021), although abnormal baseline p-ERK appears to be unique to DRD mice. The RAS/MEK/ERK signaling pathway is implicated in synaptic plasticity, as the concomitant activation of D1Rs and NMDA receptors is necessary for the induction of long-term potentiation at glutamate synapses on SPNs (Shen et al., 2008). Thus, it will be important to determine if the baseline reduction in p-ERK in DRD mice contributes to impaired plasticity, which is observed in many forms of dystonia (Gallea et al., 2018; Gilbertson et al., 2019; Porcacchia et al., 2019; Quartarone et al., 2003; Quartarone & Pisani, 2011).
D1R/D2R synergy in dystonia.
l-DOPA, an indirect agonist at D1Rs and D2Rs, increased the density of striatal PKA and p-ERK positive neurons in D1R-expressing dSPNs. It is therefore not surprising that the D1R-selective antagonist SCH 23390 attenuated the effects of l-DOPA. SCH 23390 pretreatment also attenuates the l-DOPA-induced increase in p-ERK in models of parkinsonism (Santini et al., 2009; Westin et al., 2007). The D2R-selective antagonist raclopride also attenuated the l-DOPA-induced increase in striatal PKA in dSPNs, which generally do not express D2Rs. This is consistent with previous work demonstrating a synergist effect of the combined activation of D1Rs with D2Rs on the downstream induction of immediate early genes such as c-fos and zif268 (Gerfen et al., 1995; Keefe & Gerfen, 1995; Morelli et al., 1993; Paul et al., 1992). Blockade of D2Rs also significantly reduced the l-DOPA-induced increase in ERK phosphorylation. This was unexpected because in models of parkinsonism, D2R antagonists have no effect on the l-DOPA-induced increase p-ERK (Santini et al., 2009; Westin et al., 2007), suggesting that D2R-mediated signaling is differentially affected in the DRD mouse model of dystonia. Indeed, it appears that D2R activation is required for l-DOPA-induced p-ERK induction in dSPNs as the D1R agonist SKF 81297 alone did not induce p-ERK. This contrasts with PKA, which was activated by both l-DOPA and SKF 81297. The ability of D2Rs to mediate signaling cascades in dSPNs likely occurs at the circuit level rather than direct action on dSPNs, as <2% of SPNs express both D1Rs and D2Rs (Gagnon et al., 2017). D2Rs on cholinergic interneurons mediate cholinergic neurotransmission to regulate the concentration of acetylcholine at inhibitory M4 muscarinic acetylcholine receptors located on dSPNs, thereby influencing D1R intracellular signaling (Yan et al., 1997). Additionally, D2R signaling in iSPNs are known to influence dSPN through lateral inhibition, which occurs via SPN axon collaterals (Dobbs et al., 2016; Planert et al., 2010; Taverna et al., 2008; Wilson, 2007). Activation of D2Rs on iSPNs by l-DOPA may inhibit iSPNs thereby releasing dSPNs from lateral inhibition and potentiating dSPN signaling cascades. Although the mechanisms are unclear, this cross-talk is not observed in models of parkinsonism (Darmopil et al., 2009; Westin et al., 2007) suggesting that this adaptation is unique to dystonia.
Striatal subregion-selective intracellular signaling.
