

Abstract
Objective: To investigate whether the gut microbiota differs in patients with chronic kidney disease receiving kidney transplant or hemodialysis, and to explore the relationship between the gut microbiota and clinical indicators. Methods: A total of 72 patients with kidney transplantation (RT) and 78 patients with hemodialysis (HD) admitted to the First Affiliated Hospital of Soochow University from January 2019 to October 2022 were selected as the research subjects. The V3-V4 region sequences of 16S rRNA were used for high-throughput sequencing to analyze the differences in gut microbiome between the two groups and its relationship with clinical indicators. Results: Gut microbial α diversity (Chao1, Ace, Shannon, Simpson) was significantly decreased in RT patients compared with that in HD patients (P<0.05). The relative abundance of Bacteroides, Megamonas, and Prevotella was significantly higher in HD patients than that in RT patients (P<0.05). There was a negative association between the Bacteroides and β2-microglobulin (β2-MG) (P<0.05). Lactobacillus was negatively correlated with uric acid (UA) (P<0.05). Conclusion: This study elucidates the composition and changes of the gut microbiota in RT and HD patients and its association with clinical indicators, providing a scientific basis for the regulatory mechanism of gut microbiota in the treatment of chronic kidney disease.
Keywords: Kidney transplant patients, chronic kidney disease, hemodialysis patients, gut microbiota, 16S rRNA sequencing
Introduction
The prevalence of chronic kidney disease (CKD) in Chinese adults is about 11%, and more than 30% of them are accompanied by varying degrees of renal injury [1]. In the early stage of CKD, symptomatic treatment is given, while renal transplantation and dialysis are the only methods for patients with end-stage CKD, which seriously affect the physical and mental health and quality of life of patients [2,3]. Patients with end-stage renal disease are prone to diarrhea, stomachache and other symptoms after renal transplantation or hemodialysis, which are closely related to the long-term use of hormones and immunosuppressants and abnormal metabolism of body substances [4]. In this case, the etiology, detection methods and treatment options of CKD have been studied worldwide, and the most critical research direction is the link between CKD and intestinal microecology. There are 1013-1014 microorganisms in the human gut and different bacteria in the gut jointly maintain the microecological balance in the gut, participating in human digestion, metabolism, immune regulation, energy conversion and other functions [5]. Kidney transplant patients and hemodialysis patients are prone to various infections, one of which is characterized by intestinal microbiota imbalance [6]. Gut microbiota interacts with other organs in a variety of ways, and no matter in the local or systemic system, the gut microbiota is closely related to the immune system [3,7].
In recent years, a large number of studies have shown that the clinical symptoms of patients with the end-stage renal disease after renal transplantation or hemodialysis are closely related to intestinal microbial dysbiosis [8,9]. It has been shown that after kidney transplantation the gut microbiota is altered after acute rejection [10]. Studies have shown that the relative abundance of Klebsiella, Escherichia coli, Enterobacteriaceae, Lachnospiraceae and Fusobacterium is higher in the intestines of CKD patients compared with that in healthy people [11]. Transplantation and hemodialysis treatment resulted in structural and functional changes in the intestinal flora, while its dysbiosis exacerbated the clinical symptoms [10,12,13].
In CKD, progressive decline in renal excretory function leads to the accumulation of urea and toxins in the blood. The ecological dysbiosis of the gut microbiota associated with elevated urea levels further contributes to uremia by increasing the production of intestinal toxins [14]. Certain changes in the gut microbiome have been observed in patients with CKD stage V after kidney transplantation or hemodialysis [15]. However, it is not clear about a relationship between the pattern of microbiome changes observed in patients with CKD stage V and clinical indicators. Therefore, a comprehensive reference microbiome is needed to reveal the relationship between gut microbial dysbiosis and clinical indicators in renal transplant patients and hemodialysis patients. Also, it is needed to identify the abundant microbiota in CKD patients and its possible role in the progression of the patient’s disease. In this study, we performed 16S rRNA sequencing of fecal samples from 72 kidney transplant patients (RT) and 78 hemodialysis patients (HD). We compared the differences in microbial species between the two classes of patients to obtain CKD biomarkers for patients with different treatment modalities. We also investigated the correlation between gut microbiota and clinical indicators related to liver function, renal function, and electrolytes in both RT patients and HD patients.
