

Abstract
Epipolythiodioxopiperazines (ETPs) are fungal secondary metabolites that share a 2,5‐diketopiperazine scaffold built from two amino acids and bridged by a sulfide moiety. Modifications of the core and the amino acid side chains, for example by methylations, acetylations, hydroxylations, prenylations, halogenations, cyclizations, and truncations create the structural diversity of ETPs and contribute to their biological activity. However, the key feature responsible for the bioactivities of ETPs is their sulfide moiety. Over the last years, combinations of genome mining, reverse genetics, metabolomics, biochemistry, and structural biology deciphered principles of ETP production. Sulfurization via glutathione and uncovering of the thiols followed by either oxidation or methylation crystallized as fundamental steps that impact expression of the biosynthesis cluster, toxicity and secretion of the metabolite as well as self‐tolerance of the producer. This article showcases structure and activity of prototype ETPs such as gliotoxin and discusses the current knowledge on the biosynthesis routes of these exceptional natural products.
Keywords: biosynthetic gene clusters, disulfide bridges, enzymatic reactions, fungi, toxins
Housing sulfur: Certain fungi produce diketopiperazine compounds with a transannular sulfur bridge. These so‐called epipolythiodioxopiperzines are toxic by non‐specifically interacting with intracellular proteins and contribute to fungal pathogenicity. Producer strains serve as bio‐pesticides but also question safety of food fermentation. Thus, understanding the structure and biosynthesis of these intriguing natural sulfur products is of utmost interest.
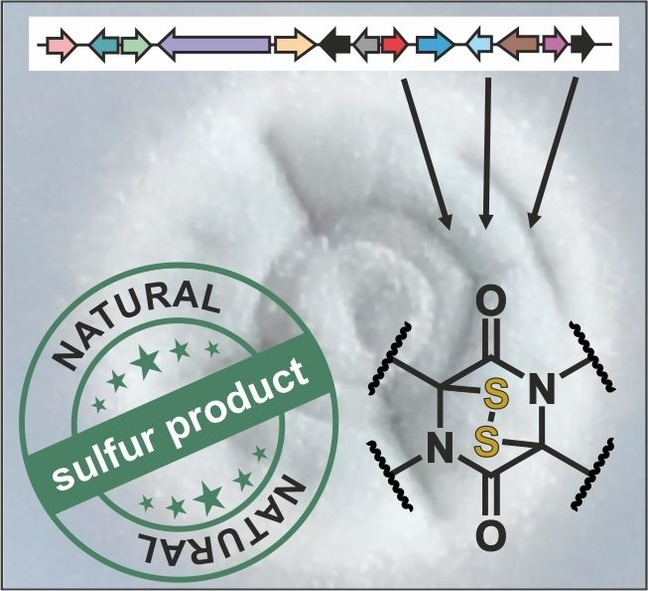
1. The Diketopiperazine Scaffold: Two Become One
Epipolythiodioxopiperazines (ETPs) are complex natural products that belong to the family of diketopiperazines (DKPs). DKPs, also known as dioxopiperazines, piperazinediones or DKP alkaloids, contain two amide linkages and according to the position of their carbonyl groups, they are designated as 2,3‐, 2,5‐ or 2,6‐isomers. 2,5‐variants are most prominent. [1] They serve as catalysts for organic synthesis, [2] are explored as self‐assembling building blocks in material science [3] and represent popular pharmacophores in medicinal chemistry. [4] As the smallest cyclodipeptide, the 2,5‐DKP framework overcomes the poor pharmacokinetic and pharmacodynamic properties of acyclic peptides, as their rigid and stable six‐membered ring structure with two hydrogen bond donors and acceptors confers enhanced resistance to proteases and better bioavailability.[ 4a , 5 ] Some even can cross the blood‐brain barrier and are considered as drug delivery systems. [6] In addition, 2,5‐DKPs feature two positions for stereochemical control and four sites for modifications to break their planar core structure.[ 4a , 7 ] All these favorable features have put 2,5‐DKPs at the forefront of medicinal chemistry efforts and led to their clinical development. For example, tadalafil (CialisTM [8] ), a 2,5‐DKP based inhibitor of phosphodiesterase‐5, has been approved for the treatment of pulmonary arterial hypertension and erectile dysfunction. Moreover, retosiban (GSK‐221149A), an oxytocin receptor antagonist, [9] is in late stage clinical evaluation for preterm labor [10] and plinabulin (NPI‐2358), a tubulin depolymerizing agent, is currently being evaluated in phase III trials for non‐small cell lung cancer. [11]
In nature, a plethora of structurally diverse and biologically active molecules, including ETPs, contain the 2,5‐DKP scaffold.[ 4a , 12 ] Although present in all kingdoms of life, microorganisms such as fungi and plant or animal venoms, are an especially rich source of compounds with a 2,5‐DKP framework. [5] The cyclic dipeptide results from head‐to‐tail condensation of two α‐amino acids either by non‐enzymatic or enzymatic processes. [13] Enzymes catalyzing this reaction in bacteria are most frequently cyclodipeptide synthases (CDPS). These small enzymes of about 30 kDa grab two aminoacyl‐tRNAs, usually used for translation, as substrates and link their amino acid cargo by two peptide bonds. [14] Besides, non‐ribosomal peptide synthetases (NRPS), large multi‐domain enzymes of more than 100 kDa, serve for cyclodipeptide production.[ 14b , 15 ] Apart from few bacterial examples, [16] these giant machineries are employed by fungi to assemble the 2,5‐DKP scaffold.
In either case, biosynthetic pathways usually combine two regular l‐amino acids, while synthetic chemistry approaches are also accessible to non‐natural building blocks such as d‐amino acids. Structural diversity and fine‐tuning of biological activities of natural DKP compounds is mostly achieved by tailoring enzymes acting up‐ or downstream of cyclodipeptide formation. For instance, hydrolases, methyl‐ and prenyltransferases, cytochrome P450 (CYP450) enzymes, cyclodipeptide oxidases or 2‐oxoglutarate‐dependent monooxygenases are known to decorate and modify the DKP skeleton. [17] The discovery of such tailoring enzymes and of CDPS,[ 14a , 18 ] the routine application of genetic code expansion tools [19] as well as the ability to engineer NRPS modules [20] fostered attempts of combinatorial biosynthetic approaches[ 17b , 21 ] and the use of non‐proteinogenic or non‐canonical amino acids to further increase the chemical space of 2,5‐DKPs. [22] Apart from tailoring enzymes, nature also evolved DKP dimers to further increase chemical diversity. These double‐decker‐like scaffolds received great attention from medicinal chemists, as the higher local concentration of pharmacophores often accounts for improved biological activity and potency. [7]
Among the myriad of DKPs known to date, the group of ETPs is by far the best characterized subclass. These monomeric or dimeric sulfur‐containing 2,5‐DKPs (for examples see Figure 1) are produced by fungi and associated with numerous biological and pharmacological activities.
Figure 1.

Chemical structures and names of prominent epipolythiodioxopiperazine (ETP) compounds. Prototype ETPs are grouped according to the type of their sulfur bridge and their higher‐order structure. In regularly bridged ETPs the disulfide moiety links the two Cα atoms of the 2,5‐diketopiperazine (2,5‐DKP) backbone. Irregularly bridged ETPs are characterized by deviations from this Cα–Cα connection and can be grouped according to the number of cycles their sulfur bridge spans. Sulfur linkages of the Cα–Cβ type only cross the DKP core (gliovirin and aspirochlorine), while epicoccin C presents with two bis‐cross‐ring bridges. In addition, dimeric ETP structures are known. They are classified according to the type of bond connecting their two units: C−C type for verticillin A and chaetocin versus C−N type for chetomin.
2. Epipolythiodioxopiperazines: Bridging the Ring
ETPs are toxic secondary metabolites, exclusively produced by terrestrial and marine fungi. However, the genetic elements required for ETP biosynthesis are discontinuously distributed among fungi. [23] Often, only few of several closely related species are ETP producers, but sometimes even distant relatives can biosynthesize the same ETP. For example, gliotoxin is produced by Aspergillus fumigatus and non‐related Trichoderma, Penicillium or Candida species, [24] but Aspergillus nidulans is a non‐producer. [25] Some ascomycetes (e. g. Saccharomyces cerevisiae, Schizosaccharomyces pombe, and Candida albicans) or basidiomycetes (e. g. Ustilago maydis and Cryptococcus neoformans) also lack the genetic information for ETP production. [23a] Both lateral gene transfer and loss of genetic material during evolution could account for this disparate spreading of ETP gene cassettes among fungi.[ 23b , 24 ] In addition, most ETP biosynthesis gene clusters have a common origin and diversified during evolution to yield the various ETP structures known to date. [23b] Although ETPs are not essential for survival, they are considered to confer fungi selective advantages under certain environmental conditions and in competition with other microorganisms. However, experimental evidence for this hypothesis is still missing. In fact, competition experiments with an ETP‐producing strain and a non‐producer did not show any selective advantage. [26]
Structurally, ETPs are compounds with a transannular di‐ or polysulfide bridge. In contrast to disulfide bonds in proteins, the sulfur atoms of ETPs adopt an eclipsed conformation. Despite being strained, the sulfide moiety is stabilized by strong n→π* interactions. [27] ETPs contain at least one aromatic amino acid and are mainly regularly (1,4) bridged, meaning that the sulfur atoms are anchored at the Cα positions of the cyclodipeptide core.[ 24 , 28 ] Yet, there are also irregularly bridged compounds, with the sulfur atoms connecting for example the Cα atom of one amino acid and the Cβ position of the second, and their number is continuously growing. [29]
As reductive agents like dithiothreitol interfere with the biological activity of ETPs, it was proposed that the di‐/polysulfide functionalities are essential for toxicity. [30] The current model of ETP action is that the oxidized form is taken up into cells, reduced by glutathione and consequently trapped. This accumulation is thought to enhance toxicity. [31] Intracellularly, ETPs are prone to cycles of reduction and oxidation, thereby producing reactive oxygen species that lead to DNA damage. [32] Conjugation to accessible Cys residues of enzymes,[ 32a , 33 ] depletion of functionally relevant zinc ions from proteins and induction of protein denaturation [34] are other mechanisms of action reported for the ETP ‘warhead’. Various studies revealed that ETPs exhibit their toxicity not by targeting a single protein or a specific pathway but rather by various non‐specific interactions of their disulfide moiety with functionally relevant intracellular biomolecules. For this reason, the biological effects reported for ETPs are diverse, ranging from antibacterial, antifungal and antiviral to antitumor or immunomodulatory activities.[ 12 , 35 ] These bioactivities initially rendered ETPs attractive for medicinal applications, but the lack of a defined target and the ability to control side effects, as well as the inherent toxicity of ETPs so far hampered clinical utility. [28] In addition, difficulties to access the compounds in large amounts either by purification or chemical synthesis and the instability of the reactive sulfur bridge hinder exploration of therapeutic applications.
In the following sections, the current knowledge on biological activities and biosynthesis routes of individual but prototype ETPs will be summarized and discussed. In focus are epidithio compounds which are characterized by a disulfide bridge spanning the 2,5‐DKP ring.
2.1. Regularly bridged ETPs
2.1.1. Gliotoxin
Gliotoxin was the first member of the ETP class that was discovered (Figure 1). Its name is derived from the wood fungus Gliocladium fimbriatum (synonym: Albifimbria/Myrothecium verrucaria [36] ), but the original source organism likely was Trichoderma viride (synonym: Trichoderma lignorium). [37] To date several Aspergillus, Penicillium, Gliocladium, Thermoascus, Candida, and Trichoderma strains are known to biosynthesize gliotoxin. Among them: the Trichoderma virens strain G‐20, which is merchandised as a bio‐pesticide under the trade name SoilGardTM (Certis, USA), [38] and Aspergillus fumigatus, the most relevant and most effective producer of gliotoxin.[ 25 , 39 ] Gliotoxin is the best characterized ETP and therefore considered as prototype.
2.1.1.1. Biological activity
Over the last decades, numerous studies probed the pleiotropic biological activities of gliotoxin, that all are assumed to be linked to the epidithio bridge. Initially, gliotoxin was shown to have anti‐viral activity [40] by interfering with viral RNA replication. [41] Moreover, it was investigated as an immunosuppressive agent in transplantation, [42] as it potently induces apoptosis of immune cells. [43] In the course of these studies, it was noted that accumulation of IκB prevents NF‐κB activation and accounts for the immunosuppression by gliotoxin. [44] After IκB has been found to be a substrate of the proteasome, [45] gliotoxin was investigated for its potential to inhibit 20S proteasome core particles and indeed found to block the chymotrypsin‐like activity. [46] However, the inhibition required high concentrations as well as oxidative conditions[ 46 , 47 ] and the mechanism of action remained elusive, although in the meanwhile the binding modes of many other natural and synthetic 20S proteasome inhibitors have been characterized by structural means. [48] In addition, gliotoxin has been reported to inhibit the proteasome from Plasmodium falciparum and has been proposed as a new antimalarial drug, [49] but still the molecular basis of proteasome inhibition remains unknown and the therapeutic potential of gliotoxin in this context was not further pursued. Altogether it is questionable, whether gliotoxin directly inhibits the 20S proteasome or whether the observed inhibition is a secondary effect of for example unspecific protein damage.
Only recently, gliotoxin and other ETPs were reported to inhibit Rpn11, a zinc‐dependent deubiquitinating enzyme and an essential component of the 19S regulatory particle of proteasomes responsible for ubiquitin‐mediated protein degradation in cells. [47] Notably, the mechanism of action involves binding to the catalytic zinc ion [47] and reminds of the finding that ETPs block the interaction of HIF1α with p300 by zinc ejection from the latter. [34b]
Besides, gliotoxin has been reported to target a number of other intracellular proteins, including farnesyltransferase, [50] geranylgeranyltransferase, [51] alcohol dehydrogenase, [32a] rabbit skeletal ryanodine receptor, [52] creatine kinase, [33a] adenine nucleotide transporter, [53] NADPH oxidase, [54] and glutaredoxin. [55] Moreover, gliotoxin triggers the release of calcium, [56] magnesium, [57] and cytochrome c from mitochondria [58] and activates the proapoptotic B‐cell lymphoma 2 (Bcl‐2) protein Bak [58a] via the c‐Jun N‐terminal kinase [59] to induce apoptosis. In this context, gliotoxin has also been reported to stimulate caspase‐3. [60] Last but not least, gliotoxin can act as an anti‐cancer agent[ 51 , 61 ] and inhibit angiogenesis.[ 61c , 62 ] All in all, gliotoxin affects many proteins and intracellular processes most likely by non‐specific interactions.
Gliotoxin is also considered as one of several virulence factors of A. fumigatus and assumed to contribute to the pathogenesis of mycoses such as invasive aspergillosis which is a deadly threat for immunocompromised patients. [63] In support of this hypothesis, gliotoxin has been identified in Aspergillus isolates of patients. [39a] However, the impact of gliotoxin on virulence of A. fumigatus appears to depend on the host and its immune status [64] as well as on the genetic and phenotypic background of the pathogen. [65] For example, both A. fumigatus and Aspergillus fischeri are able to biosynthesize gliotoxin, but only A. fumigatus is a human pathogen and the closely related A. fischeri is not. [65] In light of these findings, gliotoxin should be better considered as a defense molecule or as an anti‐oxidant [66] that provides the producer under certain conditions in its ecological niche with a selective advantage.
2.1.1.2. Structure and biosynthesis
Formation of the DKP core
Gliotoxin was discovered in 1936, but its structure was determined only in 1958. [67] At the same time, studies on the biosynthesis of gliotoxin were launched. Isotope‐labelling and feeding experiments with T. viride showed that the amino acids Phe and Ser are incorporated into gliotoxin and that Met serves as donor for the N‐methyl group. [68] In 1967, the crystal structure of gliotoxin and its absolute stereochemistry were published (Figure 1), [69] but the biosynthesis remained elusive for half a century. Only after the complete genomic sequence of A. fumigatus had become available in 2005, [70] the gene locus for the production of gliotoxin was predicted [71] based on homology to the previously identified sirodesmin biosynthesis gene cluster (see section 2.1.2.). [72] Twelve genes were assigned to the putative gliotoxin cassette and enzyme functions were predicted according to sequence comparisons. [71] In 2010, a 13th gene was shown to be essential for gliotoxin biosynthesis and hence included in the gli gene locus (Figure 2A). [66b] The putative function of the NRPS gene gliP in formation of the DKP scaffold [71] was confirmed by its disruption. [73] Cloning of the gliP gene and in vitro activity assays finally established that the A1 and A2 domains of the NRPS GliP activate l‐Phe and l‐Ser, respectively, [74] thereby confirming the initial feeding experiments. The slow release of the reaction product from the enzyme was supposed to arise from the lack of a thioesterase domain and led to the hypothesis that downstream processing of the DKP might occur while still being tethered to the NRPS. [74] Today, this on‐line tailoring seems unlikely, as many different intermediates of gliotoxin biosynthesis have been isolated as free metabolites from individual knockout strains. [75] Furthermore, the exceptional domain architecture of GliP was puzzling. Its extra condensation (C) and transfer (T) module (C2‐T3) at the C‐terminus was not expected to be required for dipeptide formation. [74] Later on however, it was noted that fungal cyclic peptides are generally synthesized by NRPS containing a terminal C domain. [77] Subsequently, in vitro and in vivo mutagenesis confirmed that instead of spontaneous cyclization, the DKP core of gliotoxin is cyclized by the action of the extra C2‐T3 unit of GliP. [78]
Figure 2.