l-DOPA-induced ERK phosphorylation was largely confined to dorsomedial striatum in DRD mice. Considering that dopamine concentrations in DRD mouse striatum are <1% of normal, it is unlikely that medial-lateral gradients of residual dopamine accounts for the difference in responsivity. However, it is known that the dorsolateral and dorsomedial striatum receive afferents from anatomically diverse regions of the cortex and subserve different functions (Balleine et al., 2009; Graybiel & Grafton, 2015). The dorsolateral striatum mediates sensorimotor function (Balleine & O’Doherty, 2010; Yin & Knowlton, 2006) and receives cortical input predominantly from sensorimotor areas (Hintiryan et al., 2016; Hunnicutt et al., 2016; McGeorge & Faull, 1989). In contrast, the dorsomedial striatum mediates goal-directed behavior and spatial learning (Devan & White, 1999; Yin et al., 2005) and receives input from associative cortices involved in movement planning and decision-making (Hintiryan et al., 2016; Hunnicutt et al., 2016; McGeorge & Faull, 1989). Indeed, ERK phosphorylation in the dorsomedial striatum is necessary for encoding and performing goal-directed actions (Girault et al., 2007). The striking pattern of ERK phosphorylation between the hyper-responsive dorsomedial striatum and the relatively unresponsive dorsolateral striatum in DRD mice, suggests that the functional divisions within the striatum are distorted. This could potentially occur through differential regulation of the synaptic strength of sensorimotor versus associative corticostriatal synapses or abnormal spatial organization of corticostriatal projections. In support of this premise, after dopamine lesions in neonatal mice, deficits in the functional connectivity of corticostriatal synapses arising from the medial prefrontal cortex are more severe than those arising from cingulate or motor cortices (Braz et al., 2015; Galinanes et al., 2011).
Dopamine receptor-mediated intracellular signaling in dystonia vs. Parkinson’s disease.
Deficits in brain dopamine can cause dystonia or Parkinson’s disease. Because the extent of dopamine deficiency is similar in both disorders in humans (Cheng et al., 2010; Furukawa et al., 1996; Ichinose et al., 1995; Ichinose et al., 1994), where dopamine concentrations are <20% in DRD and at the onset of motor signs in Parkinson’s disease, and in mice, where striatal dopamine concentration are <1% of normal in both disease models (Francardo et al., 2011; Rose et al., 2015), unique adaptations in dopamine receptor-mediated intracellular signaling responses may, at least in part, distinguish dystonia from parkinsonism. Indeed, our results in DRD mice and extensive work on models of parkinsonism suggests that striatal subregions are differentially affected in the two disorders. In contrast to the dorsomedial striatal pattern of l-DOPA-induced ERK phosphorylation in DRD mice, several studies have demonstrated that p-ERK is uniformly induced across the entire striatum in models of parkinsonism, even after chronic administration of l-DOPA (Gerfen et al., 2008; Park et al., 2014; Santini et al., 2009; Santini et al., 2012; Santini et al., 2007; Westin et al., 2007). Notably, DRD mice are also treated chronically with l-DOPA to maintain health suggesting that chronic treatment with l-DOPA per se does not instigate the unique striatal distribution of p-ERK in DRD mice. Conversely, whereas l-DOPA- and D1R agonist-induced PKA activity was pan-striatal in DRD mice, it was previously demonstrated that PKA activity is preferentially induced in the dorsolateral striatum in models of parkinsonism (Sugiyama et al., 2021). Thus, signaling cascades in striatal subregions appear to be differentially affected in models of dystonia and parkinsonism. These regional differences in combination with abnormal dopamine receptor subtype-mediated responses may contribute to the distinct signs and symptoms of these two movement disorders.
Conclusion.
While dopamine deficiency underlies both DRD and Parkinson’s disease, our results suggest that specific (mal)adaptive processes may contribute to the distinct clinical features of dystonia. In contrast to Parkinson’s disease, which occurs in the context of a mature striatum, the dopamine deficiency in DRD is present from early development (Hegarty et al., 2013), when SPNs are undergoing differentiation and migration in the embryonic brain (Knowles et al., 2021). In fact, dopamine signaling mediates the maturation of SPNs (Lieberman et al., 2018). Thus, identifying the precise temporal and spatial defects caused by early-life dopamine deficiency will be critical for identifying the specific mechanisms that drive the expression of dystonia.
Highlights.
