

Abstract
BACKGROUND:
Hereditary ARPC1B deficiency is characterized clinically by ear, skin, and lung infections, bleeding, eczema, food allergy, asthma, skin vasculitis, colitis, arthritis, short stature, and lymphadenopathy.
OBJECTIVE:
We aimed to describe the clinical, laboratory, and genetic features of six additional patients from four Mexican families.
METHODS:
We performed exome sequencing in available patients of four families with suspected actinopathy, collected their data from medical records, and reviewed the literature for other reports of patients with ARPC1B deficiency.
RESULTS:
Six patients from 4 families were included. All had recurrent infections, mainly bacterial pneumonia, and cellulitis. 67% had eczema, while 50% had food allergies, failure to thrive, hepatomegaly, and bleeding. Eosinophilia was found in all, with 84% thrombocytopenia, 67% abnormal-size platelets, and anemia. Serum levels of IgG, IgA, and IgE were highly increased in most; IgM was normal or low. T cells were decreased in 67% of patients, while B and NK cells were increased in half the patients. Two of the four probands had compound heterozygous variants. One patient was successfully transplanted. We identified 28 other patients whose most prevalent features were eczema, recurrent infections, failure to thrive, bleeding, diarrhea, allergies, vasculitis, eosinophilia, platelet abnormalities, high IgE/IgA, low T cells, and high B cells.
CONCLUSION:
ARPC1B deficiency has a variable and heterogeneous clinical spectrum, expanded by these cases to include keloid scars and Epstein-Barr virus chronic hepatitis. A novel deletion in exon 8 is shared by three unrelated families and might be the result of a founder effect.
Keywords: ARPC1B deficiency, combined immune deficiency, inborn errors of immunity, primary immunodeficiency, actinopathy, cytoskeleton, hyper-IgE, whole-exome sequencing
INTRODUCTION:
Patients with rare diseases often endure a diagnostic odyssey of multiple consults and hospitalizations before someone suspects their correct diagnoses (1). Inborn errors of immunity (IEI), previously known as primary immune deficiencies, are a group of more than 460 rare congenital diseases with increased susceptibility to infection and/or atopy, autoimmunity, malignancy, and hyper-inflammation; caused by defective proteins involved in the development or function of the immune system (2).
Actin polymerization is essential for the mobility and migration of hematopoietic cells and fibroblasts, as well as for the synapse formation, signaling, survival, and activation of immune cells (3). A group of recently described IEI with defective actin polymerization, elongation, or branching, known also as actinopathies (4), results from at least a dozen monogenic etiologies and manifest early in life (usually the first two months) as combined or syndromic defects, with atopic and hemorrhagic diatheses, susceptibility to viral and bacterial infections, an increased risk of hematologic malignancy, and inflammatory manifestations (5).
Actin-related protein 2/3 complex, subunit 1B (ARPC1B) is one of two isoforms of the first regulatory subunit of the ARP2/3 complex. Encoded in locus 7q22.1 (OMIM#604223), ARPC1B is primarily expressed in hematopoietic cells, where it promotes the branching out (initiation and remodeling) of new actin filaments from the mother thread (6). The Wiskott-Aldrich syndrome protein (WASP) binds ARPC1B and stimulates ARP2/3 complexes to nucleate branched filamentous actin networks of the cytoskeleton that control low-level constitutive signaling at rest. In B cells, the lack of ARPC1B results in increased tonic signaling with persistent B cell activation, leading to high numbers of transitional B cells (7).
Autosomal recessive ARPC1B deficiency, first reported in 2017 (8,9), is characterized clinically by ear, skin, and lung infections, bleeding, eczema, food allergy, asthma, skin vasculitis, colitis, arthritis, short stature, and lymphadenopathy (10). Previously known as Platelet abnormalities with eosinophilia and immune-mediated inflammatory disease (PLTEID, IMD 71, OMIM phenotype #617718), the main laboratory findings include leukocytosis, eosinophilia, thrombocytopenia/thrombocytosis, small or large platelets, hypergammaglobulinemia (high IgE/IgA/IgG) and serum autoantibodies (ANA, ANCA), with low CD3+ T cells and increased B cells (11). In cytotoxic lymphocytes from human patients, ARPC1B deficiency results in their inability to extend lamellipodia upon T-cell receptor (TCR) stimulation, and to assemble the immune synapse, as well as a reduced surface expression of CD8, the TCR, and Glucose transporter 1 (GLUT1, reference 11). The increased activation of B cells in response to microbe-associated molecular patterns might explain the high incidence of autoimmunity in ARPC1B-deficient patients (7).
Here, we report on six additional patients from four families. Most patients were first identified as suspected Wiskott-Aldrich syndrome (WAS) or Dedicator of cytokinesis 8 (DOCK8) deficiency. Their cases expand the already wide clinical spectrum to include keloid scars and Epstein-Barr virus (EBV) chronic hepatitis. A novel 46 base pair (bp) deletion in exon 8 is shared by three unrelated families from three states of Mexico and might result from a founder effect.
We aimed to describe the clinical, laboratory, and genetic features of six patients with suspected ARPC1B deficiency from Mexico.
METHODS:
We performed whole-exome sequencing (WES) in available members of four families with suspected actinopathy. We also reviewed the English literature for other available reports of patients with ARPC1B deficiency, querying “ARPC1B deficien* patient” on PubMed-Medline.
Patients were identified as potentially suffering actinopathies (the initial suspicion was usually WAS or DOCK8 deficiency) by their clinical immunologists in Mexico City, Monterrey, and Guadalajara. Genomic DNA was obtained from whole blood through the salting-out method. Whole- or targeted exome sequencing was performed in New York City (Genome Center), Rockville (Psomagen, Inc.), or San Francisco (Invitae, Corp.).
For the lymphoproliferative response assays, 1×10e6 peripheral blood mononuclear cells, previously stained with carboxyfluorescein diacetate succinimidyl ester (CFSE), were cultured without stimuli or with phytohemagglutinin (PHA) 10 μg/mL, Anti-CD3+CD28+ antibodies 1μL/mL, or anti-CD3 antibody, during 5 days at 37° C, under 5% CO2; afterward, cultures were harvested and CD3+ cells stained, acquired on a BD FACSAria flow cytometer, and analyzed with FlowJo v10 software (Becton, Dickinson and Co.).
To assess the protein-level consequences of two heterozygous missense variants, we used in silico tools available online: VarSome (varsome.com), gnomAD Exomes (gnomad.broadinstitute.org), Protein Data Bank (PDB)sum (www.ebi.ac.uk/pdbsum), UCSF Chimera (www.cgl.ucsf.edu), and Visual Molecular Dynamics (VMD) (ks.uiuc.edu/research/vmd/). The atomic coordinates of the human ARP2/3 are already reported (PDB code:6yw6) and were used for all the predictions herein. PDBsum server (PDBsum-EMBL-EBI) was used to analyze the protein-protein contacts. All structures were energy-minimized with Chimera software (12), and with the resulting new coordinates, the calculations were carried out. Chimera was also used for in silico mutation based on the Dunbrak backbone-dependent rotamer library (13); the rotamers with the highest probabilities were used. Electric charges and distribution were calculated with Chimera. VMD 1.9.4a48 was used to find hydrogen bonds, contacts, and clashes. Chimera and VMD were used to visualize structures and generate the supplementary images presented in this study. MEROPS database tools were used to determine the peptidases specificities.
Ethical considerations/Study approval:
Patients or their guardians consented in writing to their genetic evaluation and the publication of these case reports. The study was approved by the Institutional Review Board of the National Institute of Pediatrics (numbers 2013/049 and 2021/054).
RESULTS:
CASE REPORTS:
Patient 1.
An 11-year-old girl from a small town in the state of Veracruz, without parental consanguinity, whose elder brother had died as a newborn of intracranial bleeding. She started at the age of 2 months with severe eczema and diarrhea, the latter complicated by rectal prolapse and a recto-vesical fistula. She was diagnosed with refractory atopic dermatitis and cow’s milk protein allergy, based on a compatible history of eczema, gastroesophageal reflux, and intermittent diarrhea, as well as a clinical improvement after avoidance diet and substitution with extensively hydrolyzed formula. During her first year of life and the following years, she also developed cellulitis, ganglionic tuberculosis attributed to the Bacille Calmette-Guérin (BCG) vaccine, recurrent pneumonia, sepsis, disseminated warts, renal tubular acidosis, and a urinary tract infection caused by Mycobacterium gordonae. Although Mycobacterium bovis was not documented, the diagnosis of ganglionic tuberculosis was based on a compatible history, lymphadenitis ipsilateral to the vaccine, an excisional lymph node biopsy consistent with tuberculosis, and good response to antimycobacterial antibiotics.
On physical examination, she had short stature, severe extensive eczema, multiple plain warts, and hypopigmented spots in trunk and limbs, with cervical and axillary lymphadenopathies, and hepatosplenomegaly. Computed tomography revealed bilateral bronchiectasis (see figure 1). Isolates: Staphylococcus hominis (catheter), Pseudomonas aeruginosa (skin), Escherichia coli, Campylobacter (stools), Cytomegalovirus (PCR, blood), Mycobacterium gordonae (urine), and Candida sp (urine).
Figure 1.

Pictures and X-rays from our patients show pneumonia, bronchiectasis, keloid scarring, and skin infections.
