

Abstract
Highly immunosuppressive tumor microenvironment containing various protumoral immune cells accelerates malignant transformation and treatment resistance. In particular, tumor-associated macrophages (TAMs), as the predominant infiltrated immune cells in a tumor, play a pivotal role in regulating the immunosuppressive tumor microenvironment. As a potential therapeutic strategy to counteract TAMs, here we explore an exosome-guided in situ direct reprogramming of tumor-supportive M2-polarized TAMs into tumor-attacking M1-type macrophages. Exosomes derived from M1-type macrophages (M1-Exo) promote a phenotypic switch from anti-inflammatory M2-like TAMs toward pro-inflammatory M1-type macrophages with high conversion efficiency. Reprogrammed M1 macrophages possessing protein-expression profiles similar to those of classically activated M1 macrophages display significantly increased phagocytic function and robust cross-presentation ability, potentiating antitumor immunity surrounding the tumor. Strikingly, these M1-Exo also lead to the conversion of human patient-derived TAMs into M1-like macrophages that highly express MHC class II, offering the clinical potential of autologous and allogeneic exosome-guided direct TAM reprogramming for arming macrophages to join the fight against cancer.
Keywords: Exosome, Cancer therapy, Tumor microenvironment, Tumor-associated macrophage, Direct conversion
Graphical abstract
Highlights
-
•
Reprogramming of tumor associated macrophages exerts anti-tumor effects.
-
•
M1 macrophage-derived exosomes contain miRNAs involved in macrophage repolarization.
-
•
M1 macrophage-derived exosomes can reprogram M2 into functional M1-like macrophages.
1. Introduction
In the field of regenerative medicine, exogenous cell transplantation has been extensively studied as a strategy for generating desired functional cell types; however, the risk of teratoma formation and difficulty of transplantation limit their clinical applications [1,2]. In particular, the poor engraftment efficiency associated with transplantation methods employing injection of cells has motivated the development of breakthrough technologies capable of promoting cell fate conversion within the body [3]. Direct cell reprogramming, which directly induces the conversion of cell types from one lineage to another without bridging intermediate pluripotent steps, is a relatively simple method for producing desired functional cells [4,5]. While direct conversion circumvents the risk of tumorigenesis associated with pluripotency compared with induced pluripotent stem cell (iPSC) differentiation [6], the use of viral vectors for transporting multiple exogenous factors still poses several issues in clinical applications owing to insufficient efficiency and safety concerns [7].
Implementation of in situ direct cell reprogramming requires the safe and efficient in vivo delivery of reprogramming-inducing factors [8]. Hence, in this study, we suggest exosomes—naturally occurring vehicles capable of safely transferring cocktails of endogenous material—as an entirely new platform for in situ direct cell reprogramming. Because exosomes contain a variety of factors related to cell differentiation, growth, migration and signal transduction, they have virtually unlimited potential as cell-conversion–inducing factors [9,10]. Rather than delivering one type of inducing factor at a time, exosomes can deliver the complex signals necessary for direct cell conversion at once by simultaneously transporting biomacromolecules, such as a group of micro RNAs (miRNAs), capable of inducing synergistic direct cell reprogramming outcomes [11,12].
Exosomes are nano-sized extracellular membrane vesicles composed of a lipid bilayer that serve as integral nanocarriers of endogenous information (proteins, miRNAs, and mRNAs) obtained from their originating cells. Following the discovery of the role of exosomes as vehicles for the horizontal transfer of mRNAs or miRNAs that elicit phenotypic changes in target cells [13], several studies demonstrated applications of exosomes in cell-free therapy [14,15]. In particular, stem cell-derived exosomes have been the focus of attention as an alternative approach to stem cell-based therapy in regenerative medicine [16]. Moreover, exosomes are considered to be promising delivery vehicles with better safety profiles than those of other synthetic nanoparticles, such as viral vectors, cationic lipids, and polymer-based particles. They are also highly versatile with excellent biocompatibility [17,18].
Based on the concept of exosome-mediated in situ direct reprogramming, we here developed a simple strategy for converting M2-type tumor-associated macrophages (TAMs) to M1-type macrophages. The solid tumor microenvironment (TME) actively recruits myeloid cells, inducing widespread infiltration of macrophages [19]. However, instead of stimulating the immune system to rid the body of foreign debris, these abundant TME macrophages create a pro-tumor environment that accelerates tumor growth and angiogenesis, facilitates metastasis, and promotes immunosuppression [20]. Generally, TAMs are M2-like macrophages exhibiting an anti-inflammatory phenotype rather than M1-polarized macrophages [21]. Therefore, the polarization status of macrophages in the TME has generated considerable interest as a possible target of therapeutic approaches for suppressing pro-tumorigenic functions of macrophages.
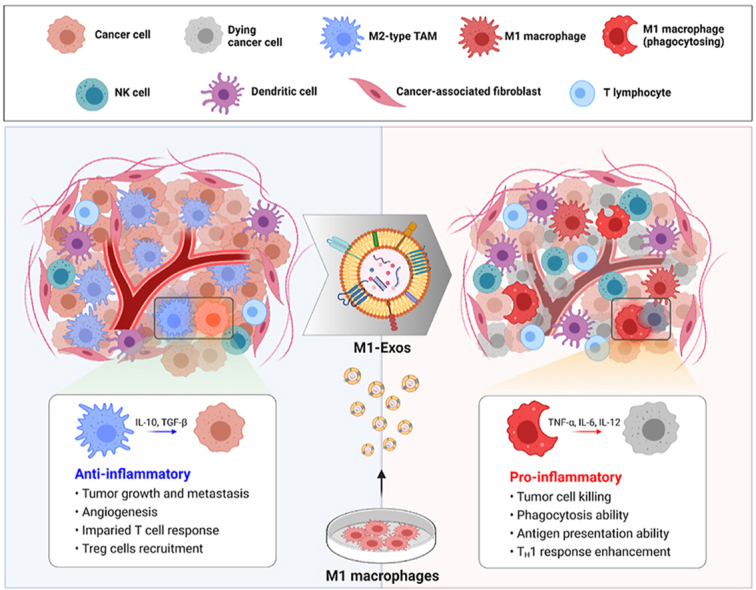
Here, we exploited the ability of exosomes derived from M1-type macrophages (M1-Exo) to redirect TAMs into tumor-killing macrophages (Fig. 1). Proteomic analyses demonstrated that gene expression profiles were similar between reprogrammed M1-type macrophages (RM1) and M1 macrophages, and that RM1 macrophages expressed co-stimulatory molecules involved in antigen-presentation, characteristic of M1 macrophages. We further found that M1-Exo contain various miRNAs that are efficient drivers of direct macrophage polarization, including miR-27a, miR-125b, miR-155, and miR-199a. In addition, macrophages reprogrammed into anti-tumorigenic M1-type showed improved phagocytosis and cross-presentation ability, and were capable of slowing tumor growth. M1-Exo were also able to induce phenotypic conversion of cancer patient-derived TAMs into macrophages with an M1-like phenotype that highly express MHC class II, suggesting their potential use for autografts or allografts. Our findings thus suggest that exosome-mediated direct reprograming of macrophages based on their central roles in cancer pathogenesis could provide a clear opportunity for cancer therapy.
Fig. 1.

Schematic representation of M1-Exo–mediated TAM reprogramming. M1-Exo are able to reprogram tumor-supporting anti-inflammatory M2-type TAMs into pro-inflammatory M1 macrophages.
2. Material and methods
2.1. Materials
Fetal bovine serum (FBS), Cell Tracker 5-chloromethylfluorescein diacetate (CMFDA), Deep Red, pHrodo Red SE, GM130 (ARC0589; MA5-35107) and EGR2 antibodies (erongr2; 17-6691-82) were purchased from Thermo Fisher Scientific Inc. RPMI-1640 and Dulbecco's Modified Eagle Medium (DMEM) were from Hyclone (Cytiva). Recombinant murine M-CSF (315-02), murine IFN-γ (315-05), and IL-4 (214-14) were purchased from Peprotech. Amicon Ultra-15 10K centrifugal filters were purchased from Merck. Nitrocellulose membranes (0.2 μm) were obtained from Bio-Rad. Lipopolysaccharide (LPS) was purchased from Sigma Aldrich. Hoechst 33342 trihydrochloride trihydrate was purchased from Life Technologies. The following antibodies were obtained from BioLegend: Brilliant Violet 650-conjugated anti-mouse F4/80 (clone BM8; 123149); fluorescein isothiocyanate (FITC)-conjugated anti-human HLA-DR (clone L243; 307603); PerCP/cyanine-conjugated anti-mouse CD11b (clone M1/70; 101228); Brilliant Violet 421-conjugated anti-mouse CD38 (clone 90; 102732); phycoerythrin (PE)-conjugated, SIINFEKL-bound anti-mouse H-2Kb (clone 25-D1.16; 141603); FITC-conjugated anti-mouse CD206 (clone C068C2, 141703), PE-conjugated anti-mouse CD86 (clone GL-1; 105007); APC-conjugated anti-mouse CD40 (clone 3/23; 124611); Brilliant Violet 650-conjugated anti-rat IgG2a; PerCP/cyanine-conjugated anti-rat IgG2b kappa; Brilliant Violet 421-conjugated anti-rat IgG2a kappa; PE-conjugated anti-mouse IgG1 kappa; FITC-conjugated anti-rat IgG2a kappa; PE-conjugated anti-rat IgG2a kappa; allophycocyanin (APC)-conjugated anti-rat IgG2a kappa; and Alexa Fluor 647-conjugated anti-mouse CD8a. PE-conjugated anti-mouse iNOS (clone CXNFT; 2082543), PE-conjugated anti-rat IgG2a kappa, and TRIzol reagent were purchased from Invitrogen. The Fc blocker, anti-CD16/CD32 (clone 2.4G2; 553142), was from BD Pharmingen. Anti-mannose receptor (ab64693), anti-CD163 (ab182422), anti-iNOS (ab15323) and anti-Tsg101 (ab83) antibodies, and Nitric Oxide Assay kit (Fluorometric) were acquired from Abcam. Anti-Alix (clone 1A12; sc53540) and anti-CD81 (sc166029) antibodies were from Santa Cruz. Anti-GAPDH antibody (MAB5718) was purchased from R&D Systems.