Dopamine-mediated intracellular signal is dysregulated in dystonia
Abnormal intracellular signaling was differentially regulated in striatal subregions
ERK phosphorylation occurred in dorsomedial striatum but not dorsolateral striatum
Dopamine-mediated intracellular signal in dystonia was distinct from Parkinsonism
Acknowledgements
We thank Alec P. Shannon for technical assistance. This work was supported by the National Institutes of Health, National Institute of Neurological Disorders and Stroke Grants R01 NS088528, R56 NS124764, F31 NS103363, T32 NS007480, T32 GM008602 and the Emory University Integrated Cellular Imaging Core.
Financial Disclosures of all authors for the preceding 12 months
K.M.R., M.A.B., Y.D., J.I., X.F., D.B., S.A.C., A.M.D., T.A.S., S.Q.B. and D.J.S. have nothing to report. D.G. is a member of the scientific advisory board for Histowiz, Inc LLC. H.A.J. serves on advisory boards or as a consultant for Addex, Allergan, CoA Therapeutics, Cavion Therapeutics, Daiichi Sankyo, Ipsen, Retrophin, Revance, and Takaha-Ene Pharmaceuticals and reports research funding from the National Institutes of Health, Cure Dystonia Now, and Revance Therapeutics, Inc. for research unrelated to this work. H.A.J. has received honoraria from the International Parkinson’s Disease and Movement Disorders Society and serves on the Scientific Advisory Boards for the Benign Essential Blepharospasm Research Foundation, the Dystonia Medical Research Foundation, the Tourette Association of America, and Tyler’s Hope for a Cure. E.J.H. is a member of the Medical and Scientific Advisory Board of the Dystonia Medical Research Foundation and reports research funding from the National Institutes of Health, United States Department of Defense and Parkinson’s Foundation.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of interest
The authors declare no conflict of interests.
Bibliography
- Alcacer C, Santini E, Valjent E, Gaven F, Girault JA, & Herve D (2012). Galpha(olf) mutation allows parsing the role of cAMP-dependent and extracellular signal-regulated kinase-dependent signaling in L-3,4-dihydroxyphenylalanine-induced dyskinesia. J Neurosci, 32(17), 5900–5910. 10.1523/JNEUROSCI.0837-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balleine BW, Liljeholm M, & Ostlund SB (2009). The integrative function of the basal ganglia in instrumental conditioning. Behav Brain Res, 199(1), 43–52. 10.1016/j.bbr.2008.10.034 [DOI] [PubMed] [Google Scholar]
- Balleine BW, & O’Doherty JP (2010). Human and rodent homologies in action control: corticostriatal determinants of goal-directed and habitual action. Neuropsychopharmacology, 35(1), 48–69. 10.1038/npp.2009.131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman BD, Hallett M, Herscovitch P, & Simonyan K (2013). Striatal dopaminergic dysfunction at rest and during task performance in writer’s cramp. Brain, 136(Pt 12), 3645–3658. 10.1093/brain/awt282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braz BY, Galinanes GL, Taravini IR, Belforte JE, & Murer MG (2015). Altered Corticostriatal Connectivity and Exploration/Exploitation Imbalance Emerge as Intermediate Phenotypes for a Neonatal Dopamine Dysfunction. Neuropsychopharmacology, 40(11), 2576–2587. 10.1038/npp.2015.104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Briscione MA, Dinasarapu AR, Bagchi P, Donsante Y, Roman KM, Downs AM, Fan X, Hoehner J, Jinnah HA, & Hess EJ (2021). Differential expression of striatal proteins in a mouse model of DOPA-responsive dystonia reveals shared mechanisms among dystonic disorders. Mol Genet Metab, 133(4), 352–361. 