Method
Study design and participants
This is a cross-sectional study. We evaluated 72 RT patients who underwent kidney transplantation and 78 HD patients who underwent hemodialysis at the First Affiliated Hospital of Soochow University from January 2019 to October 2022. RT patients’ inclusion criteria: (1) patients with an age ≥18; patients with CKD stage V who received the first living or deceased donor transplant; (2) patients survived for least 3 months after the transplant. Exclusion criteria: (1) patients who received hemodialysis or peritoneal dialysis after transplantation; (2) patients who used probiotics, biologics or prebiotics for 2 months before stool sample collection; (3) patients who were treated with antibiotics within 3 months before sampling; (4) patients who had diarrhea before sampling; (5) pregnant or nursing women; (6) patients with severe malnutrition, malignancy, severe cardiac diseases or liver diseases. HD patients’ inclusion criteria: (1) patients with an age ≥18; patients who were diagnosed with CKD stage V; (2) patients who were hemodialyzed for a minimum period of two months. Exclusion criteria: patients who had received a kidney transplant before hemodialysis. The other exclusion criteria were consistent with the (2)-(4) exclusion criteria for RT patients. Informed written consent was obtained from all participants, and the research protocols were approved by the Ethics Committee of the First Affiliated Hospital of Soochow University.
Baseline data and clinical indicators
Demographic information was collected from the hospital case system for both groups of patients. Blood samples were collected in the fasting state and frozen at -80°C until use. Blood routine, liver function, renal function, glucose metabolism, lipid metabolism and electrolytes were measured by standard methods.
Fecal microbiota DNA extraction, amplification and sequencing
The subjects collected fecal samples in the morning using stool collection kits. The fecal samples were equally divided into ~200 mg sub-samples and placed in an ultra-low temperature refrigerator at -80°C for use within 2 hours after collection. DNA extracted from the fecal samples was used to amplify the V3-V4 region of the 16S rRNA gene to determine the gut bacterial community structure. Primer set 341 F/806 R was employed to target the V3-V4 region. The amplified products were further subjected to library preparation and sequencing on the Illumina MiSeq platform as per the manufacturer’s instructions (Illumina technologies, USA).
Bioinformatics analysis
Raw data obtained from Illumina sequencers were quality-filtered using Trimmomatic to remove unqualified sequences. The filtered sequences were spliced, chimeras were removed, and a series of quality control works were performed. Then, high-quality sequences were clustered into the Operational Taxonomic Unit (OTU) using QIIME (Microbial 16s rRNA Analysis Pipeline). OTU clustering was performed on the high-quality sequences of all samples according to the similarity of 0.97, and the longest sequence in each category was selected as the representative sequence of OTU. The UCLUST classifier was used to annotate the OTU representative sequences and obtain the taxonomic information of each OTU. Each sample contained OTU and the number of sequences contained in each OTU was counted. The “vegan” package of R was used to analyze α biodiversity analysis including Chao1, Ace, Shannon and Simpson. Linear discriminant analysis LEfSe (LDA score = 4 as the cut-off value) was used to compare the microbial species with significant differences among the groups. Correlation analysis data included bacterial species with a relative abundance of more than 1% and clinical indicators with significant differences. Clustering correlation heatmap with signs was performed using the OmicStudio tools at https://www.omicstudio.cn. Pearson correlation analysis was used to analyze the correlation between microbial species and clinical indicators.
Outcomes
The primary outcome was the V3-V4 region sequences of 16S rRNA of gut microbiome. The clinical indicators can be found in Table 1.
Table 1.
