

Abstract
Background and Aims:
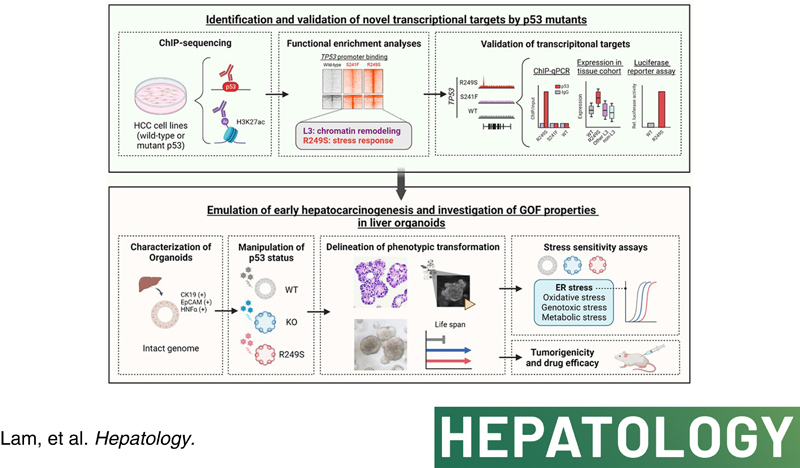
Major genomic drivers of hepatocellular carcinoma (HCC) are nowadays well recognized, although models to establish their roles in human HCC initiation remain scarce. Here, we used human liver organoids in experimental systems to mimic the early stages of human liver carcinogenesis from the genetic lesions of TP53 loss and L3 loop R249S mutation. In addition, chromatin immunoprecipitation sequencing (ChIP‐seq) of HCC cell lines shed important functional insights into the initiation of HCC consequential to the loss of tumor‐suppressive function from TP53 deficiency and gain‐of‐function activities from mutant p53.
Approach and Results:
Human liver organoids were generated from surgical nontumor liver tissues. CRISPR knockout of TP53 in liver organoids consistently demonstrated tumor‐like morphological changes, increased in stemness and unrestricted in vitro propagation. To recapitulate TP53 status in human HCC, we overexpressed mutant R249S in TP53 knockout organoids. A spontaneous increase in tumorigenic potentials and bona fide HCC histology in xenotransplantations were observed. ChIP‐seq analysis of HCC cell lines underscored gain‐of‐function properties from L3 loop p53 mutants in chromatin remodeling and overcoming extrinsic stress. More importantly, direct transcriptional activation of PSMF1 by mutant R249S could increase organoid resistance to endoplasmic reticulum stress, which was readily abrogated by PSMF1 knockdown in rescue experiments. In a patient cohort of primary HCC tumors and genome‐edited liver organoids, quantitative polymerase chain reaction corroborated ChIP‐seq findings and verified preferential genes modulated by L3 mutants, especially those enriched by R249S.
Conclusions:
We showed differential tumorigenic effects from TP53 loss and L3 mutations, which together confer normal hepatocytes with early clonal advantages and prosurvival functions.
INTRODUCTION
Hepatocellular carcinoma (HCC) has emerged as a major cause of cancer‐related deaths worldwide, ranking third in mortality rate.1 High‐throughput sequencing has provided a blueprint on the genetic landscape of HCC,2 where mutations on TERT promoter, CTNNB1, and TP53 are the major culprits driving cancer development. Recurrent mutational hotspots on TERT promoter and CTNNB1 confer dominant gain‐of‐function (GOF) activities. Conversely, different from canonical tumor suppressors, TP53 in HCC is commonly associated with concurrent loss‐of‐function (LOF) aberrations and protein‐altering missense mutations. This simultaneous occurrence can be found in approximately 60% of TP53 cases (Figure 1). Interestingly, approximately 91.2% of the TP53 missense mutations are located within the DNA binding domain (DBD; residues P92–T312). Studies have demonstrated cancer‐promoting phenotypes from p53 mutants, including enhanced cell proliferation and survival,3,4 epithelial‐mesenchymal transition,5 apoptotic evasion,6 and tumor‐initiating ability in vivo.7 Concerted efforts have revealed potential GOF mechanisms that can either ascribe to the structural alteration whereby the mutants interfere with other proteins3,4,8 or exert novel transcriptional regulation to activate downstream protumorigenic pathways,5,6 hijack chronic immune signaling,9 promote chromatin remodeling,10 and modulate alternative splicing events.11
FIGURE 1.
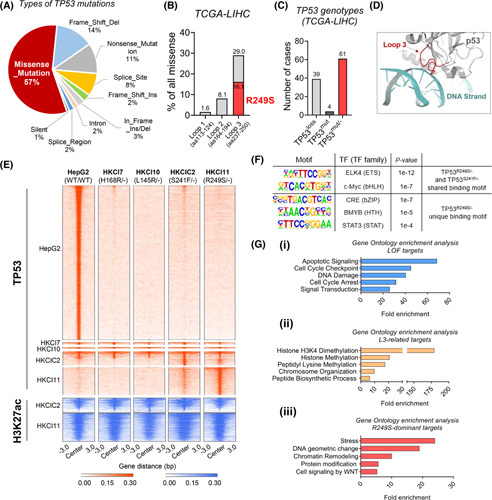
Chromatin immunoprecipitation sequencing (ChIP‐seq) of TP53 genotypes. (A) Proportion of different types of TP53 mutations in The Cancer Genome Atlas (TCGA) hepatocellular carcinoma (HCC) dataset. Missense mutations constitute 57% of all mutated cases. (B) Distribution of missense mutations within DNA binding domain Loops 1–3. (C) Incidence of TP53 copy loss, mutation, and concurrent loss and mutation in TCGA HCC dataset. (D) Structural interaction of p53 loop 3 and DNA strand as predicted by AlphaFold 2.0. (E) Heat map of genome‐wide occupancy patterns of TP53 binding and lysine H3K27 acetylation (H3K27ac) from ChIP‐seq of four TP53 mutants and wild type (WT). (F) List of DNA motifs enriched in TP53 R249S/‐ and TP53 S241F/‐. (G) Gene set enrichment analysis showed enrichment of signalings (top 5) in loss‐of‐function (LOF) targets (i), L3‐related targets (ii), and R249S‐dominant targets (iii). LIHC, Liver Hepatocellular Carcinoma, TF, transcription factor.
Cumulating evidence exemplifies on the deleterious strength of missense mutations in a contextual dependent manner, which entails the mutational prevalence, protein domains, structural motifs, and the evolutionary conservation where the mutations are located.12,13 Conceivably, regions within the TP53 DBD with a higher degree of evolutionary conservation are more likely affected by base substituting mutations. Indeed, studies have shown that structural mutations in the L3 region could substantially perturb the L3 loop, inducing high flexibility that favors alternative conformations with severe consequences for the overall protein stability and DNA binding.14,15 On the contrary, residue changes within the L1 loop (residues F113–T124) and L2 loop (residues K164–L194) do not manifest similar phenomena as that in the L3 loop, even though they are also part of the DBD.13 Given that the L3 loop (residues M237–P250) is in the closest proximity to the DNA binding surface (Figure 1D), it is plausible that structural perturbations by R249S and other direct DNA interacting residues such as Ser241 confer GOF transcriptional roles to tumor cells.
