

Summary
The implementation of next‐generation sequencing technologies has provided a sharp picture of the genetic variability in the components and regulators of the alternative pathway (AP) of the complement system and has revealed the association of many AP variants with different rare and common diseases. An important finding that has emerged from these analyses is that each of these complement‐related diseases associate with genetic variants altering specific aspects of the activation and regulation of the AP. These genotype–phenotype correlations have provided valuable insights into their pathogenic mechanisms with important diagnostic and therapeutic implications. While genetic variants in coding regions and structural variants are reasonably well characterized and occasionally have been instrumental to uncover unknown features of the complement proteins, data about complement expressed quantitative trait loci are still very limited. A crucial task for future studies will be to identify these quantitative variations and to determine their impact in the overall activity of the AP. This is fundamental as it is now clear that the consequences of genetic variants in the AP are additive and that susceptibility or resistance to disease is the result of specific combinations of genetic variants in different complement components and regulators (“complotypes”).
Keywords: complement, genetics, susceptibility to disease
1. BRIEF INTRODUCTION TO THE AMPLIFICATION NATURE OF THE AP
The complement system is a fundamental part of our innate immunity playing an essential role to fight pathogens and remove immune complexes and cell debris. Complement discriminates between self‐components and pathogens, tagging the latter for elimination by phagocytic cells or for direct destruction through cell lysis. Complement activates by three independent pathways, the classical (CP), the lectin (LP), and the alternative (AP). While activation through the CP and LP focuses C3b deposition at the location of the antigens and carbohydrates recognized by complement activating antibodies and lectins, initiation of the AP is based on the spontaneous (or protease‐mediated) activation of C3, which ends in the non‐specific deposition of C3b in all nearby surfaces. Binding of factor B (FB) to this surface‐bound C3b, and activation of the C3b‐bound FB by factor D (FD), results in the formation of surface‐bound unstable protease complexes, named AP C3‐convertase (C3bBb) that cleave C3 to generate C3b. This convertase‐generated C3b can form more AP C3‐convertase, providing the AP with the capacity to amplify exponentially. As a result, C3b and other C3‐activated molecules (iC3b and C3dg) cluster in high amounts around the surface‐bound C3‐convertase providing the ligands for the complement receptor (CR)‐mediated phagocytosis and, eventually, driving the activation of C5, which triggers inflammation and initiate the formation of the lytic membrane attack complex. 1 , 2
The lack of specificity and the amplification nature of the AP are both an advantage and a danger that requires strong regulation by a set of control proteins that collectively avoid damage to host cells and prevent the consumption of the AP components. These control mechanisms discriminate between host cells and pathogens so that the activity of complement in fighting microorganisms and removing cellular debris is not compromised. 3 , 4 , 5 , 6 , 7
In this review, I will describe the extension of AP genetic variability in normal and disease populations. I will discuss how genetic variants in components and regulators, determining differences in their activity and concentration, influence the overall activity of the AP, which results in either increased risk or protection from specific diseases. I will illustrate how the identification of disease‐associated genetic variants in AP components and, eventually, their structural and functional characterization has been instrumental to improve our molecular understanding of how AP dysregulation contributes to disease. I will describe the additive effect of AP genetic variants and how they impact on the disease risk. And, finally, I will comment on challenges involved in the identification and classification of the AP genetic variants.
2. GENETIC VARIABILITY IN GENES ENCODING AP PROTEINS IN NORMAL POPULATIONS
In recent years, the application of next‐generation sequencing (NGS) techniques to the exome sequencing of tens of thousands of normal individuals belonging to different ethnic groups has provided invaluable information of the genetic variability in all genes of the human genome. Focusing on complement, this information reveals that variability in the genes encoding proteins of the AP in the normal population, as determined by population genetic resources such as the Genome Aggregation Database (gnomAD), 8 is considerable. Table 1 summarizes this variability in the components and regulators of the AP from a total of 141 458 individuals, included in gnomAD by Jan 5, 2021. To highlight the magnitude of the numbers, the table includes only variants located in or within five base pairs of a coding exon and excludes synonymous variants. Taking factor H (FH) as an example, these data show that whilst FH presents only six variants with a minor allele frequency (MAF) >1% (common polymorphisms), there are as many as 649 variants with MAF <1% (rare variants) carried by 11 368 individuals, that is, 8% of all individuals. Similar calculations result in 3198 individuals in the database (2.3%) carrying a FH variant with a MAF <0.1%. These are significant numbers.
TABLE 1.
AP genetic variants in normal populations (n = 141 456)
| Protein | Gene location b | Size | Nonsense variants | Missense variants (Pathogenic) a | Carriers of pathogenic variants (%) | |||
|---|---|---|---|---|---|---|---|---|
| MAF > 0.01 | MAF < 0.01 | MAF > 0.01 | MAF < 0.01 | MAF < 0.01 | MAF < 0.001 | |||
| C3 | Chr 19; NC_000019.10 (6 677 704..6720650) | 1663aa | 0 | 32 | 2 (0) | 773 (503) | 2.14 | 1.49 |
| FB | Chr 6; NC_000006.12 (31 946 095..31952084) | 764aa | 0 | 31 | 6 (0) | 363 (302) | 2.63 | 0.80 |
| FI | Chr 4; NC_000004.12 (109 730 982..109801999) | 583aa | 0 | 48 | 2 (0) | 319 (199) | 1.57 | 1.06 |
| FD | Chr 19; NC_000019.10 (859 664..863641) | 246aa | 0 | 22 | 1 (0) | 213 (174) | 1.34 | 0.56 |
| P | Chr X; NC_000023.11 (47 623 282..47630305) | 469aa | 0 | 6 | 0 | 125 (69) | 0.55 | 0.16 |
| FH | Chr 1; NC_000001.11 (196 652 043..196747504) | 1231aa | 0 | 26 | 6 (0) | 623 (306) | 1.79 | 0.80 |
| MCP | Chr 1; NC_000001.11 (207 752 038..207795516) | 384aa | 0 | 30 | 1 (0) | 193 (71) | 0.37 | 0.37 |
| DAF | Chr 1; NC_000001.11 (207 321 678..207360966) | 381aa | 0 | 32 | 0 | 192 (85) | 0.48 | 0.48 |
| CR1 | Chr 1; NC_000001.11 (207 496 157..207641765) | 2039aa c | 0 | 117 | 14 (0) | 992 (559) | 5.17 | 2.76 |
| FHR‐1 | Chr 1; NC_000001.11 (196 819 731..196832189) | 330aa | 0 | 37 | 3 (0) | 204 (105) | 0.85 | 0.47 |
| FHR‐2 | Chr 1; NC_000001.11 (196 943 738..196959622) | 243aa | 0 | 27 | 2 (0) | 181 (76) | 2.62 | 0.85 |
| FHR‐3 | Chr 1; NC_000001.11 (196 774 840..196795407) | 331aa | 0 | 37 | 1 (0) | 186 (60) | 2.05 | 0.24 |
| FHR‐4 | Chr 1; NC_000001.11 (196 888 052..196918633) | 331aa d | 0 | 69 | 3 (0) | 376 (163) | 2.83 | 0.90 |
| FHR‐5 | Chr 1; NC_000001.11 (196 975 034..197009678) | 569aa | 0 | 83 | 3 (0) | 389 (162) | 5.74 | 1.18 |
Variants were considered pathogenic when their CADD PHRED C‐score > 15.
Genomic location in GRCh38.p14 Assembly.
Allotype CR1*1.
FHR‐4B.
The numbers that result from the classification of these FH variants as pathogenic or benign based on prediction algorithms like the Combined Annotation Dependent Depletion (CADD) method (https://cadd.gs.washington.edu/info) 9 , 10 are equally important. For FH, 47.1% and 47.5% of the variants with MAF < 1% and MAF < 0.1%, respectively, are predicted as pathogenic (CADD PHRED C‐score >15). This translates into 1.79% and 0.8% of individuals in the normal population being heterozygote for a potential pathogenic variant in FH with MAF < 1% and MAF < 0.1%, respectively (Table 1). As similar results are obtained for the other AP genes, we can summarize by saying that genetic variability in the AP is such that approximately 12% of people in the normal populations are heterozygous for a potential pathogenic variant with MAF < 0.1% in at least one AP component or regulator. These elevated figures may look paradoxical considering that most diseases associated with the AP genes variants are very rare diseases. However, as I will describe in this review, the rarity of these diseases is mainly determined by their association with particular genetic variants causing specific functional alterations in a complement protein, by the concurrence of a particular set of genetic variations in different complement genes and, also important, by the fact that disease‐associated AP variants are predisposition factors, which implies that the occurrence of the disease in carriers of these pathogenic variants requires non‐genetic or environmental triggers.
This picture of genetic variability in AP genes in normal populations is nevertheless incomplete because it relates only to exome sequencing and it does not include the intronic and intergenic regions where we know there are sequence variations that influence the expression of the complement proteins. These expression quantitative trait loci (eQTL) added to those affecting the activity of the AP proteins shape both the overall activity of the AP and the predisposition to complement‐related diseases.
3. RARE GENETIC VARIANTS IN THE AP AND PREDISPOSITION TO DISEASE
We have known for long time that genetic variants in the AP proteins and regulators associate with predisposition to a long list of different diseases, but it is only during the last two decades that we have started to unravel the peculiarities of these associations. These studies have identified diseases that associate with genetic variants altering specific aspects of the activation and regulation of the AP, which has provided valuable insights into the pathogenic mechanisms underlying those pathologies and occasionally they have also revealed unknown features of the complement proteins involved.
3.1. Rare loss‐of‐function mutations in complement regulators of the AP
Factor H, the key regulator of the AP, controls complement activation in the fluid phase and on cellular surfaces using distinct functional domains. FH is an abundant plasma glycoprotein composed of 20 short consensus repeats (SCR) of the complement control protein repeat (CCPR) type. The N‐terminal region (SCR1‐4) binds C3b and serves as a cofactor for Factor I (FI)‐mediated cleavage of C3b. It also accelerates the decay of the AP C3‐convertase (C3bBb). The C‐terminal region (SCR19‐20) carries separate binding sites for the thio‐ester containing (TED) domain of C3b and sialic acids, which enable FH to sense and restrain deposition of C3b in host tissues (Figure 1). 11 , 12 , 13 , 14 The mid‐region of FH (SCR5‐18) contains additional polyanion binding sites. However, their functional significance and contribution to complement regulation remain unclear. The current view is that this mid‐region plays mainly a structural role by enabling FH to bend and bind simultaneously to different sites on C3b. 11 , 13 , 15 , 16 , 17
FIGURE 1.
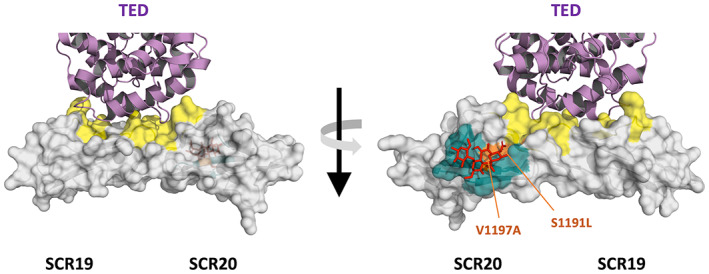
The C‐terminal region of FH. Structure of the last two SCRs of FH to illustrate the large surface area involved in the binding sites for the TED domain of C3 (in yellow) and sialic acids (in green). The TED domain (in purple) and a sialic acid molecule (in red) are depicted interacting with their respective binding sites. Because these binding sites play a crucial role in the regulation of complement activation by FH in host surfaces, the region cluster a multitude of pathogenic variants associated with aHUS, including S1191L and V1197A, the two prototypical variants that disrupt the sialic acid binding site (in orange)
Hundreds of potentially pathogenic rare FH genetic variants, most with MAF < 10−4, are present in heterozygosis in normal populations. They are distributed through the whole length of the CFH gene and include both missense and non‐sense variants. Cohorts of patients with diseases like aHUS (atypical hemolytic uremic syndrome), C3G (C3‐glomerulopathy), and AMD (age‐related macular degeneration) are enriched in these rare genetic FH variants, but the type of genetic variant and their distribution in the CFH gene varies among the different pathologies. Missense genetic variants in the C‐terminal region of FH, for example, are prototypical of aHUS. 18 , 19 , 20 , 21 Several years ago, the discovery of this association changed our understanding of the pathogenesis of aHUS revealing that this condition was not a consequence of the hypocomplementemia that characterizes many of these patients, but rather it was caused by the complement‐mediated damage to the microvascular endothelium due to a failure to regulate complement activation in host surfaces. This was a decisive paradigm shift that prompted the use of anticomplement drugs to treat the disease. 22 , 23 Further studies with these FH C‐terminal variants have also provided important structural and functional information regarding how FH interacts with the thioester domain (TED) of C3b 14 , 24 , 25 and with sialic acids 13 , 26 in cell surfaces.