Genetic elements required for gliotoxin production and current biosynthesis scheme. (A) Schematic view of the gliotoxin (gli) biosynthesis gene cluster from A. fumigatus.[ 66b , 71 ] Genes are illustrated as colored and labelled arrows. Black ones have not yet been assigned a specific function. Open reading frames within the gli locus are shaded against a gray background (left), while those outside the biosynthetic gene cluster (right) are not. Verified or predicted functions of gene products are listed. (B) Current reaction scheme for the biosynthesis of gliotoxin. The NRPS GliP is activated by the phosphopantetheinyltransferase PptA encoded outside the gli gene cluster and fuses the two starter amino acids l‐Phe (blue) and l‐Ser (red) to the 2,5‐DKP scaffold. The reaction steps until the free dithiol precursor are confirmed and fix in sequence. GliM might act as an O‐methyltransferase on a transient, yet unknown intermediate or on a shunt metabolite as indicated. The order of modifications catalyzed by GliF, GliN and GliT appears to be interchangeable. The reaction trajectory of GliF is unknown, but two potential mechanisms have been suggested. [75b] Disulfide‐bridged gliotoxin is exported outside the fungal cell by a transport protein, while bis‐thiomethylation of reduced gliotoxin by the S‐methyltransferase TmtA (also known as GtmA) encoded outside the gli gene cluster depletes oxidized gliotoxin and thereby serves to dampen expression of gliotoxin biosynthesis genes and ultimately ETP production.[ 75d , 76 ] For details see section 2.1.1.2.
As with all NRPS, the T domains have to be posttranslationally activated by transfer of a phosphopantetheinyl moiety from coenzyme A onto a conserved serine residue. The corresponding enzyme of A. fumigatus has been identified as PptA (Figure 2B). PptA primes GliP as well as other NRPS of A. fumigatus and thus serves a universal function for fungal secondary metabolism. For this reason, PptA is encoded outside the gli gene cluster (Figure 2A) and considered as a potential new drug target for fungicides. [79]
The sulfurization step
In 2011, two groups independently proposed that a bis‐hydroxylated l‐Phe‐l‐Ser‐DKP intermediate may be relevant for subsequent sulfurization.[ 75a , 75e ] Gene deletion and overexpression experiments later clarified that the CYP450 oxidase GliC hydroxylates the Cα atoms of the l‐Phe‐l‐Ser‐DKP framework.[ 75c , 80 ] The loss of two molecules of water was proposed to create the electrophiles to which subsequently glutathione (GSH) is attached by the action of the glutathione‐S‐transferase (GST) GliG.[ 75a , 75e ] This coupling of oxygenation and sulfurization reactions reminds of phase I/phase II detoxification pathways for xenobiotics [81] and is one of few examples in which GSH is used as a source of sulfur (Figure 2B). [82]
Recently, the original mechanistic proposal for activation and sulfur transfer was refined based on the crystal structure of the GliG enzyme in complex with its reaction product, the bis‐glutathionylated DKP. [83] This study revealed that GliG creates a favorable environment to selectively eliminate water from one Cα position of the dihydroxy DKP and subsequently attaches activated GSH to the resulting electrophilic carbon. Although the stereochemistry of the bis‐hydroxylated DKP is not known, the X‐ray structure of GliG implies that release of water and addition of GSH occur from the same side of the DKP. This way a mono‐glutathionylated and at the same time mono‐hydroxylated DKP is produced as an intermediate, which is processed to the bis‐glutathione adduct by repeating the SN1 reaction cascade on the other side of the DKP core. [83]
Dissecting glutathione
Upon linkage of the DKP scaffold to two GSH molecules, three sequential enzymatic reactions uncover the free thiols (Figure 2B): In a first step, the γ‐glutamyl‐cyclotransferase GliK cleaves the isopeptide bond in the glutathione moieties by release of 5‐oxoproline. [84] The functional importance of GliK for gliotoxin production in vivo has been confirmed by deletion experiments. [85] To the best of my knowledge only a single biosynthetic pathway besides gliotoxin makes use of such a reaction. Enzymatic production of the aminoglycoside antibiotic butirosin involves the γ‐glutamyl‐cyclotransferase BtrG, but like for GliK the reaction mechanism is elusive. [86]
Next, the metal‐dependent dipeptidase GliJ chops off the Gly residue of glutathione to create a bis(cysteinyl) product. [84] The crystal structure of GliJ revealed an unexpected metal promiscuity and implies that both sides of the gliotoxin precursor are processed sequentially, [87] as noted for GliG. Although many of the enzymes involved in gliotoxin biosynthesis, e. g. GliG, GliJ and GliI are homodimeric, the crystal stuctures of GliG and GliJ revealed that the active sites are too far apart from each other to enable simultaneous processing of both substrate halves.[ 83 , 87 ] Thus, the general principle appears to be consecutive rather than simultaneous tailoring of the two pseudosymmetric DKP sides.
Finally, the enzyme GliI creates the free dithiol precursor of gliotoxin. Originally annotated as a 1‐aminocyclopropane‐1‐carboxylic acid synthase, GliI acts as a pyridoxal 5’‐phosphate (PLP)‐dependent C−S bond lyase, phylogenetically related to alliinases. [88] The current mechanistic proposal is derived from that of aminotransferases and first requires aldimine formation between the α‐amino group of one Cys residue on the DKP and the cofactor. Following abstraction of the Cα proton of Cys, a ketimine is formed that subsequently undergoes β‐elimination to yield the dithiol precursor of gliotoxin and a PLP‐linked imine that spontaneously hydrolyzes to pyruvate and ammonia. [88]
All good things come in threes: establishing the three‐ring structure
Considering the structure of gliotoxin, it was tempting to speculate that Tyr could serve as a building block for or as an intermediate of biosynthesis. Yet, both hypotheses were disproven by profound isotope labelling experiments. [89] Instead, an arene oxide intermediate en route to gliotoxin was proposed. Nucleophilic attack of the epoxide by the amide backbone nitrogen atom of Phe was anticipated to trigger heterocyclization and to create the hydroxyl group as a leftover.[ 89a , 90 ] This mechanistic proposal was later on substantiated by additional feeding experiments. [89b] Another line of evidence was provided with the isolation of a gliotoxin derivative containing a spiro atom. It was speculated that oxidation of the phenyl ring to the corresponding epoxide might not be entirely stereoselective, thereby leading to two distinct diastereomers that upon nucleophilic attack could lead to gliotoxin as well as to the observed spiro compound. [91] In addition, the structures and biosynthetic pathways of three related ETPs argue in favor of the epoxidation reaction: Gliovirin contains an epoxidated phenyl ring (Figure 1; see section 2.2.1.), [92] aranotins features an oxepin ring structure [93] (Figure 1; see section 2.1.3.) that likely emerges from an arene oxide, [94] and the proposed biosynthesis route of peniciadametizines involves an epoxidated phenyl ring. [95]
Despite all these mechanistic insights, only in 2021, the enzyme oxidizing the phenylalanine side chain and establishing the pyrrolidine ring in gliotoxin was confirmed to be the membrane‐resident CYP450 monooxygenase GliF. [75b] Although the authors consider the epoxide route mechanistically reasonable, they also discuss a possible alternative that supposes a radical at the amide nitrogen of Phe, which upon closure of the pyrrolidine ring would be quenched by a water molecule and lead to the hydroxyl group at the phenyl ring (Figure 2B). [75b]
‐Methylation
About 23 % of all known S‐adenosyl‐l‐methionine (SAM)‐dependent methyltransferases alkylate nitrogen atoms. [96] While amines often serve as acceptor sites, methylation of amide nitrogen atoms is rare. Examples are however known from NRPS or PKS modules [97] and from ansamitocin, [98] omphalotin, [99] cyclo(Trp‐Trp), [100] and gliotoxin biosynthesis. In 2014, the enzyme GliN has been identified as a N‐methyltransferase involved in gliotoxin biosynthesis. Surprisingly, N‐desmethylgliotoxin, a gliotoxin variant missing the methyl group on the amide nitrogen atom of Ser, is highly instable and about 100‐fold less bioactive than the corresponding N‐methylated version. [75d] Probably, N‐methylation of gliotoxin is required for cell permeability and stability, as noted for other bioactive cyclic peptides. [101] In support of this hypothesis primary and secondary but not tertiary amines have been predicted to induce decomposition of the polysulfur compound pentathiepin. [102]
Installing the epidithio bridge
The disulfide bond of ETPs is essential for their bioactivity. Initially it was suspected from synthetic studies that air oxidation of the dithiol precursor might be sufficient to install the sulfur bridge. [103] In 2010 however, the flavin adenine dinucleotide (FAD)‐dependent oxidoreductase GliT was reported to produce gliotoxin from the corresponding dithiol (Figure 2B).[ 66b , 104 ] The crystal structure of GliT revealed a disulfide bridge at the active site that in a disulfide exchange reaction is transferred onto the substrate. [105] A transient charge‐transfer complex and a cascade of electron pair movements drain off the electrons to FAD and finally to molecular oxygen, and recover the enzyme's active site.[ 104 , 105 ] Furthermore, an A. fumigatus ΔgliT strain is severely impaired in growth and highly susceptible to exogenous gliotoxin compared to wild type, suggesting that GliT plays a crucial role in self‐resistance.[ 66b , 104 ] In agreement, in the absence of GliT, reduced gliotoxin accumulates intracellularly, conjugates to proteins and exerts its toxicity.[ 66b , 104 ]
Considering these and other results, questions about the sequence of reaction steps during gliotoxin biosynthesis arose. It appears that the initial steps of the biosynthetic route are fixed in their order, whereas later reactions (after the emergence of the dithiol intermediate) might be catalyzed randomly. GliT has been shown to oxidize reduced gliotoxin, suggesting that this reaction finishes the pathway. [104] On the other hand, it was reported that GliF [75b] and GliN [75d] act on gliotoxin intermediates featuring the epidithio bridge, implying that GliT might also work upstream of GliF and GliN, and feature relaxed substrate specificity. In this context, it is interesting to note that GliT is also able to oxidize the free thiols of reduced holomycin to yield the natural disulfide‐bridged antibiotic. [106] The different chemical structures of holomycin and gliotoxin underpin the substrate promiscuity of GliT.
Secretion of gliotoxin
To facilitate secretion of the toxin into the extracellular environment, the gli gene cluster encodes a transporter, termed GliA (Figure 2). Deletion of the gliA gene reduces the virulence of A. fumigatus and concomitantly increases its sensitivity to gliotoxin, suggesting that this translocase plays a role in pathogenicity and self‐protection. [107] In contrast to the ATP‐binding cassette (ABC) transporter encoded in the sirodesmin biosynthesis gene cluster, GliA is a major facilitator superfamily (MFS) member. [71] These efflux pumps facilitate movement of small molecules along their concentration gradient across membranes and also play a role in drug resistance. [108]
Regulation of gliotoxin biosynthesis
GliZ: The genes that are part of the gliotoxin biosynthesis gene cluster are transcriptionally co‐regulated by the zinc finger transcription factor GliZ.[ 71 , 109 ] GliZ is a distant homologue of the prominent GAL4 transcription factor of yeast [110] and a member of the zinc cluster/binuclear family that is limited to fungi. This class of regulators is hallmarked by a coiled‐coil domain required for dimerization and a CysX2CysX6CysX5‐12CysX2CysX6‐8Cys motif that coordinates two zinc ions (Zn2Cys6 domain).[ 110 , 111 ] The DNA binding site of Zn2Cys6 fingers is usually a palindromic arrangement of trinucleotides with a defined spacer in between. [112] Consistently, analysis of gli promoter sequences identified the consensus sequence TCGGN3CCGA upstream of all gli genes except for gliZ and gliA, [113] but experimental validation of this motif is currently missing.
RglT: Discovered as a GAL4‐like Zn2Cys6 transcription factor that confers resistance to oxidative stress in A. fumigatus, RglT (regulator of gliotoxin) acts upstream of GliZ. It induces expression of gliZ, gliT and gliF genes and is essential for protecting A. fumigatus against exogenous gliotoxin. In agreement, a strain deficient in RglT fails to produce disulfide‐bridged gliotoxin. [114] These observations indicate that RglT positively regulates GliT levels and thereby contributes to gliotoxin resistance and production. Notably, according to phylogenetic analyses, homologues of RglT and GliT are also present in Aspergillus species that do not produce gliotoxin and the protective function of RglT as well as the RglT‐mediated control of gliT expression are conserved. [114] However, the primary function of GliT and RglT in non ETP‐producers remains to be investigated. Notably, only very recently, KojR, another GAL4‐like Zn2Cys6 transcription factor acting upstream of RglT and controlling its expression was identified. [115]
GipA: Besides, the Cys2His2 zinc finger transcription factor GipA positively controls gliotoxin production. [116] A binding site for GipA was identified in the promoter region of gliA, close to a putative recognition site for GliZ, suggesting that both transcription factors might act interdependently to control intracellular gliotoxin levels. [116]
LaeA: Another positive regulator of gliotoxin biosynthesis and generally of secondary metabolism in A. fumigatus is LaeA. [117] It controls expression of about 10 % of the genome of A. fumigatus, [118] but its mechanism is unknown. LaeA is part of the heterotrimeric velvet complex that coordinates fungal metabolism and development in response to light. [119] Its nuclear localization and its activity as a SAM‐dependent methyltransferase implied a function as a chromatin remodeler. [117a] However so far, this putative function could not be confirmed. Instead it was noted that LaeA undergoes automethylation, but this posttranslational modification seems to be dispensable for its in vivo function. [120]
Gliotoxin: Strikingly, gliotoxin itself is also a positive regulator of gli gene cluster expression. Exogenous gliotoxin for example stimulates the expression of several gli genes in A. fumigatus. [66b] Furthermore, in a ΔgliP background, significantly reduced expression of the gli gene cluster was noted but could be restored to wild type or even higher levels by the addition of exogenous gliotoxin. [73a]
Bis(methylthio)gliotoxin: As a counterpart to the above mentioned factors that stimulate gliotoxin biosynthesis, the metabolite bis(methylthio)gliotoxin (also known as bisdethiobis(methylthio)gliotoxin; BmGT) has been discovered to attenuate gliotoxin production. [76] Bis(methylthio)gliotoxin is a biologically inactive congener of gliotoxin, produced by the S‐methyltransferase TmtA (also known as GtmA) encoded outside the gli locus.[ 75d , 76 ] TmtA uses the reduced dithiol form of gliotoxin to irreversibly methylate the sulfhydryl groups (Figure 2B). At a first glance this SAM‐dependent reaction competes with GliT‐mediated reversible oxidation of the thiols to the disulfide bridge. However, TmtA appears to have lower affinity for the dithiol substrate than GliT and in agreement moderately contributes to self‐protection of the fungus compared to GliT.[ 75d , 76 ] Nonetheless, heterologous expression of tmtA in S. cerevisiae confers resistance to exogenous gliotoxin [121] and simultaneous deletion of tmtA and gliT from A. fumigatus causes hypersensitivity to gliotoxin, [122] implying that TmtA is important as a second line of defense.
Expression of tmtA is induced by gliotoxin and leads to the conversion of reduced gliotoxin to bis(methylthio)gliotoxin, thereby depleting oxidized gliotoxin and dampening gli gene cluster expression as well as ultimately gliotoxin biosynthesis. Apart from its regulatory function, bis(methylthio)gliotoxin was considered as a potential diagnostic marker of invasive aspergillosis [123] but did not qualify. [124]
Because S‐methyltransferases are rare in nature (3 %), [96] it was of considerable interest to solve the X‐ray structure of TmtA and to understand its catalytic activity. In the end, TmtA was crystallized by two distinct groups but unfortunately without bound substrate or product.[ 122 , 125 ] The reaction cycle was modelled by computational techniques and suggested that exchange of S‐adenosyl‐l‐homocysteine (SAH) by SAM occurs ‘on the fly’ before mono(methylthio)gliotoxin is released. [125] Activity assays however indicate that both bis(methylthio)gliotoxin and mono(methylthio)gliotoxin accumulate in solution. [122] In agreement, the experimental KM value for reduced gliotoxin is almost fivefold lower than for mono(methylthio)gliotoxin, implying that reduced gliotoxin is the preferred substrate. [122] Furthermore, comparison of SAM‐ and SAH‐ bound TmtA revealed structural changes that might be relevant for catalysis. [122]
Biologically inactive, bis‐thiomethylated versions have also been described for other ETPs, including acetylaranotin (see section 2.1.3.), [28] sporidesmin (see section 2.1.4.), [28] bionectin A [126] and glionitrin A. [127] These metabolites may serve a similar regulatory function as bis(methylthio)gliotoxin. TmtA‐like enzymes have been identified in numerous species of the Ascomycota phylum and strikingly even in fungi that lack an ETP cluster such as A. nidulans.[ 75d , 76 ] It thus appears, that fungi evolved the S‐methyltransferase either for regulation of their own ETP production or for defense against exogenous ETPs. Although TmtA is unable to convert sporidesmin, [76] A. nidulans and Fusarium oxysporum are able to detoxify gliotoxin by S‐methylation. [75d] In addition, S‐methylation is a self‐protection strategy also known from bacteria, such as Streptomyces clavuligerus, the producer of the antibiotic holomycin. [128]
In summary, gliotoxin biosynthesis is a complex and fine‐tuned metabolic pathway that is controlled by numerous factors and on several levels. This review selected the most prominent and most important regulators for discussion, but many more are known [81] and may be discovered in the future, as we are currently only starting to understand the sophisticated regulatory circuits that control ETP production.
