

Abstract
Neoantigen cancer vaccines that target tumor specific mutations are emerging as a promising modality for cancer immunotherapy. To date, various approaches have been adopted to enhance efficacy of these therapies, but the low immunogenicity of neoantigens has hindered clinical application. To address this challenge, we developed a polymeric nanovaccine platform that activates the NLRP3 inflammasome, a key immunological signaling pathway in pathogen recognition and clearance. The nanovaccine is comprised of a poly(orthoester) scaffold engrafted with a small-molecule TLR7 agonist and endosomal escape peptide that facilitates lysosomal rupture and NLRP3 inflammasome activation. Upon solvent transfer, the polymer self-assembles with neoantigens to form ~50 nm nanoparticles that facilitate co-delivery to antigen-presenting cells. This polymeric activator of the inflammasome (PAI) was found to induce potent antigen-specific CD8+ T cell responses characterized by IFN-γ and GNZB secretion. Moreover, in combination with immune checkpoint blockade therapy, the nanovaccine stimulated robust anti-tumor immune responses against established tumors in EG.7-OVA, B16.F10, and CT-26 models. Results from our studies indicate that NLRP3 inflammasome activating nanovaccines demonstrate promise for development as a robust platform to enhance immunogenicity of neoantigen therapies.
Keywords: Nanovaccine, Biomaterials, Adjuvant, Inflammasome, Neoantigen therapy
Introduction
Neoantigen vaccines that target tumor-specific mutations hold enormous promise for cancer treatment.1,2 Such vaccines can be formulated with different synthetic peptides to stimulate a response against patient-specific neoantigens and afford improved safety profiles relative to chemotherapies and adoptive cell therapies. Despite extensive research toward designing neoantigen vaccines, there remains a need for improved approaches to enhance immunogenicity. This is especially true for targeting difficult to treat cancers, such as immunologically cold tumors and late-stage metastases.3 In this regard, incorporation of various immunostimulatory adjuvants has been investigated,2,4,5,6,7,8 but the limited efficacy of adjuvanted neoantigen vaccines remains a roadblock for clinical translation.9,10,11 This limited efficacy has created an urgent need to develop novel strategies that would bolster immunogenicity of neoantigen vaccines.
Among the various mechanisms to enhance immune responses, the NACHT, LRR and PYD domain-containing protein 3 (NLRP3) inflammasome could represent a valuable target.12,13 The NLRP3 inflammasome is a multi-protein complex generally formed by co-activation of Toll-Like Receptors (TLRs) and NLRP3 signaling pathways that induces Caspase-1 activation and pyroptotic cell death.14 The ability of NLRP3 inflammasome to initiate immune responses via secretion of potent immunostimulatory cytokines, IL-1β and IL-18, has been identified as a primary effector in the immunogenicity of vaccines and various therapeutics.15,16,17 More recently, studies have identified a role of the NLRP3 inflammasome in cancer immunosurveillance.18,19,20,21 For instance, Fu and co-workers reported that radiation therapy could not induce tumor destruction in absence of IL-1β secretion in inflammasome deficient mice.20 Similarly, IL-18 generated during inflammasome activation has been investigated in clinical trials as an anti-cancer therapeutic that stimulates activation of tumor-specific immune cell populations.22,23,24 These examples highlight the potential of developing inflammasome activating adjuvants in neoantigen vaccine design.
Unfortunately, the search for therapeutic inflammasome activating materials has resulted in limited success, as most known NLRP3 activators suffer from challenging chemical synthesis and formulation as well as unacceptable levels of off-target toxicity.25,26,14 In this context, Wang et al. reported the development of NLRP3 inflammasome-activating formulations for cancer immunotherapy by admixing NLRP3 activating spiky TiO2 microparticles with a TLR4 agonist, monophosphoryl lipid A (MPLA).19 The authors noted that significant therapeutic efficacy was obtained only when both the activators were used in combination. This study highlights the benefits of dual activation of TLR and NLRP3 signaling pathways for future therapeutic design. However, the development of formulations containing a mixture of activators can often be challenging due to activators’ different physicochemical properties. Specifically, rapid systemic diffusion of one or more activators from the site of injection can result in low efficacy and off-target toxicity.27,28 We are therefore interested in designing modular, single component materials that induce controlled and localized inflammasome activation.
Earlier, we reported a new class of chemically modular, NLRP3 activating peptides (TAT-P6-GWWWG) with the potential for incorporation into inflammasome-activating, therapeutic biomaterials.28 Given the importance of co-stimulation of TLR and NLRP3 signaling pathways, we designed a single component vaccine platform that leverages the advantages of co-administration of the NLRP3-activating peptide along with a TLR activator for inflammasome activation. Herein, we employed 2BXy, a potent TLR7/8 activator that induces generation of CD8+ T-cells – key effector cells in anti-tumor immunity.5,30 Finally, to localize immune stimulation at the tumor site and lymphatic organs, the material was designed to spontaneously self-assemble into ~50 nm structures.31,32 We envisioned that this NLRP3 inflammasome-activating neoantigen vaccine platform would lead to potent localized immune activation resulting in robust anti-tumor efficacy.
Results and Discussion
Synthesis and optimization of polymeric activators.
With the goal of generating an inflammasome activating immunostimulant platform, variable ratios of azido-functionalized 2BXy and azido-functionalized TAT-P6-GWWWG peptide (N3-YGRKKRRQRRR-PEG6-GWWWG) were sequentially grafted to a non-immunogenic sugar poly(orthoester) scaffold (SPOE)33 via Cu(I) catalyzed Huisgen cycloaddition chemistry (Figures 1A and S1–S8). Such a design afforded a series of amphiphilic polymers with varying ratios of 2BXy and TAT-P6-GWWWG. Owing to the polymeric nature of these immune activators, we referred to them as Polymeric Activators of Inflammasomes (PAIs). The ratio of 2BXy and TAT-P6-GWWWG peptide in the synthesized PAIs were quantified by high performance liquid chromatography (HPLC) following degradation of the pH-sensitive ortho-ester linkages with trifluoroacetic acid (Figure S8 and Table S1).