Clinical characteristics of transplant patients and dialysis patients
| Characteristics | Abbreviations | RT (n = 72) | HD (n = 78) | P |
|---|---|---|---|---|
| Male gender | - | 45 (57.69%) | 38 (52.78%) | 0.545 |
| Age (year) | - | 54.6±12.88 | 56.17±12.5 | 0.452 |
| Hypertension | - | 18 (23.08%) | 21 (29.17%) | 0.396 |
| Diabetes mellitus | - | 20 (25.64%) | 8 (11.11%) | 0.023 |
| Leukocyte (109/L) | WBC | 6.16±1.81 | 5.74±1.65 | 0.149 |
| Lymphocyte (109/L) | LY | 1.31±0.41 | 1.22±0.45 | 0.205 |
| Erythrocyte (109/L) | RBC | 3.65±0.73 | 3.46±0.93 | 0.163 |
| Hemoglobin (g/L) | HB | 113.02±21.59 | 107.44±16.81 | 0.081 |
| Platelet (109/L) | PLT | 177.1±44.97 | 160.81±44.48 | 0.027 |
| Total bilirubin (umol/L) | TBIL | 7.35±2.72 | 6.62±1.9 | 0.058 |
| Direct bilirubin (umol/L) | DBIL | 2.8±1.19 | 2.55±0.82 | 0.137 |
| Indirect bilirubin (umol/L) | I-BIL | 4.55±1.75 | 4.07±1.38 | 0.064 |
| Alanine aminotransferase | ALT | 14.26±9.4 | 13.14±3.59 | 0.332 |
| Aspartate aminotransferase | AST | 15.04±5.84 | 14.72±7.35 | 0.772 |
| Alkaline phosphatase | ALP | 75.03±34.24 | 79.28±20.99 | 0.366 |
| Total protein (g/L) | TP | 72.92±5.26 | 69.9±5.54 | <0.001 |
| Albumin (g/L) | ALB | 43.11±4.17 | 40.96±4.19 | 0.002 |
| Globulin (g/L) | GLOB | 30.15±5.74 | 29.28±6.84 | 0.4 |
| Urea (mmol/L) | UREA | 23.65±6.17 | 23.09±5.94 | 0.576 |
| Creatinine (umol/L) | CREA | 905.6±254.3 | 900.16±218.29 | 0.889 |
| Uric acid (umol/L) | UA | 444.85±68.31 | 473.11±69.19 | 0.013 |
| β2-microglobulin (mg/L) | β2-MG | 29.61±9.86 | 35.13±7.82 | <0.001 |
| Glucose (mmol/L) | GLU | 6.41±2.95 | 5.71±2.74 | 0.138 |
| Total cholesterol (mmol/L) | TCHO | 3.97±0.87 | 3.78±0.76 | 0.152 |
| Triglyceride (mmol/L) | TG | 1.64±0.8 | 1.79±0.79 | 0.254 |
| High density liptein cholesterol (mmol/L) | HDL-C | 0.97±0.23 | 0.94±0.23 | 0.397 |
| Low density liptein cholesterol (mmol/L) | LDL-C | 2.12±0.72 | 1.94±0.62 | 0.107 |
| Potassium (mmol/L) | K | 4.76±0.69 | 4.66±0.83 | 0.406 |
| Sodium (mmol/L) | Na | 138.34±2.7 | 131.46±2.96 | <0.001 |
| Chloride (mmol/L) | Cl | 102.75±3.46 | 97.29±3.31 | <0.001 |
| Calcium (mmol/L) | Ca | 2.26±0.18 | 2.4±0.2 | <0.001 |
| Phosphorus (mmol/L) | P | 1.79±0.56 | 1.97±0.53 | 0.042 |
| Transferrin Saturation (%) | TS | 31.05±10.4 | 34.4±10.23 | 0.049 |
Statistical analysis
Statistical analysis was conducted by SPSS version 23 software. The continuous variables were displayed as mean ± standard deviation. Count variables were represented by n (%). The t-test and chi-square test were performed to evaluate the correlation of baseline characteristics as appropriate. A difference of P<0.05 was considered statistically significant.
Results
Baseline data and clinical indicators of RT and HD
As shown in Table 1, there were no differences in gender, age, and history of hypertension between the HD and RT groups (P>0.05). Compared with RT, the number of platelets in HD, total protein (TP) and albumin (ALB) decreased significantly (all P<0.05). In HD patients, renal function indexes such as uric acid (UA) and β2-microglobulin (β2-MG) decreased significantly, (all P<0.05).
Comparison of gut microbial α diversity between the RT and HD
1182 OTUs were obtained from 150 samples. In the Venn diagram analysis, 634 common OTUs were identified in both groups. There were 1121 and 695 OTUs in the RT and HD groups, respectively (Figure 1A). The rarefaction curve constructed based on the number of Observed OTUs is shown in Figure 1B. The dilution curve tended to be flat, and the number of detected OTUs did not increase, indicating that the amount of measurement data was reasonable, and the sample size was sufficient. The relative abundance curve of species (Figure 1C) shows the species richness and evenness of different groups of microbial communities. There were no significant differences in species richness and evenness between RT and HD.
Figure 1.