Blind spots of the gli gene cassette: open reading frames (ORFs) coding for proteins of unknown function
Despite enormous progress in understanding the main route of gliotoxin formation, several aspects are still unaddressed. In particular, the function of the second methyltransferase encoded in the gene cluster is still unknown. GliM is a putative O‐methyltransferase and deletion of its gene interferes with gliotoxin production, [75d] suggesting an essential function. However, since the gliM knockout strain did not accumulate any stable biosynthesis intermediate, [75d] the function of GliM remains unresolved. An O‐methylated shunt product has been observed in a ΔgliG knockout strain by two independent groups (Figure 2B),[ 75a , 75e ] but its origin is unknown. The crystal structure of GliM visualizes a very flat active site pocket that may fit a non‐sulfurized gliotoxin precursor (unpublished results of E. M. Huber), implying that GliM could act upstream of GliG. It is well conceivable that GliM transiently modifies an intermediate of gliotoxin biosynthesis, [76] but experimental evidence is currently lacking.
Moreover, the function of GliH in gliotoxin biosynthesis is unclear. As a corresponding knockout strain was deficient in gliotoxin production, GliH may have an essential function in either ETP biosynthesis or secretion, whereas a function in self‐resistance to gliotoxin has been excluded. [66b]
2.1.2. Sirodesmins
2.1.2.1. Bioactivity
Sirodesmins have been first isolated as antiviral metabolites from Sirodesmium diversum (nowadays termed Coniosporium diversum) and later reported as phytotoxins produced by the plant pathogen Leptosphaeria maculans ‘brassicae’ (anamorph Phoma lingam). [129] The latter fungus infects Brassica species, such as canola and causes round whitish lesions on leaves. In the course of infection, L. maculans grows down to the stem and by killing cells, produces a black canker from which the name ‘blackleg’ disease has been inferred. Death of the plants before seed production leads to significant loss of crop yields. [130] The function of sirodesmin in blackleg disease is ambiguous. Abrogation of sirodesmin production reduces the size and number of stem lesions, but does not reduce the spots on leaves.[ 72 , 131 ] On the other hand, sirodesmin could be important for the host‐pathogen interaction, as the phytoalexin brassinin, a plant defense molecule, inhibits the production of sirodesmin. [132]
2.1.2.2. Structure and biosynthesis
A hallmark feature of sirodesmins is their spirofused tetrahydrofuran cycle (Figure 1). [133] Depending on the stereochemistry at the junction two epimers are known: sirodesmin A and sirodesmin G, also termed sirodesmin PL. [129] By isotope‐labelling and feeding experiments, the biosynthetic precursors of sirodesmin PL were identified as Tyr and Ser. [133] With the discovery of a prenylated DKP intermediate, termed phomamide, [133] assumptions about the order of cyclization and prenylation were made. [133a] Later, two additional intermediates of sirodesmin biosynthesis, phomalirazine [134] and deacetylsirodesmin PL, [134] were reported and biosynthesis pathways were proposed, [129] but no experimental proof could be provided. Only after whole genome sequencing and genome mining techniques had become available, research on sirodesmin was revitalized.
Sirodesmin PL was the first ETP for which the biosynthetic gene cluster was described in 2004 (Figure 3A). The gene cassette was spotted via a homologue of the dimethyl tryptophan synthetase gene that was identified in an expressed sequence tag library of L. maculans and later on annotated as sirD. [72] In 2021, two additional genes were identified within the sir locus, now containing 20 ORFs. [135] Most of the sirodesmin biosynthesis genes are homologous to ORFs from the gliotoxin cluster and considered as the ‘common ETP moiety’ genes. [23a] Based on the experimental work that has been done on gliotoxin biosynthesis and the corresponding enzymes, a similar reaction cascade was proposed for sirodesmin PL (Figure 3B). The sirP gene for example is the equivalent to gliP. It encodes a two‐module NRPS that establishes the DKP scaffold, as proven by genetic disruption. [72] Similar to gliotoxin biosynthesis, the gene products of sirC and sirG are expected to catalyze sulfur incorporation via the addition of GSH. In the following, the enzymes encoded by sirK, sirJ and sirI are assumed to uncover the free thiols.[ 135 , 136 ]
Figure 3.

Gene cluster and current biosynthesis scheme for sirodesmin PL production. (A) Schematic view of the sir genes forming the sirodesmin biosynthesis gene cluster in L. maculans.[ 72 , 135 ] Genes are colored according to Figure 2A. (B) Proposed reaction sequence leading to sirodesmin PL. Prominent intermediates are labelled. Putative biosynthesis intermediates produced by SirK and SirJ are not shown, as processing of the glutathione moieties likely proceeds as in gliotoxin biosynthesis. The reactions catalyzed by SirD, SirP, SirC, SirG, SirK, SirJ and SirI are likely fixed in their order. SirO might reduce the ketone group outside the 2,5‐DKP ring of phomalirazine. For details see section 2.1.2.2.
Downstream steps of sirodesmin biosynthesis likely include closure of the disulfide bridge by SirT and N‐methylation by SirN. Notably, the sir gene cluster also encodes a putative O‐methyltransferase SirM, the function of which remains unknown. Secretion of sirodesmin PL is mediated by SirA. SirA is an ABC‐transporter [71] that protects against exogenous sirodesmin and confers cross‐resistance against gliotoxin. By contrast, the corresponding efflux protein encoded in the gliotoxin biosynthesis gene cluster, GliA, is of the MFS‐type (see also section 2.1.1.2.) and does not protect from sirodesmin. [137] However, the molecular and functional differences between SirA and GliA remain to be elucidated.
Furthermore, the predicted DNA binding motif for the binuclear zinc finger transcription factor SirZ is identical to that of GliZ (TCGGN3CCGA), but the motif was found only upstream of the genes sirD, sirP, sirJ, sirT, sirB, sirQ, sirR, sirN, and sirO, despite similar transcription pattern of sir genes lacking the putative SirZ binding site. [113] This observation might indicate that other transcription factors contribute to sir gene regulation or that SirZ is promiscuous and recognizes also derivatives of the predicted consensus motif. In support of relaxed sequence specificity for SirZ, it was noted that RNA silencing of sirZ expression dampened also expression of sir genes that lack the consensus binding site. [113]
Given the complex structure of sirodesmin PL, the sir locus encodes several tailoring enzymes. Most importantly, the promiscuous prenyl transferase (SirD) catalyzes the addition of a dimethylallyl group to diverse acceptor sites, including the hydroxyl group of free l‐Tyr. [138] The resulting metabolite O‐prenyl‐l‐Tyr then serves as a building block for DKP formation, leading to the well‐known intermediate phomamide (Figure 3B). [139] Claisen rearrangement and cyclization of the prenylated Tyr may lead to the terminal five‐membered ring, but the enzyme(s) required for these reactions still remain(s) unknown. Epoxidation of the phenyl ring of Tyr by either SirB or SirE and subsequent nucleophilic attack by the amide backbone nitrogen atom of Tyr is assumed to induce intramolecular cyclization to the pyrrolidine ring structure similar to gliotoxin. Further consecutive oxidations of the Tyr remnant after the addition of water likely yield the phomalirazine intermediate (Figure 3B). A second epoxidation step, catalyzed by SirB or SirE, may trigger formation of the spiro linkage and ketone reduction by the predicted oxidoreductase SirO may set the stage for acetylation by SirH. [72] This mechanistic proposal (Figure 3B) however is incomplete and preliminary, as many of the gene products have not been assigned and proven a specific function in sirodesmin PL production and as the order of reaction steps is largely unknown. The highly similar genes sirQ, sirR and sirS for example are unusual elements of secondary metabolite gene clusters. They most likely encode nicotinamide adenine dinucleotide‐dependent epimerases that act on hydroxyl groups, [72] but their exact function awaits to be deciphered. Moreover, it its unknown which of the encoded CYP450 enzymes (SirB/SirE) is responsible for which oxidative modification. Lastly, SirX could not be assigned any potential function yet, as it shows no homology to any other protein. [135]
2.1.3. Aranotins
Aranotins are secondary metabolites produced by the fungi Arachniotus aureus and Aspergillus terreus.[ 93 , 140 ] They inhibit RNA synthesis in rhino‐, polio‐, parainfluenza and coxsackie viruses [140d] and are active against cancer cell lines. [141] Aranotins together with emethallicins and emestrins belong to a subgroup of ETPs harboring at least one seven‐membered 4,5‐dihydrooxepine ring (Figure 1). Isotope‐labelling experiments in the early 1980s surmised that two molecules of l‐Phe are condensed to build the DKP‐scaffold of aranotins, [142] but the biosynthetic route remained enigmatic. Only in 2013, the aranotin biosynthesis gene cluster was reported from A. terreus (Figure 4A) and the high homology of all nine ORFs to corresponding elements in the gliotoxin gene cassette suggested a similar biosynthetic route, which was confirmed by targeted deletion of each of the nine genes and analysis of the metabolic changes (Figure 4B). [94a] Notably, the aranotin biosynthesis gene cluster encodes two modular genes araIMG and araTC that might encode multidomain enzymes unifying the activities of the corresponding individual proteins in gliotoxin biosynthesis. [94a]
Figure 4.

Gene cluster and current biosynthesis scheme for acetylaranotin production. (A) Schematic view of the ata genes in the acetylaranotin biosynthesis gene cluster from A. terreus. [94a] Genes are colored according to Figure 2A. The cluster contains two modular genes that might encode for multifunctional enzymes: AtaTC and AtaIMG. (B) Reaction sequence proposed for the enzymatic production of acetylaranotin – a centrosymmetric ETP. Domains of predicted multifunctional enzymes required for a certain reaction are printed bold. The putative biosynthesis intermediate produced by AtaJ is not shown, as processing of the glutathione moieties likely proceeds as in gliotoxin biosynthesis. The order of reactions catalyzed by the enzymes AtaH and AtaY is interchangeable. For details see section 2.1.3.
The biosynthesis of aranotins starts with the coupling of two l‐Phe residues by the NRPS AtaP (Figure 4B). Consistent with the use of a single substrate, AtaP features only one A domain for the activation of l‐Phe. Next, the AtaC domain of AtaTC is proposed to catalyze bis‐hydroxylation and the GST domain of AtaIMG likely adds the GSH moieties. [94a] Notably, a homologue of gliK is missing in the aranotin biosynthesis gene cluster but equivalents to gliJ and gliI are present. On the one hand, the putative ataK gene could have been overlooked in the cassette, as it was the case for sirK [135] (see section 2.1.2.2.) or a cellular γ‐glutamyl‐cyclotransferase encoded outside the ETP cluster could take over this function. Strikingly, deletion of the homologous verK gene from the verticillin A locus did not completely abolish verticillin A biosynthesis, [143] suggesting that the γ‐glutamyl‐cyclotransferase in the ETP cluster might not be essential (see also section 2.3.1.), but GliK was found to be required for gliotoxin biosynthesis. [85] Irrespective of its generation, downstream action of the C−S bond lyase requires a free amine to form a Schiff base with the cofactor PLP and to initiate cleavage of the carbon‐sulfur‐bond and this amine is only uncovered by removal of the γ‐glutamyl moiety. This is at least the current mechanistic mode of action proposed for the homologue GliI. [88]
Actions of AtaJ and the AtaI domain of AtaIMG are assumed to create the epidithiol intermediate that is oxidized by the AtaT domain of the AtaTC protein. Epoxidation of the phenyl moieties by AtaF and subsequent nucleophilic attack by the amide nitrogen atoms likely lead to the pyrrolidine moieties (Figure 4B), analogously to the reaction catalyzed by GliF during gliotoxin biosynthesis. Acetylation and oxidative ring expansion are modifications absent from gliotoxin, but the enzymes installing these decorations on aranotins were identified by comparing the metabolite profiles of the single knockout strains ΔataH and ΔataY. According to these results, AtaH acts as the acetyltransferase and AtaY installs the dihydrooxepine structures, likely via epoxidation and subsequent rearrangements.[ 94b , 94c ] Both reactions are not determined in their order, [94a] but in contrast to a previous mechanistic proposal [140c] they seem to occur after pyrrolidine formation. [94a] In addition, similar to the gli gene cluster, a transporter of the major facilitator superfamily, AtaA, is encoded in the ara locus and as part of the ataIMG gene a O‐methyltransferase domain AtaM is predicted (Figure 4A). Although a sulfur‐free O‐methylated compound has been isolated from a ataIMG deletion strain, [94a] its functional relevance remains unclear. Moreover, for the gene ataL no potential function could be assigned so far. It encodes a protein that shows significant similarity to GliH. Considering that GliH is essential for gliotoxin production [66b] and that only sulfur‐free compounds could be isolated from a ΔataL strain, [94a] it is tempting to speculate that GliH and AtaL might act upstream of sulfur incorporation in a yet unknown manner.
Similar to gliotoxin, a bis(methylthio)‐variant has been reported [94a] and the corresponding S‐methyltransferase (AtaS) has been spotted outside the ara cluster. [145]
2.1.4. Sporidesmins
To date nine different sporidesmins are known, of which sporidesmin A is the most prevalent one and sporidesmin D the bis(methylthio)‐derivative of A. [28] Sporidesmin A was originally identified as the causative agent of facial eczema and liver disease in sheep and cattle predominantly in New Zealand and Australia. [146] It is produced by the fungus Pseudopithomyces chartarum (formerly named Pithomyces chartarum or Sporidesmium bakeri) that grows on grasses. [147] Once ingested by ruminants, sporidesmin A causes inflammation and necrosis in the bile ducts, resulting in a block of bile flow. As a result, degradation products of chlorophyll accumulate in the body and cause photosensitivity of the skin, leading to skin rash [146a] and ultimately to death. Originally, veterinarians treated affected animals with zinc sulfate. [148] The metal ions zinc and cadmium were later shown to protect cells from the toxic effects of sporidesmin A by chelating the ETP. [149] In addition, sporidesmin A has been found to inhibit human glutaredoxin via disulfide formation. [55]
Structurally, sporidesmins are densely functionalized, chlorinated ETPs (Figure 1).[ 69b , 150 ] They are built from l‐Trp and l‐Ala residues [151] and highly decorated with hydroxyl, methyl and methoxy groups. Only recently, the putative biosynthetic gene cluster of sporidesmin A (Figure 5A) from P. chartarum has been identified by bioinformatics tools and reported on bioRxiv. [144] It contains 21 genes, of which many products display homology to enzymes from gliotoxin (see section 2.1.1.2.), sirodesmin PL (see section 2.1.2.2.) and aspirochlorine biosynthesis (see section 2.2.2.), but validation of their biological function is still lacking. Spd20 (GliZ), Spd17 (GliP), Spd3 (GliC), Spd5 (GliG), Spd9 (GliK), Spd2 (GliJ), Spd16 (GliI), and Spd13 (GliT) are predicted to take over the same functions as their corresponding homologues from the gli gene cluster (Figure 5B). As an orthologue of AclH (see section 2.2.2.), Spd4 probably is a flavin‐dependent halogenase and Spd15 shows similarity to the acetyltransferase SirH. Four methyltransferases (Spd1, Spd7, Spd11 and Spd21) and another two CYP450 enzymes besides Spd3 (Spd8 and Spd10) are also part of the cluster. Strikingly, three transport proteins are encoded as well: one ABC (Spd6) and two MFS (Spd14 and Spd18) transporters. [144] Finally, Spd12 and Spd19 are two hypothetical proteins of unknown function (Figure 5A). Given the large number of genes in the putative cluster, experimental validation of the cassette borders and the functional involvement of the genes in sporidesmin A biosynthesis is necessary.
Figure 5.

Gene cluster and putative biosynthesis scheme for sporidesmin A production. (A) Schematic view of the predicted sporidesmin A (spd) biosynthesis gene cluster from P. chartarum according to a preprint on bioRxiv. [144] Genes are numbered sequentially and colored according to homology of their products to Gli proteins (see Figure 2A for comparison). ORFs coding for proteins of unknown function are shaded black. (B) Putative reaction sequence for the enzymatic production of sporidesmin A. The first six reaction steps are likely to occur analogously to gliotoxin. However, catalysts and order of downstream reaction steps are not predictable by bioinformatics tools. For details see section 2.1.4.
2.2. Irregularly bridged ETPs
Irregularly bridged ETPs feature a disulfide bond that is not exclusively anchored at the Cα atoms of the two amino acids building up the DKP skeleton. [29] The most prominent and best characterized representatives of this class are gliovirin (section 2.2.1.) and aspirochlorine (section 2.2.2.).