2.2. Cell culture
Murine 4T1-Luc breast cancer cells and human HT29 colon adenocarcinoma cells were cultured in RPMI-1640 (Hyclone) containing 10% FBS (Gibco) and 1% antibiotic-antimycotic (Gibco). Murine B16F10 melanoma cells were cultured in DMEM (Hyclone). Bone marrow (BM) cells, the most homogeneous macrophage source, were obtained in high yield from marrow extracted from the hind limb [22,23], and were seeded at 9 × 106 cells per plate in 150-mm culture dishes. After 24 h, the suspended cells were collected and resuspended in fresh media. Bone marrow-derived macrophages (BMDMs) were differentiated by treating bone marrow cells with 20 ng/mL of M-CSF (Peprotech). After 7 days of differentiation, adherent cells were harvested. Cells were treated for 24 h with 100 ng/mL LPS (Sigma) and 20 ng/mL IFN-γ (Peprotech) for M1 activation, and with 20 ng/mL of IL-4 for M2 activation. All cell lines were incubated at 37 °C in a humidified 5% CO2 environment.
2.3. Exosome preparation
For isolation of exosomes, BMDMs (9 × 106 cells per 150 mm dish) were differentiated by incubating with M-CSF (Peprotech). After changing the medium to serum-free RPMI-1640 (Welgene), cells were incubated for an additional 48 h. Cells, cell debris, and large vesicles were removed by serial centrifugation of cell supernatants at 300×g for 10 min, 2000×g for 10 min, and 10,000×g for 30 min in an Avanti J-E centrifuge (Beckman Coulter; fixed angle JA-20 rotor) at maximum acceleration and deceleration speed. Centrifuged supernatants were filtered using Amicon Ultra centrifugal filters (10K MWCO; Merck) and ultracentrifuged at 150,000×g for 3 h in an Optima XE-100 centrifuge (Beckman Coulter; fixed angle type 45 Ti rotor) with maximum acceleration and deceleration. Exosomes were re-suspended in autoclaved phosphate-buffered saline (PBS) containing protease inhibitor cocktail and stored at 4 °C. The concentration of exosomes was determined based on their protein concentration using a BCA Protein Assay kit (Bio-Rad).
2.4. Dynamic light scattering
The size analysis of exosomes was performed using a Zetasizer Nano XS instrument at 25 °C, and data were analyzed using the manufacturer's software.
2.5. Transmission electron microscopy
The size and shape of exosomes were analyzed using cryo-transmission electron microscopy (Cryo-TEM). Samples for Cryo-TEM analysis were prepared by first fixing exosome samples with 0.5% glutaraldehyde overnight, after which they were centrifuged at 150,000×g for 30 min, re-suspended in absolute ethanol, and placed on a lacey grid. The grid was stored in liquid nitrogen and maintained at −180 °C. Exosome membranes were vitrified using an FEI Vitrobot system (FP5350/60).
2.6. Immunoblotting
Expression of M1 or M2 markers was confirmed by first seeding 5 × 105 BMDMs on day 7 in 35 mm Petri dishes under M1 or M2 condition. After 24 h, the cells were incubated for 2 h at 4 °C with RIPA buffer with a protease inhibitor cocktail. After centrifugation at 12,000 rpm for 20 min, the supernatant was collected and protein was quantified using a BCA Protein Assay kit (Bio-Rad). 10 μg of protein was separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to nitrocellulose membranes. The membranes were washed for 2 h with 1X Tris-buffered saline containing 0.1% Tween-20 (TBST)/5% skim milk and incubated in blocking solution containing primary antibodies at 4 °C overnight. Thereafter, membranes were incubated with horseradish peroxidase (HRP)-conjugated anti-mouse or anti-rabbit peroxidase secondary antibodies at room temperature for 1 h.
2.7. MiRNA sequencing
Total RNA was prepared from M1-Exo and M2-Exo using the TRIzol reagent (Invitrogen). RNA was quantified using a NanoDrop 2000 Spectrophotometer system (Thermo Fisher Scientific), and RNA quality was examined with an Agilent 2100 bioanalyzer using the RNA 6000 Pico Chip (Agilent Technologies). Read counts mapped on differentially expressed genes and mature miRNA sequences were extracted from the alignment file using Bedtools (v2.25.0) and Bioconductor using R (version 3.2.2) statistical programming language (R development Core Team, 2016). Log2 values of read counts were applied to determine the expression levels of miRNAs. miRNA expression profiling with miRWalk 2.0 was conducted by a commercial service (e-biogen).
2.8. Cellular uptake of exosomes and in vitro cytotoxicity
Freshly isolated exosomes (100 μg) were labeled by incubating overnight with 6.8 μg of Cy5.5 NHS-ester and then washed twice with PBS. BMDMs (2 × 105 cells) were plated in 35-mm glass-bottom confocal dishes under M1 or M2 conditions. After 24 h, the medium was replaced with serum-free medium containing labeled exosomes. The cells permeabilized with 0.2% Triton X-100 for 5 min at room temperature following washing with PBS. Cell nuclei were stained by incubating with Hoechst 33342 for 5 min at room temperature. Images were analyzed using a Leica TCS SP5 confocal microscope. For in vitro cytotoxicity tests, BMDMs (2 × 104 cells) were seeded in 96-well plates containing different concentrations of exosomes. After incubating for 24 h, CCK-8 solution (20 μL) was applied to each well and cell viability was assessed using a microplate reader (SpectraMax 34; Molecular Devices).
2.9. Flow cytometry
Expression of M1 or M2 markers on the surface of exosomes was assessed by incubating 10 μg of purified exosomes with 20 μL aldehyde/sulfate latex beads (4 μm; Invitrogen) and then adjusting the final volume to 1 mL with PBS. After adding 110 μL of 1 M glycine buffer and incubating for 30 min to block free binding sites on the beads, the mixed beads were washed twice with PBS containing 0.5% BSA. Exosome-bound beads were conjugated with PE-CD86 (clone GL-1; BioLegend), APC-CD40 (clone 3/23; BioLegend) or FITC-CD206 (clone C068C2; BioLegend) antibodies by incubating for 1 h at 4 °C, then washing twice with PBS/0.5% BSA. Expression of M1 or M2 markers was confirmed by first seeding 5 × 105 BMDMs in a 35 mm Petri dish under M1 or M2 conditions. After 24 h, the cells were detached from the dish using cold Dulbecco's PBS, pre-incubated with 2 μg of purified rat anti-mouse CD16/CD32 (BD Biosciences) on ice for 25 min, and then incubated with PE-conjugated anti-CD86, APC-conjugated anti-CD40 or FITC-conjugated anti-CD206 antibodies. All data were acquired using Accuri C6 and FlowJo v8 software (BD Biosciences). For the anti-miRNA transfection, transfection was carried out at a concentration of 50 pmol/mL of anti-miRNA using lipofectamine (Thermo Fisher Scientific) according to the manufacturer's manual along with M2-Exo treatment. For analysis of macrophage populations in vivo, single-cell suspensions were prepared from tumor tissues using a mouse Tumor Dissociation Kit (#130-096-730; Miltenyi Biotec) according to the manufacturer's instructions, after which cells were filtered through a 40-μm strainer and red blood cells (RBCs) were removed using an RBC lysis buffer (#420302; BioLegend). For live cell analyses, live cells were isolated using a Dead Cell Removal Microbeads Kit (#130-090-101; Miltenyi Biotec) according to the manufacturer's protocol and then blocked with purified rat anti-mouse CD16/CD32 (Mouse BD Fc Block; BD Biosciences) in staining buffer for 15 min. Cells were stained with antibodies to the surface markers CD11b (clone M1/70; BioLegend), F4/80 (clone BM8; BioLegend), CD38 (clone 90; BioLegend) or isotype control for 30 min at 4 °C. After washing twice with staining buffer, samples were fixed with BioLegend Fixation Buffer for 45 min at room temperature and washed twice with 1X BioLegend Permeation Buffer. Intracellular proteins were stained by permeabilizing cells for 45 min at room temperature and then incubated with PE-conjugated anti-NOS2 (CXNFT; eBioscience) or isotype control. Flow cytometry was performed on cells suspended in staining buffer using a Beckman Coulter flow cytometer, and data were analyzed with FlowJo v8 software.