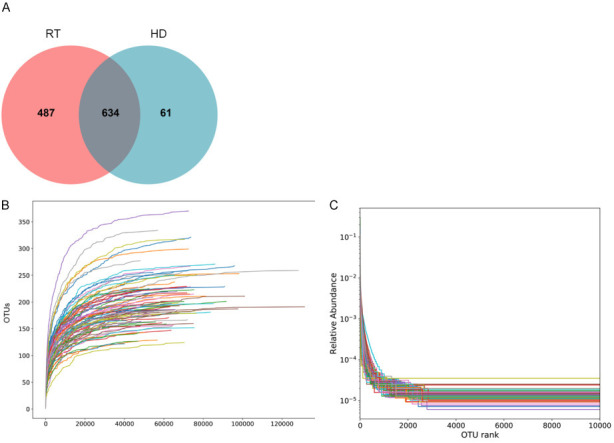
Number of OUTs for RT and HD (A). Species dilution curve (B). Species relative abundance curve (C). RT: kidney transplant patients, HD: hemodialysis patients.
The Chao1 index, Ace index, Shannon index, and Simpson index were all significantly decreased compared with HD (Figure 2), indicating that the richness of gut microbes was higher in HD than in RT.
Figure 2.
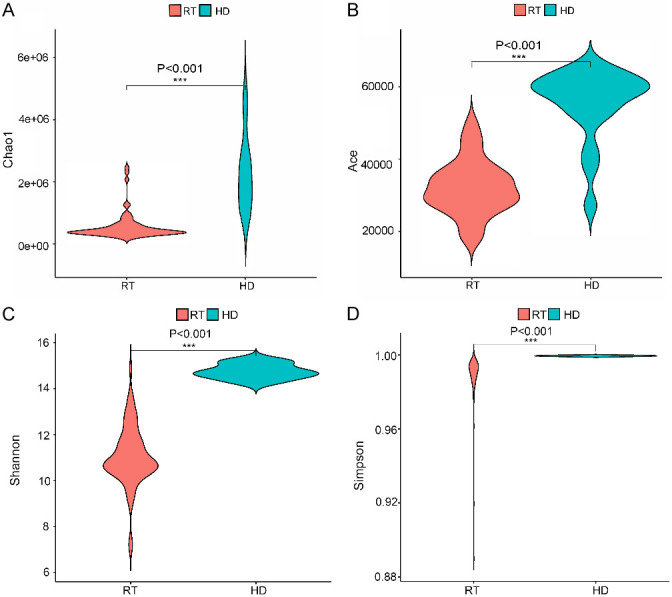
α biodiversity in RT patients and HD patients.
Analysis of species composition
The main intestinal microbiota of the RT group and HD groups were divided into three phyla: Firmicutes, Actinobacteria, and Bacteroidetes in RT (73.86%, 13.01%, 4.10%, respectively), and HD (65.55%, 2.82%, 23.92%, respectively) (Figure 3). At the family level (Figure 4A), the dominant bacterial groups in the RT group were Lachnospiraceae, Ruminococcaceae, and Bifidobacteriaceae. However, the dominant bacterial groups in the HD group were Lachnospiraceae, Bacteroidaceae, and Ruminococcaceae. In the RT group, the abundance of Bacteroidaceae was significantly decreased and Bifidobacteriaceae were significantly higher as compared to HD. With genera at the taxonomic level (Figure 4B), the top five genera with the highest abundance in the RT group were Blautia, Bifidobacterium, Collinsella, Faecalibacterium, and Roseburia. The predominant genera of HD were mainly Bacteroides, Blautia, Faecalibacterium, Roseburia, and Megamonas. The LEfSe analysis (Figure 5) revealed the specific bacteria associated with each group. In RT, the relative abundance of Bifidobacterium was significantly higher than that of HD, while the abundance of Bacteroides, Megamonas and Prevotella was significantly lower than that of HD. At the species level, Blautia massiliensis was more prevalent in the RT group, whereas Prevotella copri and Megamonas funiformis were more prevalent in the HD group.
Figure 3.
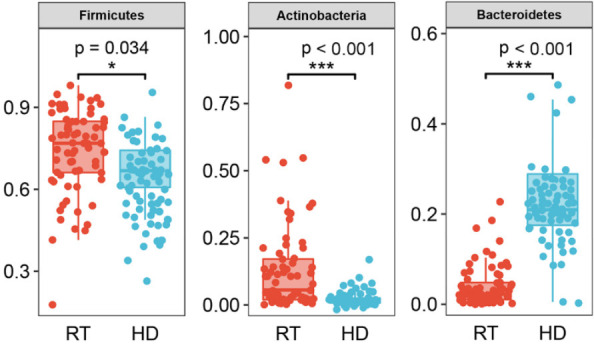
The microbiota differences at the phylum level.