10.1016/j.ymgme.2021.05.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan CS, Peterson JD, Gertler TS, Glajch KE, Quintana RE, Cui Q, Sebel LE, Plotkin JL, Shen W, Heiman M, Heintz N, Greengard P, & Surmeier DJ (2012). Strain-specific regulation of striatal phenotype in Drd2-eGFP BAC transgenic mice. J Neurosci, 32(27), 9124–9132. 10.1523/JNEUROSCI.0229-12.2012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng HC, Ulane CM, & Burke RE (2010). Clinical progression in Parkinson disease and the neurobiology of axons. Ann Neurol, 67(6), 715–725. 10.1002/ana.21995 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darmopil S, Martin AB, De Diego IR, Ares S, & Moratalla R (2009). Genetic inactivation of dopamine D1 but not D2 receptors inhibits L-DOPA-induced dyskinesia and histone activation. Biol Psychiatry, 66(6), 603–613. 10.1016/j.biopsych.2009.04.025 [DOI] [PubMed] [Google Scholar]
- DeLong MR, & Wichmann T (2007). Circuits and circuit disorders of the basal ganglia. Arch Neurol, 64(1), 20–24. 10.1001/archneur.64.1.20 [DOI] [PubMed] [Google Scholar]
- Devan BD, & White NM (1999). Parallel information processing in the dorsal striatum: relation to hippocampal function. J Neurosci, 19(7), 2789–2798. https://www.ncbi.nlm.nih.gov/pubmed/10087090 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobbs LK, Kaplan AR, Lemos JC, Matsui A, Rubinstein M, & Alvarez VA (2016). Dopamine Regulation of Lateral Inhibition between Striatal Neurons Gates the Stimulant Actions of Cocaine. Neuron, 90(5), 1100–1113. 10.1016/j.neuron.2016.04.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Downs AM, Fan X, Donsante C, Jinnah HA, & Hess EJ (2019). Trihexyphenidyl rescues the deficit in dopamine neurotransmission in a mouse model of DYT1 dystonia. Neurobiol Dis, 125, 115–122. 10.1016/j.nbd.2019.01.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francardo V, Recchia A, Popovic N, Andersson D, Nissbrandt H, & Cenci MA (2011). Impact of the lesion procedure on the profiles of motor impairment and molecular responsiveness to L-DOPA in the 6-hydroxydopamine mouse model of Parkinson’s disease. Neurobiol Dis, 42(3), 327–340. 10.1016/j.nbd.2011.01.024 [DOI] [PubMed] [Google Scholar]
- Furukawa Y, Shimadzu M, Rajput AH, Shimizu Y, Tagawa T, Mori H, Yokochi M, Narabayashi H, Hornykiewicz O, Mizuno Y, & Kish SJ (1996). GTP-cyclohydrolase I gene mutations in hereditary progressive amd dopa-responsive dystonia. Ann Neurol, 39(5), 609–617. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=8619546 [DOI] [PubMed] [Google Scholar]
- Gagnon D, Petryszyn S, Sanchez MG, Bories C, Beaulieu JM, De Koninck Y, Parent A, & Parent M (2017). Striatal Neurons Expressing D1 and D2 Receptors are Morphologically Distinct and Differently Affected by Dopamine Denervation in Mice. Sci Rep, 7, 41432. 10.1038/srep41432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galinanes GL, Braz BY, & Murer MG (2011). Origin and properties of striatal local field potential responses to cortical stimulation: temporal regulation by fast inhibitory connections. PLoS One, 6(12), e28473. 10.1371/journal.pone.0028473 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallea C, Herath P, Voon V, Lerner A, Ostuni J, Saad Z, Thada S, Solomon J, Horovitz SG, & Hallett M (2018). Loss of inhibition in sensorimotor networks in focal hand dystonia. Neuroimage Clin, 17, 90–97. 