A. Chest X-Ray with bilateral reticular infiltrates and air bronchogram without consolidation (P1). B. Chest CT scan showing central bronchiectasis of the left lower lobe, and subpleural nodules of the right lower lobe (P1). C. Bilateral interstitial infiltrates with a perihilar predominance (P2). D. Keloid scar, warts, molluscum, and multiple hypochromic lesions on the chest (P6). E. Multiple hypochromic residual lesions after 16 subcutaneous injections of interferon beta (P6). F. Post-thoracotomy monstrous scar on right posterior chest (P6). G and H. Extensive eczema, plain warts, hypertrophic scarring, and discoloration of trunk and arms (P1).
Her immunological work-up reported autoimmune hemolytic anemia (Hb 7.8g/dl, Hct 23.1%), persistent eosinophilia (1,000/mm3), mild lymphopenia and intermittent thrombocytopenia; as well as low serum complement (C4 6.3mg/dl, C3 88mg/dL); total serum IgE higher than 2,000 IU/ml (hyper-IgE), high IgG 2,278mg/dl (normal 331–1164mg/dl), high IgA 699mg/dl (normal 13–104mg/dl), and IgM 80.5–252mg/dl; (normal 40–161mg/dl). Serum autoantibodies: Antinuclear (ANA) 1:160, ENA-6 positive (Anti-Ro (57.28 U/ml), Anti-SSB/LA (59.57 U/ml), Anti Scl-70 (52.11 U/ml), Anti-Jo (43.92 U/ml), Anti-RNP (51.15 U/ml). Flow cytometry for lymphocyte subsets found CD3+CD8+ lymphopenia: 44 cell/mm3 (normal 3502,200cells/mm3), with a CD4/CD8 ratio of 1.29. Carboxyfluorescein lymphoproliferation assay (activating with PMA, PHA and CD3/CD28 antibody) was impaired when compared to a healthy control (See figure 2a, b).
Figure 2.

Flow cytometry and lymphoproliferation assays from patients 1, 2, and 5. 2A, 2C, 2D: Flow cytometry showing T, B, NK cells, and monocytes in patients P1, P2, and P5. 2B: Lymphoproliferative assay. Peripheral blood mononuclear cells 1×10e6 previously stained with CSFE were cultured without stimuli or with PHA 10 μg/mL, Anti-CD3+CD28+ antibodies 1μL/mL, or anti-CD3 antibody, during 5 days at 37° C, CO2 5%; afterward cultures were 685 harvested and CD3+ cells stained, acquired on a BD FACSAria cytometer, and analyzed with 686 Flow Jo software. Notice diminished proliferation in P1 after PHA stimulus.
ZAP70 and DOCK8 deficiencies were entertained as differential diagnoses. ZAP70 expression and Multiplex Ligation-dependent Probe Amplification (MLPA) for DOCK8 were normal; a targeted exome of 450 metabolic and immunodeficiency genes was negative. Whole-exome sequencing (WES) analysis identified a homozygous 46 base-pair deletion in exon 8 of ARPC1B (p.Glu300fsTer7). See figures 3 and 4.
Figure 3.
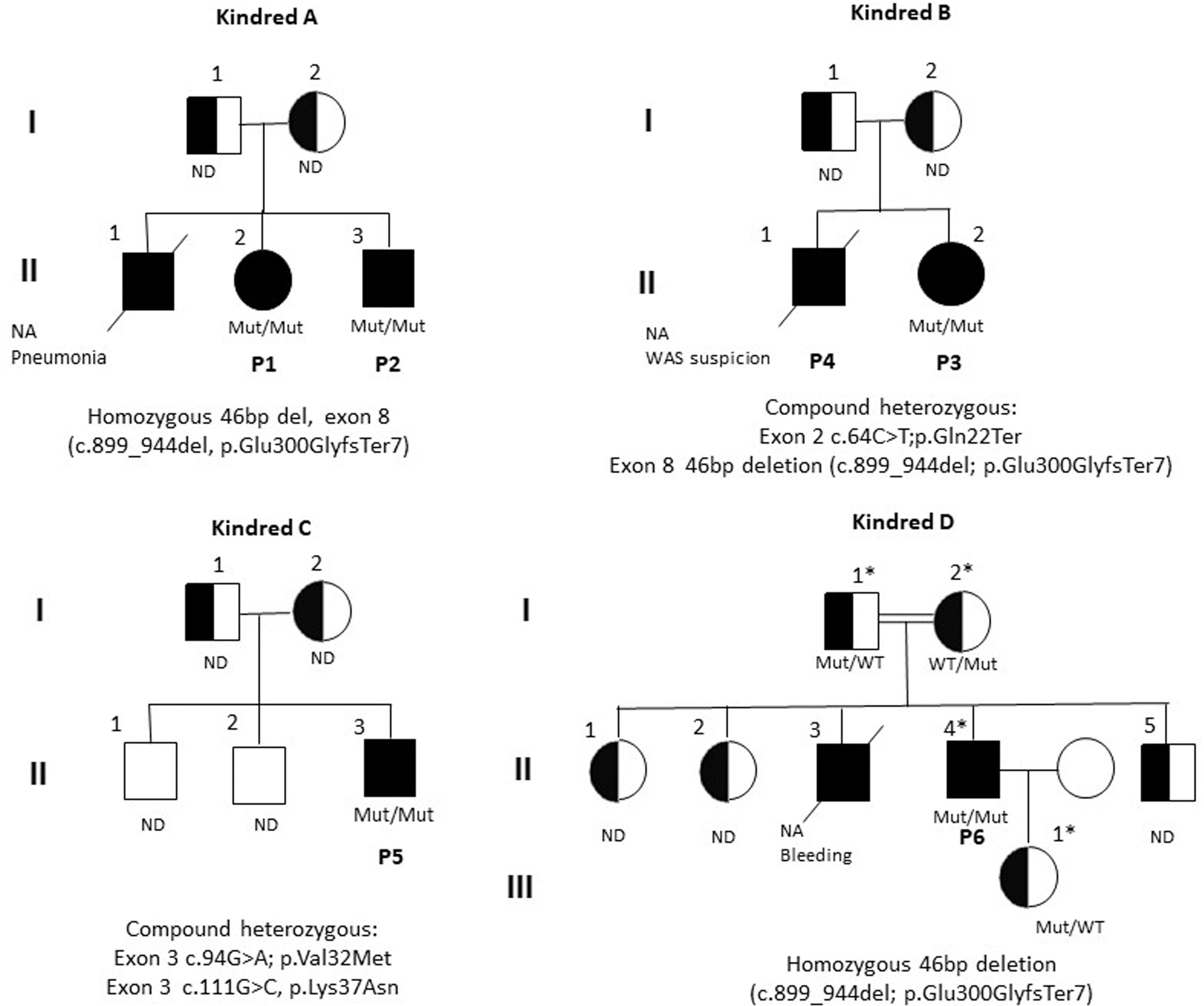
Family trees of six patients with autosomal recessive ARPC1B deficiency. Pedigree and segregation analysis of families included in the study. Squares: male subjects; circles: female subjects; black filled symbols: patients with ARPC1B deficiency (or compatible phenotype); crossed-out symbols: deceased subjects. Each generation is designated by a Roman numeral (I-III). NA: No DNA was available for testing. ND: Not done, Sanger sequencing is pending. *Invitae 407 PID gene panel.
Figure 4.

ARPC1B gene/protein structures, and variants reported to date in 34 patients with autosomal recessive ARPC1B deficiency. Nucleotide positions of variants identified in index patients and representation of amino acid changes. Red: Variants in this series; Violet: Previously reported. *P1, P2, P3, P6. ** P14, P15 ***P17, P18 ****P19, P24, P26 *****P22, P23 *****P27, P28, P29 *******P30, P31, P32, P33
The patient underwent a colostomy to treat a rectovesical fistula and received multiple antibiotic regimens for her infections. Hematopoietic stem-cell transplantation (HSCT) was attempted twice: at 2 years of age (2010) from an umbilical cord donor, and at age 4 (2012) from a haploidentical donor (her mother), without apparent engraftment. She received non-myeloablative and reduced-intensity conditioning regimes. The patient is alive and stable at age 12, despite the recurrence of cytomegalovirus infection after both transplants. She is chronically managed with intravenous immunoglobulin (IVIG), antibiotic prophylaxis, and immunosuppressants. Her condition improved considerably after HSCT, with partial chimerism of 3.99% measured in DNA from peripheral blood cells.
Patient 2.
The younger brother of patient 1, a male infant from Veracruz, started at age 1 month with eczema and one episode of rectal bleeding. At 2 months, he developed a soft palate ulcer and oral candidiasis. When he was 4 months old, bronchiolitis caused by a respiratory syncytial virus sent him to the hospital.
There, his lab work-up showed leukocytosis (17,500–33,600/mm3), severe eosinophilia (5,600–20,100/mm3), and mild thrombocytosis (467,000/mm3), with normal platelet volumes (7.5–7.9fL). Serum immunoglobulins were increased: IgG 737mg/dL (normal range 290–550mg/dL), IgA 165 (normal 20–62 mg/dl), with normal IgM (37.7mg/dL), and normal serum complement.
Lymphocyte subset counts by flow cytometry at 2 months found low CD8+ T cells, with high B and NK cells: CD3+ 2,315 cells/mm3 (normal 2500–5500 cells/mm3), CD4+ 1974 cells/mm3 (normal 1600–4000 cells/mm3), CD8+ 257 cells/mm3 (3%, normal 560–1700 cells/mm3), CD19+ 4,116 (48%, normal 300–2000 cells/mm3), CD16/56+ 1,286 cells/mm3 (15%, normal 170–1100 cells/mm3), and a CD4/CD8 ratio of 7.68. See figure 2b, 2c. ToRCH and HIV serologic screens were negative.