In contrast with the particular association of the FH C‐terminal variants with aHUS, CFH variants that impair expression of FH or eliminate the complement regulatory functions in the N‐terminal region associate with a broad spectrum of pathologies like C3G, aHUS, AMD, and IgAN (IgA nephropathy). These pathologies share a common link to complement dysregulation, but the causes that trigger the complement dysregulation and where it occurs are different among them. Later in this review, I will discuss that the final pathological outcome in a heterozygote carrier of these CFH genetic variants is impinged by the concurrence with other complement genetic variants and with environmental factors (see Section 6).
Importantly, in homozygosis or compound heterozygosis, these CFH variants that impair expression of FH or eliminate the complement regulatory functions of FH result in the complete consumption of C3 in plasma and the generation of massive amounts of activated C3 products that deposit in the kidney glomeruli, causing prototypically dense deposit disease, a rare form of C3G characterized by strong electron‐dense deposits within the glomerular basement membrane. 27 , 28 In addition, because homozygosity for these FH variants causes a secondary C3 deficiency that severely impairs opsonophagocytosis, the individuals affected by this condition are also predisposed to severe infections, particularly by encapsulated bacteria.
Genetic variants in membrane cofactor protein (MCP; CD46) that decrease the expression of this protein in the cell surfaces or reduce its regulatory activity behave like the FH C‐terminal variants described above; they do not impact significantly fluid phase complement regulation but impair the protection of self surfaces from complement damage. This is consistent with the crucial role of MCP as a cofactor in the FI‐mediated inactivation of C3b and C4b deposited on host cells. In fact, the majority of pathogenic MCP variants described to date have been found in heterozygosis during the genetic screening of aHUS patients, 29 , 30 although they are not exclusive of this disease. 31 Homozygotes (or compound heterozygotes) for MCP pathogenic variants are extremely rare. They associate with unusually severe aHUS presentations and with common variable Immunodeficiency. 32
Decay accelerating factor (DAF; CD55) regulates C3 and C5 cleavage by accelerating the decay of the AP C3/C5 convertases. DAF is a glycosylphosphatidylinositol (GPI)‐anchored membrane protein, a biochemical peculiarity that among complement proteins is only shared with CD59. The most common genetic cause associated with alterations of these two (GPI)‐anchored complement regulators are somatic mutations of the PIGA gene, which encodes a protein essential for the synthesis of the GPI anchor. When this happens in a clonal hematopoietic stem cell, it results in paroxysmal nocturnal hemoglobinuria, a rare disease that presents with hemolytic anemia, thrombosis and, eventually, bone marrow failure. 33 Recently, several homozygote carriers of non‐sense or pathogenic missense variants in the DAF gene were identified by whole‐exome sequencing of a cohort of patients with a very rare early‐onset protein‐losing enteropathy. 34 This was unexpected because individuals with a congenital DAF deficiency were known for a long time (they are the very rare Inab phenotype of the Cromer blood group 35 ), but no pathological consequence was associated with this phenotype. Consistent with the complement regulatory activities of DAF, the congenital DAF deficiencies result in complement over‐activation on cell surfaces. However, how complement dysregulation distinctively causes damage to the intestinal tissue, resulting in the protein loss enteropathy, is still unclear.
Factor I (FI) is crucial to regulate the activation of both the classical and the AP. In the presence of appropriate cofactors, it inactivates C3b and C4b. Most CFI variants described in the literature are heterozygous rare variants, mainly associated with AMD, aHUS and, occasionally, with different forms of C3G. 36 , 37 , 38 , 39 The majority are missense variants resulting in reduced plasma FI levels, but loss of functional activity has also been demonstrated for a few FI variants that express normally in plasma. 40 , 41 Heterozygotes for these pathogenic FI variants have impaired regulation of C3b which results in excessive AP activation and they normally develop diseases characterized by chronic inflammation or acute complement‐mediated tissue damage. Like carriers of partial FH deficiencies, the final pathological outcome in heterozygote carriers of CFI variants is strongly contingent on the associated genetic background. 42 Like FH, the complete FI deficiency, resulting from homozygous or compound heterozygous variants in the CFI gene, causes secondary C3 deficiencies that normally associate with recurrent infections with encapsulated microorganisms, but since C3b cannot be proteolyzed to generate iC3b and C3dg, these individuals elude the inflammation and tissue damage that characterize the FH deficiency. 28 , 43
3.2. Rare gain‐of‐function mutations in the components of the AP convertase
Activation of C3 into C3b causes huge displacement of the TED domain that exposes the reactive thioester to nucleophilic reagents and generates a new surface area in C3b containing the binding sites for FB that mediate formation of the AP pro‐convertase C3bB. Binding of FB to C3b also results in a large conformational change in FB that exposes a site that is cleaved by FD releasing the Ba fragment and yielding the active AP C3‐convertase C3bBb. 44 , 45 Modulation of the activity of the C3bBb convertase, either prolonging its half‐life on pathogen surfaces where activation must proceed (by properdin), or accelerating its spontaneous decay and inactivating C3b on host surfaces to avoid inflammation and tissue damage (by FH, MCP, DAF, CR1, and FI), is critical for the correct functioning of the AP.
Complete deficiencies of C3, FB, FD and properdin caused by homozygote (or compound heterozygote) pathogenic variants in these genes are very rare and associate with recurrent infections. 46 , 47 , 48 More interesting are, however, the genetic variants in the C3 and CFB genes that increase the functional activity of AP. These gain‐of‐function (GoF) variants have been described in association with aHUS, C3G, and AMD. 36 , 49 , 50 , 51 , 52 The functional characterization of some of the GoF variants in FB (i.e., D279 G, F286L, K323E, and K350N) has shown that they enhance formation of the C3bBb convertase or increase its resistance to accelerated decay by complement regulators (Figure 2). 49 , 51 Similarly, it has been shown that GoF variants in C3 alter the sensitivity of C3b to inactivation by FH, CR1 and MCP, and confer the AP C3 convertase resistance to accelerated decay by FH and DAF. 15 , 36 , 50 , 52 , 53 , 54 Both C3 and FB GoF variants cause hyper‐activation of the AP, which results in consumption of C3 and FB, increasing complement‐mediated inflammation and tissue damage. An important finding was that the C3 GoF variants associated with C3G affects regulation by FH and CR1 (fluid phase regulation), while the C3 GoF variants associated with aHUS affect primarily the inactivation of C3b by MCP (cell surface regulation), 54 , 55 which again illustrates that the pathogenic mechanisms of a particular disease implicates specific aspects of the activation and regulation of the AP (Figure 3). The key contribution of these GoF variants to the disease phenotype is further illustrated by the remarkably reproducible and characteristic presentation of aHUS in carriers of two C3 GoF variants (R161W and I1147T) that are relatively prevalent in Europe and Japan. 56 , 57 , 58 In a different context, the C3‐923delDG variant associated with C3G is also remarkable. It is a deletion of two amino acids (Asp923, Gly924) in the MG7 domain of C3 that makes the corresponding C3b and C3bBb convertase resistant to inactivation by FH (and CR1). Paradoxically C3‐923delDG renders C3 resistant to cleavage by the AP C3 convertase and has been crucial to identify a region in the surface of the MG7 domain that is very likely a contact surface between the C3 substrate and the C3b molecule in the AP C3 convertase and may represent a therapeutic target for inhibition of C3 activation (Figure 3). 52
FIGURE 2.

Gain‐of‐function variants in CFB. A, Crystal structure of the C3 convertase showing two representative gain‐of‐function FB variants, D279G, and K323E. The magnesium ion in the MIDAS site is represented as a yellow dot. B, Biacore analysis of the FB‐279G and FB‐323E variants. Recombinant variants and normal FB were flown with native FD over a C3b‐coated chip to form the convertase. FB‐323E convertase (orange line) formed normally and decayed with a half‐life comparable to that formed by normal FB (black line). However, whereas the normal convertase was rapidly and completely dissociated by DAF, the FB‐323E convertase was resistant to accelerated decay. The FB‐279G convertase (red line) was formed at abnormally high levels and was very stable. Like the FB‐323E convertase, the FB‐279G convertase was resistant to accelerated decay (Modified from 154 )
FIGURE 3.

Gain‐of‐function variants in C3. A, C3 variants are indicated in the crystal structure of the C3b molecule to illustrate that aHUS‐associated variants (black circles) and C3G‐associated variants (red circles) are allocated in separate regions of the C3b molecule. 52 , 53 , 155 C3b structural domains are depicted in colors. The common variant R102G (yellow circle) is located at the interface between the MG1 and TED domains. C3 variants D1115N (associated with aHUS) and C3‐923delDG (associated with C3G) are highlighted with an asterisk to indicate that they have been introduced in mice and replicate the aHUS and C3G phenotypes, respectively. 156 , 157 B, Functional analysis of the C3G‐associated C3‐923delDG and aHUS‐associated C3‐I1157T variants. SDS‐PAGE gels illustrate their resistance to FI‐mediated proteolysis using FH (C3‐923delDG) or MCP (C3‐I1157T) as a cofactor (indicated with red arrows). Experimental details are described in Martinez‐Barricarte et al. 52 , 53 Below, a model of the interaction of FH and MCP with C3b provides a structural rationalization of the differential regulation by FH and MCP. 158 , 159 C, Top view of the hypothetical complex between the C3‐convertase (C3bBb) 154 and the C3 substrate (left) with the position of the C3‐923delDG variant indicated
3.3. Rare genetic variants in the FH‐related proteins (FHRs)
Several non‐sense genetic variants the CFHR1‐5 genes, leading to FHRs deficiencies are present in normal populations (gnomAD) with MAF ranging between 10−2 and 10−4. These relative high frequencies are consistent with the high prevalence of normal individuals carrying genomic deletions of the CFHR1, CFHR3, and CFHR4 genes (see below) and suggest that loss‐of‐function variants in the FHRs may have no major pathological consequences. In contrast, rare variants in the FHRs with features of GoF variants are clearly pathogenic and illustrate additional genotype–phenotype correlations. This is the case of genetic variants in the C‐terminal region of FHR‐1 that provide it with the capacity to bind sialic acids. These variants associate specifically with aHUS because their pathological consequences are equivalent to those of the FH C‐terminal pathogenic variant. 26 , 59 , 60 FHR‐1 and FH have virtually identical C‐terminal regions, with only two amino acid differences between them (Leu290 and Ala296 in FHR‐1 are Ser1181 and Val1187 in FH). These two amino acid substitutions suffice to eliminate from FHR‐1 the capacity to bind sialic acids, which prevents that FHR‐1 hampers the complement regulatory role of FH in host surfaces. 26 aHUS‐associated variants CFHR1 Leu290Ser,Ala296Val and CFRH1 Leu290Val are pathogenic because they restore in FHR‐1 the capacity to bind sialic acids, making FHR‐1 an strong competitor of FH for binding to surface‐bound C3b and dysregulating the AP in host tissues. 26 Potential pathogenic GoF variants in FHR‐5 with increased binding to C3b and other ligands have also been described associated with aHUS, but detailed functional studies are still pending. 61
Another interesting association is that of the FHR‐1, FHR‐2, and FHR‐5 variants carrying a duplication of the N‐terminal dimerization domain with C3G. 62 , 63 , 64 , 65 , 66 , 67 , 68 The classic example is an FHR‐5 protein encoded by a CFHR5 gene with an internal duplication resulting in a duplication of SCR1 and SCR2 (FHR‐5[1–2]‐FHR‐5) that was identified in several Greek Cypriot patients with C3GN and a common ancestry (often called CFHR5 nephropathy). 62 Other FHR proteins with duplicated dimerization domains have been identified associated with C3G in small families and include: FHR‐2(1–2)‐FHR‐5, 65 FHR‐5(1–2)‐FHR‐2, 68 FHR‐1(1–3)‐FHR‐5, 67 FHR‐1(1–4)‐FHR‐1, 63 and FHR‐1(1–2)‐FHR‐1. 64 Different studies have tried to explain why these peculiar FHR variants associate with C3G. It was initially though that by forming multimeric complexes these variants would outcompete binding of FH to surface‐bound C3b (FH de‐regulation), promoting complement activation. 63 However, a competition with FH would justify better an association with aHUS than with C3G. 26 Notably, recent data generated for the FHR‐1(1–2)‐FHR‐1 variant suggest these FHRs variants dysregulate complement at C3‐opsonized surfaces by promoting complement activation and further deposition of C3‐activated fragments without interfering the binding of FH to C3b. 64 In summary, variants that confer to FHR‐1 the capacity to bind sialic acids dysregulate complement at endothelial surfaces and result in aHUS, whilst FHR mutant proteins with duplicated dimerization domains exacerbate complement activation at C3 opsonized surfaces and cause C3G. These ideas about the pathogenicity of the FHR proteins with duplicated dimerization domains may also justify the protection conferred by the deletion of the CFHR3 and CFHR1 genes (Δ CFHR3‐CFHR1 ) observed for some complement‐related diseases and entail the requisite of a previous situation generating the initial C3 deposition.