10.1016/j.nicl.2017.10.011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerfen CR, Keefe KA, & Gauda EB (1995). D1 and D2 dopamine receptor function in the striatum: coactivation of D1- and D2-dopamine receptors on separate populations of neurons results in potentiated immediate early gene response in D1-containing neurons. J Neurosci, 15(12), 8167–8176. https://www.ncbi.nlm.nih.gov/pubmed/8613751 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerfen CR, Miyachi S, Paletzki R, & Brown P (2002). D1 dopamine receptor supersensitivity in the dopamine-depleted striatum results from a switch in the regulation of ERK1/2/MAP kinase. J Neurosci, 22(12), 5042–5054. https://www.ncbi.nlm.nih.gov/pubmed/12077200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerfen CR, Paletzki R, & Worley P (2008). Differences between dorsal and ventral striatum in Drd1a dopamine receptor coupling of dopamine- and cAMP-regulated phosphoprotein-32 to activation of extracellular signal-regulated kinase. J Neurosci, 28(28), 7113–7120. 10.1523/JNEUROSCI.3952-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerfen CR, & Surmeier DJ (2011). Modulation of striatal projection systems by dopamine. Annu Rev Neurosci, 34, 441–466. 10.1146/annurev-neuro-061010-113641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbertson T, Humphries M, & Steele JD (2019). Maladaptive striatal plasticity and abnormal reward-learning in cervical dystonia. Eur J Neurosci, 50(7), 3191–3204. 10.1111/ejn.14414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Girault JA, Valjent E, Caboche J, & Herve D (2007). ERK2: a logical AND gate critical for drug-induced plasticity? Curr Opin Pharmacol, 7(1), 77–85. 10.1016/j.coph.2006.08.012 [DOI] [PubMed] [Google Scholar]
- Graybiel AM, & Grafton ST (2015). The striatum: where skills and habits meet. Cold Spring Harb Perspect Biol, 7(8), a021691. 10.1101/cshperspect.a021691 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hegarty SV, Sullivan AM, & O’Keeffe GW (2013). Midbrain dopaminergic neurons: a review of the molecular circuitry that regulates their development. Dev Biol, 379(2), 123–138. 10.1016/j.ydbio.2013.04.014 [DOI] [PubMed] [Google Scholar]
- Hintiryan H, Foster NN, Bowman I, Bay M, Song MY, Gou L, Yamashita S, Bienkowski MS, Zingg B, Zhu M, Yang XW, Shih JC, Toga AW, & Dong HW (2016). The mouse cortico-striatal projectome. Nat Neurosci, 19(8), 1100–1114. 10.1038/nn.4332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hjorth JJJ, Kozlov A, Carannante I, Frost Nylen J, Lindroos R, Johansson Y, Tokarska A, Dorst MC, Suryanarayana SM, Silberberg G, Hellgren Kotaleski J, & Grillner S (2020). The microcircuits of striatum in silico. Proc Natl Acad Sci U S A, 117(17), 9554–9565. 10.1073/pnas.2000671117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunnicutt BJ, Jongbloets BC, Birdsong WT, Gertz KJ, Zhong H, & Mao T (2016). A comprehensive excitatory input map of the striatum reveals novel functional organization. Elife, 5. 10.7554/eLife.19103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichinose H, Ohye T, Segawa M, Nomura Y, Endo K, Tanaka H, Tsuji S, Fujita K, & Nagatsu T (1995). GTP cyclohydrolase I gene in hereditary progressive dystonia with marked diurnal fluctuation. Neurosci Lett, 196(1–2), 5–8. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=7501255 [DOI] [PubMed] [Google Scholar]