Figure 4.
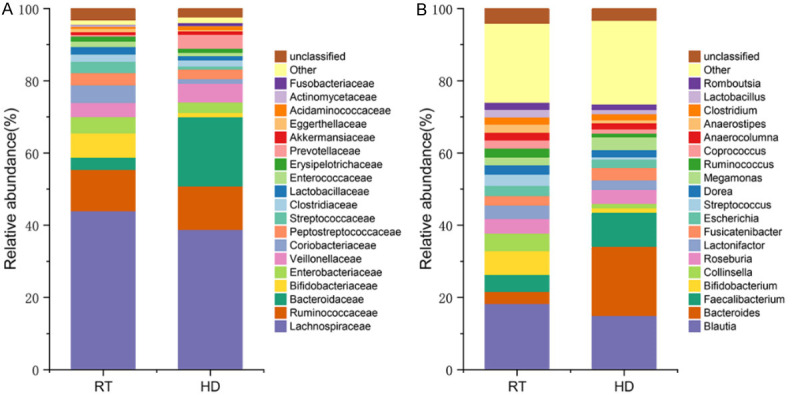
Microbiota composition at the family level (A). Microbial composition at the genus level (B).
Figure 5.

Analysis of species composition differences based on the LEfSe method.
Correlation of gut microbes with clinical indicators
We studied the association between microbial species and the clinical data from all patients, and 25 species in four phyla (Firmicutes, Actinobacteria, Bacteroidetes, and Proteobacteria) showed significant associations with the clinical data (Figure 6). β2-MG was negatively associated with Prevotella (shahii, oris), while Bacteroides (finegoldii, sylanisolvens, dorei) and Parasutterella secunda were positively associated with Bifidobacterium dentium. Phosphorus (P) was significantly negatively correlated with Anaerocolumna jejuensis. Lactobacillus (mucosae, reuteri), Klebsiella pneumoniae, Enterococcus durans, and Megasphaera micronuciformis showed a significant negative correlation with UA. Transferrin saturation (TS) was significantly and positively correlated with Ruminococcus bromii and Klebsiella pneumoniae. ALB was positively correlated with Bacteroides dorei and negatively associated with Steptococcus parasanguinis. Among the electrolyte indexes, sodium (Na) and chloride (Cl) were negatively correlated with 7 species and positively correlated with 10 species.
Figure 6.

Correlation of microbial species and clinical outcomes (β2-MG: β2-microglobulin, P: phosphorus, UA: uric acid, Ca: calcium, TS: transferrin saturation, Na: sodium, Cl: chloride, ALB: albumin, PLT: platelet, TP: total protein. *, P<0.05, **, P<0.01, ***, P<0.001).
Discussion
We first compared the gut microbiota between RT patients and HD patients. The results showed that RT patients had significantly lower alpha diversity than HD patients, and there are many reasons for this change, such as antibiotic use, dietary restrictions, constipation, and bad mood which may inhibit or promote the proliferation of certain bacteria [16]. In addition, we analyzed the correlation of microorganisms with clinical data to identify key gut microorganisms associated with RT or HD.
Our results showed a significant decrease in RT microbial diversity, which is consistent with the results [17,18]. Firmicutes are the most abundant bacterial taxa in the gut of RT patients and HD patients. A study [17] pointed out that compared with healthy people, the abundance of Firmicutes in CKD patients and RT patients decreased significantly, and the abundance of RT patients was slightly higher than that of CKD patients. Consistent results were also found in our study. LEfSe method identified that the abundance of Bacteroides, Megamonas and Prevotella in RT patients was significantly lower than that in HD patients, while the abundance of Bifidobacterium was significantly increased. Previous studies [10,13,19] have reported similar results. Shivani et al. [13] found that Bacteroides is the genus with the highest abundance of intestinal microorganisms in CKD patients, which is consistent with our research results. A study of gut microbiota in CKD patients and idiopathic nephrotic syndrome (INS) patients showed that the abundance of Megamonas in the CKD and INS groups was significantly lower than that in healthy people [19]. Another study showed that weight loss or malnutrition was associated with Megamonas, and their abundance was negatively correlated with the rate of weight loss [20]. Therefore, we hypothesized that the increased abundance of Megamonas in HD may indicate that intestinal ecology is improving. Previous reports [21,22] have shown that Prevotella is related to the progress of hemodialysis. Prevotella is a beneficial bacterium that can produce short-chain fatty acids, and its increased abundance promotes the formation of short-chain fatty acids, thus improving the clearance rate of uremic toxins [23].