WES identified the same 46bp deletion in exon 8 of ARPC1B (p.Glu300fs) as in his sister. When the patient was 8 months old, he developed a high fever and was hospitalized briefly for mild COVID-19 pneumonia (figure 1). A blood culture grew Pseudomonas aeruginosa, an RT-PCR test for SARS-CoV-2 came back positive, and he was treated with supplemental oxygen and intravenous antibiotic, without needing intensive care (reported in (13)).
Patient 3.
A 3-year-old girl from a non-consanguineous family in Monterrey, whose elder brother (P4) had died twelve years prior from suspected WAS at age 3 years. A few days after birth she presented with extensive eczema, diagnosed as atopic dermatitis, and chronically treated with topical corticosteroids. The patient tolerated the BCG and Measles-Mumps-Rubella (MMR) vaccines. She was also diagnosed with food allergy based on a clinical history of eczema exacerbated by the consumption of eggs and oranges, and positive prick tests for both foods. At age 12 months, she suffered from diarrhea and hematochezia, four episodes of bronchiolitis, and failure to thrive. Physical examination found low weight for age, severe eczema, and Cushing syndrome stigmata, without lymphadenopathy or hepatosplenomegaly. The diagnosis of exogenous Cushing syndrome was based on prolonged exposure to excess topical glucocorticoids, with facial plethora and growth deceleration, which improved and resolved after discontinuation of steroid treatment. One serum cortisol measurement in 2019 was normal (adrenocorticotropic hormone in the morning of 47.3pg/ml; normal range 0–46pg/ml).
Laboratory workup reported leukocytosis (20,400 cells/μL), eosinophilia (800–3,000 cells/μL) and thrombocytopenia (57,200–118,000 cells/μL), with mean platelet volumes ranging from 4.6 to 9.6fL; hypergammaglobulinemia: IgG 1,929mg/dL, IgA 481mg/dL, IgE 1,283 IU/ml, with marginally low IgM (43mg/dL), and increased C-reactive protein at 25mg/dL. Serum liver enzymes (GGT, ALT, AST, bilirubin) were normal. Flow cytometry for lymphocyte subsets at 1 year found low CD4+ 630 cells/mm3 (13%, normal 1300–3400 cells/mm3) and CD8+ T cells 309 cells/mm3 (normal 620–2000 cells/mm3) with a CD4/CD8 ratio of 2.03; high CD16+56+ NK cells 1329 cells/mm3 (normal 180–920 cells/mm3) and marginally high B cells CD19+ 2,406 cells/mm3 (49%, normal 720–2600 cells/mm3).
The patient started treatment with antibiotic prophylaxis, IVIG, hydrolyzed formula, and avoidance of culprit foods. WES identified a compound heterozygous genotype consisting of variants in exons 2/10 and 8/10 of ARPC1B; one of them a nonsense/splice-site variant by single nucleotide transition (c.64C>T; p.Gln22Ter), and the other a 46 bp deletion, identical to that found in patients 1 and 2. Both variants are novel and pathogenic (figures 3 and 4).
She underwent a haploidentical HSCT from her father, with a stem-cell dosage of 70×10e6 CD34+ cells/kg, a conditioning regimen with busulfan (16mg/kg/d) for 4 days, and cyclophosphamide (60mg/kg) for 2 days. As complications, she developed neutropenic colitis and cytomegalovirus (CMV) infection detected on day +20. Immune reconstitution was achieved on days +18 for neutrophils, +19 platelets, and +25 red blood cells. With stable chimerism of 99% (up from 63%) in peripheral cells after more than 7 months of follow-up, she is alive and well, sporting normal blood counts.
Patient 4.
A male patient from non-consanguineous parents in Monterrey, Mexico, the elder brother of patient 3. He started 15 days after birth with eczematous skin lesions in the malar region that became generalized and were diagnosed as severe atopic dermatitis. He had multiple relapses and exacerbations with poor response to treatment and failure to thrive. He also had several episodes of recurrent and severe infections such as pneumonia, bronchiolitis, bronchopneumonia, recurrent gastroenteritis, and orbital cellulitis. He developed septic shock in two different events, with Staphylococcus aureus and Coagulase-Negative Staphylococcus sp. isolated from blood cultures.
Laboratory work-up during infectious episodes revealed anemia, leukocytosis, neutrophilia, eosinophilia with transient thrombocytopenia, and small platelet volumes. At age 2 years, serum immunoglobulins were: high IgG 1,130 mg/dl (normal: 340–620mg/dl), IgA 426mg/dl (normal: 33–122mg/dl), and hyper-IgE (2,340 IU/ml), with low IgM 22mg/dl (normal: 48–143mg/dl). Flow cytometry for lymphocyte subsets was abnormal with CD3+ lymphopenia 1,402 cells/mm3 (normal: 2100–6200 cells/mm3), at the expense of CD4+ 626 cells/mm3 (24%, normal: 1300–3400 cells/mm3), normal CD8+ 678 cells/mm3 (normal: 620–2000 cells/mm3), for a CD/CD8 ratio of 0.92, whereas B cells were marginally high: CD19+ 2480 cells/mm3 (51%, normal: 720–2600 cells/mm3), as were NK cells: CD16+56+ 2,056 cells/mm3 (19%, normal 180720 cells/mm3).
The patient had received IVIG during the infectious episodes and then switched to monthly administration. With a provisional diagnosis of WAS, stem-cell transplantation was not performed for lack of a compatible donor. In the last pneumonia episode, he developed laryngeal edema, for which he was intubated and underwent a tracheostomy. He died at the age of three, in 2008, during that hospitalization. See Figures 3, 4, and E1.
Patient 5.
A 13-year-old male from rural Guerrero, Mexico, with two healthy elder brothers. He started at age 5 with choluria, jaundice, and right flank abdominal pain, diagnosed as chronic viral hepatitis. He also had a recent history of epistaxis, ecchymosis, and gum bleeding.
Physical examination found multiple dental cavities, cervical/inguinal lymphadenopathy, and a distended abdomen with ascites, collateral venous circulation, and hepato-splenomegaly. Laboratory workup was as follows: elevated serum liver enzymes (GGT, ALT, AST), hypercholesterolemia, hypertriglyceridemia, and elevated acute serum reactants (ferritin 819ng/ml, erythrocyte sedimentation rate 40–72mm/h), with prolonged partial thromboplastin time (PTT) and negative direct Coombs test. Serum C3 and C4 complement components were normal, while anti-nuclear autoantibodies were negative. Total serum immunoglobulins IgG 2,000mg/dL, IgA 340mg/dL and IgE 233 IU/ml were high, with normal IgM. Repeated blood counts found transient anemia and leukocytosis, with alternating thrombocytopenia and thrombocytosis, and reports of large platelet volumes; eosinophils were as high as 23% and as low as 2% at different time points. Flow cytometry for lymphocyte subsets was normal, except for very low B cells (1% or 22 cells), which was attributed to a recent regimen of rituximab.
A bone marrow aspirate found normal bone marrow cellularity and appearance of three lineages; an in-situ hybridization assay identified EBV-encoded small RNA (EBER) in the nucleus of lymphocytes from the bone marrow (figure E2). The liver biopsy reported non-specific inflammation with a monocytic infiltrate, and some eosinophils (figure E3). Quantitative polymerase chain reaction (PCR) identified 23,751 viral copies of EBV in blood and has remained positive for five consecutive years with loads above 1,000 cp/ml and up to greater than 100,000. The patient started treatment with immunosuppressants and IVIG and is currently stable. However, while awaiting a liver transplant, he was rushed to the emergency department with abrupt back pain and was found to have multiple vertebral fractures caused by osteopenia, attributable to chronic steroid treatment. Three years later, a control liver biopsy showed extensive cholestasis and veno-occlusive disease, with a diffuse lymphocytic sinusoidal infiltrate in a “string of beads” pattern with a predominance of CD8+ T cells and infrequent EBV-infected B cells (figure E3). In February 2019, the EBV genome became undetectable under treatment. In May 2021, the patient underwent an uneventful liver transplant from a cadaveric donor. The liver explant confirmed extensive veno-occlusive disease, mononucleosis pattern hepatitis, and EBV DNA positive by in situ hybridization and PCR. By the 2-month follow-up, the patient had suffered a recurrence of EBV infection, up to 200,000 cp/ml, with elevated aminotransferases. Liver biopsy of the transplant documented extensive acute EBV hepatitis without fibrosis, with positive viral latent membrane protein (LMP1) immuno-staining (figure E3). The patient was receiving oral ambulatory treatment with tacrolimus, prednisone, amlodipine, valganciclovir, and cholecalciferol. In 2022, he developed hemophagocytic lymphohistiocytosis (HLH) in the context of COVID-19 infection, and received intensive treatment with the HLH-04 protocol, despite which he died at the Children’s Hospital of Mexico (HIMFG).