Being a hotspot missense mutation distinct for HCC, TP53 R249S mutation has been shown to be associated with tumorigenic behaviors including augmentation of cell proliferation and clonogenicity in cell lines.16–18 Nonetheless, although the occurrence of TP53 mutations is believed to be an early event in HCC development,2,19 ways to directly study the early roles of p53 GOF mutants in tumor initiation are limited. In this study, to determine anew regulatory roles of p53 DBD mutants, we systematically evaluated TP53 mutations for potential gain of transcription activities by chromatin immunoprecipitation followed by high‐throughput sequencing (chromatin immunoprecipitation sequencing; ChIP‐seq) on four patient‐derived HCC cell lines that harbor concurrent TP53 loss and DBD mutation. These included two L3 loop mutations (TP53 R249S/− and TP53 S241F/− ) and two non‐L3 loop mutations (TP53 H168R/− and TP53 L145R/− ). Genome‐wide patterns of mutant p53 occupancies were compared with wild‐type (WT) p53 (HepG2) and integrated with matching ChIP‐seq of lysine H3K27 acetylation (H3K27ac) for information on the open chromatin status of active transcription. Experimental models to study human cancer initiation are scarce. Recent reports on pancreatic and colorectal cancers have shown that organoids derived from primary human cells can offer a robust system for studying cancer initiation following gene editing.20,21 This approach facilitates analysis of cancer mutations and provides an opportunity to monitor early stage tumorigenic changes. To interrogate how p53 loss and concurrent L3 loop R249S mutation contribute to HCC initiation, we established human liver organoid as our study model in clustered regularly interspaced short palinfromic repeats (CRISPR) gene editing experiments. Directly generated from human liver tissues, these liver organoids allowed recapitulation of cancer supportive phenotypes endowed in hepatocytes from knockout (KO) of TP53 (TP53 KO) and TP53 R249S and identification of protumoral changes during the initiation of human liver cancer.
MATERIALS AND METHODS
Clinical specimens
Paired HCC tumor and adjacent nontumoral liver tissues were obtained from patients who underwent curative hepatectomy at the Prince of Wales Hospital, Hong Kong. The use of these samples for research was approved by the Joint Chinese University of Hong Kong–New Territories East Cluster Clinical Research Ethics Committee (Ref. No 2020.420). Informed consent was obtained from each patient. Diagnosis of HCC was confirmed by histology.
Primary culture for liver organoids
Liver organoids from three individuals were cultured from fresh nontumoral liver tissues excised at the resection margin of surgical HCC. Nontumoral liver tissues were immersed in Transport Medium Roswell Park Memorial Institute 1640 (Gibco) supplemented with 16% FBS, 250 μg/ml Fungizone [Gibco], Penicillin‐Streptomycin (Pen‐Strep) [Gibco], and L‐Glutamine [Gibco]) were delivered to the laboratory within 20 min after collection. Tissues were minced and digested in 225 U/ml collagenase type II (Worthington) at 37°C for 10–45 min depending on consistency. Cell clusters were filtered by 100‐μm cell strainers, and approximately 5 × 105 live cells were seeded per 40 μl Matrigel (Corning). After Matrigel solidified, medium as reported by Clevers22 was applied to maintain hepatocyte culture ex vivo. The medium was slightly modified to AIM‐V (Gibco) supplemented 0.35 U/ml Pen‐Strep (Gibco), 1× L‐Glutamine (Gibco), 1× MEM Non‐Essential Amino Acids (Gibco), 1× Insulin‐Transferrin‐Selenium (Gibco), 30% (vol/vol) Wnt3a‐conditioned medium, 10% (vol/vol) R‐spondin 1 (RSpo1)‐conditioned medium (Trevigen), 1× B27 supplement (Gibco), 1× N2 supplement (Gibco), 1 mm N‐acetyl‐L‐cysteine (Sigma‐Aldrich), 10 mm Nicotinamide (Sigma‐Aldrich), 25 ng/ml Noggin (PeproTech), 100 ng/ml recombinant human EGF (PeproTech), 100 ng/ml recombinant human FGF10 (PeproTech), 25 ng/ml recombinant human HGF (PeproTech), 10 nm Leu‐15‐Gastrin I human (Sigma‐Aldrich), 10 μm Forskolin (PeproTech) and 3 μm A8301 (Tocris). Medium was changed twice a week, and organoids were passaged by TrypLE (Gibco). Frozen stocks were prepared in Recovery Cell Culture Freezing Medium (Gibco).
Lentiviral transduction of liver organoids for CRISPR KO TP53 and overexpress R249S mutant
Two single guide RNA (sgRNA) sequences spanning the intron and exon regions designed to target exon 2 (sgRNA E2) and exon 7 (sgRNA E7) of TP53 were cloned into the CRISPRv2 vector (Addgene plasmid no. 52961). To allow constitutive expression of TP53 R249S mutant, the TP53 R249S sequence was cloned into pLV‐EF1a‐IRES‐Hygro vector (Addgene plasmid no. 85134). Generation of single‐cell–derived p53 KO liver organoids was performed. Expression of endogenous WT p53 was first knocked out by cotransducing organoid cells with the two lentiCRISPRv2‐sgRNA lentiviruses as aforementioned. The organoids were treated with 1.5 μg/ml puromycin to generate p53 KO cell pool and were cultured under 1 μg/ml puromycin (Life Technologies, A1113803) for propagation. To isolate a single p53 KO clone, the p53 KO organoid pools were digested into single cells followed by sparsely seeding. After 1–2 weeks, the single‐cell–derived p53 KO organoids were large enough to be manually picked and propagated. For validation of mutations at CRISPR targeted sites, genomic DNA was isolated from each p53 KO clone, followed by polymerase chain reaction (PCR) amplification of exon 2 and exon 7 of TP53 gene using primers listed in Table S3. The PCR products were cloned into a TOPO‐TA cloning vector (Life Technologies, 450641) and transformed into bacteria. Five to ten bacterial colonies were submitted for Sanger sequencing to identify biallelic gene alterations.
To establish TP53 R249S‐overexpressed organoids, one p53 KO clone from each organoid case was transduced with pLV‐EF1a‐IRES‐Hygro‐TP53 R249S lentivirus, followed by hygromycin selection (Life Technologies, 10687010). The p53 R249S organoids were then propagated under low‐dosage hygromycin and puromycin. The presence of TP53 R249S in organoids was Sanger confirmed.
Organoid morphology and time‐lapse imaging on growth pattern
Quantitative image analysis was conducted to assess morphological changes of TP53 KO and TP53 R249S liver organoids. Cells seeded in black 96‐well culture dish were maintained in culture medium supplemented with 40 μm Calcein AM (Invitrogen) and 2 μg/ml Hoechst 33342 (Invitrogen) for 2 h at 37°C. Fluorescent images were captured by the Cellomics ArrayScan VTI (ThermoFisher Scientific) under standard culture conditions (37°C and 5% CO2). Morphology counting was conducted by two individuals based on 20 fields of each well, and only organoids larger than 50 μm were considered.
For time‐lapse live‐cell imaging, organoids were plated in a 35‐mm culture dish. On Day 4, the dish was placed into the culture chamber of the Zeiss Axio Observer Z1 Inverted Phase Contrast Fluorescence Microscope. Images were taken at a time interval of 10 min for 24 h. The images were later processed and assembled into videos using the Zen software.