We further explored the potential relationship between intestinal microbiota dysregulation and clinical indicators and found that electrolyte Na and Cl ions had the greatest impact on microbiota, showing significant correlation for almost all species. It is well known that β2-MG is a sensitive index to measure renal function decline [24]. In transplant patients, its increase indicates a rejection reaction [25]. We found that β2-MG was significantly negatively correlated with Bacteroides, which are the main players in maintaining intestinal ecology and usually play a beneficial role in the intestine [7]. However, there are limited reports on the related mechanisms of Bacteroides in the regulation of renal function. Further studies are needed to characterize the composition and functional changes related to intestinal dysbiosis related to renal transplantation and hemodialysis. In addition, this study found a significant negative correlation between UA and Lactobacillus, which is consistent with the previous reports [20,21]. Zhu et al. [26] found that kidney injury and serum urea nitrogen and creatinine were reduced in mice with oral Lactobacillus casei Zhang. Lactobacillus has a high clearance rate for uremic toxins, which can reduce proinflammatory reactions, improve the immune capacity of the kidney, and thus improve renal function [27]. The intestinal flora metabolizes proteins and produces toxins, such as amines, phenols, indole, etc. [28]. Under normal circumstances, these toxins are filtered by the kidney and discharged through excreta. However, when renal function is impaired, these toxins produced by the intestinal flora accumulate in the body and become the source of uremic toxins [29]. Limited information is available on the association of clinical indicators with microbial changes regarding functional changes in intestinal dysbiosis after renal transplantation or hemodialysis in patients with CKD.
Our study identified significant changes in microbial composition that may be associated with kidney transplants and hemodialysis-related diseases. However, there may be some limitations in this study, mainly that the sample size is insufficient to further stratify the analysis of patients with underlying diseases. In addition, long-term follow-up is needed for the next study to further clarify the dynamic changes of gut microbiota. Future research should focus on the mechanisms of interaction between intestinal flora and clinical indicators to reveal the mechanism of action of specific microbial taxa in regulating organ function in patients.
In conclusion, we report that Bacteroides, Megamonas, and Prevotella are the major taxa of differences between RT patients and HD patients. The microbiological diversity was significantly decreased in RT patients compared to HD patients. Our data suggest that Bacteroides and Lactobacillus have a certain correlation with the renal function of transplant patients and hemodialysis patients. These potential biomarkers may help physicians to conduct preventive and timely interventions in patients with CKD to avoid the risk of mortality arising from a poor prognosis.
Disclosure of conflict of interest
None.
References
- 1.Chen S, Chen L, Jiang H. Prognosis and risk factors of chronic kidney disease progression in patients with diabetic kidney disease and non-diabetic kidney disease: a prospective cohort CKD-ROUTE study. Ren Fail. 2022;44:1309–1318. doi: 10.1080/0886022X.2022.2106872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Simões-Silva L, Araujo R, Pestana M, Soares-Silva I, Sampaio-Maia B. The microbiome in chronic kidney disease patients undergoing hemodialysis and peritoneal dialysis. Pharmacol Res. 2018;130:143–151. doi: 10.1016/j.phrs.2018.02.011. [DOI] [PubMed] [Google Scholar]