In a different context, these C‐terminal and N‐terminal FHRs variants are also interesting because they originate by complex genomic rearrangements involving non‐homologous recombination and gene conversion events. These events are relatively common in the CFH‐CFHRs genomic region due to the presence of large segmental duplications 18 , 69 and are the cause of several other CFHR1‐5 structural variants that have been found in the genetic screening of patients with complement‐related diseases. The contribution of many these FHRs structural variants to the pathogenic mechanisms is still uncertain, mainly because we lack the necessary understanding of the physiological role of these proteins to perform proper functional assays and also because their identification requires special techniques and they are poorly represented in the available genetic databases of normal populations, which are generally limited to exome sequencing. As an example of the diversity of these CFHR1‐5 structural variants, Figure 4 summarizes those that we have identified in our aHUS (n = 1151) and C3G (n = 373) cohorts.
FIGURE 4.

Structural variants in the CFH‐CFHRs genomic region in the aHUS and C3G Spanish cohorts. Genomic organization of the CFH and CFHR1‐5 genes is shown at the top with exons depicted with vertical lines. The segmental duplications in this region are shown with colored boxes. Below, the structural variants are described with lines spanning the genomic region involved. Red color refers to genomic deletions and green color to duplications. Gene conversion events are shown in blue. A notation for each structural variant is shown on the left indicating the genes and exons involved. On the right are indicated the allele frequencies and the cohort in which they have been found. Pathogenic structural variants are highlighted with an asterisk
The most common structural variant described in the CFH‐CFHRs gene family is the 84 kb deletion of CFHR3 and CFHR1 (Δ CFHR3‐CFHR1 ), which has an allele frequency ranging from 2% to 51%, depending on ethnicity. 70 Δ CFHR3‐CFHR1 strongly associates with protection from AMD, IgAN, and C3G but confers risk to systemic lupus erythematosus. 64 , 71 , 72 , 73 In fact, it has been shown that the prevalence of these diseases in human populations correlates well with the allele frequencies of the Δ CFHR3‐CFHR1 polymorphism. 70 The reason for these associations is still unclear, although, as discussed above, some interesting hypotheses are emerging. Δ CFHR3‐CFHR1 is not a risk factor for aHUS, 74 , 75 as it was previously reported. 76 In homozygosis, however, is a relevant finding because it is strongly associated with the presence of auto‐antibodies against the C‐terminal region of FH, the most important acquired factor associated with the development of aHUS in children; 3%–15% in European cohorts 77 , 78 and as much as 56% of aHUS patients in India 79 have such auto‐antibodies. Δ CFHR1‐CFHR4 (deletion of CFHR1 and CFHR4) is also found with relatively high frequency in genetic screenings, but no associations with disease have been reported for this deletion. Like Δ CFHR3‐CFHR1 , finding Δ CFHR1‐CFHR4 in homozygosis or in heterozygosis with Δ CFHR3‐CFHR1 in aHUS patients should prompt the search for anti‐FH auto‐antibodies. 74
Δ CFHR3‐CFHR1 and Δ CFHR1‐CFHR4 generate by non‐homologous recombination events involving intergenic regions included in these large segmental duplications and therefore are “clean” gene deletions. Interestingly, a displacement of these non‐homologous recombination events few kb into the upstream genes, generate similar deletions but in addition creates either a CFH::CFHR1 hybrid gene or a CFHR3::CFHR4 hybrid gene. The CFH::CFHR1 hybrid gene encodes a FH protein in which the C‐terminal region has been replaced for that of FHR‐1 and it is strongly associated with aHUS 80 , 81 because this replacement eliminates the functionality of the C‐terminal region of FH. In contrast, the CFHR3::CFHR4 hybrid gene, encoding a protein in which the C‐terminal region of FHR‐3 has been replaced by that of FHR‐4, has been found in patients with aHUS, C3G, IgAN and other complement‐related diseases, but its pathogenicity is uncertain.
Although much rarer, tandem duplications of the CFHR3‐CFHR1 genes, of the CFHR1‐CFHR4 genes and duplications of just CFHR4, are also found and are interesting because increased levels of these FHRs have been reported to be risk factors for IgAN, C3G, and AMD. 64 , 82 Even rarer are a plethora of structural variants involving internal duplications and deletions in the CFHR1, CFHR3, CFHR4, and CFHR5 genes, the functional significance of which, with a few exceptions like the duplications of the FHR‐1 dimerization domain, have not been determined. Gene conversion events are also an important source of structural variants in the CFH‐CFHRs region. The variants CFH Ser1181Leu,Val1187Ile and CFHR1 Leu290Ser,Ala296Val , already discussed in this review, have generated by gene conversion events and are strongly associated with aHUS. 59 , 80
3.4. Rare genetic variants in complement receptors (CRs)
There are five distinct CRs for C3b, its degradation products iC3b, C3dg, and C3d, and for the anaphylatoxin C3a, which are called CR1 (CD35), CR2 (CD21), CR3 (CD11b/18), CR4 (CD11c/18), and C3aR. CR1 (CD35) is the cell surface receptor for the C3b fragment and in erythrocytes plays important roles in immune complex transport and phagocytosis of complement‐opsonized particles. 83 CR1 also regulates complement activation by acting as a cofactor for FI in the inactivation of surface‐bound C3b and generation of iC3b and C3dg, the ligands for CR2, CR3, and CR4. Reduced levels of CRs CR1 and CR2 due to genetic and acquired factors are associated with autoimmune disorders, infections, and other diseases (see Section 5.2). CR1 comprises a long chain of SCRs of the CCPR type arranged in long homologous repeats (LHR; each containing seven SCRs) and shows a peculiar polymorphism with four structural variants of different size (A, B, C, and D), composed of 3, 4, 5, and 6 LHRs, 9 which are also expressed at different levels on the surface of the erythrocytes. 84 , 85 Complete deficiencies of CR1 proteins have not been reported in humans and the first case of a genetic CR2 deficiency was described in 2012. 86 The patient, a 28‐year‐old man, was a compound heterozygote for deleterious mutations in CR2 and presented with recurrent infections, reduced class‐switched memory B cells, and hypogammaglobulinemia. CR3 (CD11b/CD18) binds iC3b and is found on macrophages, neutrophils, and large granular lymphocytes. CR4 (CD11c/CD18) binds iC3b and is found on neutrophils, monocytes, and macrophages. These two receptor proteins and LFA‐1 (CD11a/CD18), a very important receptor in cellular adhesion and trafficking, are known as β2 integrins. Complete deficiency of CR3 and CR4 are associated with the leukocyte adhesion deficiency type 1 (LAD‐1) syndrome, 87 a rare disease affecting one in 1 million individuals were there is a failure to synthesize CD18. No C3aR receptor deficiencies have been described in humans.
4. HAPLOTYPES IN THE RCA GENE CLUSTER
The Regulators of Complement Activation (RCA) gen cluster (Figure 5) spans 12 Mb of DNA and includes 16 complement genes. All the complement genes are in tandem within two gene groups, a telomeric 707 kb‐long DNA segment which contains the C4BPB, C4BPA, C4BPAL1, C4BPAL2, DAF(CD55), CR2(CD21), CR1(CD35), MCPL1, CR1L1, and MCP(CD46) genes and a centromeric 358 kb‐long DNA segment that contains CFH, CFHR1, CFHR2, CFHR3, CFHR4, and CFHR5. 18 , 88 These two gene groups are separated by 10. 3 Mb of DNA that contains genes that are not complement‐related and that have very diverse functions. Several common variants have been described in each of the genes included in both regions of the RCA gene cluster, but there is very strong linkage disequilibrium (LD) in region, which reduces genetic variability to a few combinations of variants that are inherited together. These combinations (haplotypes) have been described at the CFH‐CFHRs and the CR1/MCP gene regions of the RCA gene cluster and, in both regions, they show important associations with disease. 59 , 60 , 63 , 71 , 72 , 81 , 89 , 90 These RCA haplotypes may include eQTL, functional polymorphisms, and structural variants, which makes it difficult to pinpoint the variations that are ultimately responsible for the disease association. In fact, it cannot be completely excluded that the combination of different variants at various genes that characterizes a particular haplotype is relevant for the association with disease.
FIGURE 5.

The RCA gene cluster; CFH‐CFHRs haplotypes and disease associations. Genomic organization of the RCA gene cluster with a zoom on the CFH‐CFHR1‐5 gene region to illustrate the SNP composition of the main four CFH‐CFHRs haplotypes and their association with disease. aHUS, atypical Hemolytic uremic syndrome; AMD, Age‐related Macular degeneration; C3G, C3‐glomerulopathy; MD, Meningococcal Disease; SLE, Systemic Lupus Erythematosus; IgAN, IgA Nephropathy. (1) rs1061170C corresponds to FH‐402His and it confers risk for AMD. 91 (2) rs61818925G associates with increased FHR‐4 levels, which confers risk for AMD. 82 , 125 (3) rs800292A corresponds to FH‐62Ile and it is protective for AMD. 91 (4) rs1410996A associates with decreased FHR‐4 levels and it is protective for AMD. 97 (5) rs6677604A is a proxi of Δ CFHR3‐CFHR1 and it is protective for AMD, IgAN, and C3G and confers risk for SLE. 64 , 71 , 72 , 73 (6) “‐” indicates Δ CFHR3‐CFHR1 . (7) In homozygosis strongly associates with anti‐FH autoantibodies. 160 (8) rs570618, a proxi of rs1061170C, also associates with increased levels of FHR‐1, ‐2, ‐3 and ‐4. 82 , 129 , 130 (9) rs10922109A, a proxi of rs1061170C, also associates with increased levels of FHR‐1, ‐2, ‐3 and ‐4 82 , 129 , 130
4.1. The CFH‐CFHRs haplotypes
Four haplotypes, CFH‐H1, CFH‐H2, CFH‐H3, and CFH‐H4, explain more than 90% of the genetic variability at the CFH gene region and extend also into the CFHRs gene region (Figure 5). The CFH‐H1 haplotype carries the Tyr402His (rs1061170) polymorphism in SCR7 of FH and is strongly associated with the development of AMD. 91 Structural and functional studies have tried to understand the mechanism by which the 402His allele may impact AMD risk. These studies have shown that amino acid 402His is directly involved in a GAG binding site spanning SCR6‐8 of FH 92 suggesting that switching between histidine and tyrosine at this position may alter the ligand specificity resulting in failure to recruit FH to sites in the retina where complement is activated by the accumulation of endogenous compounds such as C‐reactive protein, heparan sulfates, or malondialdehyde. 92 , 93 , 94 Whilst these ideas are suggestive, it has been recently shown that the AMD‐associated CFH‐H1 haplotype extend into the CFHR4 locus and includes one or more eQTL that influences the expression levels of the FHR‐4 protein. 82 Elevated levels of FHR‐4 in the retina may overcome the regulatory activity of FH, both as a result of increased promotion of complement activation and excessive competition with FH for binding to surface‐bound C3b. Although these studies point to levels of FHR‐4 as the leading variation driving the predisposition associated with the CFH‐H1 haplotype, it remains to be determined whether a reduced activity by the FH‐402His allele is important in this context.
The CFH‐H2 haplotype is characterized by the presence of the common FH variant Val62Ile in the N‐terminal region of FH (SCR1) (rs800292). The FH‐62Ile allele was originally reported to be protective for AMD 91 and later for aHUS and other diseases. 95 The functional impact of the FH‐62Ile variant is subtle (20%–50% enhanced regulatory activities compared with FH‐62Val), 96 but the amplification nature of the complement system and the combination of this FH variant with other variants in complement components and regulators will amplify this small effect resulting in significant differences in complement activity that justify its association with disease (see Section 5.1). The CFH‐H2 haplotype includes also an eQTL (rs1410996) that associates with reduced levels of FHR‐4, 97 which again suggest that the protection effect reported for this haplotype may be the sum of distinct variants at different genes.