2.10. Proteomics analysis
M1, M2, and RM1 macrophages induced by M1-Exo were lysed in RIPA buffer with protease inhibitor cocktail. Protein concentrations were quantified using the bicinchoninic acid (BCA) method, and proteins were separated based on molecular weight using 4–12% Bis-Tris gels. Samples (100 μg) of each macrophage type were run in triplicate on the same gel. After the gel was stained with Coomassie Brilliant Blue R-250, the lane for each sample was divided into 14 slices and the proteins contained in each gel slice were subjected to tryptic digestion. Peptides were then extracted with 67% acetonitrile (ACN)/5% formic acid (FA) in water. After drying in a SpeedVac, peptides were resuspended with 20 μL of 0.4% acetic acid. For mass spectral analyses, 13.5 μL of each sample was separated using a reversed-phase Magic C18AQ column (15 cm × 75 μm) on an Eksigent MDLC system (Eksigent Technologies) coupled to an LTQ XL-Orbitrap mass spectrometer (Thermo Fisher Scientific). The operating flow rate was 350 nL/min, and the gradient conditions were as follows: 0 min, 100% buffer A (water containing 0.1% FA) and 0% buffer B (ACN containing 0.1% FA); 0–5 min, 0–8% B; 5–85 min, 8–30% B; 85–90 min, 30–70% B; 90–100 min, 70% B; 100–110 min, 70–2% B; and 100–120 min, 2% B. Survey full-scan mass spectrometry (MS) spectra (300–1800 m/z) were acquired at a resolution of 60,000. Tandem mass (MS/MS) spectra for the ten most intense ions were acquired in the ion trap with the following options: isolation width, 2 m/z; normalized collision energy, 35%; dynamic exclusion duration, 360 s. Each LC-MS/MS file was searched against the SwissProt mouse database (March 2021) containing 17077 entries using Proteome Discoverer software (version 2.4; Thermo Fisher Scientific). The search criteria were set to a mass tolerance of 15 ppm for MS1 data and 0.5 Da for MS2 data, with a fixed modification of carbamidomethylation of cysteine (+57.021 Da) and variable modification of methionine oxidation (+15.995 Da). The false discovery rate (FDR) was set to 0.01 for identification of peptides and proteins, and the proteins were identified by two or more unique peptides. The relative abundances of proteins among M1, M2, and RM1 macrophages were calculated based on peak areas using Minora algorithm-based label-free quantification in Proteome Discoverer 2.4. Statistical analyses of the dataset obtained from label-free quantification were performed using Perseus software (1.6.14.0) [24]. Normalized abundance values obtained from peak areas normalized to total peptides were log-transformed. Proteins exhibiting significant differences among M1, M2, and RM1 macrophages were detected by comparison of log2 (normalized abundance) values obtained from the three replicates of each type of macrophage samples using one-way analysis of variance (ANOVA). A p-value < 0.05 was considered to be statistically significant. For hierarchical clustering of proteins showing statistically significant changes (>2.0-fold, p-value < 0.05) in M1 and RM1 macrophages compared with M2 macrophages, normalized abundance values were first normalized using z-scores, and then both columns and rows were clustered based on Euclidean distance using the average linkage method in Perseus software (1.6.14.0). Analyses of GO functional classifications were pursued using DAVID software (http://david.abcc.ncifcrf.gov) to identify GO terms that were significantly enriched among proteins showing significant increases in M2 and RM1 macrophages relative to M2 macrophages.
2.11. Phagocytosis assay
For flow cytometry-based phagocytosis assays, BMDMs and HT29 tumor cells were first labeled with CellTracker Green (CMFDA) and CellTracker Deep Red, respectively (1 μM each), after which BMDMs were seeded at a density of 4 × 105 cells in 35 mm petri dishes and co-cultured with HT29 cells at a ratio of 1:2 for 2 h at 37 °C in a 5% CO2 environment. After incubation, intact cells were collected with cold PBS, pipetted repetitively to yield a single-cell suspension, and analyzed by flow cytometry (Accuri C6; BD Biosciences) using FlowJo v8 software. The BMDM phagocytosis percentage was calculated as phagocytosed tumor cells (double positive)/total number of BMDMs. For fluorescence microscopy-based phagocytosis analysis, BMDMs were plated at a density of 2 × 105 cells per 35 mm glass-bottom confocal dish together with pHrodo-SE–stained tumor cells at a ratio of 1:2. After co-incubation at 37 °C in a 5% CO2 environment for 2 h, cells were washed several times with PBS (pH 10) to remove unphagocytosed cancer cells. Phagocytosis in 10 randomly selected microscopic dish fields per assay was analyzed by fluorescence microscopy (Nikon), and the BMDM phagocytosis percentage was calculated as described above.
2.12. NO assay
BMDMs (5 × 105 cells) were seeded in 35 mm Petri dishes in medium containing differentiation-inducing cytokines. After culturing for 24 h, the medium was replaced with serum-free RPMI-1640 (Welgene), and cells were treated with exosomes for an additional 24 h. All supernatants were collected and cell debris was removed by filtering using a 10 kD spin column. Cell-free supernatants, diluted 20-fold, were analyzed using a Nitric Oxide Assay kit (Thermo Fisher Scientific) according to the manufacturer's protocol.
2.13. Analysis of cross-presentation
BMDMs were plated at density of 4 × 105 cells per 35 mm Petri dish in medium containing differentiation-inducing cytokines and 2 μg of OVA peptide for 24 h. The following day, cells were detached from the dish using cold DPBS. The collected cells were pre-incubated with 2 μg anti-mouse CD16/CD32 (Mouse BD Fc Block; BD Biosciences) for 25 min on ice and then stained with PE-conjugated, SIINFEKL-bound anti-H2Kb (Clone 25-D1.16; BioLegend). Data were analyzed using Accuri C6 and FlowJo v8 software.
2.14. In vivo tumor models and exosome treatment
An orthotopic mouse model was created by inoculating the fat pad of 7-week-old female BALB/c mice with 1 × 106 4T1-Luc tumor cells. Tumor dimensions were measured with calipers, and tumor volume (mm3) was calculated as (width)2 × (length) × 0.5. The mean tumor size reached 50–100 mm3 approximately 4 days after cell inoculation, and mice were treated with PBS, M1-Exo or M2-Exo, injected intratumorally on days 4, 7 and 10. All in vivo experiments were conducted in pathogen-free animal rooms following the guidelines of the Institutional Animal Care and Use Committee (IACUC) of the Korea Institute of Science and Technology (#KIST-2021-11-144).
2.15. Immunofluorescence staining
Tumor tissues were embedded in OCT compound and frozen at −80 °C. For iNOS and CD206 staining, tissues were cryo-sectioned at 15 μm, blocked by incubating with PBS/3% BSA for 1 h, and then were incubated with anti-iNOS or anti-CD206 antibody in PBS/1% BSA overnight at 4 °C. After washing three times with PBS, slides were incubated with FITC-conjugated or Percp Cy5.5-conjugated secondary antibody for 1 h at room temperature. Nuclei were stained with Hoechst 33342 for 5 min at room temperature. Images of stained slides were analyzed with a Leica fluorescence microscope.
2.16. Human TAM isolation and analysis
This prospective study was approved by the Asan Medical Center (Seoul, Republic of Korea) Institutional review board and written informed consent were obtained from all patients prior to study participation (#S2019-0794-0001). A total of 36 patients with head and neck cancer who underwent surgery at Asan Medical Center between 2019 and 2000 were included in this study. For human TAM isolation, single-cell suspensions were prepared from tumor tissues using a human tumor dissociation kit (#130-095-929; Miltenyi Biotec) according to the manufacturer's instructions, and then filtered with a 40 μm strainer. After incubating cell suspensions with RBC lysis buffer, 3 × 106 remaining cells were plated in a 100 mm standard tissue culture dish and incubated overnight at 37 °C in a 5% CO2 environment in RPMI-1640 medium containing 50 μg/mL gentamicin and 250 ng/mL M-CSF. The next day, non-adherent cells were removed by vigorous washing with DPBS, and isolated TAMs were labeled by immunostaining for HLA-DR (clone L243; BioLegend), CD163 (ab182422; Abcam), CD11b (clone M1/70; BioLegend), and CD206 (clone C068C2; BioLegend) and analyzed by flow cytometry. M1-Exo were obtained from M1-differentiated macrophages derived from the human monocytic THP-1 cell line. THP-1 cells were differentiated into macrophages by incubating for 24 h in RPMI-1640 medium containing 200 nM PMA (P8139; Sigma Aldrich). M1-polarized macrophages were obtained by further incubating THP-1 cells with 100 ng/mL LPS and 20 ng/mL IFN-γ for 48 h. For quantitative RT-PCR (RT-qPCR) analyses, total RNA was extracted with TRIzol reagent (Invitrogen) and transcribed to cDNA template using the Superscript III First-Strand Synthesis System (Invitrogen). RT-qPCR was performed in triplicated on a StepOnePlus Real-Time PCR System (Applied Biosystems) using SYBR Green PCR Master Mix (Applied Biosystems), according to the manufacturer's protocol. Data were normalized to GAPDH levels and evaluated using the ΔΔCt method.
2.17. Statistical analysis
All statistical numerical analyses were carried out using GraphPad Prism 8.0 (GraphPad Software, La Jolla, CA, USA). Multiple comparisons were performed using a one-way analysis of variance (ANOVA) with Tukey's post hoc test or two-way ANOVA with Bonferroni post hoc test. Values are expressed as means ± SD, and p-values < 0.05 were considered statistically significant. Individual p-values (*p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001) are indicated in figure legends.