Stress sensitivity assays
To assess sensitivity of TP53 KO and TP53 R249S liver organoids toward stress inducers and epigenetic drug, endoplasmic reticulum (ER) stress inducer tunicamycin (MedChemExpress, HY‐A0098) and epigenetic drug thioguanine (SelleckChem, S1774) were tested. Organoid cells were seeded in a white 96‐well culture plate at a cell density of 5000 cells per 4 μl of Matrigel. When organoids reached an average size of 100 μm, normally within 4–7 days, stress inducer–containing plain AIM‐V medium was added to the corresponding wells. After 72 h of incubation in stress inducers or drug, cell viability of organoids was assessed using CellTiter‐Glo 3D reagent as aforementioned. Data analyses were performed using GraphPad Prism 9 software, and the values of the half maximal inhibitory concentration (IC50) were calculated by applying nonlinear regression (curve fit).
Liver organoid‐derived xenograft
Organoids (3 × 106 cells in 100 μl Matrigel) with TP53 KO, TP53 R249S, or TP53 WT were subcutaneously injected into both flanks of female NOD scid gamma mice (4–8 weeks) to develop xenografts. Lesion sizes were measured once a week. All mice were sacrificed at Week 8, and lesions were embedded in paraffin for hematoxylin and eosin staining. All animal experiments were conducted under the approval of the Animal Experimentation Ethics Committee of the Chinese University of Hong Kong and adhered to the Animals (Control of Experiments) Ordinance of Hong Kong. All animals received humane care according to the NIH Guide for the Care and Use of Laboratory Animals. Experienced pathologists assessed histology for malignant changes.
Statistical analysis
Experiments were performed in triplicate, and data are plotted as mean ± SEM. Continuous data were compared with unpaired or paired Student's t test, Welch's t test, Mann–Whitney U test, or two‐way analysis of variance with Bonferroni correction when appropriate. HCS Studio Cell Analysis Software was used to quantify gated cell signal intensities of live cell fluorescence‐based images. Statistical analyses were conducted by GraphPad Prism version 9.3.1 (GraphPad) or Microsoft Excel, and p < 0.05 was considered statistically significant.
Additional experimental assays to support validations and functions are described in Supporting Methods. Supplementary videos of organoid growth on TP53 KO, TP53 R249S, and control TP53 WT as well as an example of HCC organoid growth can be downloaded and reviewed at links below:
https://www.surgery.cuhk.edu.hk/easonng/Supplementary_Video_1_Vector_Ctrl.avi
https://www.surgery.cuhk.edu.hk/easonng/Supplementary_Video_2_TP53_KO.avi
https://www.surgery.cuhk.edu.hk/easonng/Supplementary_Video_3_TP53_R249S.avi
https://www.surgery.cuhk.edu.hk/easonng/Supplementary_Video_4_HCC_organoid.avi
RESULTS
ChIP‐seq revealed GOF roles of L3 mutants on chromatin remodeling and stress responses
In HCC, TP53 is frequently altered by missense mutations, accounting for more than half of the mutation cases (Figure 1A) and mostly clustered within DBD Loops 1–3 (Figure 1B). Among all HCCs harboring TP53 mutations in The Cancer Genome Atlas dataset, approximately 60% carried a concurrent loss of TP53 and an additional protein‐altering missense mutation (TP53 missense/−) (Figure 1C). Increasing evidence suggests that mutant p53 proteins are likely a heterogeneous group of variant oncoproteins with different GOF activities.12,13 To systematically evaluate potential gain in transcriptional activation from TP53 missense mutations in HCC, we conducted data analyses of genome‐wide patterns of TP53 binding and lysine H3K27ac with chromatin immunoprecipitation followed by high‐throughput sequencing (ChIP‐seq) experiments from four TP53 mutants, including two L3 loop mutations (TP53 R249S/− and TP53 S241F/−) and two non‐L3 loop mutations (TP53 L145R/− and TP53 H168R/−). We found the genome‐wide binding of WT TP53 was mostly lost in the four mutants, irrespective of TP53 mutation sites, implying a loss of conventional TP53 transcriptional activities was common in tumors with TP53 missense mutations (Figure 1E). Notably, a set of TP53 binding peaks were exclusively identified in the L3 loop mutants when compared with WT TP53 and the two other non‐L3 loop mutants (Figure 1E). Besides, we also observed promoter‐proximal TP53 binding peaks specific for R249S mutant, rendering the possibility of additional transcriptional regulatory properties (Figure 1E). Conversely, rare unique binding peaks were observed for the non‐L3 loop mutants. Colocalization of enriched H3K27ac signals at L3 loop mutant binding regions further indicated these peaks were associated with active transcription (Figure 1E). In addition, to illustrate the potential transcription factors, we defined the core binding motifs of erythroblast transformation‐specific and basic helix‐loop‐helix family in endogenous TP53 R249S/− and TP53 S241F/− cells (Figure 1F).
To determine the functional ontologies of these TP53 mutants, we next performed pathway enrichment analysis using genes proximal to these three sets of TP53 binding peaks (i.e., WT TP53‐associated peaks, L3 loop‐associated peaks, and TP53 R249S/− dominant peaks) (Table S1). As expected, WT TP53 was mainly involved in the modulation of apoptosis, cell cycle, and DNA damage (Figure 1Gi, Table S2), which supports the tumor‐suppressive effects of TP53 in HCC.23 TP53 mutants, in contrast, exhibited distinct regulatory activities. The L3 loop mutants were bound to genes related to chromatin remodeling and biosynthetic process (Figure 1Gii, Table S2), whereas unique binding peaks found in TP53 R249S/− were enriched in other pathways, including stress responses and protein modification (Figure 1Giii, Table S2).
To corroborate ChIP‐seq findings on new binding occupancies of L3 mutants, chromatin immunoprecipitation–quantitative PCR was performed in HepG2 (TP53 WT/WT), HKCIC2 (TP53 S241F/−), and HKCI11 (TP53 R249S/−) cells to verify direct binding of TP53 mutants to transcriptional start site (TSS) regions of enriched targets. First, to affirm LOF properties of TP53 L3 mutants, targets related to intrinsic apoptotic signaling EDA2R (ectodysplasin A2 receptor), PHLDA3 (pleckstrin homology‐like domain family A member 3), and CDKN1A (cyclin‐dependent kinase inhibitor 1A) were tested. TP53 R249S/− and TP53 S241F/− did not appear to bind to these genes, whereas HepG2 TP53 WT/WT displayed enriched binding to the TSS regions (Figure 2Ai,Aii). Next, L3‐related targets RCOR3 (REST corepressor 3) and UBE2I (ubiquitin conjugating enzyme E2 I) that participated in the regulation of chromosome organization were found to show strong TSS occupancy in both TP53 R249S/− and TP53 S241F/− mutants but not TP53 WT/WT (Figure 2Bi,Bii). Additionally, R249S‐dominant targets with functions covering stress response, CLTC (clathrin heavy chain) and PSMF1 (proteasome inhibitor subunit 1), spindle organization SUN2 (Sad1 and UNC84 domain containing 2) and chromatin remodeling BPTF (bromodomain PHD finger transcription factor), BRPF1 (bromodomain and PHD finger containing 1), and SMARCD2 (SWI/SNF related, matrix associated, actin dependent regulator of chromatin, subfamily D, member 2), all displayed prominent TP53 R249S/− binding enrichment to the TSS regions compared with TP53 S241F/− and TP53 WT/WT (Figure 2Ci,Cii).
FIGURE 2.
