

Summary
Background
Interleukin‐23 inhibitors are effective and safe for treating moderate‐to‐severe plaque psoriasis.
Objectives
To evaluate the efficacy and safety of mirikizumab in adult patients with moderate‐to‐severe plaque psoriasis through 52 weeks in a phase III randomized controlled trial.
Methods
OASIS‐1 (NCT03482011) was a double‐blind, placebo‐controlled, randomized withdrawal, phase III trial. Patients (n = 530, randomized 4 : 1) received subcutaneous mirikizumab 250 mg or placebo every 4 weeks (Q4W) through week 16. Coprimary endpoints were superiority of mirikizumab vs. placebo on static Physician’s Global Assessment (sPGA; score of 0 or 1 with ≥ 2‐point improvement) and ≥ 90% improvement in Psoriasis Area and Severity Index (PASI 90, responders) at week 16. Mirikizumab responders were rerandomized (1 : 1 : 1) to mirikizumab 250 mg every 8 weeks (Q8W), mirikizumab 125 mg Q8W, or placebo Q8W through week 52. Secondary endpoints were evaluated at weeks 16 and 52. Safety was monitored in all patients.
Results
All primary and key secondary endpoints were met. At week 16, sPGA(0,1) responses were significantly greater with mirikizumab (293 of 423, 69·3%) than placebo (seven of 107, 6·5%) (P < 0·001). PASI 90 response was also greater with mirikizumab (272 of 423, 64·3%) than placebo (seven of 107, 6·5%) (P < 0·001). Significantly more patients in the mirikizumab arms achieved PASI 75 and PASI 100 (mirikizumab 349, 82·5% and 137, 32·4%; placebo 10, 9·3% and 1, 0·9%, respectively; all P < 0·001). At week 52, PASI 90, PASI 100 and sPGA(0,1) responses were mirikizumab 250Q4W/placeboQ8W (N = 91; 19%, 10%, 18%), mirikizumab 250Q4W/125Q8W (N = 90; 86%, 59%, 86%) and mirikizumab 250Q4W/250Q8W (N = 91; 86%, 60%, 82%; all P < 0·001), respectively. Rates of serious adverse events were similar across treatments (induction: mirikizumab 1·2% vs. placebo 1·9%; maintenance: mirikizumab 250Q4W/125Q8W 1%, mirikizumab 250Q4W/250Q8W 3% vs. placebo 3%). No deaths occurred.
Conclusions
Mirikizumab was superior to placebo at week 16 and maintained efficacy through week 52, with no new safety signals.
What is already known about this topic?
Interleukin (IL)‐23 is a key cytokine in the pathogenesis of psoriasis. Drugs targeting the p19 subunit of IL‐23 have recently been approved for the treatment of adult patients with moderate‐to‐severe plaque psoriasis.
Patients with moderate‐to‐severe plaque psoriasis achieved significantly greater improvements in skin measures and patient‐reported quality‐of‐life measures after 16 weeks when treated every 8 weeks with mirikizumab compared with placebo in a phase II clinical trial.
What does this study add?
Compared with placebo, mirikizumab demonstrated high levels of efficacy at week 16 in a large phase III trial; safety profiles were similar between the mirikizumab and placebo arms.
After week 16, patients maintained on doses of mirikizumab 250 mg every 8 weeks (Q8W) or 125 mg Q8W showed similar efficacy and favourable safety profiles over 52 weeks, whereas patients switched to placebo gradually lost efficacy over time.
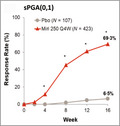
The primary efficacy endpoints were met, with significantly greater improvements in clinical response observed with mirikizumab than with placebo at the end of the induction period. sPGA(0,1) response rates were superior in the mirikizumab arm (n(%) [95% confidence interval; CI], mirikizumab: 293(69.3) [64.9‐73.7]; placebo: 7(6.5) [1.9, 11.2]; P < 0.001) compared with placebo at Week 16.
Linked Comment: L. Puig. Br J Dermatol 2022; 187:837.
Plain language summary available online
Psoriasis is a chronic, immune‐mediated, inflammatory skin disease that affects an estimated 125 million people worldwide. 1 Approximately 90% of patients with psoriasis require long‐term therapy, 2 creating a critical need for psoriasis treatments that not only clear psoriasis plaques in the short term but also demonstrate durable efficacy over time. New biologics have shown a better long‐term safety profile than conventional treatments, but the time to relapse after drug withdrawal remains a concern. 3 Notably, differences have been observed among patients in terms of maintaining response after treatment interruption. 4 , 5
Since the discovery of the key roles of interleukin (IL)‐17 and IL‐23 in the development of psoriatic disease, 6 , 7 several biologic therapies that target these cytokines and associated inflammatory pathways have been studied and approved for use in patients with plaque psoriasis. 8 , 9 , 10 , 11 , 12 , 13 , 14 , 15 Therapies selectively targeting the p19 subunit of IL‐23, such as guselkumab and risankizumab, 4 , 13 have resulted in high Psoriasis Area and Severity Index (PASI) response rates with favourable safety profiles, 4 , 13 and have shown prolonged maintenance of responses following randomized drug withdrawal. 4 , 14 , 16 This may be due to the ability of IL‐23 inhibitors to decrease resident memory T cells within the affected tissue, 17 which have been shown to be responsible for psoriasis recurrences after initial skin clearing. 18 , 19
Mirikizumab (LY3074828), a humanized, immunoglobulin G4 monoclonal antibody, specifically targets the p19 subunit of IL‐23, and has demonstrated clinical efficacy in phase II trials in psoriasis, 20 ulcerative colitis 21 and Crohn disease. 22 , 23 Mirikizumab also demonstrated efficacy up to 52 weeks, including superiority over an IL‐17 inhibitor (secukinumab), in a phase III trial (OASIS‐2, NCT03535194) for patients with moderate‐to‐severe psoriasis. 24 Here, we evaluated the efficacy and safety of mirikizumab in patients with moderate‐to‐severe plaque psoriasis through 52 weeks, including maintenance of response, time to relapse, and recapture of efficacy after treatment withdrawal in a double‐blind, placebo‐controlled, randomized withdrawal, phase III trial (OASIS‐1, NCT03482011).
Patients and methods
Study design
OASIS‐1 (NCT03482011) was a 52‐week, phase III, multicentre study with a randomized, double‐blinded, placebo‐controlled induction dosing period followed by a randomized withdrawal maintenance dosing period. This study was conducted at 69 sites across nine locations (Germany, Japan, South Korea, Mexico, Poland, Russian Federation, Taiwan, USA and Puerto Rico) and was approved by the applicable ethics review boards. All patients signed informed consent forms before undergoing study‐related procedures and administration of the investigational product. This study was conducted in accordance with the consensus ethics principles derived from international ethical guidelines, including the Declaration of Helsinki and Council for International Organizations of Medical Sciences International Ethical Guidelines, applicable International Council for Harmonisation Good Clinical Practice Guidelines, and other applicable laws and regulations. Patients were enrolled between 24 April 2018 and 16 January 2020. This study followed the CONSORT reporting guidelines.
Patients
Eligible patients were ≥ 18 years of age at the time of screening; had an investigator‐confirmed diagnosis of chronic plaque psoriasis ≥ 6 months prior to baseline; were candidates for systemic therapy and/or phototherapy; and had ≥ 10% body surface area (BSA) involvement, a static Physician’s Global Assessment (sPGA) score ≥ 3, and PASI ≥ 12 at both screening and baseline visits.