2.2.1. Gliovirin
The fungus T. virens (formerly known as Gliocladium virens) produces gliotoxin but was also discovered to be the source of another ETP termed gliovirin. [92] Later it was noted that the species T. virens can be subdivided into two distinct strains, of which the “Q” lineage accounts for gliotoxin and the “P” strain for gliovirin. [152] Gliovirin is active against oomycetes such as the plant pathogen Pythium ultimum [153] and by inhibiting the extracellular signal‐regulated kinase (ERK), it blocks synthesis of TNFα. [154]
Structurally, gliovirin is characterized by a 1,2‐oxazadecaline moiety and it was the first identified ETP with an irregularly bridged disulfide bridge (Figure 1). [92] Isotope‐labelling studies revealed that gliovirin is assembled of two l‐Phe residues. [155] In agreement, the recently identified biosynthetic gene cluster of gliovirin encodes a NRPS (Glv21) that is predicted to have a single adenylation domain (Figure 6A). While the importance of Glv21 for gliovirin production has been experimentally confirmed by deletion, the functions of many of the other 21 gene products encoded in the gene cluster are up to now only predicted based on homology to the corresponding Gli enzymes. [156] Glv8 (GliZ), Glv3 (GliC), Glv10 (GliG), Glv11 (GliJ), Glv2 (GliI), and Glv9 (GliA) likely fulfill similar functions as their counterparts from gliotoxin biosynthesis (Figure 6B). Oxidation of the free thiols might be catalyzed by either Glv4 or Glv16 (both thioredoxin reductases) and the function of GliF might be fulfilled by either Glv5 or Glv19. The additional CYP450 monooxygenases (Glv6, Glv12, and Glv14) are presumably responsible for tailoring reactions like epoxidation and hydroxylation. Furthermore, the gene cassette contains three O‐methyltransferases: Glv1 shows homology to GliM but its role in biosynthesis is unknown and Glv7 as well as Glv13 likely account for the two methoxy groups in gliovirin. Moreover, four additional genes of the glv gene cluster encode proteins with obscure function in gliovirin biosynthesis: Glv17 is a predicted aldo/keto reductase, Glv18 shares homology with permeases, Glv20 is a domain of unknown function (DUF) 1857 family member and Glv22 has been annotated as an ABC transporter transmembrane region (Figure 6A). [156] Similar to the ara locus, a gliK homologue is absent from the cluster.
Figure 6.

Gene cluster and putative biosynthesis scheme for gliovirin production. (A) Schematic view of the predicted gliovirin (glv) biosynthesis gene cluster from T. virens. [156] Genes are numbered sequentially and colored according to Figure 2A. (B) Putative biosynthesis scheme for gliovirin. DKP formation and sulfur incorporation likely occur analogously to gliotoxin (see section 2.1.1.2.). Migration of a hydroxyl group (reaction 3) has been proposed to explain how the irregular disulfide bridge is installed. [156] Recent studies on aspirochlorine (see section 2.2.2.) however suggest that the disulfide bridge is installed first and shifted later in biosynthesis. [157] What applies to gliovirin remains to be investigated.
The most intriguing structural feature of gliovirin is its disulfide moiety linking the Cα atom of one Phe residue to the Cβ atom of the second. According to the current mechanistic proposal, Glv3 hydroxylates the Cα atoms of both Phe residues and subsequent rearrangements move one hydroxyl group to the Cβ atom. [156] A spontaneous movement is unlikely, as other ETPs are devoid of this irregular bridging. For instance, acetylaranotin, that is also made of two l‐Phe residues, features a disulfide bridge connecting both Cα atoms and no Cα–Cβ linkages have been described (see also section 2.1.3.). On the other hand, converting a symmetric substrate (either the naked or the Cα‐bis‐hydroxylated Phe‐Phe‐DKP) to an asymmetric one by enzymatic action, appears unlikely. It might therefore be conceivable that the sulfur migrates after disulfide bridge formation in a manner similar to what has been observed for aspirochlorine (see section 2.2.2.). In support of this assumption, the glv gene cluster encodes a putative O‐acyltransferase (Glv15) and two thioredoxin reductases (Glv4 and Glv16). Glv15 might take over the function of AclF in aspirochlorine biosynthesis and install a transient acetylation. In agreement, an acetylated variant of gliovirin has not been reported so far. The two thioredoxin reductases (Glv4/Glv16) are potentially responsible for oxidation of the thiols and sulfur migration, respectively. Sequence analyses for either CXXC (typical of thiol oxidases like GliT, see section 2.1.1.2.) or CXXH motifs (hallmark of AclR involved in sulfur migration during aspirochlorine biosynthesis; [157] see section 2.2.2.) would help assign putative functions to these gene products.
Nonetheless, mechanistic differences between gliovirin and aspirochlorine biosynthesis are evident, because sulfur migration during aspirochlorine production is coupled to formation of a spiro center (Figure 7B) – a feature that is absent from gliovirin.
Figure 7.

Gene cluster and putative biosynthesis scheme for aspirochlorine production. (A) Schematic view of the aspirochlorine (acl) biosynthesis gene cluster from A. oryzae. [165] Genes are colored according to homology of their products to Gli proteins (see Figure 2A for comparison). (B) Reaction scheme for the biosynthesis of aspirochlorine based on current knowledge. The reaction steps starting from AlcF are experimentally verified and take place in the given order. In brief: Condensation of two l‐Phe residues yields the 2,5‐DKP skeleton, which is subsequently oxidatively modified and sulfurized. The enzymes putatively involved in these reactions are listed. Next, after transfer of an acetyl group by AlcF, the flavoprotein AlcR shifts one sulfur atom and introduces the spiro center. Two slightly different mechanisms have been discussed in literature for this reaction. [157] Here, the His147‐catalyzed production of a phenoxide intermediate is shown, but direct conversion of prespiro‐aspirochlorine to the thiirane without the help of His147 is conceivable as well. [157] For details see section 2.2.2.
All in all, the gene cluster and a first mechanistic proposal for the biosynthesis of gliovirin are available. Validation of the proposed enzymatic functions and clarification of the order of reaction steps still need to be addressed to gain insights into how the irregular disulfide bridge is established.
2.2.2. Aspirochlorine
In 1969, antifungal and antiviral activities were identified in extracts of Aspergillus oryzae and the putative substance was termed oryzachlorine. [158] Seven years later, an antibacterial and antifungal metabolite from Aspergillus tamarii was reported as A30641. [159] Furthermore, in the early 1980s, the compound aspirochlorine, was isolated from the human pathogenic fungus Aspergillus flavus [160] and the koji mold A. oryzae. [161] This compound was found to be identical to A30641 and considered a component of the initially identified oryzachlorine.[ 161 , 162 ] The fungizide activity of aspirochlorine has been attributed to selective inhibition of fungal but not bacterial or mammalian protein biosynthesis [163] and notably aspirochlorine is also effective against azole‐resistant C. albicans. [164] More recently, cytotoxic activity against mammalian cell lines was noted as well. [165]
Although aspirochlorine has not been further investigated for its biological activity, it can be assumed to be as toxic as other ETPs simply due to its disulfide moiety. For this reason, the prevalent use of A. oryzae in food technology is a potential risk factor. [166] A. oryzae is traditionally used in East Asia for brewing and food fermentation, including the production of soy sauce, miso and sake (rice wine), [167] and it has been shown that aspirochlorine is produced under such conditions. [165]
The structure and the biosynthesis of aspirochlorine are exceptional among the hundreds of ETPs known to date. Aspirochlorine is a halogenated spiro compound with an N‐methoxy amide linkage and, like in gliovirin, the disulfide bridge extends outside the DKP core connecting a Cα with a Cβ atom (Figure 1). [162] The biosynthesis of this remarkable compound however remained unexplored until recently. The structure of aspirochlorine hinted at l‐Phe and Gly as building blocks for the DKP scaffold, but this hypothesis was disproved later. First, the acl gene cluster responsible for aspirochlorine biosynthesis was identified by genome mining and experimentally verified by gene deletion experiments (Figure 7A). [165] The gene cassette encodes a transcriptional regulator (AclZ), three putative transport proteins (AclA, AclQ and AclS) and the typical set of enzymes expected for ETP production: the NRPS AclP for DKP formation; AclC (CYP450 enzyme) and AclG (glutathione‐S‐transferase) for GSH addition; AclK (γ‐glutamyl‐cyclotransferase), AclJ (dipeptidase) and AclI (aminotransferase) for GSH garbling; and AclD (thioredoxinreductase) for establishing the disulfide linkage (Figure 7).[ 157 , 165 ]
Surprisingly, the NRPS AclP was predicted to contain only a single A domain with specificity for Phe and notably the signature sequence of the A domain was identical to that of AtaP involved in acetylaranotin biosynthesis (see section 2.1.3.). Further experiments supported l‐Phe as the sole building block of aspirochlorine and showed that one Phe residue is truncated to Gly by an oxidative C−C cleavage and release of benzaldehyde. [165] Follow‐up work revealed that the CYP450 enzymes AclL and AclO install hydroxyl groups. By hydroxylating one of the nitrogen atoms of the DKP scaffold, AclO sets the stage for subsequent methylation by AclU, leading to the methoxy group in aspirochlorine. The resulting N‐alkoxy amide increases the electrophilicity of the adjacent carbonyl carbon atom [168] and facilitates non‐enzymatic retro‐aldol cleavage of the Phe side chain within the same amino acid (Figure 7B). [169] The final step of aspirochlorine biosynthesis is accomplished by AclH. The FAD‐dependent halogenase chlorinates the monophenyl precursor to yield the final natural product. [165] Notably, the order of reaction steps is fix, as the enzymes AclL, AclO and AclU favor substrates with two phenyl groups, while AclH selects for monophenyl compounds. [169] The retro‐aldol‐type fragmentation during aspirochlorine biosynthesis leads to a so far unobserved conversion of amino acids that is of significant relevance for the biological activity of the final compound. Compared to the monophenyl compounds aspirochlorine and its dechloro version, pathway intermediates with two phenyl rings showed significantly reduced fungicide activity. [169] While dechloroaspirochlorine was initially reported to have substantially less anti‐fungal and cytotoxic effects than aspirochlorine, [165] the potency in a second study was only moderately reduced. [169]
After the late biosynthesis steps had been elucidated, questions about the formation of the irregular seven‐membered disulfide ring of aspirochlorine were addressed. Revision of the gene cluster annotation revealed a previously undetected gene aclR whose product was predicted to share homology with AclD, the equivalent of GliT from gliotoxin biosynthesis. Yet, in contrast to the CXXC motif typical of thioredoxin reductases, AclR was noted to feature an unusual CXXH signature. [157] A ΔaclR knockout strain accumulated a previously unobserved metabolite (prespiro‐aspirochlorine) lacking the spiro center but instead carrying two oxymethines with one being decorated by an acetyl group that is absent from the final natural product. Further analyses revealed that AclR is an FAD‐dependent enzyme that catalyzes the conversion of prespiro‐aspirochlorine to a spiroaminal intermediate, [157] which was previously shown to be a substrate for AclL. [169] The crystal structure of AclR and mutagenesis data suggest that the unusual CXXH motif of AclR is necessary for efficient formation of the spiro center. [157] Most likely, under the release of acetate, Cys144 forms a mixed disulfide with the substrate, leading to a thiirane. Subsequent 1,2 sulfamyl migration promoted by the lone pair of the neighboring amide nitrogen is supposed to create the α,β sulfur linkage and the spiro‐fused furan ring of aspirochlorine. His147 might assist this reaction sequence (Figure 7B). [157] Notably, AclR homologues are found in several orphan biosynthesis gene clusters along with acetyltransferases, indicating that spiro‐ETPs may be more widespread than anticipated. [157] Similar to morphine [170] and vinblastine biosynthesis, [171] the acetyl group in prespiro‐aspirochlorine (introduced by the acetyltransferase AlcF) creates a better leaving group for the subsequent AlcR‐catalyzed reaction. [157]
Besides, two additional methyltransferases (AclM and AclN), another thioredoxin reductase (AclT), a dehydrogenase (AclE) and an extra CYP450 enzyme (AclB) of yet unclear function for aspirochlorine biosynthesis are encoded in the acl gene cassette (Figure 7A). While AclB might introduce hydroxyl groups in a precursor molecule of prespiro‐aspirochlorine, the need for AclM and AclN in aspirochlorine biosynthesis is unclear.
2.2.3. Brief glimpse of epicoccins
Epicoccins denote a family of sulfurized DKPs from Epicoccum nigrum of which some members are characterized by extraordinary cross‐ring sulfur bridges. Epicoccin C for example features two such unusual disulfide linkages (Figure 1), while other members of this class are characterized by either one disulfide and one monosulfur bridge or one or two monosulfur bridges. The biosynthesis of these extraordinary compounds has however not been studied yet. [172]
2.3. ETP double‐decker structures
Dimeric ETPs have been isolated from several fungi, including Verticillium, Chaetomium, Gliocladium and Penicillium species, and associated with diverse biological activities. These structurally exceptional, densely functionalized compounds act as cytotoxins, anti‐bacterial, anti‐viral or immunosuppressive agents and therefore represent attractive lead structures for drug development. Notably, these dimeric ETPs are more potent as antimicrobials compared to monomeric ETPs, [173] but their low production levels as well as many congeners hamper their careful examination.[ 143 , 174 ] Until 2012, 25 such ETP double‐decker compounds were described [175] and their number is continuously growing. The monomer units of most of these compounds are linked by a C−C (verticillins (see section 2.3.1.) and chaetocin (see section 2.3.2.)) or a C−N bond (chetomin; see section 2.3.3.) (Figure 1).
2.3.1. Verticillins
Since the discovery of verticillin A, [176] several congeners of this ETP‐dimer have been identified. [175] Verticillins are octacyclic compounds and their two tetracyclic ETP monomers are linked by a C−C bond connecting two quaternary carbon atoms. They are cytotoxic against various cancer cell lines[ 176 , 177 ] and inhibit NF‐κB dependent pathways. [175] In particular verticillin A, an ETP likely made of l‐Trp and l‐Ala, shows antimicrobial activity against Gram‐positive bacteria and has been reported as a histone methyltransferase inhibitor. [178] More recently, verticillin A has been proposed as a stimulus for fungal conidiation and a signaling molecule for morphological differentiation of fungi. [143]
After all attempts to identify the verticillin A biosynthesis gene cluster in Verticillium sp. – the genus from which the isolation of verticillin A was reported first [176] – had failed, it was proposed that Verticillium sp. likely do not produce verticillin A. Probably, verticillin A was isolated from a sample containing Verticillium sp. contaminated with a morphologically similar mycoparasite such as Clonostachys rosea. [179]
In agreement, in 2017, the ver gene cluster encoding the enzymatic machinery for verticillin production in Clonostachys rogersoniana – a parasitic fungus related to C. rosea – was published. [143] For 12 out of the 13 genes, a role in verticillin A formation could be proven by single‐gene deletions (Figure 8).[ 143 , 174 ] The cluster encodes the complete enzyme set required for ETP formation and secretion (NRPS, CYP450, GST, γ‐glutamyl‐cyclotransferase, dipeptidase, C−S bond lyase, thioredoxin reductase and transporter). Notably, the thioredoxin reductase VerT has been implied in self‐resistance to verticillin A similar as GliT from A. fumigatus. [143] Characterization of the zinc finger transcription factor VerZ showed that its expression positively correlates with the production of verticillin and direct interaction of recombinant VerZ with upstream regions of various ver genes (verA, verT‐verL, verM, verN‐verI, verC‐verP, verK, verG, verZ and verB) was confirmed. [174] Further analysis revealed the consensus motif (T/C)(C/A)(G/T)GN3CC(G/T)(A/G)(G/C), which is similar to the corresponding motif predicted for SirZ (TCGGN3CCGA). [113] Notably, the verJ gene lacks the binding motif for VerZ and consistently, VerZ does not interact with the promoter region of verJ. [174]
Figure 8.

Schematic illustration of the verticillin A (ver) biosynthesis gene cluster from C. rogersoniana. The reaction steps for the biosynthesis of verticillin A have not been reported yet. However, the gene cluster encodes the basic enzyme set for 2,5‐DKP formation (VerP), GSH addition (CYP450 monooxygenase and VerG) and truncation (VerK, VerJ, VerI) as well as thiol oxidation (VerT) along with a transporter (VerA) and a regulator (VerZ). [143] Color coding is according to Figure 2A. See also section 2.3.1.
To date, the biosynthesis of verticillin A has not been studied in detail. Yet, in an attempt to create fluorinated verticillin A derivatives, feeding experiments with 5‐F‐d/l‐Trp validated l‐Trp as a building block. [180] Considering the homology of many ver genes to their counterparts from the gliotoxin biosynthesis gene cluster, the formation of verticillin A may proceed similarly as for gliotoxin, but experimental evidence is missing and requires further investigations. Interesting about verticillin A and other double‐decker ETPs is their inverted stereochemistry of the 2,5‐DKP side chains and how and when during biosynthesis the dimer‐scaffold is installed. A putative, general reaction mechanism for dimer formation en route to double‐decker structures is discussed in section 2.3.4. and shown in Figure 11.
Figure 11.

