

Abstract
Purpose:
Assessing the risk of common, complex diseases requires consideration of clinical risk factors as well as monogenic and polygenic risks, which in turn may be reflected in family history. Returning risks to individuals and providers may influence preventive care or use of prophylactic therapies for those individuals at high genetic risk.
Methods:
To enable integrated genetic risk assessment, the eMERGE (electronic MEdical Records and GEnomics) network is enrolling 25,000 diverse individuals in a prospective cohort study across 10 sites. The network developed methods to return cross-ancestry polygenic risk scores (PRS), monogenic risks, family history, and clinical risk assessments via a Genome Informed Risk Assessment (GIRA) report and will assess uptake of care recommendations after return of results.
Results:
GIRAs include summary care recommendations for 11 conditions, education pages, and clinical laboratory reports. The return of high-risk GIRA to individuals and providers includes guidelines for care and lifestyle recommendations. Assembling the GIRA required infrastructure and workflows for ingesting and presenting content from multiple sources. Recruitment began in February 2022.
Conclusion:
Return of a novel report for communicating monogenic, polygenic, and family history based risk factors will inform the benefits of integrated genetic risk assessment for routine health care.
Keywords: polygenic risk scores, family history, monogenic risks, common variants, genotyping
Introduction:
Using genetic risk factors to identify individuals at high risk of disease promises to improve screening practices currently based on clinical risk factors and family history.1,2 Monogenic disease risks are now incorporated into screening guidelines for several cancers (breast, ovarian, colorectal) and cardiometabolic (familial hyperlipidemia) conditions.3,4 Yet, risk assessment for monogenic disease based on family history alone is suboptimal and may miss more than three-quarters of affected patients.5,6 The introduction of polygenic risk scores (PRS), reflecting the aggregate impact of many common genetic variants of small individual effect, may enable more integrated genetic risk assessment but complicates point-of-care translation. As the analytic validity of PRS is largely unknown for groups whose genetic ancestries are underrepresented in GWAS datasets, improving the generalizability of genetic risk estimation is critical to ensuring equitable implementation of genomic risk assessment across diverse populations.7–9
Implementing new and existing methods to apply complex genetic risk stratification to preventive care decisions requires new methods to communicate comprehensive genomic risk assessments to patients and providers. Such methods should integrate three components of genetic risk (family history, monogenic, and polygenic), incorporate clinical risk factors, connect genetic risk to clinical utility, and provide clear guidance on interpretation and clinical actionability. The eMERGE network developed a “genome-informed risk assessment” (GIRA) report that compiles risk information from four sources: 1) clinical data from self-report and the electronic health record (EHR); 2) family history information provided by individuals during study enrollment; 3) clinical PRS results; and 4) clinical sequencing for a limited number of monogenic risks. The GIRA report will be incorporated into individuals’ medical records and returned to both individuals and their healthcare providers. Guidance is included in the report to tailor screening tests and behavioral interventions to genetic risk that could prevent or mitigate adverse health outcomes. The network will assess whether the GIRA influences downstream healthcare utilization, behavior changes, and understanding of disease risk. This manuscript describes the network’s approach to assessing integrated risk, development of the GIRA report, and the design of the prospective cohort study aimed at examining GIRA utility for 11 complex conditions.
Materials and Methods
Network organization
The eMERGE (electronic MEdical Records and GEnomics) network is a National Human Genome Research Institute (NHGRI)-supported consortium of 10 healthcare institutions within the United States and a coordinating center (CC) responsible for data integration, report generation, and overall network logistics (Fig. 1). In its first three phases, eMERGE successfully developed tools to use EHRs for discovery and to create programs to implement genomic medicine across large cohorts. For example, eMERGE developed methods for accurately extracting phenotypes from EHRs for use within large genome-wide association studies (GWAS) and pioneered the development of phenome-wide association studies (PheWAS).10–15 eMERGE also studied the clinical implementation of sequencing across large cohorts, returning genomic results to individuals, placing results directly into EHRs, and deploying related clinical decision support.11,12,16–19
Figure 1: eMERGE network organizational structure.
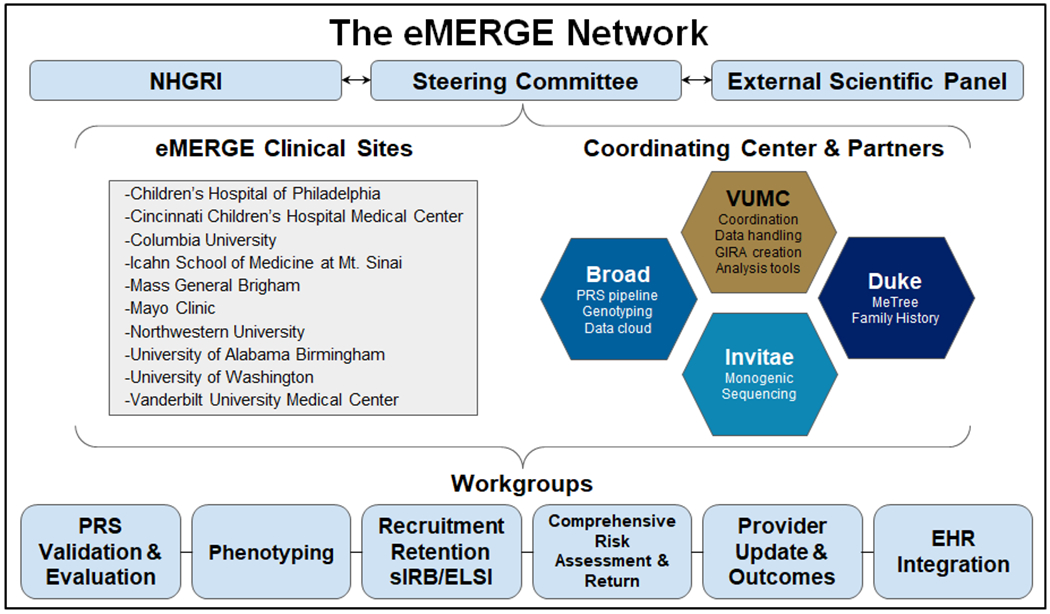
Under the guidance of the NHGRI, External Scientific Panel, and Steering committee, the ten clinical sites, coordinating center, and network partners make up six workgroups charged with developing the network protocol, risk scores, and methods as well as recruiting and returning results to 25,000 individuals. VUMC: Vanderbilt University Medical Center; PRS: Polygenic Risk Score; ELSI: Ethical Legal & Social Implications; sIRB: Single Institutional Review Board; EHR: Electronic Health Records.
To complete the study objectives of the fourth phase, the consortium has engaged three new partners to provide essential laboratory or point-of-care data collection capabilities: the Broad Institute (clinical PRS testing and reporting), Duke University (family history collection tool, MeTree), and Invitae (Invitae Corporation; clinical monogenic sequencing). The network hosts six workgroups (Supplemental Table S1) and is guided by a steering committee that meets twice monthly via teleconference and three times per year at hybrid meetings, as well as with a seven-member External Scientific Panel (ESP). Network decisions, rationale, and timelines are tracked centrally by the CC and displayed to the network via dashboards.
Study design
The eMERGE network aims to increase applicability of genomic risk prediction across populations by validating PRS in multiple ancestral groups and enrolling a prospective cohort that includes individuals who are currently underrepresented in clinical-genomic research. Six sites are committed to recruiting an “enhanced diversity cohort” with a target of 75% of individuals belonging to a racial or ethnic minority population or medically underserved, while the remainder of clinical sites will target 35%. Enrollment is not targeted to individuals with specific conditions, although individuals with prevalent conditions can be included. The network focuses on three major aims: 1) recruit 25,000 individuals (ages 3-75) from general healthcare system populations; 2) generate cross-ancestry and ancestry-adjusted PRS as the basis for reports to return risk alongside family health history, clinical, and monogenic risk; and 3) measure individual outcomes and provider behaviors and comprehension in response to receiving this information.
The Vanderbilt University Medical Center (VUMC) CC is the IRB of record (#211043) for the network’s single IRB, approved in July 2021. Exclusion criteria include inability to consent or receive results in English or Spanish, history of solid organ or bone marrow transplant, lack of a healthcare provider at the parent institution, and study personnel. The network utilizes a pre-screening survey prior to consent to ascertain eligibility. Individuals are consented electronically and then given two surveys to complete prior to providing biological samples (saliva or blood). Samples are sent to the Broad Institute for generation of polygenic risk reports and Invitae (adults only) for monogenic reports. Individuals also provide family health history data utilizing MeTree software. Data are received at the CC to generate GIRA reports (Fig. 2).
Figure 2: The GIRA report integrates multiple risk elements and is returned to individuals, providers, and the EHR.