Figure 1:
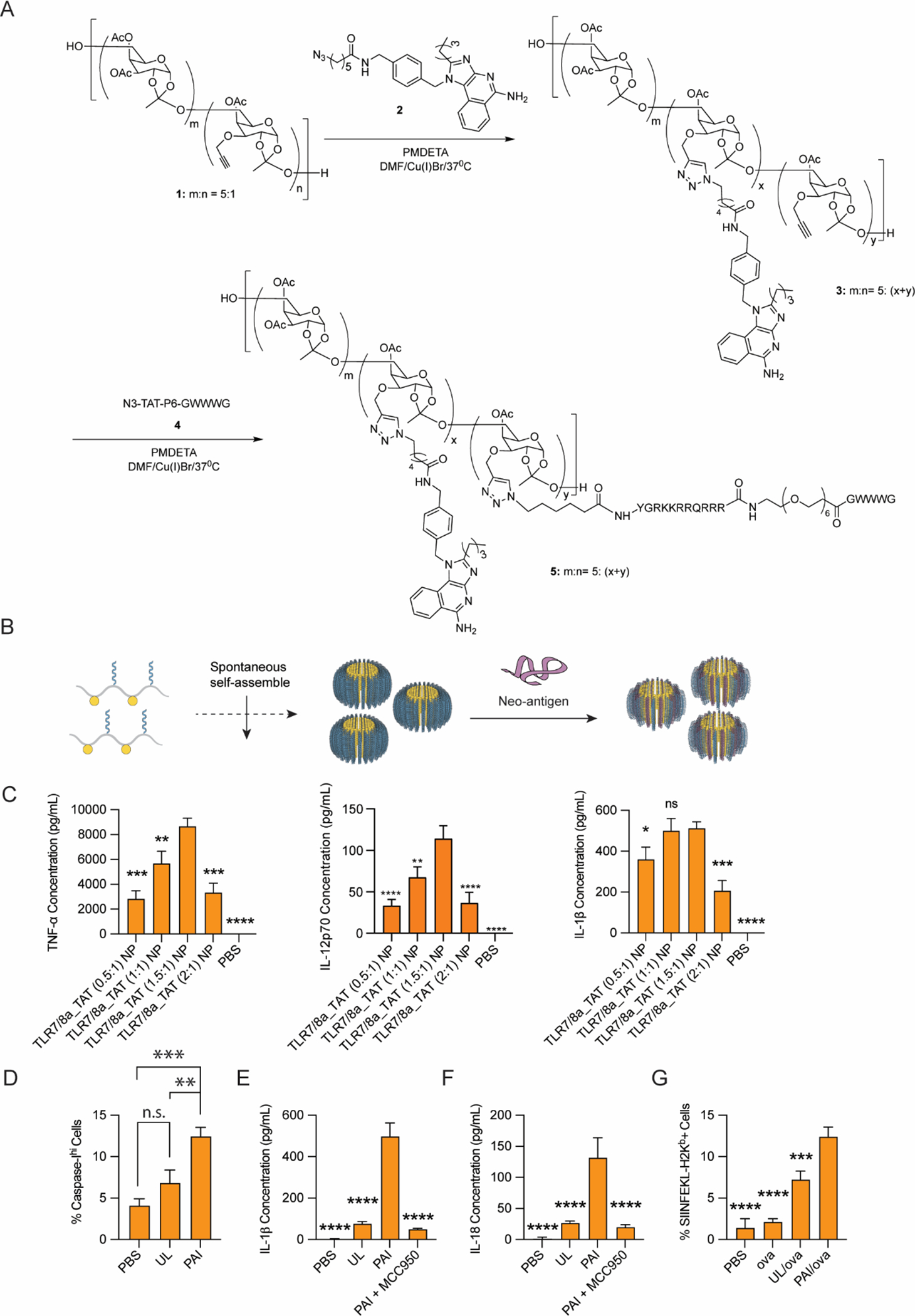
Synthesis and in vitro characterization of PAIs. (A) PAIs were synthesized via Cu(I)-mediated cycloaddition to affix a TLR7/8 agonist, 2BXy, and inflammasome activating peptide, TAT-P6-GWWWG, to the carbohydrate SPOE scaffold. (B) PAIs were assembled into nanostructures via solvent transfer from DMSO to PBS and, where appropriate, electrostatically complexed with antigen. (C) BMDCs were cultured with 25 μg/mL PAIs with various ratios of 2BXy:TAT-P6-GWWWG, and cytokine production was assayed in the supernatant after 18 h. TLR7/8a_TAT (1.5:1) NP was identified as an optimal formulation and used for all subsequent studies. (D) Caspase-1 activation was assayed in BMDCs by incubating with 25 μg/mL PAIs or unlinked controls (UL) for 18 h, staining with FAM-YVAD-FMK, and analyzing via flow cytometry. (E) IL-1β and (F) IL-18 secretion in supernatants following incubation with BMDCs for 18 h. Co-incubation of PAI with the NLRP3 inhibitor, MCC-950, results in loss of IL-1β and Il-18 production. (G) PAIs were formulated with OVA and incubated with BMDCs for 24 h, and antigen presentation was assayed via flow cytometry using the SIINFEKL:H-2Kb antibody. For all assays, n = 3, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Statistical analyses were performed using one-way ANOVA with Dunnett’s multiple comparisons test.
With these PAIs in hand, we next performed the synthesis of NPs through self-assembly (Figure 1B). The amphiphilic nature of the polymer-conjugates led to generation of well-defined nano-micelles upon solvent transfer from dimethyl sulfoxide (DMSO) to phosphate buffered saline (PBS). Depending on the ratio of 2BXy to TAT-P6-GWWWG, NPs of varying sizes were obtained as confirmed via DLS and TEM (Figure S9 and Table S2). Notably, the particles remained stable in PBS, and no significant structural or functional changes were observed when stored at 4 °C for a period of at least 8 weeks. We next investigated the ability of PAIs to elicit immune responses. Initial in vitro studies were performed to measure cytokine secretion. PAIs were incubated with bone marrow-derived dendritic cells (BMDCs), and cytokine secretion was analyzed in the supernatant (Figures 1C & Figure S10). In addition to analysis of IL-1β to measure inflammasome activation, we also investigated IL-12 and TNF-α as markers of immunotherapeutic potential. Our study indicated that a PAI with a ratio of 1.5:1 of 2Bxy and TAT-P6-GWWWG generated the highest amount of IL-1β secretion. Notably, this ratio also elicited the highest secretion of TNF-α and IL-12 – important cytokines in antigen presenting cell (APC) activation and cancer immunosurveillance. Therefore, this PAI (1.5:1) ratio was chosen for further biological studies.
PAI induces robust NLRP3 inflammasome activation.
With this encouraging initial data, we performed detailed evaluation of inflammasome activation using the selected PAI (1.5:1) (hereafter referred to as PAI) in comparison with equivalent amount of unlinked agonist mixtures (hereafter referred to as unlinked or UL) and PBS. We evaluated caspase-I enzyme activity along with secretion of IL-1β and IL-18 cytokines in BMDCs to assay inflammasome activation. Compared to the BMDCs treated with unlinked agonist, BMDCs treated with PAIs induced significantly higher caspase-I enzyme activity (Figure 1D) along with 10-fold higher IL-1β and 5-fold higher IL-18 secretion (Figure 1E–F). Additionally, to explore whether the responses are specific to the NLRP3 activation, BMDCs were co-incubated with NLRP3-specific inhibitor MCC-950 along with PAI. Co-incubation with MCC-950 resulted in a significant reduction of IL-1β and IL-18 secretion (Figure 1E–F), indicating the observed responses were specific to NLRP3 activation. Overall, these studies indicated robust NLRP3 inflammasome activation by PAI.
PAI enhances lysosomal escape and antigen cross presentation.
Results from the analysis of NLRP3 inflammasome activation demonstrated that our PAIs could induce robust IL-1β and IL-18 production. Due to the known lysosomotropic property of the TAT-P6-GWWWG peptides,28,34 we also evaluated the lysosomal rupture induced by the PAI. Lysosomal rupture and cytosolic delivery can enhance antigen presentation in APCs, leading to potent downstream adaptive immune responses.35 We thereby incubated THP-1 monocytes with PAI or an equivalent amount of unlinked agonist mixtures in addition to a fluorescent protein antigen (DQ Green BSA) to determine the efficacy of lysosomal proteolysis. Confocal microscopy analysis (Figure S11) of THP-1 cells indicated that PAI induced significant cytosolic diffusion of DQ-Green BSA, whereas DQ green BSA was mostly confined in distinct punctate lysosomes when co-incubated with PBS or unlinked agonist. These results thereby indicate that PAI formulations enhance lysosomal degradation relative to controls to allow cytosolic delivery of antigen.
With this promising observation, we then evaluated whether increased cytosolic delivery of protein antigens by PAI can enhance antigen cross-presentation. In this study, PAI formulation was incubated with BMDCs along with a model antigen, ovalbumin (OVA). After 24 h of incubation, cells were stained for MHC-I antigen presentation on the surface of DCs using an antibody against SIINFEKL:H-2Kb complexes (Figure 1G). Parallel studies were performed with unlinked agonist formulation in combination with OVA or with OVA in PBS. Flow cytometry analysis demonstrated that PAI-OVA treated cells exhibited a significantly higher level of surface SIINFEKL:H-2Kb staining compared to other treatment groups. This indicates that the PAI formulation can potentially induce higher downstream antigen-specific immune responses. These results thereby validated our hypothesis that co-activation of TLR and NLRP3 by PAI can induce potent immune activation and antigen processing by APCs.
PAI induces antigen localization and delivery.