3. Results
3.1. Isolation and characterization of exosomes from polarized macrophages
Bone marrow from mouse (C57BL/6) femurs and tibias was used as a source of cells for macrophage polarization. C57BL/6 mice are currently the most commonly used inbred strain in laboratories because they are genetically stable and phenotypically consistent and their background is well characterized [25]. They are considered a Th1-dominant mouse strain [26], and show high interferon gamma (IFN-γ) and tumor necrosis factor-alpha (TNF-α) production [27,28].
To induce polarization of macrophages, we cultured bone marrow-derived cells with monocyte colony stimulating factor (M-CSF) for ∼6 days and then stimulated them with cytokines capable of promoting differentiation into M1-or M2-type macrophages (Fig. 2A). Differentiation of bone marrow-derived macrophages (BMDMs) into each type of macrophage was confirmed by assessing expression of the M1 macrophage-specific marker inducible nitric oxide synthase (iNOS) and the M2 macrophage-specific markers arginase-1 and CD206 by Western blot analysis (Fig. 2B). After isolation of exosomes from the supernatant of activated macrophages, we characterized each type of purified macrophage-derived exosome with respect to size, morphology, and surface marker expression (Fig. 2C and D). Both M1-type macrophage-derived exosomes (M1-Exo) and M2 type macrophage-derived exosomes (M2-Exo) averaged ∼100 nm in diameter and were surrounded by a lipid bilayer membrane, as revealed by cryogenic transmission electron microscopy (Cryo-TEM). Western blot analysis clearly identified the exosome surface markers, ALIX, TSG101 and CD81, in both types of exosomes. M1-Exo and M2-Exo were found to express iNOS and arginase-1, respectively, an expression pattern reflecting that of the corresponding parental cell [29]. Note that the marker CD206 was not detected in either type of exosome, indicating that exosomal cargoes are selectively sorted into exosomes during their biogenesis [30,31].
Fig. 2.

Characterization of M1/M2-polarized macrophages, reprogrammed macrophages (RM1) and exosomes derived from differentiated macrophages. (A) Schematic diagram of the experiment for differentiation of BMDMs and preparation of exosomes. (B) Western blot analysis of polarized BMDMs using antibodies against iNOS (M1 marker) and CD206 and arginase-1 (M2 markers). (C) Size distribution of M1-Exo and M2-Exo based on DLS measurements and TEM images. (D) Western blot analysis of M1-Exo and M2-Exo using antibodies against M1 (iNOS) and M2 (CD206, arginase-1) markers. ALIX, TSG101 and CD81 were used as exosome markers. GM130 was used as a negative control. (E) FACS analysis for M1, M2 and RM1 macrophages using antibodies against M1 marker (CD86, CD40) and M2 marker (CD206).
Prior to confirming the reprogramming ability of M1-Exo, we evaluated their cellular uptake efficiency and cytotoxicity by incubating fluorescent dye-labeled M1-Exo with M2-type macrophages (Figs. S1A and B). M1-Exo were successfully taken up by M2 macrophages within 1 h and showed no cytotoxicity at any concentration in the range of 0–150 μg/mL.
3.2. M1-Exo effectively mediate in vitro reprogramming of M2 to M1 macrophages
Next, we confirmed M1-Exo–mediated direct M2-to-M1 conversion in culture. Since iNOS and arginase-1, representative markers of M1 and M2 polarized macrophages, respectively, were also detected in exosomes derived from the corresponding macrophages, we verified in vitro cell reprogramming using other markers that are not expressed in exosomes (Fig. S1C). For this purpose, the co-stimulatory molecules, CD86 and CD40, which are involved in antigen presentation, were used to detect M1 macrophages, and CD206 was used as an M2 macrophage surface marker. To determine the efficiency of macrophage reprogramming by M1-Exo, then we investigated changes in the expression of macrophage markers in M2 macrophages treated with M1-Exo using flow cytometry (Fig. 2E). This analysis showed that the distribution of cells expressing CD86 and CD40 among reprogrammed M1 macrophages (RM1) showed a shift similar to that of M1 macrophages, whereas the number of cells expressing CD206 decreased.
We further performed a proteomic analysis by mass spectrometry to compare the similarity of gene expression in detail for M1, M2, and RM1 groups. A total of 2396 proteins were identified in common among three replicate LC-MS/MS runs (Fig. 3A, Table S1). Label-free quantitative analyses of the identified proteins based on peak areas followed by statistical analysis (Table S2) showed that 909 and 508 proteins exhibited significant changes (>2.0-fold, p-value <0.05) in M1 and RM1 macrophages, respectively, compared with M2 macrophages, of which 450 were common to both M1 and RM1 groups (Fig. 3B).
Fig. 3.

Proteomic analyses of M1, M2, and RM1 macrophages. (A) Venn diagram showing the number of proteins from M1, M2, and RM1 macrophages identified by LC-MS/MS analysis following treatment with M1-Exo. Of the 3072 proteins identified in the three types of macrophages, 2830, 2738, and 2755 were identified in M1, M2, and RM1 macrophages, respectively, 2396 of which were identified in common for the three macrophage types. (B) Venn diagram showing the number of proteins that exhibited significant changes (>2.0-fold, p-value < 0.05) in M2 and RM1 macrophages relative to M2 macrophages in label-free quantitative analyses based on peak areas. A total of 909 proteins exhibited significant changes between M1 and M2 macrophages, and 508 proteins showed significant changes between RM1 and M2 macrophages, of which 450 were in common with M1/M2 changes. (C) Heat map exhibiting hierarchical clustering of 450 proteins with statistically significant changes in abundance (>2.0-fold, p-value < 0.05) for both M1 and RM1 macrophages compared with M2 macrophages. Hierarchical clustering was performed on log-transformed, normalized abundance values using Perseus software (1.6.14.0) after z-score normalization of data. Hierarchical clustering demonstrated that proteins of M1 and RM1 macrophages were clustered together, whereas those of M2 macrophage were independently clustered. (D) Top ten biological process (BP) terms over-represented among proteins exhibiting significant increases (>2.0-fold, p-value < 0.05) in M1 and RM1 macrophages relative to M2 macrophages, determined by GO enrichment analysis. Most GO BP terms were significantly enriched in proteins significantly increased in M1 and RM1 macrophages compared with M2 macrophages. Represents reprogrammed M1 macrophages obtained by treatment with M1-Exo.
Hierarchical clustering analyses of these 450 proteins showed that proteins in M1 and RM1 macrophage groups clustered together, whereas those of M2 macrophages clustered independently, suggesting that RM1 macrophages generated by treatment with M1-Exo acquired protein expression patterns similar to those of M1 macrophages (Fig. 3C). A further Gene Ontology (GO) enrichment analyses of the 596 and 374 proteins that showed significant increases (>2.0-fold, p-value <0.05) in M1 and RM1 macrophages, respectively, compared with M2 macrophages showed that seven GO biological process (BP) terms, including immune system process and response to virus, were common among the top 10 GO BP terms significantly over-represented in M1 and RM1 macrophages compared with M2 macrophages (Fig. 3D). These results imply that the RM1 macrophages generated by treatment with M1-Exo acquired functional properties similar to those of M1 macrophages. On the basis of these findings, we conclude that exosomes derived from M1 macrophages successfully reversed the polarization of M2 macrophages in culture.
3.3. RM1 macrophages gain phagocytic and antigen cross-presenting capacity
M1 macrophages are known to perform phagocytosis better than M2 macrophages and may also cross-present antigens [32,33]. Therefore, to determine whether RM1 macrophages also exert the functions of M1 macrophages, we evaluated their phagocytosis and antigen-presenting ability. To evaluate phagocytosis, we first labeled macrophages of each group with CellTracker Green CMFDA dye and co-cultured them with HT29 human colon cancer cells stained with pHrodo Red (Fig. 4A). Fluorescence microscopy images showed similarly high phagocytic capacity in M1 and RM1 macrophage groups. Quantification of these results by flow cytometric analysis of each group of fluorescence-labeled cells revealed that the percentage of phagocytosis in the RM1 group increased to 8%—a figure similar to that of the M1 group and almost twice that of the M2 group (4.4%) (Figs. 4B and S2A).
Fig. 4.

M1-Exo effectively induce in vitro reprogramming of M2 macrophages into fully functional M1 macrophages. (A) Representative microscopic images showing analysis of macrophage phagocytosis of cancer cells. BMDMs were labeled with CellTracker CMFDA, and HT29 cells were stained with pHrodo-SE. (B) FACS analysis of macrophage phagocytosis of cancer cells. BMDMs were stained with CellTracker CMFDA, and HT29 cells were labeled with CellTracker Deep Red. Data are presented as means ± S.D (n = 6). (C) Nitric oxide production by M0, M1, M2 and RM1 macrophages. Data represent means ± S.D (n = 6). (D) The percentage of H2K-b-Ova expression on M0, M1, M2 and RM1 macrophages, calculated relative to non-Ova–treated cells. Value represent means ± S.D (n = 6; p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001; one-way ANOVA with Tukey's post hoc test).
Interestingly, nitric oxide (NO) assays showed that the amount of NO production by RM1 macrophages was as high as that by M1 macrophages (Fig. 4C). These finding imply that RM1 macrophages, like M1 macrophages, produce NO and reactive oxygen intermediates (ROI), and are thus capable of killing bacteria, viruses, or tumor cells [34].