- 3.Wyld M, Morton RL, Hayen A, Howard K, Webster AC. A systematic review and meta-analysis of utility-based quality of life in chronic kidney disease treatments. PLoS Med. 2012;9:e1001307. doi: 10.1371/journal.pmed.1001307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lohia S, Vlahou A, Zoidakis J. Microbiome in chronic kidney disease (CKD): an omics perspective. Toxins (Basel) 2022;14:176. doi: 10.3390/toxins14030176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhou J, Yang C, Lei W, Yang Z, Chen J, Lin H. Exploration of the correlation between intestinal flora and peritoneal dialysis-related peritonitis. Clin Exp Nephrol. 2022;26:1030–1038. doi: 10.1007/s10157-022-02239-4. [DOI] [PubMed] [Google Scholar]
- 6.Hayat A, Walker RC, Viecelli AK, Manera KE, Jaure A, Krishnasamy R, Pascoe EM, Cho Y, Johnson DW. Range and consistency of gastrointestinal outcomes reported in peritoneal dialysis trials: a systematic review. Perit Dial Int. 2022 doi: 10.1177/08968608221126849. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 7.Zafar H, Saier MH Jr. Gut bacteroides species in health and disease. Gut Microbes. 2021;13:1–20. doi: 10.1080/19490976.2020.1848158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Jiang N, Zhang C, Feng H, Yuan J, Ding L, Fang W, Gu A, Huang J, Li N, Gu L, Ni Z, Mou S. Clinical characteristics associated with the properties of gut microbiota in peritoneal dialysis patients. Perit Dial Int. 2021;41:298–306. doi: 10.1177/0896860820976983. [DOI] [PubMed] [Google Scholar]
- 9.Crespo-Salgado J, Vehaskari VM, Stewart T, Ferris M, Zhang Q, Wang G, Blanchard EE, Taylor CM, Kallash M, Greenbaum LA, Aviles DH. Intestinal microbiota in pediatric patients with end stage renal disease: a midwest pediatric nephrology consortium study. Microbiome. 2016;4:50. doi: 10.1186/s40168-016-0195-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee JR, Magruder M, Zhang L, Westblade LF, Satlin MJ, Robertson A, Edusei E, Crawford C, Ling L, Taur Y, Schluter J, Lubetzky M, Dadhania D, Pamer E, Suthanthiran M. Gut microbiota dysbiosis and diarrhea in kidney transplant recipients. Am J Transplant. 2019;19:488–500. doi: 10.1111/ajt.14974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhou J, Yang C, Lei W, Yang Z, Chen J, Lin H, Li Q, Yuan W. Exploration of the correlation between intestinal flora and escherichia coli peritoneal dialysis-related peritonitis. BMC Nephrol. 2022;23:76. doi: 10.1186/s12882-022-02704-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Huang J, Salinas T, Westblade LF, Lee JR. The potential role of the gut microbiota in kidney transplantation. Kidney360. 2021;2:890–893. doi: 10.34067/KID.0006912020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Shivani S, Kao CY, Chattopadhyay A, Chen JW, Lai LC, Lin WH, Lu TP, Huang IH, Tsai MH, Teng CH, Wu JJ, Hsieh YH, Wang MC, Chuang EY. Uremic toxin-producing bacteroides species prevail in the gut microbiota of Taiwanese CKD patients: an analysis using the new Taiwan microbiome baseline. Front Cell Infect Microbiol. 2022;12:726256. doi: 10.3389/fcimb.2022.726256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cosola C, Rocchetti MT, Sabatino A, Fiaccadori E, Di Iorio BR, Gesualdo L. Microbiota issue in CKD: how promising are gut-targeted approaches? J Nephrol. 2019;32:27–37. doi: 10.1007/s40620-018-0516-0. [DOI] [PubMed] [Google Scholar]
- 15.Tourountzis T, Lioulios G, Fylaktou A, Moysidou E, Papagianni A, Stangou M. Microbiome in chronic kidney disease. Life (Basel) 2022;12:1513. doi: 10.3390/life12101513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lin TY, Hung SC. Association of subjective global assessment of nutritional status with gut microbiota in hemodialysis patients: a case-control study. Nephrol Dial Transplant. 2021;36:1104–1111. doi: 10.1093/ndt/gfaa019. [DOI] [PubMed] [Google Scholar]