The CFH‐H3 haplotype, originally described as a combination of CFH SNPs that confer increased risk to aHUS, 98 was later extended to include polymorphisms in the CFHR3 and CFHR1 downstream genes. 74 This extended CFH‐H3 haplotype strongly associates with risk for aHUS and carries the rs426736 variant that confers protection against meningococcal disease (MD). 99 Analysis of plasma levels of FH and FHR‐3 proteins in carriers of this extended haplotype showed slightly reduced levels of FH and twofold elevated levels of FHR‐3 compared with non‐carriers, 100 , 101 which may explain the opposite impact of this haplotype in aHUS and MD. N meningitidis recruits FH via the surface lipoprotein fHbp 102 , 103 and it has been shown that FHR‐3 competes with FH for binding to fHbp on the bacterial surface, influencing its survival in plasma. 104 Since the ability of N meningitidis to evade the host complement system is determined by the relative levels of FH and FHR‐3 on the bacterial surface, the concurrence of decreased FH levels and increased FHR‐3 plasma levels may explain the protective effect of the CFH‐H3 haplotype against MD. 101 In contrast, the elevated levels of FHR‐3 and slightly decreased FH plasma levels contributed by CFH‐H3 haplotype likely exacerbates complement dysregulation on the renal endothelial surface in carriers of other pathogenic variants, rationalizing why this haplotype increases risk of aHUS. Identification of the CFH‐H3 haplotype has diagnostic value and it is used to explain the incomplete penetrance of aHUS among carriers of rare complement pathogenic variants. 59 , 105
A common polymorphism in the CFH intron 12 (rs6677604) distinguishes haplotype CFH‐H4 and is in strong LD with Δ CFHR3‐CFHR1 . This very common polymorphism has several implications in complement‐related diseases as indicated previously in Section 3.3. Interestingly, approximately 15% of the individuals with Δ CFHR3‐CFHR1 carry that protective variant together with the also protective FH‐62Ile variant in a fifth (CFH‐H5) haplotype that has a frequency of 2% in the Spanish population.
4.2. The MCP haplotype
The strong LD in the human MCP/CD46 gene region also reduces the genetic variability within this region to a couple of SNP haplotype blocks. One of these, the MCP ggaac haplotype, is an important risk factor for aHUS, particularly in concurrence with pathogenic GoF variants in C3 or CFB. 49 , 53 , 106 The functional analysis of two SNPs included in this haplotype that are located in the CD46 promoter region demonstrated a reduced transcriptional activity compared to the prevalent MCP aaggt haplotype, which suggest MCP ggaac may associate with slightly decreased levels of CD46 in the endothelial cell surfaces. 106 In this respect, the observation that the severity and penetrance of aHUS in carriers of C3 and CFB GoF variants is influenced by the presence of the MCP aaggt risk polymorphism 49 , 53 , 58 suggest that MCP aaggt may contribute to bring the complement dysregulation caused by these variants to the endothelial cell surface. Recently, we reported the case of a carrier of a GoF CFB variant with a particularly severe presentation of aHUS and an adverse family history of disease recurrence after kidney transplantation, who had a remarkably successful isolated kidney transplant without anticomplement prophylaxis from a donor negative for the MCP ggaac risk haplotype. 107 Although the evidence is anecdotal, it may be worth exploring the idea that eluding the MCP ggaac risk haplotype may prevent the recurrence of aHUS after kidney transplantation in carriers of CFB and C3 GoF variants.
5. FUNCTIONAL POLYMORPHISMS AND EXPRESSION QUANTITATIVE TRAIT LOCI (eQTL) IN THE AP
In addition to genetic variants in AP components causing dramatic impact in the expression and/or function of the protein, which individually are very rare (MAF < 10−5) and are strongly associated with different pathologies, there are also a few relatively common AP polymorphisms (MAF > 1%) that also confer significant risk or protection from disease and have been found to cause functional or expression changes in the AP proteins. Although the changes associated with these polymorphisms are normally subtle, because of the amplification nature of the AP, these variations, alone or in combination, are functionally relevant and have an important impact in the activity of the AP.
5.1. Functional polymorphisms
I have already described two of these polymorphisms in the AP (CFH‐Y402H and CFH‐V62I) and described the functional changes that explain their associated with increased risk and protection from AMD and other diseases (Section 4.1). Other common polymorphisms that are also strongly associated with protection and increased risk for AMD are FB‐R32Q 108 and C3‐R102G. 109 Interestingly, genetic data for some of these polymorphisms associated with AMD indicate that these common variants conferring risk and protection combine to create a gradient of risk for AMD in the population. 110
Functional and structural studies have also revealed the bases for the association of the FB‐32Q and C3‐102G variants with disease. The FB‐32Q variant decreases risk from AMD because it results in a reduced activity of the AP as a consequence of the decreased interaction between the Ba fragment of FB and C3b, which impacts the formation of the AP pro‐convertase C3bB. 111 Similarly, the C3‐102G variant associates with increased risk to AMD because it is less susceptible than C3‐102R to inactivation by complement regulators, which increases the activity of the AP. 112 The biochemical and structural analyses of the C3‐R102G polymorphism are particularly interesting because they added an unanticipated complexity in complement regulation revealing that the conformational flexibility of C3b impact the interactions of complement regulators with C3b. 53 , 113 , 114 Notably, residue Arg102 at the MG1 domain of C3b (Figure 3A) is involved in a salt bridge with residue Glu1032 at the TED domain that hold together the TED domain and the MG ring in C3b, a conformation that is critical for the interaction with FH. In C3‐102G, this salt bridge is lost, altering the TED‐MG1 separation and the regulation by FH. 113 , 114
A crucial finding of these functional studies was, however, to observe that when the aggregate effect of the combination of variants conferring risk (FH‐62 V, FB‐32R, and C3‐102G) was tested experimentally, they resulted in a significant difference in complement activity vs the low‐risk combination (FH‐62I, FB‐32Q, and C3‐102R) that exceeded the subtle functional alterations of the individual complement variants (Figure 6). These high and low AP activity variant combinations should represent the extremes of a continuum in AP activity that fit very well with the additive risk effect observed at the genetic level. Individuals at the high end of complement activity should be more prone to chronic inflammation, which explains the association with AMD, whereas those with low activity may be protected from it, but likely at the cost of increased susceptibility to infection.
FIGURE 6.

Combinations of common variations in C3, FB, and FH dramatically alter AP activity. A, Figure depicts the functional analysis of the common variants in C3, FB, and FH that have been found associated with increased risk to AMD. Individually, each of these variants shows small but consistent differences in hemolytic activities, compared with the normal allele. The hemolysis assays shown here were performed in normal human serum (NHS) depleted of FH, FB, or C3 that was reconstituted with the normal or AMD‐associated variant of the corresponding protein, as described. 96 , 111 , 112 B, Additivity of the three polymorphisms was investigated by comparing hemolytic activities of the variant set promoting more AP amplification with that causing less amplification. A NHS depleted of C3, FB, and FH proteins and reconstituted with the appropriate C3, FB, and FH variants was used in these experiments as described. 112 Figure illustrates that the combination of variants C3‐102G, FB‐32R and FH‐62 V shows sixfold increased (EC50 = 50 nmol/L FB vs EC50 = 288 nmol/L FB) complement activity in these hemolytic assays compared with that of variants C3‐102R, FB‐32Q, FH‐62I. (Figure was adapted from data published in references 96 , 111 , 112 )
Since the CFH, CFB, and C3 genes segregate independently, we should expect that the prevalence of the different combinations of genotypes for the three common polymorphisms are in correspondence with their individual allele frequencies. In the Spanish population, for example, the prevalence of triple homozygotes for the alleles that associate with increased AP activity (CFH‐62VV, CFB‐32RR, and C3‐102GG) is expected to be one in 68, whereas being homozygote for the three alleles associated with reduced AP activity (CFH‐62II, CFB‐32QQ, and C3‐102RR) is only of 1 in 2928. 115 The low prevalence of the low activity combination suggests a negative selective pressure by pathogens on the variants that compose this combination and some very early observations may support this hypothesis. 116 Ironically, what may have once been a disadvantage to escape childhood infections has now become an advantage to evade AMD for an increasingly aged population in the developed world.
The conclusion that the effects of AP variants are additive, and their different combinations result in distinct AP activities, prompt us to redefine Chester Alper's early term “complotype” 117 to refer to combinations of variants in complement components and regulators that result in distinct complement activities. 111 , 112 , 115 As I will discuss in Section 6, these “complotypes” are relevant to predict disease risk and should also be of help to assist clinical decision‐making in complement‐related diseases.
5.2. Complement eQTL
Expression quantitative trait loci are genomic loci that have been associated with variations in mRNA or protein expression levels. Nowadays, there is a great interest in uncovering these eQTLs because it is thought that they likely explain many of the genetic variants located in non‐coding regions of the human genome that have been associated with disease in genome‐wide association studies (GWAS). eQTL are also the likely explanation to the large variations in the expression levels of the complement proteins in humans, but data on these eQTLs are scarce and almost limited to variations within the RCA gene cluster.
One of the first studies trying to identify the factors influencing the fivefold range of variation of the FH plasma levels in humans was performed almost 20 years ago. 118 In that study, we applied variance‐component methods 119 to a sample of 358 Spanish individuals belonging to 21 extended pedigrees in which we have recorded plasma levels of FH, environmental factors, and genotypes for a set of 363 highly informative markers distributed along the human genome. The results indicated that 62% of the FH phenotypic variance is due to genetic effects and provided suggestive evidence of three genomic regions including potential eQTL, one of them within the RCA gene cluster in 1q32. Another example of early complement eQTL are genetic variants at the CR1 locus determining the expression levels of this complement regulator on erythrocytes. 84 , 85 Nowadays, three SNPs in strong LD (rs11118133, rs3811381, and rs2274565) allow discrimination of two alleles resulting in high (CR1‐H) and low (CR1‐L) expression of CR1 in erythrocytes. 85 , 120 The CR1‐L allele is a risk factor to experience extravascular hemolysis under eculizumab treatment. 121 It has also been suggested that reduced CR1 expression on erythrocytes leading to impaired amyloid clearance is the mechanism by which the rs6656401 SNP impacts Alzheimer's disease 122 and a similar association with low CR1 expression alleles has been described in preeclampsia. 123 Other examples of complement eQTL already mentioned in this review are the SNPs in the MCP ggaac haplotype that show a reduced CD46 transcriptional activity 106 and the SNPs in the promoter and intergenic regions of the CFH, CFHR3, and CFHR4 genes that influence the plasma levels of the FH, FHR‐3, and FHR‐4 proteins. 82 , 98 , 100 , 101
Since 2005, numerous GWAS studies have been carried out trying to delineate the genetic predisposition to AMD which has resulted in the identification of numerous SNPs conferring risk or protection to the disease located in intronic or intragenic regions. As an effort to correlate disease associations with gene expression and to provide an explanation about how disease‐associated SNPs located in these non‐coding regions cause phenotypic changes, a number of recent studies have explored the contribution of these AMD‐associated variants to modulate expression of complement genes, in plasma, liver and retinal cells and tissue, 124 , 125 , 126 , 127 stablishing a correlation between some of these SNPs with complement expression levels. One of the most significant of these correlations is that of rs6677604, 128 which is protective in AMD with the protective allele in strong LD with Δ CFHR3‐CFHR1 and, obviously, correlate with decreased expression of the FHR‐1 and FHR‐3 proteins. This rs6677604 SNP may also influence expression of CFH and other CFHR genes, but this has to be confirmed. 125
Also, within the RCA gene cluster, there are variants at the CFH locus that have been associated with levels of the FH‐related proteins FHR‐1, FHR‐2, FHR‐3, and FHR‐4. For example, the AMD risk‐conferring variant rs570618[T] is associated with increased FHR‐1, FHR‐2, FHR‐3, and FHR‐4 levels, while the AMD‐protective variant rs10922109[A] is associated with decreased FHR‐1, FHR‐2, FHR‐3, and FHR‐4 levels (Figure 2). 82 , 129 , 130 Additional SNPs associated with AMD that have been shown to influence expression levels of complement proteins are the Leu9His (rs4151667) variant in the CFB gene, which is associated with reduced FB levels and it is protective for AMD, 131 , 132 and Gly119Arg (rs141853578) 133 and rs10033900 124 , 134 in the CFI gene that reduce FI levels and confers risk for AMD.
Despite these important findings, the contribution of complement protein levels to many of the complement‐related diseases remains largely unknown and, in general, data about complement eQTL are very limited. To fill in this knowledge gap, integration of high‐density genetic mapping with complement transcriptome analysis in different cells and tissues are needed. This is crucial because, as mentioned above, a comprehensive understanding of the complement eQTL in different cells and tissues may justify the large variations in the expression levels of the different complement proteins that are observed at the population level and will likely correlate different complement genetic make‐ups with distinct overall activities of the complement system, providing a better understanding of the role of complement in disease predisposition.