We also evaluated the antigen cross-presentation ability of RM1 using soluble endotoxin-free ovalbumin (OVA) peptide 257–264 (SIINFEKL) (Figs. 4D and S2B). Similar to results obtained with phagocytosis and NO production assays, RM1 macrophages showed improved antigen cross-presentation ability compared with M2 macrophages. Specifically, the cross-presentation percentage of RM1 was increased by 6.4- and 7.7-fold compared with M0 and M2, respectively, reaching a level comparable to that of M1. Taken together, our results suggest that RM1 macrophages are reprogrammed not only to express markers of M1 macrophages, but also to recapitulate their functional properties, including promoting tumor invasion, releasing ROS, and enhancing antigen-presenting ability.
3.4. Key differences in miRNA expression profile between M1-Exo and M2-Exo
Next, we investigated the factors contained within M1-Exo that drive macrophage polarization towards anti-tumorigenic M1 types. To this end, we compared miRNA expression profiles between M1-Exo and M2-Exo using miRNA sequencing (Fig. 5A). Exosome-derived miRNAs are known to play unique and essential roles in macrophage activation and polarization through exosome-mediated intercellular communication [35,36].
Fig. 5.

Analysis of exosomal miRNAs. (A) Heat map illustration of miRNA-seq data from M1-Exo and M2-Exo. The inclusion criteria were a 2-fold difference in log2 (fold-change) in either direction with a p-value < 0.05 (red, up-regulation; green, down-regulation). (B) GO and KEGG pathway analyses of miRNA target genes. The top five enriched GO terms and pathways for 18 miRNAs upregulated in M1-Exo were analyzed. (C) Efficiency of reprogramming in M2 treated with M1-Exo and anti-miRNAs.
Several pro-inflammatory miRNAs were detected in M1-Exo; in addition, miR-27a, miR-125b, miR-155 and miR-199a, which are known to be involved in macrophage activation and repolarization, were identified [[37], [38], [39], [40]]. Interestingly, it has been reported that miR-155, which is overexpressed in M1-Exo relative to M2-Exo, is not only an M1 phenotypic marker, it is also capable of reprogramming TAMs into pro-inflammatory M1 macrophages [37,41]. To determine which genes were targeted by the top 20 miRNAs (i.e., those expressed more than 2-fold higher in M1-Exo than in M2-Exo), we further performed GO and KEGG (Kyoto Encyclopedia of Genes and Genomes) pathway analyses (Fig. 5B). GO analyses showed that the identified miRNAs were significantly enriched for the BP categories of anatomical development and cell differentiation, molecular function categories of ion binding and cytoskeleton protein binding, and cellular component categories of organelle and cytoplasm. A KEGG pathway analysis revealed that the most highly correlated pathways identified were endocytosis, proteoglycans in cancer, MAPK signaling pathway, pathways in cancer, and NF-κB signaling pathway. Furthermore, to identify the key miRNAs responsible for reprogramming, miR-155 and miR-199a, the top two upregulated miRNAs in M1-Exo, were silenced with their respective anti-miRNAs during reprogramming (Fig. 5C). Clearly, inhibition of miR-155 and miR199a, which modulates the TLR4/NF-κB signaling pathway leading to pro-inflammatory properties, affected macrophage reprogramming efficiency. Macrophage reprogramming efficiency was significantly reduced in the group in which the function of both miRNAs was suppressed than in the group where each miRNA was silenced. However, inhibition of both miRNAs was not able to completely reverse polarization of macrophages, highlighting that macrophage reprogramming should be driven by a combination of factors, rather than simply one factor. Overall, these results demonstrate that M1-Exo contain not only specific miRNAs involved in cell differentiation, but also a class of miRNAs that are reported to induce macrophage activation and repolarization.
3.5. M1-Exo–induced in vivo reprogramming exerts anti-tumor effects
TAMs are central drivers of tumor progression, metastasis, and recurrence after treatment [42]. TAMs usually exhibit a pro-tumorigenic M2-like phenotype, whereas M1 macrophages exert anti-tumor functions [43]. These M1-like and M2-like TAMs coexist within tumors, and the proportion of M1/M2 subpopulations directly influences anti-tumor immune responses [44,45]. Accordingly, using a 4T1 tumor-bearing mouse model, we further investigated whether TAMs directly reprogrammed in situ by M1-Exo suppress tumor growth. A disproportionate number of TAMs from murine 4T1 breast tumor tissues are known to exhibit the M2 phenotype and are thus a potentially effective target for direct cell conversion [46,47]. Consistent with this, not only did 4T1 tumor tissues contain ∼4-fold more macrophages than B16F10 tumors (Fig. S3A), their proportion of M2 macrophages was also higher (Fig. S3B) [48].
We next examined the anti-tumor efficacy of M1-Exo administered locally into 4T1-Luc tumors in an orthotopic mouse model (Fig. 6A). Our results revealed that mice in the M1-exosome–treated group showed delayed tumor growth compared with untreated controls and M2-Exo–treated groups. Exosome treatment had no effect on body weight (Fig. S3C), implying that exosomes are relatively nontoxic. Flow cytometric analyses of tumor tissues further showed that the total macrophage population in tumor tissues of the M1-Exo–treated group was increased by ∼2-fold compared with the control groups (Fig. 6B). Exosomes contain information from the parent cells in the form of proteins, fats, nucleic acids, and bioactive substances. Thus, inflammatory factors encapsulated in M1-Exo may induce the local accumulation of macrophages [49]. Interestingly, M2-Exo treatment also slightly delayed tumor growth compared to the control group. Although it was not statistically significant, considering the result of increased macrophage population, it cannot be excluded that the immune system of BALB/c mice exerted a defense mechanism against macrophage-derived exosomes from C57BL/6 mice during allogeneic transplantation. To determine whether the observed reduction in tumor growth was attributable to TAM reprogramming by M1-Exo, we also investigated changes in macrophage subpopulations in tumor tissues. Indeed, the population of M1 macrophages (iNOS+CD38+EGR2-) in the M1-Exo–treated group was increased ∼3-fold compared with control groups, whereas the M2 population (iNOS−CD38−EGR2+) was decreased (Fig. 6C). Similar results were obtained by immunofluorescence analyses, which showed increased iNOS and decreased CD206 expression in M1-Exo–treated tumor tissues (Fig. S4).
Fig. 6.

Anti-tumor effect of M1-Exo in a 4T1 orthotopic tumor model. (A) Upper: Schedule of exosome treatment and reprogramming studies. Mice were treated intratumorally three times at 3-day intervals with 100 μg of M1-Exo or M2-Exo (or saline). Lower: Tumor growth of orthotopic 4T1 models in syngeneic mice. Data represent means ± S.D (n = 7–10 mice/group; **p < 0.01, ****p < 0.0001; two-way ANOVA with Bonferroni post hoc tests). (B) Flow cytometric analysis of single cells extracted from tumor tissues on day 13. Dot plots show the macrophage population gated using CD11b+ and F4/80+. (C) The percentage (%) of iNOS+CD38+EGR2- or iNOS−CD38−EGR2+ cells among CD11b+F4/80+ tumor tissue single cells. Each value represent means ± S.D (n = 3–4; **p < 0.01; one-way ANOVA with Tukey's post hoc tests). (D) Relative expression of TNF-α and IL-10 genes in tumor tissues of different treatment groups. All data are presented as means ± S.D (n = 6). (E) Tumor growth in the isotopic 4T1 model in syngeneic mice treated with M1-Exo and anti-PD-L1. Data represent means ± S.D (n = 3–9 mice/group; **p < 0.01, ****p < 0.0001; two-way ANOVA with Bonferroni post hoc tests). Red arrows: treatment of M1-Exo (Day 4, 7 and 10). Blue arrows: treatment of anti-PD-L1 (Day 7, 9, 11 and 13).
Exosome-mediated in situ direct macrophage reprogramming also affected on the expression level of immune cytokines. Similar to the population change of macrophage phenotypes in Fig. 6C, the pro-inflammatory cytokine TNF-α was increased in the M1-Exo treatment group, whereas the anti-inflammatory cytokine IL-10 increased in the M2-Exo treatment group (Fig. 6D). Although it is difficult to expect dramatic anti-cancer effects through targeting of TAMs alone, it has been reported that these strategies can enhance the efficacy of immune checkpoint inhibitor-based therapies. For example, the targeting CD47-SIRPα pathway has been reported to enhance the response rate of anti-PD1 therapy [50]. Similar to the results of increased total macrophage population, M1-Exo also increased the T cell population in tumor tissues, exerting an improved anticancer effect when treated in combination with anti-PD-L1 (Figs. S5 and 6E).
Collectively, these findings indicate that M1-Exo have the ability to promote anti-tumor responses, likely owing to reprogramming of pro-tumorigenic TAMs into tumor-killing macrophages as well as an increase in the number of immune cells including macrophages and T cells in tumor sites.
3.6. Cancer patient-derived TAMs can also be repolarized by M1-Exo
To determine whether M2-like macrophages within human tumors can also be repolarized by M1-Exo, we used TAMs isolated from patient-derived cancer tissues and THP-1 cells, a human monocytic cell line, as exosome sources (Fig. 7A). As expected, most TAMs isolated from tumor tissues were M2-like macrophages with low expression of MHC class-II molecules, and M1-like macrophages with high MHC class-II expression were scarce (Fig. 7B). THP-1 cells were successfully differentiated into macrophages by treatment with phorbol 12-myristate 13-acetate (PMA). The polarization of THP-1 cells into M1-type was confirmed by demonstrating the expression of CD86 and MHC class-II markers, and inflammatory factors (TNF-α, IL 6) (Fig. 7C, S6A, B), polarization markers that differ from those expressed in mouse-derived macrophages. Expression of the M2 polarization markers, CD206 and CD163, was not detected in M1-polarized THP-1 cells (Fig. 7C), consistent with previous reports [51,52].