- Ichinose H, Ohye T, Takahashi E, Seki N, Hori T, Segawa M, Nomura Y, Endo K, Tanaka H, Tsuji S, Fujita K, & Nagatsu T (1994). Hereditary progressive dystonia with marked diurnal fluctuation caused by mutations in the GTP cyclohydrolase I gene. Nat Genet, 8(3), 236–242. [DOI] [PubMed] [Google Scholar]
- Jinnah HA, & Sun YV (2019). Dystonia genes and their biological pathways. Neurobiol Dis, 129, 159–168. 10.1016/j.nbd.2019.05.014 [DOI] [PubMed] [Google Scholar]
- Keefe KA, & Gerfen CR (1995). D1–D2 dopamine receptor synergy in striatum: effects of intrastriatal infusions of dopamine agonists and antagonists on immediate early gene expression. Neuroscience, 66(4), 903–913. 10.1016/0306-4522(95)00024-d [DOI] [PubMed] [Google Scholar]
- Kim DS, Szczypka MS, & Palmiter RD (2000). Dopamine-deficient mice are hypersensitive to dopamine receptor agonists. J Neurosci, 20, 4405–4413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knappskog PM, Flatmark T, Mallet J, Ludecke B, & Bartholome K (1995). Recessively inherited L-DOPA-responsive dystonia caused by a point mutation (Q381K) in the tyrosine hydroxylase gene. Human Molecular Genetics, 4, 1209–1212. [DOI] [PubMed] [Google Scholar]
- Knowles R, Dehorter N, & Ellender T (2021). From Progenitors to Progeny: Shaping Striatal Circuit Development and Function. J Neurosci, 41(46), 9483–9502. 10.1523/JNEUROSCI.0620-21.2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lieberman OJ, McGuirt AF, Mosharov EV, Pigulevskiy I, Hobson BD, Choi S, Frier MD, Santini E, Borgkvist A, & Sulzer D (2018). Dopamine Triggers the Maturation of Striatal Spiny Projection Neuron Excitability during a Critical Period. Neuron, 99(3), 540–554 e544. 10.1016/j.neuron.2018.06.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGeorge AJ, & Faull RL (1989). The organization of the projection from the cerebral cortex to the striatum in the rat. Neuroscience, 29(3), 503–537. https://www.ncbi.nlm.nih.gov/pubmed/2472578 [DOI] [PubMed] [Google Scholar]
- Melis C, Beauvais G, Muntean BS, Cirnaru MD, Otrimski G, Creus-Muncunill J, Martemyanov KA, Gonzalez-Alegre P, & Ehrlich ME (2021). Striatal Dopamine Induced ERK Phosphorylation Is Altered in Mouse Models of Monogenic Dystonia. Mov Disord, 36(5), 1147–1157. 10.1002/mds.28476 [DOI] [PubMed] [Google Scholar]
- Morelli M, Cozzolino A, Pinna A, Fenu S, Carta A, & Di Chiara G (1993). L-dopa stimulates c-fos expression in dopamine denervated striatum by combined activation of D-1 and D-2 receptors. Brain Res, 623(2), 334–336. 10.1016/0006-8993(93)91449-3 [DOI] [PubMed] [Google Scholar]
- Nagai T, Nakamuta S, Kuroda K, Nakauchi S, Nishioka T, Takano T, Zhang X, Tsuboi D, Funahashi Y, Nakano T, Yoshimoto J, Kobayashi K, Uchigashima M, Watanabe M, Miura M, Nishi A, Kobayashi K, Yamada K, Amano M, & Kaibuchi K (2016). Phosphoproteomics of the Dopamine Pathway Enables Discovery of Rap1 Activation as a Reward Signal In Vivo. Neuron, 89(3), 550–565. 10.1016/j.neuron.2015.12.019 [DOI] [PubMed] [Google Scholar]