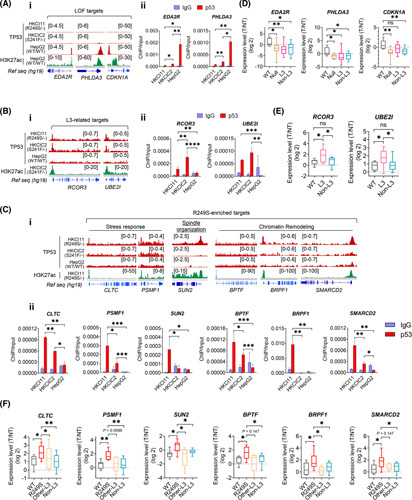
Verification of loss‐of‐function (LOF), Loop L3, and R249S‐enriched targets. (A) Chromatin immunoprecipitation sequencing (ChIP‐seq) signals for (i) LOF targets EDA2R, PHLDA3, and CDKN1A; (ii) chromatin immunoprecipitation–quantitative polymerase chain reaction (ChIP‐qPCR) validations of EDA2R and PHLDA3 in HKCI‐11 (R249S), HKCIC2 (S241F), and HepG2 (wild‐type; WT) (*p < 0.05, **p < 0.01). (B) ChIP‐seq signals for (i) L3‐related targets RCOR3 and UBE2I in HKCI‐11 (R249S), HKCIC2 (S241F), and HepG2 (WT); (ii) ChIP‐qPCR validations (**p < 0.01, ***p < 0.001, ****p < 0.0001). (C) ChIP‐seq signals for (i) R249S‐enriched targets CLTC, PSMF1, SUN2, BPTF, PSMF1, BRPF1, and SMARCD2 in HKCI‐11 (R249S), HKCIC2 (S241F), and HepG2 (WT); (ii) and ChIP‐qPCR validations (*p < 0.05, **p < 0.01, ***p < 0.001). (D) Expression of LOF targets in a cohort of patients with hepatocellular carcinoma (HCC) with various TP53 genotype statuses (*p < 0.05, **p < 0.01). Sample size: WT (n = 7), Null (n = 8), L3 (n = 14), Non‐L3 (n = 20). (E) Expression of L3‐related targets in a cohort of patients with HCC with various TP53 genotype statuses (*p < 0.05). Sample size: WT (n = 7), L3 (n = 14), Non‐L3 (n = 20). (F) Expression of R249S‐dominant targets in a cohort of patients with HCC with various TP53 genotype statuses (*p < 0.05, **p < 0.01). Sample size: WT (n = 7), R249S (n = 9), Other‐L3 (n = 5), Non‐L3 (n = 20). Data were plotted as mean ± SEM from three independent experiments in (Aii), (Bii), and (Cii). Box plots in (D)–(F) are presented as 10th to 90th percentiles. H3K27ac, H3K27 acetylation; IgG, immunoglobulin G; ns, not significant; Ref seq, reference sequence; T/NT, tumor/non‐tumor.
To substantiate augmented gene expression by L3 mutants, we performed validation in an independent primary cohort of paired HCC and adjacent nontumoral liver. These specimens were grouped according to their TP53 status verified by target‐capture sequencing24 or Sanger sequencing, which included WT (TP53 WT/WT), null (TP53 −/−), TP53 R249S, TP53 missense mutations within L3 loop (other L3), and missense mutations outside of L3 loop (non‐L3). A significant diminution on LOF targets was found in all groups except TP53 WT/WT (Figure 2D). L3 loop targets were markedly elevated in specimens with TP53 L3 loop mutations (Figure 2E). Of interest, six R249S‐dominant targets were evidently increased in cases with TP53 R249S compared with TP53 WT and other mutant groups (Figure 2F).
Generating primary liver organoids and tumorigenic induction from TP53 KO and R249S
Organoids are becoming increasingly important in cancer research for their many advantages over existing cancer models, including being physiologically relevant and predictive of outcome. Here, to understand the cancer initiating roles of TP53 KO and TP53 R249S in HCC, we developed three liver organoids, namely liver organoid 1 (Liver.Org 1), liver organoid 2 (Liver.Org 2), and liver organoid 3, from fresh nontumoral liver tissues (Figure 3A). Three liver organoids were self‐organizing with a monolayer of epithelium surrounding by a giant central lumen (Figure S1A) that resembles the ductal structure of hepatic organoids shown by other groups.25,26 High expressions of liver progenitor markers CK19 (cytokeratin 19) and EpCAM (epithelial cell adhesion molecule) and hepatic cell marker HNFα (hepatocyte nuclear factor 4 alpha) in the absence of liver cancer indicator GPC3 (glypican‐3) suggested the hepatic origin of these organoids in a nontumoral state (Figure S1B). The latter was further supported by whole‐exome sequencing, which indicated the absence of copy number variation and only random somatic nucleotide variants and insertions/deletions of no pathogenic consequences (noncosmic) (Figure S2A,B). The intact genetic background that is well‐preserved in our liver organoids suggest suitability of these models for downstream gene editing studies.
FIGURE 3.
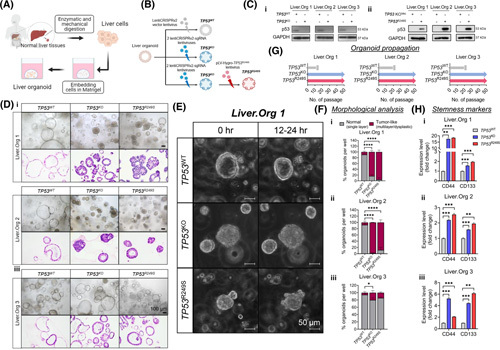
TP53 knockout (TP53 KO) and TP53 R249S liver organoids showed premalignant features. (A) Schematic workflow of primary culture for liver organoid generation. (B) Schematic diagram illustrating experimental approaches for developing TP53 wild‐type (TP53 WT), TP53 KO, and TP53 R249S organoids. (C) Protein levels of p53 in liver organoids were determined by western blot assay in (i) TP53 KO and (ii) TP53 R249S. (D) Representative brightfield and histological images of TP53 WT, TP53 KO, and TP53 R249S liver organoids from three individuals. The TP53 WT organoids grew as cystic structures, whereas TP53 KO and TP53 R249S organoids formed thickened walls or compact spheroids. Scale bar: 100 μm. (E) Representative time‐lapse images showed growth pattern of TP53 WT, TP53 KO, and TP53 R249S organoids within 12–24 h. Scale bar: 50 μm. (F) Morphological analysis indicated the proportion of single‐layered or tumor‐liked morphology in TP53 WT, TP53 KO, and TP53 R249S organoids (*p < 0.05, ****p < 0.0001). (G) Expansion potential of established liver organoids. The TP53 WT organoids stopped propagating within 20 passages, whereas both TP53 KO and TP53 R249S organoids continue to grow for more than 50 passages. (H) Expression of stemness markers, CD44 and CD133, in TP53 WT, TP53 KO, and TP53 R249S organoids (**p < 0.01, ***p < 0.001). Data were generated from three independent experiments and presented as mean ± SEM. Liver.Org 1, liver organoid 1; Liver.Org 2, liver organoid 2; Liver.Org 3, liver organoid 3; sgRNA, single guide RNA.