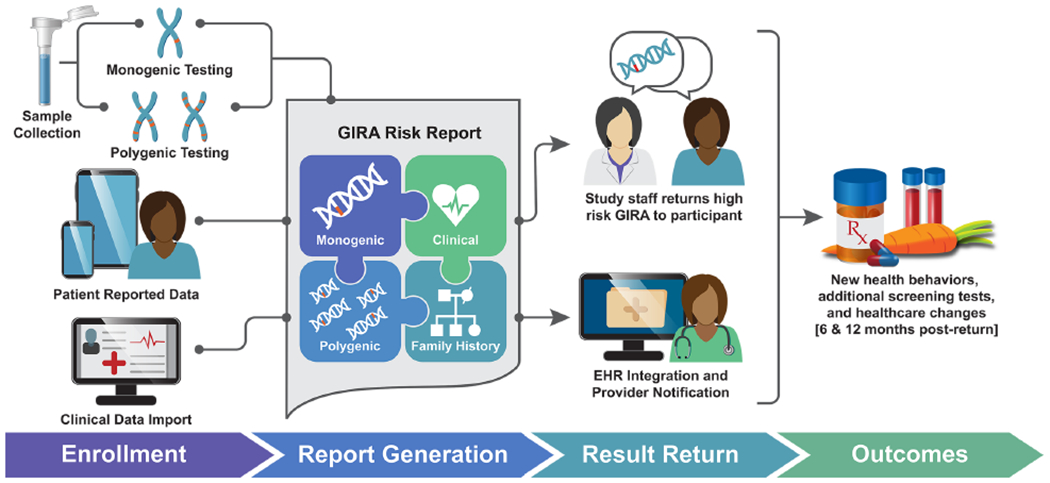
Individuals in the eMERGE study are enrolled and provide both self-reported (clinical and family history) data as well as biological samples. Polygenic risk (Broad report); monogenic risk (Invitae report); family health history (MeTree pedigree); and clinical data (from electronic health records and participant surveys) are collected and combined to create the Genome Informed Risk Assessment (GIRA) report. Results are returned along with care recommendations that can be used to guide screening recommendations and next steps for primary care providers. Outcomes are assessed to determine changes in healthcare behavior after return.
Condition selection
Conditions were selected after considering the analytic and clinical validity of the associated PRS, projected clinical actionability, and applicability of genomic risks to populations of diverse genetic ancestries. During validation studies, the network compared individuals above a selected threshold to all those below the threshold; scores with non-significant odds ratios (those with 95% confidence intervals that overlapped 1.0) in multiple genetic ancestries were deprioritized for implementation. Definitions for each selection criterion and categories of genomic risk are provided in the supplemental Glossary. The network reviewed 23 proposed conditions and selected 11 conditions for implementation (Table 1). The initial assessments included comprehensive literature reviews focused on PRS performance; the network then conducted independent secondary validations in additional clinical-genomic data sets across four genetic ancestry groups (European, Asian, African, and Hispanic). As the PRS for colorectal cancer (CRC) did not validate in either Hispanic or African ancestry sample data, the CRC PRS risk component was excluded from the GIRA. Four of the 11 conditions were determined to be pertinent clinically for individuals 3 - 17 years of age: asthma, obesity/BMI, type 1 and 2 diabetes (T1D, T2D). Asthma and T1D are only returned to those <18 years old, while obesity/BMI and T2D are returned to adults as well. Considering network ethical, legal, and social implication (ELSI) input and uncertain penetrance of monogenic genes on these four conditions, the network elected not to offer monogenic testing to individuals <18 years of age.
Table 1:
High risk criteria for Genome Informed Risk Assessment (GIRA) conditions
| Condition | Monogenic risk | Polygenic risk | Family history risk | Expected # high riska | ||
|---|---|---|---|---|---|---|
| Genes considered | P/LP Frequency | High risk threshold | Relatives | % increased risk | ||
| Pediatric only conditions (3 to 17 years at enrollment) | ||||||
| Asthma | N/A | N/A | 5% | N/A | N/A | 250 |
| Type 1 diabetes | N/A | N/A | 3% | N/A | N/A | 150 |
| Pediatric and adult conditions (3-75 years at enrollment) | ||||||
| Obesity/BMI | N/A | N/A | 3% | N/A | N/A | 750 |
| Type 2 diabetes | N/A | N/A | 2% | N/A | N/A | 500 |
| Adult only conditions (18-75 years at enrollment) | ||||||
| Atrial fibrillation | LMNA | 0.05% | 3% | Parents <75 | 5% | 610 |
| Breast cancer b,c | BRCA1; BRCA2; PALB2; PTEN; TP53; STK11 | 1.48% | 5% | 1st & 2nd degree | 11% | 348 |
| Chronic kidney disease | N/A | N/A | 2% | 1st degree | 9% | 400 |
| Colorectal cancer | EPCAM; MLH1; MSH2; MSH6; PMS2; STK11; PTEN; TP53 | 0.54% | N/A | N/A | N/A | 110 |
| Coronary heart disease | APOB; LDLR; LDLRAP1; PCSK9 | 0.89% | 5% | 1st degree | 9.8% | 1,178 |
| Hypercholesterolemia | APOB; LDLR; LDLRAP1; PCSK9 | 0.89% | 3% | N/A | N/A | 778 |
| Prostate cancer c | BRCA1; BRCA2; EPCAM; MLH1; MSH2; MSH6; PMS2 | 1.69% | 10% | 1st degree males | 9.4% | 1,169 |
Predicted ‘in person’ return for combined PRS and monogenic risk, does not account for overlap. Trigger hierarchy: Monogenic > Polygenic > Family history risk.
Breast cancer triggers from BOADICEA integrated score, 25% lifetime risk.
Breast and Prostate cancer risk returned to self-reported sex at birth of female and male, respectively. N ~ 5000 pediatric and N~ 20,000 adult individuals are expected.
Polygenic risk assessment
The network selected a genotyping panel for PRS generation based on the Global Diversity Array (GDA; Illumina, CA); the GDA balances cost and content and includes a large number of single nucleotide polymorphisms optimized for imputation performance across diverse populations. Standard imputation and analytical pipelines were implemented to support clinical reporting of the PRS, and the network developed clinical report language and a logic structure for the PRS components of the GIRA.
For each of the ten PRSs selected for clinical reporting, the threshold for high-risk status was selected by one of several methods that varied by phenotype. Thresholds were chosen to be equivalent to a clinically meaningful risk factor such as family history, or a corresponding odds ratio (OR) of ≥2 (Table 1). The Broad developed a phasing and imputation method based on the tools used by the Michigan Imputation Server20. The final PRS pipeline utilized two distinct population reference panels, the 1000 Genome project panel for imputation and parameters derived from the All of Us Research Program cohort for ancestry calibration. Imputation and PRS analytical validity were determined through a validation study in the Broad clinical laboratory that leveraged 42 samples with matched PCR-free whole genome sequences in 20 specimens with matched blood and saliva sample collection. The PRS determination pipelines were developed in the Terra cloud platform (www.terra.bio) using the Workflow Description Language (WDL), allowing the methods to be made available for research purposes in the NHGRI Genomic Data Science Analysis, Visualization, and Informatics Lab-space (AnVIL)21 platform (Supplemental Table S2).
Each condition selected underwent validation on relevant datasets based on the targeted PRS populations. The PRS pipeline performance was additionally verified on the eMERGE I-III cohorts12,22,23 by the Broad clinical laboratory to ensure the odds ratios, magnitudes, and significance could be independently verified. To accurately and equitably return scores across ancestry groups, an ancestry adjustment step was applied to the raw PRS scores, which was a derivative of the method first proposed by Khera et al. (2018)24 and includes variance correction.
Network discussions determined clinical report content. A pipeline for automated report creation was built at the Broad and includes a step for clinical laboratory staff review of results prior to release. Clinical reports are created as both PDF documents and structured data in JSON format, both of which are returned to the CC for inclusion in the GIRA and delivery to clinical sites.
Monogenic risk assessment
For a subset of conditions (Table 1), the network assessed monogenic risk with a limited panel of genes. Although funding for monogenic testing was not included in the initial eMERGE project, the network partnered with Invitae to support this testing with an in-kind contribution.
Consideration of which genes to include in monogenic testing occurred in parallel with selection of the final conditions for which PRS would be reported. The network finalized the list of genes, including the Center for Disease Control (CDC) Tier 1 conditions25 (hereditary breast and ovarian cancer syndrome, Lynch syndrome, and familial hypercholesterolemia) as well as five additional genes: TP53, STK11, PTEN, PALB226 which are important risk factors for colorectal and breast cancer, and LMNA which is associated with arrhythmias, including atrial fibrillation (Table 1). Variants of uncertain significance (VUS) will not be returned. Based on Invitae data, the network estimated a pathogenic or likely pathogenic (P/LP) result rate for this expanded panel, excluding heterozygote results from LDLRAP1, LMNA and EPCAM, of approximately 2.5%.27
Family history risk assessment
Family history is an important component of risk assessment for most conditions in this study and was therefore included as a high-risk criterion in the GIRA. MeTree, a family history application,28 is being utilized to collect variables on first and second degree relatives directly from the individuals for the conditions of interest. Family history text is displayed on all GIRA reports as contextual factors along with a pedigree. Family history is used as a high-risk return trigger for five conditions: atrial fibrillation, breast cancer (integrated into the BOADICEA29 model), chronic kidney disease, coronary artery heart disease (CHD), and prostate cancer (Table 1). The Duke team collaborated with network workgroups to customize MeTree to display only relevant eMERGE conditions and allow for a manual and automated data transfer to the CC eight weeks after individual account creation.