6. THE ADDITIVE EFFECT OF GENETIC VARIANTS IN COMPLEMENT PROTEINS; THE COMPLOTYPES
I have provided numerous examples of complement genetic variants that predispose or protect from different diseases. In this section, I will discuss that the consequences of all these genetic variants are additive and that, with few exceptions, predisposition to or protection from complement‐related disease goes beyond individual associations, being much more complex and dependent of combinations of genetic variants in different complement components and regulators. I have already introduced the term “complotype” to refer to the combination of common variants in the complement components FH, FB, and C3. Complotypes in a broader sense include the combinations of common SNPs affecting expression and activity of the different complement components with rare pathogenic variants. The CFH‐CFHRs and MCP ggaac haplotypes at the RCA gene cluster are also crucial components of these complotypes as we know that the combination of rare pathogenic variants with these haplotypes are often decisive to define the risk of protection from disease. 106
The CFH‐CFHRs locus is a major genetic factor in AMD with both risk and protective variants. 71 , 91 , 135 , 136 , 137 These variants are integrated in three CFH haplotypes: the risk CFH‐H1 haplotype (carrying the FH‐402His variant) and the protective haplotypes CFH‐H2 (carrying the FH‐62Ile variant) and CFH‐H4 (carrying the Δ CFHR3‐CFHR1 variant; see also Section 4.1). The CFH‐H3 haplotype is neutral for AMD. Interestingly, when these CFH haplotypes were analyzed for association with AMD, it was observed that the positive association of the CFH‐H1 risk haplotype disappears in heterozygosis with any of the haplotypes carrying the protective alleles FH‐62Ile or Δ CFHR3‐CFHR1. 138 , 139 In fact, the frequency of these heterozygotes is significantly decreased in AMD, indicating that the protective haplotypes are dominant over the risk haplotypes. Moreover, the risk and protective haplotypes strongly influence risk at the ARMS2/HTRA1 locus, the other major genetic component in AMD, increasing or neutralizing the risk conferred by the variant rs10490924 (ARMS2) at this locus. 139 The mechanisms associated with the susceptibility to AMD driven by the risk and protective CFH‐CFHRs haplotypes are not completely clear and as discussed in Section 4.1 may involve the combination of different factors encoded within the haplotype. The dominant effect that the Δ CFHR3‐CFHR1 variant has over the risk variants is interesting and may suggest that in the absence of the FHR‐1 and FHR‐3 proteins the complement dysregulation conferred by the risk variants is inconsequential and the disease does not manifest.
The combination of different variants in AP components and regulators also underlines the complexity of aHUS genetics. It is well documented that the presence of more than one pathogenic variant or the concurrence of a rare pathogenic variant with common risk polymorphisms influences risk to aHUS and modulates the penetrance of the disease. 98 , 106 , 140 Identification of two or more rare complement variants have been described to occur in as many as 3% of aHUS cases, which determines different clinical outcomes depending on the gene combinations. 141 The CFH‐CFHRs and MCP ggaac haplotypes have also been shown to increase the risk of aHUS in carriers of rare pathogenic variants and to exhibit interesting genotype–phenotype correlations. Thus, haplotype CFH‐H3, which is neutral in AMD is a risk haplotype for aHUS, whereas CFH‐H4 is neutral and CFH‐H1 and CFH‐H2 are protective, 98 , 106 , 142 and we have already mentioned that the MCP ggaac haplotype, alone or in combination with rare pathogenic variants, is a significant aHUS risk, 49 , 53 , 106 , 143 which is likely related to its crucial role as a membrane associated complement regulator (see Section 4.2).
To evaluate the susceptibility to aHUS in individuals carrying different loads of genetic risk factors, we have recently analyzed the penetrance of the disease in 372 relatives of aHUS patients carrying 1 or 2 rare complement pathogenic variants. 144 Our data confirmed that the main driver of aHUS in these pedigrees is the pathogenic mutation and that penetrance of the disease raises with the genetic load of risk factors (Figure 7). A detailed age‐adjusted analysis showed a relatively low aHUS penetrance of 9.6% at age 48 years for relatives carrying a single pathogenic variant that increases to 36% at age 44 years in carriers of two pathogenic variants. Notably, carrying both the MCP ggaac and the CFH‐H3 haplotypes, in addition to one or two pathogenic variants, raises the aHUS penetrance to 18.8% (age 37 years) and 100% (age 44 years), respectively (Figure 7). 144 In summary, the presence of both risk haplotypes in carriers of one or two pathogenic variants increases aHUS penetrance 2‐ to 3‐fold, whereas the absence of both risk polymorphisms reduces their risk of developing aHUS significantly.
FIGURE 7.

The genetic load of genetic risk factors determines penetrance of aHUS. aHUS is a complex disease, with additive genetic risk factors conferring predisposition to aHUS and environmental risk factors triggering disease development. Data support an inverse correlation between the genetic load of risk factors and the intensity of environmental triggers required for disease development. A, Kaplan–Meier estimations of the aHUS penetrance for different loads of genetic risk factors. B, aHUS penetrance at age 35 years and maximum aHUS penetrance in relatives of probands carrying 1 or 2 pathogenic variants and contribution of the CFH‐H3 and MCP ggaac risk haplotypes. Reproduced from 144 with permission
The addition of CFH‐CFHRs and MCP ggaa haplotypes to some rare pathogenic variants also determines the disease outcome. This is the case of CFH and CFI variants that impair expression of the protein or eliminate its complement regulatory functions, which in heterozygosis associate with different complement‐related diseases with distinct underlying pathogenic mechanisms. Notably, patients carrying these genetic abnormalities only present with one of these diseases and, within a given pedigree, carriers only develop one type of disease. 37 , 39 , 145 , 146 , 147 To investigate what determines the disease outcome, we selected heterozygote carriers (patients and relatives) of a single relatively prevalent CFH variant (FH‐1210C) causing a complete functional inactivation of FH 147 that has been found associated with aHUS, 140 early‐onset AMD, 146 and C3G. 148 As expected, disease status, determined in patients and relatives carrying the FH‐1210C variant, revealed absence of AMD phenotypes in the aHUS cohort and, vice versa, lack of renal disease in the AMD cohort. Interestingly, these findings were consistent with significant differences in the FH‐1210C‐independent overall risk for aHUS (determined by the CFH‐CFHRs and MCP ggaa haplotypes) and AMD (determined by the CFH‐CFHRs haplotypes and ARMS2‐Ala69Ser genotypes) among FH‐1210C‐carriers developing one or the other pathology. 147 In summary, these data suggest that in addition to environmental risk factors, the specific risk and protective factors for aHUS and AMD associated with the CFH‐CFHRs, MCP and ARMS2/HTRA1 loci, add‐on to determine the disease outcome in carriers of partial FH and FI deficiencies.
Along this review, I have described how genetic studies in patients with complement‐related diseases have unraveled associations between these pathologies with specific functional alterations in components and regulators of the AP. These genotype–phenotype correlations have been instrumental to unravel pathogenic mechanisms. In this section, I have presented how the different combinations resulting from genetic variability in complement proteins (“complotypes”) impact disease susceptibility. A direct application of this knowledge would be the development of statistical models for the estimation of risks to develop a particular disease based on complement genotypes. Unfortunately, complement‐related disorders are genetically complex and multifactorial, with a component that includes non‐complement genes and non‐genetic/environmental factors that are still poorly defined. It remains for future studies to acquire a precise knowledge of this component, as well as a better understanding of the complement eQTL, to provide these tests with acceptable predictive certainty.
7. IDENTIFICATION AND CLASSIFICATION OF COMPLEMENT GENE VARIANTS
Testing for genetic variants in complement genes is a routine in diseases like aHUS, where the identification of a pathogenic, or likely pathogenic, variant help to confirm diagnosis and guide short‐ and long‐term patient management. But it is also becoming habitual in several other diseases in which complement plays a role and there is suspicion of a complement genetic component. Complement genetic testing must be comprehensive and include the analysis of all types of gene variations that have been described in this review. In this respect, massive parallel sequencing (next‐generation sequencing; NGS) is a reliable, economical, and fast method to search for both nucleotide and structural variations in the whole complement gene set. There are, however, a few peculiarities in complement genes that have to be taken into account. The RCA gene cluster presents a number of segmental duplications at the CR1 and the CFH/CFHRs gene regions and, in both regions, these segmental duplications are involved in the generation of structural variants. Identification of CNVs in the RCA gene cluster is critical in the molecular diagnostic of complement‐related diseases. This can be done by bioinformatics analysis of the DNA sequence data generated by NGS, but often requires special techniques like Multiplex Ligation‐dependent Probe Amplification (MLPA). This is because the currently used NGS approaches do not easily identify all the structural variations in the RCA gene cluster. 149 Segmental duplications are also problematic for NGS because of the difficulties in assigning changes to one or the other duplicated regions. This is particularly complicated in the case of CR1, the 3′end regions of CFH and CFHR1 and, not discussed here, the C4A and C4B genes in human chromosome 6. In addition, because of these difficulties, current databases do not have a proper representation of genetic variants in these regions.
Whilst variant identification with today's methodologies should not be a problem, variant classification, however, is not trivial and it is often a barrier to the optimal medical use of genetic information. Identified variants must be classified based on their impact, but differentiating between variants that do, or do not, alter expression or function is challenging. Generalized guidelines for variant interpretation have been established by many organizations, including joint consensus recommendations released by the American College of Medical Genetics and Genomics 150 and the Association for Molecular Pathology (AMP) (ACMG/AMP) 150 ; the AMP, American Society of Clinical Oncology, 151 and College of American Pathologists (CAP) (AMP/ASCO/CAP) 151 ; and the European Society for Human Genetics (ESHG). 152 Of the available guidelines, those proposed by the ACMG/AMP have been most widely adopted. Included in these guidelines are many metrics by which to grade a variant so that it can be classified as benign, likely benign, of uncertain significance, likely pathogenic, or pathogenic. For example, validated functional studies provide strong evidence of pathogenicity (ACMG criterion PS3, strong evidence of pathogenicity). 150 But functional studies are labor‐intensive and complicated, and for these reasons variant effect is typically inferred using pathogenicity prediction algorithms and allele frequency data. Both approaches have major limitations and the level of evidence they provide is not as strong (ACMG criterion PP3, supporting evidence of pathogenicity). 150 As discussed in Section 2, the genetic variability of complement genes in the general population is such that the probability to find a pathogenic variant by chance is substantial. Therefore, an additional question relevant for the interpretation of findings in the genetic screening of patients with complement‐related diseases is whether the AP functional alterations expected from the identified variants fit the pathogenesis of the disease.
To identify the strengths and weaknesses of the current pathogenicity prediction algorithms and provide recommendations to aid the appropriate classification of novel FH variants as they are identified, we have recently characterized functionally 105 genetic variants of FH associated with aHUS. 153 These analyses indicate that rarity in normal databases can be misleading for variant classification. While it is true that pathogenic variants tent to be rarer than benign variants, 21.5% of the benign variants are absent in gnomAD. The data also identify important limitations in applying prediction algorithms to FH variants, as only 74% were classified correctly applying the standard CADD PHRED C‐score > 15. Although a differential adjustment of the prediction algorithms to accommodate the peculiarities of the distinct FH regions improves overall predictions to 85%, our final conclusion was that functional analysis of the variants remains the gold standard to provide an accurate classification.
8. CONCLUSIONS
Genetic variability in the genes encoding proteins of the AP in the normal population is considerable, with elevated numbers of rare, likely pathogenic, missense variants (MAF < 0.1%) in each of the AP proteins that, on average, affect to 2% of the individuals in the normal population per AP protein. Common missense and structural variants (MAF > 1%) are just a handful but include some with important functional implications. In contrast to this knowledge, data on complement eQTL are very limited. NGS data have also identified hundreds of genetic variants in AP components and regulators in patients with different complement‐related diseases, often following characteristic genotype–phenotype correlations that associate a disease with a specific functional alteration in the AP. I have illustrated how these genotype–phenotype correlations have been instrumental to unravel pathogenic mechanisms with important therapeutic consequences. There is also compelling evidence illustrating that the functional consequences of the genetic variants in AP proteins are additive and that the complotypes are strong determinants of disease susceptibility.
A current challenge is to have a precise understanding of the consequences of the genetic variants identified in the genetic analysis, which is very much contingent on functional assays. To identify and characterize the complement eQTL in different cells and tissues is also a crucial task for future studies. This knowledge will likely explain the variations in the expression levels of the different complement proteins that are observed at the population level and will also identify different complement genetic make‐ups with distinct overall activities of the complement system based on differences in protein expression levels. Complement‐related diseases are genetically complex and multifactorial, with the genetic component determining the individual predisposition to disease. Integrating all variables in the complement genetic component with other non‐genetic and environmental components in computational systems biology models will provide an appropriate description of how genetics influences complement dynamics and the delicate balance between complement activation and regulation. These models will be decisive to determine disease risk and for decision‐making in patients' management. Finally, and not discussed here, an intracellular role has been described for some AP proteins and it has been postulated that these newly unraveled activities are as relevant as the well stablished activities of the complement system mentioned in this review. It is yet unclear whether genetically determined differences in the activity and expression levels of the AP complement proteins have consequences for these non‐canonical intracellular complement functions.