Fig. 7.

Reprogramming of patient-derived TAMs into M1-like TAMs. (A) Schematic diagram of the experimental design and steps for reprogramming of patient-derived TAMs. (B) Flow cytometric analysis of macrophage subpopulations sorted from tumor tissues of patients. The cells were divided into two TAM subtypes based on MHC-II expression: MHC-II high (M1-like TAMs) and MHC-II low (M2-like TAMs). (C) Characterization of M1-polarized macrophages and their derived exosomes. GM130 was used as a negative control. THP-1 cells were stimulated with PMA (200 nM) and polarized with LPS (100 ng/mL) and IFN-γ (20 ng/mL). (D) Representative Western blot images showing the expression of CD206, CD163, and MHC-II in TAMs treated with 75 or 150 μg/mL M1-Exo for 24 h. (E) Quantitative presentation of Western blot data. Values are means ± SD (n = 3; *p < 0.05, **p < 0.001; one-way ANOVA with Tukey's post hoc test). (F) Flow cytometric analysis of TAMs and RM1 macrophages treated with M1-Exos (150 μg/mL).
The exosome markers, ALIX and TSG101, were observed in M1-Exo derived from M1-polarized THP-1 cells, and dynamic light scattering (DLS) also confirmed that these M1-Exo were about 90 nm in diameter (Fig. 7C). Treatment of TAMs isolated from patients' tumor tissues with M1-Exo (75 and 150 μg/mL) decreased the expression of CD206 and CD163, while significantly increasing the expression of MHC class II involved in the immune response (Fig. 7D and E). Flow cytometric analyses showed similar phenotypic changes in these TAMs, in particular revealing that M1-Exo induced the transition of low MHC class-II–expressing TAMs to high MHC class-II–expressing TAMs (Fig. 7F). Collectively, these results indicate that M1-Exo can also induce the phenotypic conversion of human-derived TAMs into M1-like macrophages that highly express MHC class II.
4. Discussion
As tumors progress, TAM abundance increases, allowing the cancer to become more aggressive and spread. TAMs are polarized toward a pro-tumorigenic and immunosuppressive M2 phenotype that lacks tumor-associated antigen specificity. Several approaches for targeting M2-macrophages, including inhibition of monocyte infiltration into solid tumors and elimination of TAMs, have been investigated as cancer therapies [8,[53], [54], [55]]. These strategies are relatively direct routes for combating tumors, but the potential for increased risk of infection and chronic inflammation associated with approved drugs that deplete TAMs pose challenges to their clinical use. These observations highlight the need for increased biological understanding of the impact of M1 macrophages in the TME on primary or metastatic tumors, underscoring the importance of such insight for the development of new strategies targeting TAMs.
In this paper, we demonstrated a simple approach for inducing TAMs to reverse course utilizing exosomes derived from M1 macrophages. Rather than supporting cancer cells, the resulting reprogrammed macrophages acquired the ability to kill cancer cells through phagocytosis. M1-Exo–induced macrophage reprogramming also enhanced the cross-presentation ability of macrophages and ultimately exerted a significant anti-tumor effect in vivo.
Given that M1-type macrophages possess pro-inflammatory activity that promotes Th1 immune responses, therapeutic strategies that target TAMs can be considered promising immunotherapy approaches. Because macrophages are not only professional antigen-presenting cells but also actively participate in the immune response through phagocytosis and clearance of cellular debris, enhancing their anti-tumoral phenotypes is closely associated with amplification of the anti-cancer immune responses of other leukocytes, such as T cells.
Current macrophage-based immunotherapeutic approaches rely mechanistically on TAMs. These TAM-targeted therapeutics include inhibition of monocyte recruitment through inhibition of CSF-1/CSF-1 or CCL2/CCR2 (agonist/receptor) signaling, induction of cancer cell phagocytosis by targeting of the CD47-SIRPα pathway, and targeted depletion of TAMs [56]. Effective anti-tumor immunotherapy has also been demonstrated through reprogramming of TAMs from the M2 phenotype towards the M1 phenotypes [57,58]. For example, activation of CD40 signaling or inhibition of the PI3Kγ pathway has been reported to polarize M2-type macrophages to the M1 phenotype [59,60]. Specifically, anti-CD40/anti-CSF-1R co-treatment was found to decrease TAMs as well as increase maturation and differentiation of pro-inflammatory macrophages, driving potent priming of effector T cells in draining lymph nodes [61]. It has also been demonstrated that inhibition of PI3Kγ synergizes with anti-PD1 therapy to promote anti-tumor immune responses that induce sustained tumor regression in mouse tumor models [62]. Despite these achievements, the fact that nanoparticle formulation or additional exosome engineering process are required to deliver bioactive agents to the TME for macrophage polarization remains a complicating factor in interpreting the observed anti-tumor effects [63,64]. The addition of engineering processes lowers the biocompatibility of the carrier and detracts from its clinical application.
In this context, the exosome-based reprogramming strategy described here represents a highly effective method for direct conversion of M2-polarized TAMs into M1-polarized TAMs. Exosomes intrinsically contain a mixture of genetic factors capable of changing cellular fates [65]. In addition, given their role in intercellular communication, exosomes can also efficiently transfer key molecules for cellular reprogramming into the targeted cells. The beauty of such a naturally derived, exosome-based reprogramming strategy is that therapeutic substances—without adverse effects—can be obtained from the patients themselves. Autologous or allogeneic transplantation strategies using human peripheral blood mononuclear cells (PBMCs)-derived M1-Exo have potential for promising anti-tumor immunotherapy in the future. Indeed, our results showed that exosomes derive from allogenic (C57BL/6) macrophages can be used to exert anti-tumor effects in tumor-bearing BALB/c mice. In this context, exosomes for cancer immunotherapy can be obtained by isolating PBMCs from the patient's own or others' blood and differentiating them into M1 macrophages. An important advantage of this allogeneic approach is that it overcomes limitations in the source of cells for transplantation, bringing this strategy one step closer to clinical application. An important advantage of this allogeneic approach is that it overcomes limitations in the source of cells for transplantation, bringing this strategy one step closer to clinical application.
5. Conclusion
Our findings demonstrate that M1-Exo–induced macrophage polarization is both very efficient and simple. The reprogrammed macrophages not only express several M1 macrophage-related markers, but also showed very similar proteomic expression patterns to that of M1-macrophages, and were functionally altered. Furthermore, we found that M1-Exos contain many miRNAs involved in macrophage differentiation and recruitment, as well as miRNAs that induce macrophage reprogramming. In situ macrophage reprogramming could be successfully performed in vivo without additional exosome engineering, and it exerted significant anti-tumor effects in 4T1 tumor bearing mice. Notably, because of their phenotypic plasticity, macrophages continuously adapt in response to the surrounding microenvironment; thus, their polarization status can change. Therefore, effective methods for inducing durable therapeutic interventions should be considered before these strategies can reach their full potential. However, the growing interest in exosomes as therapeutic entities suggests that the remaining challenges for the clinical translation of exosome-based therapeutics will ultimately be met. Our results provide evidence supporting the compelling possibility of using exosomes as therapeutic modalities for in situ direct cell reprogramming.
Ethics approval and consent to participate
This prospective study was approved by the Asan Medical Center (Seoul, Republic of Korea) Institutional review board and written informed consent were obtained from all patients prior to study participation (Nov. 1st. 2019, #S2019-0794-0001).
I confirm that I have obtained all consents required by applicable law for the publication of any personal details or images of patients, research subjects or other individuals that are used in the materials submitted to KeAi. I have retained a written copy of all such consents and I agree to provide KeAi with copies of the consents or evidence that such consents have been obtained if requested by KeAi.
CRediT authorship contribution statement
Hyosuk Kim: Conceptualization, Methodology, Investigation, Data curation, Writing – original draft, Writing – review & editing. Hyun-Ju Park: Methodology, Investigation, Formal analysis, Data curation. Hyo Won Chang: Methodology, Investigation. Ji Hyun Back: Methodology, Investigation. Su Jin Lee: Methodology, Investigation. Yae Eun Park: Methodology, Investigation. Eun Hye Kim: Methodology, Investigation. Yeonsun Hong: Methodology, Investigation. Gijung Kwak: Methodology, Investigation. Ick Chan Kwon: Conceptualization, Resources. Ji Eun Lee: Resources, Supervision. Yoon Se Lee: Methodology, Investigation. Sang Yoon Kim: Conceptualization, Visualization. Yoosoo Yang: Project administration, Supervision, Funding acquisition, Writing – review & editing. Sun Hwa Kim: Visualization, Project administration, Supervision, Funding acquisition, Writing – review & editing.
Declaration of competing interest
The authors declare no conflicts of interest.
Acknowledgments
This work was supported by the Samsung Research Funding & Incubation Center of Samsung Electronics (SRFC-MA1901-10) and the Intramural Research Program of KIST.
Footnotes
Peer review under responsibility of KeAi Communications Co., Ltd.
Supplementary data to this article can be found online at https://doi.org/10.1016/j.bioactmat.2022.07.021.
Contributor Information
Yoosoo Yang, Email: ysyang@kist.re.kr.