With the promising in-vitro results, we evaluated the in-vivo biodistribution of PAI when formulated with antigens. For this study, PAI was admixed with Alexa Fluor 647 labelled OVA (AF647-OVA) to generate PAI-AF647-OVA formulations. DLS and TEM analysis indicated that the PAI particles were stable in formulation. Identical formulations were also developed with AF647-OVA in PBS or with equivalent quantities of unlinked activators. For bio-imaging studies, the formulations were injected subcutaneously in the flank of nude mice (n=6) and biodistribution of antigen-formulations was monitored using an in-vivo imaging system (IVIS) at regular intervals (Figure 2A–C). Nude mice are commonly employed in biodistribution studies to enhance the resolution of in-vivo imaging through the skin.36 It was observed that PAI-AF647-OVA formulation induced significant antigen localization at the injection site compared to the other groups. Notably, while mice treated with free AF647-OVA in PBS or unlinked PAI formulation had undetectable levels of antigen 48 h post-injection, PAI formulations demonstrated significant antigen localization even 72 h post-injection (Figure 2C). To further explore the effect of PAI formulations, organs were isolated from a cohort of the same animals 48 h post injection and analyzed for bio-distribution of AF647-OVA. Notably, the PAI group demonstrated significant localization of PAI-AF647-OVA formulations in the draining inguinal lymph node, while detectable levels of AF647-OVA were not observed in other organs (Figure 2F). Consistent with our previous observation, antigens were also not detectable in organs isolated from groups treated with the unlinked PAI formulation or soluble AF647-OVA 48 h post-injection. These studies indicate that the PAIs formulated with antigen induce improved localization at the site of injection and draining lymph nodes compared to unlinked agonist formulation.
Figure 2:

Biodistribution of PAIs in vivo. (A-C) AF647-OVA loaded PAIs or admixed controls were injected subcutaneously in the flank of mice and imaged using an in-vivo imaging system (IVIS) at 3 h, 24 h, 48 h and 72 h post injection. (D-F) Representative images of distribution of AF-647 in lungs and heart, inguinal lymph node, spleen, liver, and kidney 48 h post injection.
PAI enhances vaccine immunogenicity and anti-tumor efficacy.
With the promising in-vitro studies and biodistribution analysis, we were motivated to evaluate the efficacy of PAI in enhancing immunogenicity of anti-tumor vaccines. Thereby, we initially performed in-vivo studies to understand immunogenicity of PAI formulated with OVA in comparison with unadjuvanted OVA or OVA formulated with molar equivalent quantities of unlinked PAI components. Mice (n=5) were injected subcutaneously with each formulation followed by a subsequent, identical boost on day 14. On day 24, blood sera were collected to analyze for antibody titers, and splenocytes were harvested to analyze antigen-specific T-cell responses (Figure 3A). It was observed that PAI significantly enhanced antibody titers compared to the mixture of unlinked components (173 ± 22%) or unadjuvanted (9362 ± 312%) formulations (Figure 3B). Additionally, analysis of splenocytes restimulated with OVA revealed that PAI formulation enhanced IFN-γ secreting CD4+ T-cell response by 113 ± 41% and IFN-γ secreting CD8+ T-cell response by 61 ± 14% relative to the unlinked formulation (Figure 3C–D). These results thereby PAI significantly enhanced antigen-specific immune responses to vaccination relative to unadjuvanted antigen or unlinked agonist formulations.
Figure 3:
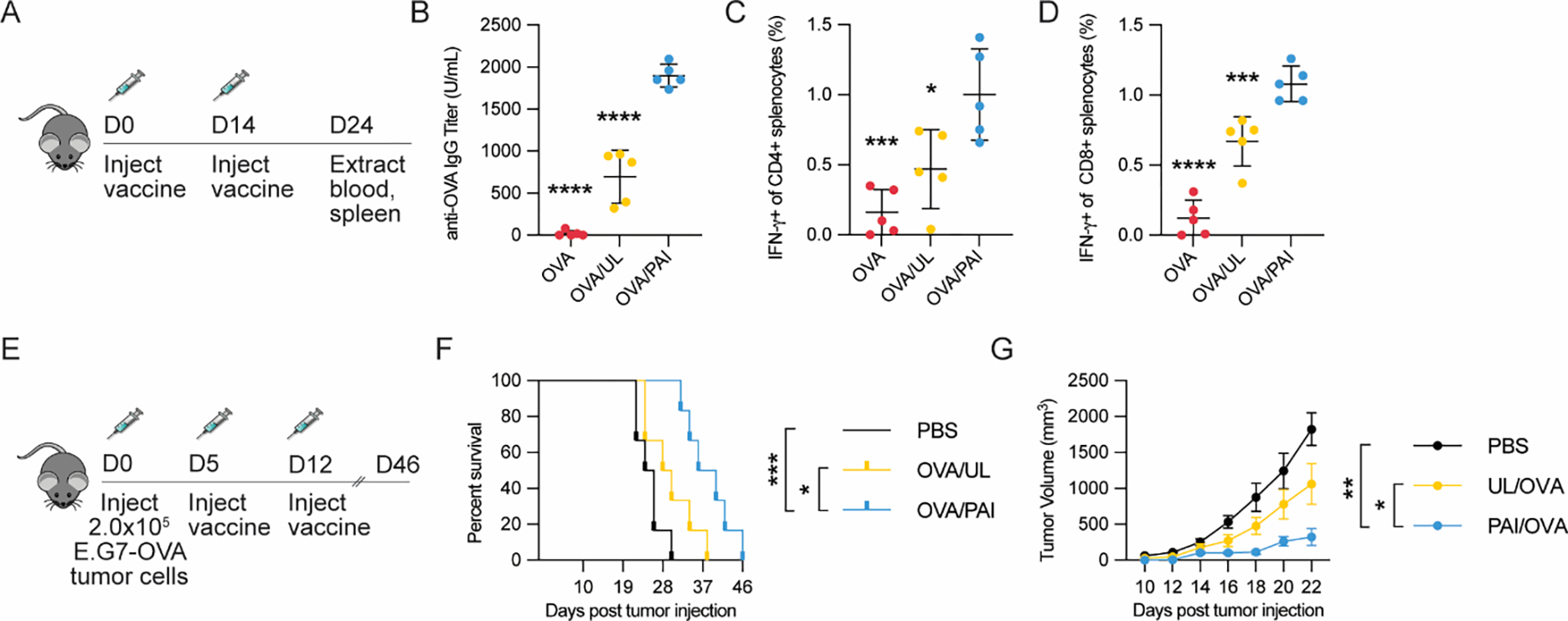
Vaccination studies of PAIs or controls formulated with OVA antigen (20 μg). (A) Study design for prophylactic vaccination model (n=5/group). (B) Serum anti-OVA IgG levels were analyzed via ELISA. (C-D) Splenocytes were analyzed for antigen-specific T-cells via intracellular cytokine staining and flow cytometry. (C) Percentage of IFN-γ secreting CD4+ splenocytes after restimulation with OVA323-339. (D) Percentage of IFN-γ secreting CD8+ splenocytes after restimulation with OVA257-264. (E) Study design for therapeutic vaccination model using a E.G7-OVA tumor (n=6/group). Mice were implanted with 2.0 × 105 E.G7-OVA cells in the flank and vaccinated with the indicated formulations 5 and 12 d after implantation. (F) Kaplan-Maier survival analysis of mice treated with various formulations. (G) Growth curves of tumors until the first mouse died (day 22). For all assays, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Statistical analyses were performed using one-way ANOVA with Dunnett’s multiple comparisons test in B-D and G and by using log-rank test with Bonferroni-correction in F.
With the promising analysis of immunogenicity PAI in the OVA vaccination studies, we next proceeded to investigate the application of PAI in enhancing antigen-specific immune responses in cancer vaccines. First, we performed studies in an E.G7-OVA lymphoma model (Figure 3E). In this model, EL4 cells that stably express OVA protein were implanted onto C57Bl6/J mice (n=6/group) in the right flank on day 0. Mice were then vaccinated subcutaneously on days 5 and 12 with OVA formulated with PAI or OVA with unlinked agonists. The control group received PBS. Mice were monitored for tumor growth and sacrificed when tumor size reached 20 mm in any linear dimension. We found that administration of PAI formulations significantly reduced tumor burden and prolonged survival compared to other formations (Figure 3F–G). Notably, animals in the PAI formulation-treated group had a median survival of 38 days – significantly higher compared to median survival of 29 days for unlinked agonist formulation and 25 days for PBS treated animals. The enhanced efficacy of PAI formulation is thereby consistent with our previous observation of enhanced antigen-specific cell-mediated immune responses of PAI formulations in OVA vaccination studies.