Clinical variables
As the study was focused on genetic risk, clinical risk factors alone do not trigger a high-risk notification. However, they are incorporated into the GIRA as contextual information for five conditions (asthma, chronic kidney disease, hypercholesterolemia, obesity, T2D); clinical data that is displayed include laboratory values, blood pressure, and ICD codes representing known diagnoses. Clinical variables are used to calculate a comprehensive risk score for two conditions (BOADICEA for breast cancer and the PCE/integrated risk score (IS) for CHD).
Data transfer & risk integration
Data use and transfer agreements for the eMERGE project were complex as they needed to encompass multiple sites, participant entered surveys, individual’s EHR data, and data generated by network partners. The CC executed data use and transfer agreements to authorize the intake and storage of identifiable data in a centralized repository and web application based on REDCap.30,31 The custom application was named R4 after the primary functions supported for the study: Recruitment, Results reporting, and Risk Reduction. This REDCap project utilizes data access groups to allow site-specific access to identifiable information and customized programming to generate individualized GIRA reports based on upstream data variables and associated standardized display text. Fig. 3 describes the data flow across the network. The CC established application programming interfaces (APIs) with the three network partners (Broad, Duke, and Invitae) to receive structured and PDF reports. GIRA also utilizes data elements from the participant surveys to generate calculated fields. Body mass index (BMI) and the pooled cohort equation (PCE) that predicts 10-year risk for a first atherosclerotic cardiovascular disease event32 were integrated directly into REDCap, and an API was established with CanRisk33 to calculate and send back BOADICEA29 scores (Breast and Ovarian Analysis of Disease Incidence and Carrier Estimation Algorithm) for breast cancer. Once all data import instruments are completed, the REDCap record is locked and the GIRA is generated for a given individual.
Figure 3: Research infrastructure for assembling risk information from site enrollment data collection, site EHRs, partner laboratories, and point-of-care tools for collecting family history.
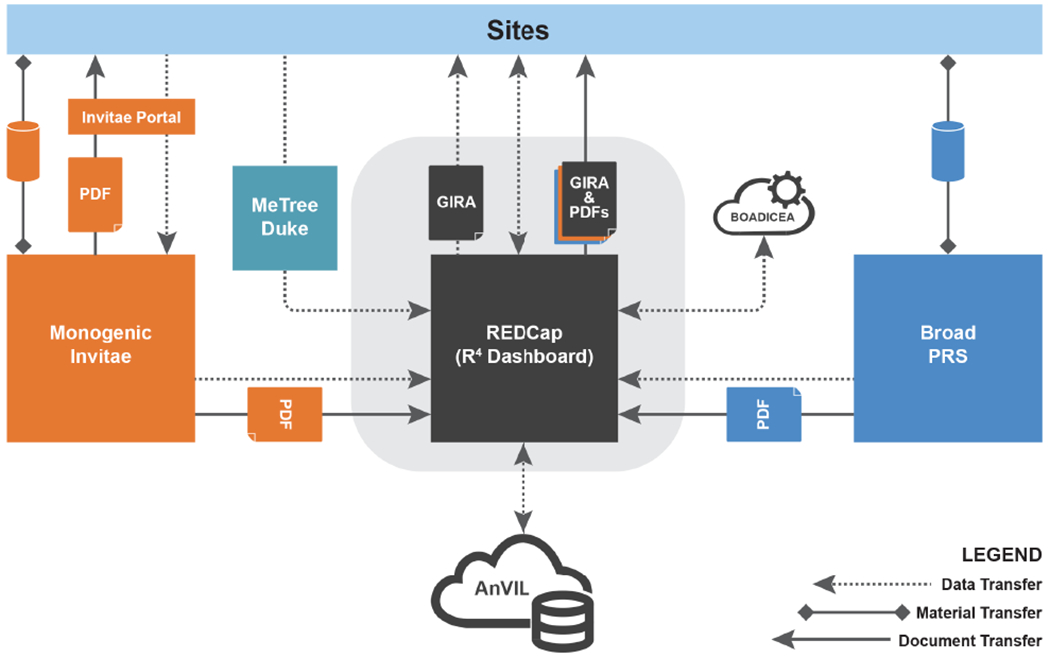
Data are transferred through APIs from partners and sites to the central VUMC housed REDCap system, R4. Custom REDCap programming was developed to generate the overall Genome Informed Risk Assessment (GIRA) report which is made available to sites along with component data and polygenic risk score (PRS) and monogenic laboratory reports.
GIRA return and outcome assessment plan
As care recommendations based on polygenic risk are not currently driven by guidelines, the network adapted existing clinical recommendations associated with risks of similar magnitude to provide evidence-based guidance to individuals and providers. Recommendations were drafted and reviewed iteratively over an eight-month period in light of feedback from study site physicians, findings from ELSI-led focus groups and interviews with physicians34, and the steering committee. The goal was to supplement the current standard of care for conditions of interest and provide individuals and providers with well-reasoned next steps. Family history and monogenic recommendations were taken directly from existing guidelines.
Individuals receiving high-risk GIRA will have a high touch ‘one-on-one’ return, during which a trained study staff member, study MD, or site genetic counselor will meet with the individual to explain the results and next steps. For scenarios when only family history risk triggers a ‘high-risk’ GIRA and there are no identified genomic risk factors, individuals will still receive a ‘high-risk’ GIRA; however, results will be returned by mail or electronically to individuals. GIRA reports are placed into the EHR and communicated to the individual’s study provider, which encompasses traditional primary care and specialists providing longitudinal care to individuals. The return methods at each site are customized based on current legal and institutional regulations. GIRA results without risk factors are deemed ‘not high-risk’; the network elected to avoid the terms ‘low risk’ in any study communications to avoid inappropriate reassurance. ‘Not high-risk’ results are returned via mail or patient portal and placed in the EHR. Educational material for patients and clinicians, as well as training of study staff about the study and its processes, were harmonized across sites.
The impact of return of results (ROR) on process and clinical outcomes will be assessed six months after ROR by EHR data and participant survey and twelve months using EHR data alone. The participant survey will examine individuals’ understanding, clarity of results, how individuals felt after receiving results, and lifestyle changes. The network will examine the following outcomes among ‘high-risk’ individuals compared to ‘not high-risk’ individuals (Supplemental Fig. S1): adoption of recommendations, uptake of risk-reducing interventions, and new diagnoses. Condition-specific analyses will be conducted that analyze prespecified process and clinical outcomes and will assess potential confounders and effect modifiers. For example, we will conduct a stratified analysis of high-risk individuals based on whether they have prevalent disease. Additionally, the propensity for high-risk individuals without disease to have risk factors that could prompt testing will be investigated with a regression discontinuity design that controls for disease related confounders. A control analysis will evaluate the contribution of an in-person, high-touch return to changes in process outcomes independent of the risk information, by assessing condition-specific outcomes among individuals who receive high-touch return for an unrelated condition.
The network estimated the overall effect size (difference in adoption of recommendations among high-risk vs. not high-risk) under conservative assumptions that the uptake of recommendations would be 20% in the high-risk group, and background rates of these recommendations in standard health care (not high-risk group) would be approximately 10%. Provider surveys will focus on perceived utility and understanding of the GIRA as well as assess if providers communicated with individuals about results. Targeted provider interviews are also planned to gain a more in depth understanding of the impact of the GIRA on care.
Results:
Enrollment began in February of 2022. As of December 2022, the 10 sites have enrolled 5671 individuals. Of those, 3688 (65%) are female at birth; 1978 (35%) are male at birth, and <1% did not indicate sex assigned at birth. Nearly half (47%) of individuals self-reported they are members of a racial or ethnic minority group and an additional 4% indicated more than one race. Hispanic ethnicity accounted for approximately 18% of enrolled individuals; see Supplement Fig. S2. Samples have begun being processed by the Broad Institute, Invitae, and family history data is currently being collected through MeTree. GIRA generation is targeted to begin in December 2022 with the first return to individuals and providers shortly thereafter.