- Park HY, Kang YM, Kang Y, Park TS, Ryu YK, Hwang JH, Kim YH, Chung BH, Nam KH, Kim MR, Lee CH, Han PL, & Kim KS (2014). Inhibition of adenylyl cyclase type 5 prevents L-DOPA-induced dyskinesia in an animal model of Parkinson’s disease. J Neurosci, 34(35), 11744–11753. 10.1523/JNEUROSCI.0864-14.2014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul ML, Graybiel AM, David JC, & Robertson HA (1992). D1-like and D2-like dopamine receptors synergistically activate rotation and c-fos expression in the dopamine-depleted striatum in a rat model of Parkinson’s disease. J Neurosci, 12(10), 3729–3742. 10.1523/JNEUROSCI.12-10-03729.1992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavon N, Martin AB, Mendialdua A, & Moratalla R (2006). ERK phosphorylation and FosB expression are associated with L-DOPA-induced dyskinesia in hemiparkinsonian mice. Biol Psychiatry, 59(1), 64–74. 10.1016/j.biopsych.2005.05.044 [DOI] [PubMed] [Google Scholar]
- Planert H, Szydlowski SN, Hjorth JJ, Grillner S, & Silberberg G (2010). Dynamics of synaptic transmission between fast-spiking interneurons and striatal projection neurons of the direct and indirect pathways. J Neurosci, 30(9), 3499–3507. 10.1523/JNEUROSCI.5139-09.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porcacchia P, Alvarez de Toledo P, Rodriguez-Baena A, Martin-Rodriguez JF, Palomar FJ, Vargas-Gonzalez L, Jesus S, Koch G, & Mir P (2019). Abnormal cerebellar connectivity and plasticity in isolated cervical dystonia. PLoS One, 14(1), e0211367. 10.1371/journal.pone.0211367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quartarone A, Bagnato S, Rizzo V, Siebner HR, Dattola V, Scalfari A, Morgante F, Battaglia F, Romano M, & Girlanda P (2003). Abnormal associative plasticity of the human motor cortex in writer’s cramp. Brain, 126(Pt 12), 2586–2596. 10.1093/brain/awg273 [DOI] [PubMed] [Google Scholar]
- Quartarone A, & Pisani A (2011). Abnormal plasticity in dystonia: Disruption of synaptic homeostasis. Neurobiol Dis, 42(2), 162–170. 10.1016/j.nbd.2010.12.011 [DOI] [PubMed] [Google Scholar]
- Ribot B, Aupy J, Vidailhet M, Mazere J, Pisani A, Bezard E, Guehl D, & Burbaud P (2019). Dystonia and dopamine: From phenomenology to pathophysiology. Prog Neurobiol, 182, 101678. 10.1016/j.pneurobio.2019.101678 [DOI] [PubMed] [Google Scholar]
- Rose SJ, Yu XY, Heinzer AK, Harrast P, Fan X, Raike RS, Thompson VB, Pare JF, Weinshenker D, Smith Y, Jinnah HA, & Hess EJ (2015). A new knock-in mouse model of l-DOPA-responsive dystonia. Brain, 138(Pt 10), 2987–3002. 10.1093/brain/awv212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santini E, Alcacer C, Cacciatore S, Heiman M, Herve D, Greengard P, Girault JA, Valjent E, & Fisone G (2009). L-DOPA activates ERK signaling and phosphorylates histone H3 in the striatonigral medium spiny neurons of hemiparkinsonian mice. J Neurochem, 108(3), 621–633. 10.1111/j.1471-4159.2008.05831.x [DOI] [PubMed] [Google Scholar]
- Santini E, Feyder M, Gangarossa G, Bateup HS, Greengard P, & Fisone G (2012). Dopamine- and cAMP-regulated phosphoprotein of 32-kDa (DARPP-32)-dependent activation of extracellular signal-regulated kinase (ERK) and mammalian target of rapamycin complex 1 (mTORC1) signaling in experimental parkinsonism. J Biol Chem, 287(33), 27806–27812. 10.1074/jbc.M112.388413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santini E, Valjent E, Usiello A, Carta M, Borgkvist A, Girault JA, Herve D, Greengard P, & Fisone G (2007). Critical involvement of cAMP/DARPP-32 and extracellular signal-regulated protein kinase signaling in L-DOPA-induced dyskinesia. J Neurosci, 27(26), 6995–7005. 10.1523/JNEUROSCI.0852-07.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen W, Flajolet M, Greengard P, & Surmeier DJ (2008). Dichotomous dopaminergic control of striatal synaptic plasticity. Science, 321(5890), 848–851. 