With these exciting findings, we evaluated the efficacy of PAI formulations to induce anti-tumor immunity against tumor-specific neoantigens in a B16.F10 melanoma model. This model has been widely investigated as a poorly immunogenic and highly aggressive murine tumor model.37 It is unresponsive to many immunotherapeutic modalities including clinically approved checkpoint blockade therapies.30,38 We wanted to test whether therapeutic cancer vaccines targeting the NLRP3 inflammasome could assist in reducing tumor burden in combination with immune checkpoint blockade therapy (ICB: treatment with anti-CTLA4 and anti-PD-L1 antibodies). For this study, PAI was formulated with multiple B16.F10 neoantigen peptides (Table S3 and Figure S12). These peptides were modified with glutamate linkers to promote adsorption with PAI in formulation. To differentiate between the effects of adjuvanticity and carrier properties of the PAI scaffold, an analogous nano-formulation (PT) was developed by grafting equivalent quantities of TAT-P6-GWWWG to the SPOE scaffold in the absence of 2BXy. For comparison, studies were also performed with ICB antibodies and with unlinked agonist formulation in combination with ICB. Finally, parallel studies were performed with NLRP3-deficient mice to investigate the role of NLRP3 inflammasome activation in the efficacy of PAI formulations. Mice (n = 7) were injected with tumor cells subcutaneously in the right flank, and vaccine formulations were injected peritumorally on days 9 and 15 (Figure 4A). ICB antibodies were administered intraperitoneally at the same time as vaccine formulations. The study was concluded at day 42 when all mice were either tumor free or sacrificed due to tumor burden (20 mm in any dimension).
Figure 4:

PAI vaccine efficacy in a B16.F10 tumor model. (A) Study design for tumor challenge model (n=7/group). Mice were implanted with 1.0 × 105 B16.F10 cells subcutaneously. They were then treated 9 and 15 d after tumor implantation with the indicated vaccine formulations peritumorally along with checkpoint blockade antibody cocktail (anti-CTLA-4 + anti-PDL-1) intraperitoneally. Tumor growth and survival were monitored in one experimental group, while in a parallel group, mice were sacrificed on day 22 to evaluate antigen-specific cellular response. (B) Growth curves of tumors until the first mouse died (day 20). (C) Kaplan-Maier survival analysis of mice treated with various formulations. (D-F) Splenocytes were stimulated ex-vivo with neoantigen peptide cocktail for 48 h, and supernatants were analyzed for (D) Granzyme B, (E) IFN-γ, and (F) IL-10 using cytometric bead array. (G) Ratio of IFN-γ to IL-10 in supernatants. For all assays, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Statistical analyses were performed using one-way ANOVA with Dunnett’s multiple comparisons test in B and D-G and by using log-rank test with Bonferroni-correction in C.
It was observed that the PAI vaccines in combination with ICB significantly reduced tumor burden and prolonged survival (Figure 4B–C). Of the mice treated with PAI + ICB, 2/7 mice achieved complete remission. Notably, treatment with the PAI vaccine improved median survival to 36 days compared to 24 days for PBS treated animals and 26 days for animals treated with ICB only. In comparison, neither the PAI-ICB combination therapy in NLRP3-deficient mice, nor the PT-ICB combination therapy improved survival compared to treatment with ICB alone. Moreover, with this treatment regime, unlinked activator formulations induced severe toxicity resulting in weight loss (>20%) and death in 7/7 mice, and hence could not be included in further studies.
To better understand the role of each formulation in the generation of antigen-specific anti-tumor responses, we performed a parallel study where splenocytes were isolated from treated animals on day 22. Splenocytes were incubated with the neoantigen cocktail for 48 h, and cytokine secretion was measured in the supernatant to quantify the antigen-specific immune response (Figure 4D–G). Remarkably, compared with other treatment groups, only the PAI formulations induced significant levels of the key apoptosis-inducing protease, Granzyme-B (GNZB) (Figure 4D), and immunostimulatory cytokine, IFN-γ (Figure 4E). In comparison, splenocytes from animals receiving ICB therapy alone did not include any detectable levels of IFN-γ or GNZB while PT treated animals were observed to induce detectable levels of IFN-γ or GNZB in only 50% of samples. Moreover, compared to ICB therapy, the PAI treated mice did not induce significantly higher levels of the immunosuppressive regulatory cytokine, IL-10 (Figure 4F), or other cytokines tested (Figure S13). We further evaluated the IFN-γ/IL-10 ratio in the supernatants – an indicator of therapeutic efficacy.39 Notably, PAI vaccines in wild-type mice demonstrated twenty-fold higher IFN-γ/IL-10 ratio compared to PAI vaccines in NLRP3-deficient, mice indicating stronger antigen-specific anti-tumor responses in presence of NLRP3 inflammasome activation (Figure 4G). Finally, restimulated splenocytes were fixed, permeabilized, and stained for intracellular IFN-γ to directly probe CD4+ and CD8+ T cell responses (Figure S14). It was observed that NLRP3-deficient mice had reduced IFN-γ+ CD8+ T cell responses, providing further evidence that the NLRP3 inflammasome drives drive the anti-tumor immune phenotype. These results suggest highly potent antigen-specific anti-tumor responses when the NLRP3-activating PAI formulation is employed as an adjuvant for therapeutic cancer neoantigen vaccines.
With our observation of significantly higher antigen-specific IFN-γ secretion by the PAI vaccine group compared to the ICB treatment group, we further analyzed for tumor infiltrating leukocytes (TILs) in isolated tumors in these groups on day 22. Immunohistochemical staining indicated the presence of significantly higher levels of CD4+ and CD8+ TILs in the PAI vaccine treated group compared to PBS or ICB treated animals (Figure S15). We also did not observe any significant increases in FoxP3+ regulatory T cells staining in all the groups, which indicates that PAI vaccines induced an immunostimulatory tumor microenvironment rather than an immunosuppressive one. Overall, these data indicate that dual TLR and NLRP3 inflammasome activation by PAI formulation can enhance antitumor efficacy and vaccine-induced protection.
With these promising findings, we were interested in performing additional studies to analyze efficacy of PAI vaccine formulations against larger and more established tumors. Hence, we performed studies in an aggressive CT-26 colon carcinoma model (Figure 5A). Here, PAI was similarly formulated with glutamate linker-modified CT-26 neoantigen peptides to promote adsorption (Table S4 and Figure S16). Vaccines were administered in mice (n = 10) peritumorally on day 13 when tumor volumes reached 150–200 mm3 followed by two additional boost injections at 18 and 23 d. As with previous experiments, ICB antibodies were administered intraperitoneally alongside vaccines, and PBS, unadjuvanted antigens, or ICB without PAI were used as controls. Animals were monitored through day 60 when all the treated animals were either observed to be tumor free or had already been sacrificed due to large tumor burden (20 mm in any linear dimension). It was observed that PAI vaccines synergized with ICB treatment leading to complete remission of tumors in 70% of treated animals on day 60 (Figure 5B–D). In contrast, ICB treatment alone or ICB+antigen only led to complete regression of tumors in just 20% of treated animals, while PBS- and antigen-treated mice had a median survival of 33 and 32 d, respectively (Figure 5B–D & Figure S17). On day 60, all surviving mice in the PAI vaccination group were rechallenged with 1.0×105 CT-26 cells and monitored for tumor recurrence until day 90. All mice rejected new tumor growth indicating development of immunological memory in the animals. These results further validated our previous observations on enhanced anti-tumor functionality of PAI-vaccine formulations and demonstrate their efficacy in a breadth of aggressive tumor models.