Patients were excluded if they had an uncontrolled or unstable health condition at screening, including, but not limited to, cerebrocardiovascular, respiratory, hepatic, renal, gastrointestinal, endocrine, haematological or neurological disease, or abnormal laboratory values. Further exclusion criteria included treatment with systemic anti‐infective medications (including oral) within 28 days of baseline, the use of anti‐tumour necrosis factor (TNF) biologics within 8 weeks of baseline, the use of anti‐IL‐17 biologics within 12 weeks of baseline, or previous use of any biologic therapy targeting IL‐12/23 (p40 subunit) or IL‐23 (p19 subunit). The complete list of inclusion and exclusion criteria is provided in Appendix S1 (see Supporting Information).
Randomization and masking
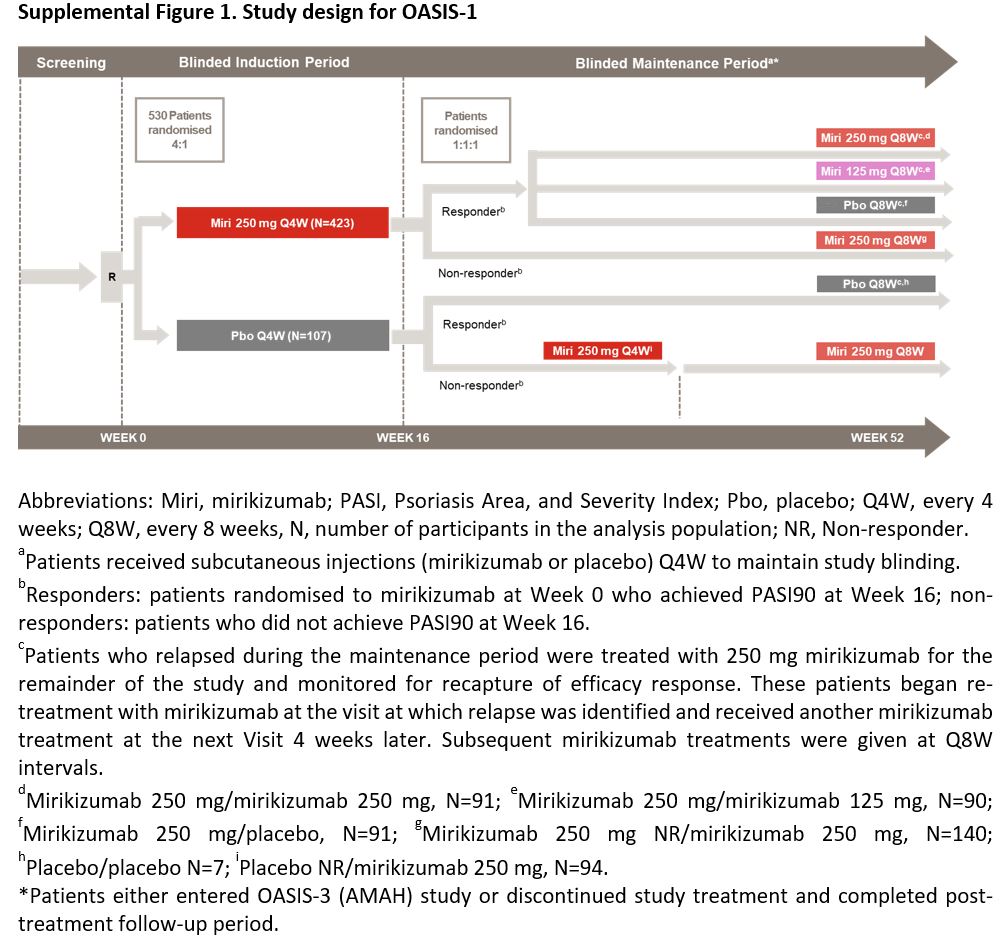
This trial consisted of a 16‐week induction period (weeks 0–16) and a 36‐week maintenance period (weeks 16–52). At week 52, patients either entered the OASIS‐3 (AMAH) study or completed a 12‐week post‐treatment follow‐up period (Figure S1; see Supporting Information). Eligible patients were randomized to a double‐blinded treatment at week 0 (visit 2). Assignment to treatment arms was determined by a computer‐generated random sequence using an interactive web‐response system. Randomization was stratified based on previous exposure to biologic psoriasis therapy (yes or no), bodyweight (< 100 kg or ≥ 100 kg) and geographical region (North America or other) for the induction period, and by bodyweight (< 100 kg or ≥ 100 kg) during the maintenance period.
At week 0, patients were randomized 4 : 1 to receive double‐blinded subcutaneous administration of mirikizumab 250 mg or matching placebo every 4 weeks (Q4W) from weeks 0 to 12. At week 16, responders to mirikizumab treatment in the induction period [defined as patients who achieved ≥ 90% improvement in PASI (PASI 90) at week 16] were randomized 1 : 1 : 1 to mirikizumab 250 mg every 8 weeks (Q8W), mirikizumab 125 mg Q8W or placebo Q8W. Patients were monitored for maintenance of response, disease relapse or rebound following treatment withdrawal, and response to retreatment following relapse. Disease relapse was defined as the loss, at any visit, of ≥ 50% of the week 16 PASI improvement from baseline. Rebound was defined as having one or more of the following: worsening of psoriasis severity over baseline sPGA score, worsening of psoriasis severity over baseline PASI score by ≥ 125%, or change in psoriasis phenotype after randomization to placebo at week 16.
Participants were enrolled by study investigators at each site, with participants and site and sponsor personnel blinded throughout. Interim safety analyses were conducted by an external data monitoring committee to review unblinded safety data.
Outcomes
The primary objective of this study was to assess whether mirikizumab was superior to placebo at week 16 as measured by the proportion of patients with sPGA(0,1) with ≥ 2‐point improvement from baseline and the proportion of patients achieving PASI 90.
Secondary efficacy outcomes during the induction period included the proportion of patients achieving PASI 75 at week 4, and the proportions of patients achieving PASI 75, PASI 100 and ≤ 1% of BSA with psoriasis involvement at week 16. Other major secondary objectives at week 16 were to assess whether mirikizumab induction dosing was superior to placebo for patient‐reported outcomes and health‐related quality‐of‐life endpoints. These were determined respectively by the proportion of patients with a Psoriasis Symptoms and Signs (PSS) score of 0 (free of itch, pain, stinging and burning) in those with a PSS score ≥ 1 at baseline, and the proportion of patients achieving a Dermatology Life Quality Index (DLQI) score of 0 or 1, with a ≥ 5‐point improvement (reduction) from baseline in patients with a baseline DLQI ≥ 5.
At week 52, secondary endpoints included the proportion of patients maintaining clinical response (PASI 90) after rerandomization at the start of the randomized withdrawal period, time to relapse, and the proportion of patients who had relapsed. In addition, rebound after treatment withdrawal and recapture after retreatment were evaluated.
Safety was evaluated in all randomized patients who received at least one dose of study treatment, by monitoring adverse events (AEs), including serious adverse events (SAEs), treatment‐emergent adverse events (TEAEs), adverse events of special interest (AESIs) and discontinuation due to AEs during the induction and maintenance periods. AESIs included infections and infestations, injection‐site reactions, cerebrocardiovascular events, malignancies, and immediate and nonimmediate hypersensitivity reactions.
Statistical analyses
Primary endpoints and other categorical efficacy outcomes were analysed using Cochran–Mantel–Haenszel tests with nonresponder imputation. Patients were considered as nonresponders if they did not meet the clinical response criteria, had missing clinical response data at the analysis assessment timepoint, or were missing postbaseline observations. The median times to loss of PASI 90 response, time to relapse, and time to regain response were evaluated for week 16 mirikizumab responders using Kaplan–Meier time‐to‐event analyses. Patients who relapsed after rerandomization during the maintenance period were considered to have entered the relapse period and were categorized as nonresponders for all subsequent visits of the maintenance efficacy analysis.