With a provisional diagnosis of susceptibility to EBV and liver failure, WES identified compound heterozygous missense variants in exon 3 of ARPC1B, one of them a polymorphism found in 5% of the population (c.111G>C, p.Lys37Asn), with pathogenicity scores of 24.1 for CADD Phred, SIFT4 0.01, and Polyphen2 0.91. The second missense variant (c.94G>A, p.Val32Met) has a minor allele frequency (MAF) of 0.000004 in gnomAD exomes, with no homozygous individuals; in silico pathogenicity scores for this variant are CADD Phred 26.7, SIFT 0, PolyPhen 0.981. Minimum significance cutoff scores (MSC with a 99% confidence interval) for ARPC1B are 7.501, 0.243, and 0.239, respectively. The two variants are close to each other in trans in exon 3 (see figures 3, 4, and E1). The co-occurrence of these two variants has not been described before, as corroborated with the gnomAD co-occurrence tool. Both amino acids, Valine (V) at position 32 and Lysine (K) at position 37, are strictly conserved through evolution in vertebrates (figure E4).
In the supplementary file “Variant effect on protein structure” (Online Repository Text) interested readers can find more details on the predicted consequences of variants V32M and K37N in ARPC1B, as interrogated in silico. See also table E1 and figures E5-E9.
Patient 6.
A male patient born to consanguineous parents (second cousins) in rural Michoacan, Mexico. One brother had died at age 3 from a lung infection. The patient started 15 days after birth with an anal abscess and fistula that required surgery at 3 months. He also suffered from skin lesions such as eczema, erythema nodosum, skin pustules, disseminated discolored plain warts, and a tendency to form hypertrophic, keloid, and monstrous scars (figure 1). In time, he developed wheezing, abdominal vasculitis, recurrent pneumonia, otitis media with subsequent hearing loss, urinary tract infections, and gastroenteritis.
Lung biopsy identified bronchiolitis obliterans (figure E2), and computed tomography of the lung revealed destruction of the right upper lobe. A right upper lobectomy was performed. Streptococcus viridans, Escherichia coli, Candida albicans, Serratia marcescens, Pseudomonas aeruginosa, and Stenotrophomonas maltophilia were isolated from lung specimens or bronchoalveolar lavage. At age 7, a fundoplication and Nissen procedure for gastroesophageal reflux disease was performed, without improvement of respiratory symptoms. On physical examination, he had multiple hypochromic scars of 3–4mm in diameter, abnormal respiratory sounds with crackles, basal hypoventilation, and disseminated purpuric lesions in different stages of resolution.
Immunological work-up reported microcytic hypochromic anemia, lymphopenia, and intermittent thrombocytopenia without eosinophilia; normal serum complement (C4 20.3mg/dl, C3 121mg/dl), and positive serum autoantibodies: Antinuclear (ANA) 1:80, with negative pANCA and mildly positive cANCA (+). Serum immunoglobulins were increased (age 11 years): IgA 968mg/dl (normal range 50–170mg/dL), IgM 420mg/dl (normal 55–155mg/dl), IgE 1031 IU/ml; except for IgG, which was low at 187 (620–1150mg/dl). Lymphocyte subsets by flow cytometry found low CD4+ 290 cells/mm3 (normal range 500–950 cells/mm3), and CD8+ 107 cells/mm3 (400–820 cells/mm3), CD4/CD8 ratio 2.71; with high B cells: 735/mm3 (50–240 cells/mm3), and normal NK cells 240/mm3 (90–350 cells/mm3).
He received treatment with thalidomide, prednisone, immunosuppressants (cyclosporine and mycophenolate mofetil), monthly IVIG since 2003, and ambulatory prophylaxis with trimethoprim/sulfamethoxazole and itraconazole. His skin lesions and keloid scars greatly improved with 16 injections of subcutaneous interferon beta (figure 1 and E2).
The genetic etiologies associated by then with Hyper-IgE syndrome: STAT3, TYK2, and DOCK8 deficiencies, were all ruled out through Sanger sequencing and WES analysis between 2009 and 2011. In 2021, a 407 gene panel identified a homozygous genotype consisting of the same novel pathogenic 46bp deletion (c.899_944del; p.Glu300GlyfsTer7). The patient is currently stable as an adult, with an infant daughter. All available family members are heterozygous for the deletion (figure 3). Their family history is negative for keloid scars, although some of his (heterozygote) relatives have complained of allergies or cutaneous viral infections, which are unlikely to be related to their known genotype.
REVIEW OF THE LITERATURE
We searched the English literature on PubMed-Medline and found 10 relevant papers (8–11,15–20) describing 28 more patients with ARPC1B deficiency. Tables I through IV summarize the prevalence of demographic and clinical features, immunological abnormalities, and mutational analysis, including the six patients in this report.
TABLE I.
Demographic and clinical features of 34 patients with actin-related protein 2/3 complex subunit 1B deficiency
| Patient | References | Origin | Sex | Onset of symptoms, mo | Age at diagnosis, y | Symptoms at onset | Consanguinity | Family history | Clinical features | Microbiological isolates | Complications | Hematopoietic stem cell transplantation | Outcome |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | This series | Mexico | F | 2 | 11 | Eczema, diarrhea, complicated by rectal prolapse and rectovesical fistula | No | Sibling to patient 2 | Eczema, food allergy, failure to thrive, diarrhea, lymphadenopathy, hepatosplenomegaly, hypopigmented spots in trunk and limbs, renal tubular acidosis and, infections (cellulitis, ganglionic tuberculosis, urinary tract infection, sepsis, disseminated warts, and recurrent pneumonia) | Staphylococcus hominis, Pseudomonas aeruginosa, Escherichia coli, Campylobacter, Mycobacterium sp, Candida sp. cytomegalovirus (CMV), Mycobacterium gordonae | Bronchiectasis, rectal prolapse and rectovesical fistula | Yes | Alive |
| 2 | This series | Mexico | M | 1 | 1.9 | Eczema and rectal bleeding | No | Sibling to patient 1 | Eczema, food allergy, soft palate ulcer, infections (oral candidiasis, bronchiolitis and pneumonia) | P aeruginosa, respiratory syncytial virus, SARS-CoV-2 | Malabsorption syndrome | No | Alive |
| 3 | This series | Mexico | M | 1 | 12 (after death) | Eczema | No | Sibling to patient 4 | Eczema, failure to thrive, hematochezia, diarrhea, hepatomegaly, infections (bronchiolitis, orbital cellulitis, sepsis, recurrent pneumonia and gastroenteritis) | Staphylococcus aureus, coagulase-negative | Laryngeal edema, orotracheal intubation and, tracheostomy | No | Dead |
| 4 | This series | Mexico | F | 1 | 3.4 | Eczema | No | Sibling to patient 3 | Eczema, food allergy, failure to thrive, hematochezia, diarrhea, recurrent bronchiolitis | ND | Cushing syndrome | Yes | Alive |
| 5 | This series | Mexico | M | 60 | 13 | Jaundice and right flank abdominal pain | No | No | Epistaxis, ecchymoses, gum bleeding, lymphadenopathy, hepatosplenomegaly, infections (chronic viral hepatitis, dental cavities) | EBV | Multiple vertebral fractures caused by osteopenia | No (had liver transplant) | Dead |
| 6 | This series | Mexico | M | 1 | 29 | Anal fistula | Yes | Brother died of lung infection at age 3 y | Eczema, erythema nodosum, skin pustule, keloids, skin and abdominal vasculitis, infections (plain warts, pneumonias, gastroenteritis, urinary tract infections) | HPV, S. viridans, E coli, Candida albicans, Serratia marcescens, P aeruginosa, Stenotrophomonas maltophilia | Chronic lung disease, bilateral hearing loss, keloid scars | No | Alive |
| 7 | Kuijpers et al (2017)8 Volpi et al (2019)10 (patient 1) | Morocco | M | 2 | 7 | Gastric bleeding | Yes | No | Eczema, food allergy with anaphylaxis, failure to thrive, gastric bleeding, diarrhea, hepatomegaly, leukocytoclastic vasculitis, infections (gastroenteritis, auricular infection, and recurrent pneumonia) | S. aureus, Salmonella typhimurium | Bronchiectasis | NR | NR |
| 8 | Brigida et al (2018)11 (patient 1) Volpi et al (2019)10 (patient 2) | Italy | M | 1 | 15 | Failure to thrive, eczema, recurring episodes of hemorrhagic enterocolitis | No | No | Eczema, failure to thrive, hemorrhagic enterocolitis, panniculitis, leukocytoclastic vasculitis, recurrent infections (complicated pneumonia, warts in hands and face) | Salmonella typhi, Staphylococcus, P aeruginosa | None described | Yes | Alive |
| 9 | Brigida et al (2018)11 (patient 2) Volpi et al (2019)10 (patient 3) | Italy | M | 2 | 5 | Macrophage activation syndrome (triggered by CMV infection), fever, polymorphic vasculitic rash, hepatosplenomegaly, and hematochezia | Yes | NR | Eczema, failure to thrive, hematochezia, lymphadenopathy, hepatosplenomegaly, leukocytoclastic vasculitis, recurrent otitis | CMV, P aeruginosa | None described | Yes | Alive |
| 10 | Brigida et al (2018)11 (patient 3) Yan et al (2005)19 | Canada | M | 2 | 24 (after death) | Infections including middle ears, venipuncture sites, and perianal areas | Yes | No | Eczema, food allergy, failure to thrive, diarrhea, lymphadenopathy, hepatosplenomegaly, infections (otitis media, perianal area infections, severe balanitis, inguinal lymphadenitis, chickenpox, pansinusitis, warts) | P aeruginosa, E coli, S aureus, chickenpox (varicella-zoster virus) | Hearing loss, learning disability | No | Died |