Putative reaction mechanisms for the dimerization of ETPs via (A) C−C and (B) C−N linkages. Reactions are assumed to be catalyzed by the iron‐loaded heme cofactor of CYP450 enzymes and exemplary shown for chaetocin. Similar reaction mechanisms have been proposed for the dimerization of DKPs in bacteria.[ 17a , 193a , 193b , 193c , 193e ] Corresponding fungal enzymes have not yet been analyzed.
2.3.2. Chaetocin
Chaetocin is structurally highly similar to verticillin A and was first isolated from the fungus Chaetomium minutum. [181] It has been reported as the first selective inhibitor of lysine‐specific histone methyltransferase SU(VAR)3‐9, [182] but follow‐up studies were rather supportive of a non‐specific inhibition of SU(VAR)3‐9 by the reactive disulfide moieties of chaetocin. [33b] In agreement, chaetocin has been shown to target a variety of proteins[ 61c , 183 ] and exerts broad anti‐tumor activity in vitro and in vivo.[ 61c , 184 ] Based on the genome sequence of Chaetomium virescens, the chaetocin biosynthesis gene cluster was predicted (Figure 9). [136] Experimental exploration of the biosynthesis steps however is precluded by the lack of a genetic manipulation system for C. virescens.[ 136 , 143 ] The biosynthesis of chaetocin is supposed to start from l‐Trp and l‐Ser and to proceed as for gliotoxin, involving the corresponding enzymes ChaP, ChaC, ChaG, ChaK, ChaJ, ChaI, ChaT and ChaN. [136] Pyrroloindole formation is proposed to be catalyzed by the CYP450 enzyme ChaB, a homologue of SirB. In addition, ChaZ is supposed to control expression of the cha gene cluster and ChaA likely acts as an ABC efflux system for chaetocin. Besides, ChaM is a putative methyltransferase but could not yet be assigned a specific function in chaetocin biosynthesis.
Figure 9.

Schematic illustration of the chaetocin (cha) biosynthesis gene cluster from C. virescens. So far, the biosynthesis of chaetocin has not been investigated, but the cha gene cluster contains the ‘common ETP moiety’ genes supposed to be required for 2,5‐DKP formation (CaP), sulfur addition (ChaC and ChaG), uncovering (ChaK, ChaJ, and ChaI) and oxidation (ChaT) as well as toxin export (ChaA) and transcriptional regulation (ChaZ). [136] Color coding is according to Figure 2A. For details see section 2.3.2.
Notably, the cha gene cluster encodes a third CYP450 enzyme, ChaE, that lacks any homologue in the sir and gli gene cassettes and is therefore assumed to create the double‐decker structure. [136] Specifically, ChaE has been proposed to install the C−C bond connecting the two tetrasubstituted β‐carbon atoms of the pyrroloindole rings via a radical mechanism [136] similar to biaryl coupling in himastatin (for details on dimerization of ETPs see section 2.3.4.). [185] Yet, failure to heterologously produce ChaE for in vitro studies prevented further investigations so far. [136]
2.3.3. Chetomin
Chetomin has been isolated as a metabolite of Chaetomium cochliodes in 1944, [186] but its unusual structure has been unraveled only in 1976. [187] Each monomer of the dimeric ETP is likely formed of l‐Trp and l‐Ser. In contrast to verticillin A and chaetocin, the monomers of chetomin are linked by a bond between the β‐pyrrolidinoindoline carbon and the indole nitrogen. Chetomin acts as an anti‐cancer agent [188] by blocking the interaction of Hif1a and p300. [34] Furthermore, it is a potent antibiotic compound and targets methicillin‐resistant Staphylococcus aureus (MRSA) with higher potency than vancomycin. [189] Chetomin is also produced by Chaetomium globosum, [190] a species that can infect human skin and nails and ultimately lead to systemic lethal mycoses in immunocompromised patients. [191]
Recently, the chetomin biosynthesis gene cluster has been identified in C. cochliodes SD‐280. [189] It comprises 18 genes, nine of which with enigmatic function in chetomin production. The predicted enzymes CheP, CheG, CheK, CheJ, CheN and CheT likely fulfill similar functions as their homologues from gliotoxin biosynthesis (Figure 10A). Moreover, cheA encodes a putative ABC transporter. CheB and CheC are CYP450 enzymes, one of which may initiate sulfur incorporation similar to GliC, while the other is supposed to create the C−N linkage and the double‐decker structure (for details on dimerization of ETPs see section 2.3.4.). [189] Until now, chetomin biosynthesis has not been analyzed experimentally, but two different putative reaction schemes were proposed based on gliotoxin biosynthesis.[ 189 , 192 ] The most recent is show in Figure 10B. [189] Future studies might concentrate on verifying the individual reaction steps and enzymes.
Figure 10.

Gene cluster and putative biosynthesis scheme for chetomin production. (A) Schematic illustration of the chetomin (che) biosynthesis gene cluster from C. cochliodes SD‐280. [189] ORFs are colored according to homology of their products to Gli proteins (see Figure 2A for comparison). Several genes of the common ETP set are present in the cluster (cheP, cheG, cheK, cheJ, cheT), but some are missing (cheI) and many are of unknown function (black ones). (B) A putative reaction sequence has been proposed based on the corresponding biosynthesis scheme for gliotoxin (see Figure 2B). [189] However, the catalysts and order of reaction steps have not yet been validated experimentally. Due to space limitations, glutathione moieties are abbreviated as ‘GS’. See section 2.3.3.
2.3.4. Dimerization of ETPs
So far, the dimerization of ETPs has not been studied, but recent work on (mostly bacterial) DKP dimerases might also provide insights into the coupling of ETPs. Known DKP dimerases are CYP450 enzymes that connect Trp‐containing 2,5‐DKP monomers via C−C and C−N bonds similar to verticillin A, chaetocin and chetomin (Figure 1). [193] According to the current mechanistic proposal a radical is generated at the N1 atom of the Trp side chain of a DKP monomer that subsequently moves to the C3 position, allowing for an intramolecular cyclization (Mannich reaction) to create a pyrroloindoline C3 radical. Radical addition to a second DKP monomer would finally yield a C−C linked dimeric scaffold (Figure 11A). [193b] Reaction of a N1 radical with another DKP monomer is thought to create the intermolecular C−N bond (Figure 11B). [193e] These radical cascades are currently favored[ 193a , 193b , 193c , 193e ] over potential other mechanisms (i. e. cationic mechanism for C−C bond formation [193a] or dimerization of two radicals as proposed for the enzymes DtpC[ 193d , 194 ] and HmtS), [185b] but further studies are required to clarify whether fungal enzymes make use of similar or other reaction mechanisms.
Moreover, bacterial DKP dimerases display fairly relaxed substrate‐ and stereo‐specificity,[ 193a , 193c ] while for fungal double‐decker ETPs no stereoisomers have been described. The chemical structure of chetomin for example reminds in part of tetratryptomycin B whose C−N linkage is installed by the bacterial DKP dimerase TtpB1. [193e] Although TtpB1 catalyzes predominantly C−N couplings, C−C dimers similar to verticillin A and chaetocin are produced to a minor extent as well. [193e] It therefore remains to be investigated, whether fungal dimerases are more selective in terms of substrate choice and product distribution or whether side products of chetomin and other double‐decker ETPs exist but have not been identified yet.
3. Final Considerations
ETPs are an ever‐growing class of fungal secondary metabolites, but often several structurally related compounds are reported. Although many of those congeners may be shunt metabolites or isolation artefacts and do not serve a biological function, some of them act as regulatory molecules as shown for the bis(methylthio)‐derivative of gliotoxin. [76] Identification of ETP biosynthesis gene clusters and analysis of the enzymatic production of ETPs will help to distinguish relevant metabolites from irrelevant ones. This strategy however is hampered by the scarcity and instability of pathway intermediates as well as orphan enzymes that are recruited from outside the ETP biosynthesis gene cluster for tailoring reactions. The enzyme TmtA for example, encoded outside the gli gene cluster, was identified to account for the production of bis(methylthio)gliotoxin.[ 75d , 76 ]
Albeit genome mining approaches significantly boostered ETP research during the last two decades, ETP production cannot be simply predicted bioinformatically. A gene cassette from Claviceps purpurea for example encodes all components required for the production of thioclapurines, but the CYP450 enzyme TcpC has altered substrate specificity leading to unusual hydroxylation and sulfurization patterns. As a result, shunt metabolites with nitrogen‐sulfur bonds are formed instead of disulfide‐linked ETPs. [195]
Moreover, to successfully study ETPs and their biosynthesis intermediates, extraction settings have to be carefully chosen to avoid artificial transformations. Under alkaline conditions for example, conversion of pretrichodermamide A to trichodermamide A and S8 was observed, suggesting that trichodermamide A is an isolation artefact. [196] Also the unique structure of the dimeric ETPs vertihemiptellide A and B, identified in the insect pathogen Verticillium hemipterigenum BCC 1449 and linked via two disulfide bonds, needs further investigations to exclude artificial dimerization of two hyalodendrin units. [197]
4. Summary and Outlook
ETPs are a structurally diverse class of fungal natural products that share at least one aromatic amino acid and a reactive sulfide moiety as a hallmark. Due to non‐selective thiol‐disulfide exchange reactions with proteins, ETPs display a myriad of bioactivities and have numerous intracellular targets. Thus, by virtue of their chemical structure, that is inevitably linked to random action, ETPs are unlikely to find application as drugs. Yet, ETPs certainly contribute to fungal virulence as well as pathogenicity and their biosynthesis pathways may be considered as potential targets of fungicides in the future. Furthermore, ETPs produced during fermentation processes are of serious concern for food safety. Therefore, understanding the biosynthesis of ETPs, their regulation and identifying potential ways to interfere with or engineer their production might open up both industrial and medical applications. In this regard, it is worth mentioning that all ETP biosynthesis pathways known to date use the same set of enzymes (NRPS, CYP450, GST, dipeptidase, lyases and oxidoreductase) to construct their sulfurized scaffold and only few tailoring enzymes are responsible for diversifying the framework and creating a specific ETP.
During the last decade much progress has been made in elucidating and studying ETP biosynthesis gene clusters and production, but there are still many knowledge gaps to be closed in the future. i) First, many gene clusters encode more proteins than one would expect to be required for the biosynthesis of the corresponding ETP. It will be interesting to analyze whether the encoded proteins really play a functional or regulatory role for ETP production. Examples from gliotoxin biosynthesis are GliH and the putative O‐methyltransferase GliM, but proteins of unknown function are encoded in all ETP biosynthesis gene clusters known to date. For instance, an O‐methyltransferase is also encoded in the ata gene cluster, although acetylaranotin production should not require an O‐methyltransferase – at least according to our current knowledge of ETP biosynthesis. In this regard, it is interesting to note that some ETP biosynthesis gene clusters encode several transport proteins of different type, the function of which in general and in ETP export is completely unknown. ii) Second, it is unclear whether Cα–Cβ bridged ETPs share a common biosynthetic principle. Specifically, it would be interesting to know whether gliovirin biosynthesis makes use of a similar mechanism for sulfur migration as aspirochlorine. Furthermore, the enzymatic reactions giving rise to epicoccin C are still elusive. iii) Last but not least, basically nothing is known about the biosynthesis of double‐decker ETPs. Compared to monomeric ETPs, the stereochemistry at the Cα atoms of the DKP rings of ETP dimers appears to be inverted during biosynthesis (Figure 1 and Figure 10B). It might therefore be worth analyzing GSTs from double‐decker ETP biosynthesis gene clusters, whether their mechanisms of action allow for stereochemistry inversion. Additional interesting aspects to study include timing of the pairing reaction during ETP biosynthesis and reaction mechanism as well as linkage and substrate selectivity of the dimerization enzymes. DKP dimerases have already been used to engineer diverse non‐natural DKP dimers[ 193b , 193c ] and ETP pairing enzymes might offer similar opportunities for sulfurized DKPs. Enzyme engineering, semisynthetic approaches [198] and feeding of non‐natural precursors [180] increase the chemical space of ETPs and enrich our understanding of enzyme reactions and biosynthetic principles. Scientific studies on these and other aspects of ETPs will carry on the long history of natural sulfur product research.
Conflict of interest
The authors declare no conflict of interest.
5.
Biographical Information
Eva M. Huber studied biochemistry at the Technical University of Munich. During her PhD thesis with Prof. Michael Groll, she solved the structure of the immunoproteasome. Postdoc studies on immunoproteasome inhibition and activation culminated in her admission to the Young Scholars’ Program of the Bavarian Academy of Sciences and Humanities (2017) and her habilitation (2021). Currently, she is a group leader at the chair of biochemistry, TUM. Her research focuses on structural biology of the proteasome, tRNA modifying enzymes and fungal virulence factors, including iron metabolism and regulation as well as biosynthesis of ETPs.

Acknowledgements
Financial support by the DFG – SFB 1309–325871075, the Hans‐Fischer‐Gesellschaft e.V. and the Young Scholars’ Program of the Bavarian Academy of Sciences and Humanities is acknowledged. Open Access funding enabled and organized by Projekt DEAL.
E. M. Huber, ChemBioChem 2022, 23, e202200341.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.
References
- 1.
- 1a. Perrotta E., Altamura M., Barani T., Bindi S., Giannotti D., Harmat N. J., Nannicini R., Maggi C. A., J. Comb. Chem. 2001, 3, 453–460; [DOI] [PubMed] [Google Scholar]
- 1b. Bojarska J., Mieczkowski A., Ziora Z. M., Skwarczynski M., Toth I., Shalash A. O., Parang K., El-Mowafi S. A., Mohammed E. H. M., Elnagdy S., AlKhazindar M., Wolf W. M., Biomol. Eng. 2021, 11; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1c. Acharya A. N., Ostresh J. M., Houghten R. A., J. Comb. Chem. 2001, 3, 612–623. [DOI] [PubMed] [Google Scholar]
- 2.
- 2a. Oku J.-i., Ito N., Inoue S., Die Makromolekulare Chemie 1982, 183, 579–586; [Google Scholar]
- 2b. Kogut E. F., Thoen J. C., Lipton M. A., J. Org. Chem. 1998, 63, 4604–4610. [Google Scholar]
- 3. Scarel M., Marchesan S., Molecules 2021, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.
- 4a. Borthwick A. D., Chem. Rev. 2012, 112, 3641–3716; [DOI] [PubMed] [Google Scholar]
- 4b. Gonzalez J. F., Ortin I., de la Cuesta E., Menendez J. C., Chem. Soc. Rev. 2012, 41, 6902–6915; [DOI] [PubMed] [Google Scholar]
- 4c. Wang Y., Wang P., Ma H., Zhu W., Expert Opin. Ther. Pat. 2013, 23, 1415–1433. [DOI] [PubMed] [Google Scholar]
- 5. Bojarska J., Wolf W. M., Proceedings 2021, 79, 10. [Google Scholar]
- 6.
- 6a.Mannkind, Diketopiperazine Salts for Drug Delivery and Related Methods. US20110008448A1 (https://patents.google.com/patent/US20110008448) 2006;
- 6b. Cornacchia C., Cacciatore I., Baldassarre L., Mollica A., Feliciani F., Pinnen F., Mini-Rev. Med. Chem. 2012, 12, 2–12; [DOI] [PubMed] [Google Scholar]
- 6c. Teixidó M., Zurita E., Malakoutikhah M., Tarragó T., Giralt E., J. Am. Chem. Soc. 2007, 129, 11802–11813; [DOI] [PubMed] [Google Scholar]
- 6d. Feni L., Jutten L., Parente S., Piarulli U., Neundorf I., Diaz D., Chem. Commun. 2020, 56, 5685–5688. [DOI] [PubMed] [Google Scholar]
- 7. Gomes N. G. M., Pereira R. B., Andrade P. B., Valentão P., Mar. Drugs 2019, 17, 551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Daugan A., Grondin P., Ruault C., Le Monnier de Gouville A. C., Coste H., Kirilovsky J., Hyafil F., Labaudiniere R., J. Med. Chem. 2003, 46, 4525–4532. [DOI] [PubMed] [Google Scholar]
- 9.
- 9a. Liddle J., Allen M. J., Borthwick A. D., Brooks D. P., Davies D. E., Edwards R. M., Exall A. M., Hamlett C., Irving W. R., Mason A. M., McCafferty G. P., Nerozzi F., Peace S., Philp J., Pollard D., Pullen M. A., Shabbir S. S., Sollis S. L., Westfall T. D., Woollard P. M., Wu C., Hickey D. M., Bioorg. Med. Chem. Lett. 2008, 18, 90–94; [DOI] [PubMed] [Google Scholar]
- 9b. Borthwick A. D., Liddle J., Med. Res. Rev. 2011, 31, 576–604. [DOI] [PubMed] [Google Scholar]
- 10. Saade G. R., Shennan A., Beach K. J., Hadar E., Parilla B. V., Snidow J., Powell M., Montague T. H., Liu F., Komatsu Y., McKain L., Thornton S., Am. J. Perinatol. 2021, 38, e309-e317. [DOI] [PubMed] [Google Scholar]
- 11. Blayney D. W., Mohanlal R., Adamchuk H., Kirtbaya D. V., Chen M., Du L., Ogenstad S., Ginn G., Huang L., Zhang Q., JAMA Netw. Open 2022, 5, e2145446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Song Z., Hou Y., Yang Q., Li X., Wu S., Mar. Drugs 2021, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mishra A. K., Choi J., Choi S. J., Baek K. H., Molecules 2017, 22. [Google Scholar]
- 14.