To mimic the TP53 R249S/− genotype, we first applied the CRISPR/Cas9 system with two single‐guided RNAs to KO TP53 in three liver organoids (Figure 3B and Figure S3A). This was followed by a corresponding TP53 R249S overexpression by pLV‐Hygro‐TP53 R249S lentivirus (Figure 3B and Figure S3B). The consequential loss and gain of TP53 protein were determined by western blot assay (Figure 3Ci,Cii), and the preserved expression of liver progenitor markers was verified by immunofluorescence (IF) staining in three organoid cases (Figures S4A, S5A, and S6A). Morphologically, WT (TP53 WT) liver organoids maintained uniformity in single‐layered compartments from both brightfield imaging and histology staining (Figure 3D). In contrast, distinct morphologies appeared in TP53 KO and R249S‐overexpressed (TP53 R249S) organoids where thickened walls invaginating into the lumens were seen and pleomorphic malignant features, including dysplasia, hyperchromasia, atypical and frequent mitosis, loss of polarity, and increased nuclear to cytoplasm ratio, presented (Figure 3D). Time‐lapse imaging of Liver.Org 1 further verified the aberrant morphological features from a change in growth pattern that emerged in TP53 KO and TP53 R249S organoids. Compared with the ductal structure in TP53 WT control organoids (Figure 3E and Video S1), we observed compact forming organoid clusters upon loss of TP53 protein (Figure 3E and Video S2). Intriguingly, organoids with R249S overexpression exhibited atypical growing with abundant bud‐shaped protrusions on the organoid surface (Figure 3E and Video S3), which strongly resembled the growth pattern of HCC organoids (Figure S7A and Video S4). By high‐throughput imaging assay, we quantified the morphological differences among different TP53 statuses. Consistent with histology, single‐layered organoids were common in TP53 WT groups, whereas tumor‐liked organoids packed with multilayer or dysplasia were widely seen in TP53 KO and TP53 R249S Liver.Org 1 and Liver.Org 2 (Figure 3F).
In addition, to assess whether loss of TP53 or gain of R249S mutation conferred growth advantages, it was noted that all TP53 WT control organoids ceased growth within 20 passages (Figure 3G). Surprisingly, both TP53 KO and TP53 R249S organoids continued to proliferate and expand in propagation, with a consistent splitting ratio of 1:4 each week for more than 50 passages (Figure 3G). Because stemness is one of the factors reported to promote cell longevity, we evaluated the expression of liver cancer stem markers, CD44 and CD133, in three liver organoids. A significant increase of CD44 and CD133 expressions was corroborated in TP53 KO and TP53 R249S organoids compared with TP53 WT control (Figure 3H), suggesting marked stimulation of cancer‐associated stemness abilities.
Modelling tumor initiation from TP53 R249S in liver organoids
Stress and cancer are interlinked, which robustly fuels tumors in both initiation and progression stages.27 The stress gene ontology (GO) category ranked top in TP53 R249S enriched targets (Figure 1Giii) intrigued us and motivated us to explore the roles of mutant R249S and cancer‐associated stress in initiating liver cancer development. We first validated expressions of TP53 LOF targeted genes and R249S‐dominant candidates that were related to stress responses and spindle organization in the edited liver organoids. In line with expressions found in primary HCC, we detected marked upregulation of R249S‐dominant targets in R249S‐overexpressed liver organoids (Figure 4A), coupled with strong diminution of LOF targets in TP53‐silenced organoids compared with control (Figure S8A). The direct transcriptional effect of R249S mutant was further determined in dual‐luciferase assay in Hep3B (TP53 −/−) cells overexpressed with TP53 WT and R249S, respectively. The promoter activities of two stress‐related R249S‐dominant targets, CLTC and PSMF1, were evidently escalated in TP53 R249S‐expressing cells compared with TP53 WT control (Figure 4B). A plausible tumorigenic initiating role was suggested from anchorage‐independent assay, which readily showed a significant presence of large compact spheroids in TP53 R249S organoids compared with spheroid clusters in TP53 KO organoids and only tiny sporadic spheroids in control organoids (Figure 4C). The tumorigenic potential of TP53 R249S was further corroborated in xenotransplantations. At the end of a 2‐month period, subcutaneous organoid‐derived xenografts (ODXs) showed increased lesion forming in R249S‐overexpressed liver organoids (37%) compared with TP53‐deficient organoids (17%) and WT control (0%) (Figure 4D). Histological examination revealed malignant features of these lesions, displaying atypical cells with glandular formation, which exhibited nuclear pleomorphism hyperchromatism, occasional vacuolated cytoplasm, and prominent nucleoli (Figure 4E). Our finding strongly indicates a malignant transformation from the concurrent loss of p53 and gain of R249S mutation.
FIGURE 4.
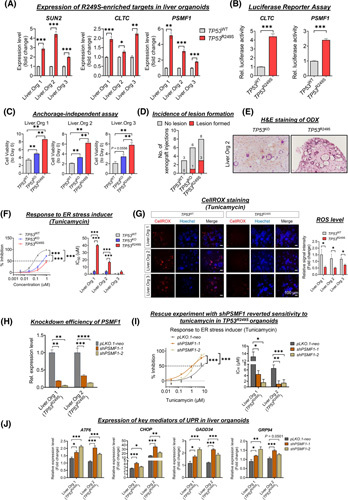
Tumor initiation in TP53 R249S organoids. (A) Expression of R249S‐enriched targets related to spindle organization and stress response in liver organoids. Increased levels of SUN2, CLTC, and PSMF1 in TP53 R249S organoids shown (*p < 0.05, **p < 0.01, ***p < 0.001). (B) Dual‐luciferase reporter assay showed promoter activities of CLTC and PSMF1 in R249S‐overexpressed Hep3B cells (***p < 0.001). (C) Induction of spheroid forming viability in TP53 wild type (TP53 WT), TP53 knockout (TP53 KO), and TP53 R249S organoids between Day 0 and 6. (D) TP53 WT, TP53 KO, and TP53 R249S organoids were subcutaneously transplanted into NSG mice. Incidence of lesions formed in each group shown. (E) Representative histological images of xenograft lesions. (F) Representative cell viability (left) and statistical analysis of the half maximal inhibitory concentration (IC50) value (right) of TP53 WT, TP53 KO, and TP53 R249S organoids after tunicamycin treatment (***p < 0.001). (G) Representative images of CellROX staining (left) and quantitative analysis of signal intensity (right) in TP53 WT and TP53 R249S organoids upon administration of tunicamycin. TP53 R249S organoids were more invulnerable to tunicamycin‐induced reactive oxygen species (ROS) levels (*p < 0.05). Scale bar: 100 μm. (H) Knockdown efficacy of PSMF1 in R249S‐expressing liver organoids (**p < 0.01, ***p < 0.001). (I) The effect of tunicamycin in PSMF1‐silenced TP53 R249S organoids. PSMF1 deficiency increased cell sensitivity to tunicamycin (left) with lower IC50 value (right) (*p < 0.05, **p < 0.01, ***p < 0.001). (J) Expression of key mediators of unfolded protein response (UPR) in liver organoids after administration of tunicamycin. Data shown as fold change between 0 and 8 h (*p < 0.05, **p < 0.01, ***p < 0.001). All data were generated from three independent experiments and presented as mean ± SEM. ER, endoplasmic reticulum; H&E, hematoxylin and eosin; Liver.Org 1, liver organoid 1; Liver.Org 2, liver organoid 2; Liver.Org 3, liver organoid 3; ODX, organoid‐derived xenograft.