A graphical multiple‐testing procedure 25 was implemented to assess the primary and major secondary objectives to control the family‐wise type I error rate at a two‐sided α‐level of 0·05. This allowed simultaneous inference of all the primary and major secondary endpoints. This study had a power of > 95% for testing superiority of mirikizumab over placebo based on a two‐sided χ2‐test with alpha of 5% on sPGA (0,1) and PASI 90, assuming sPGA(0,1) responses were 70% for the mirikizumab arm and 5% for the placebo arm and PASI 90 responses were 70% for the mirikizumab arm and 3% for the placebo arm. To account for multiple testing for the comparisons of two mirikizumab arms against the placebo arm in the maintenance period, a two‐sided χ2‐test at the 0·025 level was used. This sample size was selected to provide > 95% power to test the difference in the proportion of patients maintaining PASI 90 from week 16 after rerandomization at the start of the maintenance dosing period to week 52 between each mirikizumab dosing interval and placebo, assuming the proportions of patients maintaining PASI 90 were 80% for mirikizumab 250 mg Q8W, 70% for mirikizumab 125 mg Q8W, and 10% for placebo. Safety data were summarized by the number and percentage of patients reporting an AE.
Results
Study population
In total, 530 patients were randomized to receive placebo (n = 107) or mirikizumab 250 mg Q4W (n = 423) (Figure 1). Baseline patient demographics and disease characteristics were generally similar across treatment arms and study periods (Table 1). On average, patients were 46·3 years old, weighed 85 kg, and had been diagnosed with psoriasis for 17·6 years. There were more men in all treatment arms compared with women. On average, patients had a baseline PASI of 22·6. Patients with severe (4) and very severe (5) baseline sPGA scores accounted for 42·1% and 8·7% of the total, respectively. Mean baseline BSA involvement across all patients was 31·4%. Previous biologic therapy was reported in approximately 32·1% of patients (Table 1). Overall, 94·4% and 97·4% of patients in the placebo and mirikizumab 250 mg arms completed the induction period, respectively. The percentages of patients who completed the maintenance period until week 52 without relapse or retreatment were as follows: 50·5% mirikizumab 250 mg/placebo, 92·2% mirikizumab 250 mg/mirikizumab 125 mg, and 93·4% mirikizumab 250 mg/mirikizumab 250 mg.
Figure 1.
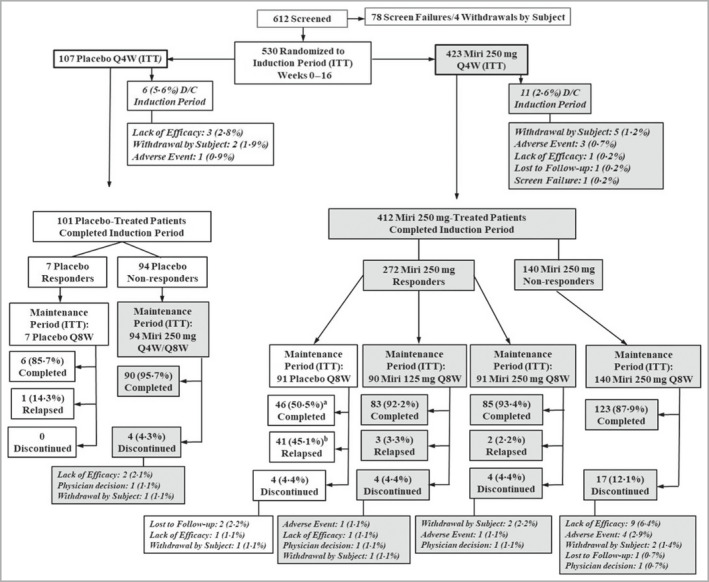
CONSORT diagram: study patient disposition during the induction and maintenance periods. D/C, discontinued; ITT, intention to treat; Miri, mirikizumab; Q4W, every 4 weeks; Q8W, every 8 weeks. aIncludes eight patients who met the relapse criteria at week 52 and then completed the maintenance period without retreatment. bThe eight patients who met the relapse criteria at week 52 and then completed the maintenance period without retreatment are not included here.
Table 1.
Baseline demographics and disease characteristics of the patients
| Study period | Induction period | Maintenance period | |||
|---|---|---|---|---|---|
| Placebo | Mirikizumab | Mirikizumab 250 mg/placebo | Mirikizumab 250 mg/mirikizumab 125 mg | Mirikizumab 250 mg/mirikizumab 250 mg | |
| Number | 107 | 423 | 91 | 90 | 91 |
| Age (years) | 45·7 (13·7) | 46·4 (13·6) | 43·8 (13·5) | 46·9 (13·3) | 45·6 (13·7) |
| Male, n (%) | 74 (69·2) | 299 (70·7) | 61 (67) | 64 (71) | 59 (65) |
| Weight (kg) | 86·5 (22·9) | 84·7 (21·0) | 83·9 (23·7) | 84·3 (20·4) | 82·8 (18·0) |
| < 100, n (%) | 87 (81·3) | 344 (81·3) | 76 (84) | 76 (84) | 76 (84) |
| ≥ 100, n (%) | 20 (18·7) | 79 (18·7) | 15 (16) | 14 (16) | 15 (16) |
| Geographical distribution, n (%) | |||||
| North America | 21 (19·6) | 84 (19·9) | 23 (25) | 16 (18) | 23 (25) |
| Other | 86 (80·4) | 339 (80·1) | 68 (75) | 74 (82) | 68 (75) |
| Prior biologic therapy, n (%) | 31 (29·0) | 139 (32·9) | 34 (37) | 26 (29) | 33 (36) |
| Psoriasis duration (years) | 17·0 (10·9) | 17·7 (11·5) | 18·9 (11·2) | 17·0 (11·8) | 17·6 (11·8) |
| Baseline psoriatic arthritis, n (%) | 8 (7·5) | 69 (16·3) | 12 (13) | 13 (14) | 15 (16) |
| sPGA, n (%) | |||||
| sPGA = 3 | 49 (45·8) | 212 (50·1) | 46 (51) | 45 (50) | 44 (48) |
| sPGA = 4 | 45 (42·1) | 178 (42·1) | 42 (46) | 41 (46) | 38 (42) |
| sPGA = 5 | 13 (12·1) | 33 (7·8) | 3 (3) | 4 (4) | 9 (10) |
| % BSA | 31·9 (19·4) | 31·3 (20·3) | 27·3 (16·1) | 31·8 (21·8) | 31·7 (21·2) |
| PASI | 23·5 (10·1) | 22·3 (9·8) | 20·7 (7·1) | 22·6 (9·7) | 22·6 (10·5) |
| PSS | 22·4 (10·7) | 23·3 (10·5) | 22·6 (11·3) | 22·4 (10·3) | 23·9 (11·0) |
| DLQI | 13 (7·0) | 14·4 (7·4) | 14·9 (7·7) | 14·0 (7·6) | 14·1 (7·3) |
Data are presented as the mean (SD) unless otherwise stated. BSA, body surface area; DLQI, Dermatology Life Quality Index; PASI, Psoriasis Area and Severity Index; PSS, Psoriasis Symptoms and Signs; sPGA, static Physician’s Global Assessment.