- 14a. Moutiez M., Belin P., Gondry M., Chem. Rev. 2017, 117, 5578–5618; [DOI] [PubMed] [Google Scholar]
- 14b. Borgman P., Lopez R. D., Lane A. L., Org. Biomol. Chem. 2019, 17, 2305–2314. [DOI] [PubMed] [Google Scholar]
- 15. Süssmuth R. D., Mainz A., Angew. Chem. Int. Ed. 2017, 56, 3770–3821; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 3824–3878. [Google Scholar]
- 16.
- 16a. King R. R., Calhoun L. A., Phytochemistry 2009, 70, 833–841; [DOI] [PubMed] [Google Scholar]
- 16b. Schultz A. W., Oh D. C., Carney J. R., Williamson R. T., Udwary D. W., Jensen P. R., Gould S. J., Fenical W., Moore B. S., J. Am. Chem. Soc. 2008, 130, 4507–4516. [DOI] [PubMed] [Google Scholar]
- 17.
- 17a. Harken L., Li S. M., Appl. Microbiol. Biotechnol. 2021, 105, 2277–2285; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17b. Giessen T. W., Marahiel M. A., Front. Microbiol. 2015, 6, 785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Lautru S., Gondry M., Genet R., Pernodet J. L., Chem. Biol. 2002, 9, 1355–1364. [DOI] [PubMed] [Google Scholar]
- 19. de la Torre D., Chin J. W., Nat. Rev. Genet. 2021, 22, 169–184. [DOI] [PubMed] [Google Scholar]
- 20. Winn M., Fyans J. K., Zhuo Y., Micklefield J., Nat. Prod. Rep. 2016, 33, 317–347. [DOI] [PubMed] [Google Scholar]
- 21. Wunsch C., Mundt K., Li S. M., Appl. Microbiol. Biotechnol. 2015, 99, 4213–4223. [DOI] [PubMed] [Google Scholar]
- 22.
- 22a. Canu N., Moutiez M., Belin P., Gondry M., Nat. Prod. Rep. 2020, 37, 312–321; [DOI] [PubMed] [Google Scholar]
- 22b. Kries H., Wachtel R., Pabst A., Wanner B., Niquille D., Hilvert D., Angew. Chem. Int. Ed. 2014, 53, 10105–10108; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 10269–10272. [Google Scholar]
- 23.
- 23a. Fox E. M., Howlett B. J., Mycol. Res. 2008, 112, 162–169; [DOI] [PubMed] [Google Scholar]
- 23b. Patron N. J., Waller R. F., Cozijnsen A. J., Straney D. C., Gardiner D. M., Nierman W. C., Howlett B. J., BMC Evol. Biol. 2007, 7, 174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gardiner D. M., Waring P., Howlett B. J., Microbiology 2005, 151, 1021–1032. [DOI] [PubMed] [Google Scholar]
- 25. Kwon-Chung K. J., Sugui J. A., Med. Mycol. 2009, 47 Suppl. 1, S97–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fitzgerald J. M., Collin R. G., Towers N. R., Lett. Appl. Microbiol. 1998, 26, 17–21. [DOI] [PubMed] [Google Scholar]
- 27. Kilgore H. R., Olsson C. R., D'Angelo K. A., Movassaghi M., Raines R. T., J. Am. Chem. Soc. 2020, 142, 15107–15115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Welch T. R., Williams R. M., Nat. Prod. Rep. 2014, 31, 1376–1404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhu M., Zhang X., Huang X., Wang H., Anjum K., Gu Q., Zhu T., Zhang G., Li D., J. Nat. Prod. 2020, 83, 2045–2053. [DOI] [PubMed] [Google Scholar]
- 30.
- 30a. Mullbacher A., Waring P., Tiwari-Palni U., Eichner R. D., Mol. Immunol. 1986, 23, 231–235; [DOI] [PubMed] [Google Scholar]
- 30b. Trown P. W., Bilello J. A., Antimicrob. Agents Chemother. 1972, 2, 261–266; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30c. Mason J. W., Kidd J. G., J. Immunol. 1951, 66, 99–106. [PubMed] [Google Scholar]
- 31. Bernardo P. H., Brasch N., Chai C. L., Waring P., J. Biol. Chem. 2003, 278, 46549–46555. [DOI] [PubMed] [Google Scholar]
- 32.
- 32a. Waring P., Sjaarda A., Lin Q. H., Biochem. Pharmacol. 1995, 49, 1195–1201; [DOI] [PubMed] [Google Scholar]
- 32b. Eichner R. D., Waring P., Geue A. M., Braithwaite A. W., Mullbacher A., J. Biol. Chem. 1988, 263, 3772–3777. [PubMed] [Google Scholar]
- 33.
- 33a. Hurne A. M., Chai C. L., Waring P., J. Biol. Chem. 2000, 275, 25202–25206; [DOI] [PubMed] [Google Scholar]
- 33b. Cherblanc F. L., Chapman K. L., Reid J., Borg A. J., Sundriyal S., Alcazar-Fuoli L., Bignell E., Demetriades M., Schofield C. J., P. A. DiMaggio, Jr. , Brown R., Fuchter M. J., J. Med. Chem. 2013, 56, 8616–8625. [DOI] [PubMed] [Google Scholar]
- 34.
- 34a. Kung A. L., Zabludoff S. D., France D. S., Freedman S. J., Tanner E. A., Vieira A., Cornell-Kennon S., Lee J., Wang B., Wang J., Memmert K., Naegeli H. U., Petersen F., Eck M. J., Bair K. W., Wood A. W., Livingston D. M., Cancer Cell 2004, 6, 33–43; [DOI] [PubMed] [Google Scholar]
- 34b. Cook K. M., Hilton S. T., Mecinovic J., Motherwell W. B., Figg W. D., Schofield C. J., J. Biol. Chem. 2009, 284, 26831–26838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Jordan T. W., Cordiner S. J., Trends Pharmacol. Sci. 1987, 8, 144–149. [Google Scholar]
- 36. Weaver M. A., Hoagland R. E., Boyette C. D., Brown S. P., J. Fungi 2021, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.
- 37a. Brian P. W., Nature 1944, 154, 667–668; [Google Scholar]
- 37b. Weindling R., Phytopath. 1941, 31, 991–1003. [Google Scholar]
- 38. Lumsden R. D., Walter J. F., Can. J. Plant Pathol. 1996, 18, 463–468. [Google Scholar]
- 39.
- 39a. Lewis R. E., Wiederhold N. P., Lionakis M. S., Prince R. A., Kontoyiannis D. P., J. Clin. Microbiol. 2005, 43, 6120–6122; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39b. Kupfahl C., Michalka A., Lass-Florl C., Fischer G., Haase G., Ruppert T., Geginat G., Hof H., Int. J. Med. Microbiol. 2008, 298, 319–327. [DOI] [PubMed] [Google Scholar]
- 40. Rightsel W. A., Schneider H. G., Sloan B. J., Graf P. R., Miller F. A., Bartz O. R., Ehrlich J., Dixon G. J., Nature 1964, 204, 1333–1334. [DOI] [PubMed] [Google Scholar]
- 41. Rodriguez P. L., Carrasco L., J. Virol. 1992, 66, 1971–1976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.
- 42a. Mullbacher A., Moreland A. F., Waring P., Sjaarda A., Eichner R. D., Transplantation 1988, 46, 120–125; [DOI] [PubMed] [Google Scholar]
- 42b. Sutton P., Moreland A., Hutchinson I. V., Müllbacher A., Transplantation 1995, 60, 900–902. [PubMed] [Google Scholar]
- 43. Sutton P., Newcombe N. R., Waring P., Mullbacher A., Infect. Immun. 1994, 62, 1192–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Pahl H. L., Krauss B., Schulze-Osthoff K., Decker T., Traenckner E. B., Vogt M., Myers C., Parks T., Warring P., Muhlbacher A., Czernilofsky A. P., Baeuerle P. A., J. Exp. Med. 1996, 183, 1829–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Palombella V. J., Rando O. J., Goldberg A. L., Maniatis T., Cell 1994, 78, 773–785. [DOI] [PubMed] [Google Scholar]
- 46. Kroll M., Arenzana-Seisdedos F., Bachelerie F., Thomas D., Friguet B., Conconi M., Chem. Biol. 1999, 6, 689–698. [DOI] [PubMed] [Google Scholar]
- 47. Li J., Zhang Y., Da Silva Sil Dos Santos B., Wang F., Ma Y., Perez C., Yang Y., Peng J., Cohen S. M., Chou T. F., Hilton S. T., Deshaies R. J., Cell Chem. Biol. 2018, 25, 1350–1358 e1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.
- 48a. Borissenko L., Groll M., Chem. Rev. 2007, 107, 687–717; [DOI] [PubMed] [Google Scholar]
- 48b. Gräwert M. A., Groll M., Chem. Commun. 2012, 48, 1364–1378. [DOI] [PubMed] [Google Scholar]
- 49. Hatabu T., Hagiwara M., Taguchi N., Kiyozawa M., Suzuki M., Kano S., Sato K., Exp. Parasitol. 2006, 112, 179–183. [DOI] [PubMed] [Google Scholar]
- 50. Van der Pyl D., Inokoshi J., Shiomi K., Yang H., Takeshima H., Omura S., J. Antibiot. 1992, 45, 1802–1805. [DOI] [PubMed] [Google Scholar]
- 51. Vigushin D. M., Mirsaidi N., Brooke G., Sun C., Pace P., Inman L., Moody C. J., Coombes R. C., Med. Oncol. 2004, 21, 21–30. [DOI] [PubMed] [Google Scholar]
- 52. Green D., Pace S. M., Hurne A. M., Waring P., Hart J. D., Dulhunty A. F., J. Membr. Biol. 2000, 175, 223–233. [DOI] [PubMed] [Google Scholar]
- 53. Orr J. G., Leel V., Cameron G. A., Marek C. J., Haughton E. L., Elrick L. J., Trim J. E., Hawksworth G. M., Halestrap A. P., Wright M. C., Hepatology 2004, 40, 232–242. [DOI] [PubMed] [Google Scholar]
- 54.
- 54a. Nishida S., Yoshida L. S., Shimoyama T., Nunoi H., Kobayashi T., Tsunawaki S., Infect. Immun. 2005, 73, 235–244; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54b. Tsunawaki S., Yoshida L. S., Nishida S., Kobayashi T., Shimoyama T., Infect. Immun. 2004, 72, 3373–3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Srinivasan U., Bala A., Jao S. C., Starke D. W., Jordan T. W., Mieyal J. J., Biochemistry 2006, 45, 8978–8987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Schweizer M., Richter C., Biochemistry 1994, 33, 13401–13405. [DOI] [PubMed] [Google Scholar]
- 57. Salvi M., Bozac A., Toninello A., Neurochem. Int. 2004, 45, 759–764. [DOI] [PubMed] [Google Scholar]
- 58.
- 58a. Pardo J., Urban C., Galvez E. M., Ekert P. G., Muller U., Kwon-Chung J., Lobigs M., Mullbacher A., Wallich R., Borner C., Simon M. M., J. Cell Biol. 2006, 174, 509–519; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58b. Suen Y. K., Fung K. P., Lee C. Y., Kong S. K., Free Radical Res. 2001, 35, 1–10. [DOI] [PubMed] [Google Scholar]
- 59. Geissler A., Haun F., Frank D. O., Wieland K., Simon M. M., Idzko M., Davis R. J., Maurer U., Borner C., Cell Death Differ. 2013, 20, 1317–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.
- 60a. Kweon Y. O., Paik Y. H., Schnabl B., Qian T., Lemasters J. J., Brenner D. A., J. Hepatol. 2003, 39, 38–46; [DOI] [PubMed] [Google Scholar]
- 60b. Zhou X., Zhao A., Goping G., Hirszel P., Toxicol. Sci. 2000, 54, 194–202. [DOI] [PubMed] [Google Scholar]
- 61.
- 61a. Chen J., Wang C., Lan W., Huang C., Lin M., Wang Z., Liang W., Iwamoto A., Yang X., Liu H., Mar. Drugs 2015, 13, 6259–6273; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61b. Kim Y.-S., Park S. J., Fish. Aquat. Sci. 2016, 19, 35; [Google Scholar]
- 61c. Reece K. M., Richardson E. D., Cook K. M., Campbell T. J., Pisle S. T., Holly A. J., Venzon D. J., Liewehr D. J., Chau C. H., Price D. K., Figg W. D., Mol. Cancer 2014, 13, 91; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61d. Ni H., Ergin M., Huang Q., Qin J. Z., Amin H. M., Martinez R. L., Saeed S., Barton K., Alkan S., Br. J. Haematol. 2001, 115, 279–286; [DOI] [PubMed] [Google Scholar]
- 61e. Izban K. F., Ergin M., Huang Q., Qin J. Z., Martinez R. L., Schnitzer B., Ni H., Nickoloff B. J., Alkan S., Mod. Pathol. 2001, 14, 297–310. [DOI] [PubMed] [Google Scholar]
- 62. Ben-Ami R., Lewis R. E., Leventakos K., Kontoyiannis D. P., Blood 2009, 114, 5393–5399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.
- 63a. Hof H., Kupfahl C., Mycotoxin Res. 2009, 25, 123–131; [DOI] [PubMed] [Google Scholar]
- 63b. Sugui J. A., Pardo J., Chang Y. C., Zarember K. A., Nardone G., Galvez E. M., Mullbacher A., Gallin J. I., Simon M. M., Kwon-Chung K. J., Eukaryotic Cell 2007, 6, 1562–1569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Spikes S., Xu R., Nguyen C. K., Chamilos G., Kontoyiannis D. P., Jacobson R. H., Ejzykowicz D. E., Chiang L. Y., Filler S. G., May G. S., J. Infect. Dis. 2008, 197, 479–486. [DOI] [PubMed] [Google Scholar]
- 65. Knowles S. L., Mead M. E., Silva L. P., Raja H. A., Steenwyk J. L., Goldman G. H., Oberlies N. H., Rokas A., mBio 2020, 11, e03361–1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.
- 66a. Owens R. A., Hammel S., Sheridan K. J., Jones G. W., Doyle S., PLoS One 2014, 9, e106942; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66b. Schrettl M., Carberry S., Kavanagh K., Haas H., Jones G. W., O′Brien J., Nolan A., Stephens J., Fenelon O., Doyle S., PLoS Pathog. 2010, 6, e1000952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Bell M. R., Johnson J. R., Wildi B. S., Woodward R. B., J. Am. Chem. Soc. 1958, 80, 1001–1001. [Google Scholar]
- 68.
- 68a. Winstead J. A., Suhadolnik R. J., J. Am. Chem. Soc. 1960, 82, 1644–1647; [Google Scholar]
- 68b. Suhadolnik R. J., Chenoweth R. G., J. Am. Chem. Soc. 1958, 80, 4391–4392. [Google Scholar]
- 69.
- 69a. Fridrichsons J., Mathieson A. M., Acta Crystallogr. 1967, 23, 439–448; [DOI] [PubMed] [Google Scholar]
- 69b. Beecham A. F., Fridrichsons J., Mathieson A. M., Tetrahedron Lett. 1966, 7, 3131–3138. [DOI] [PubMed] [Google Scholar]
- 70. Nierman W. C., Pain A., Anderson M. J., Wortman J. R., Kim H. S., Arroyo J., Berriman M., Abe K., Archer D. B., Bermejo C., Bennett J., Bowyer P., Chen D., Collins M., Coulsen R., Davies R., Dyer P. S., Farman M., Fedorova N., Fedorova N., Feldblyum T. V., Fischer R., Fosker N., Fraser A., García J. L., García M. J., Goble A., Goldman G. H., Gomi K., Griffith-Jones S., Gwilliam R., Haas B., Haas H., Harris D., Horiuchi H., Huang J., Humphray S., Jiménez J., Keller N., Khouri H., Kitamoto K., Kobayashi T., Konzack S., Kulkarni R., Kumagai T., Lafton A., Latgé J.-P., Li W., Lord A., Lu C., Majoros W. H., May G. S., Miller B. L., Mohamoud Y., Molina M., Monod M., Mouyna I., Mulligan S., Murphy L., O′Neil S., Paulsen I., Peñalva M. A., Pertea M., Price C., Pritchard B. L., Quail M. A., Rabbinowitsch E., Rawlins N., Rajandream M.-A., Reichard U., Renauld H., Robson G. D., de Córdoba S. R., Rodríguez-Peña J. M., Ronning C. M., Rutter S., Salzberg S. L., Sanchez M., Sánchez-Ferrero J. C., Saunders D., Seeger K., Squares R., Squares S., Takeuchi M., Tekaia F., Turner G., de Aldana C. R. V., Weidman J., White O., Woodward J., Yu J.-H., Fraser C., Galagan J. E., Asai K., Machida M., Hall N., Barrell B., Denning D. W., Nature 2005, 438, 1151–1156. [DOI] [PubMed] [Google Scholar]
- 71. Gardiner D. M., Howlett B. J., FEMS Microbiol. Lett. 2005, 248, 241–248. [DOI] [PubMed] [Google Scholar]
- 72. Gardiner D. M., Cozijnsen A. J., Wilson L. M., Pedras M. S., Howlett B. J., Mol. Microbiol. 2004, 53, 1307–1318. [DOI] [PubMed] [Google Scholar]
- 73.