Genome Informed Risk Assessments (GIRA)
The network created the GIRA report to concisely summarize the risk (polygenic, monogenic, family history, and clinical factors) of developing 11 common, complex conditions and to display clinical recommendations to providers and individuals. For the breast cancer and coronary heart disease conditions integrated risk scores are displayed on the GIRA (BOADICEA and Pooled Cohort Equation, respectively); for the remaining conditions, the independent risk factors are displayed along with relevant clinical factors for context). The GIRA report (Supplemental Document S1) consists of a cover page, one page summary of high-risk conditions, a breakdown of risk type (monogenic, polygenic, family history), education pages, frequently asked questions, methods, and a family history pedigree. Additionally, laboratory reports from both Invitae (for adults) and the Broad Institute are attached to the GIRA (laboratory reports not shown in supplemental information). The example GIRA includes high-risk findings for atrial fibrillation (pathogenic LMNA variant), breast cancer (BOADICEA score result > 25%), and T2D (PRS above threshold) with accompanying clinical risk factors. The frequently asked questions and education materials were included to clarify each risk type and increase overall understanding. The network gathered multiple sources of expertise including ELSI studies, physician, and potential participant feedback to develop the report34. Each GIRA report is dichotomized into high-risk’ and ‘not high-risk’ to more clearly communicate actionability as interviews with potential participants indicated a dichotomized risk was much more understandable than a quantitative risk. Details on the odds ratios and confidence intervals for the high-risk cut-off are reported stratified by ancestry in the Broad laboratory report to provide details for providers (or individuals) interested in the specifics. A table of GIRA PRS text including odds ratios can be found in Supplemental Table S3. For the two conditions utilizing integrated risk scores (breast cancer and CHD), calculated absolute risks are displayed on the GIRA itself as they are commonly utilized in health care for clinical decision making.
The network focused on PRS as the core component of genomic risk assessment. ELSI studies of study candidates were interested in receiving PRS even if they could not be fully validated in all ancestral populations, demonstrating an urgent need for developing risk assessment across populations. Monogenic and family history risk could also trigger high-risk status for many of the conditions. Table 1 displays how the different components of the GIRA trigger high-risk status. Though not all conditions use all three risk types, overall risk was generally designated utilizing the following hierarchy: monogenic risk > polygenic risk > family history. While the overall risk on the first page of the GIRA is triggered by the above hierarchy, all risk types (monogenic, polygenic, family history) are displayed on the second page of the GIRA along with risk specific text in order to provide the individual and provider with the most comprehensive information. There are two exceptions where integrated scores are available. Breast cancer for adult females at birth used a BOADICEA score; individuals with greater than or equal to 25% lifetime breast cancer risk conveyed a high-risk status. For individuals at high-risk for CHD, the PCE both with and without the PRS incorporated is displayed on reports and is intended to assist physicians with next steps while providing standard of care information. Unlike for BOADICEA, the PCE is not used to determine risk, it is displayed as context for the provider and individual.
The network modeled the expected number of high-risk return of conditions (Table 1) to determine maximum impact and sample sizes; approximately 25% (~6,200) of the 25,000 individuals are expected to receive at least one high-risk return. Estimates were primarily based on frequency of high monogenic and polygenic risk as those elements were the main components to generate a one-on-one (high touch) return for the majority of conditions. These calculations do not account for potential correlations between conditions in same individual or the small overlap between monogenic and high PRS risk.
Care recommendations
The GIRA report includes clinical recommendations for individuals receiving high-risk results. The network conducted multiple physician-led discussion groups, compiled recommendations from experts across ten clinical sites, and went through multiple iterations of the care recommendations to supply the providers with artifacts that accurately reflected clinical care based on other risk factors (clinical, family history, monogenic) in this novel risk space. The network aimed to supplement current guidelines and to provide recommendations for those with a high PRS. Table 2 summarizes the projected clinical utility and next steps recommended for each of the 11 conditions, Supplemental Table S4 provides the detailed recommendations for high-risk PRS results along with references for rationale on recommendations. Recommended actions included counseling and education as well as additional screening to consider involving laboratory tests, imaging, and referrals. Monogenic risk recommendations and family history-based recommendations were taken directly from clinical guidelines by the National Comprehensive Cancer Network (NCCN)1,2 and cardiology societies3; these recommendations are not displayed in the supplemental materials. Outcomes assessments (Table 2) are derived directly from the care recommendations.
Table 2.
Clinical utility and actionability of eMERGE return.
| Condition | Actionability | ||
|---|---|---|---|
| Clinical utility | Counseling and education | Laboratory tests and procedures | |
| Asthma | Early intervention and therapy; 4x more common in AAa | Reduce environmental triggers | Provider encounter (education) |
| Atrial fibrillation | Cost effective screening; early detection | Emphasize healthy lifestyle; assess symptoms | Test ordered (ECG; rhythm monitor) |
| Breast cancer | Enhanced screening & therapy; higher mortality in AA; BOADICEA score returned | Emphasize healthy lifestyle; screening | Imaging order (breast MRI; mammogram) |
| Chronic kidney disease | Early non-invasive screening; high prevalence >10% adults; 3.7x more common in AA | Emphasize healthy lifestyle; screening | Lab order (serum creatinine, urine albumin to creatinine ratio) |
| Colorectal cancer | Early non-invasive screening; risk varies by race & ethnicity | Early screening | Procedure order (stool test, flex sigmoidoscopy, colonoscopy, ct colonography) |
| Coronary heart disease | Non-invasive screening and intervention; highly prevalent disease; pooled cohort equation returned | Emphasize healthy lifestyle; screening | Lab order (lipid profile) imaging order (coronary CT, carotid ultrasound) |
| Hypercholesterolemia | Non-invasive, accessible screenings; disproportionate burden in AA | Emphasize healthy lifestyle; screening; medication | Lab order (lipid profile) |
| Obesity/BMI | Early intervention to modify behavior; weight in childhood is predictive of adult obesity | Emphasize healthy lifestyle; referral | Referral (weight loss or nutrition consultation) |
| Prostate cancer | Non-invasive, accessible screenings; population disparities in incidence and mortality | Shared decision making about early screening | Provider encounter; exam |
| Type 1 diabetes | Treatment dependent on distinguishing between diabetes types | Education regarding symptoms; screening | Lab order (autoantibodies) |
| Type 2 diabetes | Non-invasive screening; highly prevalent disease; racial disparities in care | Emphasize healthy lifestyle; screening | Lab order (fasting blood glucose, A1c) |
AA = African American;
Data harmonization & electronic health record integration
Multiple types of data representing the three types of genomic risk will be generated, collated, and utilized to produce the GIRA report for each individual (Fig. 3). The study aims to track how providers make use of the GIRA to inform medical decision making and care once it is incorporated into the EHR and into existing workflows for the review of medical information. Educational materials are being developed for linkage through info buttons and different versions of clinical decision support to assist clinicians receiving GIRA results. eMERGE sites primarily use the Epic EHR, with one site using Cerner. The network attempted to harmonize EHR GIRA integration methodology in advance, but discussions during monthly EHR Integration workgroup meetings indicated that some heterogeneity across sites was unavoidable. In addition to configuration differences of each EHR, sites considered the GIRA a “research report”, in that it is generated on behalf of a research study. While all sites are able to make a PDF of the GIRA accessible to providers within their EHR, site specific policies resulted in variation about where in the EHR the report was displayed or localized. In addition to the GIRA PDF, sites also had to develop integration plans for the two component genetic reports (monogenic and PRS), and had to consider whether structured data representations of any of the three reports would also be incorporated into the EHR, particularly for use in providing context-aware decision support for clinicians and individuals.35
Discussion:
This phase of the eMERGE network was established to formulate scalable methods for returning integrated genomic risk to individuals and providers, including PRS, monogenic, and family history based risks in the context of traditional clinical risks. Using a prospective cohort design, the study will determine whether returning the GIRA with recommendations and counseling for high-risk individuals is effective at increasing appropriate clinical actions to mitigate the risk of future disease or detect unrecognized disease.
Predicting the onset of common diseases or other health outcomes is essential to tailoring preventive care to individual risk. Established models focused on clinical variables for cardiovascular disease (PCE) and breast cancer (e.g., Breast Cancer Risk Assessment Tool - BRCAT) lack simultaneous consideration of monogenic and polygenic risk, and family history-based risk is inconsistently measured and applied.36,37 Developing more comprehensive tools, similar to the BOADICEA equation used in the eMERGE GIRA, will improve risk prediction and promises to guide clinical management more effectively, especially for individuals who do not know their family history of disease. Additionally, adding PRS to existing models, as has been done for the PCE, could identify an expanded group of individuals for whom primary prevention or intensified surveillance is appropriate.38 To learn how best to maximize the utility of the GIRA, the eMERGE study will both identify and address barriers to implementing health recommendations as well as assess psychosocial harms incurred from learning high-risk or overinterpreting not high-risk results.
A major obstacle the network had to overcome while developing PRS for the chosen conditions was ensuring they were applicable to the diverse range of individuals that the network aims to recruit. Sufficiently diverse GWAS data are lacking to support cross-ancestry scores for many common medical conditions, even while it has been demonstrated how genetically diverse population data are critical to the accuracy of the method.9,39,40 By limiting clinically implemented scores to those validated in multiple ancestral populations, the network aimed to increase the applicability of the risk scores.