| 11 | Brigida et al (2018)11 (patient 4) | Colombia | M | 1 | 3.5 | Gluteal abscess | Yes | Two brothers died, with similar symptoms | Eczema, failure to thrive, diarrhea, lymphadenopathy, hepatosplenomegaly, inflammatory colitis, gastrointestinal bleeding, gingival, hemorrhagic enteritis, infections (gluteal abscess, pulmonary abscess, recurrent pneumonia, bronchiolitis, sepsis) | P aeruginosa, Staphylococcus pettenkoferi | None described | No | Alive |
| 12 | Brigida et al (2018)11 (patient 5) | Morocco | F | 1 | 3 | Cold abscess in lung and recurrent pneumonias | Yes | Two siblings died, with similar symptoms | Eczema, bloody diarrhea, oral thrush, infections (cold abscess in lung and recurrent pneumonia) | NR | Bronchiectasis | NR | Alive |
| 13 | Brigida et al (2018)11 (patient 6) | Turkey | F | 1 | 11 | S awrews-positive lung and joint abscesses, bloody diarrhea, and bacterial meningitis | No | NR | Eczema, failure to thrive, bloody diarrhea, lymphadenopathy, hepatosplenomegaly, fine and brittle hair, keratotic hair lesions, big thumb and hallux, dysplastic teeth, delay of dental eruption, early release of upper gums evident since birth, microcephaly (−2 SD), psychomotor retardation. 7 y: lower extremities purpura and right knee arthritis appeared without histologic signs of vasculitis. Infections (lung and joint abscesses, and bacterial meningitis. Perianal and vulvar necrotizing ulcerations, evolving in lichenoid lesions. Epidermodysplasia verruciformis, submandibular adenitis, recurrent otorrhea, pansinusitis with osteolysis) | S aureus, actinomyces HPV, CMV | Aneurysm of left carotid sinus without calcification | NR | Alive |
| 14 | Somech et al (2017)16 (patient 1) | Israel | M | Infancy | NR | Colitis, eczema, papulovesicular lesions on scalp, severe thrombocytopenia | Yes | Sibling to patient 14 | Eczema, colitis, infections (perianal abscess, lymphadenitis, recurrent pneumonia) | CMV, adenovirus | None described | No | Died |
| 15 | Brigida et al (2018)11 (patient 7) Somech et al (2017)16 (patient 2) | Israel | M | 2 | 0.1 | Fever, eczema, purulent otitis media, bloody mucous stools, thrombocytopenia | Yes | Sibling to patient 13 | Eczema, bloody diarrhea, infections (purulent otitis media. Blood cultures positive for pneumococcus and enterococcus) | Pneumococcus and enterococcus | Severe veno-occlusive disease after bone marrow transplantation | Yes | Died |
| 16 | Kahr et al (2017)9 (patient 1) Volpi et al (2019)10 (patient 9) | South Asian | M | 1 | NR | Neck abscess and meningitis complicated by septic shock and cerebrovascular accident and thrombocytopenia | Yes | NR | Eczema, failure to thrive, bloody diarrhea, lymphadenopathy, cerebrovascular accident, leukocytoclastic vasculitis, skeletal abnormalities: mild joint limitation in elbows and wrists with slow progression to bony erosions, recurrent infections (abscesses neck, finger; meningitis, gastroenteritis, pneumonia, cellulitis, recurrent bilateral blepharitis) | S aureus, Rotavirus | Cerebrovascular accident (superior sagittal sinus thrombosis with left posterior parietal infarction), inflammatory bowel disease | No | Alive |
| 17 | Kahr et al (2017)9 (patient 2) Volpi et al (2019)10 (patient 10) | Scotland | M | 2 | NR | Extensive cradle cap, eczematous rash followed by target lesions, purpura, and swelling | No | Sibling to patient 17 | Eczema, failure to thrive, subconjunctival hemorrhage, brain vasculitis, leukocytoclastic vasculitis | NA | None described | No | Alive |
| 18 | Kahr et al (2017)9 (patient 3) Volpi et al (2019)10 (patient 11) | Scotland | M | 1 | 4 | Eczema | No | Sibling to patient 16 | Eczema, skin lesions with residual hyperpigmentation, asthma, failure to thrive, infections (pneumonia and impetigo of face) | NR | None described | No | Alive |
| 19 | Volpi et al (2019)10 (patient 4) | Nepal | M | 1 | NR | Bronchiolitis and stridor attributed to tracheomalacia | No | NR | Eczema, asthma, failure to thrive, hematemesis with wheezing, recurrent otitis media | NR | Gastritis, chronic lung disease, chronic inflammatory intestinal, hemihypertrophy | No | Alive |
| 20 | Volpi et al (2019)10 (patient 5) | Somalia | F | 1 | 10 | Generalized neonatal maculopapular rash; later, eczema | No | No | Eczema, failure to thrive, bloody diarrhea, infections (mastoiditis, recurrent skin abscesses, warts, otitis media, periodontitis with premature tooth loss, chronic CMV viremia, complicated pneumonia) | S aureus CMV, molluscum | Bronchiectasis, gut biopsies showed nonspecific inflammation | No | Alive |
| 21 | Volpi et al (2019)10 (patient 6) | Morocco | M | 1 | 11 | Recurrent bronchitis and pneumonias | Yes | No | Eczema, failure to thrive, bleeding, bloody diarrhea, infections (bronchiolitis, gastroenteritis, skin abscess, recurrent pneumonia and otitis media) | Campylobacter, P aeruginosa, Klebsiella pneumoniae, respiratory syncytial virus, molluscum | None described | Yes | Alive |
| 22 | Volpi et al (2019)10 (patient 7) | Iran | F | 2 | 4 | Umbilical bleeding and generalized maculopapular rash | Yes | NR | Eczema, food allergy, allergy to house dust mite, failure to thrive, umbilical bleeding and epistaxis, severe skin vasculitis started later in course of disease, infections (perianal abscess, fungal otitis externa, ecthyma gangrenosum, recurrent pneumonia) | NR | None described | No | NR |
| 23 | Volpi et al (2019)10 (patient 8) | Iran | F | 1 | 9 | Generalized maculopapular rash | Yes | NR | Eczema, food allergy, mites allergy, severe colitis eosinophilic, failure to thrive, rectorrhagia, infections (meningitis, perforated otitis media, purulent axillary lymphadenitis, eczema herpeticum, recurrent pneumonia) | Herpes simplex virus | Urinary tract infection with pyelocaliectasis | No | NR |
| 24 | Volpi et al (2019)10 (patient 12) | Nepal | F | 2 | 0.4 | Erosive dermatitis with purpuric and eczematous areas, ulcerative lesions: perianal, eyelid, gum, vulva, lip, and ear; poor wound healing | No | NR | Eczema, food allergy, failure to thrive, bloody diarrhea, leukocytoclastic vasculitis, infections (chronic CMV, urinary tract infection, chronic oral thrush, otitis media, periorbital cellulitis, skin infections, perianal ulcer) | Enterococcus sp., S aureus, C albicans, Pseudomonas sp., CMV | Cystic pulmonary lesion of unclear etiology and transient hepatitis | Yes | Alive |
| 25 | Volpi et al (2019)10 (patient 13) | Jordan | F | 12 | 4 | Recurrent pneumonia and otitis media | Yes | NR | Leukocytoclastic vasculitis, chronic arthritis (knees and ankles), recurrent otitis media, and pulmonary infections | NR | None described | No | Alive |
| 26 | Volpi et al (2019)10 (patient 14) | Nepal | M | S | S | Failure to thrive, eczema, lymphadenopathy with microcytic anemia and thrombocytopenia | No | NR | Eczema, failure to thrive, hemoptysis, enterorrhagia, lymphadenopathy, polyarthritis, infections (periodontal disease, recurrent otitis media and bronchiolitis) | Molluscum | Bronchiectasis | No | NR |
| 27 | Papadatou et al (2021)18 (patient 1 Backspace or II-6) | Middle East | M | 6 | 0.5 | Pneumonia | Yes | Sibling to patients 27 and 28 | Eczema, food allergy with anaphylaxis, allergic asthma, failure to thrive, autoimmune hypothyroidism, infections (perianal and skin abscesses, recurrent pneumonia and bronchiolitis) | P aeruginosa | None described | Yes | Alive |
| 28 | Papadatou et al (2021)18 (patient 2 or II-1) | Middle East | M | NR | 14 | Recurrent skin and respiratory tract infections, hypothyroidism | Yes | Sibling to patients 26 and 28 | Eczema, failure to thrive, lymphadenopathy, presence of antithyroid autoantibodies, hypothyroidism, infections (generalized lymphadenitis, skin abscesses, recurrent pneumonia) | NR | Exophthalmos | Yes | Alive |
| 29 | Papadatou et al (2021)18 (patient 3 or II-3) | Middle East | F | 120 | 10 | Failure to thrive | Yes | Sibling to patients 26 and 27 | Failure to thrive, autoimmune hypothyroidism, recurrent viral upper respiratory tract infections | NR | None described | No | Alive |
| 30 | Kopitar et al (2019)17 (patient 1) | Slovenia | NR | 1 | 27 | Eczema, thrombocytopenia, bloody diarrhea | NR | NR | Eczema, food, pollen and mites allergy, failure to thrive, enterocolitis, small vessels vasculitis, panniculitis and recurrent bronchiolitis, pneumonia, skin abscesses | NR | None described | NR | NR |
| 31 | Kopitar et al (2019)17 (patient 2) | Slovenia | NR | 1 | 24 | Eczema, thrombocytopenia, bloody diarrhea | NR | NR | Eczema, food allergy, failure to thrive, metaplasia in gastric mucosa, bowel adenoma in situ, enterocolitis, autoimmune vasculitis, infections (warts, complicated pneumonia, gastroenteritis, Candida esophagitis) | Candida | None described | NR | NR |
| 32 | Kopitar et al (2019)17 (patient 3) | Slovenia | F | 1 | 30 | Eczema, thrombocytopenia, bloody diarrhea | NR | NR | Eczema, food, mites, animal epithelia allergy, allergic asthma, failure to thrive, cervical intraepithelial neoplasia grade 2, enterocolitis, small vessels vasculitis. autoimmune vasculitis, infections (chronic leg ulceration, genital condylomata, severe warts, recurrent complicated pneumonia, skin abscess) | NR | None described | NR | NR |
| 33 | Kopitar et al (2019)17 (patient 4) | Slovenia | NR | 7 | 1.3 | Eczema and Evans syndrome | NR | NR | Mild eczema, Evans syndrome | NA | None described | NR | NR |
| 34 | Antala et al (2021)20 (patient 1) | Kenya | M | 7 | 0.33 | Hematochezia and pustular rash | Yes | Sibling died (had similar symptoms) | Pustular rash, failure to thrive, gastrointestinal bleeding | NA | Hyperplasia nodular and intestinal metaplasia | Yes | Alive |
NA, not available; NR, not reported.