- 73a. R. A. Cramer, Jr. , Gamcsik M. P., Brooking R. M., Najvar L. K., Kirkpatrick W. R., Patterson T. F., Balibar C. J., Graybill J. R., Perfect J. R., Abraham S. N., Steinbach W. J., Eukaryotic Cell 2006, 5, 972–980; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73b. Kupfahl C., Heinekamp T., Geginat G., Ruppert T., Hartl A., Hof H., Brakhage A. A., Mol. Microbiol. 2006, 62, 292–302. [DOI] [PubMed] [Google Scholar]
- 74. Balibar C. J., Walsh C. T., Biochemistry 2006, 45, 15029–15038. [DOI] [PubMed] [Google Scholar]
- 75.
- 75a. Davis C., Carberry S., Schrettl M., Singh I., Stephens J. C., Barry S. M., Kavanagh K., Challis G. L., Brougham D., Doyle S., Chem. Biol. 2011, 18, 542–552; [DOI] [PubMed] [Google Scholar]
- 75b. Scharf D. H., Chankhamjon P., Scherlach K., Dworschak J., Heinekamp T., Roth M., Brakhage A. A., Hertweck C., ChemBioChem 2021, 22, 336–339; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75c. Scharf D. H., Dworschak J. D., Chankhamjon P., Scherlach K., Heinekamp T., Brakhage A. A., Hertweck C., ACS Chem. Biol. 2018, 13, 2508–2512; [DOI] [PubMed] [Google Scholar]
- 75d. Scharf D. H., Habel A., Heinekamp T., Brakhage A. A., Hertweck C., J. Am. Chem. Soc. 2014, 136, 11674–11679; [DOI] [PubMed] [Google Scholar]
- 75e. Scharf D. H., Remme N., Habel A., Chankhamjon P., Scherlach K., Heinekamp T., Hortschansky P., Brakhage A. A., Hertweck C., J. Am. Chem. Soc. 2011, 133, 12322–12325. [DOI] [PubMed] [Google Scholar]
- 76. Dolan S. K., Owens R. A., O′Keeffe G., Hammel S., Fitzpatrick D. A., Jones G. W., Doyle S., Chem. Biol. 2014, 21, 999–1012. [DOI] [PubMed] [Google Scholar]
- 77. Gao X., Haynes S. W., Ames B. D., Wang P., Vien L. P., Walsh C. T., Tang Y., Nat. Chem. Biol. 2012, 8, 823–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Baccile J. A., Le H. H., Pfannenstiel B. T., Bok J. W., Gomez C., Brandenburger E., Hoffmeister D., Keller N. P., Schroeder F. C., Angew. Chem. Int. Ed. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.
- 79a. Allen G., Bromley M., Kaye S. J., Keszenman-Pereyra D., Zucchi T. D., Price J., Birch M., Oliver J. D., Turner G., Fungal Genet. Biol. 2011, 48, 456–464; [DOI] [PubMed] [Google Scholar]
- 79b. Johns A., Scharf D. H., Gsaller F., Schmidt H., Heinekamp T., Strassburger M., Oliver J. D., Birch M., Beckmann N., Dobb K. S., Gilsenan J., Rash B., Bignell E., Brakhage A. A., Bromley M. J., mBio 2017, 8, e01504–e01516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Chang S. L., Chiang Y. M., Yeh H. H., Wu T. K., Wang C. C., Bioorg. Med. Chem. Lett. 2013, 23, 2155–2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Dolan S. K., O′Keeffe G., Jones G. W., Doyle S., Trends Microbiol. 2015, 23, 419–428. [DOI] [PubMed] [Google Scholar]
- 82.
- 82a. Su T., Xu J., Li Y., Lei L., Zhao L., Yang H., Feng J., Liu G., Ren D., Plant Cell 2011, 23, 364–380; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82b. Czerniawski P., Bednarek P., Front. Plant Sci. 2018, 9, 1639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Scherlach K., Kuttenlochner W., Scharf D. H., Brakhage A. A., Hertweck C., Groll M., Huber E. M., Angew. Chem. Int. Ed. 2021, 60, 14188–14194; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2021, 133, 14307–14314. [Google Scholar]
- 84. Scharf D. H., Chankhamjon P., Scherlach K., Heinekamp T., Willing K., Brakhage A. A., Hertweck C., Angew. Chem. Int. Ed. 2013, 52, 11092–11095; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 11298–11301. [Google Scholar]
- 85. Gallagher L., Owens R. A., Dolan S. K., O′Keeffe G., Schrettl M., Kavanagh K., Jones G. W., Doyle S., Eukaryotic Cell 2012, 11, 1226–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Llewellyn N. M., Li Y., Spencer J. B., Chem. Biol. 2007, 14, 379–386. [DOI] [PubMed] [Google Scholar]
- 87. Marion A., Groll M., Scharf D. H., Scherlach K., Glaser M., Sievers H., Schuster M., Hertweck C., Brakhage A. A., Antes I., Huber E. M., ACS Chem. Biol. 2017, 12, 1874–1882. [DOI] [PubMed] [Google Scholar]
- 88. Scharf D. H., Chankhamjon P., Scherlach K., Heinekamp T., Roth M., Brakhage A. A., Hertweck C., Angew. Chem. Int. Ed. 2012, 51, 10064–10068; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 10211–10215. [Google Scholar]
- 89.
- 89a. Bu′Lock J. D., Ryles A. P., J. Chem. Soc. D 1970, 1404–1406; [Google Scholar]
- 89b. Johns N., Kirby G. W., J. Chem. Soc. Perkin Trans. 1 1985, 1487–1490. [Google Scholar]
- 90. Johns N., Kirby G. W., J. Chem. Soc. D 1971, 163–164. [Google Scholar]
- 91. Forseth R. R., Fox E. M., Chung D., Howlett B. J., Keller N. P., Schroeder F. C., J. Am. Chem. Soc. 2011, 133, 9678–9681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Stipanovic R. D., Howell C. R., J. Antibiot. 1982, 35, 1326–1330. [DOI] [PubMed] [Google Scholar]
- 93. Nagarajan R., Huckstep L. L., Lively D. H., DeLong D. C., Marsh M. M., Neuss N., J. Am. Chem. Soc. 1968, 90, 2980–2982. [DOI] [PubMed] [Google Scholar]
- 94.
- 94a. Guo C. J., Yeh H. H., Chiang Y. M., Sanchez J. F., Chang S. L., Bruno K. S., Wang C. C., J. Am. Chem. Soc. 2013, 135, 7205–7213; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94b. Zheng L., Wang H., Ludwig-Radtke L., Li S. M., Org. Lett. 2021, 23, 2024–2028; [DOI] [PubMed] [Google Scholar]
- 94c. Stok J. E., Chow S., Krenske E. H., Farfan Soto C., Matyas C., Poirier R. A., Williams C. M., De Voss J. J., Chem. Eur. J. 2016, 22, 4408–4412. [DOI] [PubMed] [Google Scholar]
- 95. Liu Y., Mandi A., Li X. M., Meng L. H., Kurtan T., Wang B. G., Mar. Drugs 2015, 13, 3640–3652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96. Liscombe D. K., Louie G. V., Noel J. P., Nat. Prod. Rep. 2012, 29, 1238–1250. [DOI] [PubMed] [Google Scholar]
- 97.
- 97a. Velkov T., Horne J., Scanlon M. J., Capuano B., Yuriev E., Lawen A., Chem. Biol. 2011, 18, 464–475; [DOI] [PubMed] [Google Scholar]
- 97b. Hornbogen T., Riechers S. P., Prinz B., Schultchen J., Lang C., Schmidt S., Mugge C., Turkanovic S., Sussmuth R. D., Tauberger E., Zocher R., ChemBioChem 2007, 8, 1048–1054. [DOI] [PubMed] [Google Scholar]
- 98. Wu Y., Kang Q., Shang G., Spiteller P., Carroll B., Yu T. W., Su W., Bai L., Floss H. G., ChemBioChem 2011, 12, 1759–1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.
- 99a. Ramm S., Krawczyk B., Muhlenweg A., Poch A., Mosker E., Sussmuth R. D., Angew. Chem. Int. Ed. 2017, 56, 9994–9997; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 10127–10130; [Google Scholar]
- 99b. van der Velden N. S., Kalin N., Helf M. J., Piel J., Freeman M. F., Kunzler M., Nat. Chem. Biol. 2017, 13, 833–835. [DOI] [PubMed] [Google Scholar]
- 100. Giessen T. W., von Tesmar A. M., Marahiel M. A., Biochemistry 2013, 52, 4274–4283. [DOI] [PubMed] [Google Scholar]
- 101. Chatterjee J., Gilon C., Hoffman A., Kessler H., Acc. Chem. Res. 2008, 41, 1331–1342. [DOI] [PubMed] [Google Scholar]
- 102. Brzostowska E. M., Greer A., J. Am. Chem. Soc. 2003, 125, 396–404. [DOI] [PubMed] [Google Scholar]
- 103. Amatov T., Jahn U., Angew. Chem. Int. Ed. 2014, 53, 3312–3314; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 3378–3380. [Google Scholar]
- 104. Scharf D. H., Remme N., Heinekamp T., Hortschansky P., Brakhage A. A., Hertweck C., J. Am. Chem. Soc. 2010, 132, 10136–10141. [DOI] [PubMed] [Google Scholar]
- 105. Scharf D. H., Groll M., Habel A., Heinekamp T., Hertweck C., Brakhage A. A., Huber E. M., Angew. Chem. Int. Ed. 2014, 53, 2221–2224; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 2253–2256. [Google Scholar]
- 106. Li B., Walsh C. T., Biochemistry 2011, 50, 4615–4622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Wang D. N., Toyotome T., Muraosa Y., Watanabe A., Wuren T., Bunsupa S., Aoyagi K., Yamazaki M., Takino M., Kamei K., Med. Mycol. 2014, 52, 506–518. [DOI] [PubMed] [Google Scholar]
- 108. Drew D., North R. A., Nagarathinam K., Tanabe M., Chem. Rev. 2021, 121, 5289–5335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109. Bok J. W., Chung D., Balajee S. A., Marr K. A., Andes D., Nielsen K. F., Frisvad J. C., Kirby K. A., Keller N. P., Infect. Immun. 2006, 74, 6761–6768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Hong M., Fitzgerald M. X., Harper S., Luo C., Speicher D. W., Marmorstein R., Structure 2008, 16, 1019–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111. MacPherson S., Larochelle M., Turcotte B., Microbiol. Mol. Biol. Rev. 2006, 70, 583–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112. Todd R. B., Andrianopoulos A., Fungal Genet. Biol. 1997, 21, 388–405. [DOI] [PubMed] [Google Scholar]
- 113. Fox E. M., Gardiner D. M., Keller N. P., Howlett B. J., Fungal Genet. Biol. 2008, 45, 671–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Ries L. N. A., Pardeshi L., Dong Z., Tan K., Steenwyk J. L., Colabardini A. C., Ferreira Filho J. A., de Castro P. A., Silva L. P., Preite N. W., Almeida F., de Assis L. J., Dos Santos R. A. C., Bowyer P., Bromley M., Owens R. A., Doyle S., Demasi M., Hernandez D. C. R., Netto L. E. S., Pupo M. T., Rokas A., Loures F. V., Wong K. H., Goldman G. H., PLoS Pathog. 2020, 16, e1008645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. de Castro P. A., Colabardini A. C., Moraes M., Horta M. A. C., Knowles S. L., Raja H. A., Oberlies N. H., Koyama Y., Ogawa M., Gomi K., Steenwyk J. L., Rokas A., Goncales R. A., Duarte-Oliveira C., Carvalho A., Ries L. N. A., Goldman G. H., PLoS Genet. 2022, 18, e1009965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Schoberle T. J., Nguyen-Coleman C. K., Herold J., Yang A., Weirauch M., Hughes T. R., McMurray J. S., May G. S., PLoS Genet. 2014, 10, e1004336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.
- 117a. Bok J. W., Keller N. P., Eukaryotic Cell 2004, 3, 527–535; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117b. Brakhage A. A., Nat. Rev. Microbiol. 2013, 11, 21–32. [DOI] [PubMed] [Google Scholar]
- 118. Perrin R. M., Fedorova N. D., Bok J. W., Cramer R. A., Wortman J. R., Kim H. S., Nierman W. C., Keller N. P., PLoS Pathog. 2007, 3, e50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Bayram O., Krappmann S., Ni M., Bok J. W., Helmstaedt K., Valerius O., Braus-Stromeyer S., Kwon N. J., Keller N. P., Yu J. H., Braus G. H., Science 2008, 320, 1504–1506. [DOI] [PubMed] [Google Scholar]
- 120. Patananan A. N., Palmer J. M., Garvey G. S., Keller N. P., Clarke S. G., J. Biol. Chem. 2013, 288, 14032–14045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Smith E. B., Dolan S. K., Fitzpatrick D. A., Doyle S., Jones G. W., Microb. Cell Fact. 2016, 3, 120–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122. Dolan S. K., Bock T., Hering V., Owens R. A., Jones G. W., Blankenfeldt W., Doyle S., Open Biology 2017, 7, 160292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.
- 123a. Domingo M. P., Colmenarejo C., Martinez-Lostao L., Mullbacher A., Jarne C., Revillo M. J., Delgado P., Roc L., Meis J. F., Rezusta A., Pardo J., Galvez E. M., Diagn. Microbiol. Infect. Dis. 2012, 73, 57–64; [DOI] [PubMed] [Google Scholar]
- 123b. Vidal-Garcia M., Domingo M. P., De Rueda B., Roc L., Delgado M. P., Revillo M. J., Pardo J., Galvez E. M., Rezusta A., Appl. Microbiol. Biotechnol. 2016, 100, 2327–2334. [DOI] [PubMed] [Google Scholar]
- 124. Mercier T., Resendiz Sharpe A., Waumans D., Desmet K., Lagrou K., Maertens J., Mycoses 2019, 62, 945–948. [DOI] [PubMed] [Google Scholar]
- 125. Duell E. R., Glaser M., Le Chapelain C., Antes I., Groll M., Huber E. M., ACS Chem. Biol. 2016, 11, 1082–1089. [DOI] [PubMed] [Google Scholar]
- 126. Zheng C. J., Kim C. J., Bae K. S., Kim Y. H., Kim W. G., J. Nat. Prod. 2006, 69, 1816–1819. [DOI] [PubMed] [Google Scholar]
- 127. Park H. B., Kim Y. J., Park J. S., Yang H. O., Lee K. R., Kwon H. C., J. Nat. Prod. 2011, 74, 2309–2312. [DOI] [PubMed] [Google Scholar]
- 128. Li B., Forseth R. R., Bowers A. A., Schroeder F. C., Walsh C. T., ChemBioChem 2012, 13, 2521–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129. Bulock J. D., Clough L. E., Aust. J. Chem. 1992, 45, 39–45. [Google Scholar]
- 130. Howlett B. J., Idnurm A., Pedras M. S., Fungal Genet. Biol. 2001, 33, 1–14. [DOI] [PubMed] [Google Scholar]
- 131.
- 131a. Sock J., Hoppe H. H., J. Phytopathol. 1999, 147, 169–173; [Google Scholar]
- 131b. Elliott C. E., Gardiner D. M., Thomas G., Cozijnsen A., Van De Wouw A., Howlett B. J., Mol. Plant Pathol. 2007, 8, 791–802. [DOI] [PubMed] [Google Scholar]
- 132. Soledade M., Pedras C., Taylor J. L., J. Nat. Prod. 1993, 56, 731–738. [Google Scholar]
- 133.
- 133a. Ferezou J.-P., Quesneau-Thierry A., Servy C., Zissmann E., Barbier M., J. Chem. Soc. Perkin Trans. 1 1980, 1739–1746; [Google Scholar]
- 133b. Ferezou J.-P., Quesneau-Thierry A., Barbier M., Kollmann A., Bousquet J.-F., J. Chem. Soc. Perkin Trans. 1 1980, 113–115. [Google Scholar]
- 134. Pedras M. S. C., Séguin-Swartz G., Abrams S. R., Phytochemistry 1990, 29, 777–782. [Google Scholar]
- 135. Urquhart A. S., Elliott C. E., Zeng W., Idnurm A., PLoS One 2021, 16, e0252333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136. Gerken T., Walsh C. T., ChemBioChem 2013, 14, 2256–2258. [DOI] [PubMed] [Google Scholar]
- 137. Gardiner D. M., Jarvis R. S., Howlett B. J., Fungal Genet. Biol. 2005, 42, 257–263. [DOI] [PubMed] [Google Scholar]
- 138.
- 138a. Rudolf J. D., Poulter C. D., ACS Chem. Biol. 2013, 8, 2707–2714; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138b. Bandari C., Scull E. M., Masterson J. M., Tran R. H. Q., Foster S. B., Nicholas K. M., Singh S., ChemBioChem 2017, 18, 2323–2327; [DOI] [PubMed] [Google Scholar]
- 138c. Zou H. X., Xie X., Zheng X. D., Li S. M., Appl. Microbiol. Biotechnol. 2011, 89, 1443–1451. [DOI] [PubMed] [Google Scholar]
- 139.