The network’s decisions regarding conditions included in the GIRA implementation, selection of PRS thresholds or integrated scores, and clinical recommendations were based on review of evidence, expert opinion, and the application of clinical guidelines written to address risks of comparable magnitude. However, there are currently no clinical standards for PRS implementation, and the study may reveal the strengths and weaknesses of these early decisions. We expect significant refinements will be needed within PRS development, reporting, and integration with other genetic findings for future implementations. The study will also be challenged to ensure provider and individuals understanding of the limitations and clinical significance of the GIRA results and appropriately translate the report recommendations to changes to screening practices or lifestyle. By measuring uptake of follow-up testing and recommendations in parallel with provider and individuals understanding, the study will shed light on how integrated genomic risk will be received in clinical practice.
As currently designed, the GIRA report is limited to information that is available with electronic records and laboratory reporting. Tailored preventive care plans recommended by providers at the point of care may need to consider many patient factors not included in the report, such as medical history, personal preferences, access to care, influence of social and physical environments, and the cost of care. Additionally, by limiting the recruited population to those already receiving longitudinal care at established health care institutions, the study will not reflect the full set of challenges experienced by the general population in obtaining individualized preventive care. Finally, as the report summarizes evidence within a rapidly evolving scientific field, it is likely that report redesign and updates to the underlying PRS, integrated score algorithms, and clinical recommendations will be necessary if it is proven valuable to adopt in clinical practice.
This eMERGE study aims to improve understanding of risk stratification in the field of genomics by implementing a novel integrated genomic risk with increased applicability to diverse populations. Demonstrating clinical uptake and understanding will help to establish clinical utility and facilitate additional comparative or clinical implementation studies.
Supplementary Material
Acknowledgements:
The network would like to thank the patient and physician advisory groups that helped shape study design, report content, and education materials. Due to the structure of the U01 cooperative agreement, the recruitment sites, coordinating center, and funding agency collaborated on the study design as well as collection, management, analysis, and interpretation of the data. This trial was registered with clinicaltrials.gov under identifier NCT05277116. The eMERGE Genomic Risk Assessment Network is funded by the National Human Genetic Research Institute (NHGRI) through the following grants: U01HG011172 (Cincinnati Children’s Hospital Medical Center); U01HG011175 (Children’s Hospital of Philadelphia); U01HG008680 (Columbia University); U01HG011176 (Icahn School of Medicine at Mount Sinai);U01HG008685 (Mass General Brigham); U01HG006379 (Mayo Clinic); U01HG011169 (Northwestern University); U01HG011167 (University of Alabama at Birmingham); U01HG008657 (University of Washington); U01HG011181 (Vanderbilt University Medical Center); U01HG011166 (Vanderbilt University Medical Center serving as the Coordinating Center).
Conflict of Interest:
The authors declare no conflict of interest except for the following individuals: Noura S. Abul-Husn is an employee and equity holder of 23andMe; serves as a scientific advisory board member for Allelica; received personal fees from Genentech, Allelica, and 23andMe; received research funding from Akcea; and was previously employed by Regeneron Pharmaceuticals. David R. Crosslin is a consultant for Optum Genomics and Chinook Therapeutics. Theresa Walunas has grant funding from Gilead Sciences. Lori Orlando and Tejinder Rakhra-Burris are founders of a company developing MeTree. Tara Schmidlen, Edward D. Esplin, and Eden Haverfield are employees and stockholders of Invitae. Elizabeth M. McNally has been a consultant for Avidity, Amgen, AstraZeneca, Cytokinetics, Invitae, Janssen, Pfizer, PepGen, Tenaya Therapeutics, Stealth BioTherapeutics; she is also the founder of Ikaika Therapeutics. Eimear E. Kenny received personal fees from Illumina, 23andMe, and Regeneron Pharmaceuticals, and serves as a scientific advisory board member for Encompass Bio, Foresite Labs, and Galateo Bio. Bruce Korf is an advisory board member and stockholder of GenomeMedical. Maya Sabatello is a member of the IRB of the All of Us Research Program. Emma F. Perez is a paid consultant for Allecia. Josh F. Peterson is a paid consultant for Natera.
The eMERGE Consortium:
Adam Gordon, Agboade Sobowale, Aimee Allworth, Akshar Patel, Alanna DiVietro, Alanna Strong, Alborz Sherafati, Alborz Sherfati, Alex Bick, Alexandra Miller, Alka Chandel, Alyssa Rosenthal, Amit Khera, Amy Kontorovich, Andrew Beck, Andy Beck, Angelica Espinoza, Anna Lewis, Anya Prince, Atlas Khan, Ayuko Iverson, Bahram Namjou Khales, Barbara Benoit, Becca Hernan, Ben Kallman, Ben Kerman, Ben Shoemaker, Benjamin Satterfield, Beth Devine, Bethany Etheridge, Blake Goff, Bob Freimuth, Bob Grundmeier, Brenae Collier, Brenda Mutai, Brett Harnett, Brian Chang, Brian Piening, Brittney Davis, Bruce Korf, Candace Patterson, Carmen Demetriou, Casey Ta, Catherine Hammack, Catrina Nelson, Caytie Gascoigne, Chad Dorn, Chad Moretz, Chris Kachulis, Christie Hoell, Christine Cowles, Christoph Lange, Chunhua Weng, Cindy Prows, Cole Brokamp, Cong Liu, Courtney Scherr, Crystal Gonzalez, Cynthia Ramirez, Daichi Shimbo, Dan Roden, Daniel Schaid, Dave Kaufman, David Crosslin, David Kochan, David Veenstra, Davinder Singh, Dean Karavite, Debbie Abrams, Devin Absher, Digna Velez Edwards, Eden Haverfield, Eduardo Morales, Edward Esplin, Edyta Malolepsza, Ehsan Alipour, Eimear Kenny, Elisabeth Rosenthal, Eliza Duvall, Elizabeth McNally, Elizabeth Bhoj, Elizabeth Cohn, Elizabeth Hibler, Elizabeth Karlson, Elizabeth McNally, Ellen Clayton, Emily Chesnut, Emily DeFranco, Emily Gallagher, Emily Soper, Emma Perez, Erin Cash, Eta Berner, Fei Wang, Firas Wehbe, Francisco Ricci, Frank Mentch, Gabriel Shaibi, Gail Jarvik, George Hahn, George Hripcsak, Georgia Wiesner, Gillian Belbin, Gio Davogustto, Girish Nadkarni, Haijun Qiu, Hakon Hakonarson, Hana Bangash, Hannah Beasley, Hao Liu, Heide Aungst, Hemant Tiwari, Hillary Duckham, Hope Thomas, Iftikhar Kullo, Ingrid Holm, Isabelle Allen, Iuliana Ionita-Laza, Jacklyn Hellwege, Jacob Petrzelka, Jacqueline Odgis, Jahnavi Narula, Jake Petrzelka, Jalpa Patel, James Cimino, James Meigs, James Snyder, Janet Olson, Janet Zahner, Jeff Pennington, Jen Pacheco, Jennifer Allen Pacheco, Jennifer Morse, Jeremy Corsmo, Jeritt Thayer, Jim Cimino, Jingheng Chen, Jocelyn Fournier, Jodell Jackson, Joe Glessner, Joel Pacyna, Johanna Smith, John Connolly, John Lynch, John Shelley, Jonathan Mosley, Jordan Nestor, Jordan Smoller, Jorge Alsip, Joseph Kannry, Joseph Sutton, Josh Peterson, Joshua Smith, Julia Galasso, Julia Smith, Julia Wynn, Justin Gundelach, Justin Starren, Karmel Choi, Kate Mittendorf, Katherine Anderson, Katherine Bonini, Kathleen Leppig, Kathleen Muenzen, Katie Larkin, Kelsey Stuttgen, Ken Wiley, Kenny Nguyen, Kevin Dufendach, Kiley Atkins, Konrad Sawicki, Kristjan Norland, Krzysztof Kiryluk, Laura Beskow, Laura Rasmussen-Torvik, Leah Kottyan, Li Hsu, Lifeng Tian, Lisa Mahanta, Lisa Martin, Lisa Wang, Lizbeth Gomez, Lorenzo Thompson, Lori Orlando, Lucas Richter, Luke Rasmussen, Lynn Petukhova, Lynn Seabolt, Madison O’Brien, Maegan Harden, Malia Fullerton, Margaret Harr, Mark Beasley, Marta Guindo, Martha Horike, Martha Horike-Pyne, Marwah Abdalla, Marwan Hamed, Mary Beth Terry, Mary Maradik, Matt Wyatt, Matthew Davis, Matthew Lebo, Maureen Smith, Maya del Rosario, Maya Sabatello, Meckenzie Behr, Meg Roy-Puckelwartz, Mel Habrat, Melanie Myers, Meliha Yetisgen, Merve Iris, Michael DaSilva, Michael Preuss, Michelle McGowan, Mingjian Shi, Minoli Perera, Minta Thomas, Mitch Elkind, Mohammad Abbass, Mohammad Saadatagah, Molly Hess, Molly Maradik, Nataraja “RJ” Vaitinadin, Nataraja Vaitinadin, Naveen Muthu, Neil Netherly, Niall Lennon, Ning Shang, Nita Limdi, Noah Forrest, Noheli Romero, Nora Robinson, Noura Abul-Husn, Omar Elsekaily, Ozan Dikilitas, Patricia Kovatch, Patrick Davis, Paul Appelbaum, Paul Francaviglia, Paul O’Reilly, Paulette Chandler, Pedro Caraballo, Peter Tarczy-Hornoch, Pierre Shum, Priya Marathe, Priyanka Murali, Qiping Feng, Quinn Wells, Rachel Atchley, Radhika Narla, Rene Barton, Rene Sterling, Rex Chisholm, Richard Green, Richard Sharp, Riki Peters, Rita Kukafka, Robb Rowley, Robert Freimuth, Robert Green, Robert Winter, Roger Mueller, Ruth Loos, Ryan Irvin, Sabrina Suckiel, Sajjad Hussain, Samer Sharba, Sandy Aronson, Sarah Jones, Sarah Knerr, Scott Nigbur, Scott Weiss, Sean Mooney, Shannon Terek, Sharon Aufox, Sharon Nirenberg, Shawn Murphy, Sheila O’Byrne, Shing Wang (Sam) Choi, Sienna Aguilar, ST Bland, Stefanie Rodrigues, Stephanie Ledbetter, Stephanie Rutledge, Stuart James Booth, Su Xian, Susan Brown Trinidad, Suzanne Bakken, Tara Schmidlen, Tejinder Rakhra-Burris, Teri Manolio, Tesfaye Mersha, Theresa Walunas, Thevaa Chandereng, Thomas May, Tian Ge, Todd Edwards, Tom Kaszemacher, Valentina Hernandez, Valerie Willis, Vemi Desai, Vimi Desai, Virginia Lorenzi, Vivian Gainer, Wei-Qi Wei, Wendy Chung, Wu-Chen Su, Xiao Chang, Yiqing Zhao, Yuan Luo, Yufeng Shen.