Figure 5:

PAI vaccine efficacy in a CT-26 tumor model. (A) Study design for tumor challenge model (n=10/group). Mice were implanted with 1.0 × 105 CT-26 cells subcutaneously. They were then treated 13, 18, and 23 d after tumor implantation with the indicated vaccine formulations peritumorally along with checkpoint blockade antibody cocktail (anti-CTLA-4 + anti-PDL-1) intraperitoneally. (B) Kaplan-Maier survival analysis of mice treated with various formulations. (C) Images of representative animals from PBS and PAI/ICB groups on day 33. (D) Tumor growth curves in animals treated with each of the formulations. (E) In a parallel study, mice (n=5/group) were implanted with 2.0 × 105 CT-26 cells on day 0 and treated at 13 d with the indicated vaccine and ICB treatments as in 5A. (F) Blood was collected 2 h post injection for analysis of TNF-α and IL-6. (G-I) Blood cell counts 2 d post-injection: (G) white blood cells (WBCs), (H) Lymphocytes, and (I) Thrombocytes For all experiments, *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001. Statistical analyses were performed using one-way ANOVA with Dunnett’s multiple comparisons test in F-I and by using log-rank test with Bonferroni-correction in B.
PAI reduces systemic off-target toxicity.
An important attribute of immunomodulatory therapeutics that prevents further clinical translation is unacceptable levels of off-target toxicity.40,41 With the strong in-vivo antitumor efficacy of PAI vaccines, we next evaluated off-target toxicity of PAI in the CT-26 model (Figure 5E). We evaluated systemic cytokines in the blood often resulting from diffusion of immune stimulants from the site of injection.28,42 We also evaluated correlated measures of toxicity including reduction of cellular counts including total white blood cells (WBCs), lymphocytes, and thrombocytes in blood that provide a reflection of toxicity due to diffusion of immune activators.29,30 For this study PBS, unlinked agonist formulations, PAI formulations with and without ICB, and ICB were injected 13 d post tumor injection. Analysis of serum cytokines (TNF-α and IL-6) 2 h post injection revealed that PAI resulted in no significant systemic cytokine secretion (Figure 5F) consistent with our previous observation on enhanced localization of PAI vaccines at the injection site (Figure 2). Notably, when PAI vaccines were combined with ICB therapy, no significant enhancement in levels of systemic cytokines were observed compared to ICB injection alone. In comparison, animals treated with the unlinked PAI formulation had significantly enhanced systemic cytokine secretion (Figure 5F). Consistent with our observation of systemic cytokine secretion, we observed that the PAI formulation induced significantly lower hematological cellular toxicity compared to unlinked formulations at 48 h post injection (Figures 5G–I). Additionally, when combined with ICB, the PAI formulation did not significantly enhance hematological toxicity compared to ICB treatment alone. These results suggest that the PAIs mitigate systemic toxicity compared to treatment with unlinked immune activators, possibly by localizing the immune response to the tumor site.
Conclusion
In conclusion, we designed and synthesized a novel inflammasome activating nanovaccine platform (PAI) for enhancing efficacy of neoantigen vaccines. The design incorporated a small molecule TLR7/8 activator, 2BXy, along with an inflammasome activating TAT-P6-GWWWG peptide on a carbohydrate scaffold. Upon solvent transfer, the scaffold self-assembles into stable ~50 nm PAI nanoparticles. The PAIs, when formulated with antigen, demonstrated robust NLRP3-inflammasome activation along with antigen processing in dendritic cells. They also exhibited superior localization at the injection site and draining lymph nodes, resulting in enhanced antigen specific CD8+ T cell responses with reduced systemic cytokine production. A summary of the proposed mechanism of action is provided in Figure 6. Our in-vivo vaccination studies in comparison with unlinked activators and checkpoint blockade therapy in multiple tumor models indicated that PAIs enhanced efficacy of neoantigen vaccines. PAIs also reduced off-target toxicity compared to unlinked combination of activators. This study represents a new material platform to induce robust protective immune response and, more broadly, demonstrates that localized NLRP3 inflammasome activation can be employed to enhance the efficacy of anti-cancer vaccines.
Figure 6:

Overview of the proposed mechanism of action of PAI nanovaccines based on mechanistic analyses herein. PAI nanovaccines enhance endocytosis by antigen-presenting cells on account of their ~50 nm structure. Their secondary structure localizes effects to the injection site. Upon endocytosis, they then activate endosomal TLR7 and induce lysosomal rupture to result in NLRP3 inflammasome activation and cytosolic delivery of neoantigen to afford enhanced antigen presentation on MHC-I. When nanovaccines are injected peritumorally, they induce tumor localized, neoantigen-specific CD8+ T cell responses characterized by secretion of IFN-γ and GNZB to afford tumor clearance. Created using Biorender.com.
Methods
Mice and Materials.
All reactions were conducted under dried nitrogen or argon stream. Anhydrous solvents were purchased in capped DriSolv™ bottles, used without further purification, and stored under argon. Azidohexanoic acid was purchased from Click Chemistry Tools, and PEG linkers were purchased from Quanta Biodesign. All other solvents and reagents were obtained from Sigma Aldrich or Thermo Fisher and used without further purification unless otherwise noted. All glassware was flame-dried before use. Silica gel column chromatography was performed using flash silica gel (32–63 μm). All cell culture reagents unless otherwise noted were purchased from Thermo Fisher. THP-1, B16.F10, and E.G7-OVA cells were purchased from ATCC. CT-26 cells were obtained as a gift from Prof. Jeffrey Hubbell’s laboratory. Antibodies used for flow cytometry, immunohistochemistry, and checkpoint blockade are listed with clone and vendor in the Supplementary Information. Female C57Bl/6J, Nu/J, B6.129S6-Nlrp3tm1Bhk/J, and Balb/C mice (5-week-old) were purchased from Jackson Laboratory (JAX). Mice were housed in an AAALAC accredited animal facility. All animal procedures were performed under a protocol approved by the University of Chicago Institutional Animal Care and Use Committee (IACUC). All compounds used in-vivo were tested for endotoxin prior to use. The animals were allowed to acclimatize for at least 7 days prior to experiment onset. All in-vivo experiments were conducted at least two times, and mice were randomly assigned to groups to minimize cage effects. All data unless otherwise noted are analyzed and plotted in GraphPad Prism 9.
Synthesis of Polymeric Activators of Inflammasomes (PAIs).
Synthesis of the alkyne-modified sugar poly(orthoester) (SPOE) backbone and azide modified adjuvants were synthesized as described in the Supplementary Information and characterized by proton nuclear magnetic resonance (1H-NMR) and/or mass spectrometry (MS). The synthesis of PAI was performed by sequential Cu(I) catalyzed Huisgen cycloaddition reaction with the SPOE scaffold. In a representative example, alkyne-containing SPOE (0.037 g, Mn GPC = 7.2 kDa, 0.021 mmol alkynes) and 2BXy-N3 (0.006 g, 0.013 mmol) were added to a 10 mL, flame-dried Schlenk flask in anhydrous THF (3.0 mL). After three cycles of freeze-pump-thaw, Cu(I)Br (1.0 mg, 0.0063 mmol) and N,N,N’,N’,N”-pentamethyldiethylenetriamine (4.73 mg, 0.0273 mmol) were added. The reaction was stirred at 37 °C for 12 h. The reaction mixture was purified by passing through neutral alumina and then further precipitated in diethyl ether (3×10 mL) to afford the product (2Bxy-SPOE) as a very light brown powder (0.035 g, 80%). 2Bxy-SPOE was then reacted with azido-TAT-P6-GWWWG (0.023 mg, 0.008 mmol in anhydrous DMF (3.0 mL). After three cycles of freeze-pump-thaw, CuBr (1.0 mg, 0.0063 mmol) and N,N,N’,N’,N”-pentamethyldiethylenetriamine (4.73 mg, 0.0273 mmol) were added. The reaction was stirred at 37 °C for 12 h following which the reaction mixture was dialyzed in EDTA solution followed by dialysis in deionized water. The solution was lyophilized to obtain a brown powder. PAIs with varied ratios of 2BXy:TAT-P6-GWWWG were prepared analogously by varying the molar ratios of these components.
Nanoassembly of PAIs.
PAIs were dissolved in DMSO (1.5 mL) and stirred at room temperature overnight. The solution volume was then subjected to dialysis against endotoxin-free PBS for 24 h to afford NP solution. Following this the particles were stored at 4 °C. The stability of the nanoparticles was monitored using TEM over a period of eight weeks at 4 °C.