CONFLICT OF INTEREST
I have no conflict of interest to disclose.
ACKNOWLEDGEMENTS
Many thanks to Hector Martin‐Merinero, Sheila Pinto, and Angela Ruiz for their assistance in the elaboration of this review and to Prof. José Ramón Regueiro for critically reading the final manuscript. SRdC was supported by grants from the Spanish Ministerio de Economía y Competitividad‐FEDER (PID2019‐104912RB‐I00) and Autonomous Region of Madrid (S2017/BMD‐3673).
Rodríguez de Córdoba S. Genetic variability shapes the alternative pathway complement activity and predisposition to complement‐related diseases. Immunol Rev. 2023;313:71‐90. doi: 10.1111/imr.13131
This article is part of a series of reviews covering The Alternative Pathway or Amplification Loop of Complement appearing in Volume 313 of Immunological Reviews.
DATA AVAILABILITY STATEMENT
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.
REFERENCES
- 1. Lachmann PJ. Looking back on the alternative complement pathway. Immunobiology. 2018;223:519‐523. [DOI] [PubMed] [Google Scholar]
- 2. Harrison RA. The properdin pathway: an “alternative activation pathway” or a “critical amplification loop” for C3 and C5 activation? Semin Immunopathol. 2018;40:15‐35. [DOI] [PubMed] [Google Scholar]
- 3. Ricklin D, Hajishengallis G, Yang K, Lambris JD. Complement: a key system for immune surveillance and homeostasis. Nat Immunol. 2010;11:785‐797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. de Cordoba SR, Tortajada A, Harris CL, Morgan BP. Complement dysregulation and disease: from genes and proteins to diagnostics and drugs. Immunobiology. 2012;217:1034‐1046. [DOI] [PubMed] [Google Scholar]
- 5. Holers VM. The spectrum of complement alternative pathway‐mediated diseases. Immunol Rev. 2008;223:300‐316. [DOI] [PubMed] [Google Scholar]
- 6. Ricklin D, Lambris JD. Complement in immune and inflammatory disorders: pathophysiological mechanisms. J Immunol. 2013;190:3831‐3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Sjoberg AP, Trouw LA, Blom AM. Complement activation and inhibition: a delicate balance. Trends Immunol. 2009;30:83‐90. [DOI] [PubMed] [Google Scholar]
- 8. Karczewski KJ, Francioli LC, Tiao G, et al. The mutational constraint spectrum quantified from variation in 141,456 humans. Nature. 2020;581:434‐443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kircher M, Witten DM, Jain P, O'Roak BJ, Cooper GM, Shendure J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat Genet. 2014;46:310‐315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Rentzsch P, Witten D, Cooper GM, Shendure J, Kircher M. CADD: predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019;47:D886‐D894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Morgan HP, Schmidt CQ, Guariento M, et al. Structural basis for engagement by complement factor H of C3b on a self surface. Nat Struct Mol Biol. 2011;18:463‐470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Morgan HP, Mertens HDT, Guariento M, et al. Structural analysis of the C‐terminal region (modules 18‐20) of complement regulator factor H (FH). PloS One. 2012;7:e32187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Blaum BS, Hannan JP, Herbert AP, Kavanagh D, Uhrín D, Stehle T. Structural basis for sialic acid–mediated self‐recognition by complement factor H. Nat Chem Biol. 2015;11:77‐82. [DOI] [PubMed] [Google Scholar]
- 14. Kajander T, Lehtinen MJ, Hyvärinen S, et al. Dual interaction of factor H with C3d and glycosaminoglycans in host‐nonhost discrimination by complement. Proc Natl Acad Sci USA. 2011;108:2897‐2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Sharma AK, Pangburn MK. Identification of three physically and functionally distinct binding sites for C3b in human complement factor H by deletion mutagenesis. Proc Natl Acad Sci USA. 1996;93:10996‐11001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Meri S, Pangburn MK. Discrimination between activators and nonactivators of the alternative pathway of complement: regulation via a sialic acid/polyanion binding site on factor H. Proc Natl Acad Sci USA. 1990;87:3982‐3986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Blaum BS. The lectin self of complement factor H. Curr Opin Struct Biol. 2017;44:111‐118. [DOI] [PubMed] [Google Scholar]
- 18. Perez‐Caballero D, González‐Rubio C, Gallardo ME, et al. Clustering of missense mutations in the C‐terminal region of factor H in atypical hemolytic uremic syndrome. Am J Hum Genet. 2001;68:478‐484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Manuelian T, Hellwage J, Meri S, et al. Mutations in factor H reduce binding affinity to C3b and heparin and surface attachment to endothelial cells in hemolytic uremic syndrome. J Clin Invest. 2003;111:1181‐1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Sánchez‐Corral P, Pérez‐Caballero D, Huarte O, et al. Structural and functional characterization of factor H mutations associated with atypical hemolytic uremic syndrome. Am J Hum Genet. 2002;71:1285‐1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Ferreira VP, Herbert AP, Cortés C, et al. The binding of factor H to a complex of physiological polyanions and C3b on cells is impaired in atypical hemolytic uremic syndrome. J Immunol. 2009;182:7009‐7018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gruppo RA, Rother RP. Eculizumab for congenital atypical hemolytic‐uremic syndrome. N Engl J Med. 2009;360:544‐546. [DOI] [PubMed] [Google Scholar]
- 23. Nürnberger J, Philipp T, Witzke O, et al. Eculizumab for atypical hemolytic‐uremic syndrome. N Engl J Med. 2009;360:542‐544. [DOI] [PubMed] [Google Scholar]
- 24. Bhattacharjee A, Lehtinen MJ, Kajander T, Goldman A, Jokiranta TS. Both domain 19 and domain 20 of factor H are involved in binding to complement C3b and C3d. Mol Immunol. 2010;47:1686‐1691. [DOI] [PubMed] [Google Scholar]
- 25. Lehtinen MJ, Rops AL, Isenman DE, van der Vlag J, Jokiranta TS. Mutations of factor H impair regulation of surface‐bound C3b by three mechanisms in atypical hemolytic uremic syndrome. J Biol Chem. 2009;284:15650‐15658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Martin Merinero H, Subías M, Pereda A, et al. Molecular bases for the association of FHR‐1 with atypical hemolytic uremic syndrome and other diseases. Blood. 2021;137:3484‐3494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Pickering MC, Cook HT, Warren J, et al. Uncontrolled C3 activation causes membranoproliferative glomerulonephritis in mice deficient in complement factor H. Nat Genet. 2002;31:424‐428. [DOI] [PubMed] [Google Scholar]
- 28. Rose KL, Paixao‐Cavalcante D, Fish J, et al. Factor I is required for the development of membranoproliferative glomerulonephritis in factor H‐deficient mice. J Clin Invest. 2008;118:608‐618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Richards A, Kemp EJ, Liszewski MK, et al. Mutations in human complement regulator, membrane cofactor protein (CD46), predispose to development of familial hemolytic uremic syndrome. Proc Natl Acad Sci USA. 2003;100:12966‐12971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Noris M, Brioschi S, Caprioli J, et al. Familial haemolytic uraemic syndrome and an MCP mutation. Lancet. 2003;362:1542‐1547. [DOI] [PubMed] [Google Scholar]
- 31. Liszewski MK, Atkinson JP. Complement regulator CD46: genetic variants and disease associations. Hum Genomics. 2015;9:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Fremeaux‐Bacchi V, Moulton EA, Kavanagh D, et al. Genetic and functional analyses of membrane cofactor protein (CD46) mutations in atypical hemolytic uremic syndrome. J Am Soc Nephrol. 2006;17:2017‐2025. [DOI] [PubMed] [Google Scholar]
- 33. Hill A, DeZern AE, Kinoshita T, Brodsky RA. Paroxysmal nocturnal haemoglobinuria. Nat Rev Dis Primers. 2017;3:17028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Ozen A, Comrie WA, Ardy RC, et al. CD55 deficiency, early‐onset protein‐losing enteropathy, and thrombosis. N Engl J Med. 2017;377:52‐61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Telen MJ, Hall SE, Green AM, Moulds JJ, Rosse WF. Identification of human erythrocyte blood group antigens on decay‐accelerating factor (DAF) and an erythrocyte phenotype negative for DAF. J Exp Med. 1988;167:1993‐1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Seddon JM, Yu Y, Miller EC, et al. Rare variants in CFI, C3 and C9 are associated with high risk of advanced age‐related macular degeneration. Nat Genet. 2013;45:1366‐1370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kavanagh D, Yu Y, Schramm EC, et al. Rare genetic variants in the CFI gene are associated with advanced age‐related macular degeneration and commonly result in reduced serum factor I levels. Hum Mol Genet. 2015;24:3861‐3870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Osborne AJ, Breno M, Borsa NG, et al. Statistical validation of rare complement variants provides insights into the molecular basis of atypical hemolytic uremic syndrome and C3 Glomerulopathy. J Immunol. 2018;200:2464‐2478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Tzoumas N, Kavanagh D, Cordell HJ, Lotery AJ, Patel PJ, Steel DH. Rare complement factor I variants associated with reduced macular thickness and age‐related macular degeneration in the UK biobank. Hum Mol Genet. 2022;31:2678‐2692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bienaime F, Dragon‐Durey MA, Regnier CH, et al. Mutations in components of complement influence the outcome of factor I‐associated atypical hemolytic uremic syndrome. Kidney Int. 2010;77:339‐349. [DOI] [PubMed] [Google Scholar]
- 41. Java A, Pozzi N, Schroeder MC, et al. Functional analysis of rare genetic variants in Complement factor I (CFI) in advanced age‐related macular degeneration (AMD). Hum Mol Genet. 2022. doi: 10.1093/hmg/ddac103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhang Y, Goodfellow RX, Ghiringhelli Borsa N, et al. Complement factor I variants in Complement‐mediated renal diseases. Front Immunol. 2022;13:866330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Nilsson SC, Sim RB, Lea SM, Fremeaux‐Bacchi V, Blom AM. Complement factor I in health and disease. Mol Immunol. 2011;48:1611‐1620. [DOI] [PubMed] [Google Scholar]
- 44. Gros P, Milder FJ, Janssen BJ. Complement driven by conformational changes. Nat Rev Immunol. 2008;8:48‐58. [DOI] [PubMed] [Google Scholar]
- 45. Torreira E, Tortajada A, Montes T, Rodríguez de Córdoba S, Llorca O. 3D structure of the C3bB complex provides insights into the activation and regulation of the complement alternative pathway convertase. Proc Natl Acad Sci USA. 2009;106:882‐887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Slade C, Bosco J, Unglik G, Bleasel K, Nagel M, Winship I. Deficiency in complement factor B. N Engl J Med. 2013;369:1667‐1669. [DOI] [PubMed] [Google Scholar]
- 47. Sprong T, Roos D, Weemaes C, et al. Deficient alternative complement pathway activation due to factor D deficiency by 2 novel mutations in the complement factor D gene in a family with meningococcal infections. Blood. 2006;107:4865‐4870. [DOI] [PubMed] [Google Scholar]
- 48. Linton SM, Morgan BP. Properdin deficiency and meningococcal disease—identifying those most at risk. Clin Exp Immunol. 1999;118:189‐191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. de Jorge EG, Harris CL, Esparza‐Gordillo J, et al. Gain‐of‐function mutations in complement factor B are associated with atypical hemolytic uremic syndrome. Proc Natl Acad Sci USA. 2007;104:240‐245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Fremeaux‐Bacchi V, Miller EC, Liszewski MK, et al. Mutations in complement C3 predispose to development of atypical hemolytic uremic syndrome. Blood. 2008;112:4948‐4952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Roumenina LT, Jablonski M, Hue C, et al. Hyperfunctional C3 convertase leads to complement deposition on endothelial cells and contributes to atypical hemolytic uremic syndrome. Blood. 2009;114:2837‐2845. [DOI] [PubMed] [Google Scholar]
- 52. Martinez‐Barricarte R, Heurich M, Valdes‐Cañedo F, et al. Human C3 mutation reveals a mechanism of dense deposit disease pathogenesis and provides insights into complement activation and regulation. J Clin Invest. 2010;120:3702‐3712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Martínez‐Barricarte R, Heurich M, López‐Perrote A, et al. The molecular and structural bases for the association of complement C3 mutations with atypical hemolytic uremic syndrome. Mol Immunol. 2015;66:263‐273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Chauvet S, Roumenina LT, Bruneau S, et al. A familial C3GN secondary to defective C3 regulation by Complement receptor 1 and Complement factor H. J Am Soc Nephrol. 2016;27:1665‐1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Rodríguez de Córdoba S. Complement genetics and susceptibility to inflammatory disease. Lessons from genotype–phenotype correlations. Immunobiology. 2016;221:709‐714. [DOI] [PubMed] [Google Scholar]