Next, we attempted to investigate the contributory effects of R249S‐related stress responses in tumor disposition. There are several types of stress, for instance ER, oxidative, and metabolic, that actively regulate cancer development. We administered different stress‐specific stimulators, including ER stress inducer tunicamycin, oxidative stress inducer hydrogen peroxide, genotoxic stress generated by etoposide, and metabolic stress inducer by nutrient‐deprived medium. Notably, evident tolerance in TP53 R249S was only found for tunicamycin, but not other inducers (Figure 4F and Figure S9). Given the mutual crosstalk between ER stress and reactive oxygen species (ROS) induction,28 we evaluated the ROS levels upon ER stress stimulation. CellROX Red signal was vaguely detected in the TP53 R249S organoids while clearly recognized in the control counterparts (Figure 4G), implying a homeostatic level of ROS from the R249S GOF role. To affirm association between ER stress tolerance and R249S‐dominant stress target PSMF1, we conducted rescue experiments in TP53 R249S liver organoids by knocking down PSMF1 (Figure 4H). Intriguingly, a spontaneous sensitivity to tunicamycin was restored in PSMF1‐silenced liver organoids, with IC50 values at 4‐ to 8‐fold lower than vehicle control (Figure 4I).
Because accumulation of misfolded proteins is a hallmark of ER stress,19 we also examined the expression of key sensors and mediators of unfolded protein response (UPR) in PSMF1‐downregulated organoids. Compared with vehicle control, a significant increase in ATF6 (activating transcription factor 6), GRP94 (glucose‐regulated protein 94), CHOP (C/EBP homologous protein), and GADD34 GADD (growth arrest and DNA damage‐inducible gene 34) expressions was found in PSMF1‐deficient TP53 R249S organoids when treated with tunicamycin (Figure 4J), implying the essential capacity of PSMF1 in relieving ER stress by preventing UPR activation.
A therapeutic window targeting TP53 R249S through chromatin remodeling
Considering the enriched GO terms of “Chromatin Remodeling” in R249S‐dominant targets (Figure 1Giii), we first affirmed significant upregulation of R249S‐dominant targets (BPTF, BRPF1, and SMARCD2) in TP53 R249S liver organoids (Figure 5A). As thioguanine is an epigenetic drug that inhibits DNA synthesis and methylation, we explored the pharmacological effects of thioguanine in TP53 R249S organoids in vitro. Results showed that TP53 R249S organoids were more vulnerable to thioguanine treatment compared with WT control, showing a significant increase in sensitivity by 4.5‐ to 7‐fold in terms of IC50 values (Figure 5B).
FIGURE 5.
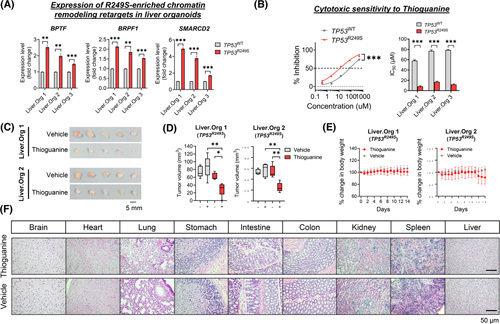
A therapeutic window in targeting TP53 R249S. (A) Expression of R249S‐enriched targets associated with chromatin remodeling in liver organoids. Upregulation of BPTF, BRPF1, and SMARCD2 were determined in TP53 R249S organoids (**p < 0.01, ***p < 0.001). (B) The effect of thioguanine in TP53 wild‐type (TP53 WT) and TP53 R249S organoids. The R249S‐sufficient organoids illustrated increased sensitivity (left) with a lower IC50 value of thioguanine compared with TP53 WT control organoids (***p < 0.001). All data generated from three independent experiments and presented as mean ± SEM. (C) Photos of TP53 R249S xenografts retrieved from mice treated with thioguanine or vehicle. (D) Tumor volume of TP53 R249S xenografts before and after thioguanine or vehicle treatments (*p < 0.05, **p < 0.01). Sample size: thioguanine (n = 5), vehicle (n = 5). (E) Body weight recorded daily in both thioguanine and vehicle treatment groups. (F) Representative histological images of healthy organs expressing TP53 WT. Scale bar: 50 μm. Data presented as mean ± SD. Liver.Org 1, liver organoid 1; Liver.Org 2, liver organoid 2.
To further underpin the in vivo efficacy of thioguanine, we established ODX models from TP53 R249S organoids of Liver.Org 1 and Liver.Org 2. Once tumor volume reached approximately 50 mm3, the mice were randomized for intraperitoneal administration with either thioguanine (1.5 mg/kg) or vehicle for 14 consecutive days. Our results showed that TP53 R249S ODXs were more susceptible to thioguanine, with marked shrinkage in tumor volume seen after 14 days of treatment compared with vehicle control counterparts (Figure 5C,D). No significant difference in body weight was recorded between the two treatment groups (Figure 5E). Normal histology was also observed in all major organs, suggesting negligible side effects from thioguanine (Figure 5F). Collectively, we demonstrated a therapeutic potential for the epigenetic drug thioguanine in targeting TP53 R249S‐associated GOF vulnerability in vitro and in vivo.
DISCUSSION
With the aid of high‐throughput next‐generation sequencing, it has become apparent that the scope of p53 mutations is much broader than previously thought. Large‐scale cancer genome projects revealed more than 80% of somatic p53 alterations are in fact missense mutations that abrogate the p53 tumor suppressor function and instead bestow mutant proteins with new GOF activities.29,30 Our ChIP‐seq findings support this notion, where we found TP53 DBD mutants showed loss of promoter binding to canonical WT targets but gained new promoter‐proximal DNA‐binding occupancies that were more evident for L3 mutants, implying exclusive transcriptional regulations from L3 mutations. TP53 R249S is an HCC‐specific hotspot mutation that is closely correlated with hepatitis B virus (HBV) infection and aflatoxin B1 exposure.12 The exceptionally high mutation frequency implies selective advantages rendered by this mutation in the context of hepatocarcinogenesis. To date, interrogation of its unique oncogenic roles at both protein and transcriptional regulatory levels had predominantly been performed in cancer cell lines. In the protein regulatory aspect, cytoplasmic p53 R249S mutant was reported to form complex with TANK‐binding protein kinase 1 or viral hepatitis B protein X to evade innate immune surveillance3 or promote HCC proliferation.18 Transcriptionally, p53 R249S could interact with other transcriptional factors, such as c‐Myc to enhance ribosomal DNA transcription,16 or direct promoter occupancy and activation of chromatin regulators.10 Nonetheless, to elucidate the GOF properties of TP53 R249S in HCC initiation, we provide in this study evidence of ER stress tolerance capacity endowed by TP53 R249S through the acquisition of new transcriptional regulatory roles.
During the multistep HCC carcinogenesis, TERT promoter mutations, which are associated with telomerase reactivation, are the earliest somatic alterations acquired during cirrhosis to sustain telomere maintenance for continuous cell proliferation.31 Previous studies demonstrated that ROS, which were frequently produced during cirrhosis and cancer progression, could jeopardize telomerase activity32 and induce telomere erosion.33 The fact that the TERT promoter mutations co‐occur with TP53 and CTNNB1 mutations would suggest that telomerase activation alone may not be sufficient for early cancer initiation and, thus, requires additional adaptation from TP53 and CTNNB1 mutations for stress alleviation and, consequently, ROS reduction. A recent study published by our group underscored the GOF impact of CTNNB1 missense mutations in curbing oxidative stress–induced senescence generated by lipid ROS and inciting immune‐excluded tumor microenvironment in NAFLD‐related HCC.24 Intriguingly, TP53 mutations are the common genetic events in HBV‐related HCC and largely considered as mutually exclusive with CTNNB1 mutations.33,34 As we uncovered ER stress endurance ability to be the putative GOF properties of TP53 R249S and upregulation of chromatin modifiers in TP53 L3 ‐bearing HCC tissues, it is plausible that HCC cells obtain different clonal advantages from either TP53 or CTNNB1 mutations for hepatocarcinogenesis depending on the etiological factors.