Efficacy: induction period
The primary efficacy endpoints were met, with significantly greater improvements in clinical response observed with mirikizumab than with placebo at the end of the induction period. sPGA(0,1) response rates were superior in the mirikizumab arm [293, 69·3%, 95% confidence interval (CI) 64·9–73·7] over placebo (7, 6·5%, 95% CI 1·9–11·2; P < 0·001) at week 16 (Figure 2a). Moreover, sPGA(0) response rates were also greater in the mirikizumab‐treated arm than placebo (33·1 vs. 0·9%, respectively; P < 0·001). At week 16, significantly more patients treated with mirikizumab achieved PASI 90 compared with placebo (272, 64·3%, 59·7–68·9 vs. 7, 6·5%, 95% CI 1·9–11·2; P < 0·001), and a significant improvement was observed in response to mirikizumab treatment as early as week 8 (Figure 2b).
Figure 2.
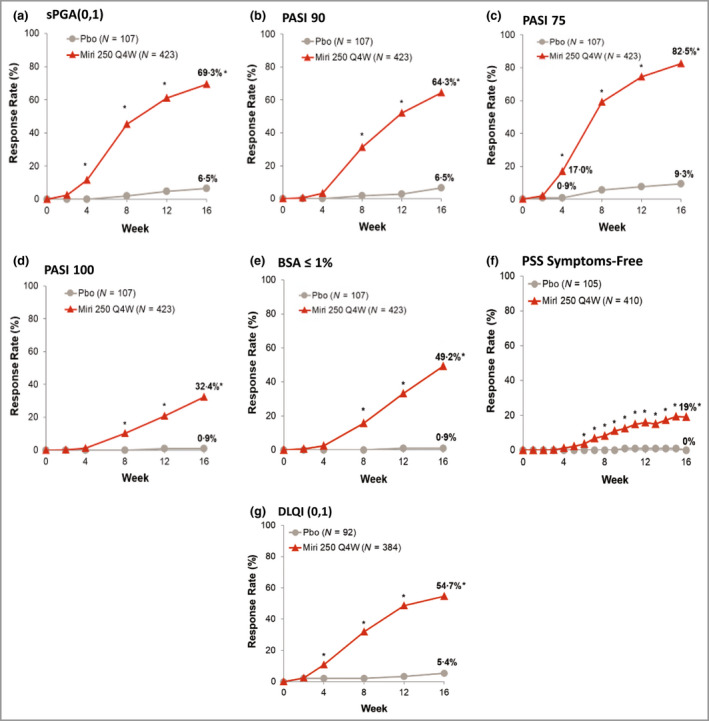
Proportion of patients achieving clinical response and improvement in patient‐reported outcomes and health‐related quality‐of‐life during the induction period. (a) Static Physician’s Global Assessment (sPGA) score of 0 or 1. (b) ≥ 90% improvement in Psoriasis Area and Severity Index (PASI 90). (c) PASI 75. (d) PASI 100. (e) Involved body surface area (BSA) ≤ 1%. (f) Psoriasis Symptoms and Signs (PSS) symptoms free. (g) Dermatology Life Quality Index (DLQI) score of 0 or 1. Miri, mirikizumab; Pbo, placebo; Q4W, every 4 weeks. *P < 0·05 for mirikizumab vs. placebo using the Cochran–Mantel–Haenszel test stratified by prior biologics, weight and region.
All major secondary efficacy objectives were also met. At week 4, a significantly greater proportion of patients in the mirikizumab arm achieved PASI 75 compared with placebo (72, 17·0%, 95% CI 13·4–20·6 vs. 1, 0·9%, 95% CI 0·0–2·8; P < 0·001) (Figure 2c). At week 16, significantly greater proportions of patients in the mirikizumab arm achieved PASI 75 (349, 82·5%, 95% CI 78·9–86·1 vs. 10, 9·3%, 95% CI 3·8–14·9; P < 0·001) and PASI 100 (137, 32·4%, 95% CI 27·9–36·8 vs. 1, 0·9%, 95% CI 0·0–2·8; P < 0·001) (Figure 2c, d). In addition, a greater proportion of patients achieved ≤ 1% BSA at week 16 (mirikizumab 208, 49·2%, 95% CI 44·4–53·9; placebo 1, 0·9%, 95% CI 0·0–2·8; P < 0·001) (Figure 2e). Additional secondary outcomes are reported on ClinicalTrials.gov.
Patient‐reported outcomes and health‐related quality of life: induction period
Improvements in DLQI and PSS scores were superior in the mirikizumab arm compared with placebo at week 16 (all P < 0·001) (Figure 2f, g). More patients in the mirikizumab arm achieved DLQI(0,1) with ≥ 5‐point improvement from baseline compared with placebo (54·7% vs. 5·4%, respectively).
Efficacy: maintenance period
At week 52, both mirikizumab arms showed superiority in PASI 90, PASI 100 and sPGA(0,1) compared with placebo withdrawal, demonstrating maintenance of response for mirikizumab week 16 responders through 52 weeks of treatment (Figure 3). Significantly more patients in the 250 mg/250 mg and 250 mg/125 mg mirikizumab arms maintained PASI 90 response compared with the placebo arm (250 mg Q4W/250 mg Q8W: 77, 85%; 250 mg Q4W/125 mg Q8W: 77, 86%; 250 mg Q4W/placebo: 17, 19%; P < 0·001 for all). The proportion of patients who achieved PASI 100 was also greater in both of the mirikizumab arms compared with the placebo arm (250 mg Q4W/250 mg Q8W: 55, 60%; 250 mg Q4W/125 mg Q8W: 53, 59%; 250 mg Q4W/placebo: 9, 10%; P < 0·001 for all). Similar response rates to those seen for PASI 90 across the treatment arms were observed in the sPGA(0,1) responders (250 mg Q4W/250 mg Q8W: 75, 82%; 250 mg Q4W/125 mg Q8W: 77, 86%; 250 mg Q4W/placebo: 16, 18%; P < 0·001 for all).
Figure 3.
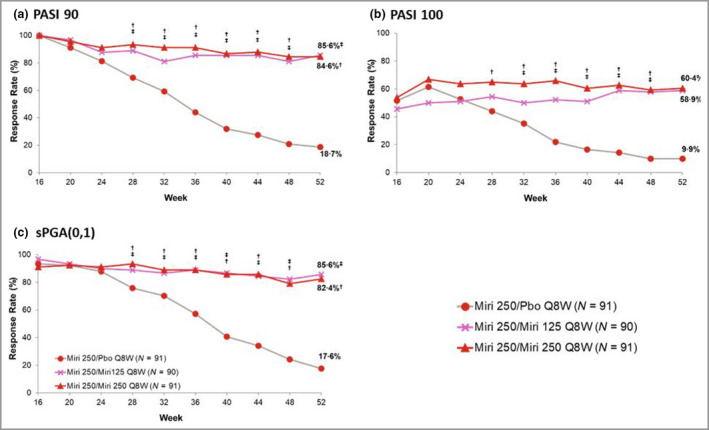
Proportion of patients maintaining or achieving (a) ≥ 90% improvement in Psoriasis Area and Severity Index (PASI 90), (b) PASI 100 or (c) static Physician’s Global Assessment (sPGA) score of 0 or 1 during the maintenance period. The time of last dose prior to randomization is week 12. Miri, mirikizumab; Pbo, placebo; Q8W, every 8 weeks. † P < 0·05 for mirikizumab 250 mg/mirikizumab 250 mg vs. mirikizumab 250 mg/placebo; ‡ P < 0·05 for mirikizumab 250 mg/mirikizumab 125 mg vs. mirikizumab 250 mg/placebo using the Cochran–Mantel–Haenszel test stratified by weight.