- 17.Ye GR, Zhou MJ, Yu LX, Ye JS, Lin Y, Shi LS. Gut microbiota in renal transplant recipients, patients with chronic kidney disease and healthy subjects. Nan Fang Yi Ke Da Xue Xue Bao. 2018;38:1401–1408. doi: 10.12122/j.issn.1673-4254.2018.12.01. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Swarte JC, Li Y, Hu S, Björk JR, Gacesa R, Vich Vila A, Douwes RM, Collij V, Kurilshikov A, Post A, Klaassen MAY, Eisenga MF, Gomes-Neto AW, Kremer D, Jansen BH, Knobbe TJ, Berger SP, Sanders JF, Heiner-Fokkema MR, Porte RJ, Cuperus FJC, de Meijer VE, Wijmenga C, Festen EAM, Zhernakova A, Fu J, Harmsen HJM, Blokzijl H, Bakker SJL, Weersma RK. Gut microbiome dysbiosis is associated with increased mortality after solid organ transplantation. Sci Transl Med. 2022;14:eabn7566. doi: 10.1126/scitranslmed.abn7566. [DOI] [PubMed] [Google Scholar]
- 19.Zhang J, Luo D, Lin Z, Zhou W, Rao J, Li Y, Wu J, Peng H, Lou T. Dysbiosis of gut microbiota in adult idiopathic membranous nephropathy with nephrotic syndrome. Microb Pathog. 2020;147:104359. doi: 10.1016/j.micpath.2020.104359. [DOI] [PubMed] [Google Scholar]
- 20.Chiu CM, Huang WC, Weng SL, Tseng HC, Liang C, Wang WC, Yang T, Yang TL, Weng CT, Chang TH, Huang HD. Systematic analysis of the association between gut flora and obesity through high-throughput sequencing and bioinformatics approaches. Biomed Res Int. 2014;2014:906168. doi: 10.1155/2014/906168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lun H, Yang W, Zhao S, Jiang M, Xu M, Liu F, Wang Y. Altered gut microbiota and microbial biomarkers associated with chronic kidney disease. MicrobiologyOpen. 2019;8:e00678. doi: 10.1002/mbo3.678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhu Y, Tang Y, He H, Hu P, Sun W, Jin M, Wang L, Xu X. Gut microbiota correlates with clinical responsiveness to erythropoietin in hemodialysis patients with anemia. Front Cell Infect Microbiol. 2022;12:919352. doi: 10.3389/fcimb.2022.919352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lin X, Liang W, Li L, Xiong Q, He S, Zhao J, Guo X, Xiang S, Zhang P, Wang H, Ying C, Yao Y, Zuo X. The accumulation of gut microbiome-derived indoxyl sulfate and p-cresyl sulfate in patients with end-stage renal disease. J Ren Nutr. 2022;32:578–586. doi: 10.1053/j.jrn.2021.09.007. [DOI] [PubMed] [Google Scholar]
- 24.García-García PM, Martín-Izquierdo E, de Basoa CM, Jarque-López A, Pérez-Suárez G, Rivero-González A, González-Posadas JM, Macía-Heras M, García-Nieto VM, Navarro-Gónzález JF. Urinary clara cell protein in kidney transplant patients: a preliminary study. Transplant Proc. 2016;48:2884–2887. doi: 10.1016/j.transproceed.2016.09.022. [DOI] [PubMed] [Google Scholar]
- 25.Woo KT, Foo MW, Choong HL, Yap HK, Lee EJ, Chan CM. Serum β2-microglobulin predicts mortality and transplant outcomes. Kidney Int. 2014;85:982. doi: 10.1038/ki.2014.7. [DOI] [PubMed] [Google Scholar]
- 26.Zhu H, Cao C, Wu Z, Zhang H, Sun Z, Wang M, Xu H, Zhao Z, Wang Y, Pei G, Yang Q, Zhu F, Yang J, Deng X, Hong Y, Li Y, Sun J, Zhu F, Shi M, Qian K, Ye T, Zuo X, Zhao F, Guo J, Xu G, Yao Y, Zeng R. The probiotic L. casei Zhang slows the progression of acute and chronic kidney disease. Cell Metab. 2021;33:1926–1942. e1928. doi: 10.1016/j.cmet.2021.06.014. [DOI] [PubMed] [Google Scholar]
- 27.Huang H, Li K, Lee Y, Chen M. Preventive effects of lactobacillus mixture against chronic kidney disease progression through enhancement of beneficial bacteria and downregulation of gut-derived uremic toxins. J Agric Food Chem. 2021;69:7353–7366. doi: 10.1021/acs.jafc.1c01547. [DOI] [PubMed] [Google Scholar]
- 28.Lv Y, Ren G, Ren X. Changes of intestinal flora and lymphocyte subsets in patients with chronic renal failure. Evid Based Complement Alternat Med. 2021;2021:4288739. doi: 10.1155/2021/4288739. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 29.Luo D, Zhao W, Lin Z, Wu J, Lin H, Li Y, Song J, Zhang J, Peng H. The effects of hemodialysis and peritoneal dialysis on the gut microbiota of end-stage renal disease patients, and the relationship between gut microbiota and patient prognoses. Front Cell Infect Microbiol. 2021;11:579386. doi: 10.3389/fcimb.2021.579386. [DOI] [PMC free article] [PubMed] [Google Scholar]