Characterization of PAIs.
Prior to nanoassembly, PAIs were characterized via gel permeation chromatography (GPC) and high-performance liquid chromatography (HPLC). Following nanoassembly, PAIs were further characterized via transmission electron microscopy (TEM) and dynamic light scattering (DLS) as described in the Supplementary Information. When loaded with neoantigen peptides, encapsulation efficiency of PAIs was again analyzed via HPLC.
Evaluation of cytokines using BMDCs.
Bone marrow-derived dendritic cells (BMDCs) were harvested from 6-week-old C57BL/6J mice (Jackson Laboratory) following previous literature protocol.29 On day 6, BMDCs were released and plated on 96-well plates at a density of 1.1×106 cells/mL (180 μL) in RPMI + 10% heat-inactivated fetal bovine serum (HI-FBS) and incubated with PAI (25 μg/mL) or equivalent quantities of unlinked activators for 18 h following which the plates were centrifuged at 400×g and the supernatants were collected. For studies with MCC-950, cells were pre-incubated with MCC-950 (1 mM) 30 mins prior to addition of PAI. IL-1β and IL-18 cytokines were measured in undiluted serum by ELISA (BioLegend ELISA MAX Deluxe kit) according to the manufacturer’s procedure and read on a Multiskan FC plate reader (Thermo Scientific) at 450 nm. All other cytokines were analyzed in 2.5x diluted serum via Mouse Inflammation CBA Kit (BD Bioscience) using a NovoCyte 3000 flow cytometer.
Evaluation of cell activation using BMDCs.
BMDCs were released and plated on 96-well plates at a density of 1.1×106 cells/mL (180 μL) and incubated with PAI (25 μg/mL) or equivalent quantities of unlinked agonists. For Caspase-1 activation, cells were incubated with PAIs for 18 h, and then washed and incubated with caspase-I substrate FAM-YVAD-FMK (FAM-Flica Caspase-I assay kit, Immunochemistry Technologies) according to the manufacturer’s protocol to identify the percentage of caspase-I positive cells. For MHC-I antigen presentation, cells were incubated with PAIs for 24 h, then washed and incubated with anti-CD16/32 antibody to block Fc receptors. The cells were subsequently stained with Zombie NIR fixable viability dye (BioLegend) and PE anti-mouse SIINFEKL:H-2Kb antibody. The percentage of live, SIINFEKL:H-2Kb positive cells were then analyzed. Flow cytometry was conducted using a NovoCyte 3000 flow cytometer, and data were analyzed using the NovoExpress software.
Evaluation of antigen processing using THP-1 cells.
THP-1 cells were plated at a density of 2×106 cells/mL (180 μL) in RPMI + 10% HI-FBS and incubated with 25 mg/mL PAI or equivalent amounts of unlinked agonists. The control group received PBS. All cells were simultaneously co-incubated with 10 mg/mL DQ Green BSA (Thermo Fisher) and various agonists for 24 h. Cells were then washed and stained with Hoechst 33342 and Lysoview 633. Fluorescent images were obtained on a Leica SP5 laser confocal microscope. Each microscopy experiment was performed twice independently. At least 5 different regions were analyzed for each sample. Images were processed with ImageJ software.
Biodistribution analysis in nude mice.
Athymic Nu/J (nude) mice (n=6) were injected subcutaneously with AF647-labelled OVA (20 μg) adjuvanted with PAI (50 μg) or with equivalent quantities of unlinked activators. AF647-labelled OVA (20 μg) was used as a control. Mice were injected subcutaneously with various formulations and fluorescence imaging was performed on an IVIS Spectrum in-vivo imaging system (PerkinElmer). 48 h post-administration of formulations, a cohort of mice from each group were euthanized, and their organs were also imaged with IVIS.
Prophylactic vaccination with OVA.
Mice (n=5) were vaccinated subcutaneously with OVA (20 μg) adjuvanted with PAI (50 μg) or with equivalent quantities of unlinked 2BXy and TAT-P6-GWWWG. The control group received unadjuvanted OVA in PBS. Mice were given an identical vaccine boost on day 14. On day 24, sera and spleens were collected from mice. Antibody titer was measured by anti-OVA IgG ELISA kit (ADI) and splenic T cell response was measured by intracellular cytokine staining (ICS). For ICS, spleens were processed into a single-cell suspension via mechanical disruption and passed through a 70 μm strainer. The splenocytes were washed with PBS and then treated with ACK lysing buffer (Gibco) for 3 min at room temperature. The single-cell suspension was washed with PBS and resuspended in RPMI. These single cell suspensions were then plated at a density of 5×106 cells/mL in 200 μL and treated with respective peptide epitopes (20 μg/mL). SIINFEKL (Invivogen) was employed as an OVA-specific CD8+ T cell epitope, and ISQAVHAAHAEINEAGR (Invivogen) was employed as an OVA-specific CD4+ T cell epitope. Following 2 h of incubation, 0.2 μL GolgiPlug (BD) was added, and the cells were stimulated for 4 h longer. Following incubation, cells were washed, stained with viability stain, stained for cell surface markers, fixed and permeabilized with Cytofix/Cytoperm kit (BD), and stained for intracellular cytokines. Samples were then analyzed on a NovoCyte 3000 flow-cytometer and analyzed using the NovoExpress software.
Immunotherapy studies with EG7.OVA lymphoma.
1 × 105 E.G7-OVA cells were injected subcutaneously into the flank of 6-week-old C57Bl/6 mice (n= 6 per group) in 100 μL of PBS. Following this OVA (20 μg) adjuvanted with PAI (50 μg) or with equivalent quantities of unlinked 2BXy and TAT-P6-GWWWG were injected subcutaneously on days 5 and 12 post tumor inoculation. The control group received PBS. The tumor size was monitored every alternate day by a singly blinded observer. Tumor volumes were measured using the equation V=1/2xLxWxW. Mice were euthanized when the tumors reached 20 mm in any linear dimension.
Immunotherapy studies with B16.F10 melanoma.
1 × 105 B16.F10 cells in 100 μL of PBS were injected subcutaneously into the flank of 6-week-old C57Bl/6J (wild type) or B6.129S6-Nlrp3tm1Bhk/J (NLRP3 knockout) mice (n= 7 per group). The tumor size was monitored on alternating days by a singly blinded observer. Tumor volumes were measured using the equation V=1/2×L×W×W. When the tumors reached a size of approx. 100 mm3 (day 9), treatment was started. Various NP (75 μg) were formulated with neoantigens (B16-M30, B16-M48, B16-M27; 20 μg each) and injected peritumorally. Neoantigens were prepared by solid-phase peptide synthesis as described in the Supplementary Information. Simultaneously, treatment groups received intraperitoneal injections of checkpoint blockade antibodies (anti-CTLA4 + anti-PD-L1, 75 μg each). Treatment was repeated on day 15. The control groups received PBS or checkpoint blockade antibodies only. Mice were euthanized when the tumors reached 20 mm in any linear dimension. A parallel study was performed with B16.F10 tumors as described to evaluate antigen-specific T cell production. This study was conducted analogously, and mice were euthanized on day 22. Tumors and spleens were collected. Tumors were smashed, strained through a 70 μm cell strainer, and treated with ACK lysing buffer (Gibco). 2 ×106 cells from each spleen were plated at a density of 2.5 M cells/mL. Cells were incubated with a cocktail of the three neoantigen peptides (5 μg/mL of each peptide) for 48 h. Cells were then centrifuged at 400 × G for 5 mins, and the supernatants were collected and analyzed using Mouse TH1/Th2/Th17 cytokine CBA kit (BD Biosciences) following manufacturer’s protocol. Granzyme B levels in the supernatant were measured by Granzyme B Mouse ELISA Kit (Thermo Fisher). Finally, tumors were extracted from mice in various treatment groups on day 22. The tissues were sectioned and fixed with 10% neutral buffered formalin solution. Samples were then washed and stored in ethanol following which they were embedded in paraffin and sectioned. Samples were then stained for CD4, CD8 and Foxp3 and imaged using an Axio Observer 7 microscope (Carl Zeiss AG) with an Axiocam 506 color camera (Carl Zeiss AG).