The median time to first loss of PASI 90 response was 20·1 weeks (95% CI 17·0–24·0) among mirikizumab 250 mg responders during the induction period who were rerandomized to placebo during the maintenance period (Figure 4). Among the 91 mirikizumab responders who were subsequently randomized to placebo, 49 patients (54%) relapsed (median time to relapse 36·1 weeks, 95% CI 32·1–37·0) (Figure 5). For relapsed patients, the median time to regain PASI 90 following retreatment after relapse was 10 weeks (95% CI 8·1–12·1). After relapse and subsequent retreatment with mirikizumab 250 mg, 19 out of 40 patients (48%) regained PASI 90 response within 16 weeks. Among patients who had the opportunity to be treated for ≥ 16 weeks (n = 8) after relapse, 88% regained PASI 90 (Table S1; see Supporting Information). No disease rebound was observed within 12 weeks after treatment withdrawal. In the maintenance treatment arms, almost none of the patients relapsed, with similar rates for the mirikizumab treatment arms (Figure 5).
Figure 4.
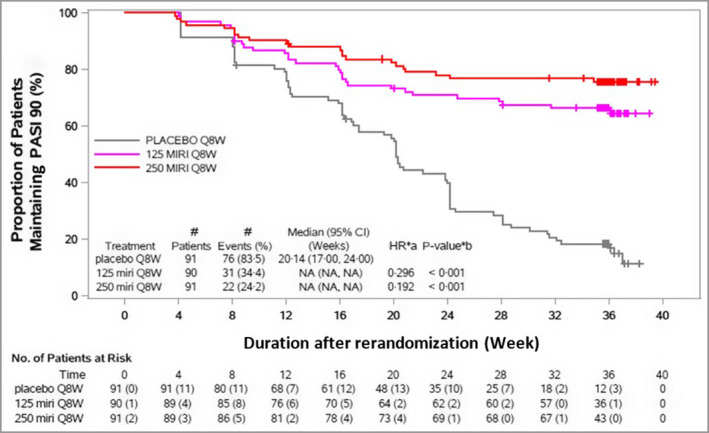
Time to first loss of ≥90% improvement in Psoriasis Area and Severity Index (PASI 90) response through week 52. Maintenance period, rerandomized maintenance intention‐to‐treat population (OASIS‐1). The time of last dose prior to randomization is week 12. CI, confidence interval; HR, hazard ratio; MIRI, mirikizumab; NA, not applicable; Q8W, every 8 weeks. The numbers of patients at risk in brackets represent the number, of those being assessed, who lost or did not maintain PASI 90. aHR stratified by bodyweight (< 100 kg or ≥ 100 kg). b P‐value (two sided): log‐rank test for comparison with placebo Q8W stratified by bodyweight (< 100 kg or ≥ 100 kg).
Figure 5.
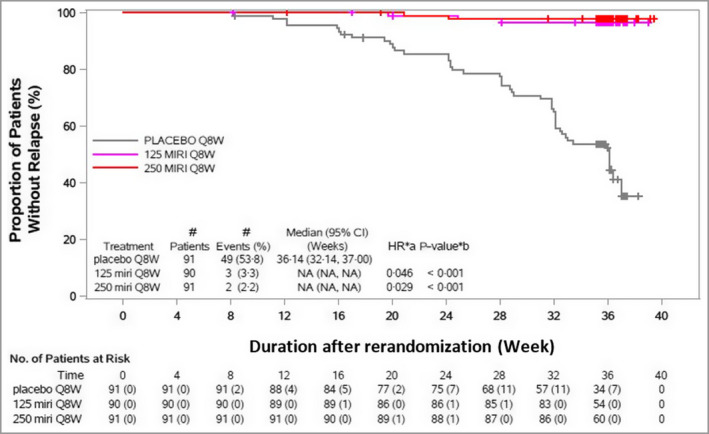
Time to relapse through week 52. Maintenance period, rerandomized maintenance intention‐to‐treat population (OASIS‐1). The time of last dose prior to randomization is week 12. CI, confidence interval; HR, hazard ratio; MIRI, mirikizumab; NA, not applicable; Q8W, every 8 weeks. The numbers of patients at risk in brackets represent the number, of those being assessed, who lost or did not maintain ≥90% improvement in Psoriasis Area and Severity Index. aHR stratified by bodyweight (< 100 kg or ≥ 100 kg). b P‐value (two sided): log‐rank test for comparison with placebo Q8W stratified by bodyweight (< 100 kg or ≥ 100 kg).
Safety
Safety: induction period
Through 16 weeks, a similar frequency of TEAEs was observed across the mirikizumab (200, 47·4%) and placebo (51, 47·7%) arms. Most TEAEs were mild or moderate in severity and the proportion of patients in each severity category was similar between the mirikizumab and placebo arms (Table 2). No deaths were reported in any treatment arm. SAEs occurred in five (1·2%) patients treated with mirikizumab and two (1·9%) of those administered placebo. A low frequency of study discontinuation due to an AE was observed across treatment arms, totalling < 1% for both arms (Table 2). The most common TEAEs (occurring in ≥ 5% of patients on mirikizumab) were injection‐site pain (22, 5·2%) and nasopharyngitis (55, 13·0%).
Table 2.
Reported treatment‐emergent adverse events (TEAEs) and serious adverse events (SAEs)
| Study period | Induction period | Maintenance period | |||
|---|---|---|---|---|---|
| Placebo | Mirikizumab | Mirikizumab 250 mg/placebo | Mirikizumab 250 mg/mirikizumab 125 mg | Mirikizumab 250 mg/mirikizumab 250 mg | |
| Number | 107 | 422 | 91 | 90 | 91 |
| Overall TEAEs | 51 (47·7) | 200 (47·4) | 50 (55) | 61 (68) | 57 (63) |
| Mild | 27 (25·2) | 114 (27·0) | 24 (26) | 36 (40) | 28 (31) |
| Moderate | 22 (20·6) | 77 (18·2) | 23 (25) | 24 (27) | 27 (30) |
| Severe | 2 (1·9) | 9 (2·1) | 3 (3) | 1 (1) | 2 (2) |
| Death | 0 | 0 | 0 | 0 | 0 |
| SAE (patients with ≥1 SAE) | 2 (1·9) | 5 (1·2) | 3 (3) | 1 (1) | 3 (3) |
| Discontinuation due to adverse events | 1 (0·9) | 3 (0·7) | 0 | 1 (1) | 1 (1) |
| TEAEs occurring in ≥5% of mirikizumab patients | |||||
| Headache | 5 (4·7) | 10 (2·4) | 2 (2) | 5 (6) | 4 (4) |
| Injection‐site pain | 5 (4·7) | 22 (5·2) | 2 (2) | 5 (6) | 6 (7) |
| Nasopharyngitis | 14 (13·1) | 55 (13·0) | 15 (16) | 13 (14) | 9 (10) |
| Upper respiratory tract infection | 0 | 16 (3·8) | 7 (8) | 5 (6) | 10 (11) |
The data are presented as n (%).
In Table 3 are listed the AESIs that occurred during the induction period. More infections and infestations occurred in patients treated with mirikizumab than those administered placebo (116, 27·5% vs. 21, 19·6%, respectively). Injection‐site reactions occurred in 28 patients in the mirikizumab arm (6·6%) vs. five patients (4·7%) in the placebo arm. One patient in the mirikizumab arm (0·2%) experienced an adjudicated and confirmed cerebrocardiovascular event, a cerebral infarction, while no cerebrocardiovascular events occurred in the placebo arm. A basal cell carcinoma was reported in one patient in the mirikizumab arm (0·2%), whereas no patients in the placebo arm experienced any malignancies. Immediate hypersensitivity reactions were observed only in patients treated with mirikizumab, including two cases of eczema and one case of allergic rhinitis. Similar proportions of patients in the mirikizumab and placebo arms had nonimmediate hypersensitivity reactions (Table 3).