- 56. Siomou E, Gkoutsias A, Serbis A, Kollios K, Chaliasos N, Frémeaux‐Bacchi V. aHUS associated with C3 gene mutation: a case with numerous relapses and favorable 20‐year outcome. Pediatr Nephrol. 2016;31:513‐517. [DOI] [PubMed] [Google Scholar]
- 57. Matsumoto T, Fan X, Ishikawa E, et al. Analysis of patients with atypical hemolytic uremic syndrome treated at the Mie university hospital: concentration of C3 p. I1157T mutation. Int J Hematol. 2014;100:437‐442. [DOI] [PubMed] [Google Scholar]
- 58. Roumenina LT, Frimat M, Miller EC, et al. A prevalent C3 mutation in aHUS patients causes a direct C3 convertase gain of function. Blood. 2012;119:4182‐4191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Goicoechea de Jorge E, Tortajada A, García SP, et al. Factor H competitor generated by gene conversion events associates with atypical hemolytic uremic syndrome. J Am Soc Nephrol. 2018;29:240‐249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Valoti E, Alberti M, Tortajada A, et al. A novel atypical hemolytic uremic syndrome‐associated hybrid CFHR1/CFH gene encoding a fusion protein that antagonizes factor H‐dependent complement regulation. J Am Soc Nephrol. 2015;26:209‐219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Westra D, Vernon KA, Volokhina EB, Pickering MC, van de Kar NC, van den Heuvel LP. Atypical hemolytic uremic syndrome and genetic aberrations in the complement factor H‐related 5 gene. J Hum Genet. 2012;57:459‐464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Gale DP, de Jorge EG, Cook HT, et al. Identification of a mutation in complement factor H‐related protein 5 in patients of Cypriot origin with glomerulonephritis. Lancet. 2010;376:794‐801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Tortajada A, Yébenes H, Abarrategui‐Garrido C, et al. C3 glomerulopathy‐associated CFHR1 mutation alters FHR oligomerization and complement regulation. J Clin Invest. 2013;123:2434‐2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Márquez‐Tirado B, Gutiérrez‐Tenorio J, Tortajada A, et al. Factor H–related protein 1 drives disease susceptibility and prognosis in C3 Glomerulopathy. J Am Soc Nephrol. 2022;33:1137‐1153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Chen Q, Wiesener M, Eberhardt HU, et al. Complement factor H‐related hybrid protein deregulates complement in dense deposit disease. J Clin Invest. 2014;124:145‐155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Medjeral‐Thomas N, Malik TH, Patel MP, et al. A novel CFHR5 fusion protein causes C3 glomerulopathy in a family without Cypriot ancestry. Kidney Int. 2014;85:933‐937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Togarsimalemath SK, Sethi SK, Duggal R, et al. A novel CFHR1‐CFHR5 hybrid leads to a familial dominant C3 glomerulopathy. Kidney Int. 2017;92:876‐887. [DOI] [PubMed] [Google Scholar]
- 68. Xiao X, Ghossein C, Tortajada A, et al. Familial C3 glomerulonephritis caused by a novel CFHR5‐CFHR2 fusion gene. Mol Immunol. 2016;77:89‐96. [DOI] [PubMed] [Google Scholar]
- 69. Díaz‐Guillén MA, Rodríguez de Córdoba S, Heine‐Suñer D. A radiation hybrid map of complement factor H and factor H‐related genes. Immunogenetics. 1999;49:549‐552. [DOI] [PubMed] [Google Scholar]
- 70. Holmes LV, Strain L, Staniforth SJ, et al. Determining the population frequency of the CFHR3/CFHR1 deletion at 1q32. PloS One. 2013;8:e60352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Hughes AE, Orr N, Esfandiary H, Diaz‐Torres M, Goodship T, Chakravarthy U. A common CFH haplotype, with deletion of CFHR1 and CFHR3, is associated with lower risk of age‐related macular degeneration. Nat Genet. 2006;38:1173‐1177. [DOI] [PubMed] [Google Scholar]
- 72. Gharavi AG, Kiryluk K, Choi M, et al. Genome‐wide association study identifies susceptibility loci for IgA nephropathy. Nat Genet. 2011;43:321‐327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Zhao J, Wu H, Khosravi M, et al. Association of genetic variants in complement factor H and factor H‐related genes with systemic lupus erythematosus susceptibility. PLoS Genet. 2011;7:e1002079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Abarrategui‐Garrido C, Martinez‐Barricarte R, Lopez‐Trascasa M, Rodriguez de Cordoba S, Sanchez‐Corral P. Characterization of complement factor H‐related (CFHR) proteins in plasma reveals novel genetic variations of CFHR1 associated with atypical hemolytic uremic syndrome. Blood. 2009;114:4261‐4271. [DOI] [PubMed] [Google Scholar]
- 75. Fakhouri F, Frémeaux‐Bacchi V. Thrombotic microangiopathy in aHUS and beyond: clinical clues from complement genetics. Nat Rev Nephrol. 2021;17:543‐553. [DOI] [PubMed] [Google Scholar]
- 76. Zipfel PF, Edey M, Heinen S, et al. Deletion of complement factor H‐related genes CFHR1 and CFHR3 is associated with atypical hemolytic uremic syndrome. PLoS Genet. 2007;3:e41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Jozsi M, Zipfel PF. Factor H family proteins and human diseases. Trends Immunol. 2008;29:380‐387. [DOI] [PubMed] [Google Scholar]
- 78. Dragon‐Durey MA, Blanc C, Marliot F, et al. The high frequency of complement factor H related CFHR1 gene deletion is restricted to specific subgroups of patients with atypical haemolytic uraemic syndrome. J Med Genet. 2009;46:447‐450. [DOI] [PubMed] [Google Scholar]
- 79. Puraswani M, Khandelwal P, Saini H, et al. Clinical and immunological profile of anti‐factor H antibody associated atypical hemolytic uremic syndrome: a Nationwide database. Front Immunol. 2019;10:1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Heinen S, Sanchez‐Corral P, Jackson MS, et al. De novo gene conversion in the RCA gene cluster (1q32) causes mutations in complement factor H associated with atypical hemolytic uremic syndrome. Hum Mutat. 2006;27:292‐293. [DOI] [PubMed] [Google Scholar]
- 81. Venables JP, Strain L, Routledge D, et al. Atypical haemolytic uraemic syndrome associated with a hybrid complement gene. PLoS Med. 2006;3:e431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Cipriani V, Lorés‐Motta L, He F, et al. Increased circulating levels of factor H‐related protein 4 are strongly associated with age‐related macular degeneration. Nat Commun. 2020;11:778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Ng YC, Schifferli JA, Walport MJ. Immune complexes and erythrocyte CR1 (complement receptor type 1): effect of CR1 numbers on binding and release reactions. Clin Exp Immunol. 1988;71:481‐485. [PMC free article] [PubMed] [Google Scholar]
- 84. Rodriguez de Cordoba S, Rubinstein P. Quantitative variations of the C3b/C4b receptor (CR1) in human erythrocytes are controlled by genes within the regulator of complement activation (RCA) gene cluster. J Exp Med. 1986;164:1274‐1283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Wilson JG, Murphy EE, Wong WW, Klickstein LB, Weis JH, Fearon DT. Identification of a restriction fragment length polymorphism by a CR1 cDNA that correlates with the number of CR1 on erythrocytes. J Exp Med. 1986;164:50‐59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Thiel J, Kimmig L, Salzer U, et al. Genetic CD21 deficiency is associated with hypogammaglobulinemia. J Allergy Clin Immunol. 2012;129:801‐810.e6. [DOI] [PubMed] [Google Scholar]
- 87. Shaw JM, al‐Shamkhani A, Boxer LA, et al. Characterization of four CD18 mutants in leucocyte adhesion deficient (LAD) patients with differential capacities to support expression and function of the CD11/CD18 integrins LFA‐1, mac‐1 and p150,95. Clin Exp Immunol. 2001;126:311‐318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Rodríguez de Córdoba S, Diaz‐Guillen M, Heine‐Suñer D. An integrated map of the human regulator of complement activation (RCA) gene cluster on 1q32. Mol Immunol. 1999;36:803‐808. [DOI] [PubMed] [Google Scholar]
- 89. Lambert JC, Heath S, Even G, et al. Genome‐wide association study identifies variants at CLU and CR1 associated with Alzheimer's disease. Nat Genet. 2009;41:1094‐1099. [DOI] [PubMed] [Google Scholar]
- 90. Francis NJ, McNicholas B, Awan A, et al. A novel hybrid CFH/CFHR3 gene generated by a microhomology‐mediated deletion in familial atypical hemolytic uremic syndrome. Blood. 2012;119:591‐601. [DOI] [PubMed] [Google Scholar]
- 91. Hageman GS, Anderson DH, Johnson LV, et al. A common haplotype in the complement regulatory gene factor H (HF1/CFH) predisposes individuals to age‐related macular degeneration. Proc Natl Acad Sci USA. 2005;102:7227‐7232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Prosser BE, Johnson S, Roversi P, et al. Expression, purification, cocrystallization and preliminary crystallographic analysis of sucrose octasulfate/human complement regulator factor H SCRs 6‐8. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2007;63:480‐483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Weismann D, Hartvigsen K, Lauer N, et al. Complement factor H binds malondialdehyde epitopes and protects from oxidative stress. Nature. 2011;478:76‐81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94. Harrison RES, Morikis D. Molecular mechanisms of macular degeneration associated with the complement factor H Y402H mutation. Biophys J. 2019;116:215‐226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95. De Córdoba SR, De Jorge EG. Translational mini‐review series on Complement factor H: genetics and disease associations of human complement factor H. Clin Exp Immunol. 2008;151:1‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Tortajada A, Montes T, Martínez‐Barricarte R, Morgan BP, Harris CL, de Córdoba SR. The disease‐protective complement factor H allotypic variant Ile62 shows increased binding affinity for C3b and enhanced cofactor activity. Hum Mol Genet. 2009;18:3452‐3461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Cipriani V, de Motta LL, He F, et al. Factor H‐related protein 4 (FHR‐4) drives complement dysregulation in age‐related macular degeneration. Invest Ophthalmol Vis Sci. 2019;60:3870. [Google Scholar]
- 98. Caprioli J, Castelletti F, Bucchioni S, et al. Complement factor H mutations and gene polymorphisms in haemolytic uraemic syndrome: the C‐257T, the A2089G and the G2881T polymorphisms are strongly associated with the disease. Hum Mol Genet. 2003;12:3385‐3395. [DOI] [PubMed] [Google Scholar]
- 99. Davila S, Wright VJ, Khor CC, et al. Genome‐wide association study identifies variants in the CFH region associated with host susceptibility to meningococcal disease. Nat Genet. 2010;42:772‐776. [DOI] [PubMed] [Google Scholar]
- 100. Bernabéu‐Herrero ME, Jiménez‐Alcázar M, Anter J, et al. Complement factor H, FHR‐3 and FHR‐1 variants associate in an extended haplotype conferring increased risk of atypical hemolytic uremic syndrome. Mol Immunol. 2015;67:276‐286. [DOI] [PubMed] [Google Scholar]
- 101. Pouw RB, Gómez Delgado I, López Lera A, et al. High complement Factor H‐Related (FHR)‐3 levels are associated with the atypical haemolytic uremic syndrome‐risk allele CFHR3*B. Front Immunol. 2018;9:848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102. Schneider MC, Exley RM, Chan H, et al. Functional significance of factor H binding to Neisseria meningitidis. J Immunol. 2006;176:7566‐7575. [DOI] [PubMed] [Google Scholar]
- 103. Madico G, Welsch JA, Lewis LA, et al. The meningococcal vaccine candidate GNA1870 binds the complement regulatory protein factor H and enhances serum resistance. J Immunol. 2006;177:501‐510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Caesar JJ, Lavender H, Ward PN, et al. Competition between antagonistic complement factors for a single protein on N. meningitidis rules disease susceptibility. eLife. 2014;3:e04008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Sansbury FH, Cordell HJ, Bingham C, et al. Factors determining penetrance in familial atypical haemolytic uraemic syndrome. J Med Genet. 2014;51:756‐764. [DOI] [PubMed] [Google Scholar]
- 106. Esparza‐Gordillo J, Jorge EG, Buil A, et al. Predisposition to atypical hemolytic uremic syndrome involves the concurrence of different susceptibility alleles in the regulators of complement activation gene cluster in 1q32. Hum Mol Genet. 2005;14:703‐712. [DOI] [PubMed] [Google Scholar]
- 107. Sánchez‐Moreno A, de la Cerda F, Rodríguez‐Barba A, et al. Is the atypical hemolytic uremic syndrome risk polymorphism in membrane cofactor protein MCPggaac relevant in kidney transplantation? A case report. Pediatr Transplant. 2021;25:e13903. [DOI] [PubMed] [Google Scholar]