Immunotherapy studies with CT-26 carcinoma.
1 × 105 CT-26 cells in 100 μL of PBS were injected subcutaneously into the flank of 6-week-old BALB/c mice (n= 10 per group). The tumor size was monitored on alternating days by a singly blinded observer. Tumor volumes were measured using the equation V =1/2×L×W×W. When the tumors reached a size of approx. 150–200 mm3 (day 13), treatment was started. PAI (100 μg) was formulated with neoantigens (CT-26-M03, CT-26-M20, CT-26-M90, CT-26-GP70; 20 μg each) and injected peritumorally. Neoantigens were prepared by solid-phase peptide synthesis as described in the Supplementary Information. Simultaneously, treatment groups received intraperitoneal injections of checkpoint blockade antibodies (anti-CTLA4 + anti-PD-L1, 75 μg each). The control groups received either PBS or antigen cocktail or checkpoint blockade antibodies only. Treatment was repeated on day 18 and day 23. Mice were euthanized when the tumors reached 20 mm in any linear dimensions. The tumors were monitored until day 60, when all surviving animals were tumor free. Remaining surviving mice in PAI groups were reinjected with 1 × 105 CT-26 cells and monitored for tumor growth until day 90.
Toxicity studies with CT-26 carcinoma.
Mice were injected with CT-26 cells and vaccinated on day 13 as described above. Plasma was collected by submandibular bleed 2 h post injection of formulations on day 13 for cytokine analysis. 2 d post injection, blood was collected from animals by submandibular bleed in EDTA coated Eppendorf tubes (Fisher Scientific). Samples were immediately analyzed for complete blood count (CBC) using a Hemavet 950 instrument (Drew Scientific). The instrument was fitted with a reagent pack obtained from the manufacturer. Prior to analysis a blank run and a quality control run (using manufacturer provided control sample) were performed to ensure optimal performance by the instrument. 20 μL of blood were injected for each analysis using a sample cycle of approximately 2 min.
Supplementary Material
Acknowledgements
Sai.M., J.S., A.M.W., E.J.M., and A.P.E.-K. acknowledge the support of NIH (U01AI124286) and DTRA (1-18-1-0052). Sam.M and W.D. acknowledge the support of NSF (CHE-1413033). A.M.W. acknowledges partial support of NIH T32 GM008720. All authors acknowledge University of Chicago’s Soft Matter Characterization, NMR, Integrated Light Microscopy, and Human Tissue Resource facilities. The authors thank Yimei Chen for assistance with TEM imaging, Dr. Vytas Bindokas for assistance with confocal microscopy, Anthony Fernald for assistance with Hemavet, Tyler Lieberthal for graphical design services, and the veterinary technicians at University of Chicago for exceptional animal care.
Abbreviations
- NLRP3
NACHT, LRR and PYD domain-containing protein 3
- TLR
Toll-Like Receptor
- MPLA
monophosphoryl lipid A
- SPOE
sugar poly(orthoester)
- PAI
polymeric activator of the inflammasome
- HPLC
high performance liquid chromatography
- DMSO
dimethyl sulfoxide
- PBS
phosphate buffered saline
- DLS
dynamic light scattering
- TEM
transmission electron microscopy
- BMDC
bone marrow derived dendritic cell
- APC
antigen presenting cell
- BSA
bovine serum albumin
- OVA
ovalbumin
- MHC
major histocompatibility complex
- IVIS
in-vivo imaging system
- ELISA
enzyme-linked immunosorbent assay
- ICB
immune checkpoint blockade
- PT
polymeric TAT peptide
- TIL
tumor infiltrating lymphocyte
- WBC
white blood cell
- PEG
poly(ethylene glycol)
- NMR
nuclear magnetic resonance
- MS
mass spectrometry
- ICS
intracellular cytokine staining
- IFN-γ
interferon gamma
- GNZB
granzyme B
Footnotes
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Additional Information
Description of Supplementary Information. Synthesis and characterization of sugar poly(orthoesters), peptides, and small molecule adjuvants. Formulation of PAIs with neoantigen peptides and encapsulation efficacy. Confocal microscopy images of cytosolic delivery, immunohistochemical analysis of tumor tissues. Additional cytokine data from in-vitro assays and restimulation studies.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Data Availability Statement
All data are available in the main text or the supplementary materials.
References
- 1.Schumacher TN; Schreiber RD, Neoantigens in cancer immunotherapy. Science 2015, 348 (6230), 69–74. [DOI] [PubMed] [Google Scholar]
- 2.Ott PA; Hu Z; Keskin DB; Shukla SA; Sun J; Bozym DJ; Zhang W; Luoma A; Giobbie-Hurder A; Peter L, An immunogenic personal neoantigen vaccine for patients with melanoma. Nature 2017, 547 (7662), 217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.van der Burg SH; Arens R; Ossendorp F; van Hall T; Melief CJ, Vaccines for established cancer: overcoming the challenges posed by immune evasion. Nature Reviews Cancer 2016, 16 (4), 219–233. [DOI] [PubMed] [Google Scholar]
- 4.Kuai R; Ochyl LJ; Bahjat KS; Schwendeman A; Moon JJ, Designer vaccine nanodiscs for personalized cancer immunotherapy. Nat Mater 2017, 16 (4), 489–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lynn GM; Sedlik C; Baharom F; Zhu Y; Ramirez-Valdez RA; Coble VL; Tobin K; Nichols SR; Itzkowitz Y; Zaidi N, Peptide–TLR-7/8a conjugate vaccines chemically programmed for nanoparticle self-assembly enhance CD8 T-cell immunity to tumor antigens. Nat Biotechnol 2020, 38 (3), 320–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li AW; Sobral MC; Badrinath S; Choi Y; Graveline A; Stafford AG; Weaver JC; Dellacherie MO; Shih T-Y; Ali OA, A facile approach to enhance antigen response for personalized cancer vaccination. Nat Mater 2018, 17 (6), 528–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gong N; Zhang Y; Teng X; Wang Y; Huo S; Qing G; Ni Q; Li X; Wang J; Ye X, Proton-driven transformable nanovaccine for cancer immunotherapy. Nat Nanotechnol 2020, 15 (12), 1053–1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ni Q; Zhang F; Liu Y; Wang Z; Yu G; Liang B; Niu G; Su T; Zhu G; Lu G, A bi-adjuvant nanovaccine that potentiates immunogenicity of neoantigen for combination immunotherapy of colorectal cancer. Sci Adv 2020, 6 (12), eaaw6071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bowen WS; Svrivastava AK; Batra L; Barsoumian H; Shirwan H, Current challenges for cancer vaccine adjuvant development. Expert Rev Vaccines 2018, 17 (3), 207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Shae D; Baljon JJ; Wehbe M; Becker KW; Sheehy TL; Wilson JT, At the bench: Engineering the next generation of cancer vaccines. J Leukoc Biol 2020, 108 (4), 1435–1453. [DOI] [PubMed] [Google Scholar]
- 11.Paston SJ; Brentville VA; Symonds P; Durrant LG, Cancer vaccines, adjuvants, and delivery systems. Front Immunol 2021, 12, 627932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Deets KA; Vance RE, Inflammasomes and adaptive immune responses. Nat Immunol 2021, 22 (4), 412–422. [DOI] [PubMed] [Google Scholar]
- 13.Hatscher L; Lehmann CH; Purbojo A; Onderka C; Liang C; Hartmann A; Cesnjevar R; Bruns H; Gross O; Nimmerjahn F, Select hyperactivating NLRP3 ligands enhance the TH1-and TH17-inducing potential of human type 2 conventional dendritic cells. Sci Signal 2021, 14 (680), eabe1757. [DOI] [PubMed] [Google Scholar]