Table 3.
Reported adverse events of special interest
| Study period | Induction period | Maintenance period | |||
|---|---|---|---|---|---|
| Placebo | Mirikizumab | Mirikizumab 250 mg/placebo | Mirikizumab 250 mg/mirikizumab 125 mg | Mirikizumab 250 mg/mirikizumab 250 mg | |
| Number | 107 | 422 | 91 | 90 | 91 |
| Infections and infestations (SOC) | 21 (19·6) | 116 (27·5) | 27 (30) | 34 (38) | 39 (43) |
| Injection‐site reactions (HLT) | 5 (4·7) | 28 (6·6) | 2 (2) | 6 (7) | 8 (9) |
| Cerebrocardiovascular events | 0 | 1 (0·2)a | 1 (1)d | 1 (1)d | 0 |
| MACE | 0 | 1 (0·2)a | 1 (1)d | 0 | 0 |
| Malignancies | 0 | 1 (0·2)b | 0 | 0 | 0 |
| Immediate hypersensitivity reaction (narrow/algorithmic) | 0 | 3 (0·7)c | 2 (2)e | 0 | 0 |
| Nonimmediate hypersensitivity reaction (narrow/algorithmic) | 4 (3·7) | 13 (3·1) | 3 (3) | 6 (7) | 3 (3) |
The data are presented as n (%). HLT, higher‐level term as defined in the Medical Dictionary for Regulatory Activities; MACE, major adverse cardiovascular event; SOC, system organ class. aAdjudicated and confirmed, preferred term: cerebral infarction. bPreferred term: basal cell carcinoma. cPreferred terms: two eczema and one rhinitis allergic. dAdjudicated and confirmed: acute myocardial infarction (mirikizumab 250 mg/placebo) and atrial fibrillation (mirikizumab 250 mg/mirikizumab 125 mg), eNarrow/algorithmic term: both urticaria.
Safety: maintenance period
Table 2 summarizes the AEs observed during the maintenance period through 52 weeks. Overall, TEAEs were more frequent in the mirikizumab 250 mg/mirikizumab 250 mg (57, 63%) and mirikizumab 250 mg/mirikizumab 125 mg (61, 68%) arms compared with the mirikizumab 250 mg/placebo arm (50, 55%) and most were either mild or moderate. No deaths occurred during this period. A low frequency of SAEs was reported in the three study arms. In total, two patients discontinued treatment due to an AE: one (1%) in the mirikizumab 250 mg/mirikizumab 250 mg arm and one (1%) in the mirikizumab 250 mg/mirikizumab 125 mg arm.
The most common TEAEs reported, respectively in the mirikizumab 250 mg/mirikizumab 250 mg, mirikizumab 250 mg/mirikizumab 125 mg, and mirikizumab 250 mg/placebo arms, were nasopharyngitis (nine, 10%; 13, 14%; 15, 16%), upper respiratory tract infection (10, 11%; five, 6%; seven, 8%), headache (four, 4%; five, 6%; two, 2%) and injection‐site pain (six, 7%; five, 6%; two, 2%).
AESIs observed during the maintenance period are summarized in Table 3. Similarly to the induction period, more patients in the mirikizumab 250 mg/mirikizumab 250 mg (39, 43%) and mirikizumab 250 mg/mirikizumab 125 mg (34, 38%) arms reported cases of infections and infestations compared with the mirikizumab 250 mg/placebo arm (27, 30%). The same pattern of cases across arms was observed for injection‐site reactions (higher‐level term), with more events in the mirikizumab 250 mg/mirikizumab 250 mg arm (eight, 9%) and mirikizumab 250 mg/mirikizumab 125 mg arm (six, 7%) than in the mirikizumab 250 mg/placebo arm (two, 2%).
Adjudicated and confirmed cerebrocardiovascular events occurred in 1% (n = 1) of patients in each of the following mirikizumab arms: the mirikizumab 250 mg/125 mg arm (atrial fibrillation) and the mirikizumab 250 mg/placebo arm (acute myocardial infarction). No cases of malignancies were present in any of the treatment arms. An immediate hypersensitivity reaction (‘in narrow/algorithmic term’) occurred in the mirikizumab 250 mg/placebo arm (two, 2%; both urticaria), whereas nonimmediate hypersensitivity reactions (‘in narrow/algorithmic term’) occurred across the three arms: 3% (n = 3) in both the mirikizumab 250 mg/mirikizumab 250 mg and mirikizumab 250 mg/placebo arms, and 7% (n = 6) in the mirikizumab 250 mg/mirikizumab 125 mg arm.
Discussion
In this phase III trial, mirikizumab demonstrated superior efficacy to placebo in the treatment of moderate‐to‐severe plaque psoriasis by achieving all primary and major secondary endpoints at weeks 16 and 52. At week 16, mirikizumab demonstrated superiority to placebo on both primary efficacy measures: sPGA(0,1) and PASI 90. Additionally, mirikizumab was superior to placebo in all patient‐reported outcomes that were evaluated. Moreover, complete disease resolution demonstrated by sPGA(0) and PASI 100 was achieved by about 33% of patients in the mirikizumab‐treated arm. At week 52, both mirikizumab doses were superior to placebo in all secondary endpoints, with similar response rates observed for both doses.
Week 52 efficacy rates, as demonstrated by maintenance of response (PASI 90) after initial response during induction, were significantly greater in responders rerandomized to continuous mirikizumab treatment than in those rerandomized to placebo at week 16. In fact, an average of 85% of patients under continuous mirikizumab treatment maintained PASI 90, compared with 19% of patients maintaining PASI 90 after treatment withdrawal. These results are consistent with data published for other IL‐23 inhibitors. 4 , 14
Here, we show that among patients who achieved PASI 90 at week 16 and were subsequently randomized to placebo, the median time to first loss of PASI 90 response was approximately 20 weeks and the median time to relapse was approximately 36 weeks. These results are aligned with other clinical trials in patients with plaque psoriasis that also assessed time to loss of response after withdrawal of IL‐23 inhibitors. 5
The overall safety profile of mirikizumab was consistent with the published safety data for other IL‐23p19 inhibitors and with previously published data for mirikizumab in phase II and III trials. Rates of TEAEs, SAEs and discontinuations due to AEs were similar across treatment arms during the induction and randomized withdrawal maintenance periods. Importantly, cases of inflammatory bowel disease and oral candidiasis, which are associated with IL‐17 inhibitors, were comparable with rates observed in the placebo arm.
The strengths of this study comprise the inclusion of patients from several countries across different geographical regions, the size of the study, the fact that it was conducted in a randomized, double‐blinded and placebo‐controlled manner, and inclusion of more than one studied dose in the maintenance phase. One limitation of this trial is the absence of an active‐comparator control.
In conclusion, mirikizumab was found to be efficacious and safe for the treatment of patients with moderate‐to‐severe psoriasis over 52 weeks in a large, randomized, placebo‐controlled study. Following an induction dose of 250 mg every 4 weeks, maintenance doses of 125 mg or 250 mg every 8 weeks from week 16 to week 52 were comparable in maintaining efficacy. Randomized withdrawal of drug eventually led to disease recurrence, albeit at a slower rate than data published with drug withdrawal of tumour necrosis factor and IL‐17 blockers. Retreatment of mirikizumab after drug withdrawal led to prompt recapture of efficacy without safety concerns.