10.1126/science.1160575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonyan K, Berman BD, Herscovitch P, & Hallett M (2013). Abnormal striatal dopaminergic neurotransmission during rest and task production in spasmodic dysphonia. J Neurosci, 33(37), 14705–14714. 10.1523/JNEUROSCI.0407-13.2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugiyama K, Kuroiwa M, Shuto T, Ohnishi YN, Kawahara Y, Miyamoto Y, Fukuda T, & Nishi A (2021). Subregion-Specific Regulation of Dopamine D1 Receptor Signaling in the Striatum: Implication for L-DOPA-Induced Dyskinesia. J Neurosci, 41(30), 6388–6414. 10.1523/JNEUROSCI.0373-21.2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Surmeier DJ, Bargas J, Hemmings HC Jr., Nairn AC, & Greengard P (1995). Modulation of calcium currents by a D1 dopaminergic protein kinase/phosphatase cascade in rat neostriatal neurons. Neuron, 14(2), 385–397. 10.1016/0896-6273(95)90294-5 [DOI] [PubMed] [Google Scholar]
- Taverna S, Ilijic E, & Surmeier DJ (2008). Recurrent collateral connections of striatal medium spiny neurons are disrupted in models of Parkinson’s disease. J Neurosci, 28(21), 5504–5512. 10.1523/JNEUROSCI.5493-07.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas GM, & Huganir RL (2004). MAPK cascade signalling and synaptic plasticity. Nat Rev Neurosci, 5(3), 173–183. 10.1038/nrn1346 [DOI] [PubMed] [Google Scholar]
- Thony B, & Blau N (2006). Mutations in the BH4-metabolizing genes GTP cyclohydrolase I, 6-pyruvoyl-tetrahydropterin synthase, sepiapterin reductase, carbinolamine-4a-dehydratase, and dihydropteridine reductase. Hum Mutat, 27(9), 870–878. 10.1002/humu.20366 [DOI] [PubMed] [Google Scholar]
- van den Heuvel LP, Luiten B, Smeitink JA, de Rijk-van Andel JF, Hyland K, Steenbergen-Spanjers GC, Janssen RJ, & Wevers RA (1998). A common point mutation in the tyrosine hydroxylase gene in autosomal recessive L-DOPA-responsive dystonia in the Dutch population. Hum Genet, 102(6), 644–646. http://www.ncbi.nlm.nih.gov/entrez/query.fcgi?cmd=Retrieve&db=PubMed&dopt=Citation&list_uids=9703425 [DOI] [PubMed] [Google Scholar]
- Walaas SI, Aswad DW, & Greengard P (1983). A dopamine- and cyclic AMP-regulated phosphoprotein enriched in dopamine-innervated brain regions. Nature, 301(5895), 69–71. 10.1038/301069a0 [DOI] [PubMed] [Google Scholar]
- Westin JE, Vercammen L, Strome EM, Konradi C, & Cenci MA (2007). Spatiotemporal pattern of striatal ERK1/2 phosphorylation in a rat model of L-DOPA-induced dyskinesia and the role of dopamine D1 receptors. Biol Psychiatry, 62(7), 800–810. 10.1016/j.biopsych.2006.11.032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson CJ (2007). GABAergic inhibition in the neostriatum. Prog Brain Res, 160, 91–110. 10.1016/S0079-6123(06)60006-X [DOI] [PubMed] [Google Scholar]
- Yan Z, Song WJ, & Surmeier J (1997). D2 dopamine receptors reduce N-type Ca2+ currents in rat neostriatal cholinergic interneurons through a membrane-delimited, protein-kinase-C-insensitive pathway. J Neurophysiol, 77(2), 1003–1015. 10.1152/jn.1997.77.2.1003 [DOI] [PubMed] [Google Scholar]
- Yin HH, & Knowlton BJ (2006). The role of the basal ganglia in habit formation. Nat Rev Neurosci, 7(6), 464–476. 10.1038/nrn1919 [DOI] [PubMed] [Google Scholar]
- Yin HH, Ostlund SB, Knowlton BJ, & Balleine BW (2005). The role of the dorsomedial striatum in instrumental conditioning. Eur J Neurosci, 22(2), 513–523. 10.1111/j.1460-9568.2005.04218.x [DOI] [PubMed] [Google Scholar]