TABLE IV.
Infection sites reported in 34 patients with actin-related protein 2/3 complex subunit 1 B deficiency
| Most frequent infections | Patients (n = 34) | Prevalence (%) |
|---|---|---|
| Abscesses | 12 | 36 |
| Warts | 8 | 24 |
| Other skin infections* | 10 | 29 |
| Pneumonia | 22 | 65 |
| Bronchiolitis | 8 | 24 |
| Otitis | 11 | 32 |
| Other respiratory tract infections† | 4 | 12 |
| Lymphadenitis | 6 | 18 |
| Gastroenteritis | 6 | 18 |
| Urinary tract infection | 3 | 9 |
| Meningitis | 3 | 9 |
Other skin infections include cellulitis (patients 1, 4. 10, 16. and 24). perianal ulcerations (patient 13), impetigo (patient 18). ecthyma gangrenosum (patient 22), eczema herpeticum (patient 23), perianal ulcer (patient 24), chronic leg ulceration (patient 32), genital condylomata (patient 32), epidermodysplasia verruciformis (patient 13), and skin infection (patient 24).
Other respiratory infections include pansinusitis (patients 10 and 13). mastoiditis (patient 20). and recurrent viral upper respiratory tract infection (patients 20 and 29).
DISCUSSION:
We described the clinical features, immunological studies, and mutational analysis of six new patients with ARPC1B deficiency from Mexico and reviewed the literature in search of all patients reported to date. In total, we have identified 34 patients with ARPC1B deficiency from the Americas, Africa, Europe, Southern Asia, and the Middle East. This is the first such report from Mexico and one of the largest case series available. All previously reported probands had homozygous variants; we identified compound heterozygous genotypes in two non-consanguineous families.
Some of the findings in our patients are unique to this series: regional BCG-itis, keloid scars, and chronic EBV hepatitis (seen in one patient each). Osteopenia, also found in one patient, might be related to chronic steroid treatment. As for ganglionic tuberculosis attributable to BCG, and keloid scars, a coincidental association cannot be excluded, although we consider the pathophysiology of ARPC1B deficiency to be compatible with these manifestations. Finally, chronic CMV hepatitis was reported in one of the patients described by Volpi et al (10), and increased susceptibility to the Herpesviridae family is common to actinopathies.
Despite having received two “failed” stem-cell transplantations, P1 showed great clinical improvement attributed to mixed chimerism, as suggested by a 3.99% donor chimerism result in DNA from peripheral blood cells. Three of four unrelated families carry the same 46bp deletion in exon 8 despite coming from distant states (Michoacan, Monterrey, and Veracruz) and being mestizos of Hispanic descent, likely a founder effect as it has not been reported in other latitudes.
Of note, in the sequence analysis of patient 5, only one of the heterozygous missense variants is exceedingly rare and likely pathogenic (V32M), while the other one (K37N) is a polymorphism with a global frequency of 5% in gnomAD but equally deleterious parameters in silico (figure E1). As these variants are found close to one another in trans, we hypothesize they combine into a deleterious biallelic haplotype. Although these substitutions did not apparently perturb the structure of the protein, two post-translational modifications might make the ARPC1B subunit prone to degradation in a mechanism originally related to the terminal marking phenomenon for natural protein turnover. The K37N variant is susceptible to being deamidated and changed to K37D, thus causing, together with phosphorylation of Thr21, a strong impairment in the structure of ARPC1B. In addition, novel negative charges generated by said modifications might highly increase the mobility at the NH-terminus.
Patient 5 has a history of bleeding diathesis and chronic EBV hepatitis complicated by liver failure and HLH, and his lab workup showed leukocytosis, eosinophilia, large platelets, and thrombocytosis/thrombocytopenia, with hyper-IgG, IgA, and IgE. Genetic heterogeneity (and clinical variability) but physiological homogeneity (24) also means that the lab results of patients with the same inborn error of immunity will often resemble each other more than their medical records do.
Keloid scars have been hypothesized to result from viral infection (25). They have also been linked to deficient production of IL-6 and IFN-gamma, with IL-6 deficient mice also developing keloid scars (26). As compared to hypertrophic scars, keloids have also been described to contain less total actin, less filamentous actin, and more globular actin per fibroblast (27–31). Interestingly, ARPC1B-deficient patients are susceptible to cutaneous viral infection, have a predominant Th2 cytokine production, and their fibroblasts are lacking proper filamentous actin fibers (12,17).
ARPC1B deficiency can be thought of as a “cross” between Wiskott-Aldrich Syndrome and DOCK8 deficiency, two other well-known “classical” actinopathies (see figure E10). In fact, roughly half of our patients were first suspected to have WAS, while the other half were thought to be DOCK8 deficiencies. We will probably keep seeing patients with “atypical” histories and a provisional diagnosis of WAS or DOCK8.
As reviewed by Seppänen, Tangye, Boztug, Kuijpers et al. (3–6), actinopathies should be suspected whenever there are eosinophilia and platelet disorders in the context of allergic and bleeding diatheses, together with infections caused by viruses, bacteria and fungi, and autoimmune manifestations that include vasculitis. Most ARPC1B-deficient patients will also have hypergammaglobulinemia (IgE, IgA, IgG) and increased B cell numbers (9,10). In contrast with DOCK8 deficiency, in which low IgM levels are frequently reported, serum IgM concentration was found normal in 16/29 (55%), low 9/29 (31%), and high 4/29 (14%) in patients with ARPC1B deficiency.
Next, we would like to study the actin content (globular and filamentous), and search for viral inclusions, in keloid tissue from our patient. To better understand the impact of variant K37N, it would be of help to study the relative abundance of ARPC1B and ARPC1A, as well as the rate of deamidation of these proteins in the B cells patient 5. To assess a founder mutation in our country, we want to sequence as many family members as possible and perform haplotype analysis.
In conclusion, patients with ARPC1B deficiency may also develop chronic EBV infection, adverse reactions to the BCG vaccine, and keloid scars. The combination of eczema, allergy, bleeding, vasculitis, viral and bacterial infections, eosinophilia, platelet abnormalities, hypergammaglobulinemia, and high B cells should raise the suspicion of ARPC1B deficiency and may be confused with other better-known actinopathies like DOCK8 or WASP deficiencies. HSCT is considered curative. Even partial T-cell chimerism may be sufficient to prevent serious infections.
Supplementary Material
TABLE II.