- 139a. Pedras M. S. C., Yu Y., Can. J. Chem. 2009, 87, 556–562; [Google Scholar]
- 139b. Kremer A., Li S. M., Microbiology 2010, 156, 278–286. [DOI] [PubMed] [Google Scholar]
- 140.
- 140a. Neuss N., Boeck L. D., Brannon D. R., Cline J. C., DeLong D. C., Gorman M., Huckstep L. L., Lively D. H., Mabe J., Marsh M. M., Molloy B. B., Nagarajan R., Nelson J. D., Stark W. M., Antimicrob. Agents Chemother. 1968, 8, 213–219; [PubMed] [Google Scholar]
- 140b. Nagarajan R., Neuss N., Marsh M. M., J. Am. Chem. Soc. 1968, 90, 6518–6519; [DOI] [PubMed] [Google Scholar]
- 140c. Neuss N., Nagarajan R., Molloy B. B., Huckstep L. L., Tetrahedron Lett. 1968, 9, 4467–4471; [Google Scholar]
- 140d. Miller P. A., Trown P. W., Fulmor W., Morton G. O., Karliner J., Biochem. Biophys. Res. Commun. 1968, 33, 219–221. [DOI] [PubMed] [Google Scholar]
- 141. Choi E. J., Park J. S., Kim Y. J., Jung J. H., Lee J. K., Kwon H. C., Yang H. O., J. Appl. Microbiol. 2011, 110, 304–313. [DOI] [PubMed] [Google Scholar]
- 142. Boente M. I. P., Kirby G. W., Robins D. J., J. Chem. Soc. 1981, 619–621. [Google Scholar]
- 143. Wang Y., Hu P., Pan Y., Zhu Y., Liu X., Che Y., Liu G., Fungal Genet. Biol. 2017, 103, 25–33. [DOI] [PubMed] [Google Scholar]
- 144. Sidhu J. S., Suresh V., Baten A., McCartney A. M., Lear G., Sprosen J. M., Oliver M. H., Forester N. T., Maclean P. H., Palevich N., Jauregui R., Voisey C. R., bioRxiv 2021, 10.1101/2021.04.29.441555 [DOI] [Google Scholar]
- 145. Sun W. W., Romsdahl J., Guo C. J., Wang C. C. C., Fungal Genet. Biol. 2018, 119, 1–6. [DOI] [PubMed] [Google Scholar]
- 146.
- 146a. Mortimer P. H., Res. Vet. Sci. 1963, 4, 166–195; [Google Scholar]
- 146b. Russell G. R., Nature 1960, 186, 788–789; [DOI] [PubMed] [Google Scholar]
- 146c. Gallagher C. H., Nature 1964, 201, 1293–1294; [DOI] [PubMed] [Google Scholar]
- 146d. Thornton R. H., Percival J. C., Nature 1959, 183, 63–63. [DOI] [PubMed] [Google Scholar]
- 147. Thornton R. H., Sinclair D. P., Nature 1959, 184, 1327–1328. [DOI] [PubMed] [Google Scholar]
- 148.
- 148a. Smith B. L., Embling P. P., Towers N. R., Wright D. E., Payne E., N. Z. Vet. J. 1977, 25, 124–127; [DOI] [PubMed] [Google Scholar]
- 148b. Towers N. R., Smith B. L., N. Z. Vet. J. 1978, 26, 199–202. [DOI] [PubMed] [Google Scholar]
- 149. Waring P., Egan M., Braithwaite A., Mullbacher A., Sjaarda A., Int. J. Immunopharmacol. 1990, 12, 445–457. [DOI] [PubMed] [Google Scholar]
- 150.
- 150a. Fridrichsons J., Mathieson A., Acta Crystallogr. 1965, 18, 1043–1052; [Google Scholar]
- 150b. Fridrichsons J., Mathieson A. M., Tetrahedron Lett. 1962, 3, 1265–1268. [Google Scholar]
- 151. Towers N. R., Wright D. E., New Zealand J. Agric. Res. 1969, 12, 275–280. [Google Scholar]
- 152. Howell C. R., Stipanovic R. D., Lumsden R. D., Biocontrol Sci. Technol. 1993, 3, 435–441. [Google Scholar]
- 153. Howell C. R., Stipanovic R. D., Can. J. Microbiol. 1983, 29, 321–324. [Google Scholar]
- 154. Rether J., Serwe A., Anke T., Erkel G., Biol. Chem. 2007, 388, 627–637. [DOI] [PubMed] [Google Scholar]
- 155. Stipanovic R. D., Howell C. R., Hedin P. A., J. Antibiot. 1994, 47, 942–944. [DOI] [PubMed] [Google Scholar]
- 156. Sherkhane P. D., Bansal R., Banerjee K., Chatterjee S., Oulkar D., Jain P., Rosenfelder L., Elgavish S., Horwitz B. A., Mukherjee P. K., ChemistrySelect 2017, 2, 3347–3352. [Google Scholar]
- 157. Tsunematsu Y., Maeda N., Sato M., Hara K., Hashimoto H., Watanabe K., Hertweck C., J. Am. Chem. Soc. 2021, 143, 206–213. [DOI] [PubMed] [Google Scholar]
- 158. Kato A., Saeki T., Suzuki S., Ando K., Tamura G., J. Antibiot. 1969, 22, 322–326. [PubMed] [Google Scholar]
- 159. Berg D. H., Massing R. P., Hoehn M. M., Boeck L. D., Hamill R. L., J. Antibiot. 1976, 29, 394–397. [DOI] [PubMed] [Google Scholar]
- 160. Sakata K., Masago H., Sakurai A., Takahashi N., Tetrahedron Lett. 1982, 23, 2095–2098. [Google Scholar]
- 161. Sakata K., Kuwatsuka T., Sakurai A., Takahashi N., Tamura G., Agric. Biol. Chem. 1983, 47, 2673–2674. [Google Scholar]
- 162. Sakata K., Maruyama M., Uzawa J., Sakurai A., Lu H., Clardy J., Tetrahedron Lett. 1987, 28, 5607–5610. [Google Scholar]
- 163. Monti F., Ripamonti F., Hawser S. P., Islam K., J. Antibiot. 1999, 52, 311–318. [DOI] [PubMed] [Google Scholar]
- 164. Klausmeyer P., McCloud T. G., Tucker K. D., Cardellina, 2nd J. H., Shoemaker R. H., J. Nat. Prod. 2005, 68, 1300–1302. [DOI] [PubMed] [Google Scholar]
- 165. Chankhamjon P., Boettger-Schmidt D., Scherlach K., Urbansky B., Lackner G., Kalb D., Dahse H. M., Hoffmeister D., Hertweck C., Angew. Chem. Int. Ed. 2014, 53, 13409–13413; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2014, 126, 13627–13631. [Google Scholar]
- 166. Frisvad J. C., Moller L. L. H., Larsen T. O., Kumar R., Arnau J., Appl. Microbiol. Biotechnol. 2018, 102, 9481–9515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.
- 167a. Barbesgaard P., Heldt-Hansen H. P., Diderichsen B., Appl. Microbiol. Biotechnol. 1992, 36, 569–572; [DOI] [PubMed] [Google Scholar]
- 167b. Machida M., Yamada O., Gomi K., DNA Res. 2008, 15, 173–183; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167c. Ito K., Matsuyama A., J. Fungi 2021, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168. Sato T., Chida N., Org. Biomol. Chem. 2014, 12, 3147–3150. [DOI] [PubMed] [Google Scholar]
- 169. Tsunematsu Y., Maeda N., Yokoyama M., Chankhamjon P., Watanabe K., Scherlach K., Hertweck C., Angew. Chem. Int. Ed. 2018, 57, 14051–14054; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 14247–14250. [Google Scholar]
- 170. Lenz R., Zenk M. H., J. Biol. Chem. 1995, 270, 31091–31096. [DOI] [PubMed] [Google Scholar]
- 171. Caputi L., Franke J., Farrow S. C., Chung K., Payne R. M. E., Nguyen T. D., Dang T. T., Soares Teto Carqueijeiro I., Koudounas K., Duge de Bernonville T., Ameyaw B., Jones D. M., Vieira I. J. C., Courdavault V., O′Connor S. E., Science 2018, 360, 1235–1239. [DOI] [PubMed] [Google Scholar]
- 172.
- 172a. Zhang Y., Liu S., Che Y., Liu X., J. Nat. Prod. 2007, 70, 1522–1525; [DOI] [PubMed] [Google Scholar]
- 172b. Guo H., Sun B., Gao H., Chen X., Liu S., Yao X., Liu X., Che Y., J. Nat. Prod. 2009, 72, 2115–2119; [DOI] [PubMed] [Google Scholar]
- 172c. Wang J. M., Ding G. Z., Fang L., Dai J. G., Yu S. S., Wang Y. H., Chen X. G., Ma S. G., Qu J., Xu S., Du D., J. Nat. Prod. 2010, 73, 1240–1249. [DOI] [PubMed] [Google Scholar]
- 173. Dong J. Y., He H. P., Shen Y. M., Zhang K. Q., J. Nat. Prod. 2005, 68, 1510–1513. [DOI] [PubMed] [Google Scholar]
- 174. Guo Z., Hao T., Wang Y., Pan Y., Ren F., Liu X., Che Y., Liu G., Microbiology 2017, 163, 1654–1663. [DOI] [PubMed] [Google Scholar]
- 175. Figueroa M., Graf T. N., Ayers S., Adcock A. F., Kroll D. J., Yang J., Swanson S. M., Munoz-Acuna U., Carcache de Blanco E. J., Agrawal R., Wani M. C., Darveaux B. A., Pearce C. J., Oberlies N. H., J. Antibiot. 2012, 65, 559–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176. Katagiri K., Sato K., Hayakawa S., Matsushima T., Minato H., J. Antibiot. 1970, 23, 420–422. [DOI] [PubMed] [Google Scholar]
- 177. Chu M., Truumees I., Rothofsky M. L., Patel M. G., Gentile F., Das P. R., Puar M. S., Lin S. L., J. Antibiot. 1995, 48, 1440–1445. [DOI] [PubMed] [Google Scholar]
- 178. Paschall A. V., Yang D., Lu C., Choi J. H., Li X., Liu F., Figueroa M., Oberlies N. H., Pearce C., Bollag W. B., Nayak-Kapoor A., Liu K., J. Immunol. 2015, 195, 1868–1882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179. Schenke D., Böttcher C., Lee J., Scheel D., J. Antibiot. 2011, 64, 523–524. [DOI] [PubMed] [Google Scholar]
- 180. Amrine C. S. M., Long J. L., Raja H. A., Kurina S. J., Burdette J. E., Pearce C. J., Oberlies N. H., J. Nat. Prod. 2019, 82, 3104–3110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181. Hauser D., Weber H. P., Sigg H. P., Helv. Chim. Acta 1970, 53, 1061–1073. [DOI] [PubMed] [Google Scholar]
- 182. Greiner D., Bonaldi T., Eskeland R., Roemer E., Imhof A., Nat. Chem. Biol. 2005, 1, 143–145. [DOI] [PubMed] [Google Scholar]
- 183.
- 183a. Tibodeau J. D., Benson L. M., Isham C. R., Owen W. G., Bible K. C., Antioxid. Redox Signaling 2009, 11, 1097–1106; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183b. Song X., Zhao Z., Qi X., Tang S., Wang Q., Zhu T., Gu Q., Liu M., Li J., Oncotarget 2015, 6, 5263–5274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.
- 184a. Isham C. R., Tibodeau J. D., Jin W., Xu R., Timm M. M., Bible K. C., Blood 2007, 109, 2579–2588; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184b. Jiang H., Li Y., Xiang X., Tang Z., Liu K., Su Q., Zhang X., Li L., Eur. J. Pharmacol. 2021, 910, 174459. [DOI] [PubMed] [Google Scholar]
- 185.
- 185a. Ma J., Wang Z., Huang H., Luo M., Zuo D., Wang B., Sun A., Cheng Y. Q., Zhang C., Ju J., Angew. Chem. Int. Ed. 2011, 50, 7797–7802; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 7943–7948; [Google Scholar]
- 185b. D′Angelo K. A., Schissel C. K., Pentelute B. L., Movassaghi M., Science 2022, 375, 894–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186. Waksman Selman A., Bugie E., J. Bacteriol. 1944, 48, 527–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187. McInnes A. G., Taylor A., Walter J. A., J. Am. Chem. Soc. 1976, 98, 6741–6741. [DOI] [PubMed] [Google Scholar]
- 188.
- 188a. Min S., Wang X., Du Q., Gong H., Yang Y., Wang T., Wu N., Liu X., Li W., Zhao C., Shen Y., Chen Y., Wang X., Cancer Biol. Ther. 2020, 21, 698–708; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188b. Yano K., Horinaka M., Yoshida T., Yasuda T., Taniguchi H., Goda A. E., Wakada M., Yoshikawa S., Nakamura T., Kawauchi A., Miki T., Sakai T., Int. J. Oncol. 2011, 38, 365–374; [DOI] [PubMed] [Google Scholar]
- 188c. Viziteu E., Grandmougin C., Goldschmidt H., Seckinger A., Hose D., Klein B., Moreaux J., Br. J. Cancer 2016, 114, 519–523; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188d. Dewangan J., Srivastava S., Mishra S., Pandey P. K., Divakar A., Rath S. K., Biochem. Biophys. Res. Commun. 2018, 495, 1915–1921; [DOI] [PubMed] [Google Scholar]
- 188e. Staab A., Loeffler J., Said H. M., Diehlmann D., Katzer A., Beyer M., Fleischer M., Schwab F., Baier K., Einsele H., Flentje M., Vordermark D., BMC Cancer 2007, 7, 213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189. Zhao P., Liu H., Wu Q., Meng Q., Qu K., Yin X., Wang M., Zhao X., Qi J., Meng Y., Xia X., Zhang L., Appl. Microbiol. Biotechnol. 2022, 106, 3093–3102. [DOI] [PubMed] [Google Scholar]
- 190. Xu G. B., He G., Bai H. H., Yang T., Zhang G. L., Wu L. W., Li G. Y., J. Nat. Prod. 2015, 78, 1479–1485. [DOI] [PubMed] [Google Scholar]
- 191.
- 191a. Mohd Tap R., Sabaratnam P., Ahmad N. A., Abd Razak M. F., Hashim R., Ahmad N., Mycopathologia 2015, 180, 137–141; [DOI] [PubMed] [Google Scholar]
- 191b. Shi D., Lu G., Mei H., de Hoog G. S., Zheng H., Liang G., Shen Y., Li T., Liu W., Med. Mycol. Case Rep. 2016, 13, 12–16; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191c. Hubka V., Mencl K., Skorepova M., Lyskova P., Zalabska E., Med. Mycol. 2011, 49, 724–733. [DOI] [PubMed] [Google Scholar]
- 192. Welch T. R., Williams R. M., Tetrahedron 2013, 69, 770–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.
- 193a. Shende V. V., Khatri Y., Newmister S. A., Sanders J. N., Lindovska P., Yu F., Doyon T. J., Kim J., Houk K. N., Movassaghi M., Sherman D. H., J. Am. Chem. Soc. 2020, 142, 17413–17424; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193b. Sun C., Luo Z., Zhang W., Tian W., Peng H., Lin Z., Deng Z., Kobe B., Jia X., Qu X., Nat. Commun. 2020, 11, 6251; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193c. Tian W., Sun C., Zheng M., Harmer J. R., Yu M., Zhang Y., Peng H., Zhu D., Deng Z., Chen S. L., Mobli M., Jia X., Qu X., Nat. Commun. 2018, 9, 4428; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193d. Saruwatari T., Yagishita F., Mino T., Noguchi H., Hotta K., Watanabe K., ChemBioChem 2014, 15, 656–659; [DOI] [PubMed] [Google Scholar]
- 193e. Liu J., Xie X., Li S. M., Chem. Commun. 2020, 56, 11042–11045; [DOI] [PubMed] [Google Scholar]
- 193f. Yu H., Li S. M., Org. Lett. 2019, 21, 7094–7098; [DOI] [PubMed] [Google Scholar]
- 193g. Liu J., Liu A., Hu Y., Nat. Prod. Rep. 2021, 38, 1469–1505. [DOI] [PubMed] [Google Scholar]
- 194. Kishimoto S., Sato M., Tsunematsu Y., Watanabe K., Molecules 2016, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195. Dopstadt J., Neubauer L., Tudzynski P., Humpf H. U., PLoS One 2016, 11, e0158945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196. Seephonkai P., Kongsaeree P., Prabpai S., Isaka M., Thebtaranonth Y., Org. Lett. 2006, 8, 3073–3075. [DOI] [PubMed] [Google Scholar]
- 197. Isaka M., Palasarn S., Rachtawee P., Vimuttipong S., Kongsaeree P., Org. Lett. 2005, 7, 2257–2260. [DOI] [PubMed] [Google Scholar]
- 198. Amrine C. S. M., Huntsman A. C., Doyle M. G., Burdette J. E., Pearce C. J., Fuchs J. R., Oberlies N. H., ACS Med. Chem. Lett. 2021, 12, 625–630. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing is not applicable to this article as no new data were created or analyzed in this study.