Given the interesting fact that TP53 copy‐loss coexisting with allelic mutation is highly frequent in HCC tumors, a major unanswered question is the need for such genetic pressure to mutate a TP53 allele in HCCs rather than losing both alleles. The answer must be that mutation of TP53, rather than its simple loss, endows cancer cells with advantageous properties allowing tumors to become more aggressive. We leveraged on liver organoid models derived from human liver tissues to emulate the early stages of hepatocarcinogenic changes by manipulating the TP53 status for TP53 R249S and TP53 KO. Both p53 KO and R249S organoids showed distinct phenotypic changes, morphogenesis that resembles tumor budding, prolonged in vitro life span, and enhanced stemness compared with WT. Surprisingly, p53 R249S further showed enhanced stemness capacities aggravating tumor‐initiating ability in mice models over p53 deficiency. Histological examination showed bona fide HCC in TP53 R249S xenograft lesions. Taken together, our results underscore loss of TP53 in driving major growth pattern changes and stemness in cancer initiation, whereas R249S mutation exacerbates the stemness capacity to elicit neoplastic transformation through GOF transcription activities.
The unique GOF feature of ER stress alleviation by p53 R249S mutation could perhaps explain why this mutation was particularly selected along HCC evolution. Being the predominant cell type in the liver, hepatocytes require the extensive usage of ER to produce secretory proteins, lipid metabolism, and detoxification functions, bestowing cells with susceptibility toward ER stress.35,36 When the ER is accumulated with misfolded or unfolded proteins, UPR is triggered. Of note, risk factors of HCC, for instance alcohol consumption, HBV/hepatitis C virus infection, NAFLD, and aflatoxin B1, could all instigate ER stress.37,38 It is known that UPR activation reduces unfolded protein load to maintain cellular homeostasis, but prolonged activation evokes stress‐induced apoptosis.35 Therefore, the ability of p53 R249S mutant to alleviate ER stress is exceptionally beneficial for tumor cells to restore ER proteostasis and escape ER stress–mediated cell death. We showed that mutant R249S could directly activate transcription of stress‐related PSMF1, and silencing PSMF1 could reinstate sensitivity to ER stress in p53 R249S organoids by potentially impeding proteasomal degradation of misfolded or unfolded peptides. Furthermore, functional enrichment analyses shed insight into a therapeutic window of tumors harboring p53 R249S. Based on the defined GO chromatin remodeling, organoids with p53 R249S showed higher sensitivity toward the epigenetic drug thioguanine, which targets the chromatin modifier. In sum, our studies uncovered the unique transcription regulations of L3 loop mutants, in particular, stress tolerance pathways asserted by TP53 R249S mutant conferred a distinct advantage of tumorigenicity in vitro and in vivo. Concurrent TP53 loss and R249S mutation confer differential oncogenic properties from each genomic event, in which the combined bestows clonal advantages to premalignant liver cells in the hepatocarcinogenesis.
AUTHOR CONTRIBUTIONS
Yin Kau Lam, Jianqing Yu, and Xiaofan Ding analyzed and interpreted data and prepared and wrote the manuscript. Hao Huang and Xin Wang performed the bioinformatics analysis of ChIP‐seq. Yin Kau Lam, Jianqing Yu, Alissa M. Wong, and Mingjing Xu prepared the samples, performed organoid cultures, and conducted CRISPR biological experiments, ChIP‐seq validations, IF, western blot, luciferase assays, and animal experimentations. Howard H. Leung, Anthony W. Chan, and Kelvin K. Ng provided liver specimens, associated clinical annotations, and experimental histology support. Nathalie Wong conceived and supervised the study and prepared and wrote the manuscript.
ACKNOWLEDGMENTS
We thank the Core Utilities of Cancer Genomics and Pathobiology (CUHK) for providing the facilities that supported this research. The authors also thank Main He, Matthew Man, Simon You, and Cario Lo for technical assistance.
FUNDING INFORMATION
This work was supported by the Hong Kong Research Grants Council Area of Excellence Scheme (Ref. AoE/M‐401/20), Research Impact Fund (Ref. R4017‐18) and internal funds from CUHK. This research is also supported in part by a National Cancer Institute fund of the National Institutes of Health (No. R01CA229836).
CONFLICTS OF INTEREST
Nothing to report.
ACCESSION CODE
ChIP‐seq data of HCC cell lines and whole‐exome sequencing data of organoids are available in EBI database under accession no. EGAS00001005779.
Supplementary Material
Footnotes
Funding information National Cancer Institute, Grant/Award Number: R01CA229836; Research Grants Council, University Grants Committee, Grant/Award Number: Area of Excellence Scheme (Ref. AoE/M-401/20) and Research Impact Fund (Ref. R4017-18)
Abbreviations: ChIP‐seq, chromatin immunoprecipitation sequencing; DBD, DNA binding domain; ER, endoplasmic reticulum; GO, gene ontology; GOF, gain‐of‐function; H3K27ac, H3K27 acetylation; HBV, hepatitis B virus; HCC, hepatocellular carcinoma; KO, knockout; Liver.Org 1, liver organoid 1; Liver.Org 2, liver organoid 2; LOF, loss‐of‐function; ODX, organoid‐derived xenograft; PCR, polymerase chain reaction; PSMF1, proteasome inhibitor subunit 1; ROS, reactive oxygen species; TSS, transcriptional start site; UPR, unfolded protein response; WT, wild‐type.
Yin Kau Lam and Jianqing Yu contributed equally to this work.
Supplemental Digital Content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's website, www.hepjournal.com.