Sun Hwa Kim, Email: sunkim@kist.re.kr.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Hentze H., Soong P.L., Wang S.T., Phillips B.W., Putti T.C., Dunn N.R. Teratoma formation by human embryonic stem cells: evaluation of essential parameters for future safety studies. Stem Cell Res. 2009;2(3):198–210. doi: 10.1016/j.scr.2009.02.002. [DOI] [PubMed] [Google Scholar]
- 2.Lee M.O., Moon S.H., Jeong H.C., Yi J.Y., Lee T.H., Shim S.H., Rhee Y.H., Lee S.H., Oh S.J., Lee M.Y., Han M.J., Cho Y.S., Chung H.M., Kim K.S., Cha H.J. Inhibition of pluripotent stem cell-derived teratoma formation by small molecules. Proc. Natl. Acad. Sci. U. S. A. 2013;110(35):E3281–E3290. doi: 10.1073/pnas.1303669110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liu G., David B.T., Trawczynski M., Fessler R.G. Advances in pluripotent stem cells: history, mechanisms, technologies, and applications. Stem Cell Rev. Rep. 2020;16(1):3–32. doi: 10.1007/s12015-019-09935-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fang L., El Wazan L., Tan C., Nguyen T., Hung S.S.C., Hewitt A.W., Wong R.C.B. Potentials of cellular reprogramming as a novel strategy for neuroregeneration. Front. Cell. Neurosci. 2018;12:460. doi: 10.3389/fncel.2018.00460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grath A., Dai G. Direct cell reprogramming for tissue engineering and regenerative medicine. J. Biol. Eng. 2019;13:14. doi: 10.1186/s13036-019-0144-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Srivastava D., DeWitt N. In vivo cellular reprogramming: the next generation. Cell. 2016;166(6):1386–1396. doi: 10.1016/j.cell.2016.08.055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shao L., Wu W.S. Gene-delivery systems for iPS cell generation. Expet Opin. Biol. Ther. 2010;10(2):231–242. doi: 10.1517/14712590903455989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xiang X., Wang J., Lu D., Xu X. Targeting tumor-associated macrophages to synergize tumor immunotherapy. Signal Transduct. Targeted Ther. 2021;6(1):75. doi: 10.1038/s41392-021-00484-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dai J., Su Y., Zhong S., Cong L., Liu B., Yang J., Tao Y., He Z., Chen C., Jiang Y. Exosomes: key players in cancer and potential therapeutic strategy. Signal Transduct. Targeted Ther. 2020;5(1):145. doi: 10.1038/s41392-020-00261-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Weng Z., Zhang B., Wu C., Yu F., Han B., Li B., Li L. Therapeutic roles of mesenchymal stem cell-derived extracellular vesicles in cancer. J. Hematol. Oncol. 2021;14(1):136. doi: 10.1186/s13045-021-01141-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gam R., Sung M., Prasad Pandurangan A. Experimental and computational approaches to direct cell reprogramming: recent advancement and future challenges. Cells. 2019;8(10) doi: 10.3390/cells8101189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Li M.A., He L. microRNAs as novel regulators of stem cell pluripotency and somatic cell reprogramming. Bioessays. 2012;34(8):670–680. doi: 10.1002/bies.201200019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Valadi H., Ekström K., Bossios A., Sjöstrand M., Lee J.J., Lötvall J.O. Exosome-mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat. Cell Biol. 2007;9(6):654–659. doi: 10.1038/ncb1596. [DOI] [PubMed] [Google Scholar]
- 14.Herrmann I.K., Wood M.J.A., Fuhrmann G. Extracellular vesicles as a next-generation drug delivery platform. Nat. Nanotechnol. 2021;16(7):748–759. doi: 10.1038/s41565-021-00931-2. [DOI] [PubMed] [Google Scholar]
- 15.Shi H., Wang M., Sun Y., Yang D., Xu W., Qian H. Exosomes: emerging cell-free based therapeutics in dermatologic diseases. Front. Cell Dev. Biol. 2021;9 doi: 10.3389/fcell.2021.736022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yin L., Liu X., Shi Y., Ocansey D.K.W., Hu Y., Li X., Zhang C., Xu W., Qian H. Therapeutic advances of stem cell-derived extracellular vesicles in regenerative medicine. Cells. 2020;9(3) doi: 10.3390/cells9030707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liang Y., Duan L., Lu J., Xia J. Engineering exosomes for targeted drug delivery. Theranostics. 2021;11(7):3183–3195. doi: 10.7150/thno.52570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.van der Koog L., Gandek T.B., Nagelkerke A. Liposomes and extracellular vesicles as drug delivery systems: a comparison of composition, pharmacokinetics, and functionalization. Adv. Healthc. Mater. 2021 doi: 10.1002/adhm.202100639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Binnewies M., Roberts E.W., Kersten K., Chan V., Fearon D.F., Merad M., Coussens L.M., Gabrilovich D.I., Ostrand-Rosenberg S., Hedrick C.C., Vonderheide R.H., Pittet M.J., Jain R.K., Zou W., Howcroft T.K., Woodhouse E.C., Weinberg R.A., Krummel M.F. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat. Med. 2018;24(5):541–550. doi: 10.1038/s41591-018-0014-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.DeBerardinis R.J., Chandel N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016;2(5) doi: 10.1126/sciadv.1600200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Pathria P., Louis T.L., Varner J.A. Targeting tumor-associated macrophages in cancer. Trends Immunol. 2019;40(4):310–327. doi: 10.1016/j.it.2019.02.003. [DOI] [PubMed] [Google Scholar]
- 22.Wang C., Yu X., Cao Q., Wang Y., Zheng G., Tan T.K., Zhao H., Zhao Y., Wang Y., Harris D. Characterization of murine macrophages from bone marrow, spleen and peritoneum. BMC Immunol. 2013;14:6. doi: 10.1186/1471-2172-14-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhao Y.L., Tian P.X., Han F., Zheng J., Xia X.X., Xue W.J., Ding X.M., Ding C.G. Comparison of the characteristics of macrophages derived from murine spleen, peritoneal cavity, and bone marrow. J. Zhejiang Univ. - Sci. B. 2017;18(12):1055–1063. doi: 10.1631/jzus.B1700003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tyanova S., Temu T., Sinitcyn P., Carlson A., Hein M.Y., Geiger T., Mann M., Cox J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods. 2016;13(9):731–740. doi: 10.1038/nmeth.3901. [DOI] [PubMed] [Google Scholar]
- 25.Sherborne A.L., Thom M.D., Paterson S., Jury F., Ollier W.E., Stockley P., Beynon R.J., Hurst J.L. The genetic basis of inbreeding avoidance in house mice. Curr. Biol. 2007;17(23):2061–2066. doi: 10.1016/j.cub.2007.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Watanabe H., Numata K., Ito T., Takagi K., Matsukawa A. Innate immune response in Th1- and Th2-dominant mouse strains. Shock. 2004;22(5):460–466. doi: 10.1097/01.shk.0000142249.08135.e9. [DOI] [PubMed] [Google Scholar]
- 27.Jovicic N., Jeftic I., Jovanovic I., Radosavljevic G., Arsenijevic N., Lukic M.L., Pejnovic N. Differential immunometabolic phenotype in Th1 and Th2 dominant mouse strains in response to high-fat feeding. PLoS One. 2015;10(7) doi: 10.1371/journal.pone.0134089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Santos J.L., Andrade A.A., Dias A.A., Bonjardim C.A., Reis L.F., Teixeira S.M., Horta M.F. Differential sensitivity of C57BL/6 (M-1) and BALB/c (M-2) macrophages to the stimuli of IFN-gamma/LPS for the production of NO: correlation with iNOS mRNA and protein expression. J. Interferon Cytokine Res. 2006;26(9):682–688. doi: 10.1089/jir.2006.26.682. [DOI] [PubMed] [Google Scholar]
- 29.Zubkova E., Evtushenko E., Beloglazova I., Osmak G., Koshkin P., Moschenko A., Menshikov M., Parfyonova Y. Analysis of MicroRNA profile Alterations in extracellular vesicles from mesenchymal stromal cells overexpressing stem cell factor. Front. Cell Dev. Biol. 2021;9 doi: 10.3389/fcell.2021.754025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.O'Brien K., Breyne K., Ughetto S., Laurent L.C., Breakefield X.O. RNA delivery by extracellular vesicles in mammalian cells and its applications. Nat. Rev. Mol. Cell Biol. 2020;21(10):585–606. doi: 10.1038/s41580-020-0251-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Villarroya-Beltri C., Baixauli F., Gutierrez-Vazquez C., Sanchez-Madrid F., Mittelbrunn M. Sorting it out: regulation of exosome loading. Semin. Cancer Biol. 2014;28:3–13. doi: 10.1016/j.semcancer.2014.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Muntjewerff E.M., Meesters L.D., van den Bogaart G. Antigen cross-presentation by macrophages. Front. Immunol. 2020;11:1276. doi: 10.3389/fimmu.2020.01276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hirayama D., Iida T., Nakase H. The phagocytic function of macrophage-enforcing innate immunity and tissue homeostasis. Int. J. Mol. Sci. 2017;19(1) doi: 10.3390/ijms19010092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ley K. M1 means kill; M2 means heal. J. Immunol. 2017;199(7):2191–2193. doi: 10.4049/jimmunol.1701135. [DOI] [PubMed] [Google Scholar]