Author contributions
Andrew Blauvelt: Investigation (lead); methodology (lead); supervision (lead); visualization (lead); writing – review and editing (lead). Alexa B. Kimball: Formal analysis (equal); investigation (equal); methodology (equal); writing – review and editing (equal). Matthias Augustin: Conceptualization (equal); investigation (equal); validation (equal); writing – review and editing (equal). Yukari Okubo: Resources (equal); writing – review and editing (equal). Michael Witte: Conceptualization (lead); data curation (lead); formal analysis (lead); investigation (lead); methodology (lead); project administration (lead); resources (lead); supervision (lead); validation (lead); visualization (lead); writing – original draft (lead); writing – review and editing (lead). Claudia Rodriguez Capriles: Conceptualization (equal); data curation (equal); formal analysis (equal); investigation (equal); methodology (equal); project administration (equal); resources (equal); supervision (equal); validation (equal); visualization (equal); writing – original draft (equal); writing – review and editing (equal). Angelina Sontag: Project administration (equal); resources (equal); supervision (equal); writing – review and editing (equal). Vipin Arora: Conceptualization (equal); data curation (equal); formal analysis (lead); methodology (equal); resources (equal); software (equal); supervision (equal); validation (equal); visualization (equal); writing – original draft (equal); writing – review and editing (equal). Olawale Osuntokun: Conceptualization (lead); data curation (equal); formal analysis (equal); funding acquisition (equal); investigation (equal); methodology (equal); project administration (equal); resources (equal); supervision (equal); validation (equal); visualization (equal); writing – original draft (equal); writing – review and editing (equal). Bruce Strober: Investigation (equal); methodology (equal); supervision (equal); writing – review and editing (equal).
Funding sources
This work was supported by Eli Lilly and Company. The funder of the study was involved in the study design, data collection, data analysis and data interpretation. The study funder provided funding for writing support and editorial assistance with manuscript preparation. Medical writing support was provided by Laura de Ugarte Corbalan.
Conflicts of interest
A.B. has served as a scientific adviser and/or clinical study investigator for AbbVie, Abcentra, Aligos, Almirall, Amgen, Arcutis, Arena, Aslan, Athenex, Boehringer Ingelheim, Bristol Myers Squibb, Dermavant, EcoR1, Eli Lilly and Company, Evommune, Forte, Galderma, Incyte, Janssen, Landos, LEO, Novartis, Pfizer, Rapt, Regeneron, Sanofi Genzyme, Sun Pharma, UCB Pharma and Vibliome. A.B.K. is a consultant and investigator for AbbVie, Janssen, Eli Lilly, Novartis, UCB and Target RWE; is an investigator for Bristol Meyers Squibb; is a consultant for Amgen, LEO, Meiji Seiki Pharma, Ventyx and Moonlake; has received fellowship funding from Janssen and AbbVie; has served previously on the board of directors and was past president of the International Psoriasis Council; and has served on the board of directors of Almirall. M.A. has served as a consultant, lecturer and researcher for, and/or has received research grants from companies manufacturing drugs for psoriasis, including AbbVie, Almirall, Amgen, Bayer, Biogen, Boehringer Ingelheim, Bristol Myers Squibb, Celgene, Centocor, Dermira, Eli Lilly, Genzyme, Hexal, Incyte, Janssen, LEO, Medac, MSD, Mylan B.V., Novartis, Pfizer, Regeneron, Sandoz and UCB. Y.O. reports grants from Sun Pharma Japan Ltd, Maruho Co, Ltd, Eisai Co, Ltd and Torii Pharmaceutical Co, Ltd; has received consulting fees from Boehringer Ingelheim, UCB Japan Co, Ltd, and Eli Lilly Japan KK; has received payment for lectures from Kyowa Kirin Co, Ltd, Novartis Pharma KK, Eli Lilly Japan KK, AbbVie GK, UCB Japan Co, Ltd, Janssen Pharmaceutical KK, Taiho Pharmaceutical Co, Ltd, Maruho Co, Ltd and Amgen Inc.; and has participated on a data safety monitoring board or advisory board for Boehringer Ingelheim, UCB Japan Co, Ltd and Eli Lilly Japan KK. M.M.W., C.R.C, A.S., V.A. and O.O. are a full‐time employees and stockholders of Eli Lilly and Company. B.S. has served as a consultant (honoraria) for AbbVie, Almirall, Amgen, Arcutis, Arena, Aristea, Asana, Boehringer Ingelheim, Immunic Therapeutics, Bristol Myers Squibb, Connect Biopharma, Dermavant, Eli Lilly, EPI Health, Evelo Biosciences, Janssen, LEO, Maruho, Meiji Seika Pharma, Mindera Health, Novartis, Ono, Pfizer, UCB Pharma, Sun Pharma, Regeneron, Sanofi Genzyme, Union Therapeutics, Ventyxbio and vTv Therapeutics; is a stockholder of Connect Biopharma and Mindera Health; has served as a speaker for AbbVie, Eli Lilly, Janssen, Regeneron and Sanofi Genzyme; has been a co‐scientific director (consulting fee) for CorEvitas (formerly Corrona) Psoriasis Registry; has been an investigator for Dermavant, AbbVie, CorEvitas Psoriasis Registry, Dermira, Cara and Novartis; and has been editor in chief (honorarium) of the Journal of Psoriasis and Psoriatic Arthritis.
Ethics statement
This study was conducted at 69 sites across nine locations (Germany, Japan, South Korea, Mexico, Poland, Russian Federation, Taiwan, USA and Puerto Rico) and was approved by the applicable ethics review boards. All patients signed informed consent forms before undergoing study‐related procedures and administration of the investigational product. This study was conducted in accordance with consensus ethics principles derived from international ethical guidelines, including the Declaration of Helsinki and Council for International Organizations of Medical Sciences International Ethical Guidelines, applicable International Council for Harmonisation Good Clinical Practice Guidelines, and other applicable laws and regulations.
Supporting information
Appendix S1 Inclusion and exclusion criteria.
Figure S1 Design for OASIS‐1.
{kind=link}
Table S1 Patients regaining ≥90% improvement in Psoriasis Area and Severity Index after relapse and retreatment (rerandomized relapsed population).
Acknowledgments
The authors wish to thank the study participants, investigators and trial staff for their participation in the OASIS‐1 study. Writing support was provided by Laura de Ugarte Corbalan, PhD, medical review was provided by Cem Kayhan, MD, statistical support review was provided by Lu Zhang, and editorial assistance was provided by Roheena Deshmukh, all employees of Eli Lilly and Company. Manuscript revision was provided by Dana Schamberger, MA, an employee of Syneos Health.
Plain language summary available online
Data availability
Lilly provides access to all individual participant data collected during the trial, after anonymization, with the exception of pharmacokinetic or genetic data. Data are available to request 6 months after the indication studied has been approved in the USA and EU and after primary publication acceptance, whichever is later. No expiration date of data requests is currently set once data are made available. Access is provided after a proposal has been approved by an independent review committee identified for this purpose and after receipt of a signed data sharing agreement. Data and documents, including the study protocol, statistical analysis plan, clinical study report, and blank or annotated case report forms, will be provided in a secure data sharing environment. For details on submitting a request, see the instructions provided at www.vivli.org.