Footnotes
Publisher's Disclaimer: This is a PDF file of an article that has undergone enhancements after acceptance, such as the addition of a cover page and metadata, and formatting for readability, but it is not yet the definitive version of record. This version will undergo additional copyediting, typesetting and review before it is published in its final form, but we are providing this version to give early visibility of the article. Please note that, during the production process, errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Ethics Declaration
The eMERGE Genomic Risk Assessment study was approved by a central institutional review board at Vanderbilt University Medical Center (#211043) and acknowledged by the local institutional review boards under reliance agreements. Informed consent is required from all enrolled individuals.
Contributor Information
Jodell E. Linder, Vanderbilt Institute for Clinical and Translational Research, Vanderbilt University Medical Center, Nashville, TN
Aimee Allworth, Department of Medicine (Medical Genetics), University of Washington Medical Center, Seattle, WA.
Harris T. Bland, Department of Biomedical Informatics, Department of Medicine, Vanderbilt University Medical Center, Nashville, TN
Pedro J. Caraballo, Department of Internal Medicine and Department of Quantitative Health Sciences, Mayo Clinic, Rochester, MN
Rex L. Chisholm, Center for Genetic Medicine, Northwestern University, Chicago, IL
Ellen Wright Clayton, Center for Biomedical Ethics, Vanderbilt University Medical Center, Nashville, TN.
David R. Crosslin, Division of Biomedical Informatics and Genomics, John W. Deming Department of Medicine, Tulane University School of Medicine, New Orleans, LA
Ozan Dikilitas, Department of Internal Medicine, Department of Cardiovascular Medicine, Mayo Clinician Investigator Training Program, Mayo Clinic, Rochester, MN.
Alanna DiVietro, Vanderbilt Institute for Clinical and Translational Research, Vanderbilt University Medical Center, Nashville, TN.
Edward D. Esplin, Invitae, San Francisco, CA
Sophie Forman, Vanderbilt Institute for Clinical and Translational Research, Vanderbilt University Medical Center, Nashville, TN.
Robert R. Freimuth, Department of Artificial Intelligence and Informatics, Mayo Clinic, Rochester, MN
Adam S. Gordon, Department of Pharmacology & Center for Genetic Medicine, Northwestern University, Chicago, IL
Richard Green, Department of Biomedical Informatics and Medical Education, University of Washington Medical Center, Seattle, WA.
Maegan V. Harden, Broad Institute of MIT and Harvard, Cambridge, MA
Ingrid A. Holm, Division of Genetics and Genomics and Manton Center for Orphan Diseases Research, Boston Children’s Hospital, Department of Pediatrics, Harvard Medical School, Boston, MA
Gail P. Jarvik, Departments of Medicine (Medical Genetics) and Genome Science, University of Washington Medical Center, Seattle, WA
Elizabeth W. Karlson, Division of Rheumatology, Inflammation and Immunity, Department of Medicine, Brigham and Women’s Hospital, Boston, MA
Sofia Labrecque, Vanderbilt Institute for Clinical and Translational Research, Vanderbilt University Medical Center, Nashville, TN.
Niall J. Lennon, Broad Institute of MIT and Harvard, Cambridge, MA
Nita A. Limdi, Department of Neurology, The Heersink School of Medicine, University of Alabama at Birmingham, Birmingham, AL
Kathleen F. Mittendorf, Vanderbilt-Ingram Cancer Center, Vanderbilt University Medical Center, Nashville, TN
Shawn N. Murphy, Department of Neurology, Massachusetts General Hospital, Boston, MA
Lori Orlando, Center for Applied Genomics & Precision Medicine, Duke University, Durham, NC.
Cynthia A. Prows, Divisions of Human Genetics and Patient Services, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH
Luke V. Rasmussen, Department of Preventive Medicine, Northwestern University, Chicago, IL
Laura Rasmussen-Torvik, Department of Preventive Medicine, Northwestern University, Chicago, IL.
Robb Rowley, Division of Genomic Medicine, National Human Genome Research Institute, Bethesda, MD.
Konrad Teodor Sawicki, Department of Cardiology and Center for Genetic Medicine, Northwestern University, Chicago, IL.
Tara Schmidlen, Invitae, San Francisco, CA.
Shannon Terek, Center for Applied Genomics, Children’s Hospital of Philadelphia, Philadelphia, PA.
David Veenstra, Department of Pharmacy, University of Washington, Seattle, WA.
Digna R. Velez Edwards, Division of Quantitative Science, Department of Obstetrics and Gynecology, Department of Biomedical Sciences, Vanderbilt University Medical Center, Nashville, TN.
Devin Absher, Kaiser Permanente, Oakland, CA.
Noura S. Abul-Husn, The Institute for Genomic Health, Department of Medicine, Department of Genetics and Genomic Sciences, Icahn School of Medicine at Mount Sinai, New York, NY
Jorge Alsip, UAB Medicine Enterprise, Birmingham, AL.
Hana Bangash, Department of Cardiovascular Medicine, Mayo Clinic, Rochester, MN.
Mark Beasley, Department of Biostatistics, School of Public Health, University of Alabama at Birmingham, Birmingham, AL.
Jennifer E. Below, Department of Medicine, Division of Genetic Medicine, Vanderbilt Genetics Institute, Vanderbilt University Medical Center, Nashville, TN
Eta S. Berner, Department of Health Services Administration, University of Alabama at Birmingham, Birmingham, AL
James Booth, Department of Emergency Medicine, The Heersink School of Medicine, University of Alabama at Birmingham, Birmingham, AL.
Wendy K. Chung, Departments of Pediatrics and Medicine, Columbia University Irving Medical Center New York, NY
James J. Cimino, Department of Medicine, Division of General Internal Medicine and the Informatics Institute, The Heersink School of Medicine, University of Alabama at Birmingham, Birmingham, AL
John Connolly, Center for Applied Genomics, Children’s Hospital of Philadelphia, Philadelphia, PA.
Patrick Davis, Department of Biomedical Informatics and Medical Education, University of Washington Medical Center, Seattle, WA.
Beth Devine, Department of Pharmacy, University of Washington, Seattle, WA.
Stephanie M. Fullerton, Department of Bioethics and Humanities, University of Washington School of Medicine, Seattle, WA
Candace Guiducci, Broad Institute of MIT and Harvard, Cambridge, MA.
Melissa L. Habrat, Department of Biomedical Informatics and Medical Education, University of Washington Medical Center, Seattle, WA
Heather Hain, Center for Applied Genomics, Children’s Hospital of Philadelphia, Philadelphia, PA.