- 35.Kwon Y., Kim M., Kim Y., Jung H.S., Jeoung D. Exosomal MicroRNAs as mediators of cellular interactions between cancer cells and macrophages. Front. Immunol. 2020;11:1167. doi: 10.3389/fimmu.2020.01167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Essandoh K., Li Y., Huo J., Fan G.C. MiRNA-mediated macrophage polarization and its potential role in the regulation of inflammatory response. Shock. 2016;46(2):122–131. doi: 10.1097/SHK.0000000000000604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cai X., Yin Y., Li N., Zhu D., Zhang J., Zhang C.Y., Zen K. Re-polarization of tumor-associated macrophages to pro-inflammatory M1 macrophages by microRNA-155. J. Mol. Cell Biol. 2012;4(5):341–343. doi: 10.1093/jmcb/mjs044. [DOI] [PubMed] [Google Scholar]
- 38.Chaudhuri A.A., So A.Y., Sinha N., Gibson W.S., Taganov K.D., O'Connell R.M., Baltimore D. MicroRNA-125b potentiates macrophage activation. J. Immunol. 2011;187(10):5062–5068. doi: 10.4049/jimmunol.1102001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Jia Y., Zheng Z., Xue M., Zhang S., Hu F., Li Y., Yang Y., Zou M., Li S., Wang L., Guan M., Xue Y. Extracellular vesicles from albumin-induced tubular epithelial cells promote the M1 macrophage phenotype by targeting klotho. Mol. Ther. 2019;27(8):1452–1466. doi: 10.1016/j.ymthe.2019.05.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhang M., Sheng S., Zhang W., Zhang J., Zhang Z., Zhang M., Hatch G.M., Chen L. MiR27a promotes the development of macrophage-like characteristics in 3T3-L1 preadipocytes. Int. J. Biol. Sci. 2018;14(11):1599–1609. doi: 10.7150/ijbs.26274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Curtale G., Rubino M., Locati M. MicroRNAs as molecular switches in macrophage activation. Front. Immunol. 2019;10:799. doi: 10.3389/fimmu.2019.00799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mantovani A., Marchesi F., Malesci A., Laghi L., Allavena P. Tumour-associated macrophages as treatment targets in oncology. Nat. Rev. Clin. Oncol. 2017;14(7):399–416. doi: 10.1038/nrclinonc.2016.217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zhao H., Wu L., Yan G., Chen Y., Zhou M., Wu Y., Li Y. Inflammation and tumor progression: signaling pathways and targeted intervention. Signal Transduct. Targeted Ther. 2021;6(1):263. doi: 10.1038/s41392-021-00658-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Duan Z., Luo Y. Targeting macrophages in cancer immunotherapy. Signal Transduct. Targeted Ther. 2021;6(1):127. doi: 10.1038/s41392-021-00506-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vitale I., Manic G., Coussens L.M., Kroemer G., Galluzzi L. Macrophages and metabolism in the tumor microenvironment. Cell Metabol. 2019;30(1):36–50. doi: 10.1016/j.cmet.2019.06.001. [DOI] [PubMed] [Google Scholar]
- 46.Schiavoni G., Gabriele L., Mattei F. The tumor microenvironment: a pitch for multiple players. Front. Oncol. 2013;3:90. doi: 10.3389/fonc.2013.00090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Park O., Choi E.S., Yu G., Kim J.Y., Kang Y.Y., Jung H., Mok H. Efficient delivery of tyrosinase related protein-2 (TRP2) peptides to lymph nodes using serum-derived exosomes. Macromol. Biosci. 2018;18(12) doi: 10.1002/mabi.201800301. [DOI] [PubMed] [Google Scholar]
- 48.Luo Y., Zhou H., Krueger J., Kaplan C., Lee S.H., Dolman C., Markowitz D., Wu W., Liu C., Reisfeld R.A., Xiang R. Targeting tumor-associated macrophages as a novel strategy against breast cancer. J. Clin. Invest. 2006;116(8):2132–2141. doi: 10.1172/JCI27648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cheng L., Wang Y., Huang L. Exosomes from M1-polarized macrophages potentiate the cancer vaccine by creating na pro-inflammatory microenvironment in the lymph node. Mol. Ther. 2017;25(7):1665–1675. doi: 10.1016/j.ymthe.2017.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chao M.P., Weissman I.L., Majeti R. The CD47-SIRPalpha pathway in cancer immune evasion and potential therapeutic implications. Curr. Opin. Immunol. 2012;24(2):225–232. doi: 10.1016/j.coi.2012.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Martinez F.O., Helming L., Milde R., Varin A., Melgert B.N., Draijer C., Thomas B., Fabbri M., Crawshaw A., Ho L.P., Ten Hacken N.H., Cobos Jimenez V., Kootstra N.A., Hamann J., Greaves D.R., Locati M., Mantovani A., Gordon S. Genetic programs expressed in resting and IL-4 alternatively activated mouse and human macrophages: similarities and differences. Blood. 2013;121(9):e57–69. doi: 10.1182/blood-2012-06-436212. [DOI] [PubMed] [Google Scholar]
- 52.Orecchioni M., Ghosheh Y., Pramod A.B., Ley K. Macrophage polarization: different gene signatures in M1(LPS+) vs. Classically and M2(LPS-) vs. Alternatively activated macrophages. Front. Immunol. 2019;10:1084. doi: 10.3389/fimmu.2019.01084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cassetta L., Kitamura T. Targeting tumor-associated macrophages as a potential strategy to enhance the response to immune checkpoint inhibitors. Front. Cell Dev. Biol. 2018;6:38. doi: 10.3389/fcell.2018.00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Han S., Wang W., Wang S., Yang T., Zhang G., Wang D., Ju R., Lu Y., Wang H., Wang L. Tumor microenvironment remodeling and tumor therapy based on M2-like tumor associated macrophage-targeting nano-complexes. Theranostics. 2021;11(6):2892–2916. doi: 10.7150/thno.50928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lee C., Jeong H., Bae Y., Shin K., Kang S., Kim H., Oh J., Bae H. Targeting of M2-like tumor-associated macrophages with a melittin-based pro-apoptotic peptide. J. Immunother. Cancer. 2019;7(1):147. doi: 10.1186/s40425-019-0610-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lin Y., Xu J., Lan H. Tumor-associated macrophages in tumor metastasis: biological roles and clinical therapeutic applications. J. Hematol. Oncol. 2019;12(1):76. doi: 10.1186/s13045-019-0760-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Gunassekaran G.R., Poongkavithai Vadevoo S.M., Baek M.C., Lee B. M1 macrophage exosomes engineered to foster M1 polarization and target the IL-4 receptor inhibit tumor growth by reprogramming tumor-associated macrophages into M1-like macrophages. Biomaterials. 2021;278 doi: 10.1016/j.biomaterials.2021.121137. [DOI] [PubMed] [Google Scholar]
- 58.Nie W., Wu G., Zhang J., Huang L.L., Ding J., Jiang A., Zhang Y., Liu Y., Li J., Pu K., Xie H.Y. Responsive exosome nano-bioconjugates for synergistic cancer therapy. Angew Chem. Int. Ed. Engl. 2020;59(5):2018–2022. doi: 10.1002/anie.201912524. [DOI] [PubMed] [Google Scholar]
- 59.Komohara Y., Fujiwara Y., Ohnishi K., Takeya M. Tumor-associated macrophages: potential therapeutic targets for anti-cancer therapy. Adv. Drug Deliv. Rev. 2016;99(Pt B):180–185. doi: 10.1016/j.addr.2015.11.009. [DOI] [PubMed] [Google Scholar]
- 60.Mantovani A., Allavena P. The interaction of anticancer therapies with tumor-associated macrophages. J. Exp. Med. 2015;212(4):435–445. doi: 10.1084/jem.20150295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wiehagen K.R., Girgis N.M., Yamada D.H., Smith A.A., Chan S.R., Grewal I.S., Quigley M., Verona R.I. Combination of CD40 agonism and CSF-1R blockade reconditions tumor-associated macrophages and drives potent antitumor immunity. Cancer Immunol. Res. 2017;5(12):1109–1121. doi: 10.1158/2326-6066.CIR-17-0258. [DOI] [PubMed] [Google Scholar]
- 62.Kaneda M.M., Messer K.S., Ralainirina N., Li H., Leem C.J., Gorjestani S., Woo G., Nguyen A.V., Figueiredo C.C., Foubert P., Schmid M.C., Pink M., Winkler D.G., Rausch M., Palombella V.J., Kutok J., McGovern K., Frazer K.A., Wu X., Karin M., Sasik R., Cohen E.E., Varner J.A. PI3Kgamma is a molecular switch that controls immune suppression. Nature. 2016;539(7629):437–442. doi: 10.1038/nature19834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Ding J., Lu G., Nie W., Huang L.L., Zhang Y., Fan W., Wu G., Liu H., Xie H.Y. Self-activatable photo-extracellular vesicle for synergistic trimodal anticancer therapy. Adv. Mater. 2021;33(7) doi: 10.1002/adma.202005562. [DOI] [PubMed] [Google Scholar]
- 64.Wang P., Wang H., Huang Q., Peng C., Yao L., Chen H., Qiu Z., Wu Y., Wang L., Chen W. Exosomes from M1-polarized macrophages enhance paclitaxel antitumor activity by activating macrophages-mediated inflammation. Theranostics. 2019;9(6):1714–1727. doi: 10.7150/thno.30716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Chen H., Wang L., Zeng X., Schwarz H., Nanda H.S., Peng X., Zhou Y. Exosomes, a new star for targeted delivery. Front. Cell Dev. Biol. 2021;9 doi: 10.3389/fcell.2021.751079. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.