- 108. Gold B, Merriam JE, Zernant J, et al. Variation in factor B (BF) and complement component 2 (C2) genes is associated with age‐related macular degeneration. Nat Genet. 2006;38:458‐462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Maller JB, Fagerness JA, Reynolds RC, Neale BM, Daly MJ, Seddon JM. Variation in complement factor 3 is associated with risk of age‐related macular degeneration. Nat Genet. 2007;39:1200‐1201. [DOI] [PubMed] [Google Scholar]
- 110. Maller J, George S, Purcell S, et al. Common variation in three genes, including a noncoding variant in CFH, strongly influences risk of age‐related macular degeneration. Nat Genet. 2006;38:1055‐1059. [DOI] [PubMed] [Google Scholar]
- 111. Montes T, Tortajada A, Morgan BP, Rodríguez de Córdoba S, Harris CL. Functional basis of protection against age‐related macular degeneration conferred by a common polymorphism in complement factor B. Proc Natl Acad Sci USA. 2009;106:4366‐4371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Heurich M, Martínez‐Barricarte R, Francis NJ, et al. Common polymorphisms in C3, factor B, and factor H collaborate to determine systemic complement activity and disease risk. Proc Natl Acad Sci USA. 2011;108:8761‐8766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113. Rodriguez E, Nan R, Li K, Gor J, Perkins SJ. A revised mechanism for the activation of complement C3 to C3b: a molecular explanation of a disease‐associated polymorphism. J Biol Chem. 2015;290:2334‐2350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. López‐Perrote A, Harrison RES, Subías M, et al. Ionic tethering contributes to the conformational stability and function of complement C3b. Mol Immunol. 2017;85:137‐147. [DOI] [PubMed] [Google Scholar]
- 115. Harris CL, Heurich M, Rodríguez de Córdoba S, Morgan BP. The complotype: dictating risk for inflammation and infection. Trends Immunol. 2012;33:513‐521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. de Vries RR, Meera Khan P, Bernini LF, van Loghem E, van Rood JJ. Genetic control of survival in epidemics. J Immunogenet. 1979;6:271‐287. [DOI] [PubMed] [Google Scholar]
- 117. Alper CA, Awdeh Z, Raum D, Yunis EJ. Complement genes of the major histocompatibility complex (complotypes), extended haplotypes and disease markers. Biochem Soc Symp. 1986;51:19‐28. [PubMed] [Google Scholar]
- 118. Esparza‐Gordillo J, Soria JM, Buil A, et al. Genetic and environmental factors influencing the human factor H plasma levels. Immunogenetics. 2004;56:77‐82. [DOI] [PubMed] [Google Scholar]
- 119. Almasy L, Blangero J. Multipoint quantitative‐trait linkage analysis in general pedigrees. Am J Hum Genet. 1998;62:1198‐1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Subías Hidalgo M, Yébenes H, Rodríguez‐Gallego C, et al. Functional and structural characterization of four mouse monoclonal antibodies to complement C3 with potential therapeutic and diagnostic applications. Eur J Immunol. 2017;47:504‐515. [DOI] [PubMed] [Google Scholar]
- 121. Rondelli T, Risitano AM, de Latour RP, et al. Polymorphism of the complement receptor 1 gene correlates with the hematologic response to eculizumab in patients with paroxysmal nocturnal hemoglobinuria. Haematologica. 2014;99:262‐266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Brubaker WD, Crane A, Johansson JU, et al. Peripheral complement interactions with amyloid beta peptide: erythrocyte clearance mechanisms. Alzheimers Dement. 2017;13:1397‐1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123. Feinberg BB, Jack RM, Mok SC, Anderson DJ. Low erythrocyte complement receptor type 1 (CR1, CD35) expression in preeclamptic gestations. Am J Reprod Immunol. 2005;54:352‐357. [DOI] [PubMed] [Google Scholar]
- 124. Acar IE, Willems E, Kersten E, et al. Semi‐quantitative multiplex profiling of the Complement system identifies associations of Complement proteins with genetic variants and metabolites in age‐related macular degeneration. J Pers Med. 2021;11:1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125. Strunz T, Grassmann F, Gayán J, et al. A mega‐analysis of expression quantitative trait loci (eQTL) provides insight into the regulatory architecture of gene expression variation in liver. Sci Rep. 2018;8:5865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126. Orozco LD, Chen HH, Cox C, et al. Integration of eQTL and a single‐cell atlas in the human eye identifies causal genes for age‐related macular degeneration. Cell Rep. 2020;30:1246‐1259.e6. [DOI] [PubMed] [Google Scholar]
- 127. Ratnapriya R, Sosina OA, Starostik MR, et al. Retinal transcriptome and eQTL analyses identify genes associated with age‐related macular degeneration. Nature Genet. 2019;51:606‐610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128. Spencer KL, Hauser MA, Olson LM, et al. Deletion of CFHR3 and CFHR1 genes in age‐related macular degeneration. Hum Mol Genet. 2008;17:971‐977. [DOI] [PubMed] [Google Scholar]
- 129. Lorés‐Motta L, van Beek AE, Willems E, et al. Common haplotypes at the CFH locus and low‐frequency variants in CFHR2 and CFHR5 associate with systemic FHR concentrations and age‐related macular degeneration. Am J Hum Genet. 2021;108:1367‐1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130. Cipriani V, Tierney A, Griffiths JR, et al. Beyond factor H: the impact of genetic‐risk variants for age‐related macular degeneration on circulating factor‐H‐like 1 and factor‐H‐related protein concentrations. Am J Hum Genet. 2021;108:1385‐1400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131. Smailhodzic D, Klaver CCW, Klevering BJ, et al. Risk alleles in CFH and ARMS2 are independently associated with systemic complement activation in age‐related macular degeneration. Ophthalmology. 2012;119:339‐346. [DOI] [PubMed] [Google Scholar]
- 132. Hecker LA, Edwards AO, Ryu E, et al. Genetic control of the alternative pathway of complement in humans and age‐related macular degeneration. Hum Mol Genet. 2010;19:209‐215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133. van de Ven JP, Nilsson SC, Tan PL, et al. A functional variant in the CFI gene confers a high risk of age‐related macular degeneration. Nat Genet. 2013;45:813‐817. [DOI] [PubMed] [Google Scholar]
- 134. Fagerness JA, Maller JB, Neale BM, Reynolds RC, Daly MJ, Seddon JM. Variation near complement factor I is associated with risk of advanced AMD. Eur J Hum Genet. 2009;17:100‐104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135. Klein RJ, Zeiss C, Chew EY, et al. Complement factor H polymorphism in age‐related macular degeneration. Science. 2005;308:385‐389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Edwards AO, Ritter R III, Abel KJ, Manning A, Panhuysen C, Farrer LA. Complement factor H polymorphism and age‐related macular degeneration. Science. 2005;308:421‐424. [DOI] [PubMed] [Google Scholar]
- 137. Haines JL, Hauser MA, Schmidt S, et al. Complement factor H variant increases the risk of age‐related macular degeneration. Science. 2005;308:419‐421. [DOI] [PubMed] [Google Scholar]
- 138. Martinez‐Barricarte R, Recalde S, Fernández‐Robredo P, et al. Relevance of complement factor H‐related 1 (CFHR1) genotypes in age‐related macular degeneration. Invest Ophthalmol Vis Sci. 2012;53:1087‐1094. [DOI] [PubMed] [Google Scholar]
- 139. Pappas CM, Zouache MA, Matthews S, et al. Protective chromosome 1q32 haplotypes mitigate risk for age‐related macular degeneration associated with the CFH‐CFHR5 and ARMS2/HTRA1 loci. Hum Genomics. 2021;15:60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140. Martinez‐Barricarte R, Pianetti G, Gautard R, et al. The complement factor H R1210C mutation is associated with atypical hemolytic uremic syndrome. J Am Soc Nephrol. 2008;19:639‐646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141. Bresin E, Rurali E, Caprioli J, et al. Combined complement gene mutations in atypical hemolytic uremic syndrome influence clinical phenotype. J Am Soc Nephrol. 2013;24:475‐486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142. Pickering MC, de Jorge EG, Martinez‐Barricarte Ŕ, et al. Spontaneous hemolytic uremic syndrome triggered by complement factor H lacking surface recognition domains. J Exp Med. 2007;204:1249‐1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143. Le Clech A, Simon‐Tillaux N, Provôt F, et al. Atypical and secondary hemolytic uremic syndromes have a distinct presentation and no common genetic risk factors. Kidney Int. 2019;95:1443‐1452. [DOI] [PubMed] [Google Scholar]
- 144. Arjona E, Huerta A, Goicoechea de Jorge E, Rodríguez de Córdoba S. Familial risk of developing atypical hemolytic‐uremic syndrome. Blood. 2020;136:1558‐1561. [DOI] [PubMed] [Google Scholar]
- 145. Wong EKS, Kavanagh D. Diseases of complement dysregulation—an overview. Semin Immunopathol. 2018;40:49‐64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146. Raychaudhuri S, Iartchouk O, Chin K, et al. A rare penetrant mutation in CFH confers high risk of age‐related macular degeneration. Nat Genet. 2011;43:1232‐1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147. Recalde S, Tortajada A, Subias M, et al. Molecular basis of factor H R1210C association with ocular and renal diseases. J Am Soc Nephrol. 2016;27:1305‐1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148. Servais A, Frémeaux‐Bacchi V, Lequintrec M, et al. Primary glomerulonephritis with isolated C3 deposits: a new entity which shares common genetic risk factors with haemolytic uraemic syndrome. J Med Genet. 2007;44:193‐199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149. García‐Fernández J, Vilches‐Arroyo S, Olavarrieta L, Pérez‐Pérez J, Rodríguez de Córdoba S. Detection of genetic rearrangements in the regulators of Complement activation RCA Cluster by high‐throughput sequencing and MLPA. Methods Mol Biol. 2021;2227:159‐178. [DOI] [PubMed] [Google Scholar]
- 150. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17:405‐423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151. Li MM, Datto M, Duncavage EJ, et al. Standards and guidelines for the interpretation and reporting of sequence variants in cancer: a joint consensus recommendation of the Association for Molecular Pathology, American Society of Clinical Oncology, and College of American Pathologists. J Mol Diagn. 2017;19:4‐23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152. Matthijs G, Souche E, Alders M, et al. Guidelines for diagnostic next‐generation sequencing. Eur J Hum Genet. 2016;24:2‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153. Martín Merinero H, Zhang Y, Arjona E, et al. Functional characterization of 105 factor H variants associated with aHUS: lessons for variant classification. Blood. 2021;138:2185‐2201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154. Rodríguez de Córdoba S, Harris CL, Morgan BP, Llorca O. Lessons from functional and structural analyses of disease‐associated genetic variants in the complement alternative pathway. Biochim Biophys Acta. 2011;1812:12‐22. [DOI] [PubMed] [Google Scholar]
- 155. Schramm EC, Roumenina LT, Rybkine T, et al. Mapping interactions between complement C3 and regulators using mutations in atypical hemolytic uremic syndrome. Blood. 2015;125:2359‐2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156. Smith‐Jackson K, Yang Y, Denton H, et al. Hyperfunctional complement C3 promotes C5‐dependent atypical hemolytic uremic syndrome in mice. J Clin Invest. 2019;129:1061‐1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157. Subias Hidalgo M, Alonso Riano M, Juana‐Lopez L, Tisseires V, Lopez‐Rios J, Rodriguez‐de‐Cordoba S. eds.Mouse C3‐Glomerulopathy models based on gain‐of‐function C3 mutations. Mol Immunol. 2019;114:457‐458. [Google Scholar]
- 158. Wu J, Wu YQ, Ricklin D, Janssen BJ, Lambris JD, Gros P. Structure of complement fragment C3b‐factor H and implications for host protection by complement regulators. Nat Immunol. 2009;10:728‐733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159. Persson BD, Schmitz NB, Santiago C, et al. Structure of the extracellular portion of CD46 provides insights into its interactions with complement proteins and pathogens. PLoS Pathog. 2010;6:e1001122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160. Jozsi M, Licht C, Strobel S, et al. Factor H autoantibodies in atypical hemolytic uremic syndrome correlate with CFHR1/CFHR3 deficiency. Blood. 2008;111:1512‐1514. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing not applicable to this article as no datasets were generated or analysed during the current study.