Hakon Hakonarson, Center for Applied Genomics, Children’s Hospital of Philadelphia, Philadelphia PA.
Margaret Harr, Center for Applied Genomics, Children’s Hospital of Philadelphia, Philadelphia, PA.
Eden Haverfield, Invitae, San Francisco, CA.
Valentina Hernandez, Mountain Park Health Center, Phoenix, AZ.
Christin Hoell, Department of Obstetrics and Gynecology & Center for Genetic Medicine, Northwestern University, Chicago, IL.
Martha Horike-Pyne, Department of Medicine (Medical Genetics), University of Washington Medical Center, Seattle, WA.
George Hripcsak, Department of Biomedical Informatics, Columbia University Irving Medical Center, New York, NY.
Marguerite R. Irvin, Department of Epidemiology, School of Public Health, University of Alabama at Birmingham, Birmingham, AL
Christopher Kachulis, Broad Institute of MIT and Harvard, Cambridge, MA.
Dean Karavite, Department of Biomedical and Health Informatics, Children’s Hospital of Philadelphia, Philadelphia, PA.
Eimear E. Kenny, The Institute for Genomic Health, Department of Medicine, Department of Genetics and Genomic Sciences, Icahn School of Medicine at Mount Sinai, New York, NY
Atlas Khan, Division of Nephrology, Department of Medicine, Columbia University Irving Medical Center, New York, NY.
Krzysztof Kiryluk, Division of Nephrology, Department of Medicine, Columbia University Irving Medical Center, New York, NY.
Bruce Korf, Department of Genetics, Heersink School of Medicine, University of Alabama at Birmingham, Birmingham, AL.
Leah Kottyan, Center for Autoimmune Genomics and Etiology, Division of Human Genetics, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH.
Iftikhar J. Kullo, Department of Cardiovascular Medicine, Mayo Clinic, Rochester, MN
Katie Larkin, Broad Institute of MIT and Harvard, Cambridge, MA.
Cong Liu, Department of Biomedical Informatics, Columbia University Irving Medical Center, New York, NY.
Edyta Malolepsza, Broad Institute of MIT and Harvard, Cambridge, MA.
Teri A. Manolio, Division of Genomic Medicine, National Human Genome Research Institute, Bethesda, MD
Thomas May, Elson S. Floyd College of Medicine, Washington State University, Vancouver, WA.
Elizabeth M. McNally, Center for Genetic Medicine, Northwestern University, Chicago, IL
Frank Mentch, Center for Applied Genomics, Children’s Hospital of Philadelphia, Philadelphia, PA.
Alexandra Miller, Department of Cardiovascular Medicine, Mayo Clinic, Rochester, MN.
Sean D. Mooney, Department of Biomedical Informatics and Medical Education, University of Washington Medical Center, Seattle, WA
Priyanka Murali, Department of Medicine (Medical Genetics), University of Washington Medical Center, Seattle, WA.
Brenda Mutai, Department of Medicine (Medical Genetics), University of Washington Medical Center, Seattle, WA.
Naveen Muthu, Department of Biomedical and Health Informatics, Children’s Hospital of Philadelphia, Philadelphia, PA.
Bahram Namjou, Center for Autoimmune Genomics and Etiology, Division of Human Genetics, Cincinnati Children’s Hospital Medical Center, Cincinnati, OH.
Emma F. Perez, Department of Medicine, Brigham and Women’s Hospital, Boston, MA, USA, Mass General Brigham Personalized Medicine, Boston, MA
Megan J. Puckelwartz, Department of Pharmacology & Center for Genetic Medicine, Northwestern University, Chicago, IL
Tejinder Rakhra-Burris, Center for Applied Genomics & Precision Medicine, Duke University, Durham, NC.
Dan M. Roden, Departments of Medicine, Pharmacology, and Biomedical Informatics, Vanderbilt University Medical Center, Nashville, TN
Elisabeth A. Rosenthal, Department of Medicine (Medical Genetics), University of Washington Medical Center, Seattle, WA
Seyedmohammad Saadatagah, Department of Cardiovascular Medicine, Mayo Clinic, Rochester, MN.
Maya Sabatello, Division of Nephrology, Department of Medicine & Division of Ethics, Department of Medical Humanities and Ethics, Columbia University Irving Medical Center, New York, NY.
Dan J. Schaid, Department of Quantitative Health Sciences, Mayo Clinic, Rochester, MN
Baergen Schultz, Division of Genomic Medicine, National Human Genome Research Institute, Bethesda, MD.
Lynn Seabolt, Vanderbilt Institute for Clinical and Translational Research, Vanderbilt University Medical Center, Nashville, TN.
Gabriel Q. Shaibi, Center for Health Promotion and Disease Prevention, Arizona State University, Phoenix, AZ
Richard R. Sharp, Biomedical Ethics Program, Department of Quantitative Health Science, Mayo Clinic, Rochester, MN
Brian Shirts, Department of Laboratory Medicine and Pathology, University of Washington Medical Center, Seattle, WA.
Maureen E. Smith, Department of Cardiology & Center for Genetic Medicine, Northwestern University, Chicago, IL
Jordan W. Smoller, Department of Psychiatry and Center for Genomic Medicine, Massachusetts General Hospital, Boston, MA
Rene Sterling, Division of Genomics and Society, National Human Genome Research Institute, Bethesda, MD.
Sabrina A. Suckiel, The Institute for Genomic Health, Department of Medicine, Icahn School of Medicine at Mount Sinai, New York, NY
Jeritt Thayer, Department of Biomedical and Health Informatics, Children’s Hospital of Philadelphia, Philadelphia, PA.
Hemant K. Tiwari, Department of Biostatistics, School of Public Health, University of Alabama at Birmingham, Birmingham, AL
Susan B. Trinidad, Department of Bioethics and Humanities, University of Washington School of Medicine, Seattle, WA
Theresa Walunas, Department of Medicine & Center for Health Information Partnerships, Northwestern University, Chicago, IL.
Wei-Qi Wei, Department of Biomedical Informatics, Department of Medicine, Vanderbilt University Medical Center, Nashville, TN.
Quinn S. Wells, Division of Cardiovascular Medicine, Department of Medicine, Vanderbilt University School of Medicine
Chunhua Weng, Department of Biomedical Informatics, Columbia University Irving Medical Center, New York, NY.
Georgia L. Wiesner, Division of Genetic Medicine, Department of Medicine, Vanderbilt-Ingram Cancer Center, Vanderbilt University School of Medicine, Nashville, TN
Ken Wiley, Division of Genomic Medicine, National Human Genome Research Institute, Bethesda, MD.
Josh F. Peterson, Department of Biomedical Informatics, Department of Medicine, Vanderbilt University Medical Center, Nashville, TN.
Data Availability:
The de-identified individual data that underlie the results reported in this article (including text, tables, figures, and appendices) will be made for noncommercial, academic purposes upon request. De-identified data derived from individuals enrolled in the study will be made available on the AnVIL platform (https://anvil.terra.bio/) at periodic intervals over the course of the study as it is generated.