References
- 1. Parisi R, Symmons DP, Griffiths CE et al. Global epidemiology of psoriasis: a systematic review of incidence and prevalence. J Invest Dermatol 2013; 133:377–85. [DOI] [PubMed] [Google Scholar]
- 2. Griffiths CE, Barker JN. Pathogenesis and clinical features of psoriasis. Lancet 2007; 370:263–71. [DOI] [PubMed] [Google Scholar]
- 3. Wang X‐Y, Zhang C‐L, Wang W‐H. Time to relapse after treatment withdrawal for different biologics used to treat plaque psoriasis. Chin Med J (Engl) 2020; 133:2998–3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Blauvelt A, Leonardi CL, Gooderham M et al. Efficacy and safety of continuous risankizumab therapy versus treatment withdrawal in patients with moderate to severe plaque psoriasis: a phase 3 randomized clinical trial. JAMA Dermatol 2020; 156:649–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Reich K, Armstrong AW, Foley P et al. Efficacy and safety of guselkumab, an anti‐interleukin‐23 monoclonal antibody, compared with adalimumab for the treatment of patients with moderate to severe psoriasis with randomized withdrawal and retreatment: results from the phase III, double‐blind, placebo‐ and active comparator‐controlled VOYAGE 2 trial. J Am Acad Dermatol 2017; 76:418–31. [DOI] [PubMed] [Google Scholar]
- 6. Fitch E, Harper E, Skorcheva I et al. Pathophysiology of psoriasis: recent advances on IL‐23 and Th17 cytokines. Curr Rheumatol Rep 2007; 9:461–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hawkes JE, Yan BY, Chan TC et al. Discovery of the IL‐23/IL‐17 signaling pathway and the treatment of psoriasis. J Immunol 2018; 201:1605–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lebwohl MG, Gordon KB, Gallo G et al. Ixekizumab sustains high level of efficacy and favourable safety profile over 4 years in patients with moderate psoriasis: results from UNCOVER‐3 study. J Eur Acad Dermatol Venereol 2020; 34:301–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Leonardi C, Reich K, Foley P et al. Efficacy and safety of ixekizumab through 5 years in moderate‐to‐severe psoriasis: long‐term results from the UNCOVER‐1 and UNCOVER‐2 phase‐3 randomized controlled trials. Dermatol Ther (Heidelb) 2020; 10:431–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Warren RB, Blauvelt A, Poulin Y et al. Efficacy and safety of risankizumab vs. secukinumab in patients with moderate‐to‐severe plaque psoriasis (IMMerge): results from a phase III, randomized, open‐label, efficacy‐assessor‐blinded clinical trial. Br J Dermatol 2021; 184:50–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Reich K, Warren RB, Iversen L et al. Long‐term efficacy and safety of tildrakizumab for moderate‐to‐severe psoriasis: pooled analyses of two randomized phase III clinical trials (reSURFACE 1 and reSURFACE 2) through 148 weeks. Br J Dermatol 2020; 182:605–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Gordon KB, Blauvelt A, Papp KA et al. Phase 3 trials of ixekizumab in moderate‐to‐severe plaque psoriasis. N Engl J Med 2016; 375:345–56. [DOI] [PubMed] [Google Scholar]
- 13. Reich K, Armstrong AW, Langley RG et al. Guselkumab versus secukinumab for the treatment of moderate‐to‐severe psoriasis (ECLIPSE): results from a phase 3, randomized controlled trial. Lancet 2019; 394:831–9. [DOI] [PubMed] [Google Scholar]
- 14. Gordon KB, Strober B, Lebwohl M et al. Efficacy and safety of risankizumab in moderate‐to‐severe plaque psoriasis (UltIMMa‐1 and UltIMMa‐2): results from two double‐blind, randomised, placebo‐controlled and ustekinumab‐controlled phase 3 trials. Lancet 2018; 392:650–61. [DOI] [PubMed] [Google Scholar]
- 15. Puig L, Lebwohl M, Bachelez H et al. Long‐term efficacy and safety of brodalumab in the treatment of psoriasis: 120‐week results from the randomized, double‐blind, placebo‐ and active comparator‐controlled phase 3 AMAGINE‐2 trial. J Am Acad Dermatol 2020; 82:352–9. [DOI] [PubMed] [Google Scholar]
- 16. Gordon KB, Armstrong AW, Foley P et al. Guselkumab efficacy after withdrawal is associated with suppression of serum IL‐23‐regulated IL‐17 and IL‐22 in psoriasis: VOYAGE 2 study. J Invest Dermatol 2019; 139:2437–46. [DOI] [PubMed] [Google Scholar]
- 17. Mehta H, Mashiko S, Angsana J et al. Differential changes in inflammatory mononuclear phagocyte and T‐cell profiles within psoriatic skin during treatment with guselkumab vs. secukinumab. J Invest Dermatol 2021; 141:1707–18. [DOI] [PubMed] [Google Scholar]
- 18. Cheuk S, Wikén M, Blomqvist L et al. Epidermal Th22 and Tc17 cells form a localized disease memory in clinically healed psoriasis. J Immunol 2014; 192:3111–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Mashiko S, Edelmayer RM, Bi Y et al. Persistence of inflammatory phenotype in residual psoriatic plaques in patients on effective biologic therapy. J Invest Dermatol 2020; 140:1015–25. [DOI] [PubMed] [Google Scholar]
- 20. Reich K, Rich P, Maari C et al. Efficacy and safety of mirikizumab (LY3074828) in the treatment of moderate‐to‐severe plaque psoriasis: results from a randomized phase II study. Br J Dermatol 2019; 181:88–95. [DOI] [PubMed] [Google Scholar]
- 21. Sandborn WJ, Ferrante M, Bhandari BR et al. Efficacy and safety of mirikizumab in a randomized phase 2 study of patients with ulcerative colitis. Gastroenterology 2020; 158:537–49. [DOI] [PubMed] [Google Scholar]
- 22. Sands B, Sandborn W, Peyrin‐Biroulet L et al. 1003 – efficacy and safety of mirikizumab (LY3074828) in a phase 2 study of patients with Crohn’s disease. Gastroenterology 2019; 156:S216. [DOI] [PubMed] [Google Scholar]
- 23. Sands B, Peyrin‐Biroulet L, Kierkus J et al. Efficacy and safety of mirikizumab in a randomized phase 2 study of patients with Crohn’s disease. Gastroenterology 2022; 162:495–508. [DOI] [PubMed] [Google Scholar]
- 24. Papp KA, Warren RB, Green L et al. Efficacy and safety of mirikizumab versus secukinumab and placebo in the treatment of moderate‐to‐severe psoriasis: 52‐week results from OASIS‐2, a multicenter, randomized, double‐blind study. J Clin Aesthet Dermatol 2021; 14 (5 Suppl. 1):S8–30.34055192 [Google Scholar]
- 25. Bretz F, Posch M, Glimm E et al. Graphical approaches for multiple comparison procedures using weighted Bonferroni, Simes, or parametric tests. Biom J 2011; 53:894–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1 Inclusion and exclusion criteria.
Figure S1 Design for OASIS‐1.
Table S1 Patients regaining ≥90% improvement in Psoriasis Area and Severity Index after relapse and retreatment (rerandomized relapsed population).
Data Availability Statement
Lilly provides access to all individual participant data collected during the trial, after anonymization, with the exception of pharmacokinetic or genetic data. Data are available to request 6 months after the indication studied has been approved in the USA and EU and after primary publication acceptance, whichever is later. No expiration date of data requests is currently set once data are made available. Access is provided after a proposal has been approved by an independent review committee identified for this purpose and after receipt of a signed data sharing agreement. Data and documents, including the study protocol, statistical analysis plan, clinical study report, and blank or annotated case report forms, will be provided in a secure data sharing environment. For details on submitting a request, see the instructions provided at www.vivli.org.