- 14.Swanson KV; Deng M; Ting JP-Y, The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol 2019, 19 (8), 477–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sharp FA; Ruane D; Claass B; Creagh E; Harris J; Malyala P; Singh M; O’Hagan DT; Pétrilli V; Tschopp J, Uptake of particulate vaccine adjuvants by dendritic cells activates the NALP3 inflammasome. Proc Nat Acad Sci 2009, 106 (3), 870–875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Seydoux E; Liang H; Cauwelaert ND; Archer M; Rintala ND; Kramer R; Carter D; Fox CB; Orr MT, Effective combination adjuvants engage both TLR and inflammasome pathways to promote potent adaptive immune responses. J Immunol 2018, 201 (1), 98–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tahtinen S; Tong A-J; Himmels P; Oh J; Paler-Martinez A; Kim L; Wichner S; Oei Y; McCarron MJ; Freund EC, IL-1 and IL-1ra are key regulators of the inflammatory response to RNA vaccines. Nat Immunol 2022, 23 (4), 532–542. [DOI] [PubMed] [Google Scholar]
- 18.Bae JY; Lee S-W; Shin Y-H; Lee J-H; Jahng JW; Park K, P2X7 receptor and NLRP3 inflammasome activation in head and neck cancer. Oncotarget 2017, 8 (30), 48972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wang J; Chen H-J; Hang T; Yu Y; Liu G; He G; Xiao S; Yang B.-r.; Yang C; Liu F, Physical activation of innate immunity by spiky particles. Nat Nanotechnol 2018, 13 (11), 1078–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Han C; Godfrey V; Liu Z; Han Y; Liu L; Peng H; Weichselbaum RR; Zaki H; Fu Y-X, The AIM2 and NLRP3 inflammasomes trigger IL-1–mediated antitumor effects during radiation. Sci Immunol 2021, 6 (59), eabc6998. [DOI] [PubMed] [Google Scholar]
- 21.Dupaul-Chicoine J; Arabzadeh A; Dagenais M; Douglas T; Champagne C; Morizot A; Rodrigue-Gervais IG; Breton V; Colpitts SL; Beauchemin N, The Nlrp3 inflammasome suppresses colorectal cancer metastatic growth in the liver by promoting natural killer cell tumoricidal activity. Immunity 2015, 43 (4), 751–763. [DOI] [PubMed] [Google Scholar]
- 22.Robertson MJ; Mier JW; Logan T; Atkins M; Koon H; Koch KM; Kathman S; Pandite LN; Oei C; Kirby LC, Clinical and biological effects of recombinant human interleukin-18 administered by intravenous infusion to patients with advanced cancer. Clin Cancer Res 2006, 12 (14), 4265–4273. [DOI] [PubMed] [Google Scholar]
- 23.Zhou T; Damsky W; Weizman O-E; McGeary MK; Hartmann KP; Rosen CE; Fischer S; Jackson R; Flavell RA; Wang J, IL-18BP is a secreted immune checkpoint and barrier to IL-18 immunotherapy. Nature 2020, 583 (7817), 609–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ma Z; Li W; Yoshiya S; Xu Y; Hata M; El-Darawish Y; Markova T; Yamanishi K; Yamanishi H; Tahara H, Augmentation of Immune Checkpoint Cancer Immunotherapy with IL18. Clin Cancer Res 2016, 22 (12), 2969–2980. [DOI] [PubMed] [Google Scholar]
- 25.Wei S; Ma W; Zhang B; Li W, NLRP3 inflammasome: a promising therapeutic target for drug-induced toxicity. Front Cell Dev Biol 2021, 9, 634607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Weiss AM; Hossainy S; Rowan SJ; Hubbell JA; Esser-Kahn AP Immunostimulatory Polymers as Adjuvants, Immunotherapies, and Delivery Systems, Macromolecules 2022, 55, 16, 6913–6937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhao BG; Vasilakos JP; Tross D; Smirnov D; Klinman DM, Combination therapy targeting toll like receptors 7, 8 and 9 eliminates large established tumors. J Immunother Cancer 2014, 2 (1), 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Manna S; Maiti S; Shen J; Du W; Esser-Kahn AP, Pathogen-like Nanoassemblies of Covalently Linked TLR Agonists Enhance CD8 and NK Cell-Mediated Antitumor Immunity. ACS Cent Sci 2020, 6 (11), 2071–2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Manna S; Howitz WJ; Oldenhuis NJ; Eldredge AC; Shen J; Nihesh FN; Lodoen MB; Guan Z; Esser-Kahn AP, Immunomodulation of the NLRP3 inflammasome through structure-based activator design and functional regulation via lysosomal rupture. ACS Cent Sci 2018, 4 (8), 982–995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nuhn L; De Koker S; Van Lint S; Zhong Z; Catani JP; Combes F; Deswarte K; Li Y; Lambrecht BN; Lienenklaus S, Nanoparticle-Conjugate TLR7/8 Agonist Localized Immunotherapy Provokes Safe Antitumoral Responses. Adv Mater 2018, 30 (45), 1803397. [DOI] [PubMed] [Google Scholar]
- 31.Black M; Trent A; Kostenko Y; Lee JS; Olive C; Tirrell M, Self-assembled peptide amphiphile micelles containing a cytotoxic T-cell epitope promote a protective immune response in vivo. Adv Mater 2012, 24 (28), 3845–3849. [DOI] [PubMed] [Google Scholar]
- 32.Reddy ST; Rehor A; Schmoekel HG; Hubbell JA; Swartz MA, In vivo targeting of dendritic cells in lymph nodes with poly (propylene sulfide) nanoparticles. J Control Release 2006, 112 (1), 26–34. [DOI] [PubMed] [Google Scholar]
- 33.Maiti S; Manna S; Shen J; Esser-Kahn AP; Du W, Mitigation of hydrophobicity-induced immunotoxicity by sugar poly (orthoesters). J Am Chem Soc 2019, 141 (11), 4510–4514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lönn P; Kacsinta AD; Cui X-S; Hamil AS; Kaulich M; Gogoi K; Dowdy SF, Enhancing endosomal escape for intracellular delivery of macromolecular biologic therapeutics. Sci Rep 2016, 6 (1), 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shen H; Ackerman AL; Cody V; Giodini A; Hinson ER; Cresswell P; Edelson RL; Saltzman WM; Hanlon DJ, Enhanced and prolonged cross-presentation following endosomal escape of exogenous antigens encapsulated in biodegradable nanoparticles. Immunology 2006, 117 (1), 78–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kang M; Jordan V; Blenkiron C; Chamley LW, Biodistribution of extracellular vesicles following administration into animals: A systematic review. J Extracell Vesicles 2021, 10 (8), e12085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Wang J; Saffold S; Cao X; Krauss J; Chen W, Eliciting T cell immunity against poorly immunogenic tumors by immunization with dendritic cell-tumor fusion vaccines. J Immunol 1998, 161 (10), 5516–5524. [PubMed] [Google Scholar]
- 38.Ishihara J; Ishihara A; Sasaki K; Lee SS-Y; Williford J-M; Yasui M; Abe H; Potin L; Hosseinchi P; Fukunaga K, Targeted antibody and cytokine cancer immunotherapies through collagen affinity. Sci Trans Med 2019, 11 (487), eaau3259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Giunta EF; Barra G; De Falco V; Argenziano G; Napolitano S; Vitale P; Zanaletti N; Terminiello M; Martinelli E; Morgillo F, Baseline IFN-γ and IL-10 expression in PBMCs could predict response to PD-1 checkpoint inhibitors in advanced melanoma patients. Sci Rep 2020, 10 (1), 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Engel AL; Holt GE; Lu H, The pharmacokinetics of Toll-like receptor agonists and the impact on the immune system. Expert Rev Clin Pharmacol 2011, 4 (2), 275–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Van Herck S; De Geest BG, Nanomedicine-mediated alteration of the pharmacokinetic profile of small molecule cancer immunotherapeutics. Acta Pharmacol Sin 2020, 41 (7), 881–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moser B; Steinhardt R; Escalante-Buendia Y; Boltz D; Barker K; Cassaidy B; Rosenberger M; Yoo S; McGonnigal B; Esser-Kahn A, Increased vaccine tolerability and protection via NF-κB modulation. Sci Adv 2020, 6 (37), eaaz8700. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data are available in the main text or the supplementary materials.