Genetic features of 34 patients with inherited actin-related protein 2/3 complex subunit 1B deficiency
| Patient | References | Actin-related protein 2/3 complex subunit 1B gene variant (NM_005720.3) | Zygosity | Exon | Protein consequence |
|---|---|---|---|---|---|
| 1 | This report | c.899_944del (46bp) AGCGCTTCCAGAACCTGGA CAAGAAGGCGA GCTCCGAGGGTGGCAC | Homozygous | Exon 8 | p.Glu300GlyfsTer31 |
| 2 | This report | c.899_944del (46bp) AGCGCTTCCAGAACCTGGA CAAGAAGGCGA GCTCCGAGGGTGGCAC | Homozygous | Exon 8 | p.Glu300GlyfsTer31 |
| 3 | This report | c.64C>T / c.899_944del(46bp) AGCGCTTCCAGAACCTG GACAAGAAGGCGA GCTCCGAGGGTGGCAC | Compound heterozygous |
Exon 2/Exon 8 of 10 | p.Gln22Ter / p.Glu300GlyfsTer31 |
| 4 | This report | NA (brother to patient 4) | Homozygous | Exons 2 and 8 | NA (brother to patient 3) |
| 5 | This report | c.94G>A/111G>C | Compound heterozygous |
Exon 3 | p.Val32Met/Lys37Asn |
| 6 | This report | c.899_944del(46bp) AGCGCTTCCAGAACCTG GACAAGAAGGCGA GCTCCGAGGGTGGCAC | Homozygous | Exon 8 | p.Glu300GlyfsTer31 |
| 7 | Kuijpers et al (2017)8 Volpi et al (2019)10 (patient 1) | c.491_495delinsCCTGCCC | Homozygous | Exon 7 | p.Phe164SerfsTer31 |
| 8 | Brigida et al (2018)11 (patient 1) Volpi et al (2019)10 (patient 2) | c.64+1G>C | Homozygous | Donor splice site of intron 2 | NA |
| 9 | Brigida et al (2018)11 (patient 2) Volpi et al (2019)10 (patient 3) | c.622G>T | Homozygous | Exon 6 | p.Val208Phe |
| 10 | Brigida et al (2018)11 (patient 3) Yan et al (2005)19 | c.1087dup | Homozygous | Exon 10 | p.Glu363GlyfsTer95 |
| 11 | Brigida et al (2018)11 (patient 4) | c.258G>A | Homozygous | Exon 4 | p.Trp86Ter |
| 12 | Brigida et al (2018)11 (patient 5) | c.318del | Homozygous | Exon 4 | p.Asn107ThrfsTer13 |
| 13 | Brigida et al (2018)11 (patient 6) | c.64+1G>A | Homozygous | Donor splice site of intron 2 | NA |
| 14 | Somech et al (2017)16 (patient 1) | c.G623del-TC | Homozygous | Exon 6 | p.Val208ValfsTer20 |
| 15 | Brigida et al (2018)11 (patient 7) Somech et al (2017)16 (patient 2) | c.G623del-TC | Homozygous | Exon 6 | p.Val208ValfsTer20 |
| 16 | Kahr et al (2017)9 (patient 1) Volpi et al (2019)10 (patient 9) | c.269_270dupCT | Homozygous | Exon 4 | p.Val91TrpfsTer30 |
| 17 | Kahr et al (2017)9 (patient 2) Volpi et al (2019)10 (patient 10) | c.314C>T | Homozygous | Exon 4 | p.Ala105Val |
| 18 | Kahr et al (2017)9 (patient 3) Volpi et al (2019)10 (patient 11) | c.314C>T | Homozygous | Exon 4 | p.Ala105Val |
| 19 | Volpi et al (2019)10 (patient 4) | c.64+2T>A | Homozygous | Donor splice site of intron 2 | NA |
| 20 | Volpi et al (2019)10 (patient 5) | c.392+2T>C | Homozygous | Donor splice site of intron 4 | NA |
| 21 | Volpi et al (2019)10 (patient 6) | c.311G>C | Homozygous | Exon 4 | p.Trp104Ser |
| 22 | Volpi et al (2019)10 (patient 7) | c.897_910delCGAGCGCTTCCAGA | Homozygous | Exon 10 | p.Glu300ProfsTer153 |
| 23 | Volpi et al (2019)10 (patient 8) | c.897_910delCGAGCGCTTCCAGA | Homozygous | Exon 10 | p.Glu300ProfsTer153 |
| 24 | Volpi et al (2019)10 (patient 12) | c.64+2T>A | Homozygous | Donor splice site of intron 2 | NA |
| 25 | Volpi et al (2019)10 (patient 13) | c.708–1G>A | Homozygous | Splice site acceptor of exon 6 | NA |
| 26 | Volpi et al (2019)10 (patient 14) | c.64+2T>A | Homozygous | Donor splice site of intron 2 | NA |
| 27 | Papadatou et al (2021)18 (patient 1 or II-6) | c.783G>A | Homozygous | Exon 7, last nucleotide position | p.Ala261Ala loss of donor splice site |
| 28 | Papadatou et al (2021) (patient 2 or II-1) | c.783G>A | Homozygous | Exon 7, last nucleotide position | p.Ala261Ala loss of donor splice site |
| 29 | Papadatou et al (2021)18 (patient 3 or II-3) | c.783G>A | Homozygous | Exon 7, last nucleotide position | p.Ala261Ala loss of donor splice site |
| 30 | Kopitar et al (2019)17 (patient 1) | c.265A>C | Homozygous | Exon 4 | p.Thr89Pro |
| 31 | Kopitar et al (2019)17 (patient 2) | c.265A>C | Homozygous | Exon 4 | p.Thr89Pro |
| 32 | Kopitar et al (2019)17 (patient 3) | c.265A>C | Homozygous | Exon 4 | p.Thr89Pro |
| 33 | Kopitar et al (2019)17 (patient 4) | c.265A>C | Homozygous | Exon 4 | p.Thr89Pro |
| 34 | Antala et al (2021)20 (patient 1) | NA | Homozygous | Splice site | NA |
NA, not available.
TABLE III.
Prevalence of most common attributes found in human patients with autosomal recessive actin-related protein 2/3 complex subunit 1B deficiency
| Clinical or laboratory features | Prevalence (n = 34) | Prevalence (%) |
|---|---|---|
| General manifestations | ||
| Eczema | 30 | 88 |
| Recurrent infections | 31 | 91 |
| Failure to thrive | 26 | 76 |
| Bleeding disorders* | 25 | 74 |
| Diarrhea | 16 | 47 |
| Allergy (food and or pollen allergy) | 13 | 38 |
| Vasculitis† | 13 | 38 |
| Lymphadenopathy | 9 | 26 |
| Other skin lesions‡ | 12 | 36 |
| Hepatomegaly | 8 | 24 |
| Colitis and or enterocolitis | 7 | 21 |
| Splenomegaly | 6 | 18 |
| Asthma | 4 | 12 |
| Autoimmune hypothyroidism | 3 | 9 |
| Complications | ||
| Bronchiectasis | 5 | 15 |
| Hearing loss | 2 | 6 |
| Learning disability | 2 | 6 |
| Arthritis | 2 | 6 |
| Chronic lung disease | 2 | 6 |
| Vertebral fractures | 1 | 3 |
| Cushing syndrome | 1 | 3 |
| Laboratory features | ||
| Thrombocytopenia | 23/33 | 70 |
| Abnormal platelet size§ | 12/23 | 52 |
| Hypereosinophilia | 22/24 | 92 |
| Hyper IgG | 11/30 | 37 |
| Hyper IgA | 20/30 | 67 |
| Hyper IgE | 31/32 | 97 |
| Low CD3+ (cells/mL) | 20/33 | 61 |
| Low CD4+ (cells/mL) | 19/33 | 56 |
| Low CD8+ (cells/mL) | 20/33 | 61 |
| High CD19+ (cells/mL) | 21/33 | 64 |
| Low natural killer (cells/mL) | 11/33 | 33 |
Bleeding disorders include gastrointestinal bleeding (patients 2–4, 7–9, 11–13, 15, 16, 19–21, 23, 24, 26, 30–32, and 34); bleeding of gums (patients 5, 11, and 12) and umbilical stump (patient 22); epistaxis (patients 5 and 22); hemoptysis (patient 26); ecchymosis (patient 5); and subconjunctival hemorrhage (patient 17).
Vasculitis includes brain (patient 1), leukocytoclastic (patients 7–9, 16, 17, 24, and 25), polymorphic vasculitis rash (patient 9), skin vasculitis (patients 6 and 22), and small vessels (patients 31 and 32).
Other skin lesions include erythema nodosum (patient 6), skin pustules (patient 6), panniculitis (patients 8 and 30), papulovesicular lesions (patient 14), lesions with residual hyperpigmentation (patient 18), maculopapular rash (patients 20, 22, and 23), warts (patients 1, 6, 8, 10, 20, 31, and 32), and pustular rash (patient 34).
Abnormal platelet size: large in patient 5 (small in other patients).
HIGHLIGHTS:
1. What is already known about this topic?
ARPC1B deficiency is a combined immune defect with short stature, skin infection, allergic and bleeding diatheses, vasculitis, leukocytosis, eosinophilia, platelet abnormalities, hypergammaglobulinemia with elevated B cells, and risk of malignancy. Stem-cell transplantation has been curative.
2. What does this article add to our knowledge?
Six new patients from four Mexican families expand the clinical spectrum to include keloid scars and Epstein-Barr virus chronic hepatitis. All four gene variants are novel, including two compound heterozygous and a potential founder effect.
3. How does this study impact current management guidelines?
One of our patients developed liver failure. Two other patients underwent stem cell transplantation and improved greatly. A high index of suspicion and multidisciplinary engagement are required for correct, early diagnosis and management of ARPC1B-deficient patients.
ACKNOWLEDGMENTS:
Dr. Eduardo Liquidano Perez worked on an early version of the manuscript. Dr. Mario E. Cruz Muñoz critically reviewed the manuscript for clarity and content. Dr. Francisco J. Espinosa-Rosales provided medical care and follow-up for patient 6. Funding for whole-exome sequencing provided by Conacyt (Consejo Nacional de Ciencia y Tecnología), Innovation Stimuli Program 2018 and Frontier science 2019 (grant number #10869). We thank the Mexican Foundation for Children with Primary Immunodeficiencies (FUMENI, AC) for support with the shipment of blood DNA samples, and postgraduate financial support. Our thanks are also due to doctors Capucine Picard, Satoshi Okada, and Carmen Alaez-Verson, who performed assays and analyses that ruled out differential diagnoses before 2016.
Funding:
Conacyt (Frontier Science) grant CF-2019 #10869.
The study was supported in part by a grant from the St. Giles Foundation, The Rockefeller University, Institut National de la Santé et de la Recherche Médicale (INSERM), University of Paris, the National Institute of Allergy and Infectious Diseases (P01AI061093), the National Center for Research Resources, the National Center for Advancing Sciences of the National Institutes of Health (UL1TR001866), the French National Research Agency (ANR) under the “Investments for the Future” program (ANR-10-IAHU-01), the Integrative Biology of Emerging Infectious Diseases Laboratory of Excellence (ANR-10-LABX-62-IBEID) and the French Foundation for Medical Research (FRM) (EQU201903007798).