REFERENCES
- 1.Sung H, Ferlay J, Siegel RL, Laversanne M, Soerjomataram I, Jemal A, et al. Global Cancer Statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin. 2021;71:209–49. [DOI] [PubMed] [Google Scholar]
- 2.Schulze K, Imbeaud S, Letouze E, Alexandrov LB, Calderaro J, Rebouissou S, et al. Exome sequencing of hepatocellular carcinomas identifies new mutational signatures and potential therapeutic targets. Nat Genet. 2015;47:505–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ghosh M, Saha S, Bettke J, Nagar R, Parrales A, Iwakuma T, et al. Mutant p53 suppresses innate immune signaling to promote tumorigenesis. Cancer Cell. 2021;39:494–508.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Morrison CD, Chang JC, Keri RA, Schiemann WP. Mutant p53 dictates the oncogenic activity of c‐Abl in triple‐negative breast cancers. Cell Death Dis. 2017;8:9–e2899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dong P, Karaayvaz M, Jia N, Kaneuchi M, Hamada J, Watari H, et al. Mutant p53 gain‐of‐function induces epithelial–mesenchymal transition through modulation of the miR‐130b–ZEB1 axis. Oncogene. 2012;32:3286–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Monteith JA, Mellert H, Sammons MA, Kuswanto LA, Sykes SM, Resnick‐Silverman L, et al. A rare DNA contact mutation in cancer confers p53 gain‐of‐function and tumor cell survival via TNFAIP8 induction. Mol Oncol. 2016;10:1207–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee MK, Teoh WW, Phang BH, Tong WM, Wang ZQ, Sabapathy K. Cell‐type, dose, and mutation‐type specificity dictate mutant p53 functions in vivo. Cancer Cell. 2012;22:751–64. [DOI] [PubMed] [Google Scholar]
- 8.Xu J, Reumers J, Couceiro JR, De Smet F, Gallardo R, Rudyak S, et al. Gain of function of mutant p53 by coaggregation with multiple tumor suppressors. Nat Chem Biol. 2011;7:285–95. [DOI] [PubMed] [Google Scholar]
- 9.Rahnamoun H, Lu H, Duttke SH, Benner C, Glass CK, Lauberth SM. Mutant p53 shapes the enhancer landscape of cancer cells in response to chronic immune signaling. Nat Commun. 2017;8:1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhu J, Sammons MA, Donahue G, Dou Z, Vedadi M, Getlik M, et al. Gain‐of‐function p53 mutants co‐opt chromatin pathways to drive cancer growth. Nature. 2015;525:206–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Escobar‐Hoyos LF, Penson A, Kannan R, Cho H, Pan CH, Singh RK, et al. Altered RNA splicing by mutant p53 activates oncogenic RAS signaling in pancreatic cancer. Cancer Cell. 2020;38:198–211.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Wang H, Liao P, Zeng SX, Lu H. It takes a team: a gain‐of‐function story of p53‐R249S. J Mol Cell Biol. 2019;11:277–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kotler E, Shani O, Goldfeld G, Lotan‐Pompan M, Tarcic O, Gershoni A, et al. A systematic p53 mutation library links differential functional impact to cancer mutation pattern and evolutionary conservation. Mol Cell. 2018;71:178–90.e8. [DOI] [PubMed] [Google Scholar]
- 14.Joerger AC, Ang HC, Fersht AR. Structural basis for understanding oncogenic p53 mutations and designing rescue drugs. Proc Natl Acad Sci U S A. 2006;103:15056–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Joerger AC, Fersht AR. The tumor suppressor p53: from structures to drug discovery. Cold Spring Harb Perspect Biol. 2010;2:a000919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liao P, Zeng SX, Zhou X, Chen T, Zhou F, Cao B, et al. Mutant p53 gains its function via c‐Myc activation upon CDK4 phosphorylation at serine 249 and consequent PIN1 binding. Mol Cell. 2017;68:1134–46.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fei Q, Shang K, Zhang J, Chuai S, Kong D, Zhou T, et al. Histone methyltransferase SETDB1 regulates liver cancer cell growth through methylation of p53. Nat Commun. 2015;6:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gouas DA, Shi H, Hautefeuille AH, Ortiz‐Cuaran SL, Legros PC, Szymanska KJ, et al. Effects of the TP53 p.R249S mutant on proliferation and clonogenic properties in human hepatocellular carcinoma cell lines: interaction with hepatitis B virus X protein. Carcinogenesis. 2010;31:1475–82. [DOI] [PubMed] [Google Scholar]
- 19.Llovet JM, Kelley RK, Villanueva A, Singal AG, Pikarsky E, Roayaie S, et al. Hepatocellular carcinoma. Nat Rev Disease Primers. 2021;7:1–28. [DOI] [PubMed] [Google Scholar]
- 20.Li X, Nadauld L, Ootani A, Corney DC, Pai RK, Gevaert O, et al. Oncogenic transformation of diverse gastrointestinal tissues in primary organoid culture. Nat Med. 2014;20:769–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Matano M, Date S, Shimokawa M, Takano A, Fujii M, Ohta Y, et al. Modeling colorectal cancer using CRISPR‐Cas9‐mediated engineering of human intestinal organoids. Nat Med. 2015;21:256–62. [DOI] [PubMed] [Google Scholar]
- 22.Clevers H. Modeling development and disease with organoids. Cell. 2016;165:1586–97. [DOI] [PubMed] [Google Scholar]
- 23.Bieging KT, Mello SS, Attardi LD. Unravelling mechanisms of p53‐mediated tumour suppression. Nat Rev Cancer. 2014;14:359–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wong AM, Ding X, Wong AM, Xu M, Zhang L, Leung HH, et al. Unique molecular characteristics of NAFLD‐associated liver cancer accentuate β‐catenin/TNFRSF19‐mediated immune evasion. J Hepatol. 2022;77:410–23. [DOI] [PubMed] [Google Scholar]
- 25.Artegiani B, van Voorthuijsen L, Lindeboom RGH, Seinstra D, Heo I, Tapia P, et al. Probing the tumor suppressor function of BAP1 in CRISPR‐engineered human liver organoids. Cell Stem Cell. 2019;24:927–43.e6. [DOI] [PubMed] [Google Scholar]
- 26.Broutier L, Mastrogiovanni G, Verstegen MM, Francies HE, Gavarro LM, Bradshaw CR, et al. Human primary liver cancer‐derived organoid cultures for disease modeling and drug screening. Nat Med. 2017;23:1424–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Eckerling A, Ricon‐Becker I, Sorski L, Sandbank E, Ben‐Eliyahu S. Stress and cancer: mechanisms, significance and future directions. Nat Rev Cancer. 2021;21:767–85. [DOI] [PubMed] [Google Scholar]
- 28.Lin Y, Jiang M, Chen W, Zhao T, Wei Y. Cancer and ER stress: mutual crosstalk between autophagy, oxidative stress and inflammatory response. Biomed Pharmacother. 2019;118:109249. [DOI] [PubMed] [Google Scholar]
- 29.Liu Y, Chen C, Xu Z, Scuoppo C, Rillahan CD, Gao J, et al. Deletions linked to TP53 loss drive cancer through p53‐independent mechanisms. Nature. 2016;531:471–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Giacomelli AO, Yang X, Lintner RE, McFarland JM, Duby M, Kim J, et al. Mutational processes shape the landscape of TP53 mutations in human cancer. Nat Genet. 2018;50:1381–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Nault JC, Ningarhari M, Rebouissou S, Zucman‐Rossi J. The role of telomeres and telomerase in cirrhosis and liver cancer. Nat Rev Gastroenterol Hepatol. 2019;16:544–58. [DOI] [PubMed] [Google Scholar]
- 32.Moon DO, Kang SH, Kim KC, Kim MO, Choi YH, Kim GY. Sulforaphane decreases viability and telomerase activity in hepatocellular carcinoma Hep3B cells through the reactive oxygen species‐dependent pathway. Cancer Lett. 2010;295:260–6. [DOI] [PubMed] [Google Scholar]
- 33.Opresko PL, Fan J, Danzy S, Wilson DM, Bohr VA. Oxidative damage in telomeric DNA disrupts recognition by TRF1 and TRF2. Nucleic Acids Res. 2005;33:1230–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Laurent‐Puig P, Legoix P, Bluteau O, Belghiti J, Franco D, Binot F, et al. Genetic alterations associated with hepatocellular carcinomas define distinct pathways of hepatocarcinogenesis. Gastroenterology. 2001;120:1763–73. [DOI] [PubMed] [Google Scholar]
- 35.Liu X, Green RM. Endoplasmic reticulum stress and liver diseases. Liver Res. 2019;3:55–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Reibe S, Febbraio MA. Relieving ER stress to target NASH‐driven hepatocellular carcinoma. Nat Rev Endocrinol. 2019;15:73–4. [DOI] [PubMed] [Google Scholar]
- 37.Dara L, Ji C, Kaplowitz N. The contribution of endoplasmic reticulum stress to liver diseases. Hepatology. 2011;53:1752–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Park W, Park MY, Song G, Lim W. Exposure to aflatoxin B1 attenuates cell viability and induces endoplasmic reticulum‐mediated cell death in a bovine mammary epithelial cell line (MAC‐T). Toxicol In Vitro. 2019;61:104591. [DOI] [PubMed] [Google Scholar]