References
- 1.Daly MB, Pilarski R, Yurgelun MB, et al. NCCN Guidelines Insights: Genetic/Familial High-Risk Assessment: Breast, Ovarian, and Pancreatic, Version 1.2020: Featured Updates to the NCCN Guidelines. J Natl Compr Canc Netw. 2020; 18(4):380–391. doi: 10.6004/jnccn.2020.0017 [DOI] [PubMed] [Google Scholar]
- 2.Gupta S, Provenzale D, Regenbogen SE, et al. NCCN Guidelines Insights: Genetic/Familial High-Risk Assessment: Colorectal, Version 3.2017. J Natl Compr Canc Netw.2017; 15(12): 1465–1475. doi: 10.6004/jnccn.2017.0176 [DOI] [PubMed] [Google Scholar]
- 3.Grundy SM, Stone NJ, Bailey AL, et al. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA Guideline on the Management of Blood Cholesterol: A Report of the American College of Cardiology/American Heart Association Task Force on Clinical Practice Guidelines. Circulation. 2019;139(25):e1082–e1143. doi: 10.1161/CIR.0000000000000625 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hattersley AT, Greeley SAW, Polak M, et al. ISPAD Clinical Practice Consensus Guidelines 2018: The diagnosis and management of monogenic diabetes in children and adolescents. Pediatr Diabetes. 2018;19 Suppl 27:47–63. doi: 10.1111/pedi.12772 [DOI] [PubMed] [Google Scholar]
- 5.Murray MF, Khoury MJ, Abul-Husn NS. Addressing the routine failure to clinically identify monogenic cases of common disease. Genome Med. 2022;14(1):60. doi: 10.1186/s13073-022-01062-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Grzymski JJ, Elhanan G, Morales Rosado JA, et al. Population genetic screening efficiently identifies carriers of autosomal dominant diseases. Nat Med. 2020;26(8):1235–1239. doi: 10.1038/s41591-020-0982-5 [DOI] [PubMed] [Google Scholar]
- 7.Need AC, Goldstein DB. Next generation disparities in human genomics: concerns and remedies. Trends Genet TIG. 2009;25(11):489–494. doi: 10.1016/j.tig.2009.09.012 [DOI] [PubMed] [Google Scholar]
- 8.Landry LG, Ali N, Williams DR, Rehm HL, Bonham VL. Lack Of Diversity In Genomic Databases Is A Barrier To Translating Precision Medicine Research Into Practice. Health Aff Proj Hope. 2018;37(5):780–785. doi: 10.1377/hlthaff.2017.1595 [DOI] [PubMed] [Google Scholar]
- 9.Khera AV, Chaffin M, Zekavat SM, et al. Whole-Genome Sequencing to Characterize Monogenic and Polygenic Contributions in Patients Hospitalized With Early-Onset Myocardial Infarction. Circulation. 2019;139(13):1593–1602. doi: 10.1161/CIRCULATIONAHA.118.035658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McCarty CA, Chisholm RL, Chute CG, et al. The eMERGE Network: A consortium of biorepositories linked to electronic medical records data for conducting genomic studies. BMC Med Genomics. 2011;4:13. doi: 10.1186/1755-8794-4-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gottesman O, Kuivaniemi H, Tromp G, et al. The Electronic Medical Records and Genomics (eMERGE) Network: past, present, and future. Genet Med. 2013;15(10):761–771. doi: 10.1038/gim.2013.72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.eMERGE Consortium. Lessons from the eMERGE Network: Balancing genomics in discovery and in practice. Hum Genet Genomics Adv. Published online In press. [Google Scholar]
- 13.Denny JC, Ritchie MD, Basford MA, et al. PheWAS: demonstrating the feasibility of a phenome-wide scan to discover gene-disease associations. Bioinforma Oxf Engl. 2010;26(9):1205–1210. doi: 10.1093/bioinformatics/btq126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ritchie MD, Verma SS, Hall MA, et al. Electronic medical records and genomics (eMERGE) network exploration in cataract: several new potential susceptibility loci. Mol Vis. 2014;20:1281–1295. [PMC free article] [PubMed] [Google Scholar]
- 15.Dumitrescu L, Ritchie MD, Denny JC, et al. Genome-wide study of resistant hypertension identified from electronic health records. PloS One. 2017;12(2):e0171745. doi: 10.1371/journal.pone.0171745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Crawford DC, Crosslin DR, Tromp G, et al. eMERGEing progress in genomics—the first seven years. Appl Genet Epidemiol. 2014;5:184. doi: 10.3389/fgene.2014.00184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Crosslin DR, Robertson PD, Carrell DS, et al. Prospective participant selection and ranking to maximize actionable pharmacogenetic variants and discovery in the eMERGE Network. Genome Med. 2015;7(1):67. doi: 10.1186/s13073-015-0181-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fossey R, Kochan D, Winkler E, et al. Ethical Considerations Related to Return of Results from Genomic Medicine Projects: The eMERGE Network (Phase III) Experience. J Pers Med. 2018;8(1). doi: 10.3390/jpm8010002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Williams JL, Chung WK, Fedotov A, et al. Harmonizing Outcomes for Genomic Medicine: Comparison of eMERGE Outcomes to ClinGen Outcome/Intervention Pairs. Healthc Basel Switz. 2018;6(3). doi: 10.3390/healthcare6030083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Das S, Forer L, Schönherr S, et al. Next-generation genotype imputation service and methods. Nat Genet. 2016;48(10):1284–1287. doi: 10.1038/ng.3656 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schatz MC, Philippakis AA, Afgan E, et al. Inverting the model of genomics data sharing with the NHGRI Genomic Data Science Analysis, Visualization, and Informatics Lab-space. Cell Genomics. 2022;2(1):100085. doi: 10.1016/j.xgen.2021.100085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.eMERGE Consortium. Harmonizing Clinical Sequencing and Interpretation for the eMERGE III Network. Am J Hum Genet. Published online August 20, 2019. doi: 10.1016/j.ajhg.2019.07.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Stanaway IB, Hall TO, Rosenthal EA, et al. The eMERGE genotype set of 83,717 subjects imputed to ~40 million variants genome wide and association with the herpes zoster medical record phenotype. Genet Epidemiol. Published online October 8, 2018. doi: 10.1002/gepi.22167 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Khera AV, Chaffin M, Aragam KG, et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat Genet. 2018;50(9):1219–1224. doi: 10.1038/s41588-018-0183-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tier 1 Genomics Applications and their Importance to Public Health | CDC. Published March 16, 2021. Accessed June 15, 2022. https://www.cdc.gov/genomics/implementation/toolkit/tier1.htm [Google Scholar]
- 26.Breast Cancer Association Consortium, Dorling L, Carvalho S, et al. Breast Cancer Risk Genes - Association Analysis in More than 113,000 Women. N Engl J Med. 2021;384(5):428–439. doi: 10.1056/NEJMoa1913948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Haverfield EV, Esplin ED, Aguilar SJ, et al. Physician-directed genetic screening to evaluate personal risk for medically actionable disorders: a large multi-center cohort study. BMC Med. 2021;19(1):199. doi: 10.1186/s12916-021-01999-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Orlando LA, Buchanan AH, Hahn SE, et al. Development and validation of a primary care-based family health history and decision support program (MeTree). N C Med J. 2013;74(4):287–296. [PMC free article] [PubMed] [Google Scholar]
- 29.Lee A, Mavaddat N, Wilcox AN, et al. BOADICEA: a comprehensive breast cancer risk prediction model incorporating genetic and nongenetic risk factors. Genet Med. 2019;21(8):1708–1718. doi: 10.1038/s41436-018-0406-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Harris PA, Taylor R, Thielke R, Payne J, Gonzalez N, Conde JG. Research electronic data capture (REDCap)--a metadata-driven methodology and workflow process for providing translational research informatics support. J Biomed Inform. 2009;42(2):377–381. doi: 10.1016/j.jbi.2008.08.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Harris PA, Taylor R, Minor BL, et al. The REDCap consortium: Building an international community of software platform partners. J Biomed Inform. 2019;95:103208. doi: 10.1016/j.jbi.2019.103208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Goff DC, Lloyd-Jones DM, Bennett G, et al. 2013 ACC/AHA Guideline on the Assessment of Cardiovascular Risk. Circulation. 2014;129(25_suppl_2):S49–S73. doi: 10.1161/01.cir.0000437741.48606.98 [DOI] [PubMed] [Google Scholar]
- 33.Lee A, Mavaddat N, Cunningham AP, et al. Enhancing the BOADICEA cancer risk prediction model to incorporate new data on RAD51C, RAD51D, BARD1, updates to tumour pathology and cancer incidences | medRxiv. medRxiv. doi: 10.1101/2022.01.27.22269825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lewis ACF, Perez EF, Prince AER, et al. Patient and provider perspectives on polygenic risk scores: implications for clinical reporting and utilization. Genome Med. 2022;14(1):114. doi: 10.1186/s13073-022-01117-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cook DA, Teixeira MT, Heale BS, Cimino JJ, Del Fiol G. Context-sensitive decision support (infobuttons) in electronic health records: a systematic review. J Am Med Inform Assoc JAMIA. 2017;24(2):460–468. doi: 10.1093/jamia/ocw104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ginsburg GS, Wu RR, Orlando LA. Family health history: underused for actionable risk assessment. Lancet Lond Engl. 2019;394(10198):596–603. doi: 10.1016/S0140-6736(19)31275-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Polubriaginof F, Tatonetti NP, Vawdrey DK. An Assessment of Family History Information Captured in an Electronic Health Record. AMIA Annu Symp Proc AMIA Symp. 2015;2015:2035–2042. [PMC free article] [PubMed] [Google Scholar]
- 38.Aragam KG, Dobbyn A, Judy R, et al. Limitations of Contemporary Guidelines for Managing Patients at High Genetic Risk of Coronary Artery Disease. J Am Coll Cardiol. 2020;75(22):2769–2780. doi: 10.1016/j.jacc.2020.04.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wojcik GL, Graff M, Nishimura KK, et al. Genetic analyses of diverse populations improves discovery for complex traits. Nature. 2019;570(7762):514–518. doi: 10.1038/s41586-019-1310-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Graham SE, Clarke SL, Wu KHH, et al. The power of genetic diversity in genome-wide association studies of lipids. Nature. 2021;600(7890):675–679. doi: 10.1038/s41586-021-04064-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The de-identified individual data that underlie the results reported in this article (including text, tables, figures, and appendices) will be made for noncommercial, academic purposes upon request. De-identified data derived from individuals enrolled in the study will be made available on the AnVIL platform (https://anvil.terra.bio/) at periodic intervals over the course of the study as it is generated.