ABBREVIATIONS:
- ARPC1B
Actin Related Protein 2/3 Complex Subunit 1B
- BCG
Bacille Calmette-Guérin
- CMV
Cytomegalovirus
- DNA
Deoxyribonucleic acid
- HSCT
Hematopoietic Stem Cell Transplant
- DOCK8
Dedicator of cytokinesis 8
- EBER
EBV-encoded small RNAs
- EBV
Epstein-Barr virus
- HIMFG
Children’s Hospital of Mexico “Dr. Federico Gómez”
- IEI
Inborn errors of immunity
- LMP1
latent membrane protein 1
- PCR
polymerase chain reaction
- PMA
phorbol myristate acetate
- PHA
phytohemagglutinin
- RNA
ribonucleic acid
- WAS
Wiskott-Aldrich syndrome
- WES
whole-exome sequencing
- MLPA
Multiplex Ligation-dependent Probe Amplification
- IVIG
intravenous immunoglobulin
- PTT
Partial Thromboplastin Time
Footnotes
Ethical/IRB Approval statement: Patients or their guardians consented in writing to their genetic evaluation and the publication of these case reports. The study was approved by the Institutional Review Board of the National Institute of Pediatrics (numbers 2013/049 and 2021/054).
Code availability: Not applicable.
Consent for publication: Consent for publication was given by the patients’ family members.
DECLARATIONS
Conflicts of interest: The authors declare no competing interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Availability of dataset and material:
Not applicable. All data and further information are available upon request.
REFERENCES:
- 1.Ronicke S, Hirsch MC, Türk E, Larionov K, Tientcheu D, Wagner AD. Can a decision support system accelerate rare disease diagnosis? Evaluating the potential impact of Ada DX in a retrospective study. Orphanet J Rare Dis. 2019. Mar 21;14(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sullivan KE, Stiehm ER (eds). Stiehm’s Immune Deficiencies [Internet]. 2nd ed. Sullivan KE, Stiehm R, editors. Stiehm’s Immune Deficiencies. New York: Elsevier; 2020 [cited 2021 Feb 17]. 1332 p. [Google Scholar]
- 3.Sprenkeler EGG, Webbers SDS, Kuijpers TW. When Actin is Not Actin’ like It Should: A New Category of Distinct Primary Immunodeficiency Disorders. Journal of Innate Immunity. S. Karger AG; 2020. p. 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Seppänen MRJ. Novel cytoskeletal mutations with immunodeficiency: Why is the raven like a writing desk? J Allergy Clin Immunol. 2018;142(5):1444–6. [DOI] [PubMed] [Google Scholar]
- 5.Tangye SG, Bucciol G, Casas-Martin J, Pillay B, Ma CS, Moens L, et al. Human inborn errors of the actin cytoskeleton affecting immunity: way beyond WAS and WIP. Immunol Cell Biol. 2019;97(4):389–402. [DOI] [PubMed] [Google Scholar]
- 6.Dupré L, Boztug K, Pfajfer L. Actin Dynamics at the T Cell Synapse as Revealed by Immune-Related Actinopathies. Front Cell Dev Biol. 2021;9(June). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Leung G, Zhou Y, Ostrowski P, Mylvaganam S, Boroumand P, Mulder DJ, et al. ARPC1B binds WASP to control actin polymerization and curtail tonic signaling in B cells. JCI Insight [Internet]. 2021. Dec 8;6(23). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kuijpers TW, Tool ATJ, van der Bijl I, de Boer M, van Houdt M, de Cuyper IM, et al. Combined immunodeficiency with severe inflammation and allergy caused by ARPC1B deficiency. J Allergy Clin Immunol. 2017. Jul 1;140(1):273–277.e10. [DOI] [PubMed] [Google Scholar]
- 9.Kahr WHA, Pluthero FG, Elkadri A, Warner N, Drobac M, Chen CH, et al. Loss of the Arp2/3 complex component ARPC1B causes platelet abnormalities and predisposes to inflammatory disease. Nat Commun 2017. Apr 3;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Volpi S, Cicalese MP, Tuijnenburg P, Tool ATJ, Cuadrado E, Abu-Halaweh M, et al. A combined immunodeficiency with severe infections, inflammation, and allergy caused by ARPC1B deficiency. J Allergy Clin Immunol. 2019. Jun 1;143(6):2296–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brigida I, Zoccolillo M, Cicalese MP, Pfajfer L, Barzaghi F, Scala S, et al. T-cell defects in patients with ARPC1B germline mutations account for combined immunodeficiency. Blood. 2018;132(22):2362–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Randzavola LO, Strege K, Juzans M, Asano Y, Stinchcombe JC, Gawden-Bone CM, et al. Loss of ARPC1B impairs cytotoxic T lymphocyte maintenance and cytolytic activity. J Clin Invest. 2019. Dec 2;129(12):5600–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shapovalov MV, Dunbrack RL Jr. A smoothed backbone-dependent rotamer library for proteins derived from adaptive kernel density estimates and regressions. Structure 2011; 19(6): 844–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pettersen EF; Goddard TD; Huang CC; Couch GS; Greenblatt DM; Meng EC; Ferrin TE, UCSF Chimera--a visualization system for exploratory research and analysis. J Comput Chem 2004; 25(13): 1605–12. [DOI] [PubMed] [Google Scholar]
- 15.Castano‐Jaramillo LM, Yamazaki‐Nakashimada MA, Scheffler Mendoza SC, Bustamante‐Ogando JC, Espinosa‐Padilla SE, Lugo Reyes SO. A male infant with COVID‐19 in the context of ARPC1B deficiency. Eigenmann P, editor. Pediatr Allergy Immunol [Internet]. 2021. Jan 2;32(1):199–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Somech R, Lev A, Lee YN, Simon AJ, Barel O, Schiby G, et al. Disruption of Thrombocyte and T Lymphocyte Development by a Mutation in ARPC1B . J Immunol. 2017;199(12):4036–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kopitar AN, Markelj G, Oražem M, Blazina Š, Avčin T, Ihan A, et al. Flow cytometric determination of actin polymerization in peripheral blood leukocytes effectively discriminate patients with homozygous mutation in ARPC1B from asymptomatic carriers and normal controls. Front Immunol. 2019;10(JULY):1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Papadatou I, Marinakis N, Botsa E, Tzanoudaki M, Kanariou M, Orfanou I, et al. Case Report: A Novel Synonymous ARPC1B Gene Mutation Causes a Syndrome of Combined Immunodeficiency, Asthma, and Allergy With Significant Intrafamilial Clinical Heterogeneity. Front Immunol. 2021;12(February):1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yan SR, Bortolussi R, Issekutz TB, Issekutz AC. Increased chemoattractant induced neutrophil oxidative burst, accelerated apoptosis, and dysregulated tyrosine phosphorylation associated with lifelong bacterial infections. Clin Immunol. 2005;117(1):36–47. [DOI] [PubMed] [Google Scholar]
- 20.Antala S, Whitehead B, Melin-Aldana H, Bass LM. ARPC1B Mutation Manifesting as Recurrent Hematemesis With Metaplasia. JPGN Reports. 2021. Aug;2(3):e095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vadlamudi RK, Li F, Barnes CJ, Bagheri-Yarmand R, Kumar R. p41-Arc subunit of human Arp2/3 complex is a p21-activated kinase-1-interacting substrate. EMBO Rep 2004; 5(2):154–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de la Mora-de la Mora I, Torres-Larios A, Enríquez-Flores S, Méndez ST, Castillo-Villanueva A, Gómez-Manzo S, López-Velázquez G, Marcial-Quino J, Torres-Arroyo A, García-Torres I, Reyes-Vivas H, Oria-Hernández J. Structural effects of protein aging: terminal marking by deamidation in human triosephosphate isomerase. PLoS One 2015; 10(4): e0123379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Rawlings ND, Bateman A. How to use the MEROPS database and website to help understand peptidase specificity. Protein Sci 2021; 30(1):83–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Notarangelo LD, Bacchetta R, Casanova JL, & Su HC Human Inborn Errors of Immunity: an Expanding Universe. Science Immunol 2020; 5(49). 10.1126/sciimmunol.abb1662 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alonso PE, Rioja LF, Pera C. Keloids: A viral hypothesis. Med Hypotheses. 2008. Jan 1;70(1):156–66. [DOI] [PubMed] [Google Scholar]
- 26.Ghazizadeh M, Tosa M, Shimizu H, Hyakusoku H, Kawanami O. Functional implications of the IL-6 signaling pathway in keloid pathogenesis. J Invest Dermatol. 2007;127(1):98–105. [DOI] [PubMed] [Google Scholar]
- 27.Chipev CC, Simon M. Phenotypic differences between dermal fibroblasts from different body sites determine their responses to tension and TGFβ1. BMC Dermatol. 2002;2:1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nangole FW, Agak GW. Keloid pathophysiology: fibroblast or inflammatory disorders? JPRAS Open. 2019;22:44–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Shih B, Bayat A. Genetics of keloid scarring. Arch Dermatol Res. 2010;302(5):319–39. [DOI] [PubMed] [Google Scholar]
- 30.Mukhopadhyay A, Sui YC, Lim IJ, Phillips DJ, Phan TT. The role of the activin system in keloid pathogenesis. Am J Physiol - Cell Physiol. 2007;292(4):1331–8. [DOI] [PubMed] [Google Scholar]
- 31.Shih B, Garside E, McGrouther DA, Bayat A. Molecular dissection of abnormal wound healing processes resulting in keloid disease. Wound Repair Regen. 2010;18(2):139–53. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Not applicable. All data and further information are available upon request.